
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed electrochemical C(sp3)–H acetoxylation of alcohol derivatives with an exo-directing group†
Daisuke
Ogawa
a,
Ayumu
Sasaki
a,
Takuya
Kochi
 a and
Fumitoshi
Kakiuchi
*ab
a and
Fumitoshi
Kakiuchi
*ab
aDepartment of Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: kakiuchi@chem.keio.ac.jp
bJST, CREST, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 2nd September 2024
Abstract
Palladium-catalyzed electrochemical C(sp3)–H acetoxylation to prepare 1,2-diol derivatives was achieved using oxime ethers as exo-directing groups. Various substrates containing alkoxy groups with a methyl branch at the α-position as well as norbornan-2-ol derivative were acetoxylated only using a catalytic amount of Pd(OAc)2 along with NaOAc/Ac2O/AcOH under anodic oxidation conditions.
Introduction
Combining metal-catalyzed C–H bond cleavage with electrochemical oxidation has become a widely-used strategy for environmentally-benign organic synthesis, because both technologies can contribute to reducing the amounts of reactive reagents and inevitable by-products.1,2 One of the challenges in electrochemical reactions, in search of greener synthesis, is avoiding the use of excessive mediators and supporting electrolytes other than the actual substrates directly involved in the transformation. One of the strategies to achieve this goal in metal-catalyzed electrochemical C–H functionalization is to utilize the cheap functionalizing agents that also work as a supporting electrolyte. In this case, the redox process with an electrode may proceed for either the substrate or the metal catalyst. In this context, our group has developed some C–H chlorination and bromination reactions3 of aromatic compounds using combinations of palladium catalysts with platinum electrodes, and anodic oxidation of halide ions is considered to produce halogen species at high oxidation states (Fig. 1a).4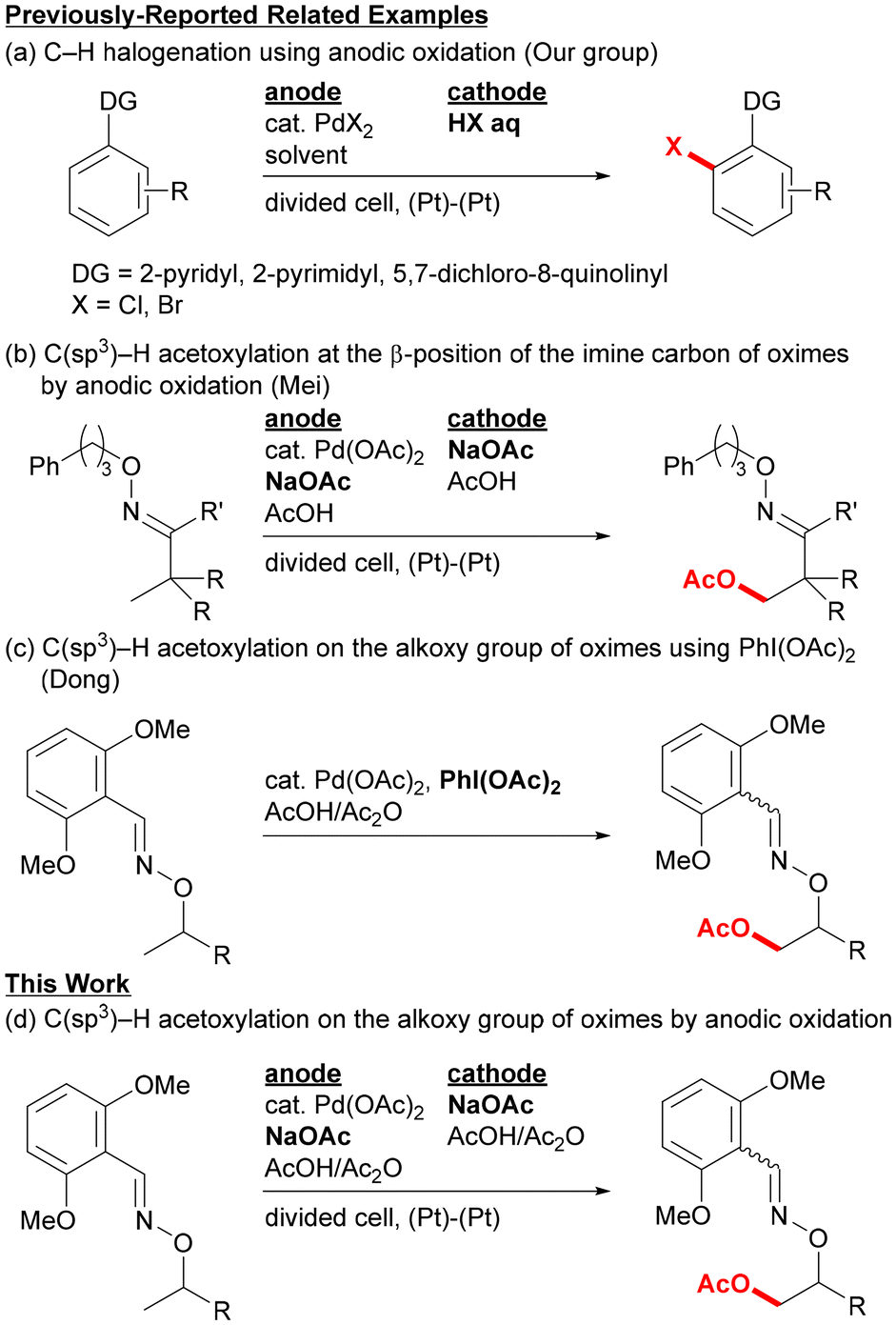 | ||
| Fig. 1 Palladium-catalyzed C–H oxidative functionalizations. | ||
In contrast to the metal-catalyzed electrochemical C–H functionalization of aromatic compounds, far fewer reports have been made on those of unreactive C(sp3)–H bonds.5–7 For example, Mei and co-workers reported in 2017 on the palladium-catalyzed electrochemical C(sp3)–H oxidation to introduce oxygen functionalities at the β-position of the imine carbon.5 In this reaction, β-hydroxy imine derivatives were obtained as products, and the reaction was considered to proceed via direct anodic oxidation of electron-rich palladium complexes formed by chelation-assisted C–H bond cleavage (Fig. 1b). Sanford's group also reported on a similar oxime-directed β-oxygenation of the imine carbon as well as benzylic C–H oxidation using several nitrogen directing groups.6 Inspired by these examples, we envisioned that the use of the metal-catalyzed electrochemical C–H functionalization technique for Dong's system reported in 2012,8 in which oxime ethers were used as exo-directing groups (Fig. 1c), would provide an efficient method for synthesis of 1,2-diol derivatives.
Here we report the palladium-catalyzed electrochemical C(sp3)–H acetoxylation to prepare 1,2-diol derivatives using oxime ethers as exo-directing groups (Fig. 1d). Various substrates containing alkoxy groups with a methyl branch at the α-position were acetoxylated at the methyl group only using a catalytic amount of Pd(OAc)2 along with NaOAc/Ac2O/AcOH under anodic oxidation conditions. Acetoxylation at a methine carbon was also possible for a norbornan-2-ol derivative.
Results and discussion
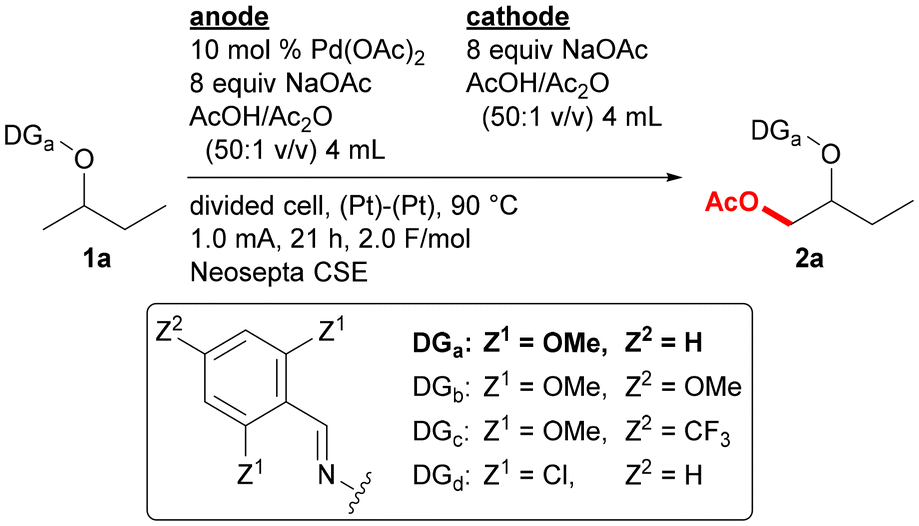
The optimized reaction conditions for the reaction of oxime ether 1a to form acetoxylation product 2a are as follows (Table 1): the reaction was conducted using an H-type divided cell equipped with two platinum electrodes, and two chambers were separated by a cation exchange membrane, Neosepta CSE. While substrate 1a and 10 mol% of Pd(OAc)2 was added to the anodic chamber, NaOAc, used as both an electrolyte and a substrate, and a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of AcOH and Ac2O was introduced to both chambers. The reaction was conducted with a 1.0 mA constant current at 90 °C for 21 h. For the optimization of the reaction conditions, the yield of product 2a was determined by adding the NMR yield of the (E)-isomer with the yield of (Z)-isomer calculated based on the GC ratio of the (E)/(Z)-isomers. Under the optimized reaction conditions, product 2a was formed in 72% yield with an (E)/(Z)-ratio of 3.0 (entry 1). Some byproducts, such as 2,6-dimethoxybenzaldehyde and -benzonitrile, were structurally determined, but no oxidation was observed at the 3- or 4-position of the 2-butoxy group. Reducing the amount of Pd(OAc)2 to 5.0 mol% decreased the yield to 38% (entry 2). Substrate 1a contains a 2,6-dimethoxyphenyl group in the directing group. While the use of 2,4,6-trimethoxyphenyl group (1b) provided the corresponding product 2b in a similar yield (entry 3), the substrate containing a 2,6-dimethoxy-4-trifluoromethylphenyl (1c) or a 2,6-dichlorophenyl (1d) group gave the corresponding product in a lower yield (entries 4 and 5). Extension of the reaction time to 24 h also reduced the yield, probably due to decomposition of the product (entry 6). With keeping the same electronic equivalent at 2.0 F mol−1, the electric current value was varied, but the reactions at 0.5 mA (43 h) and 1.5 mA (14 h) gave 2a in lower yields (entries 7 and 8). Several cation exchange membranes, such as Nafion 117, 324, and 966, and a sand core were also examined but provided product 2a in lower yields (entries 9–12). When AcOH was used as a sole solvent instead of AcOH/Ac2O (50:1 v/v), the yield slightly lowered to 60% (entry 13), probably because Ac2O promotes the regeneration of Pd(OAc)2 with avoiding catalyst decomposition.9 The desired reaction did not proceed without using Pd(OAc)2 or an electric current (entries 14 and 15). The use of an undivided cell instead of a divided cell reduced the product yield to 35%, and a black deposit, presumed to be palladium black, was observed on the cathode (entry 16). Under the reaction conditions for entry 1, product 2a was isolated in 64% yield (entry 1).
:1 mixture of AcOH and Ac2O was introduced to both chambers. The reaction was conducted with a 1.0 mA constant current at 90 °C for 21 h. For the optimization of the reaction conditions, the yield of product 2a was determined by adding the NMR yield of the (E)-isomer with the yield of (Z)-isomer calculated based on the GC ratio of the (E)/(Z)-isomers. Under the optimized reaction conditions, product 2a was formed in 72% yield with an (E)/(Z)-ratio of 3.0 (entry 1). Some byproducts, such as 2,6-dimethoxybenzaldehyde and -benzonitrile, were structurally determined, but no oxidation was observed at the 3- or 4-position of the 2-butoxy group. Reducing the amount of Pd(OAc)2 to 5.0 mol% decreased the yield to 38% (entry 2). Substrate 1a contains a 2,6-dimethoxyphenyl group in the directing group. While the use of 2,4,6-trimethoxyphenyl group (1b) provided the corresponding product 2b in a similar yield (entry 3), the substrate containing a 2,6-dimethoxy-4-trifluoromethylphenyl (1c) or a 2,6-dichlorophenyl (1d) group gave the corresponding product in a lower yield (entries 4 and 5). Extension of the reaction time to 24 h also reduced the yield, probably due to decomposition of the product (entry 6). With keeping the same electronic equivalent at 2.0 F mol−1, the electric current value was varied, but the reactions at 0.5 mA (43 h) and 1.5 mA (14 h) gave 2a in lower yields (entries 7 and 8). Several cation exchange membranes, such as Nafion 117, 324, and 966, and a sand core were also examined but provided product 2a in lower yields (entries 9–12). When AcOH was used as a sole solvent instead of AcOH/Ac2O (50:1 v/v), the yield slightly lowered to 60% (entry 13), probably because Ac2O promotes the regeneration of Pd(OAc)2 with avoiding catalyst decomposition.9 The desired reaction did not proceed without using Pd(OAc)2 or an electric current (entries 14 and 15). The use of an undivided cell instead of a divided cell reduced the product yield to 35%, and a black deposit, presumed to be palladium black, was observed on the cathode (entry 16). Under the reaction conditions for entry 1, product 2a was isolated in 64% yield (entry 1).
|
|
|||
|---|---|---|---|
| Entry | Variation from optimized conditions | Yieldb (%) | E/Zc |
|
a Reaction conditions: 1a (0.4 mmol, anode), Pd(OAc)2 (0.04 mmol, anode), NaOAc (3.2 mmol each, both chambers), AcOH/AC2O (50:1 v/v, 4 mL each, both chambers), an H-type divided cell with two platinum electrodes and a Neosepta CSE membrane, 90 °C, 1.0 mA, 21 h, 2.0 F mol−1.
b Combined yields of (E)- and (Z)-isomers calculated based on the NMR yield of (E)-isomer and the GC ratio of (E)/(Z)-isomers.
c GC ratios of (E)/(Z)-isomers.
d Numbers in parentheses are the yield and the (E)/(Z)-ratio of the isolated product.
e 1.5 mA, 14 h (2.0 F mol−1).
|
|||
| 1 | None | 72 (64)d | 3.0/1 (3.9/1)d |
| 2 | 5 mol% of Pd(OAc)2 | 38 | 4.0/1 |
| 3 | DGb instead of DGa [1b] | 72 [2b] | 3.6/1 |
| 4 | DGc instead of DGa [1c] | 47 [2c] | 3.8/1 |
| 5 | DGd instead of DGa [1d] | 33 [2d] | 0.6/1 |
| 6 | 24 h, 2.2 F/mol | 64 | 3.8/1 |
| 7 | 0.5 mA, 43 h (2.0 F mol−1) | 61 | 3.3/1 |
| 8 | 1.5 mA, 14 h (2.0 F mol−1) | 59 | 3.2/1 |
| 9e | Nafion 117 instead of Neosepta CSE | 51 | 4.5/1 |
| 10e | Nafion 324 instead of Neosepta CSE | 52 | 4.3/1 |
| 11e | Nafion 966 instead of Neosepta CSE | 49 | 4.4/1 |
| 12 | Sand core instead of Neosepta CSE | 53 | 4.1/1 |
| 13 | AcOH only instead of AcOH/Ac2O (50:1 v/v) |
60 | 4.1/1 |
| 14 | Without Pd(OAc)2 | nd | — |
| 15 | Without electric current | 2 | 2.7/1 |
| 16 | Undivided cell | 35 | 3.8/1 |
Substrate scope was examined by incorporating various types of alkoxy groups onto the nitrogen atom of the 2,6-dimethoxybenzylideneamino group and by isolating the products (Fig. 2). Various substrates containing α-branched ethoxy groups were first investigated. The reaction of substrate 1e bearing 1-pentyl group proceeded to afford the corresponding product 2e in 51% yield. For the reaction of methyl-substituted substrate 1f, monoacetoxylation product 2f was obtained in 59% yield, while only a trace amount of the corresponding diacetoxylation product was deteced by GCMS analysis. Substrates bearing branched alkyl groups such as isobutyl (1g), isopropyl (1h), and cyclohexyl (1i) groups at the α-position were also monoacetoxylated to provide products 2g–2i in 60–66% yields. While only 21% yield of product 2j was obtained by the reaction of a substrate with no branch at the α-position (1j), the reaction of substrates with α,α-dibranched ethoxy group such as tert-butoxy (1k) and 1-methylcyclohexyloxy (1l) groups gave monoacetoxylation products 2k and 2l in 44 and 40% yields, respectively. While the reaction of a substrate with a phenyl group at the α-position (1m) mostly provided C(sp2)–H acetoxylation product 2m′ with only a small amount of the desired C(sp3)–H acetoxylation product 2m, benzyl-substituted substrate 1n was selectively converted to C(sp3)–H acetoxylation product 2n in 50% yield. Substrates having a functional group were also examined. The reaction of substrate 1o bearing a 1-methoxypropan-2-yl group offered glycerine derivative 2o with three different protecting groups in 41% yield, acetoxylation products were not detected for the reaction of substrates containing an alkene moiety (1p and 1q). The reaction of substrate without a methyl substituent on the α-carbon of the alkoxy group is much more challenging, and no acetoxylation product was obtained for the reaction of substrates derived from 3-pentyl and cyclopentyl alcohols (1r and 1s, respectively). However, the acetoxylation of a mixture of endo/exo-norbornan-2-ol derivative (1t) proceeded at the 1-position to give the corresponding product 2t in 30% yield as a mixture of stereoisomers (Scheme 1).
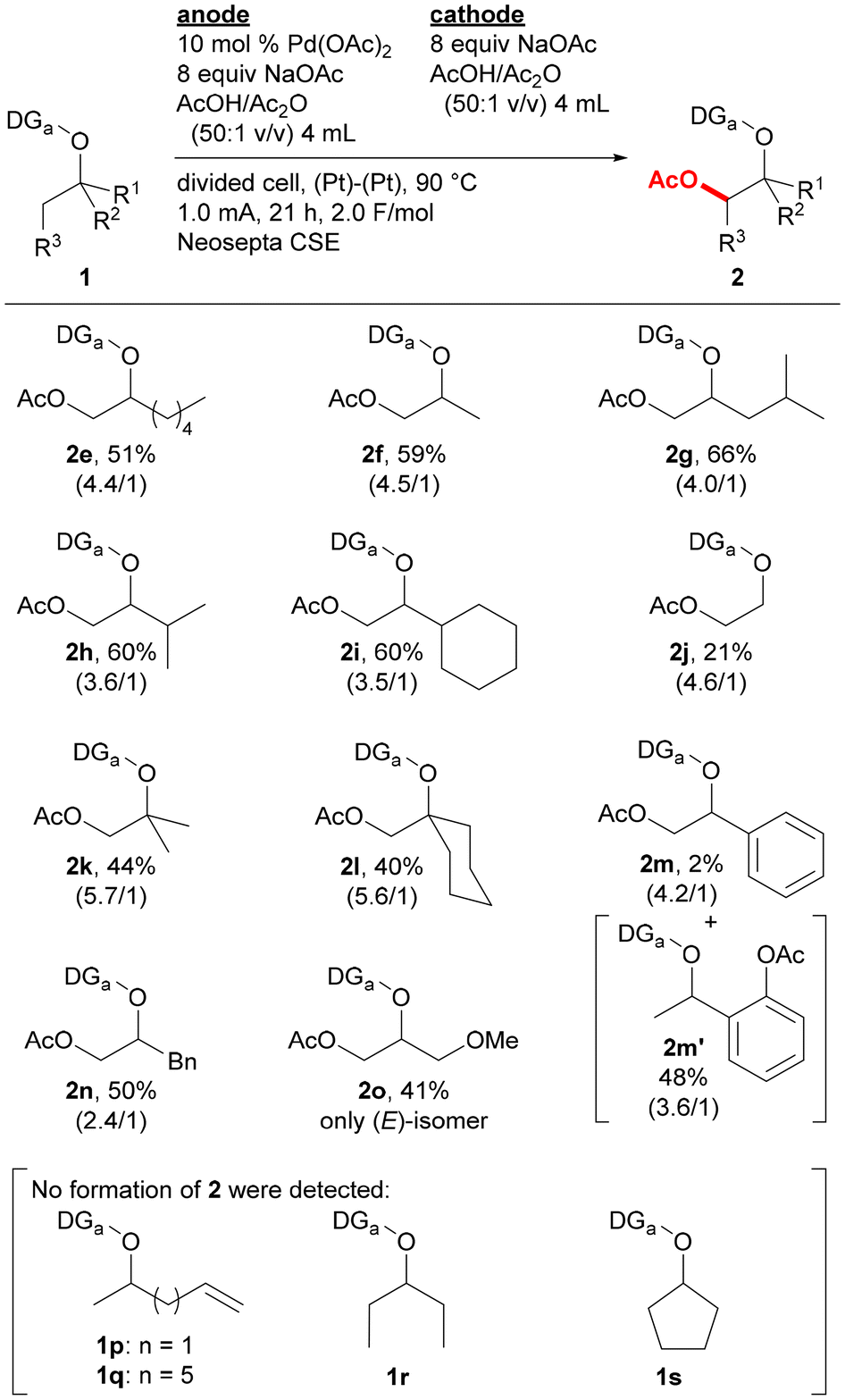 | ||
| Fig. 2 Substrate scope. Reaction conditions: 1 (0.4 mmol, anode), Pd(OAc)2 (0.04 mmol, anode), NaOAc (3.2 mmol each, both chambers), AcOH/AC2O (50:1 v/v, 4 mL each, both chambers), an H-type divided cell with two platinum electrodes and a Neosepta CSE membrane, 90 °C, 1.0 mA, 21 h, 2.0 F mol−1. Isolated yields are presented, and E/Z ratios are shown in parentheses. | ||
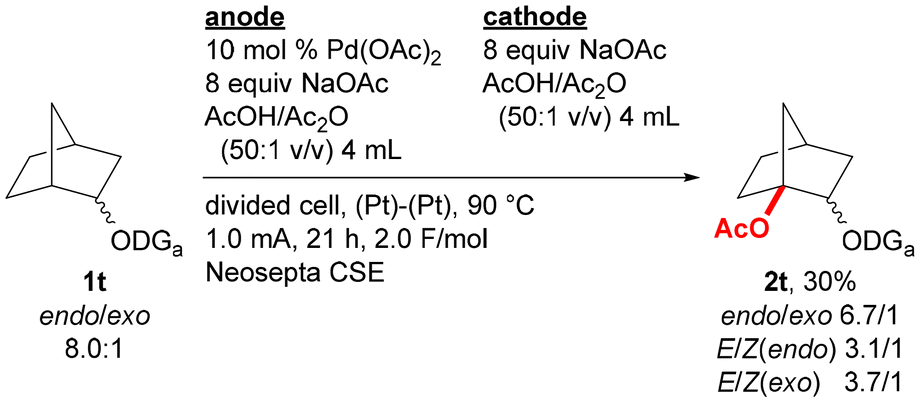 | ||
| Scheme 1 Reaction of norbornan-2-ol derivative 1t. | ||
In order to gain an understanding of the mechanism, the reaction of 1a was conducted in AcOD (Scheme 2a). The yield, determined in the same way as those in Table 1, was comparable to the reaction in AcOH (Table 1, entry 1), and no deuterium incorporation was detected for either product 2a or recovered 1a by 2H NMR. Therefore, the C–H bond cleavage by the concerted metalation-deprotonation (CMD) mechanism is considered to be irreversible in this case. The reaction using NaOCOCD3 was also conducted, and 1H NMR analysis suggested that the amount of the product possessing a CD3COO group instead of an AcO group in the isolated product 2a is ca. 5%, which is nearly equal to the proportion of the CD3CO group in the total amount of the acetyl group in the reaction system (Scheme 2b). Therefore, the acetate ion in the NaOAc should be rapidly exchanged with AcOH during the reaction.
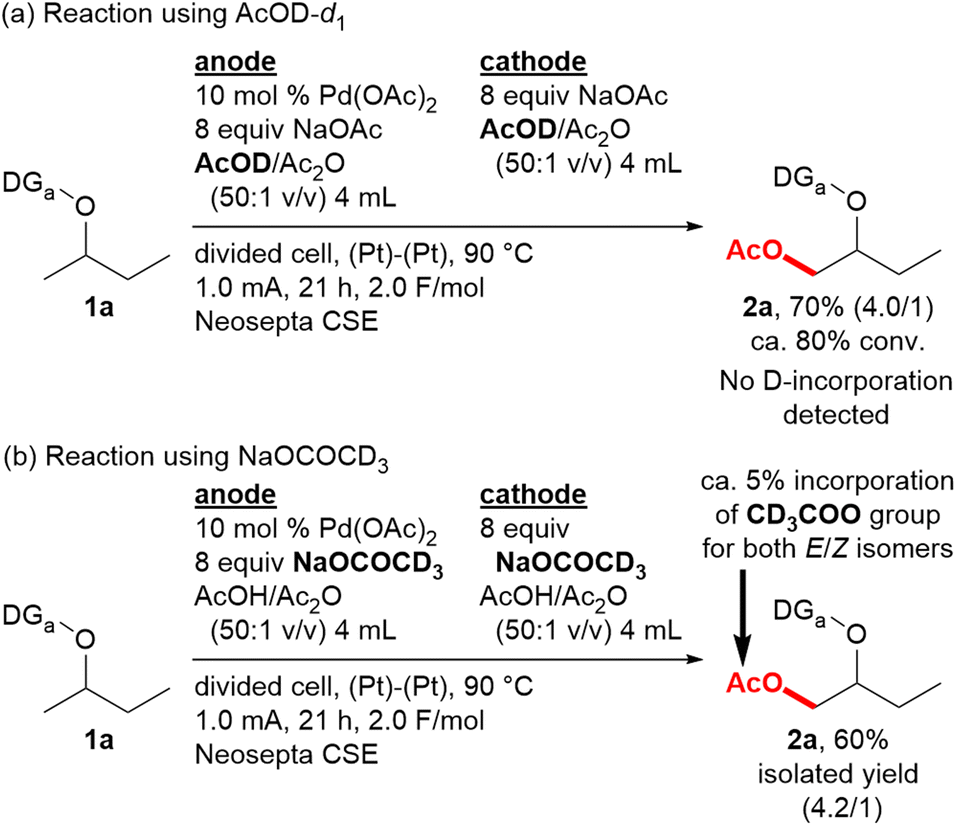 | ||
| Scheme 2 Deuterium-labelling experiments. | ||
A proposed catalytic cycle of this reaction is described in Fig. 3, though the exact number of acetates on the palladium center would be highly speculative. Substrate coordination to the palladium center, followed by C–H bond cleavage via a CMD mechanism, gives a palladium(II) metallacycle species. Anodic oxidation of the palladium complex then occurs to provide a high-oxidation-state palladium species, which undergoes rapid reductive elimination to form a C–O bond. Ligand exchange with 1a provides product 2a.
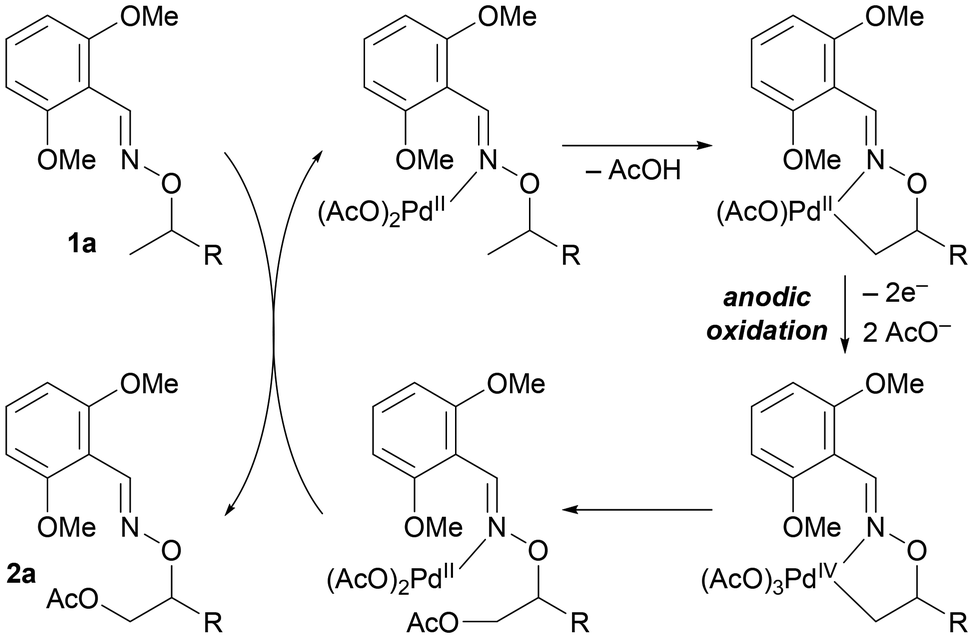 | ||
| Fig. 3 A proposed catalytic cycle. | ||
Conclusions
Palladium-catalyzed electrochemical C(sp3)–H acetoxylation of alcohol derivatives was achieved using an oxime as an exo-directing group to prepare 1,2-diol derivatives. Various substrates containing alkoxy groups with a methyl branch at the α-position were acetoxylated at the methyl group only using Pd(OAc)2 catalyst and NaOAc in Ac2O/AcOH under anodic oxidation conditions. The acetoxylation of a norbornan-2-ol derivative also proceeded at the 1-position.Experimental
A general procedure for the palladium-catalyzed electrochemical acetoxylation
The electrochemical oxidation was carried out in an H-type divided cell equipped with two platinum electrodes (1.7 × 1.7 cm2) and a cation-exchange membrane. The anodic chamber was charged with a solution of oxime ether 1 (0.4 mmol, 1 equiv.), NaOAc (262.5 mg, 3.2 mmol, 8 equiv.), and palladium acetate (8.98 mg, 0.04 mmol, 10 mol%) in AcOH/Ac2O (4 mL, 50:1 v/v). The cathodic chamber was charged with a solution of NaOAc (262.5 mg, 3.2 mmol, 8 equiv.) in AcOH/Ac2O (4 mL, 50:1 v/v). After the mixture in the anodic chamber was stirred for 15 min at 90 °C, an electric field was applied at room temperature under a 1.0 mA constant current condition for 21 h. After the reaction, the mixtures in both of the chambers were transferred to a flask through a funnel. The electrodes and H-type divided cell were washed with AcOEt. The mixture was mixed with an aqueous solution of NaHCO3 and was extracted with AcOEt. The combined organic portions were washed with brine. The resulting solution was dried over MgSO4 and concentrated. The acetoxylation product 2 was isolated by flash column chromatography. The E/Z ratios of the products were determined based on the integral ratios of the 1H NMR signals corresponding to the acetyl group.
:1) of the crude material afforded 2a in 64% yield (75.6 mg, 0.26 mmol) as a colorless oil. The analytical data for this product are in good agreement with those reported in literature.8
:1) of the crude material afforded 2e in 51% yield (69.0 mg, 0.20 mmol) as a colorless oil. The analytical data for this product are in good agreement with those reported in literature.8
:1) of the crude material afforded 2f in 59% yield (66.2 mg, 0.24 mmol) as a colorless oil. The analytical data for this product are in good agreement with those reported in literature.8
:1) of the crude material afforded 2g in 66% yield (85.3 mg, 0.26 mmol) as a colorless oil. The analytical data for this product are in good agreement with those reported in literature.8
:1) of the crude material afforded 2h in 60% yield (74.2 mg, 0.24 mmol) as a colorless oil. The analytical data for this product are in good agreement with those reported in literature.8
:1) of the crude material afforded 2i in 60% yield (84.1 mg, 0.24 mmol) as a colorless oil. The analytical data for this product are in good agreement with those reported in literature.8
:1) of the crude material afforded 2k in 44% yield (52.0 mg, 0.18 mmol) as a colorless oil. The analytical data for this product are in good agreement with those reported in literature.8
:1) of the crude material afforded 2l in 40% yield (54.5 mg, 0.16 mmol) as a colorless oil. The analytical data for this product are in good agreement with those reported in literature.8
:1) of the crude material afforded 2m and 2m′ in 50% combined yield (70.2 mg, 0.20 mmol, 2m:2m′ = 1:20) as a colorless oil. 1H NMR (400 MHz, CDCl3): [2m′] Major isomerδ 8.45 (s, 1H), 7.54–7.53 (m, 1H), 7.32–7.29 (m, 1H), 7.27–7.25 (m, 1H), 7.27–7.21 (m, 2H), 7.08–7.05 (m, 1H), 6.54 (d, J = 6.8 Hz, 2H), 5.55 (q, J = 6.6 Hz, 1H), 3.79 (s, 6H), 2.32 (s, 3H), 1.59 (d, J = 6.3 Hz, 3H). Minor isomer (*distinct resonances) δ 6.54 (d, J = 8.6 Hz, 1H), 5.44 (q, J = 6.6 Hz, 1H), 3.80 (s, 6H), 2.24 (s, 3H), 1.44 (d, J = 6.3 Hz, 3H); the analytical data for 2m′ are in good agreement with those reported in literature.10
:1) of the crude material afforded 2n in 50% yield (71.6 mg, 0.20 mmol) as a colorless oil. 1H NMR (400 MHz, CDCl3): Major isomerδ 8.48 (s, 1H), 7.31–7.12 (m, 6H), 6.57 (d, J = 8.7 Hz, 2H), 4.70–4.64 (m, 1H), 4.29 (dd, J = 11.5, 4.1 Hz, 1H), 4.19 (dd, J = 11.5, 5.4 Hz, 1H), 3.85 (s, 6H), 3.18 (dd, J = 14.2, 5.9 Hz, 1H), 2.98 (dd, J = 14.2, 7.3 Hz, 1H), 2.08 (s, 3H). Minor isomerδ 7.47 (s, 1H), 7.31–7.12 (m, 6H), 6.54 (d, J = 8.3 Hz, 2H), 4.63–4.57 (m, 1H), 4.17 (dd, J = 11.5, 5.4 Hz, 1H), 4.11 (dd, J = 11.5, 5.0 Hz, 1H), 3.75 (s, 6H), 3.04 (dd, J = 13.9, 6.3 Hz, 1H), 2.88 (dd, J = 13.9, 6.8 Hz, 1H), 2.02 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): Major isomerδ 171.0, 159.0, 144.8, 137.6, 130.9, 129.5, 128.4, 126.3, 109.4, 104.1, 80.8, 64.4, 56.0, 37.0, 20.9. Minor isomerδ 170.8, 157.8, 144.3, 137.5, 130.5, 129.6, 128.2, 126.2, 109.4, 103.5, 80.9, 64.4, 55.6, 37.1, 20.9; IR (NaCl, mixed isomers): 3087 w, 3062 w, 3026 w, 3005 w, 2940 m, 2839 w, 1739 s, 1595 s, 1471 s, 1432 m, 1367 w, 1305 w, 1282 w, 1256 s, 1113 s, 1039 m, 944 m, 895 w, 778 m, 748 w, 734 w, 702 m; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C20H23NO5Na+ 380.1468, found 380.1472.
:1) of the crude material afforded 2o in 41% yield (53.4 mg, 0.17 mmol) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 8.50 (s, 1H), 7.27 (t, J = 8.3 Hz, 1H), 6.60 (d, J = 8.3 Hz, 2H), 4.61–4.56 (m, 1H), 4.42–4.33 (m, 2H), 3.84 (s, 6H), 3.72–3.65 (m, 2H), 3.41 (s, 3H), 2.09 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 171.0, 159.1, 145.2, 131.0, 109.2, 104.1, 79.2, 71.2, 63.3, 59.4, 56.0, 20.1; IR (NaCl, mixed isomers): 2938 w, 2840 w, 1740 s, 1596 m, 1471 s, 1434 m, 1368 w, 1344 w, 1307 w, 1257 s, 1208 m, 1114 s, 1045 m, 956 m, 881 w, 779 m, 731 w, 633 w; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C15H21NNaO6+ 334.1261, found 334.1263.
:1) of the crude material afforded 2t in 30% yield (39.3 mg, 0.12 mmol) as colorless oil. The analytical data for this product are in good agreement with those reported in literature.8
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST CREST grant number JPMJCR20R1.References
-
(a) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086 CrossRef CAS
; (b) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179 CrossRef CAS
- A seminal work on electrochemical C–H functionalization using a catalytic amount of a mediator: C. Amatore, C. Cammoun and A. Jutand, Adv. Synth. Catal., 2007, 349, 292 CrossRef CAS
-
(a) F. Kakiuchi and T. Kochi, Chem. Rec., 2021, 21, 2320 CrossRef CAS PubMed
- Other related examples of our electrochemical directed C–H functionalizations:
(a) H. Aiso, T. Kochi, H. Mutsutani, T. Tanabe, S. Nishiyama and F. Kakiuchi, J. Org. Chem., 2012, 77, 7718 CrossRef CAS PubMed
- Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293 CrossRef CAS
- A. Shrestha, M. Lee, A. L. Dunn and M. S. Sanford, Org. Lett., 2018, 20, 204 CrossRef CAS PubMed
- Metal-catalyzed non-directed electrochemical C(sp3)–H azidations:
(a) L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693 CrossRef CAS PubMed
- Z. Ren, F. Mo and G. Dong, J. Am. Chem. Soc., 2012, 134, 16991 CrossRef CAS
- R. Giri, J. Liang, J.-G. Lei, J.-J. Li, D.-H. Wang, X. Chen, I. C. Naggar, C. Guo, B. M. Foxman and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 7420 CrossRef CAS PubMed
- Z. Ren, J. E. Schulz and G. Dong, Org. Lett., 2015, 17, 2696 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01241k |
| This journal is © The Royal Society of Chemistry 2024 |