
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparative scale Achmatowicz and aza-Achmatowicz rearrangements catalyzed by Agrocybe aegerita unspecific peroxygenase†
Balázs
Pogrányi
a,
Tamara
Mielke
a,
Alba
Díaz Rodríguez
b,
Jared
Cartwright
c,
William P.
Unsworth
 *a and
Gideon
Grogan
*a
*a and
Gideon
Grogan
*a
aDepartment of Chemistry, University of York, York YO10 5DD, UK. E-mail: william.unsworth@york.ac.uk; gideon.grogan@york.ac.uk
bGSK Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire, SG1 2NY, UK
cDepartment of Biology, University of York, York YO10 5DD, UK
First published on 15th July 2024
Abstract
The unspecific peroxygenase (UPO) from Agrocybe aegerita (rAaeUPO-PaDa-I-H) is an effective and practical biocatalyst for the oxidative expansion of furfuryl alcohols/amines on a preparative scale, using the Achmatowicz and aza-Achmatowicz reaction. The high activity and stability of the enzyme, which can be produced on a large scale as an air-stable lyophilised powder, renders it a versatile and scalable biocatalyst for the preparation of synthetically valuable 6-hydroxypyranones and dihydropiperidinones. In several cases, the biotransformation out-performed the analogous chemo-catalysed process, and operates under milder and greener reaction conditions.
Introduction
The oxidative rearrangement of furyl-alcohols to dihydropyranones – generally termed the Achmatowicz reaction1 – is one of the most important and widely used furan transformations in organic chemistry (e.g.1 → 3, Scheme 1).2,3 Most synthetic Achmatowicz reactions rely on the use of strong stoichiometric chemical oxidants4,5 such as mCPBA, which in this case promotes the reaction via an initial epoxidation (to form 2) and subsequent rearrangement as shown (Scheme 1). Various other Achmatowicz-type methods are also known using other oxidants, e.g. employing electrochemistry,6 singlet oxygen7 and photocatalysis.8 Apart from the latter, catalytic methods performed under ambient conditions are rare.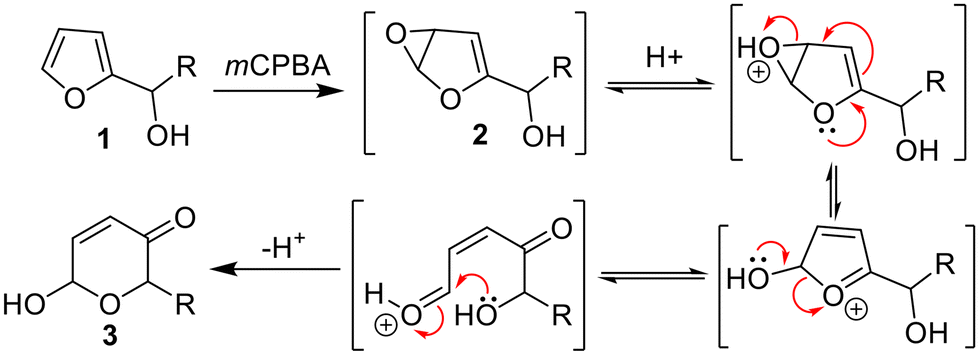 | ||
| Scheme 1 The Achmatowicz reaction promoted by meta-chloroperbenzoic acid. | ||
Biocatalysis offers significant advantages compared with chemical methods, to enable milder and more sustainable Achmatowicz reactions (Scheme 2). The first enzymatic Achmatowicz reaction reported was developed by Beifuss et al., and is based on the use of laccase from Trametes versicolor and hydroxy-TEMPO as a redox mediator (e.g.4 → 5a, Scheme 2A).9 After this, Deska et al. reported that C. fumago chloroperoxidase (CfuCPO), catalyzes the Achmatowicz reaction on a range of substrates and with moderate enantioselectivity.10 A subsequent study by the same group utilized CfuCPO, assisted by glucose oxidase (GOx), for the generation of peracid from an acetic acid buffer, for Achmatowicz reactions of the type 4 → 5b (Scheme 2B).11 The authors also demonstrated that C. antarctica lipase B (CAL-B) can also be used to generate peracetic acid from the acetate present in the buffer and the added H2O2 (e.g.4 → 5c; Scheme 2C).11 Each of these reactions should proceed through an initial oxidation of the furan and subsequent rearrangement, via a mechanism similar to that depicted in Scheme 1.
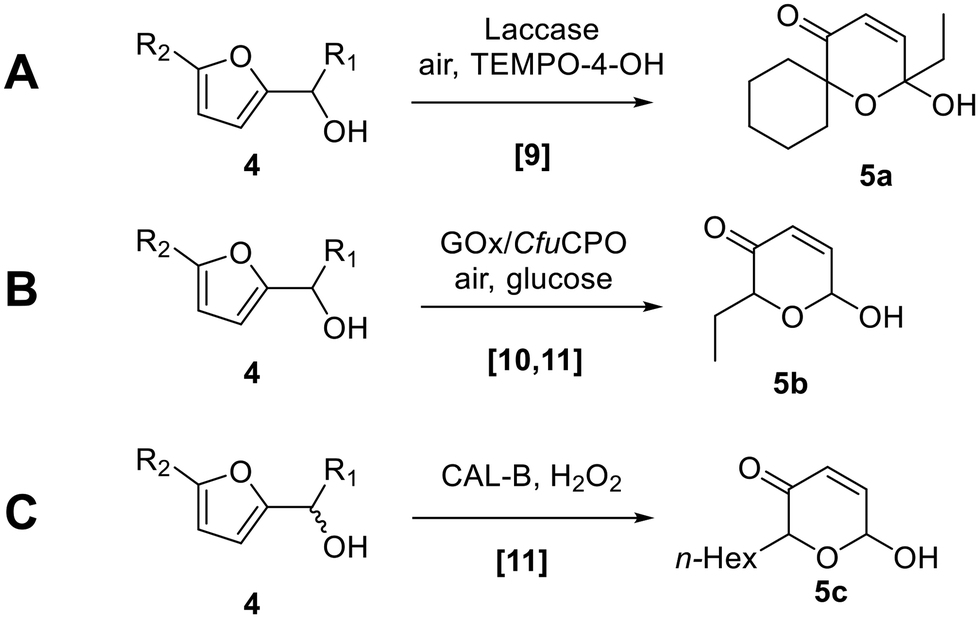 | ||
| Scheme 2 Biocatalytic Achmatowicz protocols. | ||
The aza-Achmatowicz reaction for the transformation of aza-analogues such as 6 into products 8 is another important synthetic transformation, with a general reaction depicted in Scheme 3A.12 Biocatalytic aza-Achmatowicz reactions are rare however, although Hollman and co-workers have reported successful examples using the vanadium-dependent peroxidase from C. inaequalis (CinVPO) in the presence of H2O2 and KBr in aqueous media with ethanol.13 In this case, the reactive oxidant is likely to be hypohalous acid generated biocatalytically from KBr (Scheme 3B). Notably, reactions such as the formation of 5d were performed at up to 100s milligram scale, although relatively limited scope was demonstrated to date. The CAL-B-mediated in situ peracetic acid generation system developed by the Deska group can also be used to perform aza-Achmatowicz reactions in limited cases.11
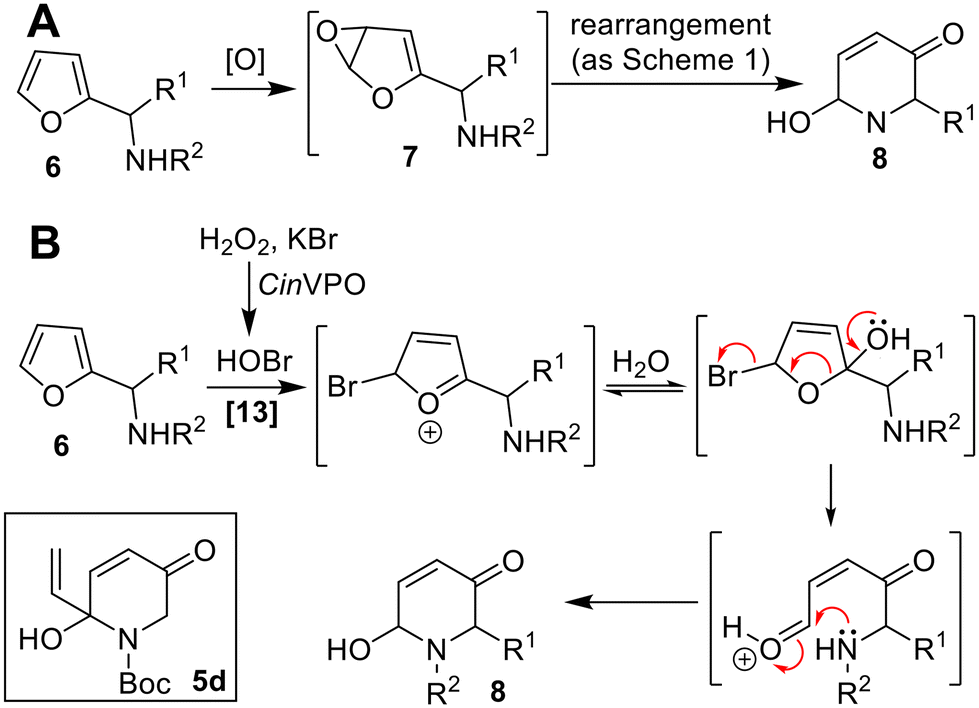 | ||
| Scheme 3 Biocatalytic aza-Achmatowicz reaction. | ||
Unspecific Peroxygenases (UPOs) have recently been shown to possess catalytic characteristics bridging those of cytochromes P450 (P450s) and haloperoxidases such as CfuCPO, as well as exhibiting superior stability and catalytic activity.14–17 In contrast to P450s however, these heme dependent enzymes are able to catalyze oxygenation reactions at the expense of only hydrogen peroxide as the external oxidant. These attributes, coupled with advances in heterologous expression,18–20 improved protocols for protein engineering21–23 and also methods for gradual hydrogen peroxide delivery that reduce oxidative stress on the enzyme,24–26 have established them as attractive biocatalysis for scalable oxygenation reactions. We reasoned therefore that UPOs could also be good biocatalysts for both Achmatowicz and aza-Achmatowicz reactions initiated by epoxidation, especially as UPOs have been shown previously to be capable of promoting the epoxidation of a range of alkenes aliphatic and alicyclic alkenes27 and styrene derivatives.28 Therefore, in this study, both reaction classes were explored, using a variant of the PaDa-I mutant of Agrocybe aegerita UPO originally described by Alcalde and co-workers,18,19 which was expressed in Pichia pastoris using a modified vector and previously used by our groups as rAaeUPO-PaDa-I-H.29–32 Our studies confirm that rAaeUPO-PaDa-I-H is an effective biocatalyst for both transformations (Scheme 4). Both reaction series gave high synthetic yields and were readily scaled-up. The UPO reactions show an improved substrate scope over CfuCPO-catalyzed reactions, with 23 preparative scale reactions (up to 1 g scale) successfully demonstrated and further expand our knowledge of the ever-growing promiscuous activity of UPOs.
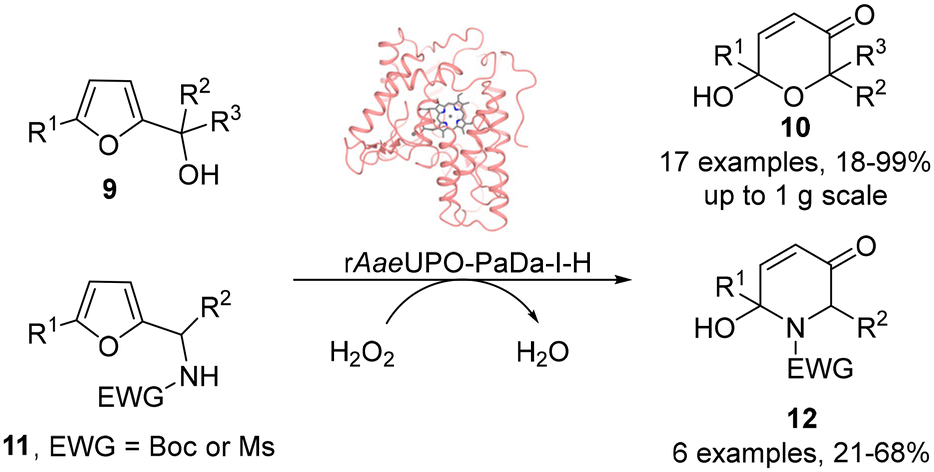 | ||
| Scheme 4 Preparative scale Achmatowicz and aza-Achmatowicz rearrangements catalyzed by rAaeUPO-PaDa-I-H in this study. | ||
Results and discussion
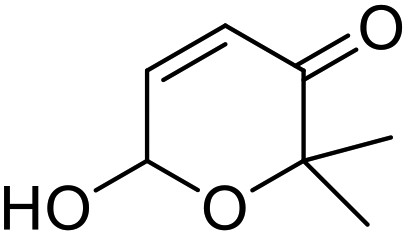
rAaeUPO-PaDa-I-H was expressed and lyophilized using our reported method.29 The 2-furylcarbinol substrates 9a–v used in this study were prepared using standard methods, with full synthetic details described in the ESI (ESI, section 7†). Synthetic standards for the Achmatowicz products 10a–i were also prepared from the 2-furylcarbinols via oxidation with N-bromosuccinimide (NBS) or potassium hypobromite (ESI, section 7†).We started by examining the reactions of 2-furylcarbinols 9a–i on small scale. The substrates (10 mM) were reacted with H2O2 (3.3 mM in a single portion, or 10 mM added in three portions at 10 min intervals) in the presence of rAaeUPO-PaDa-I-H, on 1 mL scale in a pH 5.5 buffer solution, with 10% t-BuOH as co-solvent at RT33 (Table 1). The reaction extracts were analysed by GC-FID. The results of biotransformations of the first substrates tested were somewhat disappointing; for example, the simplest furyl-carbinol 9a (entry 1), was not converted into the Achmatowicz product 10a and a complex mixture of side products was obtained. Substitution with an ethyl group on the furan 5-position (9b) resulted in a simpler product mixture, but aldehyde 13 was the only product identified (entries 2 and 3). 2-Furylcarbinol 9c was not oxidized by rAaeUPO-PaDa-I-H (entry 4). In this regard, rAaeUPO-PaDa-I-H is similar to CPO as that enzyme was also reported not to convert a 2,5-disubstituted substrate in the original report.10
|
|
|||
|---|---|---|---|
| Entry | Substrate | [H2O2]/mM | Producta |
| Reaction conditions: 10 mM substrate, 4.4 U mL−1rAaeUPO or 20 U mL−1 CPO, 1 mL total volume, 10% v/v tBuOH, 50 mM sodium citrate buffer (pH 5.5) at RT.33 Reactions were extracted with 2 × 1 mL EtOAc, dried over MgSO4 and analysed by GC-FID (HP-5MS column).a Yields based on analysis by GC-FID.b H2O2 was added dropwise by a syringe pump over 7 h. Control reactions, to which no enzyme was added, gave no conversion.34 | |||
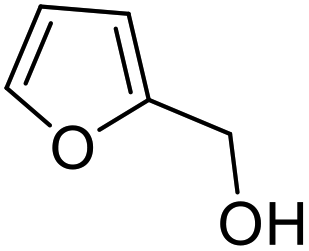
|
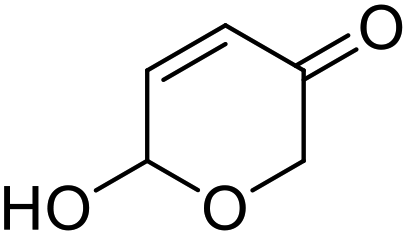
|
||
| 1 | 9a | 3.3 | 10a, 0% |
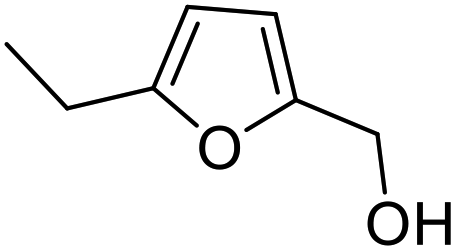
|
|
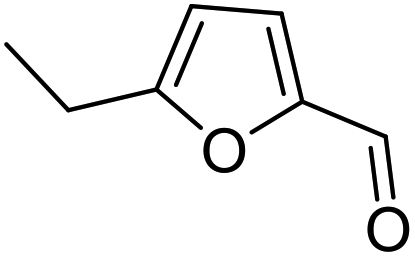
||
| 2 | 9b | 3.3 | 13, 29% |
| 3 | 10 | 13, 85% | |
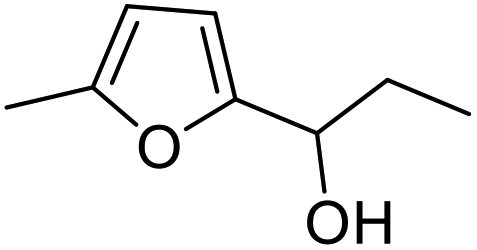
|
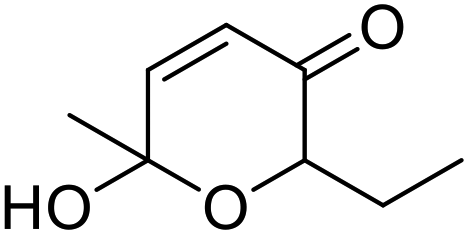
|
||
| 4 | 9c | 3.3 | 10c, 0% |
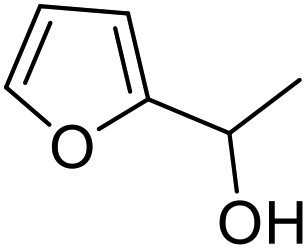
|
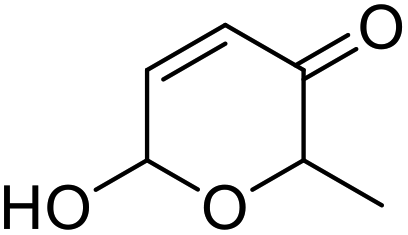
|
||
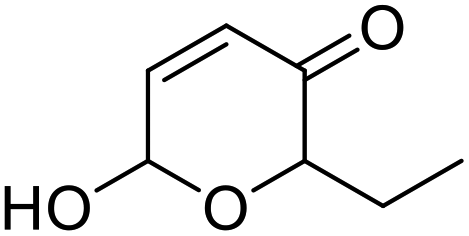
| 5 | 9d | 3.3 | 10d, 13% |
| 6 | 10 | 10d, 80% | |

|
|
||
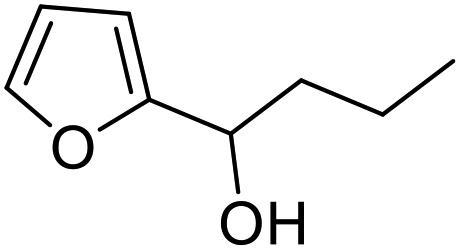
| 7 | 9e | 3.3 | 10e, 20% |
| 8 | 10 | 10e, 59% | |
|
|
||
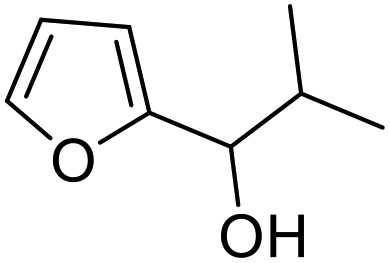
| 9 | 9f | 3.3 | 10f, 27% |
| 10 | 10 | 10f, 70% | |
|
|
||
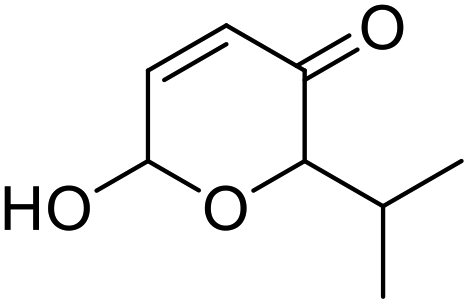
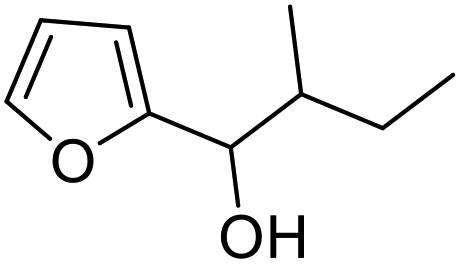
| 11 | 9g | 3.3 | 10g, 21% |
| 12 | 10 | 10g, 59% | |
|
|
||
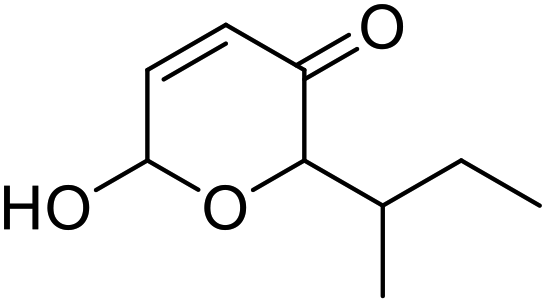
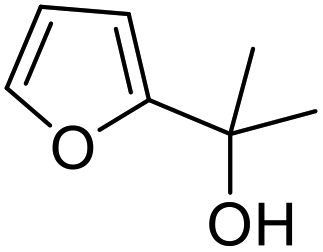
| 13 | 9h | 3.3 | 10h, 16% |
| 14 | 10 | 10h, 47% | |
|
|
||
| 15 | 9i | 3.3 | 10i, 30% |
| 16 | 10 | 10i, 84%b | |
However, more promisingly, rAaeUPO-PaDa-I-H successfully converted monosubstituted 2-furylcarbinols 9d–g into the analogous pyranones 10d–g (entries 5–16; the products were positively identified using authentic standards). This initial screen showed that less bulky furyl-carbinols are transformed more readily by the enzyme, with similar results obtained in published studies using CfuCPO as the biocatalyst.11 It is also noteworthy that, using the same H2O2 addition method, in our hands CfuCPO failed to convert any of 2-furylcarbinols 9d–i at 3.3 mM and 10 mM H2O2 loading (data not shown). This may be due to the instability of CfuCPO in the presence of the comparatively higher peroxide loadings used, and serves as a useful illustration of the practicality and robustness of rAaeUPO-PaDa-I-H. The >50% conversions observed in several cases show that the UPO is capable of converting both enantiomers of the chiral 2-furylcarbinols tested, at least to some degree. This observation, coupled to near-zero specific rotation measurements for the products (vide infra), suggests that achieving an enantioselective rAaeUPO-PaDa-I-H-catalysed Achmatowicz reaction through kinetic resolution of racemic 2-furylcarbinols is unlikely to be feasible using this enzyme.
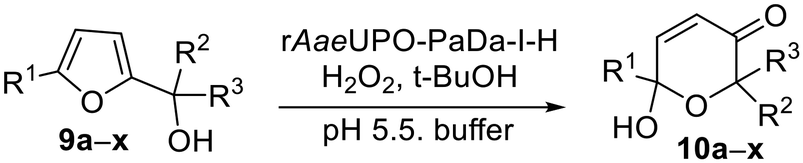
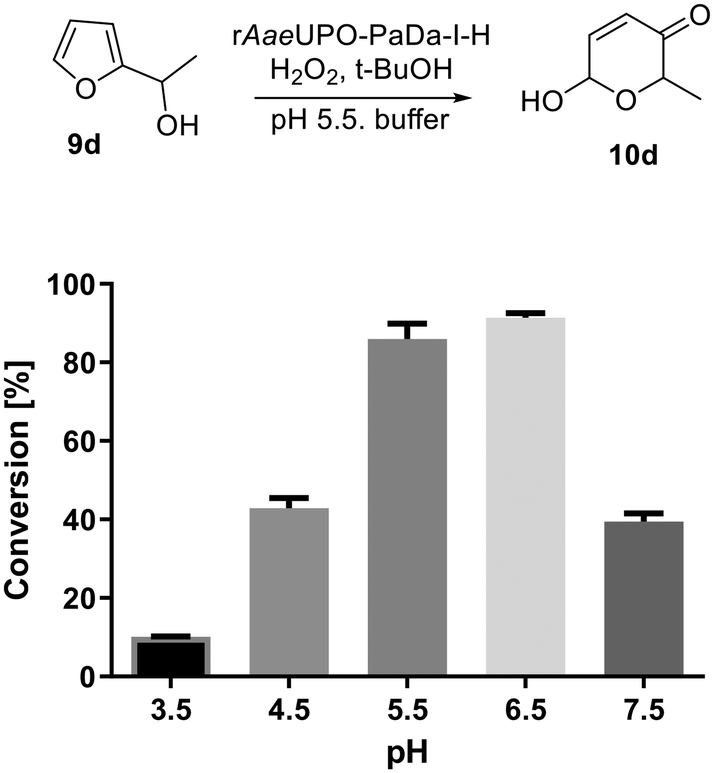
Next, we set out to optimize the reaction conditions prior to increasing the reaction scale. First, a pH screen was carried out using 1-(2-furyl)-ethanol 9d. A pH between 5.0 and 7.0 was found to be optimal for its conversion into 10d (Fig. 1), hence a pH 5.5 citrate buffer was taken forward and used in the preparative reactions.24 The effect of different co-solvents was also examined, with acetone identified as the best co-solvent based solely on conversion, especially at higher concentrations (Fig. 2).
 | ||
| Fig. 1 pH profile of the rAaeUPO-PaDa-I-H catalysed transformation of 9d into 10d. Reactions were performed in 150 μL total volume, extracted with 2 × 200 μL EtOAc and analysed by GC-FID on a HP-5MS column (30 m) at RT.33 Citrate buffers were used between pH 3.50 and 5.50 (50 mM) and above this KPi buffers (50 mM) were employed. Error bars show ±one standard deviation from the mean. | ||
 | ||
| Fig. 2 Effect of co-solvent on the rAaeUPO-PaDa-I-H catalysed transformation of 9d into 10d. Tests were performed in 150 μL total volume in sodium citrate buffer (pH = 5.50, 50 mM) at 10 mM substrate concentration at RT.33 Reactions were extracted with 2 × 200 μL EtOAc after 15 min and the extract was directly analysed by GC-FID (30 m HP-5MS column). Error bars indicate ±one standard deviation. MeCN – acetonitrile; DMF – dimethylformamide; DMSO – dimethylsulfoxide. | ||
However, t-BuOH performed similarly at 10% and 20% (v/v), and it was later found that product purification in the preparative reactions was simpler from reactions containing t-BuOH compared with those containing acetone. Therefore, t-BuOH was preferred for the preparative studies that follow (vide infra, Scheme 5).
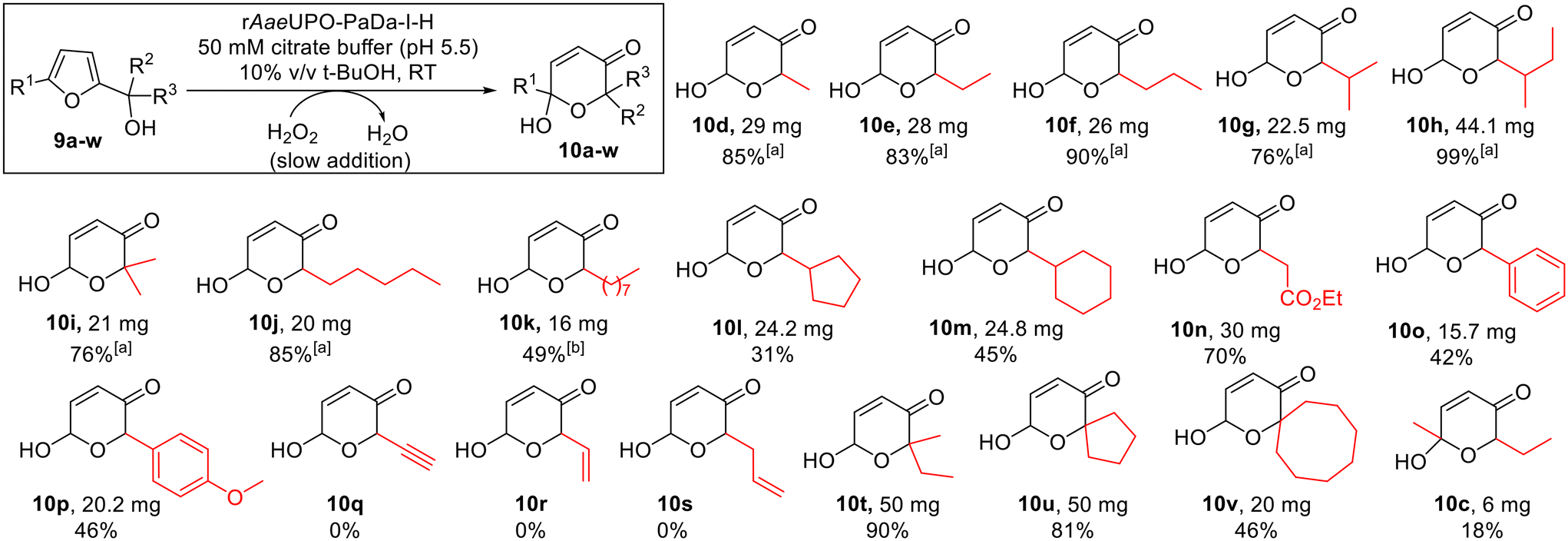 | ||
Scheme 5 Achmatowicz reactions promoted by rAaeUPO-PaDa-I-H and H2O2. Reaction conditions as shown in the box above (see ESI section 7† for more detail). 10 mM substrate concentration was used, with H2O2 supplied via syringe pump at 5 mM h−1 (2 h reaction time in total) at RT.33a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) No chromatographic purification was needed in these cases. bPerformed with 10 v/v % MeCN in place of t-BuOH. No chromatographic purification was needed in these cases. bPerformed with 10 v/v % MeCN in place of t-BuOH. | ||
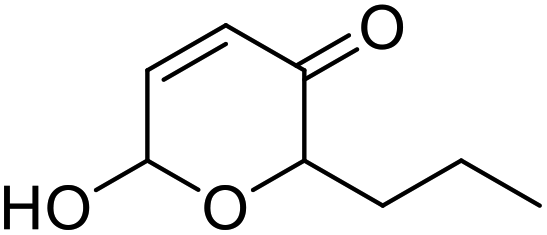
The optimised reaction conditions were then tested at preparative scale. A range of monosubstituted 2-furylcarbinols 9d–v was explored (Scheme 5), with a syringe pump used to control slow H2O2 addition at a rate of 5 mM h−1, at 10 mM substrate concentration. In all cases the reactions were run for 2 h at room temperature. Pleasingly, the preparative results for 2-furylcarbinols 9d–i were in line with the small scale experiments, with pyranones 10d–i all being formed and isolated in good to excellent yields. Longer chain homologues 10j and 10k were also prepared in the same way. In the case of 10k the co-solvent was switched to acetonitrile, as the pyranone product was found to be too poorly soluble when using t-BuOH. Notably, such bulky substrates were not reported to be transformed by CfuCPO in previous studies.11
Products containing bulkier cyclic hydrocarbon substituents (10l and 10m) and aryl (10o and 10p) were also obtained in acceptable yields, while ester-containing pyranone 10n was isolated in 70% yield. Conversely, alkyne and alkene containing starting materials 9q–9s were not converted into the expected products (10q–s), with unreacted 2-furylcarbinol starting material recovered in each case and no epoxidation of the double bond detected during the reactions. We postulated that this may be a consequence of enzyme inhibition by these substrates, and indeed, this notion was supported by the observation that 2-furylcarbinols 9r and 9s significantly reduced the conversion of 9e into 10e in inhibition studies (ESI section 4, Fig. S1†). However, a mechanism for this inhibition was not clear from these experiments. Bulky tertiary alcohol containing starting materials were well-tolerated, with pyranones 10t–v isolated in good yields using the standard protocol. Finally, pyranone 10c was obtained from 2,5-disubstituted starting material 9c, although the isolated yield was modest in this case (18%). This example is notable as 9c was not converted in the initial small-scale screen. This highlights the synthetic advantages that can be realised by using the slow H2O2 addition method.
Notably, in our hands, these biocatalytic Achmatowicz reactions using rAaeUPO-PaDa-I-H tended to give similar or superior yields to those obtained for the analogous reactions promoted by N-bromosuccinimide (NBS, ESI section 5†). The biocatalytic method also generally gave rise to much cleaner reaction mixtures; indeed, column chromatographic purification was not required in several cases. As expected, all of the pyranone products formed from chiral alcohols were obtained as mixtures of diastereoisomers (dr between 1:1 and 3:1; see ESI†). The dr of the products produced by the enzymatic method was the same as in the material synthesised using NBS, strongly indicating that these are simply thermodynamic outcomes associated with the relative energies of the two equilibrating diastereoisomers. It is therefore very unlikely that other UPOs would affect the dr of the products formed. However, it remains possible that the application of sequence diverse UPOs or engineered enzymes may be capable of delivering enantiomerically enriched products, via the kinetic resolution of chiral starting materials.
UPO-catalyzed aza-Achmatowicz reactions
To our knowledge, there have been few reports of biocatalytic aza-Achmatowicz reactions reported in the literature,11,13 and those examples relied on the formation of a diffusible oxidants by either lipase-catalysed perhydrolysis of acetate11 or the vanadium-dependent peroxidase from Curvularia inequalis (CinVPO).13 Based on our results with the standard Achmatowicz reaction, we reasoned that the active site of rAaeUPO-PaDa-I-H may permit the binding and direct oxidation of aza-Achmatowicz substrates within the enzyme active site. To probe this, we challenged the enzyme with a series of N-protected furan-2-ylmethanamine substrates (11a–f, Scheme 6A), on preparative scale, using the method established for the Achmatowicz reaction of 2-furylcarbinols (ESI section 5†).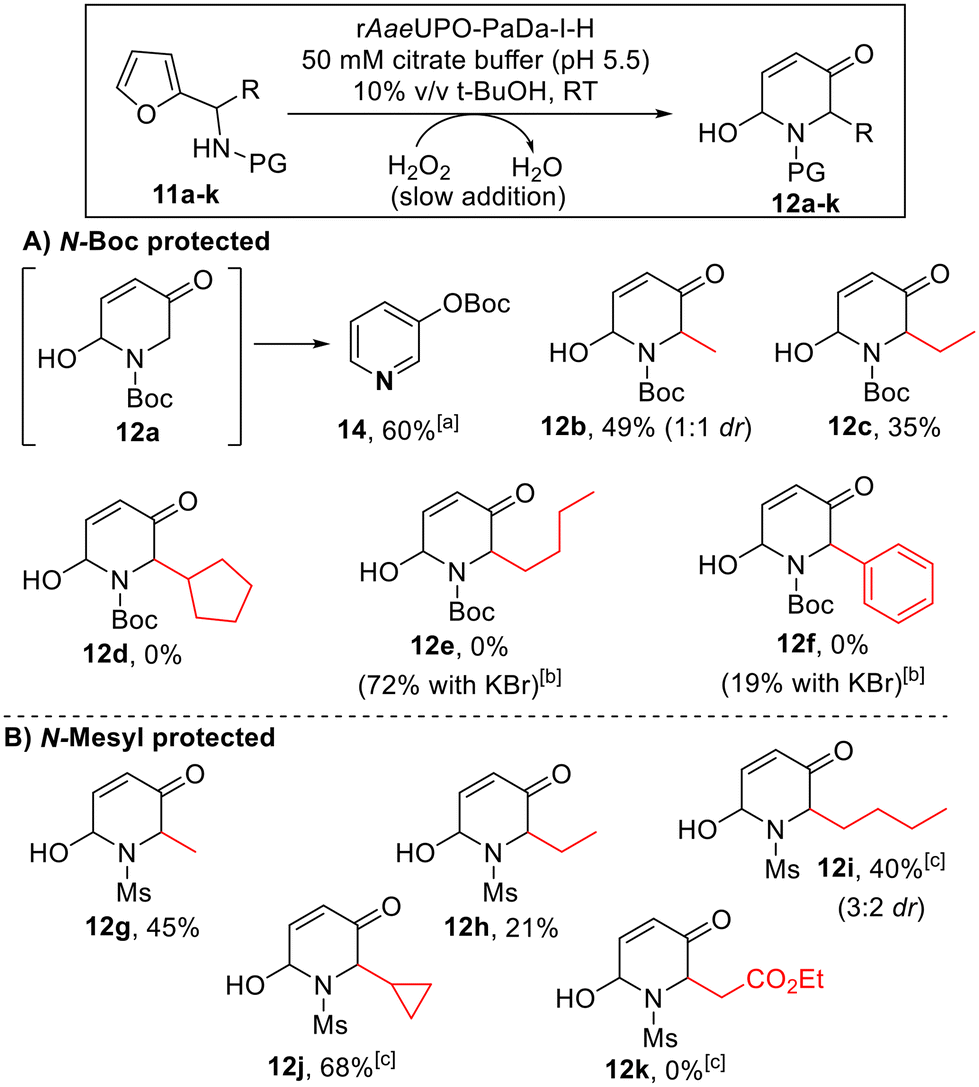 | ||
| Scheme 6 Aza-Achmatowicz reactions promoted by rAaeUPO-PaDa-I-H and H2O2. Reaction conditions as shown in the box above. 10 mM substrate concentration was used, with H2O2 supplied using a syringe pump at a rate of 5 mM h−1 at RT.33a12a could not be isolated, due to its propensity to rearrange into tert-butyl pyridine-3-yl carbonate product 14. bKBr (100 mM) was added to the buffer. c1 mM h−1 H2O2 addition rate was used. | ||
Substrates bearing a tert-butyloxycarbonyl (Boc) protecting group in the amine (11a–f) were explored first (Scheme 3A). Pleasingly, the smaller substrates in this series were accepted by rAaeUPO-PaDa-I-H. However, in the case of the smallest substrate in this series (11a, R = H), while we believe that the aza-Achmatowicz took place, the product 12a could not isolated as it was observed to be unstable and to convert into pyridine 14, which was isolated in 60% yield. Pleasingly, substituted analogues 12b and 12c were more stable, and using the standard method, the expected aza-Achmatowicz products were obtained in acceptable yields. To the best of our knowledge, these represent the first biocatalytic aza-Achmatowicz reactions to be performed without using diffusible oxidants. Substrate 11c apparently represents the limit of the N-Boc series with respect to size however, as bulkier substrates 11d–f were not converted into the respective products 12d–f. This result was not wholly surprising, especially considering the additional steric imposition of the Boc protecting group.
Nonetheless, in these more challenging cases, we found that a biocatalytic aza-Achmatowicz can still be achieved, demonstrated by the successful synthesis of 12e and 12f, by simply adding KBr to the reaction medium. Presumably, these modified examples operate via enzyme-catalysed oxidation of the added bromide to form hypobromite, which then diffuses from the enzyme active site to promote the oxidative rearrangement. This discovery is useful in expanding the scope the method to include bulkier substrates. However, this is be balanced against the strong likelihood that the oxidation takes place outside of the UPO active site when KBr is used, and hence the opportunity for the enzyme to impart stereocontrol on the reaction is lost.
N-Mesyl protected substrates 11g–k (Scheme 6B) were also examined, in the hope that the switch to a smaller protecting group would allow more flexibility to incorporate bulkier substituents. Although yields from substrates 11g and 11h gave results comparable to analogous N-Boc protected substrates 11b and 11c, in the case of substrate 11i, this was transformed with 40% yield, whereas the NBoc-protected analog 11e was not. No conversion was observed when trying to convert the larger ester containing substrate 11k however. In most cases (12b and 12i being the exceptions) the products were isolated as single diastereoisomers. It is not always clear whether the 2,6-cis or 2,6-trans diastereoisomer will predominate in aza-Achmatowicz reactions, although it is relatively common for there to be a thermodynamic preference for one diastereoisomer.2 Based on specific rotation measurements, all products 12b–j appear to be racemic, indicating little or no kinetic resolution of the chiral amine precursors 11b–j, as was the case for the 2-furylcarbinol substrates.
Finally, having demonstrated the feasibility of both the rAaeUPO-PaDa-I-H-catalyzed Achmatowicz and aza-Achmatowicz reactions, we wanted to further demonstrate the scalability of the protocol by performing the oxidation of 9e on a 1 g scale (Scheme 7). To do this, the concentration of the substrate was raised radically to 400 mM. The enzyme tolerated this change well, but it was necessary to switch the co-solvent to acetone better solubilise 9e. The use of acetone as co-solvent introduces the possibility of unwanted dimethyldioxirane (DMDO – potentially explosive) being formed in the reaction, although the slow addition of H2O2 and the quick enzymatic reaction both minimise this risk. Under these conditions, 0.92 g of the product 10e was isolated, which corresponds to 82% isolated yield. This transformation showcases well the robustness of rAaeUPO-PaDa-I-H under challenging biocatalytic conditions.
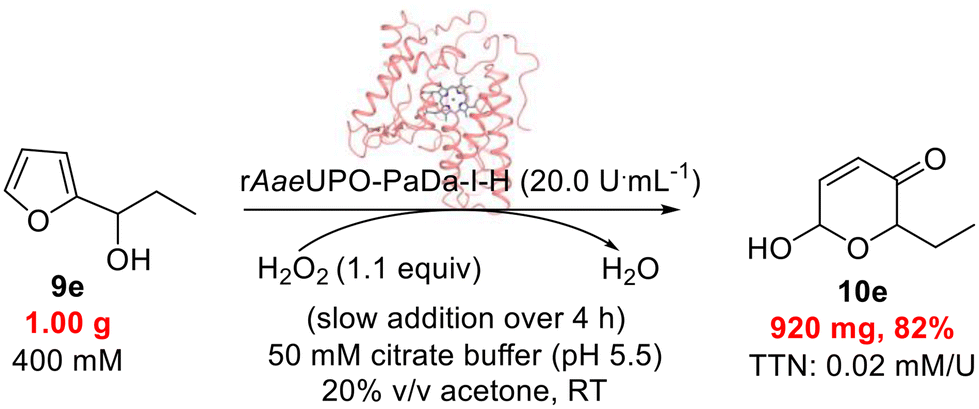 | ||
| Scheme 7 rAaeUPO-PaDa-I-H catalyzed oxidation of 9e on a 1 g scale at RT.33 | ||
Conclusions
We have demonstrated that rAaeUPO-PaDa-I-H is an easy-to-use catalyst for Achmatowicz and aza-Achmatowicz reactions. A simple, slow-addition H2O2 delivery method has been established for preparative scale reactions, and the lyophilized enzyme is practical, easy to use and is stable for years when stored at −20 °C. The transformations are performed under mild reaction conditions, and are generally highly chemoselective – the biocatalytic method often out-performed the standard chemo-promoted method (NBS), and in several cases afforded products that were sufficiently clean that column chromatography was not needed. The scope of the transformations is relatively broad, with substrate specificity primarily influenced by steric factors. Notably, the scalability of the rAaeUPO-PaDa-I-H catalyzed Achmatowicz reaction was further demonstrated by performing the oxidation of 1-(2-furyl)propan-1-ol 9e at 1 g scale, with the desired product 10e isolated in 82% yield. This study expands the reaction scope of UPOs and provides further substantial evidence of practicality and ever-growing practical synthetic utility as green oxidants.Author contributions
All authors contributed to the design and execution of experiments. BP, WPU and GG wrote the manuscript, with contributions also from ADR.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the industrial affiliates of the Centre of Excellence for Biocatalysis, Biotransformation and Biocatalytic Manufacture (CoEBio3) for studentship awards to B. P. and T. M.References
- O. Achmatowicz, P. Bukowski, B. Szechner, Z. Zwierzchowska and A. Zamojski, Tetrahedron, 1971, 27, 1973–1996 CrossRef CAS.
- A. K. Ghosh and M. Brindisi, RSC Adv., 2016, 6, 111564–111598 RSC.
- L. Liang, L.-D. Guo and R. Tong, Acc. Chem. Res., 2022, 55, 2326–2340 CrossRef CAS PubMed.
- M. P. Georgiadis, E. A. Couladouros, M. G. Polissiou, S. E. Filippakis, D. Mentzafos and A. Terzis, J. Org. Chem., 1982, 47, 3054–3058 CAS.
- T. Shono and Y. Matsumura, Tetrahedron Lett., 1976, 17, 1363–1364 CrossRef.
- X. Liu, B. Li, G. Han, X. Liu, Z. Cao, D.-e. Jiang and Y. Sun, Nat. Commun., 2021, 12, 1868 CrossRef CAS.
- G. C. M. Lee, E. T. Syage, D. A. Harcourt, J. M. Holmes and M. E. Garst, J. Org. Chem., 1991, 56, 7007–7014 CrossRef CAS.
- M. B. Plutschack, P. H. Seeberger and K. Gilmore, Org. Lett., 2017, 19, 30–33 CrossRef CAS PubMed.
- C. Asta, D. Schmidt, J. Conrad, B. Förster-Fromme, T. Tolasch and U. Beifuss, RSC Adv., 2013, 3, 19259–19263 RSC For a review on laccase mediated organic transformations, see: J. Jayakumar, D. Priyadarshini, A. Parthasarathy and S. R. Reddy, Asian J. Org. Chem., 2022, 12, e202200564 CrossRef CAS.
- D. Thiel, D. Doknić and J. Deska, Nat. Commun., 2014, 5, 5278 CAS.
- D. Thiel, F. Blume, C. Jäger and J. Deska, Eur. J. Org. Chem., 2018, 2717–2725 CrossRef CAS.
- F. van der Pijl, F. L. van Delft and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2015, 4811–4829 CrossRef CAS.
- E. Fernández-Fueyo, S. H. H. Younes, S. v. Rootselaar, R. W. M. Aben, R. Renirie, R. Wever, D. Holtmann, F. P. J. T. Rutjes and F. Hollmann, ACS Catal., 2016, 6, 5904–5907 Search PubMed.
- M. Hofrichter, H. Kellner, M. J. Pecyna and R. Ullrich, in Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450, ed. E. G. Hrycay and S. M. Bandiera, Springer International Publishing, Cham, 2015, pp. 341–368 Search PubMed.
- Y. Wang, D. Lan, R. Durrani and F. Hollmann, Curr. Opin. Chem. Biol., 2017, 37, 1–9 CrossRef PubMed.
- M. Hobisch, D. Holtmann, P. Gomez de Santos, M. Alcalde, F. Hollmann and S. Kara, Biotechnol. Adv., 2021, 51, 107615 CrossRef CAS.
- D. T. Monterrey, A. Menés-Rubio, M. Keser, D. Gonzalez-Perez and M. Alcalde, Curr. Opin. Green Sustainable Chem., 2023, 100786 CrossRef CAS.
- P. Molina-Espeja, E. Garcia-Ruiz, D. Gonzalez-Perez, R. Ullrich, M. Hofrichter and M. Alcalde, Appl. Environ. Microbiol., 2014, 80, 3496–3507 Search PubMed.
- P. Molina-Espeja, S. Ma, D. M. Mate, R. Ludwig and M. Alcalde, Enzyme Microb. Technol., 2015, 73–74, 29–33 CrossRef CAS PubMed.
- J. Carro, A. González-Benjumea, E. Fernández-Fueyo, C. Aranda, V. Guallar, A. Gutiérrez and A. T. Martínez, ACS Catal., 2019, 9, 6234–6242 CrossRef CAS.
- P. Molina-Espeja, M. Caňellas, F. J. Plou, M. Hofrichter, F. Lucas, V. Guallar and M. Alcalde, ChemBioChem, 2016, 17, 341–349 CrossRef CAS.
- J. Münch, J. Soler, N. Hünecke, D. Homann, M. Garcia-Borràs and M. J. Weissenborn, ACS Catal., 2023, 13, 8963–8972 CrossRef.
- P. Gomez de Santos, I. Mateljak, M. D. Hoang, S. J. Fleishman, F. Hollmann and M. Alcalde, J. Am. Chem. Soc., 2023, 145, 3443–3453 CrossRef CAS.
- Y. Ni, E. Fernández-Fueyo, A. Gomez Baraibar, R. Ullrich, M. Hofrichter, H. Yanase, M. Alcalde, W. J. H. van Berkel and F. Hollmann, Angew. Chem., Int. Ed., 2016, 55, 798–801 CrossRef CAS.
- S. J. Freakley, S. Kochius, J. van Marwijk, C. Fenner, R. J. Lewis, K. Baldenius, S. S. Marais, D. J. Opperman, S. T. L. Harrison, M. Alcalde, M. S. Smit and G. J. Hutchings, Nat. Commun., 2019, 10, 4178 CrossRef PubMed.
- E. Romero, M. J. Johansson, J. C. Cartwright, G. Grogan and M. A. Hayes, Angew. Chem., Int. Ed., 2022, 61, e202207831 CrossRef CAS PubMed.
- P. M. Kinne, R. Ullrich, G. Kayser and M. Hofrichter, Enzyme Microb. Technol., 2013, 52, 370–376 CrossRef.
- M. C. R. Rauch, F. Tieves, C. E. Paul, I. W. C. E. Arends, M. Alcalde and F. Hollmann, ChemCatChem, 2019, 11, 4519–4523 CrossRef CAS PubMed.
- H. E. Bonfield, K. Mercer, A. Diaz-Rodriguez, G. C. Cook, B. S. J. McKay, P. Slade, G. M. Taylor, W. X. Ooi, J. D. Williams, J. P. M. Roberts, J. A. Murphy, L. Schmermund, W. Kroutil, T. Mielke, J. Cartwright, G. Grogan and L. J. Edwards, ChemPhotoChem, 2020, 4, 45–51 CrossRef CAS.
- L. Schmermund, S. Reischauer, S. Bierbaumer, C. K. Winkler, A. Diaz-Rodriguez, L. J. Edwards, S. Kara, T. Mielke, J. Cartwright, G. Grogan, B. Pieber and W. Kroutil, Angew. Chem., Int. Ed., 2021, 60, 6965–6969 CrossRef CAS.
- B. Pogrányi, T. Mielke, A. Díaz-Rodríguez, J. Cartwright, W. P. Unsworth and G. Grogan, Angew. Chem., Int. Ed., 2023, 63, e202214759 Search PubMed.
- B. Melling, T. Mielke, A. C. Whitwood, T. J. C. O'Riordan, N. Mulholland, J. Cartwright, W. P. Unsworth and G. Grogan, Chem. Catal., 2024, 100889 CrossRef CAS.
- Throughout this manuscript, ‘RT’ refer to the ambient temperature in out laboratory, which is typically ≈21 °C.
- No reaction occurred in a control reaction in which 9d was reacted under the same conditions but lacking the enzyme; this confirms that the oxidation does not simply result from the generation of a peracid oxidant derived from the citrate buffer and H2O2.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00939h |
| This journal is © The Royal Society of Chemistry 2024 |