
A relay ring-closing metathesis/Diels–Alder approach to sugar-derived pluramycin-hybrids†‡
Ajad
Singh
 and
Krishna P.
Kaliappan
*
and
Krishna P.
Kaliappan
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai-400076, India. E-mail: kpk@chem.iitb.ac.in
First published on 19th July 2024
Abstract
Herein, we present a general approach for synthesizing pluramycin hybrids, which are analogous to the pluramycinone carbocyclic skeleton. This method involves a sequence of relay ring-closing enyne metathesis, Diels–Alder and oxidative aromatization reactions to synthesize pluramycinone-sugar hybrids. As part of our ongoing research, we have successfully synthesized two pluramycin hybrid analogues by carefully monitoring the late-stage oxidative aromatization steps, which depend on the stereo-orientation of the Diels–Alder cycloadduct at the C-4 center. The undesired ring-opening product can also serve as a C-glycoside analog, providing a versatile convergent route to access both types of hybrids and highlighting the significance of this strategy.
Introduction
Natural products have historically provided crucial insights into biological processes due to their intricate chemical makeup and have been comprehensively investigated in relation to their biosynthesis and mechanisms of action. It is worth noting that numerous natural products have proven to be valuable substances or starting compounds in various industries, including fragrances, crop science, and pharmaceuticals.In fact, over 40% of recently approved drugs are either natural products or derived from them.1 However, many traditional drugs have become ineffective due to multidrug resistance (MDR), posing a significant challenge in the field. To overcome this obstacle, scientists have made significant progress in identifying new synthetic compounds with enhanced therapeutic properties. Among the various strategies for rapidly synthesizing a diverse range of molecules, the hybrid approach has emerged as one of the methods, as it combines structural features from different classes of compounds to produce distinct compounds with modified or unprecedented biological activities in a short time frame.2,3 The drive for the creation of these hybrids has been derived from nature, which employs mixed biosynthesis pathways to produce various bioactive natural products (Fig. 1),4–6 for example, an intricate indole alkaloid, such as strychnine (1), which is derived from the amino acid tryptophan and the monoterpenic precursor loganin,5a,b and ansa antibiotics, such as rifamycins (3)5c,d in which the aromatic core is derived from shikimate and the ansa chain from polyketides. On the other hand, glycoproteins, chlorophyll-a (2), vitamin E (4), and vinblastine (5) (Fig. 1) are notable examples of naturally occurring hybrid molecules, where different segments of the molecule have diverse biosynthetic origins but are firmly bonded together to form a functional unit.5,6 Drawing on this natural process, synthetic chemists have designed and synthesized several hybrid compounds comprising both natural and unnatural elements with various biological functions. These hybrid molecules, existing as dimers or polymers, may exhibit a behaviour that is distinct from those of their monomeric counterparts.
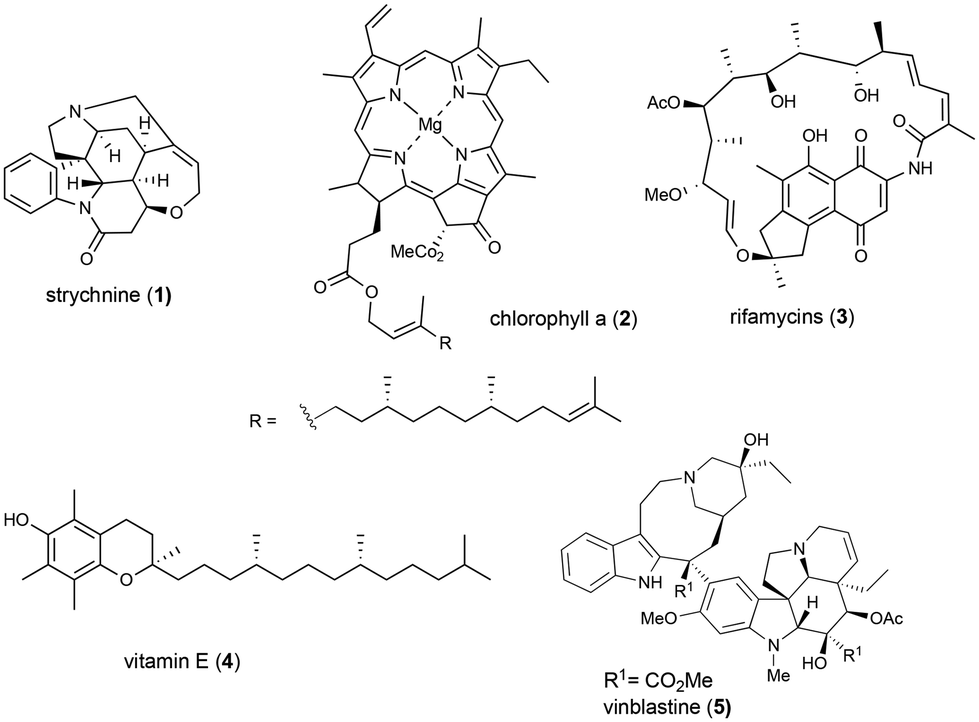 | ||
| Fig. 1 Representative examples of hybrid natural products derived from biosynthetic pathways. | ||
Anthrapyran antibiotics constitute a diverse range of natural products obtained from various terrestrial and marine Streptomyces sp. strains as secondary metabolites. These compounds feature a distinct 4H-anthra[1,2-b]-pyran-4,7,12-trione nucleus (Fig. 2) and exhibit versatile and potent biological activities, making them appealing synthetic targets (6–10).7a,c–e Apart from their significant biological activity profile, it has been found that these families of natural products display prominent chemical instability. They are sensitive to heat and decompose when exposed to temperatures exceeding 60 °C.7b
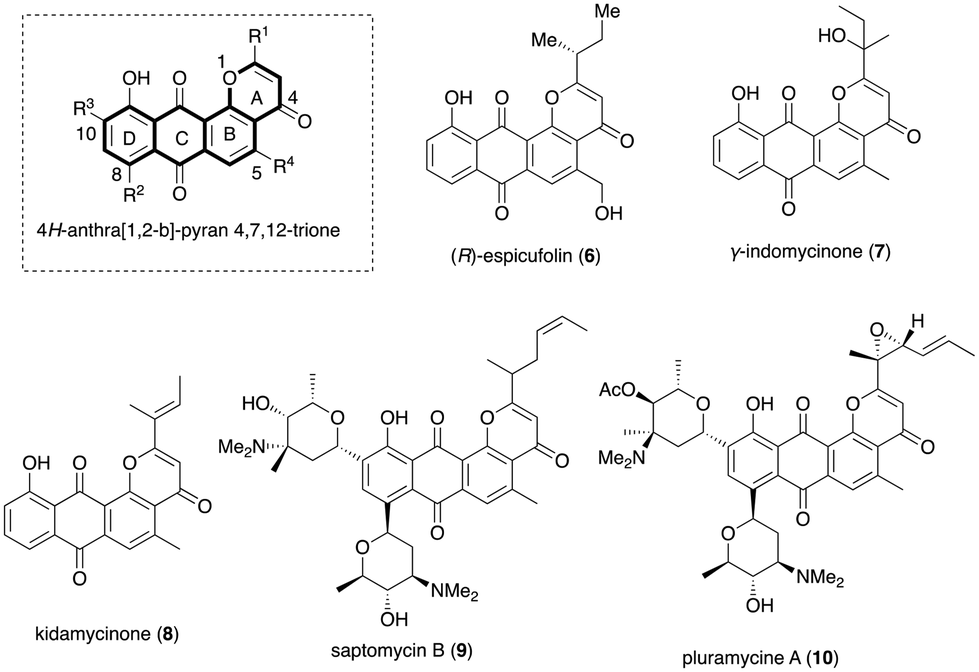 | ||
| Fig. 2 Selected examples of pluramycin/pluramycinone natural products. | ||
In addition, a considerable portion of these compounds was discovered to be unstable when exposed to ultraviolet light or sunlight.7b,c They also exhibit high cytotoxicity and low solubility issues. Due to these factors, they have not been investigated much in clinical trials despite exhibiting widespread biological activities, including antitumor properties. Given our laboratory's interest in the total synthesis of biologically active natural products and their analogues, we have designed and synthesised several interesting classes of hybrid molecules derived from sugar as the chiral backbone.8 In this work, we report a general approach by exploring the RRCEM (relay ring-closing enyne metathesis) as a key step to access the pluramycinone family of natural product analogues.
Results and discussion
The synthesis of hybrid natural products using chemical methods offers a means for rapidly accessing diverse structural compounds ready for pharmacological testing. Our strategy includes a sequential relay ring-closing enyne metathesis/Diels–Alder approach to synthesize the required tetracyclic core skeleton 3,4-dihydro-2H-anthra[1,2-b]-pyran-7,12-dione, as depicted in Scheme 1.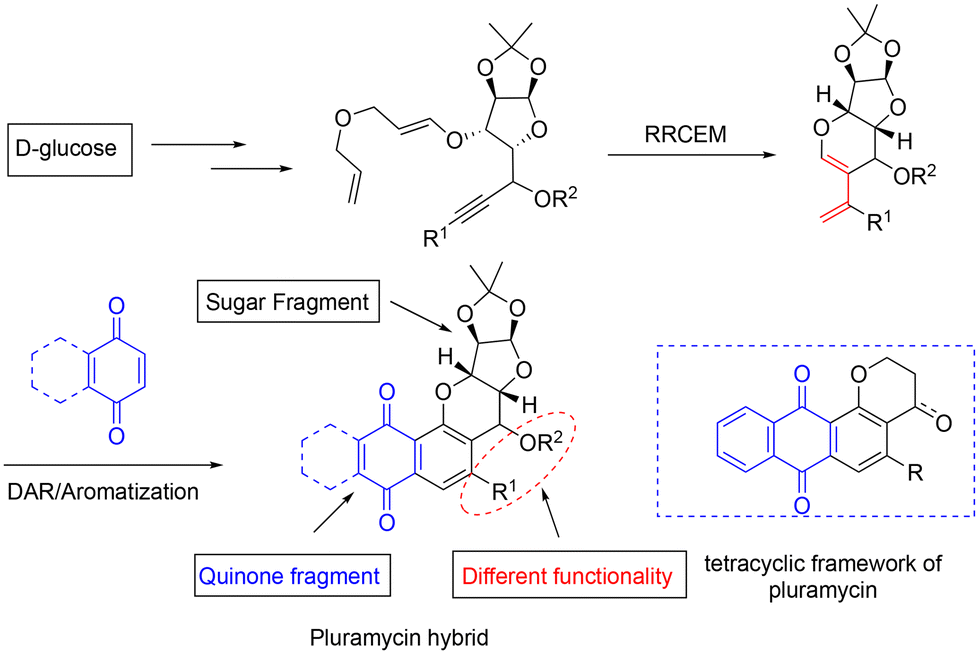 | ||
| Scheme 1 Relay ring-closing enyne metathesis/Diels–Alder approach. | ||
Retrosynthetic analysis of diene 11
We anticipated that the chiral diene 11 could be accessed by a relay ring-closing enyne metathesis (RRCEM) from the enyne precursor 12, which, in turn, could be accessed in three steps from alkyne 13via a diastereoselective oxa-Michael addition with ethyl propiolate and the reduction of the resultant ester to primary alcohol followed by O-allylation reaction. Alcohol 13 could be synthesized in two steps from alkynol 14via protection of alkynol and removal of the PMB group. The alkyne functionality on alcohol 14 could be installed by diastereoselective nucleophilic addition of the terminal alkyne into the known aldehyde 15, which in turn could be prepared from commercially available D-glucose in four steps (Scheme 2) by following known literature procedure (see the ESI‡).9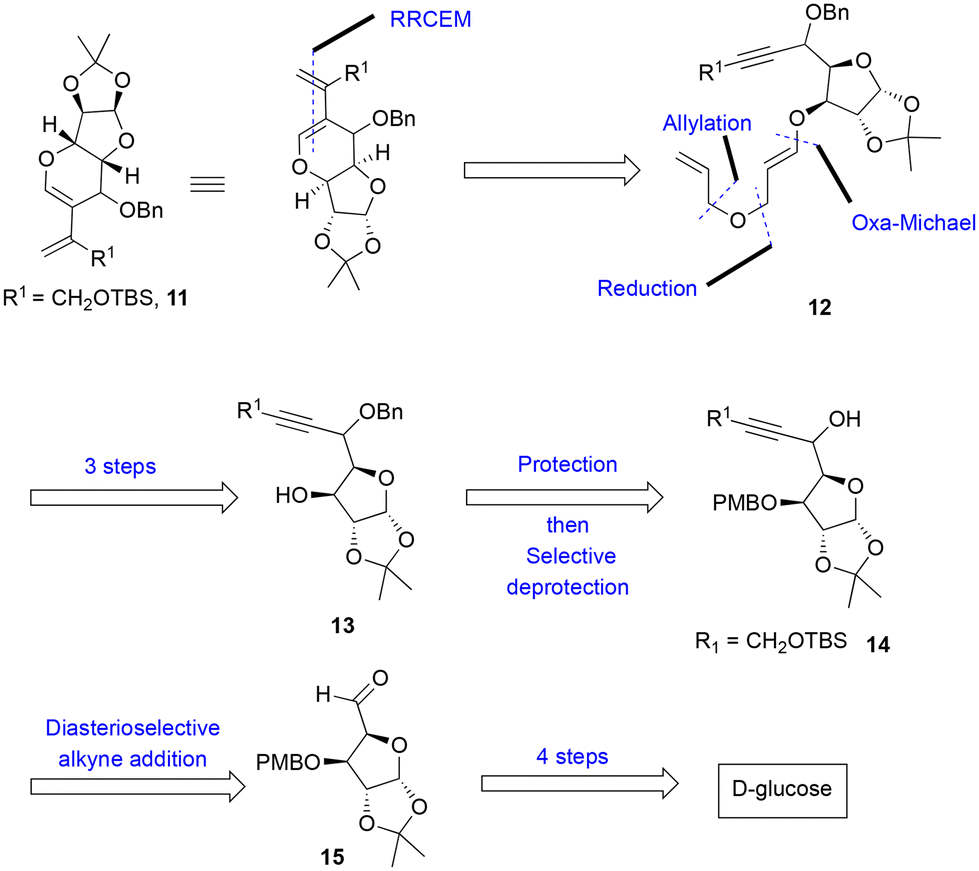 | ||
| Scheme 2 Retrosynthetic analysis for diene 11. | ||
Our synthesis began with the incorporation of the alkyne functionality into the known aldehyde 15 through a diastereoselective nucleophilic addition reaction. The addition of the lithium anion of TMS acetylene 16 with n-BuLi resulted in only 24% yield of the addition product 17 (Scheme 3). However, when TBDPS-protected propargylic alcohol 18 was employed under the same conditions, alcohol 14 was obtained in 46% yield as a single isomer. Attempts to use an in situ generated organocerium reagent (prepared from anhydrous cerium chloride and n-BuLi) did not improve the yield and diastereoselectivity of the product. So, we decided to move further with alcohol 14 obtained in a moderate yield. Attempts to protect the alcohol as MOM ether under different conditions did not give a good yield of the expected product. Thus, efforts were redirected towards using a less sterically hindered ether-protecting group, such as benzyl, which was smoothly incorporated by using benzyl bromide with NaH in DMF to provide the desired benzyl ether product 20 (Scheme 4).
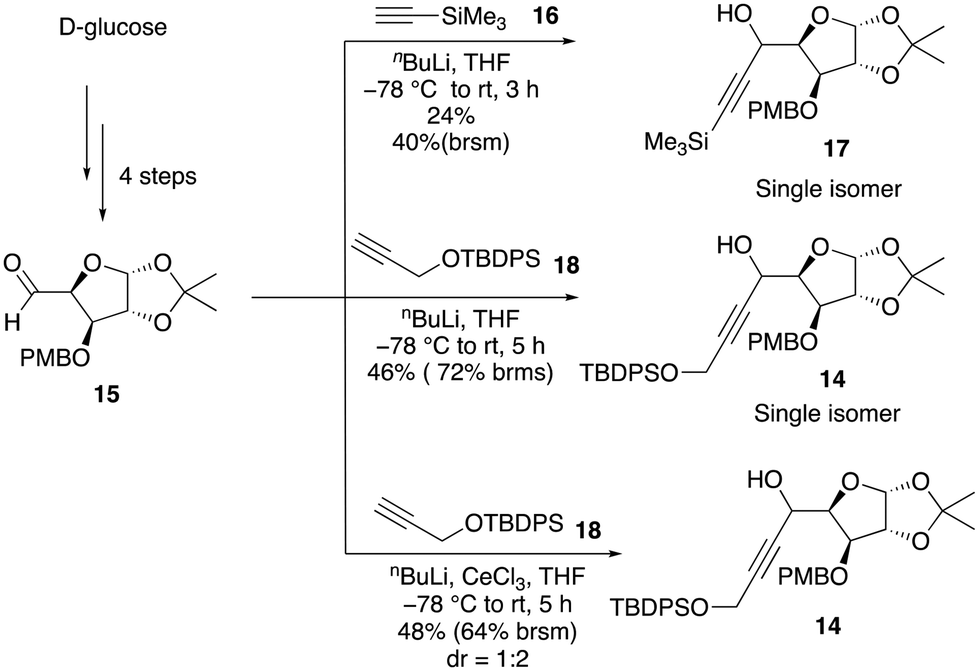 | ||
| Scheme 3 Synthesis of propargylic alcohols 17 and 14. | ||
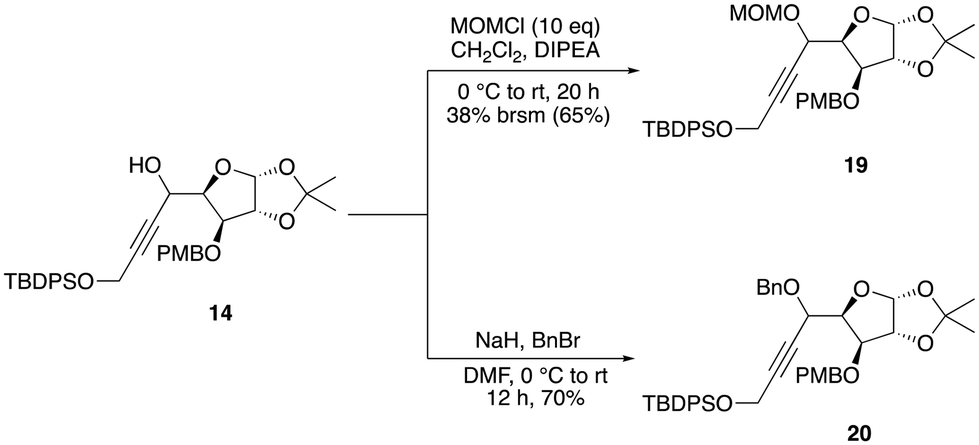 | ||
| Scheme 4 Synthesis of compounds 19 and 20. | ||
The next objective was to selectively remove the PMB group of 20, which was easily achieved by treating with DDQ to provide alcohol 13 in 70% yield. The oxa-Michael addition reaction of 13 with ethyl propiolate in the presence of N-methylmorpholine (NMM) delivered ester 21, which was immediately reduced with DIBAL-H to furnish the allylic alcohol 22 in 85% yield (Scheme 5).
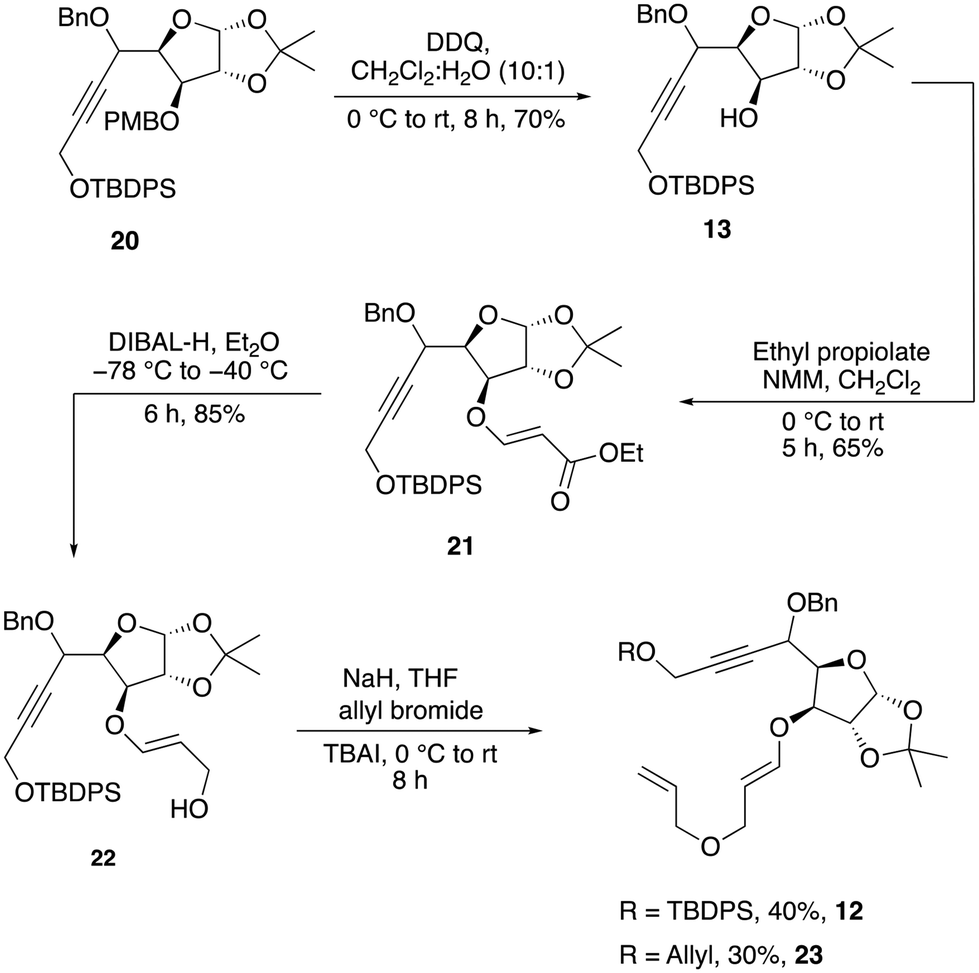 | ||
| Scheme 5 Synthesis of the enyne precursor 12. | ||
When 22 was subjected to O-allylation by treating it with NaH and allyl bromide, the key enyne precursor 12 was obtained only in 40% yield. It was observed that a minor undesired double O-allylated product, 23, was formed during this reaction. This unwanted by-product resulted from the deprotection of the TBDPS group under basic conditions followed by a second allylation (Scheme 5). After multiple attempts to prevent the formation of the unwanted product 23 using various bases, including NaH, potassium tert-butoxide and solvents such as DMF and THF, we were unsuccessful in controlling the formation of 23. The low yield and decomposition of the starting materials appeared to have forced us to abandon this route.
At this stage, it became evident that a stable protecting group was crucial to overcome this problem to obtain a substantial quantity of the enyne precursor product. Consequently, we chose to replace the TBDPS protecting group with the robust benzyl group. Subsequently, we performed nucleophilic addition of the benzyl-protected propargylic alcohol 24 to aldehyde 15, which resulted in the formation of a mixture of diastereomers 25 and 26 (dr = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), with a combined yield of 80% (Scheme 6). The conversion of the minor isomer 25 into the major isomer 26 was achieved exclusively through sequence of oxidation and reduction reactions. Initially, the minor diastereomer was oxidized using DMP oxidation, which resulted in the formation of an alkynone product, which was subsequently reduced using various reducing reagents with the aim of obtaining a single isomer of alcohol 26 (see the ESI‡) (Scheme 6). Among the various options, diisobutylaluminium hydride (DIBAL-H), a sterically hindered electrophilic hydride donor, was found to be much more efficient and predominantly produced a single isomer of alcohol 26. The major isomer of 26 was then protected as benzyl ether 27 in 72% yield. Now, the chemo-selective removal of the PMB group with DDQ proceeded smoothly to afford alcohol 28 in 80% yield. The oxa-Michael addition of alcohol 28 with ethyl propiolate in the presence of N-methylmorpholine furnished the conjugate ester 29 in 68% yield. Reduction of 29 with DIBAL-H afforded the allylic alcohol 30, which, upon O-allylation using NaH and allyl bromide, afforded the O-alkylated product 31 in good yield via two steps. Having sufficient quantity of the enyne precursor 31 in hand, we proceeded to carry out the first key step (relay ring-closing enyne metathesis) with the Grubbs’ second-generation catalyst (G-II). To our delight, this reaction worked well to afford the desired diene product 32 in 80% yield within 30 min (Scheme 7).
:2), with a combined yield of 80% (Scheme 6). The conversion of the minor isomer 25 into the major isomer 26 was achieved exclusively through sequence of oxidation and reduction reactions. Initially, the minor diastereomer was oxidized using DMP oxidation, which resulted in the formation of an alkynone product, which was subsequently reduced using various reducing reagents with the aim of obtaining a single isomer of alcohol 26 (see the ESI‡) (Scheme 6). Among the various options, diisobutylaluminium hydride (DIBAL-H), a sterically hindered electrophilic hydride donor, was found to be much more efficient and predominantly produced a single isomer of alcohol 26. The major isomer of 26 was then protected as benzyl ether 27 in 72% yield. Now, the chemo-selective removal of the PMB group with DDQ proceeded smoothly to afford alcohol 28 in 80% yield. The oxa-Michael addition of alcohol 28 with ethyl propiolate in the presence of N-methylmorpholine furnished the conjugate ester 29 in 68% yield. Reduction of 29 with DIBAL-H afforded the allylic alcohol 30, which, upon O-allylation using NaH and allyl bromide, afforded the O-alkylated product 31 in good yield via two steps. Having sufficient quantity of the enyne precursor 31 in hand, we proceeded to carry out the first key step (relay ring-closing enyne metathesis) with the Grubbs’ second-generation catalyst (G-II). To our delight, this reaction worked well to afford the desired diene product 32 in 80% yield within 30 min (Scheme 7).
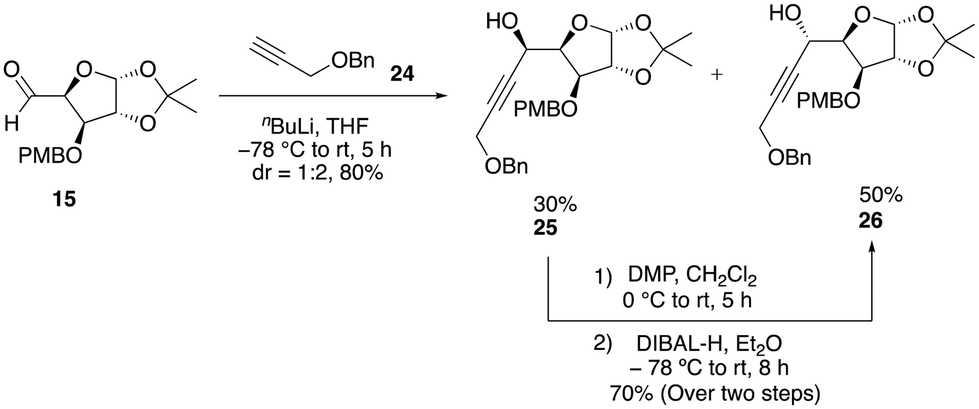 | ||
| Scheme 6 Synthesis of alcohols 25 and 26. | ||
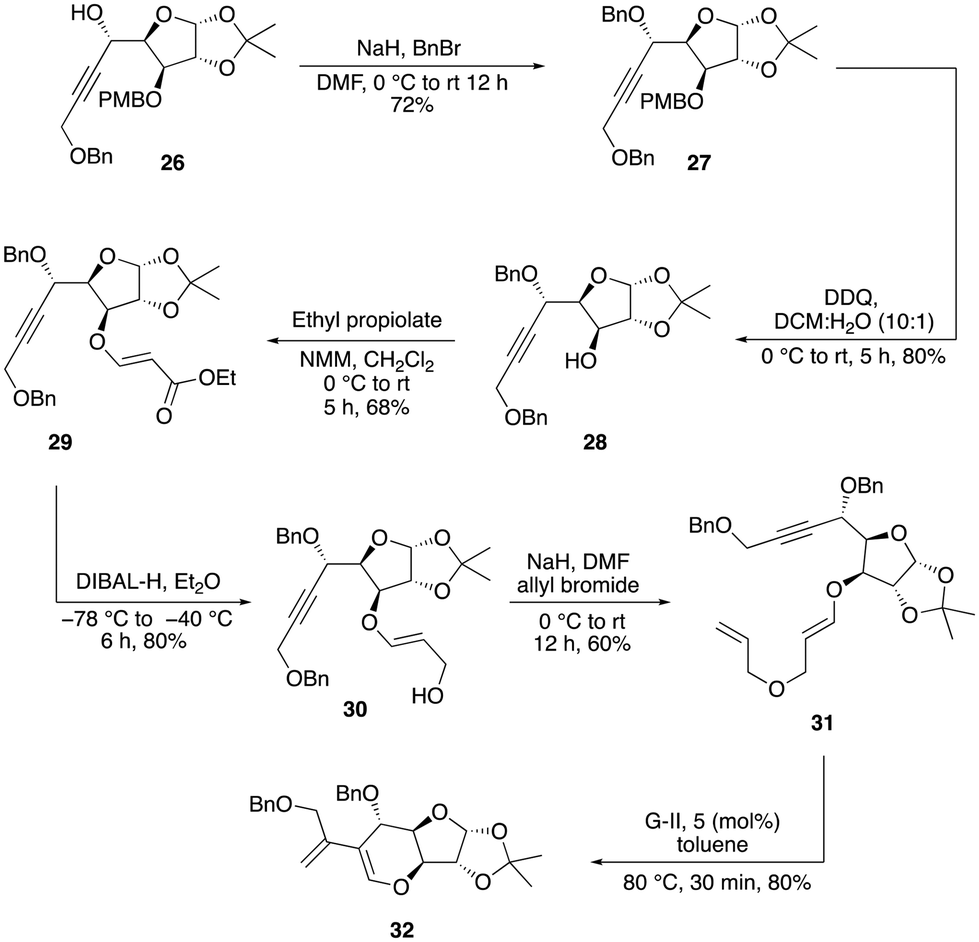 | ||
| Scheme 7 Synthesis of diene 32. | ||
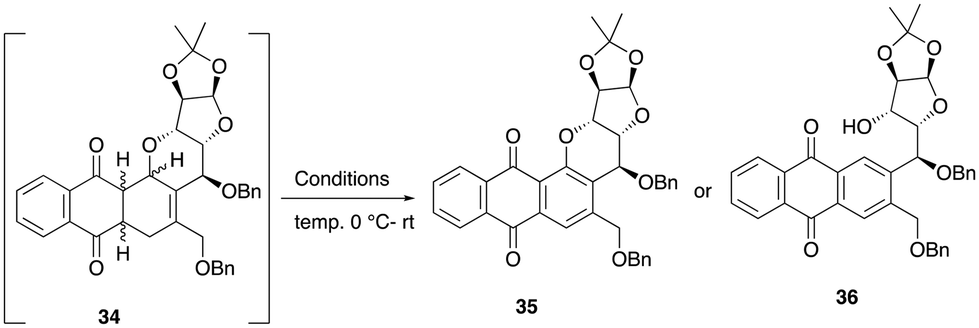
With diene 32 in hand, we next focused our efforts on the sequential Diels–Alder-aromatization reaction by exposing it to various quinone dienophiles to obtain the tetracyclic framework of pluramycin hybrids. Initially, diene 32 was heated in toluene at 100 to 110 °C with commercially available 1,4-naphthoquinone 33, which resulted in a crude cycloadduct 34 in 24 h. However, when this crude product was subjected to aromatization under air and the SiO2-Et3N reagent mixture, contrary to our expected tetracycle 35, we observed the formation of the ring-cleaved aromatized product 36 (Scheme 8). The plausible mechanism for ring opening product is shown in Scheme 9.
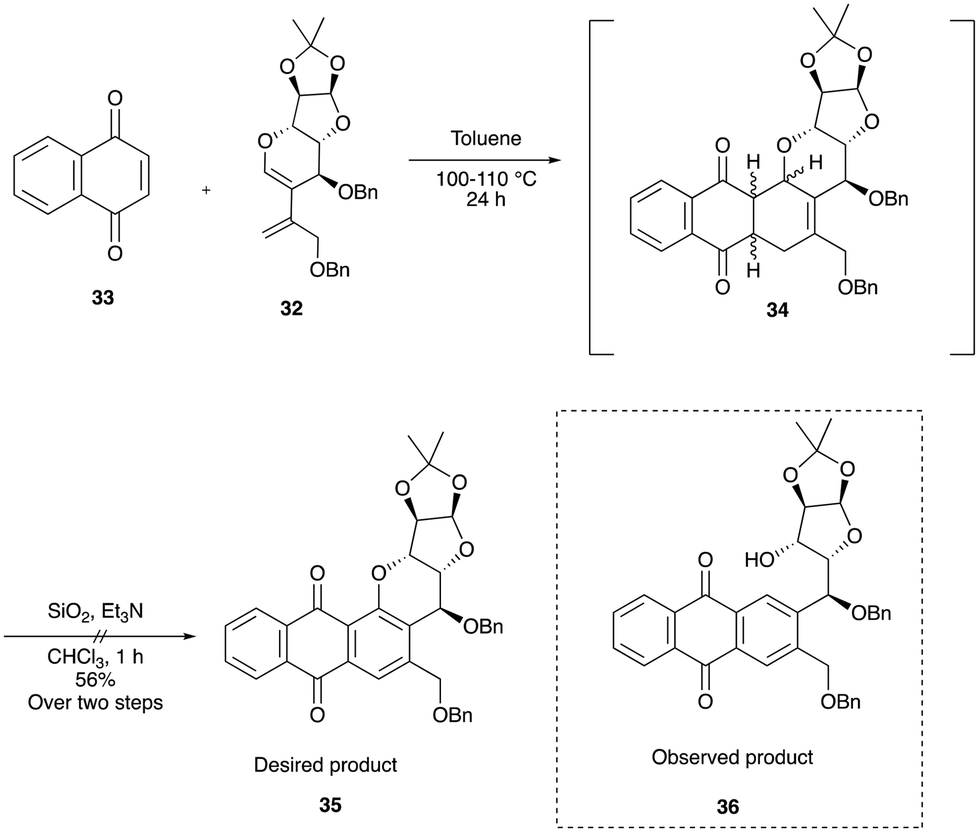 | ||
| Scheme 8 Attempted conditions for tetracycle 35. | ||
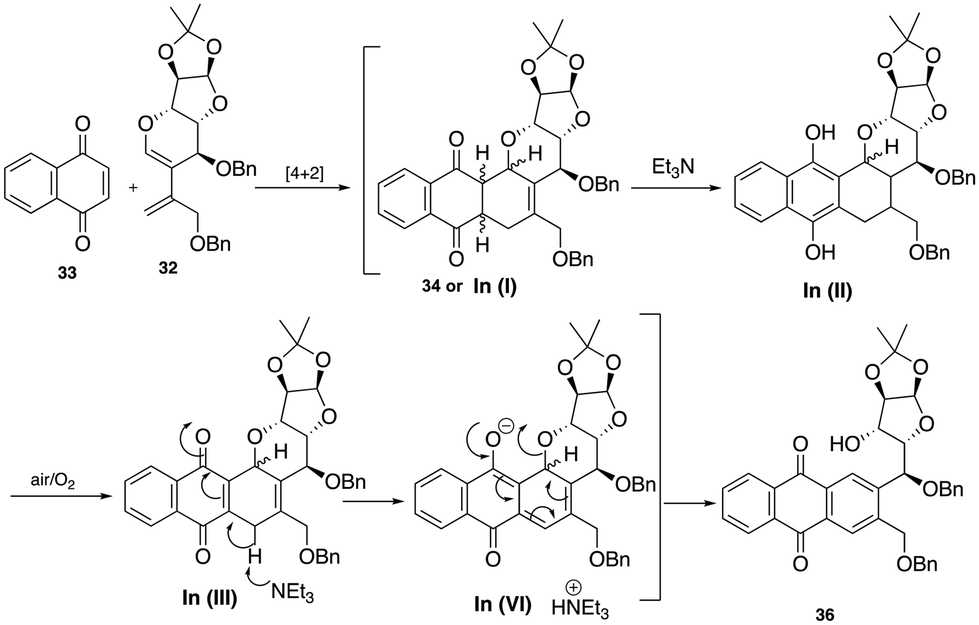 | ||
| Scheme 9 Plausible reaction mechanism for the aromatization of cycloadduct 34. | ||
The formation of Cycloadduct intermediate 34 was confirmed by high resolution mass spectrometry (HRMS) data (see the ESI‡) and at this stage we realized that the Diels–Alder reaction proceeded smoothly, but further optimization is required for the aromatization step.
The use of milder bases, such as Et3N (entry 1) and K2CO3 (entry 2), did not yield the desired aromatic tetracyclic product 35; instead, it provided the ring-cleaved product 36 exclusively. At this point, it became apparent that basic conditions may not be suitable for obtaining the desired product, as C–O bond cleavage is more facile under basic conditions due to its better-leaving group capability. As a result, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)10 was used to provide the expected tetracyle, but this reagent also unfortunately provided a complex mixture (entry 3, Table 1). Since direct aromatization of cycloadduct 34 was unsuccessful under various conditions, we anticipated that prior functionalization of the cyclohexene intermediate 34, before aromatization, might facilitate the process to provide the desired product 35. Thus, attempts were made to form the bromohydrin intermediate via a bromonium ion (entry 4, Table 1), but this reaction also led to a complex mixture. Inspired by the seminal work done by Krohn and co-workers,11a we subjected the crude cycloadduct 34 to hydroxylation by exposing it to a catalytic amount of osmium tetroxide along with the co-oxidant N-methylmorpholine-N-oxide (NMO) (entry 5, Table 1).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Reaction conditions | Time | Result | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Isolated overall yield. NR = no reaction. b Only the starting material remains. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Et3N, SiO2, CHCl3 | 1 h | 36 (56%)a | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | K2CO3, MeOH | 2 h | 36 (50%)a | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | DDQ, CH2Cl2 | 12 h | Complex mixture | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | NBS, THF:H2O (5:1) then Et3N, CH2Cl2 |
8 h | Complex mixture | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | OsO4, NMO, acetone:H2O (5:1) |
24 h | 36 (45%)a | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | m-CPBA, NaHCO3, CH2Cl2 | 24 h | NRb | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The NMR analysis of the crude mixture revealed that a product with ring cleavage and aromatization was formed during the hydroxylation step. We presumed that the in situ generation of N-methylmorpholine would have facilitated the elimination process by means of a base-induced elimination process. Recently, Collet and co-workers reported a strategy wherein the Diels–Alder cycloadduct intermediate was subjected to a one-pot epoxidation followed by aromatization to furnish the aromatic tetracyclic natural product.11b However, our attempts to use similar conditions to synthesize the anthrapyran analogue from the cyclohexene intermediate 34 with m-CPBA in dichloromethane (entry 6) were unsuccessful.12 Further, when we carried out the Diels–Alder reaction of 32 with a known and activated brominated dienophile 37, the required crude cycloadduct intermediate was obtained in 15 h. However, the addition of a triethyl amine base resulted in the immediate formation of the C–O bond cleavage product 39 in 50% yield over two steps (Scheme 10). Efforts to epoxidize the cyclohexene adducts 38 were unsuccessful despite repeated attempts. Furthermore, our efforts to isolate the cyclohexene intermediate 38 using silica gel or neutral alumina column chromatography were unsuccessful, as it is seems to be unstable during purification and hence immediately subjected to aromatization conditions after the consumption of diene 32 (Scheme 10).
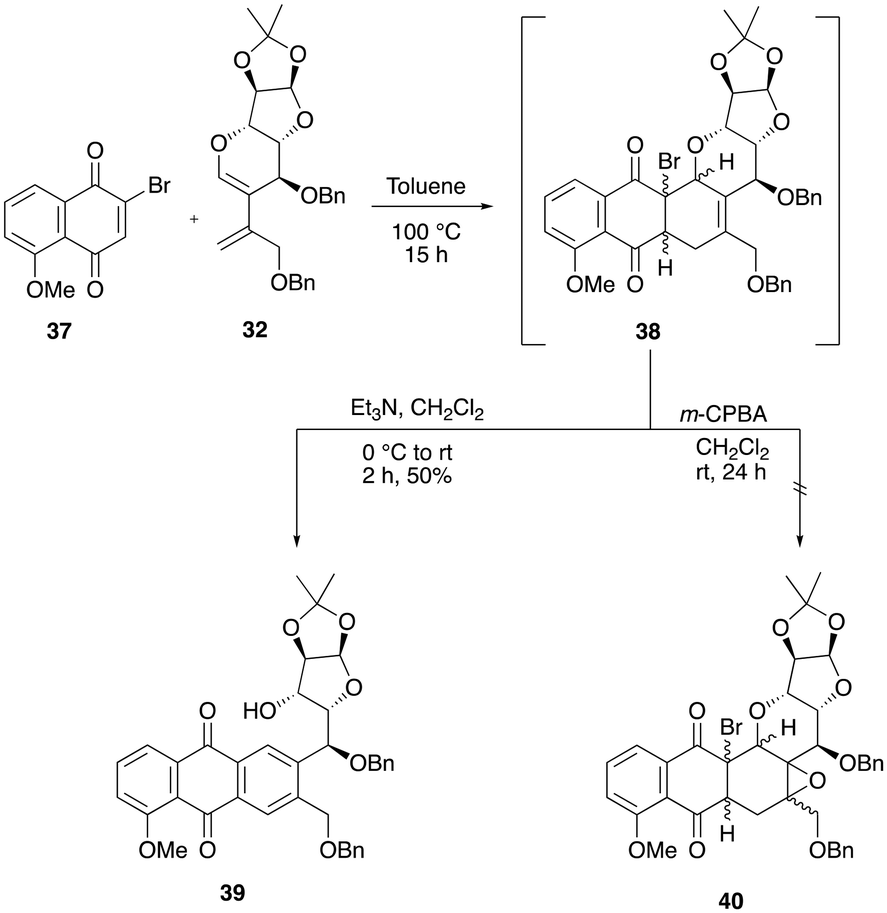 | ||
| Scheme 10 Attempted aromatization conditions with tetracycle 38. | ||
We presumed that the presence of two bulky benzyloxy groups around the cyclohexene double bond of 38 might be blocking the epoxidation from both faces of the alkene. As a result, we redirected our attention towards synthesizing an epimer of diene 32 to address this issue.
Following a similar reaction sequence, diene 41 was successfully synthesized from alcohol 25 in 6 steps in an overall yield of 32% (Scheme 11). At this stage, the stereochemical orientation of the newly formed stereocenter in the synthesis of 25 and 26 (Scheme 6) was confirmed by NOE NMR analysis of 41 (see the ESI‡), which also resolves the relative configuration of the key intermediate 32 and all compounds derived further.
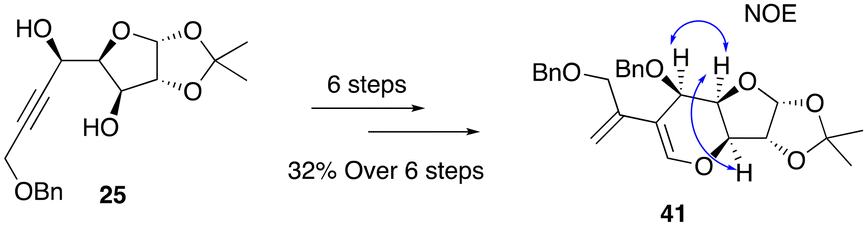 | ||
| Scheme 11 Synthesis of the minor diene isomer 41 from alcohol 25. | ||
Synthesis of hybrid 42
When the minor diastereomer of diene, i.e.41, was subjected to Diels–Alder reaction conditions with the commercially available 1,4-naphthaquinone 33, the crude cycloadduct product was delivered within 24 h. The crude cyclohexene adduct was then directly used for epoxidation with m-CPBA in DCM. After 12 h, to our delight, TLC showed complete consumption of the starting materials, and we were pleased to observe the formation of the epoxide product. After consumption of the starting material, the crude epoxide was then directly treated with excess triethylamine in the same pot under an open-air atmosphere, resulting in the desired tetracyclic analogue 42 in an overall yield of 45% over three steps (Scheme 12).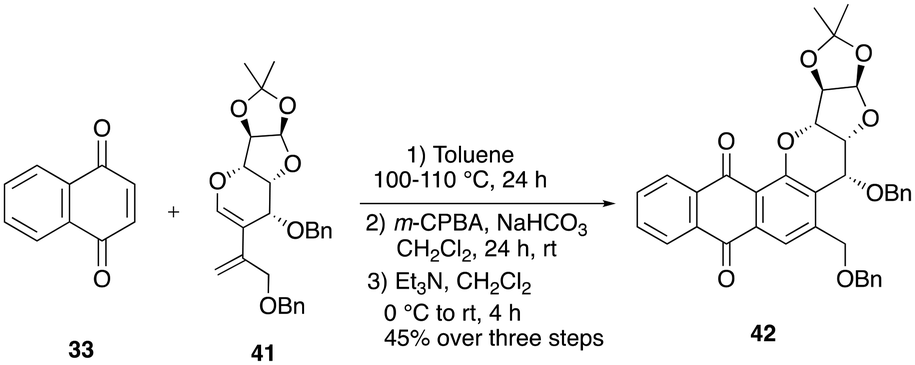 | ||
| Scheme 12 Synthesis of the pluramycin-sugar hybrid 42. | ||
The plausible reaction mechanism for the aromatization via epoxide ring opening reaction is depicted in Scheme 13.
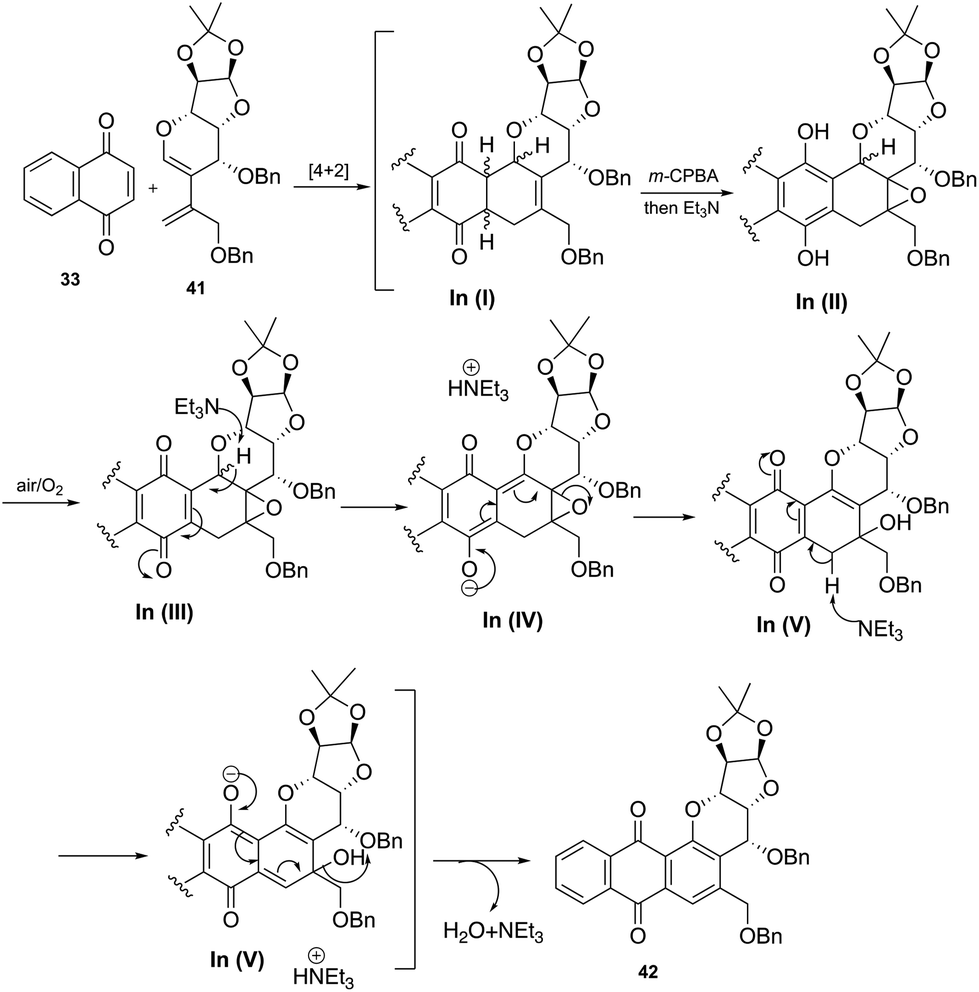 | ||
| Scheme 13 Plausible reaction mechanism for the aromatization step. | ||
Synthesis of hybrid 45
Given the biological importance of juglone 43 and the presence of hydroxyl groups in numerous natural products from the pluramycin family, we were motivated to create a sugar hybrid analog of juglone. To achieve this aim, juglone 43 was subjected to optimized Diels–Alder reaction conditions with diene 41 in toluene at 100 to 110 °C, resulting in the formation of the cycloadduct intermediate 44 within 20 h. The crude cycloadduct mixture was dried under vacuum and re-dissolved in dichloromethane and then treated with m-CPBA, and it led to the formation of a crude epoxide mixture. This mixture was then immediately subjected to aromatization within the same pot by adding an excess of Et3N to the reaction mixture in an open-air atmosphere, resulting in the desired pluramycin-sugar hybrid 45 in 50% overall yield over three steps (Scheme 14).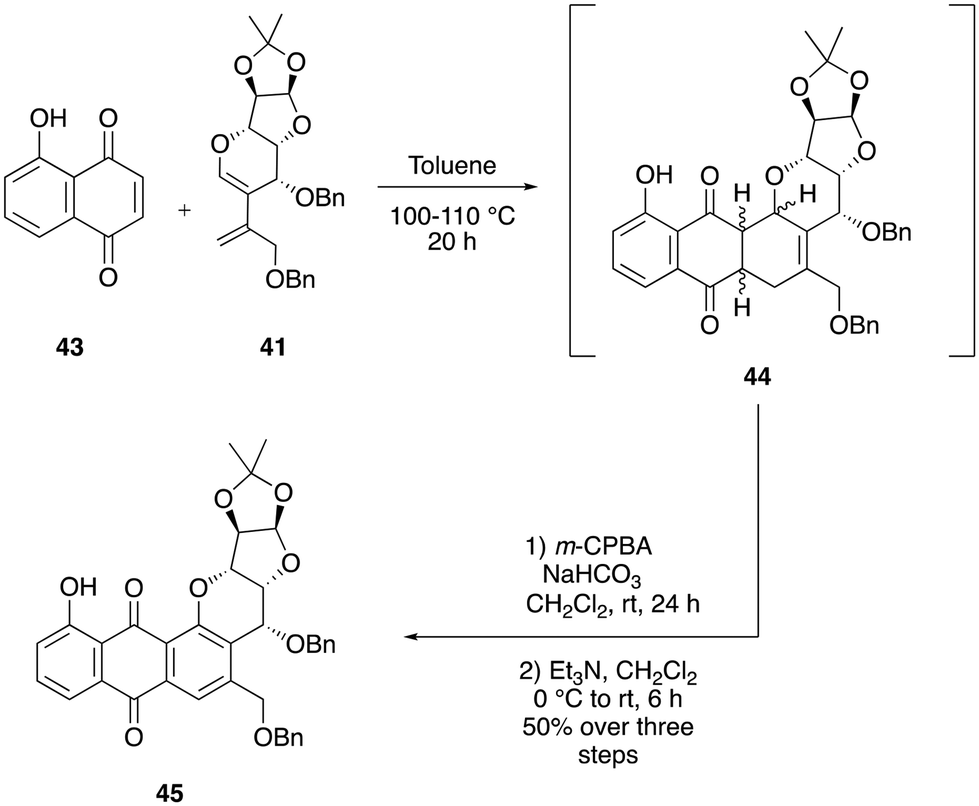 | ||
| Scheme 14 Synthesis of the pluramycin-sugar hybrid 45. | ||
However, when the same sequence was attempted with the major diene 32, the C–O bond cleaved product 46 was obtained in 40% yield over three steps (Scheme 15).
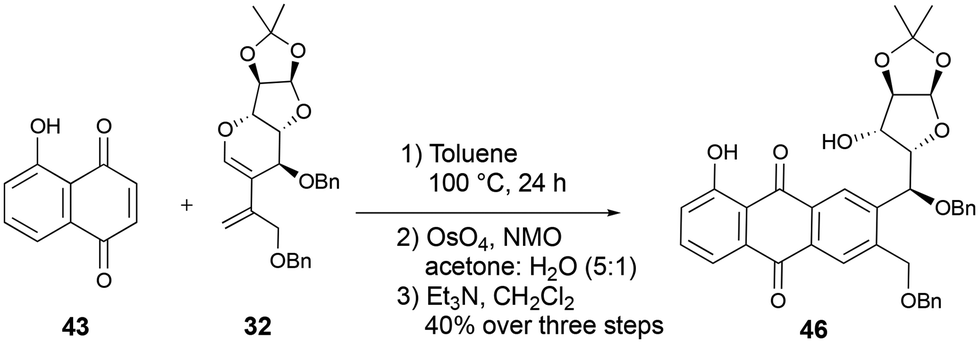 | ||
| Scheme 15 Synthesis of the sugar-quinone hybrid 46. | ||
In this study, our attempts to isolate any of the cycloadducts seem challenging, as they either lead to the ring-opening product or the decomposition of the product. Therefore, we decided to subject them immediately to subsequent steps after their formation.
Conclusions
In conclusion, we have developed a simple convergent strategy for the synthesis of a new class of sugar-derived pluramycin hybrids, which share similarity to the pluramycinone carbocyclic skeleton. This approach involves key steps, namely sequential relay ring-closing enyne metathesis (RRCEM) and Diels–Alder and oxidative aromatization reactions. This methodology opens up interesting possibilities for the synthesis of such hybrids by using diverse sugar units and quinones as starting materials. Ongoing studies in our laboratory will apply this methodology to synthesize various simpler analogues of pluramycinone natural products. In this study, we have successfully synthesized two hybrid molecules, 42 and 45, exhibiting an oxa-tetracyclic framework of pluramycin natural products by successfully controlling the challenging late-stage aromatization reaction with a minor isomer of diene 41 in overall 14 longest linear steps (LLS). In addition to the inherent stability of C-glycosidic natural products in vitro, the late-stage C–O bond cleavage products derived from the major diene 32 could also serve as novel C-glycoside analogues of sugar-quinone hybrids. Hence, these efforts have led to a divergent route for the synthesis of both types of analogues and have potential for further discovery. Efforts to synthesize other different sugar-derived quinone-hybrid molecules and study their biological activities are currently underway in our laboratory.Experimental section
General information
Unless otherwise noted, all starting materials and reagents were obtained from commercial suppliers and used after further purification. THF was distilled from sodium benzophenone ketyl and toluene from sodium. DCM and DMF were freshly distilled from calcium hydride. All solvents for routine isolation of products and chromatography were of reagent grade and glass distilled. Reaction flasks were dried in an oven at 130 °C for 12 h. Air- and moisture-sensitive reactions were performed under an argon/UHP nitrogen atmosphere. Column chromatography was performed using silica gel (100–200 mesh size) with the indicated solvents. Thin-layer chromatography (TLC) was conducted with Merck silica gel 60 F254 percolated plates (0.25 mm) and visualized with UV, KMnO4, and ceric ammonium molybdate. IR spectra were recorded with PerkinElmer Spectrum One and JASCO V-570 spectrophotometers. Mass spectra were obtained with a Bruker ESI-QTOF spectrometer recorded on Bruker Maxis Impact S No. 282001.0008 and Agilent spectrometers. 1H NMR spectra were recorded on Bruker 400 MHz and Bruker 500 MHz spectrometers and are reported in ppm using a solvent as an internal standard (CDCl3 at 7.26 ppm). Proton-decoupled 13C NMR spectra were recorded on Bruker 400 MHz and Bruker 500 MHz spectrometers and have been reported using a solvent as an internal standard (CDCl3 at 77.2 ppm). Optical rotations were measured at 20 °C using an Autopol IV polarimeter.Experimental data and characterization of products
:4 EtOAc/hexane) to obtain 17 (0.064 g, 24% yield, 40% brsm) as a single isomer. Physical appearance: Sticky yellow liquid; Rf: 0.6 (30% EtOAc/hexane × 2); IR (neat, cm−1): 3468, 2957, 2348, 1613, 1515, 1466, 1375, 1302, 1251, 1215, 1165, 1121, 1077, 1029, 845, 760; [α]20D: −42.3 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.27 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 8.5 Hz, 2H), 5.94 (d, J = 3.6, 1H), 4.71 (d, J = 8.5 Hz, 1H), 4.57–4.47 (m, 3H), 4.30 (dd, J = 8.4 Hz, 3.6 Hz, 1H), 4.07 (d, J = 3.3 Hz, 1H), 3.79 (s, 3H), 2.55 (br, 1H), 1.51 (s, 3H), 1.31(s, 3H), 0.17(s, 9H); 13C NMR (100 MHz, CDCl3): δ 159.4, 129.4, 129.2, 128.5, 113.8, 112.18, 105.6, 102.5, 90.7, 83.9, 82.4, 72.9, 62.4, 55.2, 26.8, 26.3, −0.24; HRMS (ESI-QTOF): [M + Na]+ calcd for C21H30NaO6Si 429.1709; found 429.1704.
:45 EtOAc/hexane) to obtain 14 (0.18 g, 46% yield, 72% brsm) as a single isomer. Physical appearance: Sticky yellow liquid; Rf: 0.5 (30% EtOAc/hexane × 2); IR (neat, cm−1): 3467, 2933, 2858, 2345, 1613, 1514, 1472, 1428, 1374, 1302, 1215, 1165, 1112, 1075, 1019, 890, 825, 755, 704, 614, 506; [α]20D: −37.4 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.71–7.69 (m, 4H), 7.43–7.36 (m, 6H), 7.23 (d, J = 8.6 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 5.95 (d, J = 3.6 Hz, 1H), 4.70 (d, J = 7.8 Hz, 1H), 4.57 (d, J = 3.6 Hz, 1H), 4.53 (d, J = 10.9 Hz, 1H), 4.44 (d, J = 10.9 Hz, 1H), 4.35 (d, J = 1.6 Hz, 1H), 4.34(d, J = 1.6 Hz, 1H), 4.23 (dd, J = 8.3 Hz, 3.2 Hz, 1H), 4.01(d, J = 3.2 Hz, 1H), 3.75 (s, 3H), 2.42 (br, 1H), 1.52 (s, 3H), 1.33 (s, 3H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 159.5, 135.7, 135.7, 133.0, 130.0, 129.7, 129.3, 127.9, 113.9, 112.3, 105.7, 84.4, 83.8, 82.6, 82.2, 82.0, 72.7, 62.0, 55.3, 52.7, 27.0, 26.8, 26.5, 19.2; HRMS (ESI-QTOF):[M + Na]+ calcd for C35H42NaO7Si 625.2596; found 625.2592.
:5 EtOAc/hexane) to obtain 19 (0.062 g, 38% yield, 65% brsm). Physical appearance: Colourless oily liquid; Rf: 0.5 (25% EtOAc/hexane); IR (neat, cm−1): 2932, 2858, 2057, 1737, 1613, 1587, 1471, 1428, 1374, 1302, 1250, 1214, 1163, 1111, 1076, 1030, 946, 917, 824, 757, 704, 624, 505; [α]20D: −1.7 (c = 1.00, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.68 (d, J = 7.5 Hz, 4H), 7.44–7.36 (m, 6H), 7.23 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.6 Hz, 2H), 5.95 (d, J = 3.6 Hz, 1H), 4.86 (d, J = 6.7 Hz, 1H), 4.66 (dd, J = 6.7 Hz, 8.8 Hz, 2H), 4.53 (d, J = 3.6 Hz, 1H), 4.50 (d, J = 10.8 Hz, 1H), 4.45 (d, J = 10.8 Hz, 1H), 4.35–4.31(m, 3H), 4.02 (d, J = 3.1 Hz, 1H), 3.75 (s, 3H), 3.39 (s, 3H), 1.63 (br, 1H), 1.52 (s, 3H), 1.32 (s, 3H), 1.03 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 159.3, 135.5, 135.5, 132.8, 132.8, 129.8, 126.7, 129.2, 127.7, 113.7, 111.9, 105.7, 94.2, 84.6, 82.5, 82.2, 81.8, 80.5, 72.5, 65.3, 55.6, 55.2, 52.5, 26.9, 26.6, 26.3, 19.1; HRMS (ESI-QTOF): [M + Na]+ calcd for C37H46O8Si 669.2842 found 669.2854.
:20 EtOAc/hexane) to obtain the benzylated compound 20 (70%, 0.245 g). Physical appearance: Sticky colourless liquid; Rf: 0.8 (20% EtOAc/hexane); IR (neat, cm−1): 3071, 3015, 2933, 2858, 1612, 1588, 1514, 1471, 1427, 1374, 1303, 1251, 12161164, 1174, 1112, 1078, 1035, 936, 910, 823, 758, 703, 667, 614, 505; [α]20D: −2.3 (c = 1.00, CHCl3); 1H NMR (500 MHz, CDCl3)δ 7.73 (d, J = 7.2 Hz, 4H), 7.46–7.43 (m, 2H), 7.41–7.38 (m, 6H), 7.35–7.32 (m, 2H), 7.28 (d, J = 7.1 Hz, 1H), 7.24 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.6 Hz, 2H), 5.99 (d, J = 3.7 Hz, 1H), 5.78 (d, J = 12.1 Hz, 1H), 4.60 (d, J = 12.1 Hz, 1H), 4.55–4.47 (m, 4H), 4.42 (dd, J = 3.1 Hz, 8.5 Hz, 1H), 4.38–4.37 (m, 2H), 4.07 (d, J = 3.21 Hz, 1H), 3.77 (s, 3H), 1.54 (s, 3H), 1.34 (s, 3H), 1.08 (s, 9H); 13C NMR(125 MHz, CDCl3): δ 159.2, 137.7, 135.5, 135.4, 132.8, 132.8, 129.8, 129.5, 129.3, 128.1, 127.9, 127.7, 127.7, 127.4, 113.6, 111.8, 105.8, 85.0, 82.4, 82.3, 81.7, 81.0, 72.4, 70.7, 68.6, 55.1, 52.5, 26.8, 26.6, 26.3, 19.1; HRMS (ESI-QTOF): [M + K]+ calcd for C42H48KO7Si 731.2797; found 731.2801.
:1) cooled to 0 °C was added 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (0.19 g, 0.85 mmol) and the solution was stirred at 25 °C for 8 h. The reaction mixture was quenched with sat. NaHCO3 (20 mL) and the mixture was extracted with EtOAc (20 mL × 3). The organic layer was washed with water and brine solution, dried over Na2SO4, and then concentrated under reduced pressure to obtain the crude product, which was further purified by silica gel column chromatography (1:4 EtOAc/hexane) to obtain alcohol 13 (70%, 0.138 g). Physical appearance: Sticky colourless liquid; Rf: 0.2 (30% EtOAc/hexane); IR (neat, cm−1): 3463, 2932, 2858, 1961, 1471, 1454, 1428, 1374, 1313, 1259, 1216, 1163, 1112, 1070, 1013, 956, 887, 854, 823, 754, 702, 614, 702, 614, 505; [α]20D: +36.5 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.76 (d, J = 6.8 Hz, 4H), 7.48–7.41 (m, 6H), 7.37–7.31 (m, 5H), 6.0 (d, J = 3.5 Hz, 1H), 4.79 (d, J = 11.7 Hz, 1H), 4.57(d, J = 11.8 Hz, 1H), 4.49–4.42 (m, 4H), 4.28–4.25 (m, 2H), 2.66 (br, 1H), 1.52 (s, 3H), 1.34 (s, 3H), 1.10 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 136.9, 135.4, 132.8, 129.8, 128.3, 128.1, 127.7, 127.7, 113.7, 111.7, 105.2, 86.2, 84.681.6, 80.3, 75.7, 70.6, 67.9, 52.5, 26.7, 26.5, 26.2, 19.0; HRMS (ESI-QTOF): [M + Na]+ calcd for C34H40NaO6Si 595.2476; found 595.2486.
:10 EtOAc/hexane) to obtain ester 21 (0.25 g, 65%). Physical appearance: Sticky colourless liquid; Rf: 0.6 (20% EtOAc/hexane); IR (neat, cm−1): 2933, 2861, 1712, 1624, 1515, 1463, 1371, 1329, 1253, 1217, 1136, 1039, 966, 852, 824, 757, 702, 610, 505; [α]20D: +14.5 (c = 1.00, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.72–7.69 (m, 4H), 7.45–7.38 (m, 6H), 7.38–7.37 (m, 3H), 7.33 (t, J = 7.6 Hz, 2H), 7.30 (d, J = 12.7 Hz, 1H), 5.96 (d, J = 3.81 Hz, 1H), 5.30 (d, J = 12.7 Hz, 1H), 4.90 (d, J = 11.8 Hz, 1H), 4.55 (d, J = 11.8 Hz, 1H), 4.50 (d, J = 3.8 Hz, 1H), 4.45–4.42 (m, 2H), 4.43–4.37 (m, 3H), 4.14(qt, J = 7.1 Hz, 2H), 1.32 (s, 3H), 1.25 (t, J = 7.9 Hz, 3H), 1.07 (s, 9H); 13C NMR (125 MHz, CDCl3): δ 166.9, 160.0, 137.4, 135.5, 135.5, 132.8, 132.8, 129.8, 128.2, 127.9, 127.7, 127.7, 127.6, 112.4, 105.5, 98.8, 86.0, 83.6, 81.4, 81.2, 79.7, 70.7, 67.6, 59.9, 52.4, 26.6, 26.6, 26.3, 19.0, 14.2; HRMS (ESI-QTOF): [M + K]+ calcd for C39H46KO8Si 709.2594; found 709.2592.
:3 EtOAc/hexanes) to obtain the allylic alcohol 22 (128 mg, 85%). Physical appearance: Sticky colourless oil; Rf: 0.4 (25% EtOAc/hexanes); IR (neat, cm−1): 3485, 2932, 2861, 1961, 1889, 1825, 1667, 1651, 1455, 1428, 1373, 1306, 1260, 1216, 1165, 1023, 939, 854, 739, 702, 613, 506; [α]20D: + 79.9 (c = 0.50, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.75–7.69 (m, 4H), 7.48–7.26 (m, 11H), 6.31 (d, J = 12.7 Hz, 1H), 5.96 (d, J = 3.8 Hz, 1H), 5.13 (dt, J = 12.7, 7.3 Hz, 1H), 4.76 (d, J = 11.8 Hz, 1H), 4.56 (dd, J = 10.1, 8.0 Hz, 2H), 4.47–4.30 (m, 5H), 3.97 (d, J = 7.2 Hz, 2H), 1.54 (s, 3H), 1.32 (s, 3H), 1.08 (s, 9H); 13C NMR (100 MHz, CDCl3)δ 147.9, 137.7, 135.7, 135.6, 133.0, 132.9, 130.0, 128.4, 128.1, 127.9, 127.9, 127.7, 112.3, 105.8, 105.7, 85.6, 82.3, 81.7, 81.6, 80.5, 70.8, 68.1, 60.3, 52.7, 26.9, 26.8, 26.5, 19.3; HRMS (ESI-QTOF): [M + K]+ calcd for C37H44KO7Si 667.2488; found 667.2487
:7 EtOAc/hexane) to obtain the RRCEM precursor 12 (42 mg, 40%) and the minor compound 23 (31 mg, 30%).
Data for compound (12). Physical appearance: Sticky colourless oil; Rf: 0.6 (20% EtOAc/hexane); IR (neat, cm−1): 3077, 2932, 2867, 1653, 1456, 1429, 1374, 1261, 1217, 1164, 1114, 1070, 1020, 936, 856, 826, 741, 703, 614, 506; [α]20D: + 18.7 (c = 0.5, CHCl3); 1H NMR (400 MHz, CDCl3)δ 7.71 (dt, J = 8.0, 1.8 Hz, 4H), 7.45–7.35 (m, 8H), 7.35–7.27 (m, 3H), 6.30 (d, J = 12.7 Hz, 1H), 5.95 (d, J = 3.8 Hz, 1H), 5.93–5.83 (m, 1H), 5.25 (dd, J = 17.2, 1.6 Hz, 1H), 5.17 (dd, J = 10.4, 1.4 Hz, 1H), 5.05 (dt, J = 12.7, 7.3 Hz, 1H), 4.76 (d, J = 11.9 Hz, 1H), 4.60–4.52 (m, 2H), 4.45–4.39 (m, 2H), 4.36 (s, 2H), 4.33 (d, J = 2.6 Hz, 1H), 3.91 (dd, J = 4.0, 2.8 Hz, 2H), 3.82 (d, J = 7.2 Hz, 2H), 1.53 (s, 3H), 1.32 (s, 3H), 1.07 (s, 9H); 13C NMR (100 MHz, CDCl3)δ 148.4, 135.7, 135.7, 134.9, 133.1, 130.1, 128.5, 128.2, 127.9, 127.9, 127.7, 117.3, 112.4, 105.9, 102.9, 85.7, 82.4, 81.8, 81.6, 80.4, 70.9, 70.8, 68.1, 67.4, 52.7, 26.9, 26.8, 26.5, 19.3; HRMS (ESI-QTOF): [M + Na]+ calcd for C40H48NaO7Si 691.3063; found 691.3062.
Data for compound (23). Physical appearance: Sticky colourless oil; Rf: 0.7 (30% EtOAc/hexane); IR (neat, cm−1): 3079, 2930, 2866, 1663, 1450, 1429, 1374, 1261, 1217, 1164, 1113, 1072, 1010, 932, 944, 856
826, 826, 741, 703, 645, 614, 502; [α]20D: +26.7 (c = 0.5, CHCl3); 1H NMR (500 MHz, CDCl3)δ 7.44–7.26 (m, 5H), 6.38 (d, J = 12.7 Hz, 1H), 5.95 (d, J = 3.7 Hz, 1H), 5.95–5.83 (m, 2H), 5.30 (d, J = 13.5 Hz, 1H), 5.27 (d, J = 14.6 Hz, 1H), 5.22 (d, J = 10.4 Hz, 1H), 5.19 (d, J = 10.4 Hz, 1H), 5.14–5.06 (m, 1H), 4.82 (d, J = 11.9 Hz, 1H), 4.65 (d, J = 11.9 Hz, 1H), 4.56 (d, J = 3.7 Hz, 1H), 4.50–4.42 (m, 2H), 4.41 (d, J = 2.4 Hz, 1H), 4.19 (s, 2H), 4.03 (d, J = 5.5 Hz, 2H), 3.95 (d, J = 5.6 Hz, 2H), 3.88 (d, J = 7.3 Hz, 2H), 1.51 (s, 3H), 1.31 (s, 3H); 13C NMR (100 MHz, CDCl3)δ 148.4, 137.7, 134.8, 134.0, 128.4, 128.1, 127.8, 118.0, 117.35, 112.4, 105.1, 102.9, 82.3, 81.8, 81.5, 71.2, 70.8, 70.7, 68.2, 67.4, 57.4, 26.9, 26.5; HRMS (ESI-QTOF): [M + Na]+ calcd for C27H34NaO7 493.2206; found 493.2208.
:45 EtOAc/hexane) to obtain alcohols 26 (2.21 g, 50%) and 25 (1.34 g, 30%).
Data for major diastereomers (26). Physical appearance: Sticky yellow liquid; Rf: 0.5 (30% EtOAc/hexane × 2); IR (neat, cm−1): 3467, 2933, 2858, 2345, 1613, 1514, 1472, 1428, 1374, 1302, 1215, 1165, 1112, 1075, 1019, 890, 825, 755, 704, 614, 506; [α]20D: −37.4 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3)δ 7.36–7.28 (m, 5H), 7.26 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H), 5.97 (d, J = 3.6 Hz, 1H), 4.78 (d, J = 8.1 Hz, 1H), 4.61 (dd, J = 7.4, 3.8 Hz, 2H), 4.57 (s, 2H), 4.48 (d, J = 11.2 Hz, 1H), 4.31 (dd, J = 8.0, 3.3 Hz, 1H), 4.19 (s, 2H), 4.08 (d, J = 3.3 Hz, 1H), 2.54 (brs, 1H), 1.51 (s, 3H), 1.33 (s, 3H); 13C NMR (100 MHz, CDCl3)δ 159.6, 137.5, 129.7, 129.2, 128.6, 128.16 128.0, 114.0, 112.4, 105.7, 83.8, 83.7, 82.6, 82.1, 81.9, 72.5, 71.8, 62.0, 57.5, 55.4, 27.0, 26.5; HRMS (ESI-QTOF): [M + Na]+ calcd for C26H30NaO7 477.1884; found 477.1888.
Data for minor diastereomers (25). Physical appearance: Sticky yellow liquid; Rf: 0.6 (30% EtOAc/hexane ×2); IR (neat, cm−1): 3461, 2989, 2932, 2323, 1720, 1610, 1513, 1454, 1380, 1354, 1300, 1254, 1219, 1164, 1073, 1029, 888, 839, 756, 699, 666, 636, 590, 517; [α]20D: −45.80 (c = 2.00, CHCl3); 1H NMR (400 MHz, CDCl3)δ 7.34–7.27 (m, 5H), 7.25 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.6 Hz, 2H), 6.01 (d, J = 3.8 Hz, 1H), 4.80–4.73 (m, 1H), 4.63 (d, J = 11.1 Hz, 1H), 4.59 (d, J = 3.8 Hz, 1H), 4.56 (s, 2H), 4.49 (d, J = 11.1 Hz, 1H), 4.29–4.22 (m, 2H), 4.16 (d, J = 1.4 Hz, 2H), 3.75 (s, 3H), 3.46 (d, J = 9.2 Hz, 1H), 1.48 (s, 3H), 1.31 (s, 3H); 13C NMR (100 MHz, CDCl3)δ 159.8, 137.5, 130.0, 128.6, 128.2, 128.0, 114.2, 112.1, 105.7, 85.1, 83.4, 82.0, 81.8, 81.1, 72.5, 71.8, 61.5, 57.5, 55.4, 27.0, 26.4; HRMS (ESI-QTOF): [M + Na]+ calcd for C26H30NaO7 477.1884; found 477.1867.
:10 EtOAc/hexane) to obtain the benzylated compound 27 (72%, 4.35 g). Physical appearance: Sticky colourless liquid; Rf: 0.7 (20% EtOAc/hexane); IR (neat, cm−1): 3071, 3015, 2933, 2858, 1612, 1588, 1514, 1471, 1427, 1374, 1303, 1251, 1216, 1164, 1174, 1112, 1078, 1035, 936, 910, 823, 758, 703, 667, 614, 505; [α]20D: −35.3 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3)δ 7.45–7.26 (m, 10H), 7.18 (d, J = 8.6 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 5.97 (d, J = 3.6 Hz, 1H), 4.90 (d, J = 11.3 Hz, 1H), 4.69–4.61 (m, 3H), 4.61–4.42 (m, 4H), 4.38 (dd, J = 9.1, 3.0 Hz, 1H), 4.31 (d, J = 1.4 Hz, 2H), 4.09 (d, J = 2.9 Hz, 1H), 3.78 (s, 3H), 1.53 (s, 3H), 1.34 (s, 3H); 13C NMR (100 MHz, CDCl3)δ 139.2, 131.5, 123.8, 123.3, 77.2, 77.0, 76.7, 59.1, 39.4, 26.2, 25.5, 17.5, 16.1; HRMS (ESI-QTOF): [M + Na]+ calcd for C33H36NaO7 567.2354; found 567.2353.
:1) cooled to 0 °C was added DDQ (2.82 g, 12.4 mmol) and the solution was stirred at 25 °C for 5 h. The reaction mixture was quenched with sat. NaHCO3 (20 mL) which was added at 0 °C, and the mixture was extracted with ethyl acetate (20 mL × 3). The organic layer was washed with water, followed by brine, dried over NaSO4, and then concentrated to obtain the crude product. The crude product was purified through silica gel column chromatography (1:3 EtOAc/hexane) to obtain alcohol 28 (80%, 1.5 g). Physical appearance: Sticky colourless liquid; Rf: 0.25 (30% EtOAc/hexane); IR (neat, cm−1): 3540, 2932, 2858, 1961, 1471, 1454, 1428, 1374, 1313, 1259, 1216, 1163, 1112, 1070, 1013, 956, 887, 854, 823, 754, 702, 614, 702, 614, 505; [α]20D: +38.5 (c = 1.00, CHCl3); 1H NMR (500 MHz, CDCl3)δ 7.46–7.26 (m, 10H), 6.02 (d, J = 3.6 Hz, 1H), 4.90 (d, J = 11.8 Hz, 1H), 4.69 (d, J = 11.8 Hz, 1H), 4.63 (s, 2H), 4.56 (d, J = 6.2 Hz, 1H), 4.50 (d, J = 3.6 Hz, 1H), 4.40–4.33 (m, 2H), 4.27 (d, J = 1.3 Hz, 2H), 1.53 (s, 3H), 1.34 (s, 3H); 13C NMR (125 MHz, CDCl3)δ 137.0, 136.9, 128.4, 128.3, 128.1, 128.0, 127.9, 127.8, 111.8, 105.3, 84.7, 83.8, 81.8, 81.6, 77.2, 77.0, 76.7, 75.8, 71.7, 71.0, 68.1, 57.2, 26.7, 26.2; HRMS (ESI-QTOF): [M + Na]+ calcd for C25H28NaO6 447.1774; found 447.1778.
:10 EtOAc/hexane) to obtain ester 29 (0.82 g, 68%). Physical appearance: Sticky colourless liquid; Rf: 0.5 (20% EtOAc/hexanes); IR (neat, cm−1): 3032, 2933, 2861, 1712, 1624, 1515, 1463, 1371, 1329, 1253, 1217, 1136, 1039, 966, 852, 824, 757, 702, 610, 505; [α]20D: +24.5 (c = 1.00, CHCl3); 1H NMR (400 MHz, CDCl3)δ 7.51 (d, J = 12.7 Hz, 1H), 7.43–7.23 (m, 10H), 5.99 (d, J = 3.8 Hz, 1H), 5.37 (d, J = 12.7 Hz, 1H), 4.84 (d, J = 11.9 Hz, 1H), 4.66 (d, J = 11.9 Hz, 1H), 4.54 (ddd, J = 10.3, 6.2, 2.3 Hz, 5H), 4.47 (t, J = 4.7 Hz, 1H), 4.29–4.18 (m, 2H), 4.14 (q, J = 7.1 Hz, 2H), 1.53 (s, 3H), 1.32 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3)δ 166.9, 160.1, 137.3, 137.1, 128.3, 128.2, 127.9, 127.8, 127.6, 112.5, 105.5, 98.9, 83.9, 83.6, 81.5, 81.4, 81.3, 77.3, 77.0, 76.6, 71.6, 71.0, 67.8, 59.9, 57.1, 26.6, 26.2, 14.2; HRMS (ESI-QTOF): [M + Na]+ calcd for C30H34NaO8 545.2146; found 545.2146.
:25 EtOAc/hexanes) to obtain the allylic alcohol 30 (585 mg, 80%). Physical appearance: sticky colourless oil; Rf: 0.3 (25% EtOAc/hexane); IR (neat, cm−1): 3485, 3021, 2932, 2861, 1632, 1455, 1428, 1373, 1306, 1260, 1216, 1165, 1023, 939, 854, 739, 702, 613, 506; [α]20D: +79.9 (c = 0.50, CHCl3); 1H NMR (400 MHz, CDCl3)δ 7.52–7.15 (m, 10H), 6.37 (d, J = 12.7 Hz, 1H), 5.98 (d, J = 3.8 Hz, 1H), 5.16 (dd, J = 7.1, 5.6 Hz, 1H), 4.84 (d, J = 11.9 Hz, 1H), 4.67 (d, J = 11.9 Hz, 1H), 4.58 (s, 2H), 4.56 (d, J = 3.8 Hz, 1H), 4.50 (d, J = 2.8 Hz, 2H), 4.43 (d, J = 2.4 Hz, 1H), 4.22 (d, J = 0.7 Hz, 2H), 3.95 (d, J = 7.2 Hz, 2H), 1.53 (s, 3H), 1.32 (s, 3H); 13C NMR (100 MHz, CDCl3)δ 147.8, 137.6, 137.2, 128.5, 128.4, 128.1, 128.0, 127.7, 112.3, 105.8, 105.7, 83.1, 82.4, 82.1, 81.9, 81.6, 71.7, 71.1, 68.3, 60.1, 57.3, 26.8, 26.4; HRMS (ESI-QTOF): [M + Na]+ calcd for C28H32NaO7 503.2040; found 503.2040.
:9 EtOAc/hexane) to obtain the RRCEM precursor 31 (300 mg, 60%). Physical appearance: sticky colourless oil; Rf: 0.5 (20% EtOAc/hexane); IR (neat, cm−1): 3077, 2932, 2867, 1653, 1456, 1429, 1374, 1261, 1217, 1164, 1114, 1070, 1020, 936, 856, 826, 741, 703, 614, 506; [α]20D: +28.7 (c = 0.5, CHCl3); 1H NMR (500 MHz, CDCl3)δ 7.41 (d, J = 7.3 Hz, 2H), 7.39–7.30 (m, 7H), 7.28 (d, J = 7.3 Hz, 1H), 6.39 (d, J = 12.7 Hz, 1H), 5.97 (d, J = 3.8 Hz, 1H), 5.89 (ddd, J = 22.8, 10.8, 5.6 Hz, 1H), 5.25 (dd, J = 17.2, 1.4 Hz, 1H), 5.17 (d, J = 10.4 Hz, 1H), 5.11 (dt, J = 12.9, 7.3 Hz, 1H), 4.84 (d, J = 11.9 Hz, 1H), 4.67 (d, J = 11.9 Hz, 1H), 4.57 (s, 3H), 4.49 (d, J = 3.0 Hz, 2H), 4.43 (d, J = 2.5 Hz, 1H), 4.22 (s, 2H), 3.92 (d, J = 5.7 Hz, 2H), 3.83 (d, J = 7.3 Hz, 2H), 1.53 (s, 3H), 1.32 (s, 3H); 13C NMR (125 MHz, CDCl3)δ 148.4, 137.7, 137.4, 134.8, 128.6, 128.4, 128.1, 128.1, 128.0, 127.8, 117.2, 112.4, 105.9, 102.9, 83.2, 82.1, 82.1, 81.9, 81.1, 71.7, 71.2, 70.8, 68.3, 67.3, 57.4, 26.9, 26.5; HRMS (ESI-QTOF): [M + Na]+ calcd for C31H36NaO7 543.2368; found 543.2353.
:12 EtOAc/hexane) to obtain the corresponding 1,3-diene 32 (132 mg, 80%). Physical appearance: Sticky colourless oil; Rf: 0.6 (20% EtOAc/hexane); [α]20D: +21.82 (c = 1.5, CHCl3); IR (neat, cm−1): 3077, 2939, 2867, 1634, 1450, 1429, 1374, 1261, 1217, 1164, 1114, 1070, 1020, 936, 856, 826, 741, 703, 614, 506; 1H NMR (500 MHz, CDCl3)δ 7.39–7.27 (m, 10H), 6.86 (s, 1H), 5.89 (d, J = 3.6 Hz, 1H), 5.12 (s, 1H), 5.07 (s, 1H), 4.71 (d, J = 11.5 Hz, 1H), 4.69 (d, J = 3.6 Hz, 1H), 4.63 (d, J = 11.5 Hz, 1H), 4.54–4.51 (m, 1H), 4.48 (d, J = 0.8 Hz, 2H), 4.39 (d, J = 2.0 Hz, 1H), 4.38 (d, J = 1.5 Hz, 1H), 4.21–4.11 (m, 2H), 1.54 (s, 3H), 1.36 (s, 3H); 13C NMR (100 MHz, CDCl3)δ 143.6, 140.2, 138.3, 138.0, 128.5, 128.4, 128.0, 127.9, 127.7, 112.4, 111.7, 109.9, 104.8, 83.8, 74.8, 74.24, 72.0, 71.6, 71.4, 66.8, 26.8, 26.3; HRMS (ESI-QTOF): [M + Na]+ calcd for C27H30NaO6 473.1935; found 473.1929.
:5 EtOAc/hexane), affording the aromatized product 36 (60 mg, 56%). Physical appearance: Yellow-orange powder solid; Rf: 0.4 (30% EtOAc/hexane × 2); IR (KBr) cm−1: 3432, 3077, 2932, 2867, 1456, 1429, 1374, 1261, 1217, 1164, 1114, 1070, 1020, 936, 856, 826, 741, 703, 614, 506; [α]20D: +20.7 (c = 0.5, CHCl3); 1H NMR (500 MHz, CDCl3)δ 8.61 (s, 1H), 8.35–8.28 (m, 2H), 8.23 (s, 1H), 7.84–7.79 (m, 2H), 7.40–7.20 (m, 10H), 6.03 (d, J = 3.7 Hz, 1H), 5.11 (d, J = 5.8 Hz, 1H), 4.55 (d, J = 2.5 Hz, 1H), 4.54–4.51 (m, 2H), 4.49 (dd, J = 5.5, 2.6 Hz, 1H), 4.48 (dd, J = 11.3, 4.3 Hz, 2H), 4.42 (d, J = 3.7 Hz, 1H), 4.36 (d, J = 12.0 Hz, 1H), 3.86 (d, J = 2.3 Hz, 1H), 1.43 (s, 3H), 1.27 (s, 3H); 13C NMR (125 MHz, CDCl3)δ 182.7, 182.5, 144.0, 143.2, 141.7, 137.0, 136.5, 134.3, 134.2, 133.6, 133.4, 132.9, 128.9, 128.6, 128.5, 128.3, 128.2, 128.1, 128.0, 128.0, 127.3, 127.2, 111.7, 105.0, 84.9, 82.5, 77.3, 77.0, 76.7, 75.4, 73.2, 71.3, 69.4, 26.7, 26.1; HRMS (ESI-QTOF): [M + Na]+ calcd for C37H34NaO8 629.2254; found 629.2252.
:5 EtOAc/hexane), affording the aromatized product 39 (35 mg, 50%). Physical appearance: Yellow-orange sticky solid; Rf: 0.5 (30% EtOAc/hexane); IR (KBr, cm−1): 3528, 3423, 2922, 2847, 1752, 1674, 1605, 1591, 1446, 1216, 1163, 1147, 1020, 976, 790, 668; [α]20D: +85.40 (c = 0.4, CHCl3); 1H NMR (500 MHz, CDCl3)δ 8.52 (s, 1H), 8.29 (s, 1H), 7.95 (d, J = 7.4 Hz, 1H), 7.70 (t, J = 8.0 Hz, 1H), 7.37–7.31 (m, 5H), 7.28–7.24 (m, 2H), 7.22 (dd, J = 7.4, 5.2 Hz, 3H), 5.80 (d, J = 3.5 Hz, 1H), 5.12 (d, J = 7.2 Hz, 1H), 4.75 (d, J = 12.4 Hz, 1H), 4.57 (s, 2H), 4.52 (dd, J = 12.2, 7.4 Hz, 2H), 4.44 (d, J = 3.5 Hz, 1H), 4.31 (s, 1H), 4.24–4.18 (m, 2H), 4.03 (s, 3H), 1.31 (s, 3H), 1.21 (s, 3H); 13C NMR (125 MHz, CDCl3)δ 183.2, 182.4, 160.5, 144.1, 143.7, 137.7, 137.3, 135.9, 135.2, 134.6, 132.5, 128.8, 128.6, 128.4, 128.2, 128.1, 128.0, 128.0, 125.8, 121.6, 120.0, 118.1, 111.8; HRMS (ESI-QTOF): [M + K]+ calcd for C38H36O9 675.1962; found 675.1962.
:1) and treated with OsO4 (1 mol%) and N-methylmorpholine N-oxide (26.3 μL, 0.24 mmol) at room temperature. The reaction mixture was then allowed to stir for 8 h. After the consumption of the starting material, the reaction temperature was brought to 0 °C, and an excess of Et3N (0.5 mL) was added in a drop-wise manner and stirred at rt for 12 h. After completion of the reaction, the solvent was evaporated, and the residue was purified using silica gel column chromatography (1:4 EtOAc/hexane), affording compound 46 (20 mg, 40%). Physical appearance: Yellow-orange sticky solid; Rf: 0.5 (20% EtOAc/hexane × 2); IR (neat) cm−1: 3427, 2922, 2857, 1674, 1639, 1603, 1456, 1354, 1291, 1212, 1166, 1070, 1015, 914, 885, 863, 760, 699, 607; [α]20D: +34.48 (c = 1.5, CHCl3); 1H NMR (400 MHz, CDCl3)δ 12.62 (s, 1H), 8.63 (s, 1H), 8.23 (s, 1H), 7.84 (d, J = 7.5 Hz, 1H), 7.70 (t, J = 8.0 Hz, 1H), 7.43–7.37 (m, 3H), 7.36–7.29 (m, 7H), 7.27–7.22 (m, 2H), 6.05 (d, J = 3.7 Hz, 1H), 5.14 (d, J = 5.5 Hz, 1H), 4.56 (d, J = 11.9 Hz, 3H), 4.52–4.47 (m, 3H), 4.44 (d, J = 3.8 Hz, 1H), 4.39 (d, J = 12.0 Hz, 1H), 4.18 (d, J = 3.1 Hz, 1H), 3.92 (s, 1H), 1.44 (s, 3H), 1.30 (s, 4H); 13C NMR (100 MHz, CDCl3)δ 188.1, 182.1, 162.7, 144.3, 142.4, 137.0, 136.9, 136.6, 133.5, 133.4, 133.1, 129.1, 128.8, 128.7, 128.4, 128.3, 128.3, 128.2, 128.1, 124.7, 119.7, 116.3, 111.8, 105.1, 85.1, 82.4, 75.7, 73.4, 71.5, 69.5, 26.9, 26.3; HRMS (ESI-QTOF): [M + Na]+ calcd for C37H34NaO9 645.2047; found 645.2047.
:10 EtOAc/hexane) to obtain the benzylated compound S4 (80%, 3.3 g). Physical appearance: Sticky yellow liquid; Rf: 0.7 (25% EtOAc/hexane); IR (neat, cm−1): = 3461, 2989, 2932, 2227, 1720, 1610, 1584, 1513, 1454, 1380, 1354, 1300, 1254, 1164, 1073, 1029, 839, 756, 699, 666, 636, 590, 517; [α]20D: +31.53 (c = 0.5, CHCl3); 1H NMR (400 MHz, CDCl3)δ 7.45–7.26 (m, 10H), 7.18 (d, J = 8.6 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 5.97 (d, J = 3.6 Hz, 1H), 4.90 (d, J = 11.3 Hz, 1H), 4.68–4.61 (m, 3H), 4.58 (t, J = 7.7 Hz, 2H), 4.50 (d, J = 11.3 Hz, 1H), 4.46 (d, J = 11.4 Hz, 1H), 4.37 (dd, J = 9.1, 2.9 Hz, 1H), 4.30 (d, J = 1.3 Hz, 2H), 4.09 (d, J = 2.9 Hz, 1H), 3.78 (s, 3H), 1.53 (s, 3H), 1.34 (s, 3H); 13C NMR (100 MHz, CDCl3)δ 159.4, 137.7, 137.6, 129.7, 129.2, 128.5, 128.4, 128.3, 128.0, 127.9, 127.8, 113.9, 112.0, 105.3, 83.9, 83.2, 82.3, 81.3, 80.8, 72.1, 71.4, 70.9, 66.6, 57.6, 55.3, 26.9, 26.4; HRMS (ESI-QTOF): [M + Na]+ calcd for C33H36NaO7 567.2354; found 567.2353.
:1) cooled to 0 °C was added DDQ (4.72 g, 17.5 mmol) and the solution was stirred at 25 °C for 4 h. The reaction mixture was quenched with sat. NaHCO3 (30 mL) and the mixture was extracted with ethyl acetate (30 mL × 3). The organic layer was washed with water followed by brine, dried over Na2SO4, and then concentrated to obtain the crude product, which was further purified through silica gel column chromatography (1:35 EtOAc/hexane) to obtain the alcohol S5 (90%, 2.1 g). Physical appearance: Sticky colorless liquid; Rf: 0.2 (30% EtOAc/hexane); IR (neat, cm−1): 3471, 3089, 2988, 2931, 2861, 2237, 1957, 1723, 1630, 1606, 1496, 1454, 1375, 1354, 1315, 1216, 1164, 1081, 1020, 947, 884, 858, 750, 699, 638, 605, 540, 520; [α]20D: −52.56 (c = 3.0, CHCl3); 1H NMR (400 MHz, CDCl3)δ 7.39–7.28 (m, 10H), 5.98 (d, J = 3.6 Hz, 1H), 4.87 (d, J = 11.7 Hz, 1H), 4.65–4.59 (m, 4H), 4.50 (d, J = 3.6 Hz, 1H), 4.45 (d, J = 2.2 Hz, 1H), 4.28 (dd, J = 6.5, 2.1 Hz, 3H), 1.68 (br s, 1H), 1.50 (s, 3H), 1.32 (s, 3H); 13C NMR (100 MHz, CDCl3)δ 137.3, 136.6, 128.8, 128.6, 128.4, 128.3, 128.1, 112.0, 105.3, 84.9, 84.4, 81.9, 81.1, 75.5, 71.9, 71.6, 67.8, 57.5, 27.0, 26.3; HRMS (ESI-QTOF): [M + K]+ calcd for C25H28KO6 463.1482; found 463.1481.
:12 EtOAc/hexane) to obtain the ester S6 (0.92 g, 75%). Physical appearance: Sticky colourless liquid; Rf: 0.5 (20% EtOAc/hexanes); IR (neat, cm−1): 3030, 2980, 2850, 1713, 1645, 1626, 1496, 1454, 1374, 1324, 1285, 1260, 1216, 1194, 1166, 1138, 1067, 1048, 1024, 949, 907, 884, 853, 755, 699, 667, 644, 519; [α]20D: +87.41 (c = 1.5, CHCl3); 1H NMR (400 MHz, CDCl3)δ 7.42–7.27 (m, 10H), 5.93 (d, J = 3.7 Hz, 1H), 5.33 (d, J = 12.2 Hz, 1H), 4.80 (d, J = 12.2 Hz, 1H), 4.65 (s, 2H), 4.54 (d, J = 3.8 Hz, 1H), 4.48 (t, J = 7.2 Hz, 2H), 4.43–4.33 (m, 2H), 4.29 (d, J = 1.4 Hz, 2H), 4.23–4.13 (m, 2H), 1.51 (s, 3H), 1.31 (s, 3H), 1.28 (s, 3H); 13C NMR (100 MHz, CDCl3)δ 167.2, 160.6, 137.4, 136.8, 128.7, 128.5, 128.5, 128.3, 128.24, 128.0, 112.6, 105.2, 99.0, 83.6, 83.0, 82.9, 82.1, 80.5, 71.6, 70.9, 65.5, 60.1, 57.5, 26.7, 26.4, 14.4; HRMS (ESI-QTOF): [M + K]+ calcd for C30H34KO8 561.1851; found 561.1851.
:3 EtOAc/hexanes) to obtain the allylic alcohol S7 (1.6 g, 90%). Physical appearance: Sticky colourless oil; Rf: 0.35 (20% EtOAc/hexane); IR (neat, cm−1): 3475, 3064, 2989, 2935, 2345, 1876, 1808, 1670, 1652, 1496, 1454, 1375, 1310, 1218, 1167, 1107, 1073, 1025, 941, 854, 755, 699, 638, 517; [α]20D: −48.37 (c = 5.0, CHCl3); 1H NMR (500 MHz, CDCl3)δ 7.45–7.22 (m, 10H), 6.32 (d, J = 12.6 Hz, 1H), 5.96–5.85 (m, 2H), 5.27 (d, J = 17.2 Hz, 1H), 5.19 (d, J = 10.4 Hz, 1H), 5.16–5.05 (m, 1H), 4.82 (d, J = 11.3 Hz, 1H), 4.64 (s, 2H), 4.58 (d, J = 3.3 Hz, 1H), 4.49 (d, J = 11.3 Hz, 1H), 4.44 (d, J = 9.4 Hz, 1H), 4.40–4.33 (m, 2H), 4.28 (s, 2H), 3.95 (d, J = 5.3 Hz, 2H), 3.87 (d, J = 7.0 Hz, 2H), 1.51 (s, 3H), 1.31 (s, 3H); 13C NMR (125 MHz, CDCl3)δ 148.7, 137.5, 137.4, 134.9, 128.6, 128.5, 128.4, 128.0, 117.2, 112.4, 105.3, 102.7, 83.5, 83.3, 82.1, 81.1, 80.8, 77.2, 71.6, 71.0, 70.8, 67.3, 66.0, 57.6, 26.9, 26.5; HRMS (ESI-QTOF): [M + Na]+ calcd for C28H32NaO7 503.2040; found 503.2040.
:10 EtOAc/hexane) to obtain the RRCEM precursor S8 (240 mg, 70%). Physical appearance: Sticky colourless oil; Rf: 0.6 (20% EtOAc/hexane); IR (neat, cm−1): 3031, 2938, 2850, 2345, 1725, 1493, 1452, 1386, 1216, 1163, 1097, 1072, 1017, 946, 883, 885, 756, 699, 670, 521; [α]20D: 14.63 (c = 0.6, CHCl3); 1H NMR (500 MHz, CDCl3)δ 7.41–7.27 (m, 10H), 6.32 (d, J = 12.6 Hz, 1H), 5.95–5.87 (m, 2H), 5.27 (d, J = 17.2 Hz, 1H), 5.19 (d, J = 10.4 Hz, 1H), 5.16–5.07 (m, 1H), 4.82 (d, J = 11.3 Hz, 1H), 4.64 (s, 2H), 4.58 (d, J = 3.3 Hz, 1H), 4.49 (d, J = 11.3 Hz, 1H), 4.44 (d, J = 9.4 Hz, 1H), 4.40–4.33 (m, 2H), 4.28 (s, 2H), 3.95 (d, J = 5.3 Hz, 2H), 3.87 (d, J = 7.0 Hz, 2H), 1.51 (s, 3H), 1.31 (s, 3H); 13C NMR (125 MHz, CDCl3)δ 148.6, 137.4, 137.2, 134.7, 128.4, 128.3, 128.2, 127.8, 117.1, 112.2, 105.1, 102.6, 83.3, 83.1, 81.9, 80.9, 80.6, 71.4, 70.8, 70.6, 67.2, 65.8, 57.4, 26.7, 26.3; HRMS (ESI-QTOF): [M + Na]+ calcd for C31H36NaO7 543.2312; found 543.2312.
:12 EtOAc/hexane) to obtain the corresponding 1,3-diene 41 (160 mg, 85%). Physical appearance: Sticky colourless oil; Rf: 0.5 (20% EtOAc/hexane); [α]20D: +21.82 (c = 1.5, CHCl3); IR (neat, cm−1): 3037, 2983, 2928, 1725, 1637, 1496, 1454, 1380, 1379, 1283, 1264, 1215, 1164, 1082, 1024, 890, 865, 843, 756, 699, 666, 604; 1H NMR (400 MHz, CDCl3)δ 7.40–7.26 (m, 10H), 6.63 (s, 1H), 6.03 (d, J = 3.7 Hz, 1H), 5.20 (d, J = 1.4 Hz, 1H), 5.07 (d, J = 1.4 Hz, 1H), 4.76 (d, J = 11.5 Hz, 1H), 4.69 (dd, J = 3.7, 0.9 Hz, 1H), 4.64 (dd, J = 5.0, 2.3 Hz, 1H), 4.59 (s, 1H), 4.53 (d, J = 17.7 Hz, 1H), 4.45 (d, J = 25.8 Hz, 1H), 4.38 (d, J = 7.8 Hz, 1H), 4.24 (d, J = 12.2 Hz, 1H), 4.06 (d, J = 12.3 Hz, 1H), 1.54 (s, 3H), 1.36 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 142.1, 140.4, 138.3, 137.9, 128.4, 128.4, 128.1, 128.0, 127.7, 127.7, 113.5, 112.7, 112.3, 104.8, 83.6, 78.4, 72.3, 72.3, 71.7, 70.4, 67.6, 26.8, 26.4; HRMS (ESI-QTOF): [M + Na]+ calcd for C27H30NaO6 473.1894; found 473.1893.
:4 EtOAc/hexane), affording the aromatized product 42 (38 mg, 45%). Physical appearance: Yellow-orange powder solid; Rf: 0.5 (30% EtOAc/hexane); IR (KBr, cm−1): 3055, 2926, 2857, 2349, 1729, 1673, 1589, 1455, 1381, 13224, 1265, 1216, 1163, 1094, 1024, 890, 755, 717, 700; [α]20D: −26.94 (c = 1.00 CHCl3); 1H NMR (400 MHz, CDCl3)δ 8.23 (d, J = 7.4 Hz, 2H), 8.11 (s, 1H), 7.80–7.70 (m, 2H), 7.40–7.30 (m, 10H), 6.08 (d, J = 3.8 Hz, 1H), 5.16–5.11 (m, 1H), 5.02 (d, J = 4.8 Hz, 1H), 4.86–4.81 (m, 1H), 4.77 (d, J = 11.0 Hz, 1H), 4.74 (s, 1H), 4.72 (d, J = 9.6 Hz, 1H), 4.64 (d, J = 13.5 Hz, 1H), 4.57 (d, J = 11.0 Hz, 1H), 4.54 (s, 2H), 1.56 (s, 3H), 1.41 (s, 3H); 13C NMR (100 MHz, CDCl3)δ 183.1, 181.9, 154.9, 145.9, 137.8, 137.1, 135.1, 134.7, 134.4, 133.5, 132.6, 128.7, 128.5, 128.3, 128.0, 128.0, 127.3, 126.8, 126.7, 120.5, 113.0, 105.3, 84.7, 79.4, 73.1, 72.5, 71.3, 70.5, 69.0, 27.1, 26.6. HRMS (ESI-QTOF): [M + H]+ calcd for C37H33O8 605.2170; found 605.2163.
:5 EtOAc/hexane), affording the aromatized product 45 (35 mg, 50%). Physical appearance: Yellow sticky solid; Rf: 0.5 (20% EtOAc/hexane); IR (KBr, cm−1): 3502, 3040, 2926, 2864, 1672, 1638, 1592, 1512, 1454, 1372, 1318, 1262, 1161, 1098, 1023, 890, 836, 756, 699, 670; [α]20D: −56. 80 (c = 0.65, CHCl3); 1H NMR (500 MHz, CDCl3)δ 12.96 (s, 1H), 8.10 (s, 1H), 7.79–7.73 (m, 1H), 7.61 (t, J = 7.9 Hz, 1H), 7.40–7.27 (m, 11H), 6.09 (d, J = 3.8 Hz, 1H), 5.12 (d, J = 3.8 Hz, 1H), 5.01 (d, J = 4.6 Hz, 1H), 4.85–4.80 (m, 1H), 4.74 (d, J = 11.4 Hz, 2H), 4.69 (d, J = 13.5 Hz, 1H), 4.62 (d, J = 13.5 Hz, 1H), 4.55 (d, J = 11.2 Hz, 1H), 4.53 (s, 2H), 3.81 (s, 1H), 1.56 (s, 3H), 1.42 (s, 3H); 13C NMR (125 MHz, CDCl3)δ 188.2, 182.4, 162.6, 155.4, 146.6, 137.7, 137.0, 136.0, 134.7, 132.8, 128.7, 128.4, 128.3, 128.1, 128.0, 127.2, 124.9, 120.8, 120.0, 119.0, 117.2, 113.2, 105.3, 84.9, 79.8, 77.4, 77.2, 76.9, 73.2, 72.9, 71.4, 70.3, 69.0, 27.2, 26.8; HRMS (ESI-QTOF): [M + Na]+ calcd for C37H32NaO9 643.1894; found 643.1894.
Data availability
All of the data related to new compounds pertaining to this manuscript are available in the ESI.‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. P. K. acknowledges the financial support from the Science and Engineering Research Board, New Delhi, through a research grant (CRG/2022/001698). A. S. thanks the Indian Institute of Technology Bombay for the fellowship. The Indian Institute of Technology Bombay is gratefully acknowledged for providing all the necessary infrastructure and spectral facilities.References
- (a) J. Stetter and F. Lieb, Angew. Chem., 2000, 112, 1792–1812 ( Angew. Chem. , 2000 , 39 , 1724–1744 ) CrossRef; (b) G. M. Cragg, D. J. Newman and K. M. Snader, J. Nat. Prod., 1997, 60, 52–60 CrossRef CAS PubMed; (c) G. A. Cordell, Phytochemistry, 2000, 55, 463–480 CrossRef CAS PubMed; (d) A. Zeeck, G. Bach and H. Terlau, Internist, 2001, 42, 1422–1427 CrossRef CAS PubMed; (e) D. J. Newman, G. M. Cragg and K. M. Snader, J. Nat. Prod., 2003, 66, 1022–1037 CrossRef CAS PubMed; (f) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed; (g) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed; (h) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed; (i) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- Selected reviews for hybrid natural products and their analogues: (a) G. Mehta and V. K. Singh, Chem. Soc. Rev., 2002, 31, 324–334 RSC; (b) L. F. Tietze, H. P. Bell and S. Chandrasekhar, Angew. Chem., Int. Ed., 2003, 42, 3996–4028 CrossRef CAS PubMed; (c) R. J. M. Goss, S. Shankar and A. A. Fayad, Nat. Prod. Rep., 2012, 29, 870–889 RSC; (d) K. Palanichamy and K. P. Kaliappan, Pure Appl. Chem., 2013, 85, 1185–1202 CrossRef CAS; (e) P. Borah and P. Chowdhury, Curr. Drug Ther., 2017, 12, 3–22 CrossRef CAS; (f) S. Choudhary, P. K. Singh, H. Verma and H. Singh, Eur. J. Med. Chem., 2018, 151, 62–97 CrossRef CAS PubMed; (g) H. M. S. Kumar, L. Herrmann and S. B. Tsogoeva, Bioorg. Med. Chem. Lett., 2020, 30, 127514 CrossRef PubMed; (h) N. Micale, M. S. Molonia, A. Citarella, F. Cimino, A. Saija, M. Cristani and A. Speciale, Molecules, 2021, 26, 4665 CrossRef CAS PubMed; (i) I. Mancini, J. Vigna, D. Sighel and A. Defant, Molecules, 2022, 27, 4948 CrossRef CAS PubMed; (j) E. Sflakidou, G. Leonidis, E. Foroglou, C. Siokatas and V. Sarli, Molecules, 2022, 27, 6632 CrossRef CAS PubMed.
- (a) L. R. Reddy, J. F. Fournier, B. V. S. Reddy and E. J. Corey, J. Am. Chem. Soc., 2005, 127, 8974–8976 CrossRef CAS PubMed; (b) Y. Matsuya, T. Kawaguchi, K. Ishihara, K. Ahmed, Q.-L. Zhao, T. Kondo and H. Nemoto, Org. Lett., 2006, 8, 4609–4612 CrossRef CAS PubMed; (c) I. Paterson, G. J. Naylor and A. E. Wright, Chem. Commun., 2010, 46, 261–263 RSC.
- (a) D. J. Aberhart, Y. S. Chen, P. De Mayo and J. B. Stothers, Tetrahedron, 1965, 21, 1417–1432 CrossRef CAS; (b) B. Franck and G. Baumann, Chem. Ber., 1966, 99, 3863–3874 CrossRef CAS; (c) G. R. Pettit, M. Inoue, Y. Kamano, D. L. Herald, C. Arm, C. Dufresne, N. D. Christie, J. M. Schmidt, D. L. Doubek and T. S. Krupa, J. Am. Chem. Soc., 1988, 110, 2006–2007 CrossRef CAS; (d) M. A. F. Jalal, M. B. Hossain, D. J. Robeson and D. van der-Helm, J. Am. Chem. Soc., 1992, 114, 5967–5971 CrossRef CAS; (e) K. Matsuzaki, N. Tabata, H. Tomoda, Y. Iwai, H. Tanaka and S. Omura, Tetrahedron Lett., 1993, 34, 8251–8254 CrossRef CAS; (f) H. Shiozawa, T. Kagasaki, T. Kinoshita, H. Haruyama, H. Domon, Y. Utsui, K. Kodama and S. Takahashi, J. Antibiot., 1993, 46, 1834–1842 CrossRef CAS PubMed; (g) Y. F. Hallock, J. H. Cardellina II, M. Schaffer, G. Bringmann, G. Francois and M. R. Boyd, Bioorg. Med. Chem. Lett., 1998, 8, 1729–1734 CrossRef CAS PubMed.
- (a) P. Sensi, Rev. Infect. Dis., 1983, 5, S402–S406 CrossRef CAS PubMed; (b) H. G. Floss, Nat. Prod. Rep., 1997, 14, 433–452 RSC; (c) J. Bonjoch and D. Solé, Chem. Rev., 2000, 100, 3455–3482 CrossRef CAS PubMed; (d) H. Benke, G. Dagny, C. Lorenzo, S. Prashant, C. E. R. López, M. O. Kaileen, N. J. Hernández, V. Grabe and S. E. O'Connor, Nature, 2022, 607, 617–622 CrossRef PubMed.
- (a) G. Blasko, G. A. Cordell, A. Brossi and M. Suffness, Isolation, structure elucidation, and biosynthesis of the indole alkaloids of Catharanthus, in The Alkaloids, Academic Press, New York, 1990, vol. 37, pp. 1–76 Search PubMed; (b) Z. Bánóczi, A. Gorka-Kereskényi, J. Reményi, E. Orbán, L. Hazai, N. Tökési, J. Oláh, J. Ovádi, Z. Béni, V. Háda, C. Szántay, F. Jr Hudecz, G. Kalaus and C. Szántay, Bioconjugate Chem., 2010, 21, 1948–1955 CrossRef PubMed; (c) Z. Bánóczi, A. Keglevich, I. Szabó, I. Ranđelović, Z. Hegedüs, F. L. Regenbach, P. Keglevich, Z. Lengyel, Á. G-Kereskényi, Z. Dubrovay, V. Háda, Á. Szigetvári, C. Szántay Jr, L. Hazai, J. Tóvári and F. Hudecz, J. Pept. Sci., 2018, 24, 3118 CrossRef PubMed; (d) S. Mayer, P. Keglevich, A. Keglevich and L. Hazai, Curr. Org. Chem., 2021, 25, 1224–1234 CrossRef CAS.
- (a) K. Maeda, T. Takeuchi, K. Nitta, K. Yagashita, R. Utahara, T. Osata, M. Ueda, S. Kondo, Y. Okami and H Umezawa, J. Antibiot., 1956, 9, 75–81 CAS; (b) S. K. Gonda, K. M. Byrne, P. K. Herber, Y. Tondeur, D. Liberato and B. D. Hilton, J. Antibiot., 1984, 37, 1344–1356 CrossRef CAS PubMed; (c) A. Fredenhagen and U. Sequin, J. Antibiot., 1985, 38, 236–241 CrossRef PubMed; (d) K. Eckardt, in Quinones and Other Carbocyclic Antitumor Antibiotics in Antitumor Compounds of Natural Origin: Chemistry and Biology, ed. A. Aszalos, CRC Press, Boca Raton, 1981, vol. II Search PubMed; (e) U. Sequin, The Antibiotics of the Pluramycin Group (4H-Anthra [1,2-b] pyran Antibiotics), in Prog. Chem. Org. Nat. Prod, ed. L. Zechmeister, Springer, Wien, New York, 1986, vol. 50, pp. 58–122 Search PubMed.
- (a) K. P. Kaliappan and V. Ravikumar, Org. Biomol. Chem., 2005, 3, 848–851 RSC; (b) A. V. Subrahmanyam, K. Palanichamy and K. P. Kaliappan, Chem. – Eur. J., 2010, 16, 8545–8556 CrossRef CAS PubMed; (c) K. Palanichamy and K. P. Kaliappan, Pure Appl. Chem., 2013, 85, 1185–1202 CrossRef CAS; (d) A. A. Sayyad and K. P. Kaliappan, Eur. J. Org. Chem., 2017, 2017, 5055–5065 CrossRef CAS.
- S. Ghosh, M. F. Hossain, C. K. Malik and S. Maity, Tetrahedron, 2010, 66, 9159–9164 CrossRef CAS.
- (a) P. Muller, Helv. Chim. Acta, 1973, 56, 1243–1251 CrossRef; (b) W. Zhang, H. Ma, L. Zhou, Z. Sun, Z. Du, H. Miao and J. Xu, Molecules, 2008, 13, 3236–3245 CrossRef CAS PubMed; (c) L. Zhai, R. Shukla and R. Rathore, Org. Lett., 2009, 11, 3474–3477 CrossRef CAS PubMed; (d) L.-Y. Chen, S.-R. Li, P.-Y. Chen, H.-C. Chang, T.-P. Wang, I.-L. Tsai and E.-C. Wanga, Arkivoc, 2010, 11, 64–76 Search PubMed; (e) O. R. Luca, T. Wang, S. J. Konezny, V. S. Batista and R. H. Crabtree, New J. Chem., 2011, 35, 998–999 RSC; (f) Y. Li, D. Hong, Y. Zhu, P. Lu and Y.-G. Wang, Tetrahedron, 2011, 67, 8086–8091 CrossRef CAS; (g) X. Li, C. Li, B. Yin, C. Li, P. Liu, J. Li and Z. Shi, Chem. – Asian J., 2013, 8, 1408–1411 CrossRef CAS PubMed.
- (a) K. Krohn and J. Micheel, Tetrahedron, 1998, 54, 4827–4838 CrossRef CAS; (b) M. Pantin, D. Zon, R. Vomiandry, R. Foulgoc, D. Sissouma, A. Guingant and S. Collet, Tetrahedron Lett., 2015, 56, 2110–2112 CrossRef CAS.
- It was observed that in the epoxidation attempts of the major diene isomer 32 using m-CPBA in DCM, 32 did not react at all, even after adding an excess of reagents and extending the reaction time.
Footnotes |
| † Dedicated with respect to Prof. Sukh Dev on the occasion of his 100th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01049c |
| This journal is © The Royal Society of Chemistry 2024 |