
Deciphering substitution effects on reductive hydroalkoxylation of alkynyl aminols for stereoselective synthesis of morpholines and 1,4-oxazepanes: total synthesis of tridemorph and fenpropimorph†‡
Santosh J.
Gharpure
 *,
Deepika
Kalita
,
Shipra
Somani
and
Juhi
Pal
*,
Deepika
Kalita
,
Shipra
Somani
and
Juhi
Pal
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai – 400076, India. E-mail: sjgharpure@iitb.ac.in
First published on 18th June 2024
Abstract
Acid catalysed reductive etherification of N-propargyl amino alcohols for the stereoselective synthesis of cis-2,5/2,6-disubstituted morpholines and cis-2,6/2,7-disubstituted oxazepanes has been developed. Mechanistic studies revealed that terminal alkynols gave morpholines via a 6-exo-dig hydroalkoxylation–isomerization–reduction cascade. Interestingly, an alkyne hydration–cyclization–reduction sequence is found to be involved in the formation of oxazepanes from alkyl substituted internal alkynols. The strategy was used as a key step in the total synthesis of fungicides tridemorph and fenpropimorph.
1,4-Heterocycles have attracted considerable attention because of their ubiquity in natural products and bioactive molecules. Among them, substituted morpholines are not only present in agrochemicals but also can act as property-enhancing functional groups in drug discovery.1 This is due to the well-balanced lipophilic–hydrophilic profile, relatively weak basicity, and chair-like flexible conformation of morpholines. For example, tridemorph (1) and fenpropimorph (2), introduced by BASF, Germany, act as cereal fungicides, especially for the control of powdery mildews (Fig. 1).2 Sch 50911 (3) is a selective GABA B antagonist, which works as an anticonvulsant.3 Similarly, the 1,4-oxazepane unit is found in naturally occurring alkaloids, such as dopamine D4 receptor ligand 4 used for the treatment of neurological disorders and oxazepane 5, a peripheral-selective noradrenaline reuptake inhibitor.4 Besides this, they also have a wide range of applications in enantioselective synthesis as chiral organocatalysts, chiral auxiliaries, and chiral templates in the synthesis of α-hydroxy acids and oxa-cycles. Not surprisingly, various methodologies have been developed for the synthesis of morpholine and oxazepane derivatives.
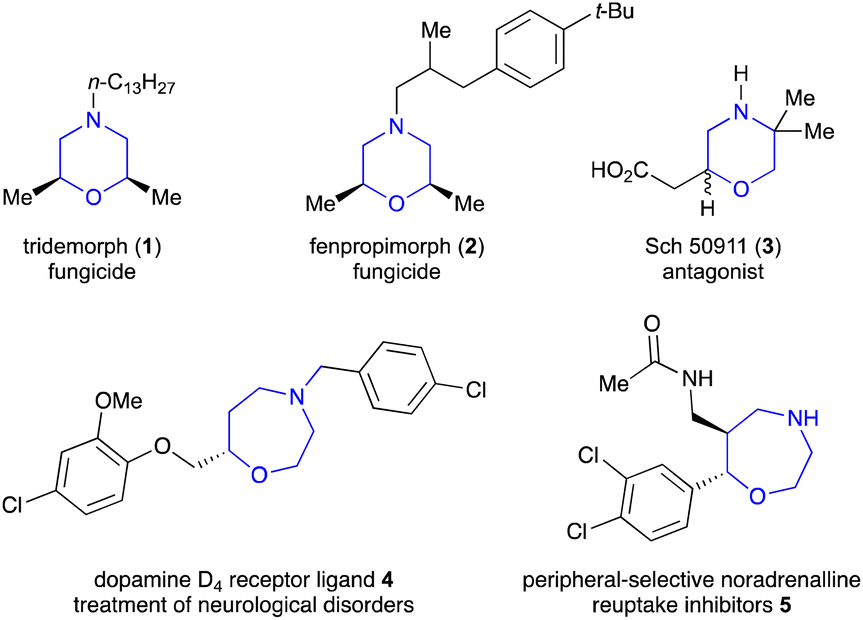 | ||
| Fig. 1 Bioactive molecules having morpholine and 1,4-oxazepane cores. | ||
However, there are very few reports on the synthesis of 1,4-heterocycle derivatives using alkynes. In this context, the Huang group demonstrated gold(I)-catalyzed 6-exo-dig cyclization to afford morpholine and piperazine derivatives (Scheme 1, eqn (1)).5 Recently, Panda et al. reported the synthesis of 3,4-dihydro-2H-1,4 oxazines via Ag(I) promoted intramolecular cyclization.6 During the exploration of platinum catalysed domino cycloisomerization–hydroalkoxylation reactions of alkyne-diol derivatives for the synthesis of fused bicyclic acetals, the Ley group highlighted the potential regioselectivity problems in such transformations (Scheme 1, eqn (2)).7 Similar potential regioselectivity issues were observed independently by Van der Eycken (for morpholine vs. oxazepine) and Schreiber (for oxazepanone vs. oxazocenones).8 Despite the importance of these 1,4-heterocycles, common strategies which can give access to both morpholines as well as oxazepanes in a highly diastereoselective manner are uncommon. In this context, we have developed a Lewis acid mediated, highly diastereoselective synthesis of morpholines as well as oxazepanes via reductive etherification of keto alcohols.9 Further, TMSOTf mediated reductive etherification of aryl substituted N-alkynyl amino alcohols was shown to furnish oxazepanes, albeit with moderate to poor diastereoselectivity (Scheme 1, eqn (3)).10 The reaction was found to be extremely sluggish with alkyl substituted alkynes under the conditions employed. In continuation of our program directed towards synthesis of oxa- and aza-cycles, herein, we disclose the reductive etherification of N-protected alkynols for the stereoselective synthesis of cis-2,5/2,6-disubstituted morpholines and cis-2,6/2,7-disubstituted oxazepanes. We also utilise the developed method for the total synthesis of fungicides tridemorph (1) and fenpropimorph (2).
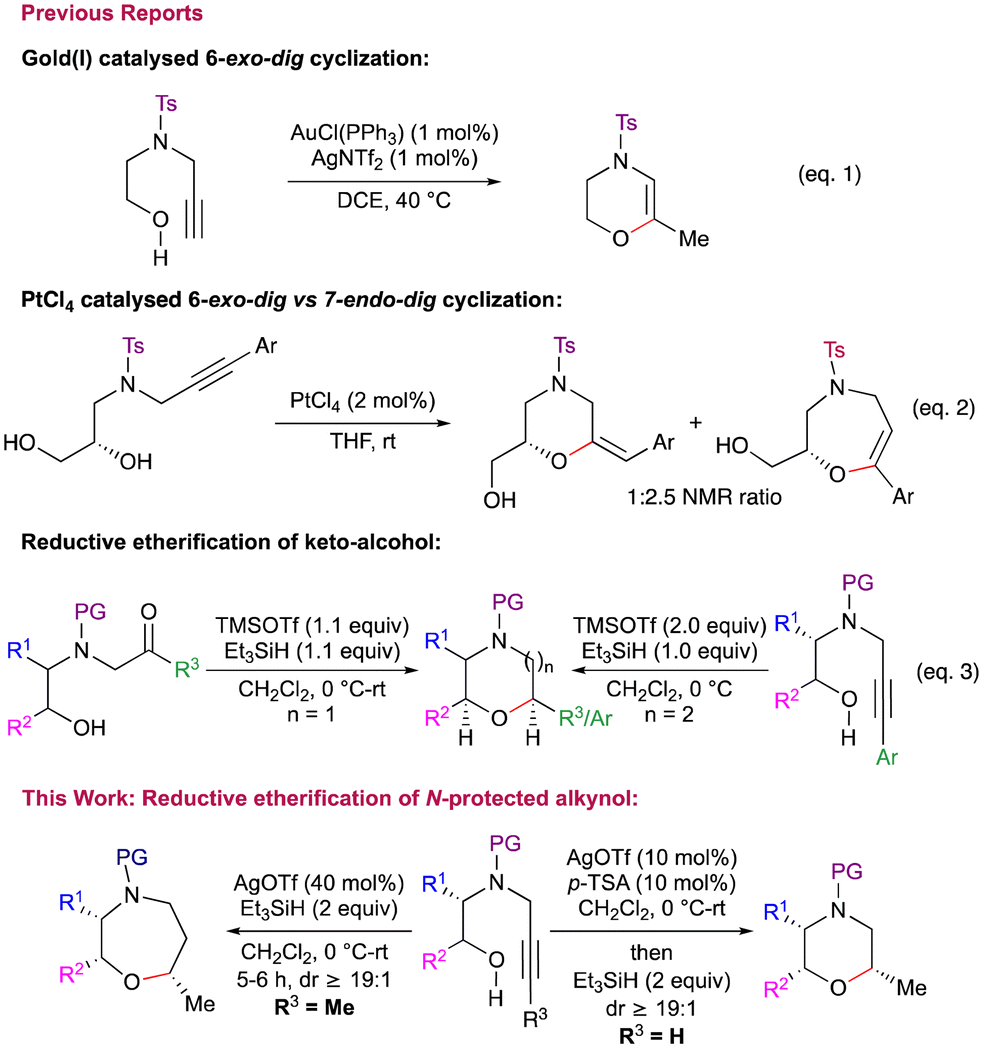 | ||
| Scheme 1 Synthesis of morpholines using alkynes. | ||
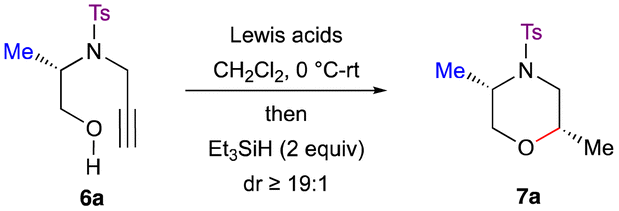
Our study commenced with the reaction of N-protected alkynyl amino alcohol 6a in the presence of TMSOTf (4.0 equiv.) and Et3SiH (2.0 equiv.) in CH2Cl2 at 0 °C–rt, but no formation of the product 7a was observed (Table 1, entry 1). Next, N-protected alkynyl amino alcohol 6a was subjected to reductive etherification in the presence of Cu(OTf)2 (1.0 equiv.) and Et3SiH (2.0 equiv.) in CH2Cl2 at 0 °C–rt, and the reaction failed to give morpholine 7a (Table 1, entry 2). When alkynol 6a was treated with Ag(OTf) (0.4 equiv.) and Et3SiH (2.0 equiv.), the reaction successfully furnished morpholine 7a in excellent yield and diastereoselectivity (dr ≥ 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Table 1, entry 3). To reduce the loading of the Lewis acid, when p-TSA (0.1 equiv.) was used as an additive with Ag(OTf) (0.1 equiv.), the reaction indeed yielded the morpholine 7a in excellent yield and diastereoselectivity (Table 1, entry 4). Further lowering the equivalents of Ag(OTf) resulted in a decreased yield of the product 7a (Table 1, entry 5). Changing the solvent from dichloromethane to 1,2-dichloroethane, toluene or acetonitrile led to reduced yields (Table 1, entries 6–8). The reaction failed to furnish morpholine 7a when p-TSA (1.0 equiv.) alone was used as the catalyst (Table 1, entry 9). Changing the additive to MsOH (0.1 equiv.) failed to improve the yield of morpholine 7a (Table 1, entry 10). Other Lewis acids, such as In(OTf)3, Bi(OTf)3 and Sc(OTf)3 gave the desired product 7a (Table 1, entries 11–13) albeit in poor yield.
:1) (Table 1, entry 3). To reduce the loading of the Lewis acid, when p-TSA (0.1 equiv.) was used as an additive with Ag(OTf) (0.1 equiv.), the reaction indeed yielded the morpholine 7a in excellent yield and diastereoselectivity (Table 1, entry 4). Further lowering the equivalents of Ag(OTf) resulted in a decreased yield of the product 7a (Table 1, entry 5). Changing the solvent from dichloromethane to 1,2-dichloroethane, toluene or acetonitrile led to reduced yields (Table 1, entries 6–8). The reaction failed to furnish morpholine 7a when p-TSA (1.0 equiv.) alone was used as the catalyst (Table 1, entry 9). Changing the additive to MsOH (0.1 equiv.) failed to improve the yield of morpholine 7a (Table 1, entry 10). Other Lewis acids, such as In(OTf)3, Bi(OTf)3 and Sc(OTf)3 gave the desired product 7a (Table 1, entries 11–13) albeit in poor yield.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Acidsa | Equiv. | Additive (equiv.) | Solvent | Time (h) | Yieldb (%) (4a) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a All reactions were carried out using alkynol 1 (0.20 mmol), with addition of acid at 0 °C–rt in dry solvent (3 ml). b Yield of isolated product after purification by silica gel column chromatography. c Starting material recovered. d Gram scale yield of 7a 83%. e No reaction was observed even at reflux, only SM was recovered. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | TMSOTf | 4.0 | — | CH2Cl2 | 72 | —c | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Cu(OTf)2 | 1.0 | — | CH2Cl2 | 72 | —c | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Ag(OTf) | 0.4 | — | CH2Cl2 | 4 | 94 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Ag(OTf) | 0.1 | p-TSA (0.1) | CH2Cl2 | 4 | 95d | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Ag(OTf) | 0.05 | p-TSA (0.1) | CH2Cl2 | 48 | 52 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Ag(OTf) | 0.1 | p-TSA (0.1) | (CH2Cl)2 | 6 | 87 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Ag(OTf) | 0.1 | p-TSA (0.1) | Toluene | 24 | 40 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | Ag(OTf) | 0.1 | p-TSA (0.1) | CH3CN | 72 | 70 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | — | — | p-TSA (1.0) | CH2Cl2 | 72 | 0e | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | Ag(OTf) | 0.1 | MsOH (0.1) | CH2Cl2 | 24 | 30 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | In(OTf)3 | 0.1 | — | CH2Cl2 | 72 | Trace | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | Bi(OTf)3 | 0.1 | — | CH2Cl2 | 48 | 23 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | Sc(OTf)3 | 0.1 | p-TSA (0.1) | CH2Cl2 | 24 | 22 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
With the optimal conditions in hand, we next explored the scope of the reaction (Scheme 2). To begin with, the effect of the N-protecting groups on the efficiency of the reductive etherification was investigated. Thus, various protecting group containing alkynols 6b–e were subjected to the optimised conditions. N-Sulfonylated alkynols 6b and c worked well to furnish morpholines 7b and c under these optimised conditions. On the other hand, L-alanine derived Bn and Cbz protected alkynols 6d and e failed to give the desired products 7d and e, even after increasing the catalyst loading (1 equiv. of both Ag(OTf) and p-TSA). Next, Lewis acid driven reductive etherification was employed for the enantiospecific synthesis of morpholine derivatives. L-Leucine, L-valine, L-phenylalanine and L-phenyl glycine derived alkynyl amino alcohols 6f–i upon treatment with Ag(OTf)/p-TSA/Et3SiH also gave the morpholine derivatives 7f–i in an enantiospecific manner in good to excellent yield with excellent diastereoselectivity. Racemic alkynyl amino alcohols 6j–l containing substituents Me, Cy and CH2OBn α to the oxygen, afforded the corresponding morpholines 7j–l in good yield. The reaction is amenable to ‘gram scale’ synthesis as demonstrated for the product 7a. The structure and stereochemistry of the products 7 were assigned based on spectral data, including NOE, and was also unambiguously confirmed by single crystal X-ray diffraction studies on morpholine 7c.11
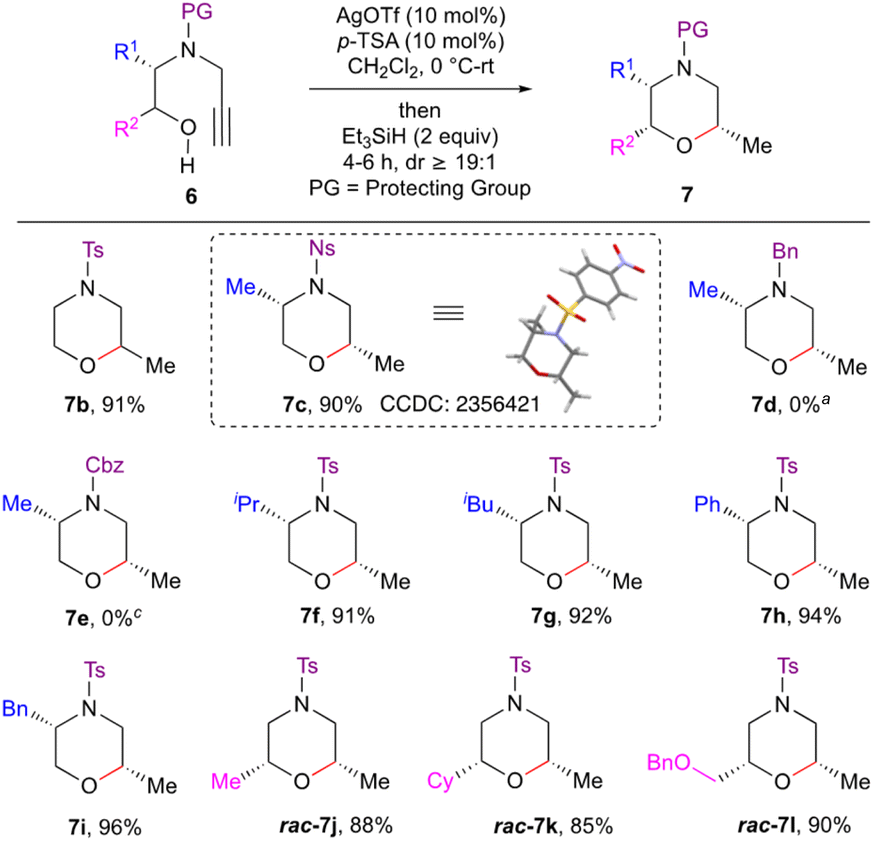 | ||
| Scheme 2 Stereoselective synthesis of highly substituted morpholines. Compounds 7c–i are synthesized in an enantiospecific manner. Isolated yields. dr was measured by 1H NMR on the crude reaction mixture. aStarting material recovered. | ||
The significance of the methodology was further demonstrated by the synthesis of 1,4-oxazepanes (Scheme 3) by employing alkyl substituted internal alkynols 8 as the starting substrates. The enantiopure L-alanine, L-leucine, L-valine, L-phenylalanine and L-phenyl glycine derived alkynyl amino alcohols 8a–d and 8f were reacted with Et3SiH (2.0 equiv.) in the presence of Ag(OTf) under the optimized reaction conditions, leading to the formation of 1,4-oxazepanes 9a–d and 9f in good yield and diastereoselectivity (dr ≥ 19:1). In the case of Ns protected alkynol 8e, formation of 1,4-oxazepane 9e was observed in good yield. Racemic substrates having substituents like Me and Cy, α to the oxygen, gave the corresponding 1,4-oxazepanes 9i and j in good yield. The structure and stereochemistry of the product 9 were assigned based on spectral data, including NOE, and was also unambiguously confirmed by single crystal X-ray diffraction studies on the oxazepane derivative 9f.11
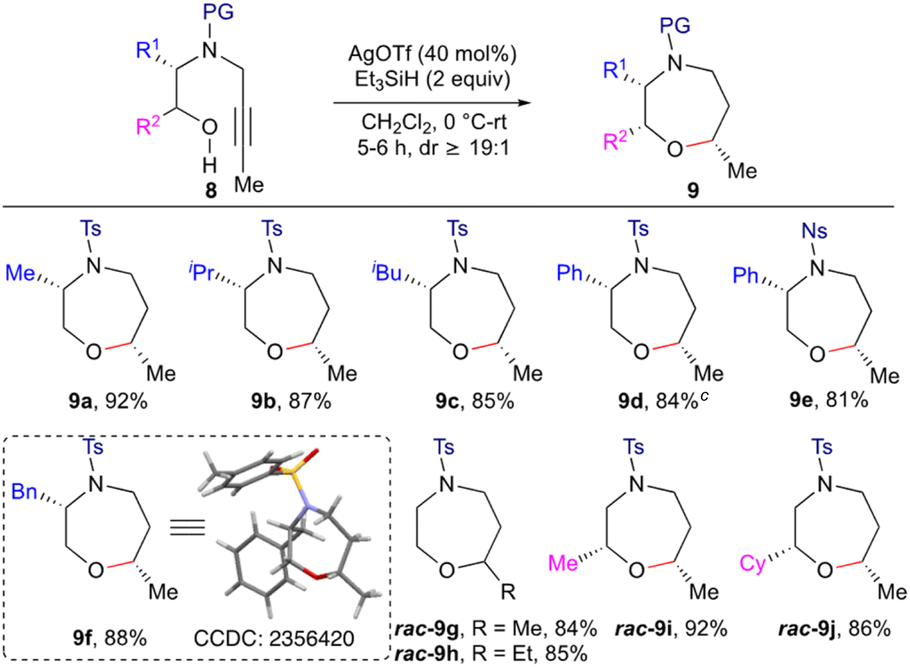 | ||
| Scheme 3 Stereoselective synthesis of substituted 1,4-oxazepanes. Compounds 9a–f are synthesized in an enantiospecific manner. Isolated yields. dr was measured by 1H NMR on the crude reaction mixture. aGram scale yield of 9d 79%. | ||
Mechanistically, the formation of the morpholines 7 and 1,2-oxazepanes 9 can be explained as follows (Scheme 4). N-Protected terminal alkynyl amino alcohol 6j on treatment with Ag(OTf) underwent 6-exo-dig cyclization followed by isomerisation to generate intermediate 10. Intermediate 10 on protonation generated oxonium ion Int-A, which on reduction with silane gave morpholine rac-7j (Scheme 4A). To prove the intermediacy of the 3,4-dihydro-oxazine 10, the reaction course starting from N-protected terminal alkynyl amino alcohol 6j to the morpholine rac-7j was monitored by react-IR. After the initiation of the reaction, the appearance of the absorption band at 1600 cm−1 corresponding to O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration denotes the formation of 3,4-dihydro-oxazine 10 (Scheme 4B). To corroborate this observation, the reaction was monitored by 1H NMR spectroscopy as well. Treatment of terminal alkynyl amino alcohol 6j with Ag(OTf)/p-TSA resulted in the disappearance of the singlet at 2.16 ppm and the appearance of a singlet at 5.81 ppm corresponding to CHC(O)Me for 3,4-dihydro-oxazine 10. Addition of Et3SiH further led to the disappearance of the singlet at 5.81 ppm and appearance of a multiplet at 3.71–3.63 ppm for morpholine rac-7j (Scheme 4C). On the other hand, hydration of alkyl substituted alkynyl amino alcohol 8d in the presence of Ag(OTf) furnished keto-alcohol 11. Keto alcohol 11 reacted further to generate oxonium ion intermediate Int-B, which on reduction with silane yielded 1,2-oxazepane 9a (Scheme 4A). The reaction course starting from internal alkynyl amino alcohol 8d to the 1,2-oxazepane 9a was monitored by react-IR. After the initiation of the reaction, the appearance of the absorption band at 1715 cm−1 corresponding to CO stretching vibration gives credence to the formation of keto-alcohol 11 (Scheme 4B). Along with this, when an NMR study of the reductive etherification of internal alkynyl amino alcohol 8a was carried out, in the presence of Ag(OTf)/Et3SiH, disappearance of the triplet at 1.75 ppm and appearance of a singlet at 1.97 ppm denoted the formation of keto-alcohol 11. Moreover, disappearance of the singlet at 1.97 ppm and appearance of a multiplet at 3.77–3.73 ppm signified the generation of 1,2-oxazepane 9a (Scheme 4C). The stereochemical outcome of these reactions can be rationalised based on the model proposed in our earlier study. The substituent on the cyclic oxonium ion intermediate Int-A/Int-B occupies a pseudo-equatorial orientation to reduce steric interaction and delivery of the incoming hydride nucleophile from the axial direction results in a chair conformation for the 1,4-heterocyclic products.9 Encouraged by this result, to further probe the regioselectivity, we decided to explore the reactivity of N-alkynylated diols. When terminal N-alkynylated diol 12 was subjected to reductive etherification, instead of the formation of the morpholine, spiro-ketal 14 was obtained. On the other hand, reaction of internal N-alkynylated diol 13 under the optimised conditions gave spiro-ketal 15 in place of the oxazepane (Scheme 4D). In both the cases, the intermediate oxonium ion was trapped by the internal hydroxy group as a nucleophile, rather than the silane. This is in contrast to our earlier studies where we obtained reductive etherification products while using TMSOTf as the catalyst.9,10 The acetal opening followed by reduction in the present cases is perhaps precluded due to the mild Lewis acidic nature of Ag(OTf). These outcomes suggest that the terminal alkyne follows 6-exo-dig hydroalkoxylation whereas the internal alkyne follows a 7-endo-dig path exclusively.
C stretching vibration denotes the formation of 3,4-dihydro-oxazine 10 (Scheme 4B). To corroborate this observation, the reaction was monitored by 1H NMR spectroscopy as well. Treatment of terminal alkynyl amino alcohol 6j with Ag(OTf)/p-TSA resulted in the disappearance of the singlet at 2.16 ppm and the appearance of a singlet at 5.81 ppm corresponding to CHC(O)Me for 3,4-dihydro-oxazine 10. Addition of Et3SiH further led to the disappearance of the singlet at 5.81 ppm and appearance of a multiplet at 3.71–3.63 ppm for morpholine rac-7j (Scheme 4C). On the other hand, hydration of alkyl substituted alkynyl amino alcohol 8d in the presence of Ag(OTf) furnished keto-alcohol 11. Keto alcohol 11 reacted further to generate oxonium ion intermediate Int-B, which on reduction with silane yielded 1,2-oxazepane 9a (Scheme 4A). The reaction course starting from internal alkynyl amino alcohol 8d to the 1,2-oxazepane 9a was monitored by react-IR. After the initiation of the reaction, the appearance of the absorption band at 1715 cm−1 corresponding to CO stretching vibration gives credence to the formation of keto-alcohol 11 (Scheme 4B). Along with this, when an NMR study of the reductive etherification of internal alkynyl amino alcohol 8a was carried out, in the presence of Ag(OTf)/Et3SiH, disappearance of the triplet at 1.75 ppm and appearance of a singlet at 1.97 ppm denoted the formation of keto-alcohol 11. Moreover, disappearance of the singlet at 1.97 ppm and appearance of a multiplet at 3.77–3.73 ppm signified the generation of 1,2-oxazepane 9a (Scheme 4C). The stereochemical outcome of these reactions can be rationalised based on the model proposed in our earlier study. The substituent on the cyclic oxonium ion intermediate Int-A/Int-B occupies a pseudo-equatorial orientation to reduce steric interaction and delivery of the incoming hydride nucleophile from the axial direction results in a chair conformation for the 1,4-heterocyclic products.9 Encouraged by this result, to further probe the regioselectivity, we decided to explore the reactivity of N-alkynylated diols. When terminal N-alkynylated diol 12 was subjected to reductive etherification, instead of the formation of the morpholine, spiro-ketal 14 was obtained. On the other hand, reaction of internal N-alkynylated diol 13 under the optimised conditions gave spiro-ketal 15 in place of the oxazepane (Scheme 4D). In both the cases, the intermediate oxonium ion was trapped by the internal hydroxy group as a nucleophile, rather than the silane. This is in contrast to our earlier studies where we obtained reductive etherification products while using TMSOTf as the catalyst.9,10 The acetal opening followed by reduction in the present cases is perhaps precluded due to the mild Lewis acidic nature of Ag(OTf). These outcomes suggest that the terminal alkyne follows 6-exo-dig hydroalkoxylation whereas the internal alkyne follows a 7-endo-dig path exclusively.
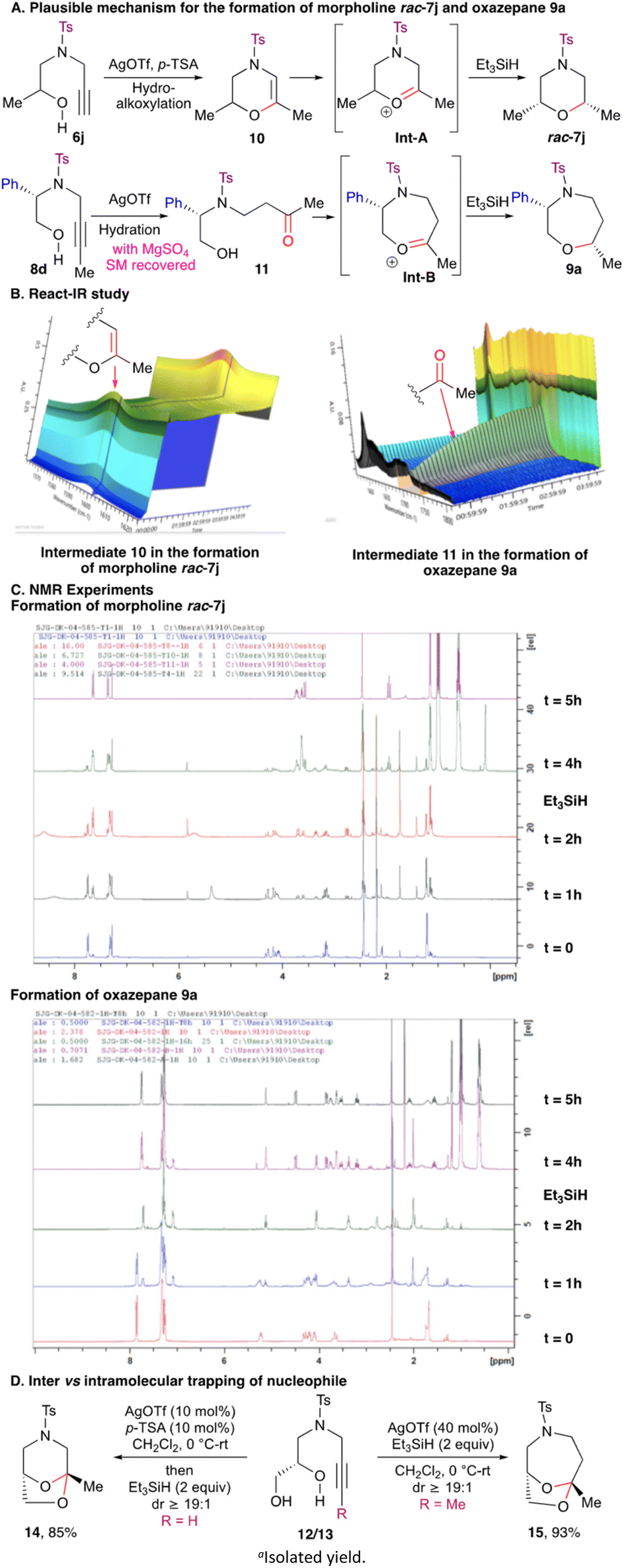 | ||
| Scheme 4 Mechanistic investigation for the stereoselective synthesis of substituted morpholine and 1,4-oxazepanes. Isolated yields. | ||
After successfully demonstrating the scope of the strategy, we turned our attention to applying this strategy for the total synthesis of tridemorph (1) and fenpropimorph (2) (Scheme 5). The tosyl group of the morpholine rac-7j (cf.Scheme 2) was deprotected using sodium naphthalide to give the product 16 in good yield. Further imine formation with aldehydes 17 and 18 and reduction by NaBH4 in the same pot furnished tridemorph (1) and fenpropimorph (2), respectively.
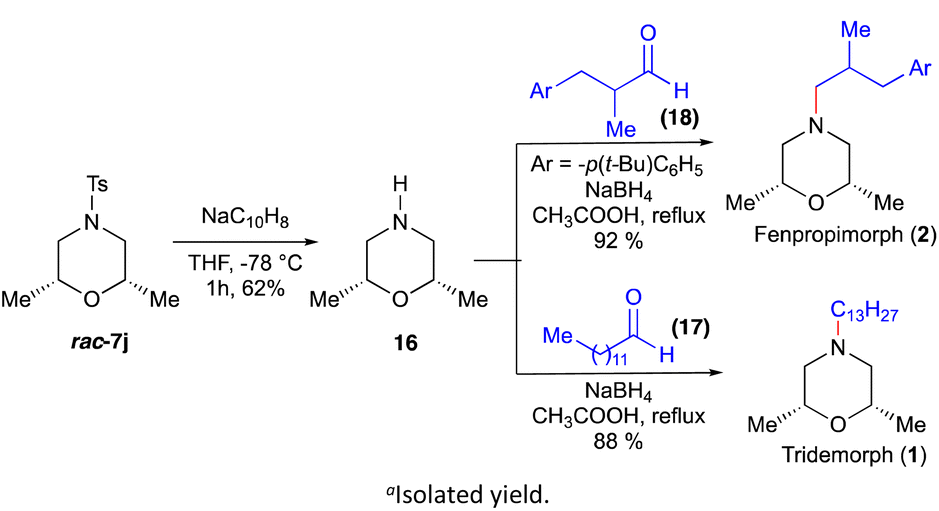 | ||
| Scheme 5 Total synthesis of fungicides, tridemorph and fenpropimorph. Isolated yields. | ||
Conclusions
In conclusion, we have developed a reductive etherification-based strategy for the synthesis of diversely substituted morpholines and 1,4-oxazepanes in a highly diastereoselective manner. cis-2,5/2,6-Morpholines and cis-2,6/2,7-1,4-oxazepanes derived from enantiopure amino alcohols were prepared with good yields and excellent diastereoselectivities by utilizing this strategy. The developed strategy was also successfully applied in the stereoselective total synthesis of fungicides, tridemorph and fenpropimorph.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank SERB, New Delhi for financial support. We thank Mr Darshan Mhatre of the X-ray facility of the Department of Chemistry, IIT Bombay, for collecting the crystallographic data and IRCC, IIT Bombay for funding central facilities. We are grateful to the Prime Minister’s Research Fellowship (PMRF) for providing a fellowship to D. K., UGC, New Delhi to S. S. and CSIR, New Delhi and IRCC, IIT Bombay to J. P.References
- (a) A. Kumari and R. K. Singh, Bioorg. Med., 2020, 96, 103578 CAS; (b) X. Tang, L. Lei, A. Liao, W. Sun, J. Zhang and J. Wu, J. Agric. Food Chem., 2023, 71, 13197 CrossRef CAS PubMed.
- (a) D. J. Fisher, Pestic. Sci., 1974, 5, 219 CrossRef CAS; (b) S. A. Forsyth, H. Q. Nimal Gunaratne, C. Hardacre, A. Mckeown and D. W. Rooney, Org. Process Res. Dev., 2006, 10, 94 CrossRef CAS.
- D. J. Blythin, S.-C. Kuo, H.-J. Shue, A. T. Mcphail, R. W. Chapman, W. Kreutner, C. Rizzo, H. S. She and R. West, Bioorg. Med. Chem. Lett., 1996, 6, 1529 CrossRef CAS.
- (a) K. Ishimoto, K. Yamaguchi, A. Nishimoto, M. Murabayashi and T. Ikemoto, Org. Process Res. Dev., 2017, 21, 2001 CrossRef CAS; (b) T. Yukawa, Y. Nakada, N. Sakauchi, T. Kamei, M. Yamada, Y. Ohba, I. Fujimori, H. Ueno, M. Takiguchi, M. Kuno, I. Kamo, H. Nagakawa, Y. Fujioka, T. Igari, Y. Ishichi and T. Tsukamato, Bioorg. Med. Chem. Lett., 2016, 24, 3716 CrossRef CAS PubMed.
- (a) L.-F. Yao, Y. Wang and K.-W. Huang, Org. Chem. Front., 2015, 2, 721 RSC; (b) J. Barluenga, A. Fernandez, A. Dieguez, F. Rodriguez and F. J. Fananas, Chem. – Eur. J., 2009, 15, 11660 CrossRef CAS PubMed; (c) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed; (d) A. Fürstner, Chem. Soc. Rev., 2009, 38, 320 RSC; (e) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed.
- A. Kumar, S. Bhattacherjee and G. Panda, ARKIVOC, 2022, vi, 239 Search PubMed.
- (a) A. Dieguez-Vazquez, C. C. Tzschucke, W. Y. Lam and S. V. Ley, Angew. Chem., Int. Ed., 2008, 47, 209 CrossRef CAS PubMed; (b) E. Jiménez-Núňez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef PubMed; (c) V. P. Demertzidou and A. L. Zografos, Org. Biomol. Chem., 2016, 14, 6942 RSC; (d) C. I. Stathakis, P. L. Gkizisa and A. L. Zografos, Nat. Prod. Rep., 2016, 33, 1093 RSC; (e) V. Michelet, P. Y. Toullec and J.-P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268 CrossRef CAS PubMed; (f) L. Sotorríos, V. P. Demertzidou, A. L. Zografos and E. Gómez-Bengoa, Org. Biomol. Chem., 2019, 17, 5112 RSC.
- (a) A. A. Peshkov, A. A. Nechaev, O. P. Pereshivko, J. L. Goeman, J. Van der Eycken, V. A. Peshkov and E. V. Van der Eycken, Eur. J. Org. Chem., 2015, 4190 CrossRef CAS; (b) S. S. Scully, S.-L. Zheng, B. K. Wagner and S. L. Schreiber, Org. Lett., 2015, 17, 418 CrossRef CAS PubMed.
- (a) S. J. Gharpure and J. V. K. Prasad, J. Org. Chem., 2011, 76, 10325 CrossRef CAS PubMed; (b) S. J. Gharpure and J. V. K. Prasad, Eur. J. Org. Chem., 2013, 2076 CrossRef CAS; (c) S. J. Gharpure, A. Dandela, J. V. K. Prasad and P. S. Rao, Eur. J. Org. Chem., 2015, 86 CrossRef CAS.
- S. J. Gharpure, D. S. Vishwakarma and S. K. Nanda, Org. Lett., 2017, 19, 6534 CrossRef CAS PubMed.
- CCDC 23564217(c) and 23564209(f) contain the supplementary crystallographic data for this paper.†.
Footnotes |
| † Dedicated with profound respect to Prof. Sukh Dev on his 100th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and characterization. CCDC 2356421 and 2356420. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00855c |
| This journal is © The Royal Society of Chemistry 2024 |