
Visible-light promoted oxidative annulation of 2-naphthols with phenylglyoxal monohydrates toward hydroxy-naphthofuranone and its derivatives†
Vijayakumar
Hemamalini
a,
Markabandhu
Shanthi
a,
Bhaskaran
Shankar
 c,
Rambabu
Dandela
b,
Karuppaiah
Perumal
a and
Subburethinam
Ramesh
*a
c,
Rambabu
Dandela
b,
Karuppaiah
Perumal
a and
Subburethinam
Ramesh
*a
aDepartment of Chemistry, School of Chemical and Biotechnology, SASTRA Deemed University, Thanjavur 613 401, Tamil Nadu, India. E-mail: rameshsbdu@gmail.com
bDepartment of Industrial and Engineering Chemistry, Institute of Chemical Technology, Indian Oil Odisha Campus, Samantapuri, Bhubaneswar, Odisha 751013, India
cDepartment of Chemistry, Thiagarajar College of Engineering, Madurai 625 015, Tamil Nadu, India
First published on 6th June 2024
Abstract
A highly efficient and innovative method involving base-mediated oxidative annulation between 2-naphthols and phenylglyoxal monohydrate under visible light irradiation has been successfully developed. This method leads to the formation of oxygen-containing heterocyclic compounds, particularly hydroxy-naphthofuranone derivatives, encompassing a unique quaternary carbon center. An X-ray diffraction study has unambiguously confirmed the structure of one such derivative. In particular, water molecules in this reaction serve various functions as a solvent, reagent, and additive, with the conversion of the process found to be influenced by the volume of water present. This atom-economical approach demonstrates tolerance for different substituents in both phenylglyoxal monohydrate and 2-naphthol, enabling the synthesis of a variety of naphthofuranones in satisfactory to good yields. The formation of a naphthofuranium cationic intermediate under acidic circumstances enables the formation of C–C or C–O bonds with a wide range of aromatic or alcoholic nucleophilic partners. Furthermore, the identification and generation of pinacol-type starting precursors from these naphthofuranone derivatives enable the synthesis of highly regioselective naphthofuran derivatives.
Introduction
A range of biologically active compounds containing a furanone structure have been identified, displaying a diverse array of biological activities.1 These oxygen-containing heterocyclic compounds are classified based on the position of the oxygen atom and the carbonyl group, falling into three categories: 3(2H)-furanone, 2(5H)-furanone, and 2(3H)-furanone.2 Found in numerous natural products and biologically active substances, 3(2H)-furanone derivatives demonstrate various activities, including anti-tuberculosis, antioxidant, analgesic, antiulcer, anticonvulsant, anticancer, antibacterial, antifungal, and anti-inflammatory properties (Fig. 1, 1–5).3 Naphthofuranone, furanone and naphthalene-fused structures, found not only in secondary metabolites but also in their derivatives, exhibit significant biological activities. Moreover, several fused furan derivatives, naphthofurans and benzofurans in particular, are widely known for their remarkable biological properties and presence in natural products.4 Almost thirty medications containing benzofuran have been approved by the US Food and Drug Administration (USFDA).5 Furthermore, naphthofurans have recently received a lot of attention due to their potential efficacy in the design and synthesis of therapeutics for cancer treatment,6 Alzheimer's disease inhibitors,7 modulators of human protein kinase,8 and nuclear receptors,9 and a variety of other biological activities10 (Fig. 1, 6–9).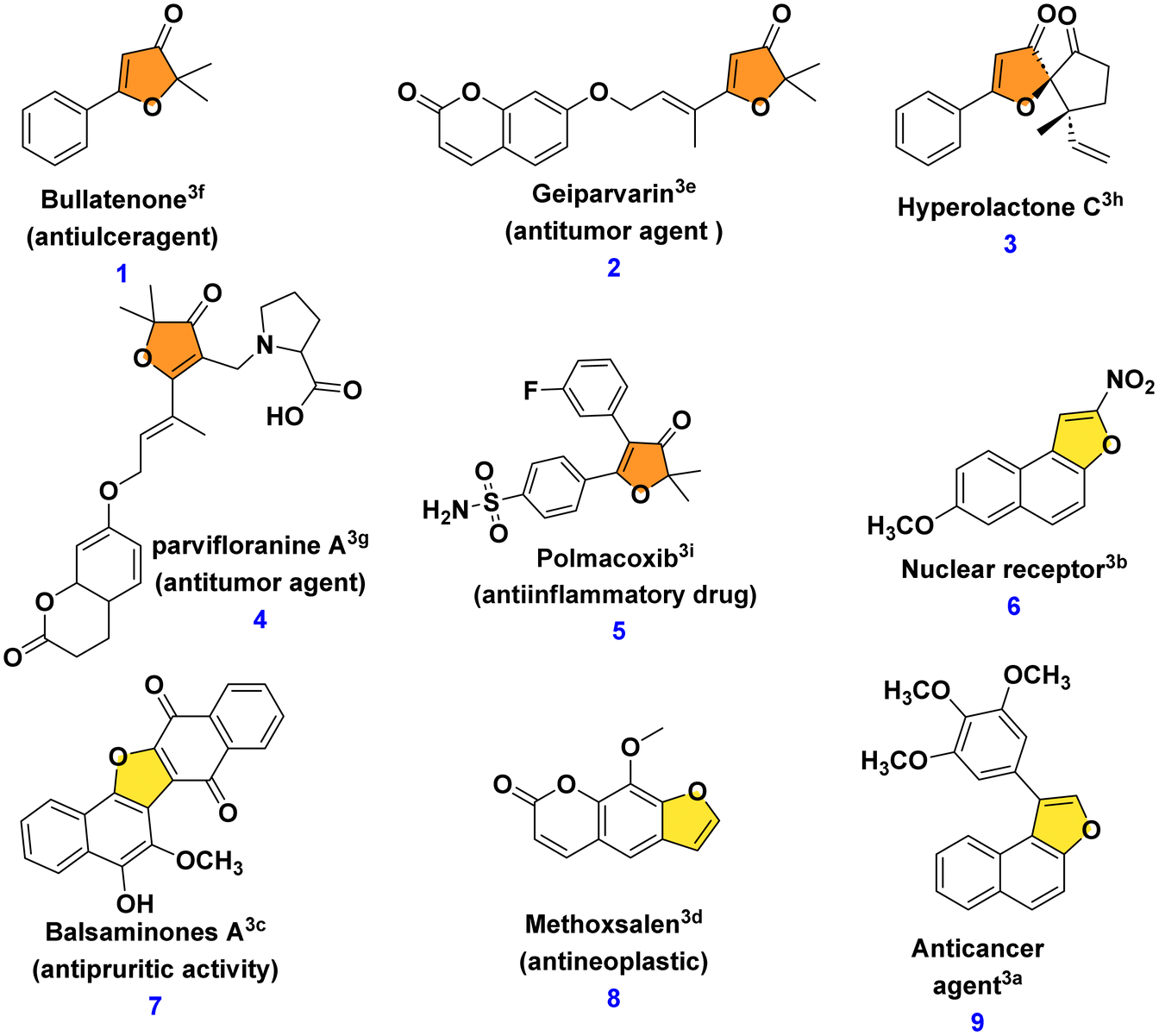 | ||
| Fig. 1 Examples of bioactive molecules containing furanone and naphthofurans. | ||
It was hypothesised that 10a, an alpha hydroxy naphthofuranone molecule, would be an appropriate precursor for functionalization as it can easily eliminate a water molecule in the presence of an acid and would generate naphtho[2,1-b]furan-3-ium 11a intermediate. Furthermore, electrophilic substitution reactions using various aromatic and aliphatic nucleophiles can be performed on the in situ generated naphtho[2,1-b]furan-3-ium carbocation 11a for the preparation of functionalized naphthofuranone 12a. When reduced, the alpha-functionalized naphthofuranone 12a yields naphthofuranol 13a, which is classified as a pinacol-type precursor. Subsequently, the pinacol-type precursor 13a would be subjected to an acidic rearrangement reaction, which would produce a regioselective naphthofuran derivative denoted as 14a. The presence of a furan oxygen atom was expected to be the main driving force for this rearrangement (Scheme 1).
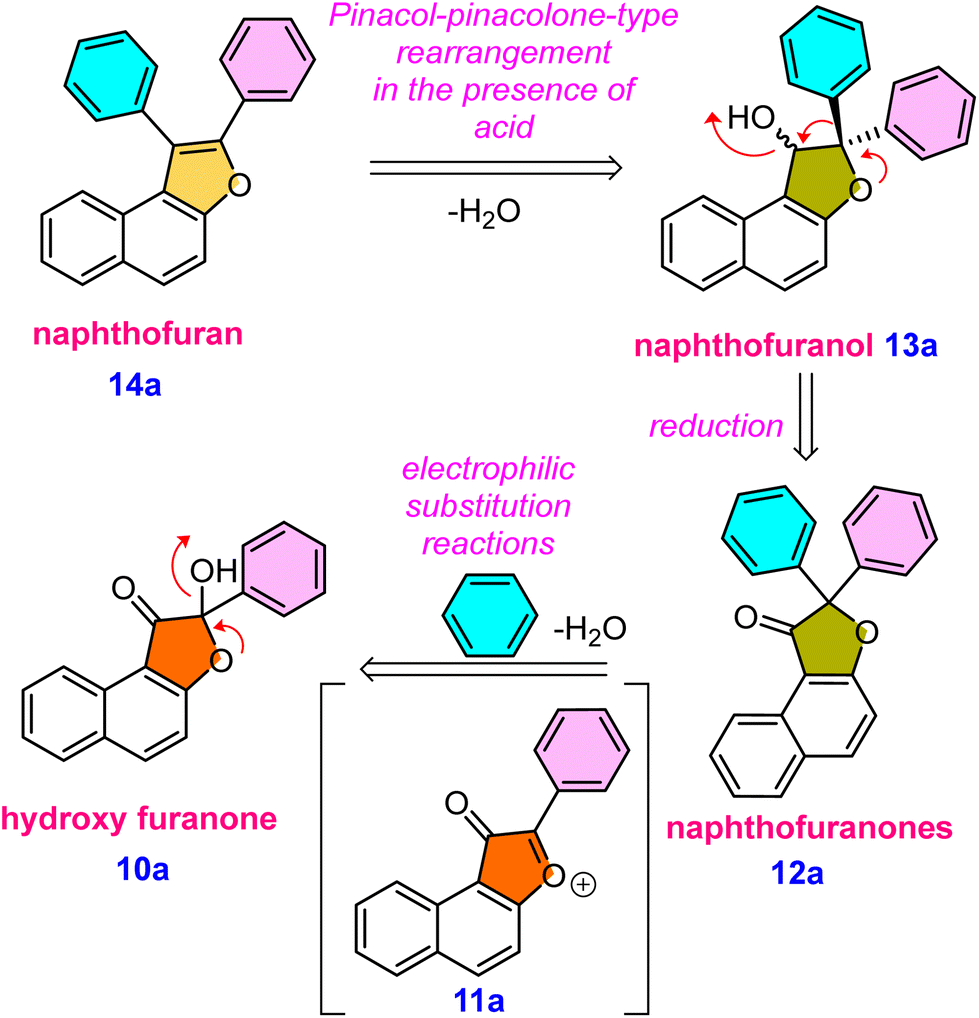 | ||
| Scheme 1 Retrosynthetic analysis of the synthesis of naphthofuran 14a from hydroxy-naphthofuranone 10a. | ||
In this context, our goal was to synthesize hydroxy-naphthofuranone 10 and then convert it into naphthofuran 14. However, the synthesis of hydroxy-naphthofuranone remains highly limited, with no literature reports for its conversion into naphthofuran 14. Ahmed et al.11 described a microwave-assisted approach for synthesising hydroxy-naphthofuranone 10a using copper salts under microwave conditions (eqn (a), Scheme 2). Anxin Wu et al.12 recently described the selective oxidative coupling of 2-naphthol (15a) and methyl ketone 17a under iodine/DMSO oxidative conditions (eqn (b), Scheme 2), a method also explored by Chun Cai et al.13 utilizing a copper catalyst (eqn (b), Scheme 2). In addition, Meng-Yang Chang et al.14 developed a unique method for synthesizing naphthofuranone derivative 20 by light and iodine-mediated dearylacetylative dimerization (eqn (c), Scheme 2). However, these methods have several disadvantages, such as high temperatures, dependence on metal catalysts, and limited possibilities for further synthetic modifications of the furanone molecules. We intended to use a metal-free organophotocatalyst triggered by visible light15 to synthesize hydroxy-naphthofuranones, which would then be used to synthesize different naphthofuranone and naphthofuran derivatives. To the best of our knowledge, there are no reports on the synthesis of naphthofuran from naphthofuranone. The reported methodologies for naphthofuran synthesis rely primarily on metal-mediated C–H activation. The most often used technique involves cyclizing prefunctionalized naphthols, such as 2-alkenyl naphthol, which is typically aided by the addition of a transition metal catalyst.16 Many methods have been described for preparing these scaffolds via base-promoted cyclizations, Lewis or Brønsted acid catalysis, or transition metal catalysis.17 Here, we demonstrate our unique collective strategies for coupling 2-naphthol 15a and phenylglyoxal monohydrate 16a under visible light irradiation to yield a wide range of hydroxy-naphthofuranones 10a–10s and subsequent modification for the synthesis of naphthofuranone 12a–12s, alkoxy-naphthofuranone 12t–12v and naphthofuran derivatives 14a–14h (eqn (d), Scheme 2).
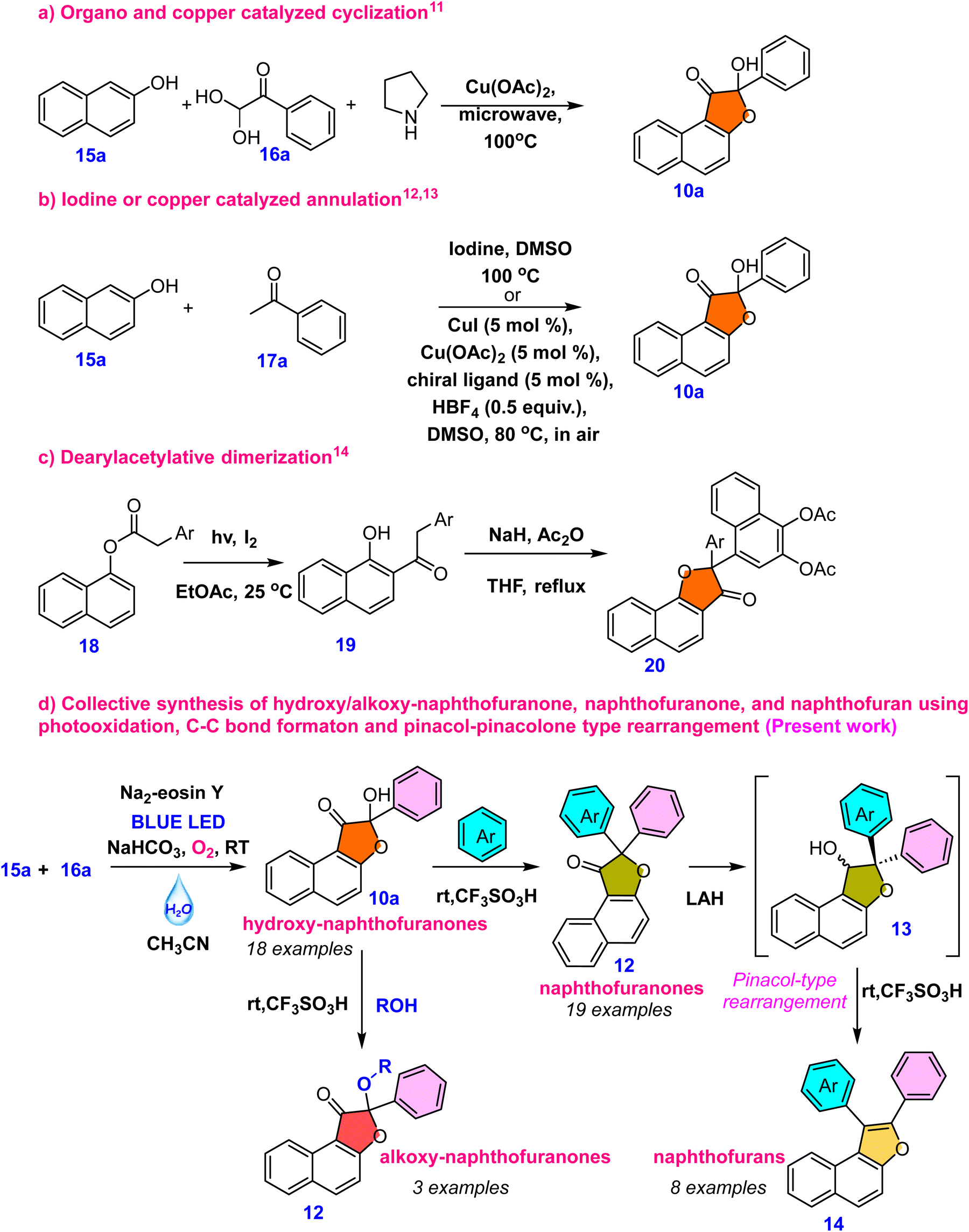 | ||
| Scheme 2 Various synthetic approaches for the synthesis of hydroxy-naphthofuranone and our synthetic plan for various naphthofuranone and furan derivatives. | ||
Initially, 3 W blue LED light was used to optimize the reaction between 2-naphthol 15a and phenylglyoxal monohydrate 16a in acetonitrile solvent with a mild base and Na2–eosin Y under a nitrogen atmosphere (entry 1, Table 1). This resulted in 20% yield of naphtho-3(2H)-furanone 10a, which has a unique quaternary center. Single crystal X-ray analysis and 1H & 13C NMR spectroscopic studies provided unambiguous confirmation of product 10a's structure.20 The ORTEP structure of 10a is provided in Table 1. In order to facilitate the solubility of the inorganic base, we next added water (14 equiv.) to the reaction mass and found that the yield of 10a improved to 35% (entry 2, Table 1). The requirement of base for this reaction was found out from entry 3 as no product 10a formed in the absence of base (entry 3, Table 1). Consequently, further reactions proceeded in the presence of a base and an additive. As expected, when the reaction was performed under an oxygen atmosphere, the yield of 10a was improved (entry 4, Table 1). To emphasize the crucial role of water as an additive, further optimization reactions were focused on water equivalents (entries 5–7, Table 1). These experiments led to the conclusion that the presence of 50 equivalents of water in acetonitrile solvent favored the desired product with a 81% yield (entry 6, Table 1). Later optimization studies concentrated on trying out several solvents, including DMSO, DMF, ethanol, and methanol; acetonitrile gave the maximum yield of the product (entries 6 and 8–11, Table 1). Next, we concentrated on examining the effect of other bases on our reaction, such as NaOH, Na2CO3, and K2CO3. Among these, NaHCO3 appeared to be the superior base compared to others (entries 6 and 12–14, Table 1). Subsequently, we screened various photocatalysts, but observed no enhancement in the product yield (entries 6 and 15–17, Table 1). Different light sources were also assessed, emphasizing the importance of blue LED illumination (entries 6, 18 and 19, Table 1).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Base | Solvent | Additive | Catalyst | Conditions | Atm | Yielda (%) |
| a Isolated yield; reaction conditions: 0.5 mmol of 15a, 0.6 mmol of 16a, 0.5 mmol of NaHCO3, 10 mol% of Na2–eosin Y, 25 mmol of H2O and acetonitrile as a solvent (0.5 mL) in O2 atm under a 3 W blue LED. | |||||||
| 1 | NaHCO3 | ACN | — | Na2–eosin Y | Blue light | N2 | 20 |
| 2 | NaHCO3 | ACN | H2O (14 equiv.) | Na2–eosin Y | Blue light | N2 | 35 |
| 3 | — | ACN | H2O (14 equiv.) | Na2–eosin Y | Blue light | N2 | ND |
| 4 | NaHCO3 | ACN | H2O (14 equiv.) | Na2–eosin Y | Blue light | O2 | 61 |
| 5 | NaHCO3 | ACN | H2O (30 equiv.) | Na2–eosin Y | Blue light | O2 | 71 |
| 6 | NaHCO 3 | ACN | H 2 O (50 equiv.) | Na 2 –eosin Y | Blue light | O 2 | 81 |
| 7 | NaHCO3 | — | H2O (70 equiv.) | Na2–eosin Y | Blue light | O2 | 39 |
| 8 | NaHCO3 | Ethanol | H2O (50 equiv.) | Na2–eosin Y | Blue light | O2 | 63 |
| 9 | NaHCO3 | Methanol | H2O (50 equiv.) | Na2–eosin Y | Blue light | O2 | 66 |
| 10 | NaHCO3 | DMF | H2O (50 equiv.) | Na2–eosin Y | Blue light | O2 | 54 |
| 11 | NaHCO3 | DMSO | H2O (50 equiv.) | Na2–eosin Y | Blue light | O2 | 51 |
| 12 | NaOH | ACN | H2O (50 equiv.) | Na2–eosin Y | Blue light | O2 | 53 |
| 13 | Na2CO3 | ACN | H2O (50 equiv.) | Na2–eosin Y | Blue light | O2 | 61 |
| 14 | K2CO3 | ACN | H2O (50 equiv.) | Na2–eosin Y | Blue light | O2 | 59 |
| 15 | NaHCO3 | ACN | H2O (50 equiv.) | Eosin-Y | Blue light | O2 | 81 |
| 16 | NaHCO3 | ACN | H2O (50 equiv.) | Rose bengal | Blue light | O2 | 73 |
| 17 | NaHCO3 | ACN | H2O (50 equiv.) | Rhodamine B | Blue light | O2 | 73.5 |
| 18 | NaHCO3 | ACN | H2O (50 equiv.) | Na2–eosin Y | Pink light | O2 | 60 |
| 19 | NaHCO3 | ACN | H2O (50 equiv.) | Na2–eosin Y | Green light | O2 | 51 |
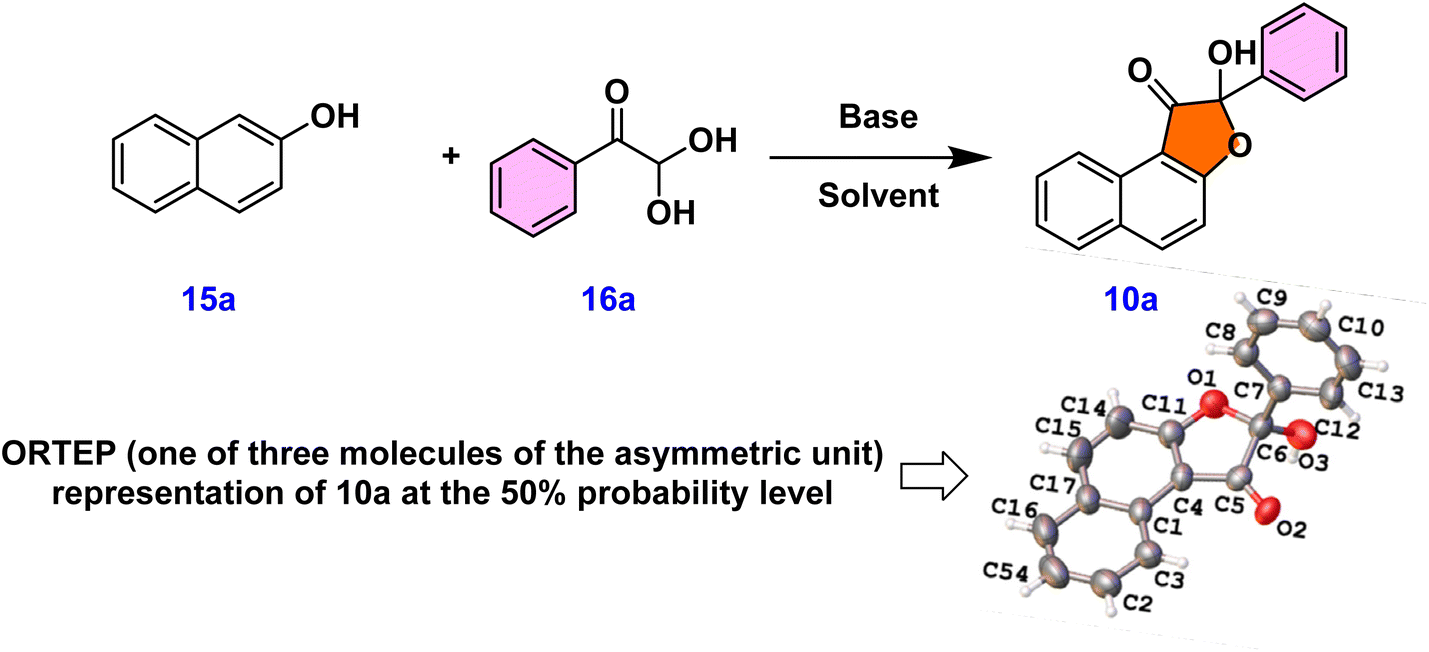
These results indicated that the reaction between 15a and 16a in the presence of NaHCO3 base, with water as an additive, in an oxygen environment under 3 W blue LED light, and with Na2–eosin Y photocatalyst were the optimum conditions for the formation of the hydroxy-naphthofuranone product 10a (entry 6, Table 1).
Having established the standard reaction conditions as outlined in entry 6 of Table 1, our next objective was to examine the substrate scope of the reactions employing derivatives of phenylglyoxal monohydrate 16a–16o and 2-naphthol 15a–15e, as depicted in Scheme 3. All derivatives of 2-naphthol and phenyl glyoxal monohydrates converted into the corresponding products 10b–10s, as anticipated, with good results. The introduction of halogen substituents on the phenyl ring of phenyl glyoxal monohydrates, such as 4-F, 4-Cl, and 4-Br, caused decreased yields of products 10b–10d in comparison with that of 10a. On the other hand, a 52% lowering of yield was determined when phenyl glyoxal monohydrate's phenyl ring had electron-withdrawing groups such as –NO2 (10e). Similarly, the yield of product 10f decreased by 69% when a 4-CF3 group was incorporated into the phenylglyoxal moiety. When 2,4-dichloro substituents were present on phenyl glyoxal monohydrate, a moderate yield of 10i (77%) was produced. However, oxoaldehyde substituted with electron-donating groups such as 4-CH3 and 4-OCH3 gave the desired product in better yields of 68% (10g) and 69% (10h), respectively. The presence of electron-donating or electron-withdrawing groups did not appear to have a significant impact on the yield. A study into steric effects through ortho-chloro and ortho-methyl substitution of phenylglyoxal revealed minimal hindrance, providing 73% (10k) and 81% (10l) yields, respectively. The introduction of a methoxy group at the meta-position of the phenyl ring resulted in a yield of 69% for 10j. Additionally, the reaction utilizing heterocyclic glyoxal derivatives 16m and 16n provided products 10m and 10r with a moderate yield of 69% and 73%, respectively. Then, 2-naphthyl oxoaldehyde subsequently provided the desired product 10s with a yield of 70%. The scope of the reaction was then investigated using other 2-naphthol derivatives. When 2-naphthol was substituted with 7-OCH3, the yield of 10o enhanced to 81%. 2-Naphthol derivatives substituted with bromo, including 3-bromo and 6-bromo-2-naphthol compounds, provided yields of 77% and 56% for 10q and 10n, respectively. For 2-naphthol, the inclusion of a 6-CN substituent delivered 10p with a 55% yield. It is noteworthy that the presence of a cyano group was not tolerated under the conditions reported in the literature.12 This finding is most likely explained by the stability of –CN in such circumstances or by an insufficient activation of the naphthol derivative with an electron-withdrawing –CN group.
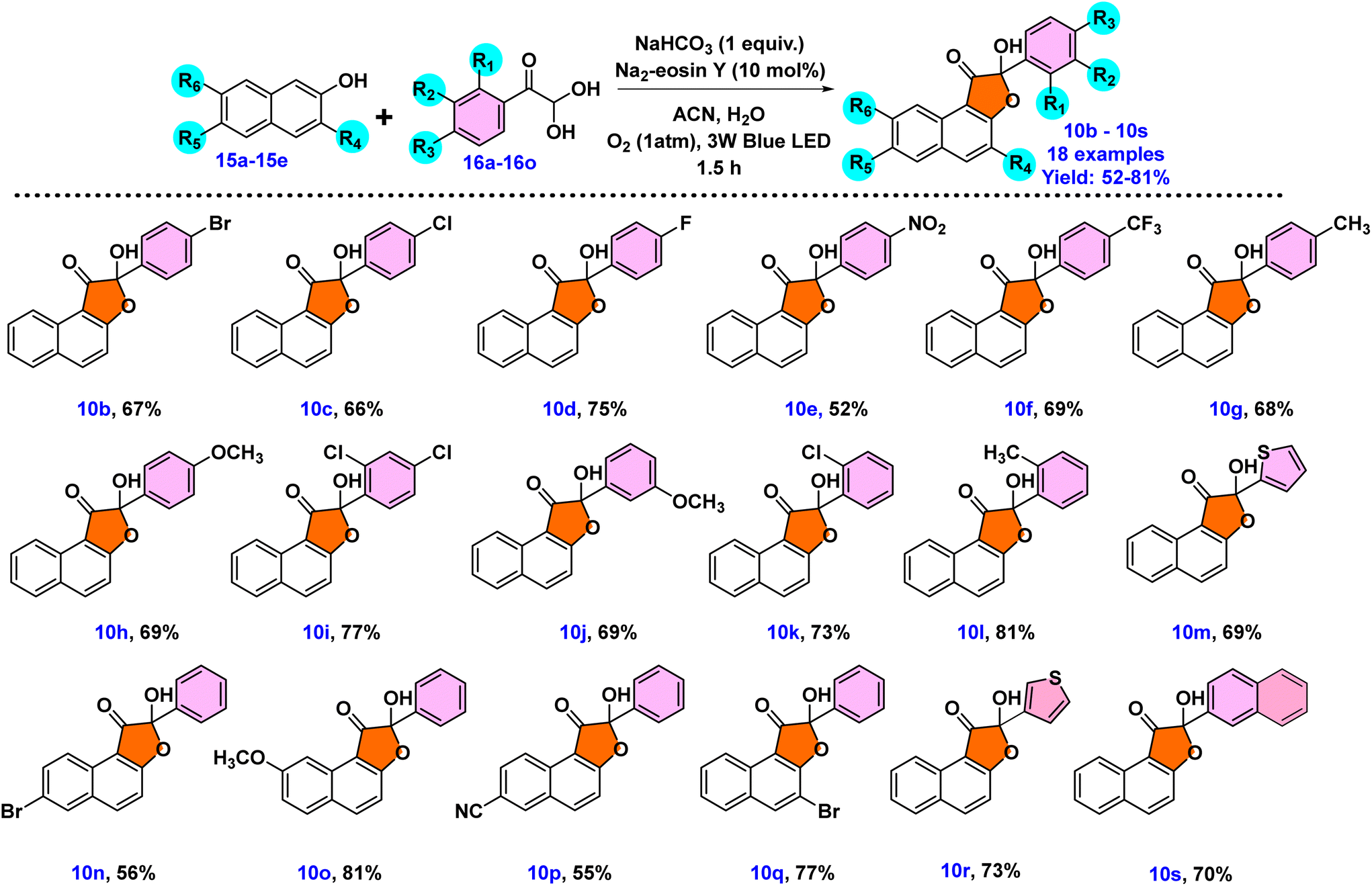 | ||
| Scheme 3 Substrate scope for the synthesis of hydroxy-naphthofuranone derivatives 10b–10s using phenylglyoxal monohydrates 16a–16o and 2-naphthols 15a–15e. Reaction conditions: 0.5 mmol of 15a, 0.6 mmol of 16a, 0.5 mmol of NaHCO3, 10 mol% of Na2–eosin Y, 25 mmol of H2O and acetonitrile as a solvent (0.5 mL) in O2 atm under a 3 W blue LED. | ||
Investigating the synthesis of naphthofuranones 12a–12v from hydroxy-naphthofuranone derivatives 10 was our next objective. Because the hydroxyl group in 10a is a poor leaving group due to the formation of a strong conjugate base, we hypothesized that protonation of this hydroxyl group with acid would generate a hydronium ion, a much better leaving group. The hydroxyl group was protonated and a water molecule was eliminated, as expected when triflic acid was used as the acid source. Consequently, the electrophilic substituted product 12b was obtained in 73% yield by utilizing the in situ formed furanium cation 11a with a xylene molecule. Afterward, we adopted the same strategy to synthesize several naphthofuranone derivatives, 12a, 12c–12v. We attained yields ranging from 55% to 98% by employing triflic acid along with appropriate aromatic/alcoholic nucleophilic partners (Scheme 4). p-Xylene facilitated the synthesis of several naphthofuranone derivatives 12c–12h when employing different hydroxy-naphthofuranone derivatives. Notably, the presence of a methoxy group in compound 12f decreased the yield to 67% compared to those of compounds 12c–12e and 12g–12h, probably due to the methoxy group stabilizing the in situ formed furanium cation.
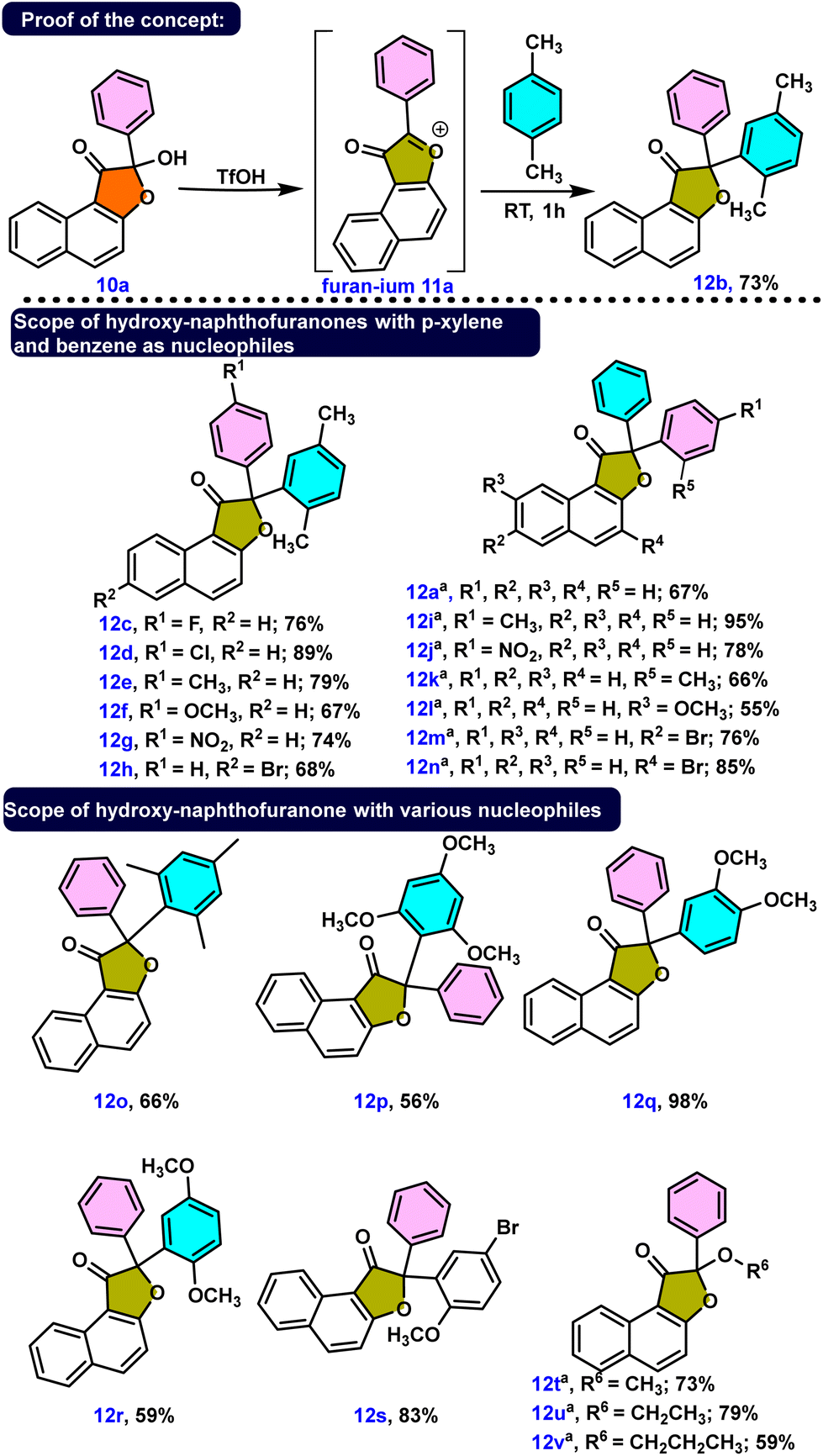 | ||
Scheme 4 (a) Proof of concept and (b) synthesis of naphthofuranones 12a–12v by using various hydroxy-naphthofuranone derivatives along with various nucleophiles. Unless otherwise noted: reaction conditions: 0.5 mmol of 10a, 1 mmol of nucleophile and 0.5 mmol of triflic acid in a closed reaction vial at room temperature. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction temperature is 70 °C. Reaction temperature is 70 °C. | ||
On the other hand, we found slower reactivity than for xylene when benzene was used as the nucleophilic partner for the synthesis of 12a, 12i–12n. Because benzene is less nucleophilic than xylene, the reaction necessitated slightly higher temperatures to produce compounds 12a, 12i–12n in good to outstanding yields. We then investigated some other aromatic nucleophiles, leading to predicted compounds 12o–12s in a very selective manner. Likewise employing different alcohol derivatives, we effectively produced alkoxy naphthofuranones 12t–12v under acidic conditions, as shown in Scheme 4.
Our hypothesis aimed to reduce the keto functionality in 12i to yield the pinacol-type precursor 13b, followed by an acid-catalyzed rearrangement to produce naphthofuran derivative 14b. To test this hypothesis, we first reduced compound 12i using LAH, resulting in a mixture of diastereomeric alcohols 13b with a dr ratio of 1:1 as measured by 1H NMR spectroscopy. Following a simple workup, the diastereomeric mixture 13b underwent further acid-catalyzed rearrangement. As predicted, the naphthofuran derivative 14b was produced via a smooth rearrangement of the pinacol-type precursor 13b, giving 95% yield with high regioselectivity (Scheme 5). Electronic effects provide a rationale for the migration of the 4-methyl substituted phenyl ring over the phenyl ring to the electron-deficient carbon.
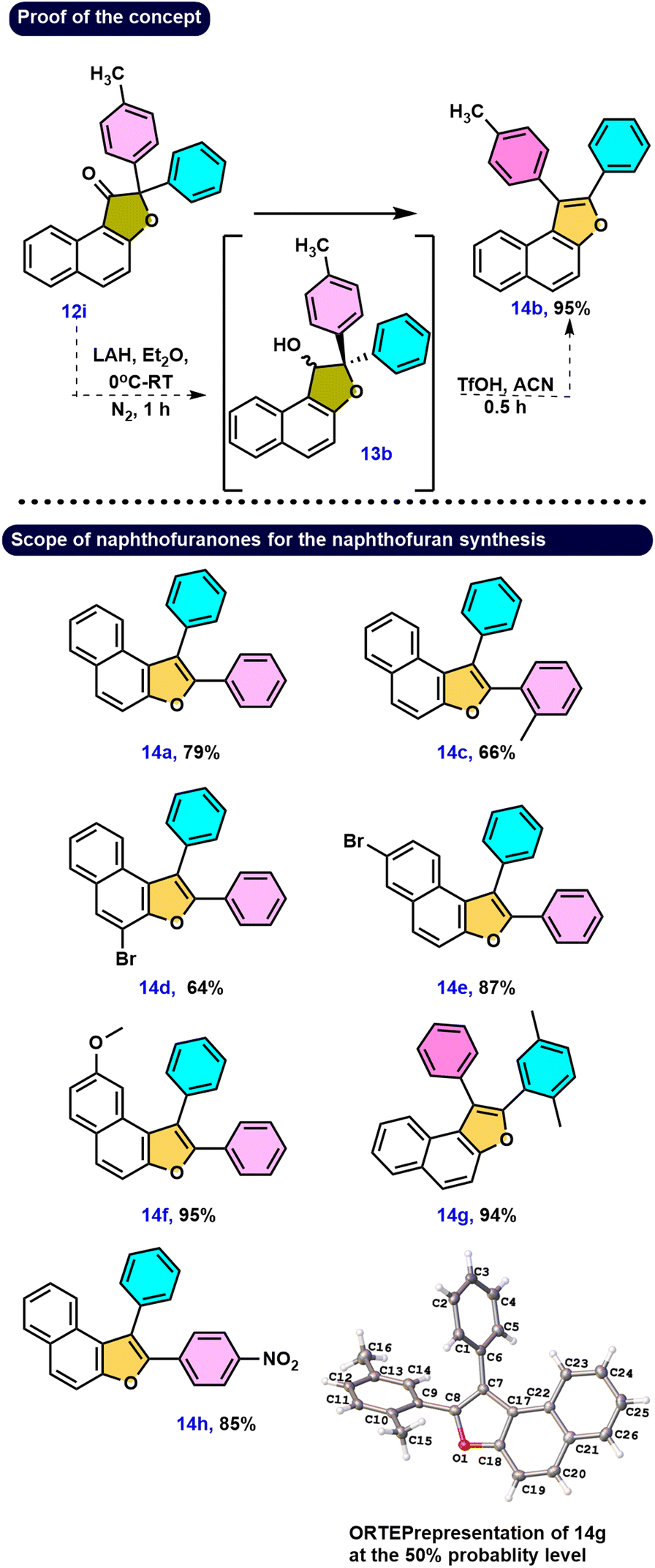 | ||
| Scheme 5 Synthesis of naphthofurans 14a–14h using various naphthofuranone derivatives 12a, 12b, 12i–12n. Reaction conditions: 0.5 mmol of 12a, 1.25 mmol of LAH, and ether as a solvent (0.5 mL) at 0 °C in N2 atm, then 0.5 mmol of 13a, 0.5 mmol of triflic acid, and acetonitrile (0.5 mL) as a solvent. | ||
More specifically, the carbocation would be stabilized more successfully by the +I activity of the methyl-substituted phenyl ring than by the phenyl ring alone. Furthermore, investigation of this type of rearrangement was done on the bromo- and methoxy-substituted groups on the naphthalene core in naphthofuranones 12l–12n. Following the reduction step, the alcoholic compounds yielded reasonable to outstanding yields of the respective naphthofuran derivatives 14d–14f under acidic conditions. These findings support two essential insights: first, the generation of a carbocation because of only one product, and second, the prioritization of migration of a more nucleophilic aromatic partner. Compound 12b upon reduction and then treatment with acid gives 14g, with the migration of the benzene ring instead of the p-xylene group, and the regioselectivity of the compound 14g structure was unambiguously confirmed by single crystal XRD.20 The ORTEP structure of 14g is provided in Scheme 5. Similarly, compound 12j, as predicted, underwent a reduction reaction, and treatment with acid generated only one naphthofuran 14h, with migration taking place toward the benzene partner instead of the nitroaryl group. The strongly electron-withdrawing –NO2 group is probably responsible for this behavior since it might not be able to stabilize the carbocation enough. Compound 12k, which included benzene and ortho-substituted benzene, was reduced/rearranged to provide compound 14c with excellent regioselectivity. Here, the phenyl-migrated compound 14c is produced in excellent yield due to the predominance of steric factors.
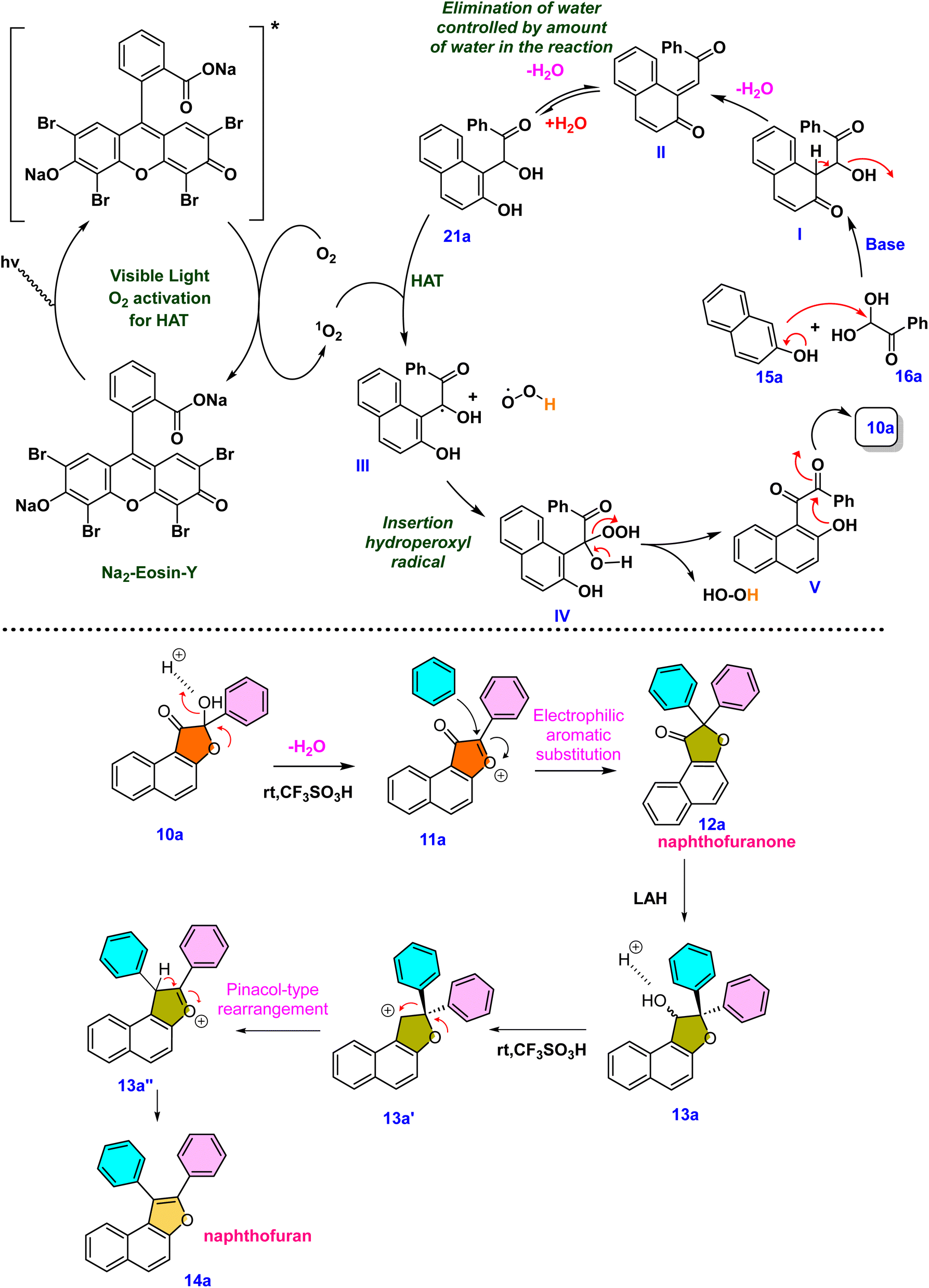
Control experiments were then carried out to provide insight into the possible reaction path. Under standard conditions, the reaction between 2-naphthol 15a and phenylglyoxal monohydrate 16a was carried out without light or a catalyst. This, as expected, resulted in a decrease in the product yield, highlighting the need for light and a photocatalyst (eqn (1), Scheme 6). Furthermore, the reaction did not proceed under standard conditions in the absence of a base, emphasizing the necessity of a base (eqn (2), Scheme 6). Intriguingly, under standard conditions and in the absence of water, our desired product was generated in a low yield of 20% (eqn (3), Scheme 6). To further understand the role that oxygen plays in the process, more control tests were conducted. As a result, the experiment was carried out under a nitrogen atmosphere rather than an oxygen atmosphere using standard conditions, which led to a lower product yield and illustrated the need for oxygen (eqn (4), Scheme 6). The reactive intermediate 21a was subsequently isolated, and its structure was confirmed using single XRD20 and HRMS techniques to deduce the mechanism (eqn (6), Scheme 6). The expected product 10a was obtained by subjecting the isolated intermediate 21a to standard reaction conditions (eqn (7), Scheme 6). Scheme 7 elaborates on a plausible mechanism by putting together information from the relevant literature18 and control experiment results.
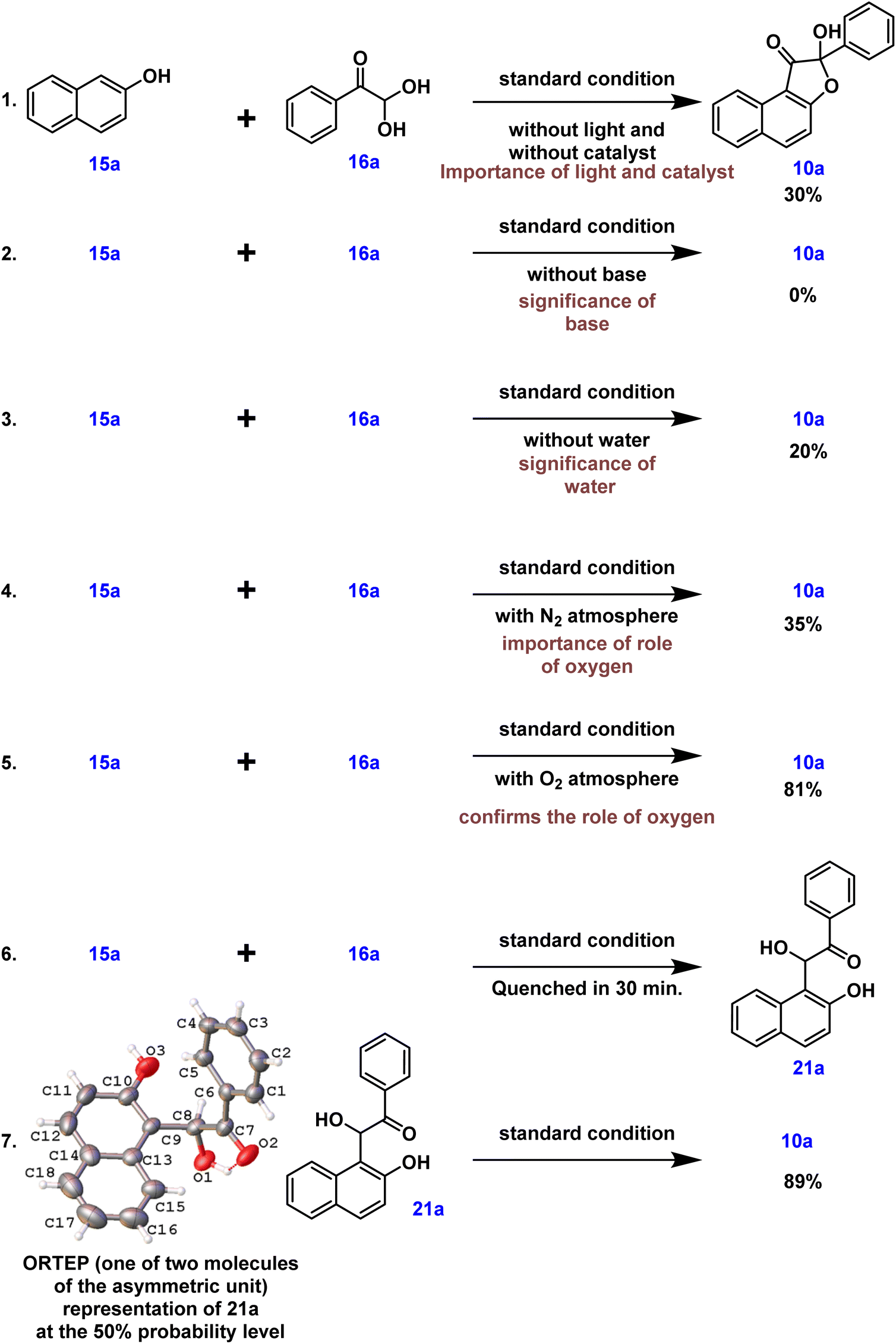 | ||
| Scheme 6 Control experiments. | ||
 | ||
| Scheme 7 Plausible mechanism. | ||
Based on the control experiments and various substrate scope in each stage, a possible mechanism is proposed in Scheme 7. The compounds 15a and 16a reacted together and produced an intermediate I under basic conditions. The intermediate I eliminates water and generates an intermediate II. The existence of this intermediate emphasizes the vital importance of water. Water can be added to promote the generation of the hydrated intermediate 21a or II. High-resolution mass spectrometry and single XRD analysis confirm the structure of the intermediate 21a.20 The ORTEP structure of 21a is provided in Scheme 6. The organic dye Na2–eosin Y is excited when visible light is absorbed, and this energy is then transferred to ground-state triplet oxygen species (3O2). The excited state of the photocatalyst returns to its ground state (Na2–eosin Y) because of this energy transfer, and triplet oxygen changes into singlet oxygen (1O2).19 The reactive intermediate undergoes a hydrogen atom transfer process facilitated by photoactivated oxygen species, providing hydroxyl radical species III and hydroperoxyl radical species. These intermediates can be converted into compound IV by an insertion reaction. In the end, the desired product 10a is formed by an intramolecular cyclization that occurs when the oxygen atom attacks the β-carbonyl group present in the intermediate V. Surprisingly, protonation of –OH happens under acidic circumstances, causing the lone pair of oxygen to induce electron movement and eliminate a water molecule. The resultant furanium 11a ion is unstable, but it is possible to regenerate the single oxygen atom by inserting incoming nucleophiles. Naphthofuranone 12a is further reduced to yield the pinacol-type precursor 13a, which is then subjected to an acid-catalyzed pinacol-type rearrangement reaction to produce the furan derivative 14avia transition intermediates 13a′ and 13a′′ (Scheme 7).
Conclusion
In conclusion, this work highlights a unique approach for synthesizing hydroxy-naphthofuranone using visible light irradiation. Water functions as both a reagent and an additive, playing an important role in the process. Furthermore, our approach was effective in synthesising naphthofuranone and naphthofuran from hydroxy-naphthofuranone derivatives, which represents a unique addition to the field. Importantly, all reactions were carried out under gentle settings, without the use of harsh chemicals or high temperatures. The hypothesised mechanism is consistent with the observed substrate scope and control experiments. Notably, the process produces high yields of different hydroxy-naphthofuranone derivatives and has a wide tolerance for substituents. Our complete synthesis methodology shows great promise for effectively generating target molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. R. sincerely thanks SERB, Government of India, New Delhi, for financial support under the SERB – Research Scientist Program (Grant No. SB/SRS/2022-23/78/CS) and thanks SASTRA Deemed University for Professor TRR research grant. V. H. gratefully acknowledges SASTRA Deemed University for the teaching assistantships. The authors gratefully acknowledge the DST-FIST grant (SR/FST/CS-I/2018/65) to SCBT, SASTRA Deemed University, for the NMR facility.References
- (a) P. Nimnual, K. Norseeda, B. Akkachairin, J. Tummatorn, P. Laohapaisan, N. Supantanapong, P. Chuangsoongnern, C. Thongsornkleeb, S. Sittihan, S. Ruchirawat and W. Rodphon, Asian J. Org. Chem., 2018, 7, 932–945 CrossRef CAS; (b) M. Miura, T. Tsuda, T. Satoh and M. Nomura, Chem. Lett., 1997, 26, 1103–1104 CrossRef; (c) H. He, C. Qi, X. Hu, L. Ouyang, W. Xiong and H. Jiang, J. Org. Chem., 2015, 80, 4957–4965 CrossRef CAS PubMed; (d) A. R. Gomez, R. O. Torres-Ochoa, R. Gamez-Montano, Q. Wang and J. Zhu, Org. Lett., 2020, 22, 7030–7033 CrossRef PubMed.
- V. K. Omanakuttan, J. John and H. Hopf, Eur. J. Org. Chem., 2020, 2021, 163–201 CrossRef.
- (a) V. Srivastava, A. S. Negi, J. K. Kumar, U. Faridi, B. S. Sisodia, M. P. Darokar, S. Luqman and S. P. S. Khanuja, Bioorg. Med. Chem. Lett., 2006, 16, 911–914 CrossRef CAS PubMed; (b) V. E. Borisenko, S. A. Krekov, M. Yu. Fomenko, A. Koll and P. Lipkovski, J. Mol. Struct., 2008, 882, 9–23 CrossRef CAS; (c) J. Padwal, W. Lewis and C. J. Moody, J. Org. Chem., 2011, 76, 8082–8087 CrossRef CAS PubMed; (d) J. Maenpaa, R. Juvonen, H. Raunio, A. Rautio and O. Pelkonen, Biochem. Pharmacol., 1994, 48, 1363–1369 CrossRef CAS PubMed; (e) P. J. Jerris and A. B. Smith, J. Org. Chem., 1981, 46, 577–585 CrossRef CAS; (f) J. McK. R. Woollard, N. B. Perry, R. T. Weavers and J. W. van Klink, Phytochemistry, 2008, 69, 1313–1318 CrossRef CAS PubMed; (g) Q. Shou, L. K. Banbury, D. E. Renshaw, J. E. Smith, X. He, A. Dowell, H. J. Griesser, M. Heinrich and H. Wohlmuth, J. Nat. Prod., 2013, 76, 1384–1387 CrossRef CAS PubMed; (h) D. M. Hodgson, D. Angrish, S. P. Erickson, J. Kloesges and C. H. Lee, Org. Lett., 2008, 10, 5553–5556 CrossRef CAS PubMed; (i) Y.-H. Kang, H. J. Lee, C. J. Lee and J.-S. Park, Biomol. Ther., 2019, 27, 503–513 CrossRef CAS PubMed.
- (a) M. Tian, Y. Peng and J. Zheng, Drug Metab. Dispos., 2022, 50(5), 655–670 CrossRef CAS PubMed; (b) B. Z. Mahmoud, M. K. Adel, O. S. Moustafa and R. M. Zaki, J. Chem. Res., 2006, 2006, 748–752 CrossRef; (c) T. Dao-Huy, M. Haider, F. Glatz, M. Schnürch and M. D. Mihovilovic, Eur. J. Org. Chem., 2014, 8119–8125 CrossRef CAS PubMed.
- A. Radadiya and A. Shah, Eur. J. Org. Chem., 2015, 97, 356–376 CAS.
- V. Srivastava, A. S. Negi, J. K. Kumar, U. Faridi, B. S. Sisodia, M. P. Darokar, S. Luqman and S. P. S. Khanuja, Bioorg. Med. Chem. Lett., 2006, 16, 911–914 CrossRef CAS PubMed.
- (a) A. Kumar, G. Srivastava, S. Srivastava, S. Verma, A. S. Negi and A. Sharma, J. Mol. Model, 2017, 23, 1–16 CrossRef CAS PubMed; (b) D. Goyal, A. Kaur and B. Goyal, J. Med. Chem., 2018, 13, 1275–1299 CAS.
- G. Cozza, S. Sarno, M. Ruzzene, C. Girardi, A. Orzeszko, Z. Kazimierczuk, G. Zagotto, E. Bonaiuto, M. Luisa and L. A. Pinna, Biochim. Biophys. Acta, 2013, 1834, 1402–1409 CrossRef CAS PubMed.
- R. Le Guével, F. Oger, A. Lecorgne, Z. Dudasova, S. Chevance, A. Bondon, P. Barath, G. Simonneaux and G. Salbert, Bioorg. Med. Chem. Lett., 2009, 17, 7021–7030 CrossRef PubMed.
- (a) P.-T. Phan, T.-T. Nguyen, H.-N. Nguyen, B.-K. Le, T. Vu, D. Tran and T.-A. Pham, Molecules, 2017, 22, 687 CrossRef PubMed; (b) T. M. Kadayat, S. Banskota, P. Gurung, G. Bist, T. Bahadur, A. Shrestha, J.-A. Kim and E.-S. Lee, Eur. J. Org. Chem., 2017, 137, 575–597 CAS; (c) A. Baldisserotto, M. Demurtas, I. Lampronti, D. Moi, G. Balboni, S. Vertuani, S. Manfredini and V. Onnis, Eur. J. Org. Chem., 2018, 156, 118–125 CAS; (d) T. Wakabayashi, N. Tokunaga, K. Tokumaru, T. Ohra, N. Koyama, S. Hayashi, R. Yamada, M. Shirasaki, Y. Inui and T. Tsukamoto, J. Med. Chem., 2016, 59, 5109–5114 CrossRef CAS PubMed.
- N. Battini, S. Battula, R. Ranjith Kumar and Q. N. Ahmed, Org. Lett., 2015, 17, 2992–2995 CrossRef CAS PubMed.
- Q. Gao, X. Wu, S. Liu and A. Wu, Org. Lett., 2014, 16, 1732–1735 CrossRef CAS PubMed.
- T. Deng, H. Wang and C. Cai, Eur. J. Org. Chem., 2015, 2015, 1569–1574 CrossRef CAS.
- M.-Y. Chang, K.-T. Chen, Y.-T. Hsiao and S.-M. Chen, J. Org. Chem., 2020, 85, 3605–3616 CrossRef CAS PubMed.
- (a) A. Bosveli, T. Montagnon, D. Kalaitzakis and G. Vassilikogiannakis, Org. Biomol. Chem., 2021, 19, 3303–3317 RSC; (b) T. C. Sherwood, H.-Y. Xiao, R. G. Bhaskar, E. M. Simmons, S. Zaretsky, M. Rauch, R. R. Knowles and G. Murali, J. Org. Chem., 2019, 84, 8360–8379 CrossRef CAS PubMed; (c) W. H. García-Santos, J. Ordóñez-Hernández, M. Farfán-Paredes, H. M. Castro-Cruz, N. A. Macías-Ruvalcaba, N. Farfán and A. Cordero-Vargas, J. Org. Chem., 2021, 86, 16315–16326 CrossRef PubMed; (d) T. C. Sherwood, N. Li, A. N. Yazdani and G. Murali, J. Org. Chem., 2018, 83, 3000–3012 CrossRef CAS PubMed; (e) R. Saritha, S. Babiola Annes and S. Ramesh, RSC Adv., 2021, 11, 14079–14084 RSC.
- (a) V. Kameshwara Rao, G. M. Shelke, R. Tiwari, K. Parang and A. Kumar, Org. Lett., 2013, 15, 2190–2193 CrossRef PubMed; (b) C. Sreenivasulu, K. Reddy and G. Satyanarayana, Org. Chem. Front., 2017, 4, 972–977 RSC; (c) B. Wang, Q. Zhang, J. Luo, Z. Gan, W. Jiang and Q. Tang, Molecules, 2019, 24, 2187 CrossRef PubMed; (d) Q. Zhang, J. Luo, B. Wang, X. Xiao, Z. Gan and Q. Tang, Tetrahedron Lett., 2019, 60, 1337–1340 CrossRef CAS; (e) J. Huang, W. Wang, H.-Y. He, L. Jian, H.-Y. Fu, X.-L. Zheng, H. Chen and R.-X. Li, J. Org. Chem., 2017, 82, 2523–2534 CrossRef CAS PubMed; (f) K. Nakanishi, T. Sasamori, K. Kuramochi, N. Tokitoh, T. Kawabata and K. Tsubaki, J. Org. Chem., 2014, 79, 2625–2631 CrossRef CAS PubMed; (g) N. Chalotra, I. H. Shah, S. Raheem, M. Ahmad Rizvi and B. Ali Shah, J. Org. Chem., 2021, 86, 16770–16784 CrossRef CAS PubMed; (h) S. W. Youn and J. I. Eom, Org. Lett., 2005, 7, 3355–3358 CrossRef CAS PubMed.
- (a) C. Moldoveanu, I. Mangalagiu, D. L. Isac, A. Airinei and G. Zbancioc, Molecules, 2018, 23, 1968–1968 CrossRef PubMed; (b) Q. Chen, P. Jiang, M. Guo and J. Yang, Molecules, 2018, 23, 2450 CrossRef PubMed.
- (a) D. G. Ho, R. Gao, J. Celaje, H.-Y. Chung and M. Selke, Science, 2003, 302, 259–262 CrossRef CAS PubMed; (b) Y. Zhang, C. Ye, S. Li, A. Adam Ding, G. Gu and H. Guo, RSC Adv., 2017, 7, 13240–13243 RSC.
- M. DeRosa, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- (i) The CCDC number for 10a = 2350169; unit cell parameters: (a) 10.989(7), (b) 11.463(8), (c) 17.763(13), space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . (ii) The CCDC number for 21a = 2350170; unit cell parameters: (a) 8.7791(11), (b) 24.495(3), (c) 13.0852(17), space group P21/c. (iii) The CCDC number for 14g = 2360763; unit cell parameters: (a) 8.0311(5), (b) 10.4014(6), (c) 11.3139 (7), space group P21.
. (ii) The CCDC number for 21a = 2350170; unit cell parameters: (a) 8.7791(11), (b) 24.495(3), (c) 13.0852(17), space group P21/c. (iii) The CCDC number for 14g = 2360763; unit cell parameters: (a) 8.0311(5), (b) 10.4014(6), (c) 11.3139 (7), space group P21.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal information for 10a, 21a and 14g, general procedure, NMR data and 1H, 13C and HRMS analysis reports. CCDC 2350169, 2350170 and 2360763. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00693c |
| This journal is © The Royal Society of Chemistry 2024 |