
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of charges on the hybridization kinetics and thermal stability of PNA duplexes†
Miguel
López-Tena
and
Nicolas
Winssinger
 *
*
Department of Organic Chemistry, NCCR Chemical Biology, Faculty of Science, University of Geneva, 1211 Geneva, Switzerland. E-mail: nicolas.winssinger@unige.ch
First published on 19th June 2024
Abstract
Peptide nucleic acid (PNA) is a prominent artificial nucleic acid mimetic and modifications at the γ-position of the peptidic backbone are known to further enhance the desirable properties of PNA in terms of duplex stability. Here, we leveraged a propargyl ether modification at this position for late stage functionalization of PNA to obtain positively charged (cationic amino and guanidinium groups), negatively charged (anionic carboxylate and alkyl phosphonate groups) and neutral (PEG) PNAs to assess the impact of these charges on DNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PNA and PNA:PNA duplex formation. Thermal stability analysis findings concurred with prior studies showing PNA:DNA duplexes are moderately more stable with cationic PNAs than anionic PNAs at physiological salt concentrations. We show that this effect is derived predominantly from differences in the association kinetics. For PNA:PNA duplexes, anionic PNAs were found to form the most stable duplexes, more stable than neutral PNA:PNA duplexes.
:PNA and PNA:PNA duplex formation. Thermal stability analysis findings concurred with prior studies showing PNA:DNA duplexes are moderately more stable with cationic PNAs than anionic PNAs at physiological salt concentrations. We show that this effect is derived predominantly from differences in the association kinetics. For PNA:PNA duplexes, anionic PNAs were found to form the most stable duplexes, more stable than neutral PNA:PNA duplexes.
Introduction
Peptide nucleic acid (PNA) was first reported three decades ago1,2 and the field continues to draw attention from the community by virtue of its exceptional properties (for recent examples, see ref. 3–16). It is an artificial mimetic of DNA or RNA wherein the phosphoribosyl backbone has been replaced by a peptidic backbone. The high stability of PNA:DNA duplexes was attributed to the lack of repulsive interactions between the negatively charged DNA and the neutral PNA, compared to a DNA:DNA duplex.1 The higher stability of the PNA:DNA or PNA:RNA duplexes relative to DNA or RNA homoduplexes has inspired many biomedical applications including in diagnostics and therapeutics17,18 in addition to their use as encoding or biosupramolecular tags for assembly and hybridization circuitry.19–21 Extensive SAR studies have been performed to further optimise PNA properties.22 Notably, alternative nucleobases23–30 have been advanced for improved affinity and cellular uptake; conformationally restricted backbones31–35 have been shown to enhance duplex stability significantly; and substitution of the achiral 2-aminoethyl glycine has been used to tune and improve the properties of PNAs. In particular, substitution at the γ-position of the 2-aminoethyl glycine backbone has been found to enhance hybridization to DNA by chirality-induced preorganization (Fig. 1A).36,37 Examples with a broad array of substituents have been reported.38–51
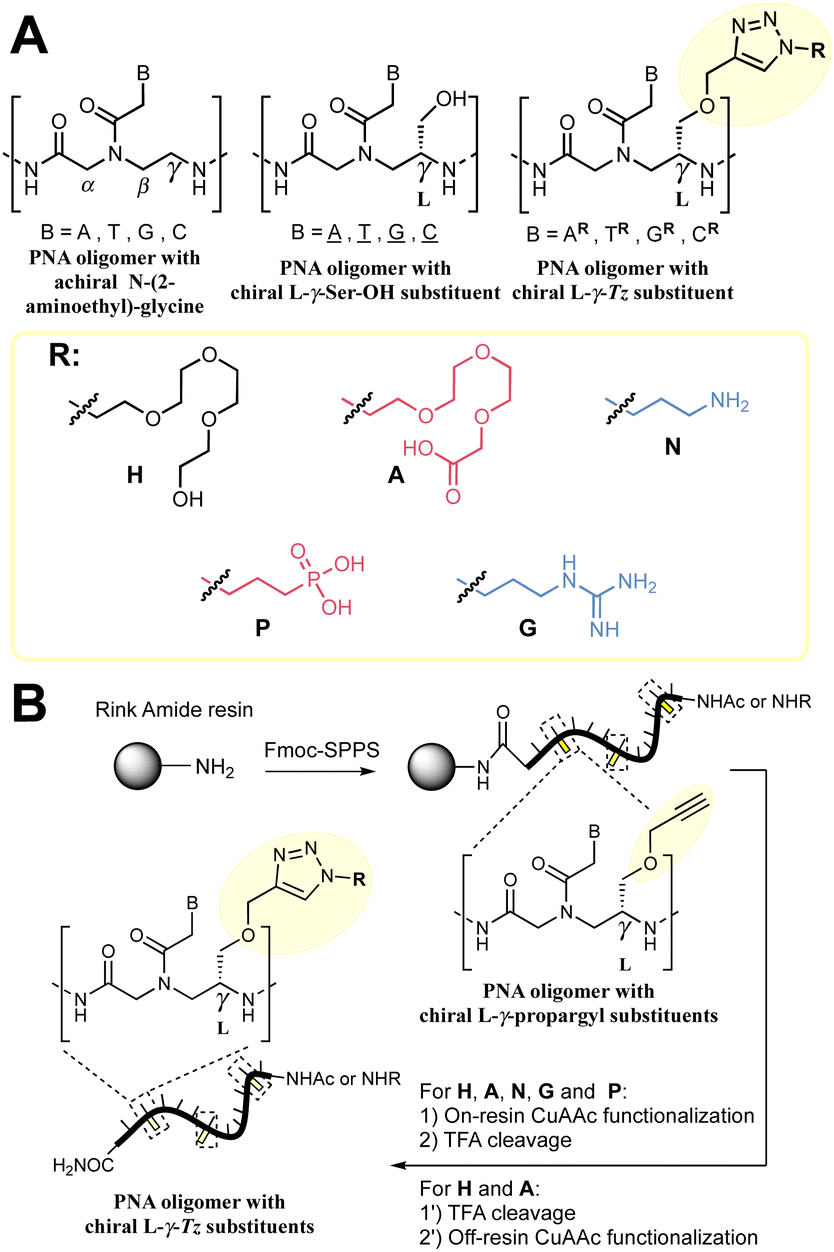 | ||
| Fig. 1 (A) Summary of the nomenclature of PNA structures used in this work together with the γ-Tz modifications. (B) Workflow followed for the synthesis γ-Tz PNAs on or off-resin. | ||
We recently reported the synthesis of a γ-modified PNA with a propargyl ether side chain to facilitate late-stage functionalization of PNA oligomers by CuAAC (γ-Tz, Fig. 1B);52 however, the impact of the ensuing triazole functionality on the hybridization properties was not studied in detail. Herein, we report this analysis and revisit the impact of charges on duplex stability with a comparison of PNAs bearing cationic, anionic, and neutral modifications using thermal stability and kinetics of hybridization for both PNA:DNA and PNA:PNA duplexes.52
Results and discussion
We set out to compare the affinity of PNAs bearing one to four γ-modifications covering different physicochemical space (Fig. 1A), a PEG chain as a hydrophilic neutral modification (H), the same PEG chain terminating with a carboxylic acid (A) rather than a hydroxyl group, a phosphonic acid (P), an amino group (N) and a guanidino group (G). The different PNAs were synthesised by traditional Fmoc-based SPPS and functionalized by CuAAc prior to cleavage from the resin (Fig. 1B, the sequence of each PNA is shown in the figure it is reported in, see Fig S1 for the explicit structure of each PNA†). Alternatively, the functionalisation could also be performed after cleavage from the resin (performed for H and A, Fig. 1B). Both approaches afforded the desired product as a single major product. Traditionally, the melting temperatures of hybridization duplexes are measured via the change in absorption related to π → π* transitions of the nucleobases (260 nm) which shifts upon duplex formation due to the hydrogen-bond network. However, in our experience, this technique does yield inflection points that are as sharp with PNA as with DNA. We opted to include a fluorophore (FITC) or quencher (Dabcyl) on the PNA (introduced during the SPPS, see the ESI for full synthetic details†) to make use of FRET to measure the melting temperature (Fig. 2). This allows multiple measurements to be performed in parallel using a qPCR instrument and facilitates Tm measurements at lower concentrations. As shown in Fig. 2B, this approach yielded sharp inflection points for Tm assignment. Comparison between Tm measurements with the fluorophore on the PNA and quencher on the DNA or vice versa afforded nearly identical curves suggesting that nonspecific interactions between the fluorophore or quencher and the strand it is attached to are negligible (Fig. 2B). Comparison of Tm measurements in PBS pH 7.4 (NaCl 137 mM, KCl 2.7 mM, NaHPO4 10 mM, KH2PO4 1.8 mM) across different modifications (H, N, G, A, P) showed that all modifications afforded higher duplex stability relative to an unmodified (achiral) PNA, corroborating the generality of the benefits of γ-modifications36,37 (Fig. 2C). The effect was additive and PNAs bearing four γ-modifications afforded higher duplex stability than PNAs bearing a single γ-modification. The PNAs with cationic side chains (N, G) had the largest benefit and the PNAs with negative side chains (A, P) had the smallest gain. These results corroborate the prior study reported by Heemstra and coworkers46 comparing the impact of three types of modification at the γ-position using PNA synthetically derived from alanine, lysine and aspartic acid. The data reported herein shows that the nature of the cation or anion does not have a profound impact (N vs. G or A vs. P) on this trend. Importantly, a thorough analysis of the impact of a mismatch showed that the gain in duplex stability does not come at the detriment of selectivity; the penalty for the single base pair mismatch (ΔTm) ranged from −24.0 to −24.2 °C in achiral PNA, −24.0 to −26 °C for H1 (1 sidechain modification) and −24.8 to −25.4 °C for G1 (Fig. S2†). Collectively, the data shows that while a negative charge on the PNA is detrimental to the DNA:PNA duplex stability, as could be expected on the basis of electrostatic repulsion,1,46 the effect is inferior to the benefit conferred by the preorganisation afforded by the γ-modification. However, the impact of charges had not been investigated for PNA:PNA duplexes. As expected based on the chirality-induced preorganization of γ-modified PNAs, higher duplex stability was observed with increasing numbers of modifications (Fig. 2C). Surprisingly, the largest benefit was observed for the anionic PNAs (A and P), with the duplex bearing four carboxylate groups (A4) on each strand gaining 9.9 °C in duplex stability relative to the achiral PNA and 3.7 °C relative to the PEG-modified PNA (H4, Fig. 2C). While this observation does not mirror what is seen with DNA:PNA, the density of charges on PNA is significantly lower that on DNA where an anion is present on every position of the oligonucleotide vs. four positions out of twelve in the PNA duplexes studied here.
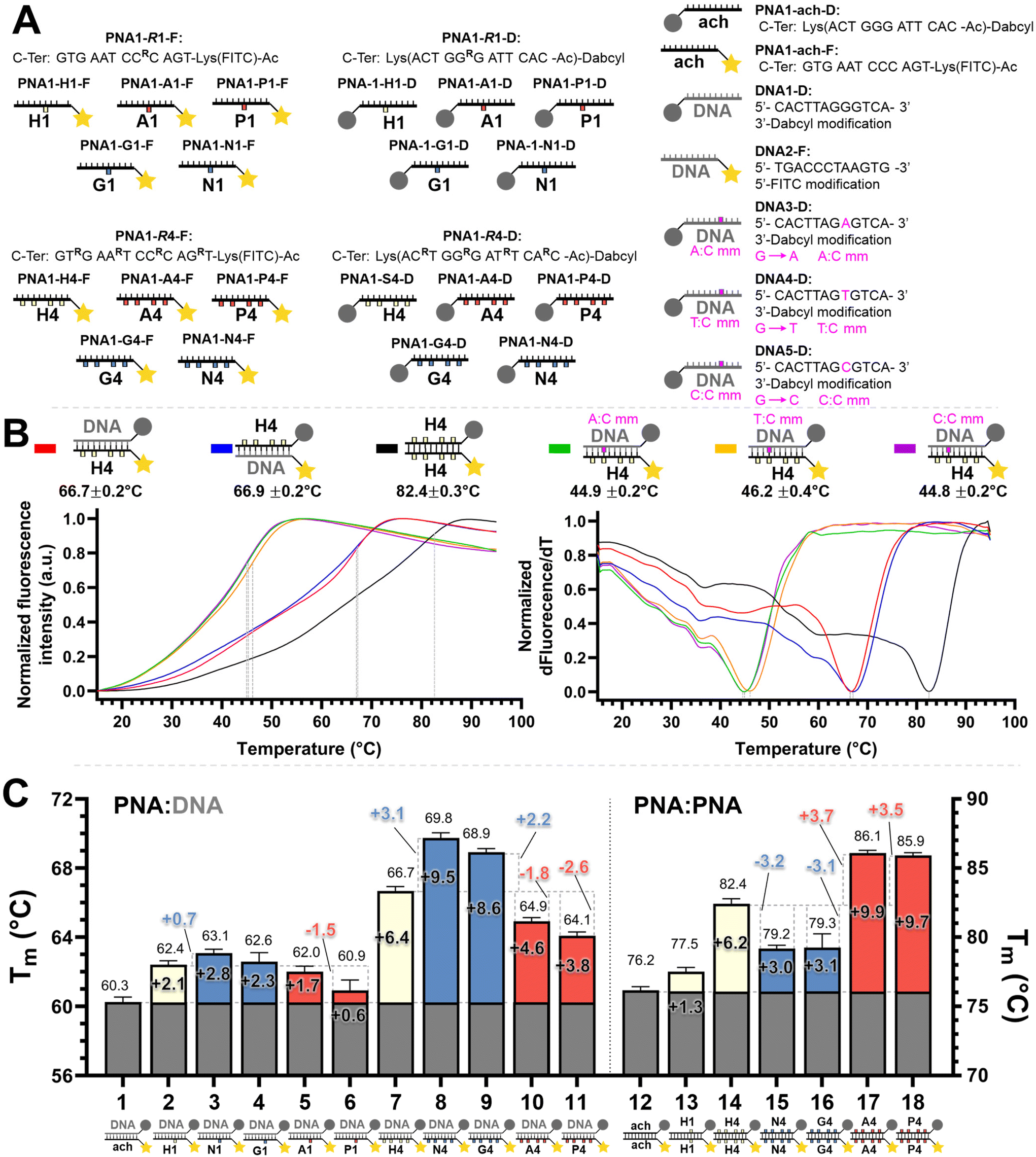 | ||
| Fig. 2 (A) Summary of the oligonucleotide sequences used for FRET melting temperature measurements. (B) Example of normalized melting profiles for the PNA1-H4-F and PNA1-H4-D series, including the normalized first derivative of the melting profiles versus temperature. Dotted lines indicate the inflexion points of the curves taken as the melting temperature (Tm) for each duplex. The colour legend is indicated next to the duplex. Conditions: 0.5 μM each oligonucleotide strand in 1× PBS pH 7.4 buffer. (C) PNA:DNA and PNA:PNA duplex melting temperatures. The colours in the bars denote the charge with the following legend: white: neutral, blue: positive, red: negative, as in Fig. 1. Conditions: 0.5 μM each oligonucleotide strand in 1× PBS pH 7.4 buffer. Further details of the explicit structures and protocols are available in the ESI.† Data presented as the average of the individual Tm values obtained from 2 independent samples measured in triplicate (n = 6) and presented as the mean ± 95% CI (z = 1.96). | ||
We next investigated the kinetics of hybridization which is an important consideration in the designed hybridization-based circuitry13,21,53–58 and had not been thoroughly examined with γ-modified PNAs. As shown in Fig. 3, the affinities calculated from the kinetic data follow the same trends as observed for the Tm, namely a PNA with three γ-modified residues forms a more stable duplex with DNA than the corresponding PNA with a single modification (H3 vs. H1); cationic PNAs form more stable duplexes with DNA than anionic PNAs (N3 or G3 vs. A3 or P3). A PNA with a single nucleotide mismatch had no measurable affinity under these assay conditions. Despite the overall higher duplex stability of PNA:DNA duplexes relative to DNA:DNA duplexes, the rate of hybridization (kon) is overall slower. Surprisingly, the benefit of γ-modification in duplex stability is not reflected in the rate of association (kon) as could have been anticipated based on the chirality-induced preorganization that it confers (PNA2-ach vs. PNA2-H1 & H3) but rather it results in a slower rate of dissociation (koff). However, when comparing the effect of charges at the γ-position on the hybridization kinetics, the charge on the PNA impacts the association kinetics more (6-fold difference between P3 vs. G3) than the dissociation (2-fold difference between P3 vs. G3). The faster kon of cationic PNA is in-line with the findings of Windsor and co-workers29 but a direct comparison should be treated with caution because the charges were incorporated on the nucleobase (achiral backbone) in this prior study rather than at the γ-position of the backbone as in the present study.
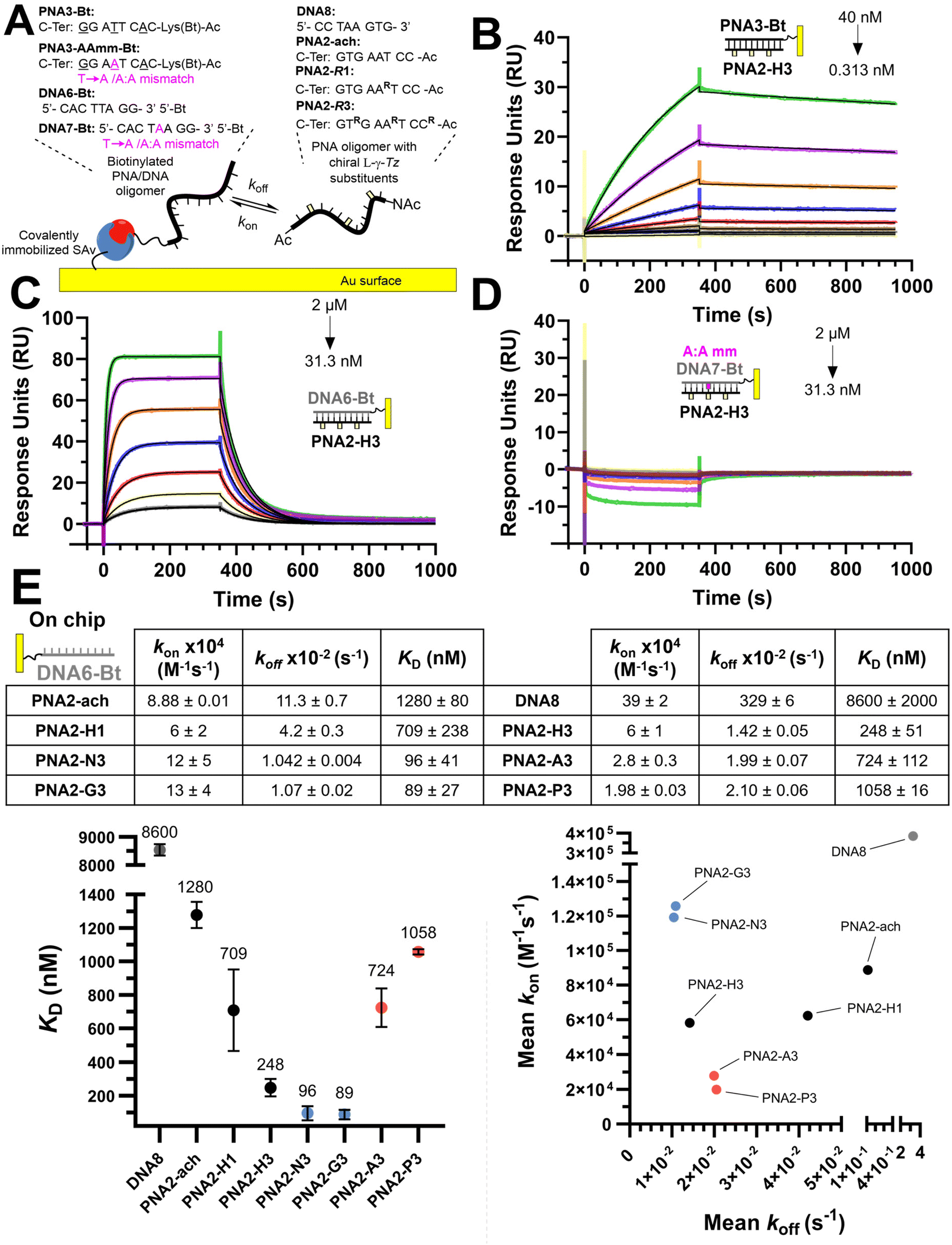 | ||
| Fig. 3 (A) Illustration of the experimental SPR set-up used for the oligonucleotide hybridization experiments. (B) SPR association and dissociation curves (coloured) with curves fitted to a 1:1 binding model (black lines) for PNA2-H3 and PNA3-Bt immobilized on the chip in a 2-fold dilution series from 40 nM to 0.313 nM. (C) SPR association and dissociation curves (coloured) with curves fitted to a 1:1 binding model (black lines) for PNA2-H3 and DNA6-Bt immobilized on the chip in a 2-fold dilution from 2 μM to 3.13 nM. (D) SPR association and dissociation curves (coloured) with curves fitted to a 1:1 binding model (black lines) for PNA2-H3 and DNA7-Bt (Tamm) immobilized on the chip in a 2-fold dilution from 2 μM to 3.13 nM. (E) SPR dissociation equilibrium constants (KD), association rates (ka) and dissociation rates (kd) for PNAs for DNA6-Bt immobilized on the chip. Data presented as the average of duplicate measurements (n = 2) and reported as the mean ± 95% CI (z = 1.96). Plotted error bars represent the standard deviation (SD). | ||
The same kinetic analysis for PNA:PNA duplexes (Fig. 4) showed that the rate of hybridization (kon) for the formation of a PNA:PNA duplex is comparable to that of a PNA:DNA duplex with a neutral achiral PNA (8 × 104 M−1 s−1 at 25 °C). The better duplex stability of PNA:PNA vs. PNA:DNA arises from the slower dissociation kinetics (1.74 × 10−4 s−1vs. 11.3 × 10−2 s−1). However, the presence of charges on one strand has negligible impact on hybridization when the other complementary strand is neutral. This comparison further supports the assertion that the differences in the kinetics of hybridization of charged PNAs to DNA is due to electrostatic interactions between the oligomers (i.e. slower kon for anionic PNA vs. cationic PNA). While γ-modifications improve the duplex stability of both PNA:DNA and PNA:PNA duplexes, it is important to note that the DNA:DNA duplex has the fastest association kinetics, despite the inherent repulsive interaction of the negative charges present in this duplex. This suggests that, while γ-modifications on PNA are beneficial, they do not preorganize the single strand oligomer in the duplex conformation as well as the phosphoryl ribose backbone does.
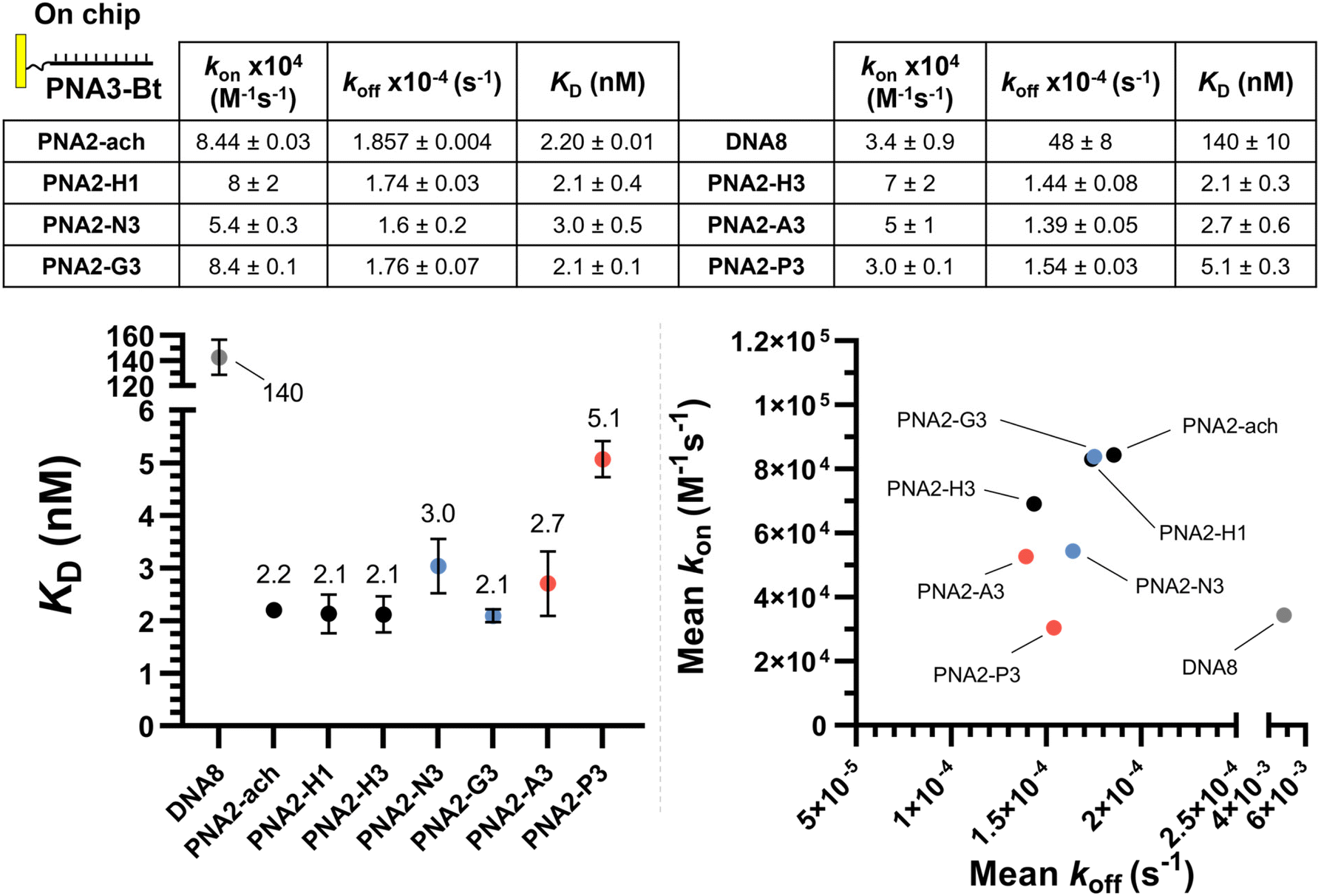 | ||
| Fig. 4 SPR dissociation equilibrium constants (KD), association rates (ka) and dissociation rates (kd) for PNAs for PNA3-Bt immobilized on the chip. Data presented as the average of duplicate measurements (n = 2) and reported as the mean ± 95% CI (z = 1.96). Plotted error bars represent the standard deviation (SD). | ||
We next compared the hybridization kinetics of PNA containing pseudo-complementary C–G (G-clamp and N-7 methyl guanine)28 in combination with γ-modifications (Fig. S4†). The pseudo-complementary nucleobases are cationic, thus their inclusion on the achiral PNA backbone without γ-modifications results in a cationic PNA. Concurring with our previous report,28 the combination of these nucleobases significantly improves the stability of a PNA:DNA duplex (KD 48.1 nM vs. 1280 nM). This improvement comes from both a faster association kinetics (4-fold) and slower dissociation kinetics (6-fold). Addition of γ-modifications resulted in the same affinity trend as before (neutral modifications lead to better duplex stability; negative charges slow down the association kinetics). It is interesting to note that in this case, the γ-modification with a neutral group (PNA2-pc-H3) led to slower association kinetics than the achiral backbone (PNA2-pc-ach).
Conclusions
The data reported provides a detailed landscape of the impact of modifications at the γ-position of PNA oligomers on hybridization stability and kinetics. This analysis was facilitated by the use of late-stage functionalization of the γ-position (γ-Tz modifications) with various side chains enabling a comparison across different charge statuses of PNA oligomers in their hybridization to DNA or PNA. All tested γ-Tz modifications were beneficial in duplex stability for both PNA:DNA and PNA:PNA duplexes. While a negatively charged PNA forms a less stable duplex with DNA than a positively charged PNA due to electrostatic interactions, the detriment of the negative charges is outweighed by the benefit of γ-modifications. The lower affinity of negatively charged PNA towards DNA comes predominantly from slower association kinetics. The higher stability of duplexes involving PNA comes from slower dissociation kinetics relative to the DNA:DNA duplex. The fact that γ-modified PNA hybridizes to DNA slower than DNA despite the lack of electrostatic repulsion suggests that further improvements in preorganization are possible.
Experimental section
General methods
All reagents and solvents for the organic synthesis were purchased from commercial sources and were used without further purification. Fmoc-Rink-Amide PEG AM resin for peptide synthesis was obtained from Iris Biotech. DNA oligonucleotides were bought from Eurogentec with the desired modifications and used without any further purification. HPLC purification was performed with an Agilent Technologies 1260 infinity HPLC using a ZORBAX 300SB-C18 column (9.4 × 250 mm). LC-MS spectra were recorded on a DIONEX Ultimate 3000 UHPLC (conditions for elution gradient: 0 min, A:B = 100:0; 4 min, A:B = 10:90; solution A: 0.01% aqueous TFA solution; solution B, 0.01% TFA in HPLC grade acetonitrile; flow rate: 0.750 mL min−1) with a Thermo LCQ Fleet Mass Spectrometer System using a PINNACLE DB C18 column (1.9 μm, 50 × 2.1 mm) operated in positive mode. All the LC-MS spectra were measured by electrospray ionization (ESI), linear gradient 0 to 100%. MALDI-TOF mass spectra were measured using a Bruker Daltonics Autoflex spectrometer operated in positive mode. The samples were analysed using a 2,5-dihydroxybenzoic acid (DHB) matrix. NMR spectra were acquired at the University of Geneva NMR platform (https://www.unige.ch/sciences/chiorg/nmr/) using either a 500 MHz Avance III Bruker NMR spectrometer equipped with a helium-cooled cryogenic 5 mm DCH 13C–1H/D Bruker probe, a 400 MHz Avance III HD NanoBay spectrometer equipped with a N2 Prodigy cryogenic 5 mm CPP BB(F)–H–D probe or a 300 MHz Avance III, HD NanoBay spectrometer, equipped with a 5 mm PA BBO, BB(F)–H–D probe. All 1H and 13C59 experiments were internally referenced with respect to either DMSO-d6 or CDCl3 solvent signals and acquired at 298 K. Retention times (RT) are given in minutes. Thin layer chromatography (TLC) was performed on plates of silica precoated with 0.25 mm Kieselgel 60 F254 from Merck. Flash chromatography was performed using silica gel SiliaFlash® P60 (230–400 mesh) from Silicycle. Automated solid-phase synthesis was carried out on an Intavis AG Multipep RS instrument. Concentrations of the PNA and DNA stocks were measured with a NanoDropRM 2000c at λ = 260 nm. High-resolution mass spectra (HRMS) were obtained on a Xevo G2 Tof spectrometer (ionization mode: ESI positive polarity; mobile phase: MeOH 100 μl min−1). All statistical data were calculated using GraphPad Prism and are presented as the mean value ± SD unless otherwise noted.
Surface plasmon resonance (SPR) measurements
SPR experiments were performed on a Biacore T200 instrument (GE Healthcare) at 25 °C in PBS-P+ buffer (10× stock from Cytiva Life Sciences, 28995084). Streptavidin was chemically immobilized via NHS/EDC coupling on a CM5 sensor chip (Cytiva Life Sciences, 29104988). Immobilization was performed following the standard Cytiva protocol (amine coupling of ligand to Biacore sensor chips) using streptavidin at 20 μg mL−1, contact time of 600 s and flow rate of 10 μL min−1. Subsequently, PNA3-Bt/PNA3-AAmm-Bt/DNA6-Bt/DNA7-Bt were independently immobilized on the previous streptavidin coated chip. Prior to immobilization, the two channels were conditioned with 1 M NaCl in 50 mM NaOH. After stabilization, the biotinylated compound was flowed over one of the flow cells of the sensor chip at a concentration of 50 nM solution in PBS-P+ at a flow rate of 10 μL min−1 with a response unit (RU) target of 100. The system (not including the flow cells) was washed with 50% isopropanol in 1 M NaCl and 50 mM NaOH after each ligand injection. Kinetic measurements consisted of injections (association 350 seconds, dissociation 650 seconds, flow rate: 30 μL min−1) of decreasing concentrations of PNAs (2-fold cascade dilutions from the starting concentration). The chip was regenerated between cycles by one injection of regeneration solution (50 mM NaOH) for 10 seconds at a flow rate of 20 μL min−1, followed by a 10 second stabilization period. Binding was measured as resonance units over time after blank subtraction, and the data interpreted using the Biacore T200 software, version 3.2. All measurements were repeated in duplicate for PNAs versus PNA3-Bt/DNA6-Bt and once for PNAs vs. PNA3-AAmm-Bt/DNA7-Bt. The KD values were calculated based on steady-state affinity (1:1 binding).
FRET melting temperature measurements
FRET melting temperature measurements were performed in a parallel fashion in Hard-Shell® 96-Well PCR Plates, low profile, thin wall, skirted, black/white (BioRad, #HSP9665) using a CFX Connect Real-Time PCR Detection System. 50 μL 0.5 μM of each oligomer in 1× PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4) was placed in each well. Each individual sample was annealed to 95 °C for 3 min before gradual cooling to 15 °C over the course of 2 h, then gradual heating up to 95 °C at a rate of 0.5 °C per min while monitoring the FITC fluorescence emission signal (excitation: 450–490 nm, emission: 515–530 nm). Then, samples were kept at 95 °C for 3 min before again gradual cooling to 15 °C over the course of 2 h, followed by gradual heating up to 95 °C at a rate of 0.5 °C per min while monitoring the FITC fluorescence emission signal. This iteration was repeated once more with a total of three measurements per individual sample, each sample was done in duplicate. For each individual curve, the first derivative of the fluorescence emission of FITC versus temperature was calculated, and the temperature with the minimum value of the first derivative was assigned as the melting temperature (Tm), being the inflexion point of the melting curve. Data were presented as the average of the 6 individual Tm values obtained from 2 independent samples measured in triplicate and presented as the mean ± 95% CI (z = 1.96).Click on/off-resin after SPPS
After Fmoc-SPPS, the Fmoc-Rink-Amide PEG AM resin (0.33 mmol g−1, 1.65 μmol) was cleaved with TFA for 2 h at room temperature. The filtrates were added to cold ether and following centrifugation, the ethereal was layer discarded. The resulting pellet was air dried. Then, it was dissolved in DMSO/H2O 1:1 and the crude propargyl PNA concentration was assessed by absorption. Typically, 100 μL 2 mM crude PNA in DMSO/H2O 1:1 (0.2 μmol), was diluted with 57 μL DMSO. Then, 41 μL of the above Cu-click mixture [2 μmol THPTA, 2 μmol CuSO4, 4 μmol NaAsc] was added and vortexed before adding 2 μL 100 mM in DMSO of either Az-H or Az-A (2 μmol) making a total volume of 200 μL. The mixture was left overnight at room temperature before HPLC purification by direct injection of the crude oligomers.
Synthesis protocols for compounds Az-H/A/N/G/P
1H NMR (400 MHz, CDCl3): δ 3.87 (s, 1H), 3.70–3.66 (m, 2H), 3.64–3.60 (m, 12H), 3.60–3.53 (m, 2H), 3.35 (t, J = 5.1, 5.1 Hz, 2H).
1H NMR (400 MHz, CDCl3): δ 9.69 (br. s, 1H), 4.15 (s, 2H), 3.73–3.61 (m, 10H), 3.35 (t, J = 5.2, 5.2 Hz, 2H).
R f: 0.70 in 1/3 EtOAc/pentane. UV inactive and stains yellow with KMnO4 stain. HR-MS/TOF-MS-ES+: m/z expected for [M + Na]+: 223.1171, m/z found: 223.1181. 1H NMR (300 MHz, CDCl3): δ 4.66 (br. s, 1H), 3.35 (t, J = 6.7 Hz, 2H), 3.26–3.15 (m, 2H), 1.76 (p, J = 6.7 Hz, 2H), 1.44 (s, 9H). 13C NMR (75 MHz, CDCl3): δ 156.1, 79.6, 49.3, 38.2, 29.4, 28.5.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and in the ESI.† The raw data have been deposited on Zenodo (https://doi.org/10.5281/zenodo.11257281).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Sofia Barluenga for all the HR-MS measurements and Millicent Dockerill for the assistance through the SPR measurements. This work was supported, in part, by the Swiss National Science Foundation (219316).References
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 566–568 CrossRef CAS PubMed.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CrossRef CAS PubMed.
- M. Lyu, L. G. Kong, Z. L. Yang, Y. T. Wu, C. E. McGhee and Y. Lu, J. Am. Chem. Soc., 2021, 143, 9724–9728 CrossRef CAS PubMed.
- G. C. Gavins, K. Gröger, M. D. Bartoschek, P. Wolf, A. G. Beck-Sickinger, S. Bultmann and O. Seitz, Nat. Chem., 2021, 13, 15 CrossRef CAS PubMed.
- P. M. Sabale, M. Imiolek, P. Raia, S. Barluenga and N. Winssinger, J. Am. Chem. Soc., 2021, 143, 18932–18940 CrossRef CAS PubMed.
- I. Todkari, M. K. Gupta and K. N. Ganesh, Chem. Commun., 2022, 58, 4083–4086 RSC.
- N. G. Economos, E. Quijano, K. E. W. Carufe, J. D. R. Perera and P. M. Glazer, Nucleic Acids Res., 2022, 50, e59 CrossRef CAS PubMed.
- P. Picchetti, S. Volpi, M. Rossetti, M. D. Dore, T. Trinh, F. Biedermann, M. Neri, A. Bertucci, A. Porchetta, R. Corradini, H. Sleiman and L. De Cola, J. Am. Chem. Soc., 2023, 145, 22896–22902 CrossRef CAS PubMed.
- Y. Altrichter, P. Bou-Dib, C. Kuznia and O. Seitz, J. Pept. Sci., 2023, 29, e3477 CrossRef CAS PubMed.
- H. C. Zheng, V. Clausse, H. Amarasekara, S. J. Mazur, I. Botos and D. H. Appella, JACS Au, 2023, 3, 1952–1964 CrossRef CAS PubMed.
- S. N. Oyaghire, E. Quijano, J. D. R. Perera, H. K. Mandl, W. M. Saltzman, R. Bahal and P. M. Glazer, Cell Rep. Phys. Sci., 2023, 4, 101584 CrossRef PubMed.
- Y. F. Liang, S. Willey, Y. C. Chung, Y. M. Lo, S. Q. Miao, S. Rundell, L. C. Tu and D. Bong, Nat. Commun., 2023, 14, 2987 CrossRef CAS PubMed.
- M. Dockerill, D. J. Ford, S. Angerani, I. Alwis, L. J. Dowman, J. Ripoll-Rozada, R. E. Smythe, J. S. T. Liu, P. J. B. Pereira, S. P. Jackson, R. J. Payne and N. Winssinger, Nat. Biotechnol., 2024 DOI:10.1038/s41587-024-02209-z.
- O. Tepper, D. H. Appella, H. C. Zheng, R. Dzikowski and E. Yavin, ACS Sens., 2024, 9, 1458–1464 CrossRef CAS PubMed.
- L. De Paepe, E. Cadoni, A. Manicardi and A. Madder, Methods, 2023, 218, 210–223 CrossRef CAS PubMed.
- E. Cadoni, F. Pennati, P. Muangkaew, J. Elskens, A. Madder and A. Manicardi, RSC Chem. Biol., 2022, 3, 1129–1143 RSC.
- P. E. Nielsen, Acc. Chem. Res., 1999, 32, 624–630 CrossRef CAS.
- J. Saarbach, P. M. Sabale and N. Winssinger, Curr. Opin. Chem. Biol., 2019, 52, 112–124 CrossRef CAS PubMed.
- S. Barluenga and N. Winssinger, Acc. Chem. Res., 2015, 48, 1319–1331 CrossRef CAS PubMed.
- C. Zambaldo, S. Barluenga and N. Winssinger, Curr. Opin. Chem. Biol., 2015, 26, 8–15 CrossRef CAS PubMed.
- E. E. Watson, S. Angerani, P. M. Sabale and N. Winssinger, J. Am. Chem. Soc., 2021, 143, 4467–4482 CrossRef CAS PubMed.
- A. Das and B. Pradhan, Chem. Biol. Drug Des., 2021, 97, 865–892 CrossRef CAS PubMed.
- C. Ausín, J. A. Ortega, J. Robles, A. Grandas and E. Pedroso, Org. Lett., 2002, 4, 4073–4075 CrossRef PubMed.
- S. A. Thadke, J. D. R. Perera, V. M. Hridya, K. Bhatt, A. Y. Shaikh, W. C. Hsieh, M. S. Chen, C. Gayathri, R. R. Gil, G. S. Rule, A. Mulcherjee, C. A. Thornton and D. H. Ly, Biochemistry, 2018, 57, 2094–2108 CrossRef CAS PubMed.
- S. A. Thadke, V. M. Hridya, J. D. R. Perera, R. R. Gil, A. Mukherjee and D. H. Ly, Commun. Chem., 2018, 1, 79 CrossRef PubMed.
- M. Hibino, Y. Aiba and O. Shoji, Chem. Commun., 2020, 56, 2546–2549 RSC.
- S. S. Moriya, H. Shibasaki, M. Kohara, K. Kuwata, Y. Imamura, Y. Demizu, M. Kurihara, A. Kittaka and T. Sugiyama, Bioorg. Med. Chem. Lett., 2021, 39, 127850 CrossRef CAS PubMed.
- M. López-Tena, L. Farrera-Soler, S. Barluenga and N. Winssinger, JACS Au, 2023, 3, 449–458 CrossRef PubMed.
- F. Podlaski, S. Cornwell, K. Y. Wong, B. Mckittrick, J. H. Kim, D. R. Jung, Y. Jeon, K. B. Jung, P. Tolias and W. T. Windsor, ACS Omega, 2023, 8, 33426–33436 CrossRef CAS PubMed.
- M. Lopez-Tena, S. K. Chen and N. Winssinger, Bioconjugate Chem., 2023, 34, 111–123 CrossRef CAS PubMed.
- M. Cantin, R. Schutz and C. J. Leumann, Tetrahedron Lett., 1997, 38, 4211–4214 CrossRef CAS.
- R. Schütz, M. Cantin, C. Roberts, B. Greiner, E. Uhlmann and C. Leumann, Angew. Chem., Int. Ed., 2000, 39, 1250–1253 CrossRef.
- M. Hollenstein and C. J. Leumann, J. Org. Chem., 2005, 70, 3205–3217 CrossRef CAS PubMed.
- V. Clausse, H. C. Zheng, H. Amarasekara, M. Kruhlak and D. H. Appella, Nucleic Acids Res., 2022, 50, 10839–10856 CrossRef CAS PubMed.
- C. Suparpprom and T. Vilaivan, RSC Chem. Biol., 2022, 3, 648–697 RSC.
- J. I. Yeh, B. Shivachev, S. Rapireddy, M. J. Crawford, R. R. Gil, S. Du, M. Madrid and D. H. Ly, J. Am. Chem. Soc., 2010, 132, 10717–10727 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, S. Rapireddy, B. M. Frezza, C. Gayathri, R. R. Gil and D. H. Ly, J. Am. Chem. Soc., 2006, 128, 10258–10267 CrossRef CAS PubMed.
- E. A. Englund and D. H. Appella, Org. Lett., 2005, 7, 3465–3467 CrossRef CAS PubMed.
- M. C. de Koning, L. Petersen, J. J. Weterings, M. Overhand, G. A. van der Marel and D. V. Filippov, Tetrahedron, 2006, 62, 3248–3258 CrossRef CAS.
- E. A. Englund and D. H. Appella, Angew. Chem., Int. Ed., 2007, 46, 1414–1418 CrossRef CAS PubMed.
- F. Totsingan, T. Tedeschi, S. Sforza, R. Corradini and R. Marchelli, Chirality, 2009, 21, 245–253 CrossRef CAS PubMed.
- B. Sahu, V. Chenna, K. L. Lathrop, S. M. Thomas, G. Zon, K. J. Livak and D. H. Ly, J. Org. Chem., 2009, 74, 1509–1516 CrossRef CAS PubMed.
- S. Pensato, M. Saviano, N. Bianchi, M. Borgatti, E. Fabbri, R. Gambari and A. Romanelli, Bioorg. Chem., 2010, 38, 196–201 CrossRef CAS PubMed.
- B. Sahu, I. Sacui, S. Rapireddy, K. J. Zanotti, R. Bahal, B. A. Armitage and D. H. Ly, J. Org. Chem., 2011, 76, 5614–5627 CrossRef CAS PubMed.
- C. Avitabile, L. Moggio, G. Malgieri, D. Capasso, S. Di Gaetano, M. Saviano, C. Pedone and A. Romanelli, PLoS One, 2012, 7, e35774 CrossRef CAS PubMed.
- H.-A. Tajmir-Riahi, N. T. S. De Costa and J. M. Heemstra, PLoS One, 2013, 8, e58670 CrossRef PubMed.
- D. R. Jain and K. N. Ganesh, J. Org. Chem., 2014, 79, 6708–6714 CrossRef CAS PubMed.
- Y. Kirillova, N. Boyarskaya, A. Dezhenkov, M. Tankevich, I. Prokhorov, A. Varizhuk, S. Eremin, D. Esipov, I. Smirnov and G. Pozmogova, PLoS One, 2015, 10, e0140468 CrossRef PubMed.
- I. Sacui, W. C. Hsieh, A. Manna, B. Sahu and D. H. Ly, J. Am. Chem. Soc., 2015, 137, 8603–8610 CrossRef CAS PubMed.
- J. Saarbach, D. Masi, C. Zambaldo and N. Winssinger, Bioorg. Med. Chem., 2017, 25, 5171–5177 CrossRef CAS PubMed.
- P. Bhingardeve, B. R. Madhanagopal and K. N. Ganesh, J. Org. Chem., 2020, 85, 13680–13693 CrossRef CAS PubMed.
- M. López-Tena, E. E. Watson, P. Romanens and N. Winssinger, Helvetica, 2023, 106, e202300110 CrossRef.
- D. Chang, E. Lindberg and N. Winssinger, J. Am. Chem. Soc., 2017, 139, 1444–1447 CrossRef CAS PubMed.
- W. C. Hsieh, G. R. Martinez, A. Wang, S. F. Wu, R. Chamdia and D. H. Ly, Commun. Chem., 2018, 1, 89 CrossRef.
- K. T. Kim, S. Angerani, D. L. Chang and N. Winssinger, J. Am. Chem. Soc., 2019, 141, 16288–16295 CrossRef CAS PubMed.
- S. Angerani and N. Winssinger, J. Am. Chem. Soc., 2020, 142, 12333–12340 CrossRef CAS PubMed.
- K. T. Kim, S. Angerani and N. Winssinger, Chem. Sci., 2021, 12, 8218–8223 RSC.
- I. Dhami, S. A. Thadke and D. H. Ly, J. Org. Chem., 2022, 87, 13873–13881 CrossRef CAS PubMed.
- R. Binette, M. Desgagne, C. Theaud and P. L. Boudreault, Molecules, 2022, 27, 2788 CrossRef CAS PubMed.
- N. Khiar, M. P. Leal, R. Baati, C. Ruhlmann, C. Mioskowski, P. Schultz and I. Fernández, Chem. Commun., 2009, 4121–4123, 10.1039/b904717d.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization data. See DOI: https://doi.org/10.1039/d4ob00887a |
| This journal is © The Royal Society of Chemistry 2024 |