
cisPro stabilization in prolyl carbamates influenced by tetrel bonding interactions†
Shreya Banerjeea,
Shama Tumminakattia,
Sudip Ghosha,
Vamsee K. Voora b and
Erode N. Prabhakaran*a
b and
Erode N. Prabhakaran*a
aDepartment of Chemistry, Indian Institute of Science, Bangalore, Karnataka – 560012, India. E-mail: eprabhak@iisc.ac.in
bDepartment of Chemical Sciences, Tata Institute of Fundamental Research, Mumbai, 400005, India
First published on 23rd October 2024
Abstract
NMR spectral and theoretical analyses of homologous prolyl carbamates reveal subtle charge transfer tetrel bonding interactions (TBIs), selectively stabilizing their cisPro rotamers. These TBIs involve C-terminal-amide to N-terminal carbamate carbonyl–carbonyl (n → π* type) followed by intra-carbamate (n → σ* type) charge transfer interactions exclusively in the cisPro motif. The number of TBIs and hence the cisPro stability increase with increasing number of Cβ groups at the carbamate alcohol. Increasing solvent polarities also increase the relative cisPro carbamate stabilities.
1 Introduction
Interactions influencing cisPro stability in prolyl carbamates are much less understood unlike those in prolyl amides. We know from earlier studies of homologous prolyl amides that their cisPro relative stabilities and populations decrease compared to transPro, as the number of methyl substituents on the amide acyl Cα increases (Fig. 1a).1,2 This is due to increasing steric clashes involving the Cα-substituents, which destabilize the cisPro conformer more than the transPro conformer.1,3,4 In addition to sterics, several local short-range electronic interactions, such as C7 H-bond,5 C10 H-bond6 and C5 O⋯C′ interactions,7 also naturally favour their transPro stabilities (see the ESI, S2†) and reduce their relative cisPro populations. Hence the cisPro rotamers of prolyl amides have few stabilizing interactions although rare assistance from the local sequence8–13 (see the ESI, S2†) has been observed.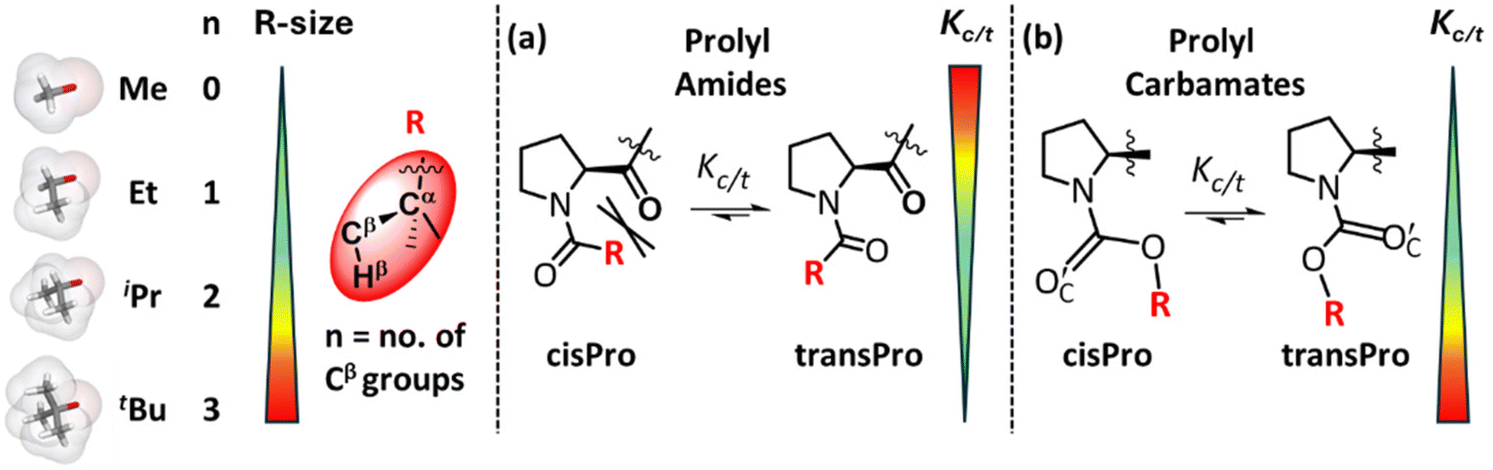 | ||
| Fig. 1 Effect of increasing size of R (R = –Cα(CβH3)nHβ3−n; Me → tBu) on Kc/t (equilibrium constant) for cis–trans isomerism of prolyl amides (a) and carbamates (b). | ||
A similar investigation of stereoelectronic interactions governing cisPro stability in prolyl carbamates has long been lacking. We know that in carbamates, the N–C sigma bonds are largely locked in a plane due to the resonance effect at the N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group14,15 (Fig. 1b). The CO2R group has also been shown to be locked in a plane with a predominantly cisoid geometry (Fig. 2a) due to stabilization by n → π* (ref. 16) or q → σ* (ref. 17) interactions from the carbamate carbonyl O′C lone pair donor (D) to the π* or σ* acceptors (A) in the phenyl and alkyl substituents respectively on Cα of the R group. Hence, primarily the O–Cα and Cα–Cβ σ-bonds in the R group (Fig. 1) are free to rotate. Charge transfer interactions such as n → π* and n → σ* that occur from an electron-donating Lewis base to a covalently bonded C atom acting as a Lewis acid have been referred to as tetrel bonding interactions18,19 (TBIs).
O group14,15 (Fig. 1b). The CO2R group has also been shown to be locked in a plane with a predominantly cisoid geometry (Fig. 2a) due to stabilization by n → π* (ref. 16) or q → σ* (ref. 17) interactions from the carbamate carbonyl O′C lone pair donor (D) to the π* or σ* acceptors (A) in the phenyl and alkyl substituents respectively on Cα of the R group. Hence, primarily the O–Cα and Cα–Cβ σ-bonds in the R group (Fig. 1) are free to rotate. Charge transfer interactions such as n → π* and n → σ* that occur from an electron-donating Lewis base to a covalently bonded C atom acting as a Lewis acid have been referred to as tetrel bonding interactions18,19 (TBIs).
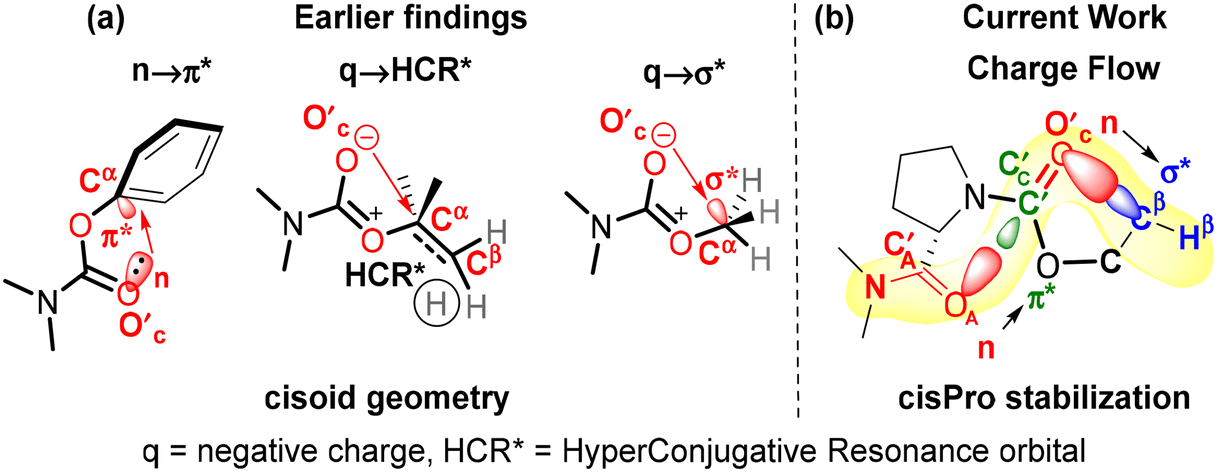 | ||
| Fig. 2 (a) Earlier reports of n → π*,16 q → HCR* (ref. 17) and q → σ* (ref. 17) interactions stabilizing the cisoid conformation of carbamates, (b) current findings of carbonyl–carbonyl (n → π* type) and intra-carbamate (n → σ* type) tetrel bonding interactions stabilizing cisPro. | ||
Further investigations into stereoelectronic interactions in carbamates are essential, given the prevalence of carbamates in several bioactive peptides,20 drugs,21 materials22 and fertilizers.23 The pharmacokinetic properties of amide-based drugs, most notably their in vivo stability and bioavailability, have been improved by using their structural analogues – carbamates which have higher metabolic stability and cell permeability.24 Additionally, it is possible to modulate the biological activity of carbamates by varying the substituents at the amino and carboxyl termini,25,26 making them an integral structural and/or functional part of many drugs,27 enzyme inhibitors,28 and enzyme mimetics.29
Current systematic studies involve the synthesis and NMR spectral and theoretical analyses of homologous prolyl carbamates with increasing steric bulk on the R group (Fig. 1b) and their comparison with the corresponding homologous reference pyrrolidine models. These studies reveal an anomaly where cisPro carbamate relative stabilities improve with increasing number of methyl substituents on the Cα of the R group. This is an inverse steric effect unlike that observed in amides (Fig. 1). DFT and NBO calculations were performed at varying C′C–OC–Cα–Cβ (τ) torsions (Fig. 8a) which reveal the optimum geometries at which a relay of TBIs are observed predominantly in the cisPro carbamate rotamers. These TBIs also stabilize the cisPro conformer relative to the transPro conformer. Solvent-polarity dependent studies show the improvement of this stabilization with increasing solvent polarities.
2 Results
The spectral signatures of charge transfer TBIs are weak and cannot be observed directly in the spectra of model compounds. For this reason, it is conventional to identify these interactions through increasing/decreasing trends of observables in homologues. Prolyl carbamate homologues 1–4 (Fig. 3a) were synthesized using standard solution phase synthetic protocols (see the ESI, S3 and S4†). The equilibrium constant for the transPro → cisPro isomerism (Kc/t) (10 mM in CDCl3, 298 K) was determined as the ratio of the two HαPro integrals in their 1H NMR spectra. For the prolyl carbamate 1, Kc/t (0.48) is 3.6 times that of the corresponding prolyl amide 11 (0.14). This is not due to the lack of the transPro stabilizing C7 γ-turn H-bond in 1, because Kc/t of 1 is 4-fold greater than that of 9 (0.12) which also lacks such an H-bond. The relatively lesser unfavorable steric clashes in the cisPro rotamer of 1 due to the replacement of the bulky acyl CH3 in 9 with a less bulky oxygen atom in 1 cannot entirely explain such a remarkable increase in Kc/t either.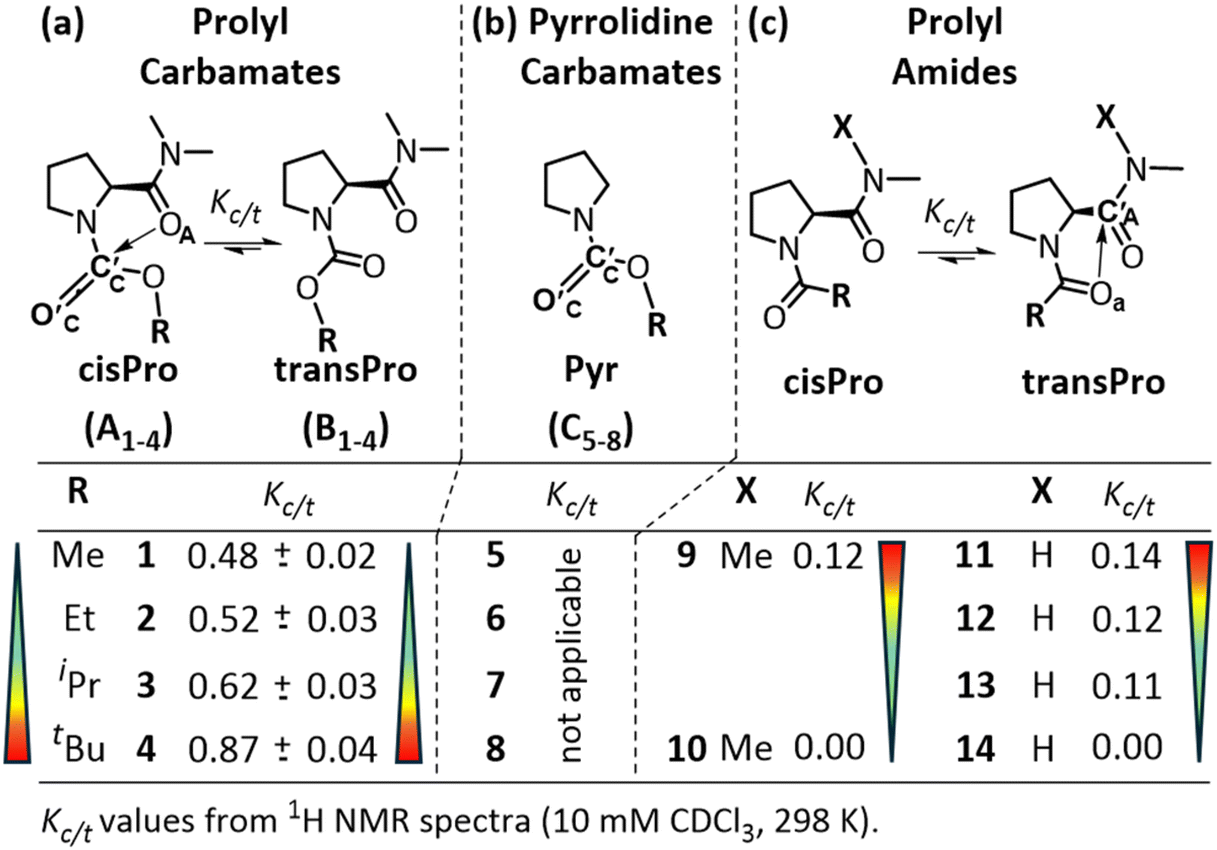 | ||
| Fig. 3 cis/trans isomerism in homologous (a) prolyl carbamates (1–4), (b) corresponding pyrrolidine carbamates (5–8) and (c) prolyl amides (9–14). With increasing bulk at R, the equilibrium constant for transPro → cisPro isomerism (Kc/t) increases from 1 to 4, unlike from 9 to 10 and 11 to 14, where they decrease.1 | ||
This is because, whereas Kc/t values decrease to zero for both 10 and 14 (containing the sterically bulky tBu substituent), it increases for 4 (compared to 1). In fact, along the homologous carbamates 1–4, their Kc/t values increased from 0.48 to 0.87 with increasing steric bulk at Cα of the alcohol of carbamate (Fig. 3a). In contrast, the Kc/t values of prolyl amides 9–10 and 11–14 show an opposite trend upon homologation1 (Fig. 3c).
In pyrrolidine carbamates 5–8 (Fig. 3b), the presence of n → σ* type TBIs, from the carbamate carbonyl oxygen donor (D) to its alcohol Cβ acceptor (A) (O′C → Cβ), has been noted.17,30,31 This increases the electrophilicity of the carbamate carbonyl carbon (C′C) and decreases the nucleophilicity of its carbonyl oxygen, compared to the corresponding amide carbonyl. We hypothesized that the prolyl carbamate carbonyl in 1–4 could act as an electron acceptor from amide oxygen (C′C ← OA) in cisPro rotamers (Fig. 3a). This would be in the reverse direction compared to 9–14 (Fig. 3c) where the oxygens of prolyl amides (Oa) are the electron donors (Oa → C′A), in the transPro rotamers.7
Natural Bond Orbital (NBO) analyses indicate the presence of orbital overlap interactions (1.45 kcal mol−1, 1.75 kcal mol−1) from two of the C–H σ orbitals in the Z N–CH3 group to the σ* of the amide N–C′A pseudo double bond in the cisPro isomer of 2 (Fig. 4a). A weaker n → σ* interaction (0.78 kcal mol−1) is observed in the opposite direction – from the np lone pair32 (Fig. 4b) of the amide OA to the σ* of the third C–H in the Z N–CH3 group. Similar interactions are absent in the transPro isomer of 2. These interactions (observed in 3 and 4 as well) indicate an overall accumulation of negative charge on the amide carbonyl carbon (C′A) exclusively in the cisPro carbamate conformers.
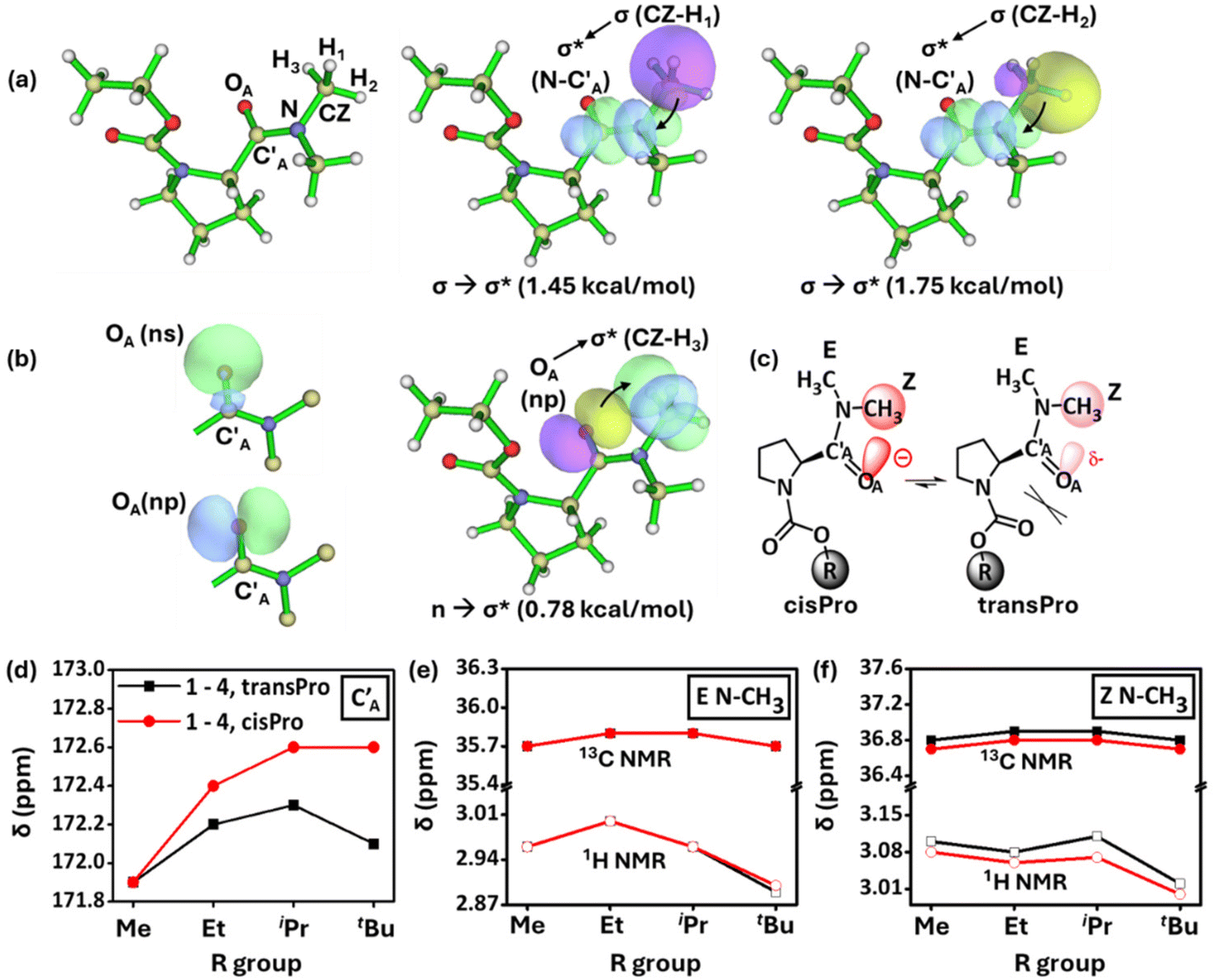 | ||
| Fig. 4 (a) and (b) NBO overlap diagrams of 2 showing interaction between Z N–CH3 and the amide group in the cisPro isomer. ns and np orbitals of amide carbonyl OA; (c) ChemDraw rendition showing polarization along the amide carbonyl in the cisPro rotamer; plots of (d) 13C δ ppm of amide carbonyl C′A; (e and f) 1H, 13C δ ppm for the E and Z N–CH3 in 1–4, versus the R group of carbamates. | ||
Investigation of the 13C NMR spectrum of 1 (10 mM, CDCl3) showed identical chemical shifts for the amide carbonyl carbon (C′A) in both the cisPro and transPro rotamers (Fig. 4c and d). However, the signals in cisPro started to become increasingly more downfield shifted from the transPro signals in 2–4 (Fig. 4d), which contain the methyl substituents on Cα with their numbers increasing from one to three respectively. Such shifts occur despite the energy minimized structures of 1–4 (minimized using the B3LYP 6-311G(**)33 basis set in Gaussian 09W34), showing that the amide carbonyl oxygens are not directly interacting with the carbamate Cα (OA⋯Cα ≥ 4.25 ± 0.15 Å), where the homologation occurs remotely (see the ESI, S9.3†).
Steric effects from homologation cannot cause such distal electronic changes. The 1H and 13C nuclei of Z N–CH3 in the cisPro rotamers of 1–4, concomitantly, selectively get upfield shifted compared to those of transPro, unlike the E N–CH3 protons where there are no such shifts (Fig. 4e and f). Clearly there is an incremental build-up of greater negative charge at the amide oxygen (OA) selectively in the cisPro rotamer, as the number of Cβ-substituents on Cα increases at the remote alcohol group of carbamate from 1–4.
This negative charge on OA is transferred to the carbamate C′CO′C through n → π* type TBIs (Fig. 5). The charge accumulation on the carbamate CO (C′C, O′C) in 1–4 was calculated from the DFT-optimized structures using the B3LYP 6-311G(**)33 basis set in Gaussian 09W.34 With the increase in the number of Cα-substituents in 1–4, there is an increase in negative charge on O′C (Fig. 5b) and a corresponding increase in the positive charge on C′C exclusively in the cisPro isomer. Note that there is no direct orbital overlap between the amide and carbamate groups (Fig. 5a), which indicates that this charge transfer is a through-space effect. There is no such charge transfer observed in the transPro isomer (Fig. 5b and c) where the carbamate C′CO′C dipole remains similar in 1–4. Although the C′AOA⋯C′CO′C distance is longer (3.29 ± 0.03 Å) than the Bürgi–Dunitz distances necessary for n → π* orbital overlap,35 in both cisPro and transPro, the angle of incidence ∠OA⋯C′CO′C is within that prescribed trajectory (105.3°–133.7°)36 exclusively in cisPro (130.5° ± 0.52°) unlike in transPro (91.80° ± 0.36°).
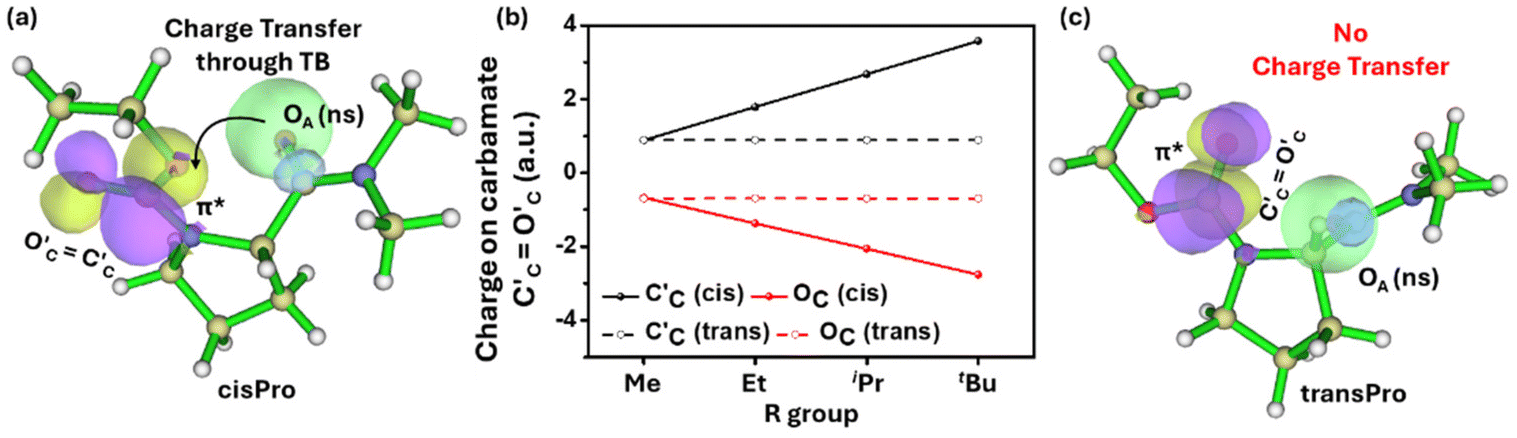 | ||
| Fig. 5 NBO diagrams in the cisPro (a) and transPro (c) isomers of 2 showing the presence of charge transfer from amide OA to carbamate C′CO′C selectively in cisPro. (b) Plot of net charge on carbamate carbonyl in cisPro and transPro (C′C, O′C) as a function of increasing size of the R group in carbamates (Me to tBu, 1–4). | ||
Consistent with the results of the NBO analyses, there is upfield shifting of the charge acceptor carbamate C′C 13C NMR chemical shifts in 1–4 selectively in cisPro, compared to their transPro (Fig. 6a and b) and their corresponding reference pyrrolidine models 5–8 (Δδ = 0.8 ± 0.2 ppm) where there is no C-terminal amide group to act as a charge donor. The 13C NMR signals of transPro carbamate C′C in 1–4 were nearly identical to those of pyrrolidine models 5–8 (Δδ = 0.2 ± 0.1 ppm). Hence the selective subtle shielding and increase in electron density of cisPro carbamate C′C is due to carbonyl amide (C′AOA) → carbonyl carbamate (C′CO′C) (C′AOA⋯C′CO′C) charge transfer TBIs – which are largely absent in transPro.
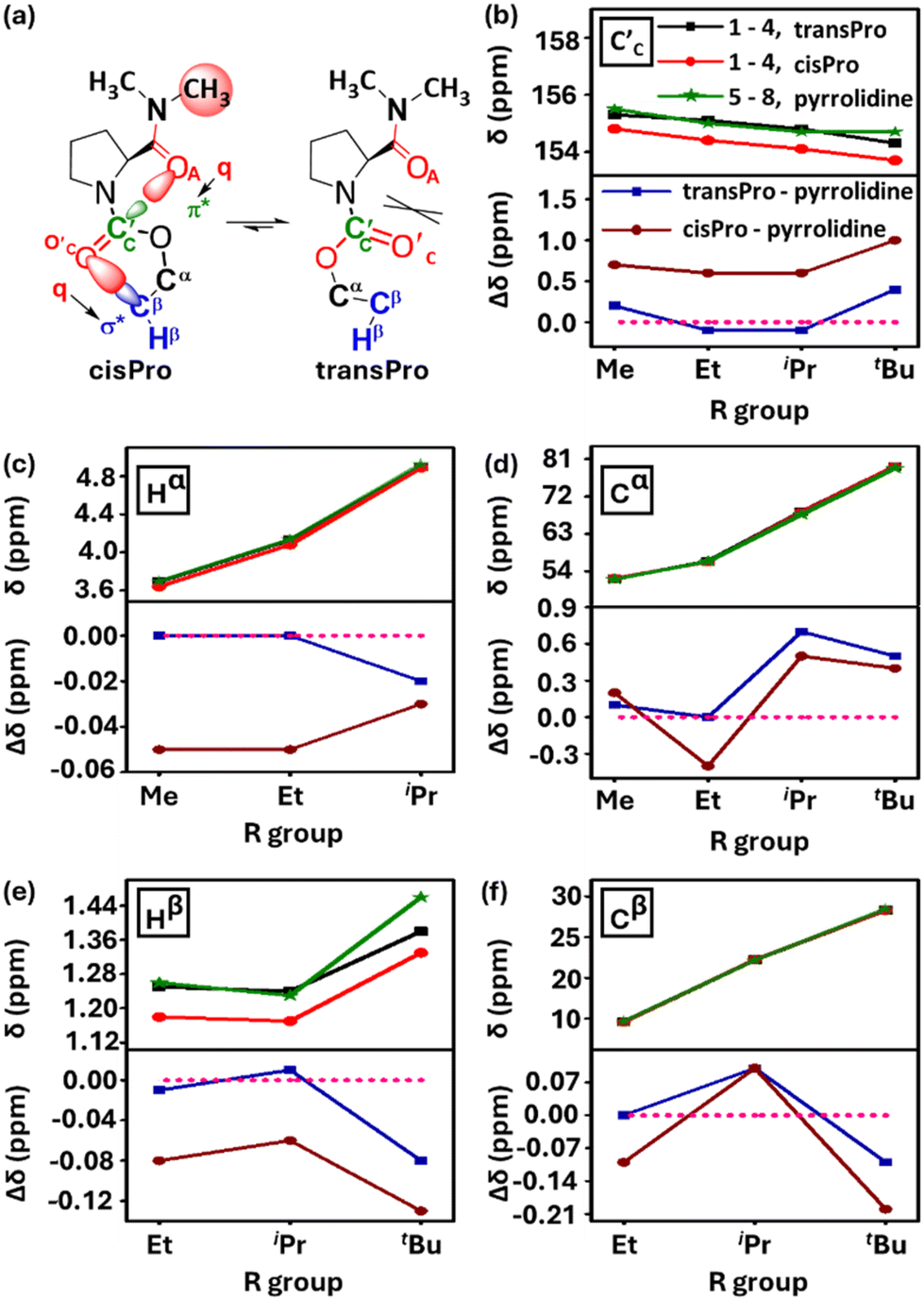 | ||
| Fig. 6 (a) ChemDraw images highlighting the Hα, Hβ, Cα, Cβ nuclei in carbamate alcohol; (b–f) plots of 13C and 1H NMR (10 mM, CDCl3, 298 K) chemical shifts (δ ppm) for carbamate carbonyl C′C and for Hα, Hβ, Cα, Cβ of carbamate alcohol in 1–4 and 5–8 versus the R group of carbamates and their difference of chemical shifts from transPro & cisPro rotamers (1–4) to pyrrolidine (5–8) (Δδ ppm). | ||
These relatively small Δδ ppm values observed for C′ and N–CH3 are reliable and indicate perturbations from the charge relay interactions and are not resulting from mere conformational anisotropies. This is because the Δδ ppm values of the prolyl ring protons (Hβ, Hγ) and carbons (Cβ, Cγ) between the cisPro and transPro rotamers of 1–4 are ∼0.00 ppm (see the ESI, S6.6 and S6.7†). However, for the Cα, Hα and Cβ, Hβ of the Pro ring, which directly interact with the different charge environments in cisPro and transPro rotamers, the Δδ ppm values are non-zero (Fig. 6c–f). Additionally, concentration dependent FT-IR and NMR studies (see the ESI, S7†) showed no variance at all in any of the vibrational bands and chemical shifts of any of the nuclei, indicating that the observed spectral shifts are not due to intermolecular associations either.
NBO analyses7,32 of the energy minimized (using the B3LYP 6-311G(**)33 basis set in Gaussian 09W34) structures of 1–4 showed no orbital overlaps (zero energies) between the lone pair of amide oxygen and π* of carbamate C′C in the cisPro or transPro rotamers (see the ESI, S9.4†). The shortest possible OA⋯C′C distances, derived from energy minimized structures, are also longer (3.29 ± 0.03 Å) than the Bürgi–Dunitz distances necessary for n → π* orbital overlap interactions.7,35 The weak C′AOA⋯C′CO′C interactions in cisPro evidenced by the NMR spectral markers thus involve charge transfer TBIs and not orbital overlaps (n → π*). In transPro, the two carbonyls are not oriented suitably to favour such charge transfer, but their oxygens are close (3.49 ± 0.03 Å) to experience Pauli repulsions (Fig. 6a). Hence similar TBIs are unavailable in transPro, due to which its spectral shifts are similar to those of the pyrrolidine models 5–8 that lack the charge donor amide group.
The FT-IR spectra (10 mM, CHCl3) showed a net decrease in carbamate CO stretching frequency and a net increase in amide CO stretching frequency from 1–4 (see the ESI, S6.8†). The relatively low correlation between the stretching frequencies (χ2 = 0.74) is expected since cisPro, where most of the subtle spectral variations occur, is the minor rotamer. However, the slope of the observed correlation suggests about a two-fold increase in the carbamate CO stretch concomitant to a unit decrease in the amide CO stretch, as the number of σ* acceptors increases (Fig. 7b), which is another spectral observable for the charge transfer C′AOA⋯C′CO′C interactions.
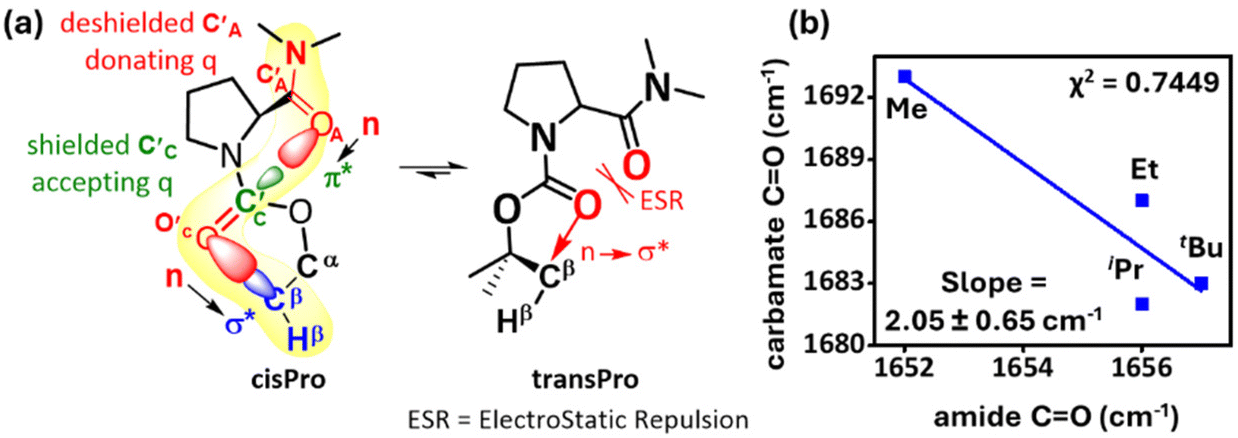 | ||
| Fig. 7 (a) Summary of spectral observations revealing charge transfer through carbonyl–carbonyl and intra-carbamate TBIs at cisPro carbamates. ESR interactions perturb transPro carbamates; (b) correlation plot between amide and carbamate FT-IR CO stretch bands of donor (x-axis) /acceptor (y-axis) carbonyls. | ||
The consequence of the greater negative charge build-up at the carbamate oxygen (O′C) selectively in the cisPro rotamers of 1–4 is seen in the upfield shifts of their carbamate ester Hα and Cα NMR signals (compared to both transPro and pyrrolidine 5–8). Notably, the Hβ and Cβ of cisPro also shift upfield (Fig. 6c–f). The formation of intramolecular 5- and 6-membered H-bonds of the type O⋯H–C is unlikely, as it would have caused upfield shifts in Hα, Hβ and downfield shifts in Cα, Cβ due to polarization of the C–H bond.37 On the other hand, perturbation of the intra-carbamate TBIs along O → Cβ–Hβ by the charge transfer from the carbamate oxygen (Fig. 7a) is more consistent with the observed shifts. Indeed, DFT optimized structures of the cisPro isomer of 1–4 showed only O → Cβ–Hβ and no O → Cα–Hα/Cα–Cβ interactions (Fig. 8).
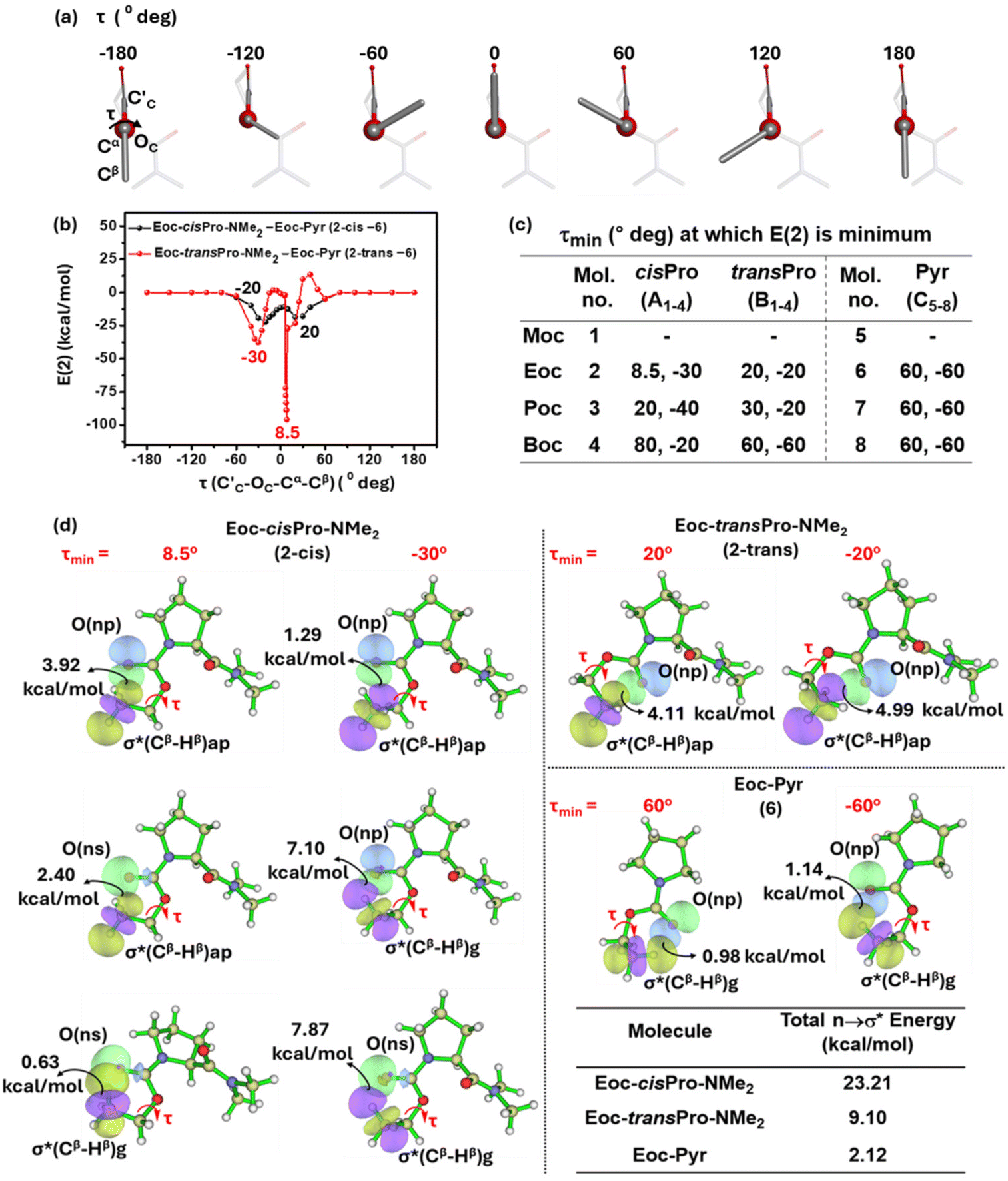 | ||
| Fig. 8 (a) Rotational scan of energy over the entire range of τ (C′C–OC–Cα–Cβ). (b) Variation of normalized second-order perturbation energy E(2) with τ in Eoc-cisPro-NMe2 and Eoc-transPro-NMe2 (2). (c) Table of τmin values of 1–4, 5–8. (d) NBO overlap diagrams showing the energy of each interaction. The net energies are tabulated. (Moc methyloxycarbonyl, Eoc ethyloxycarbonyl, Poc isopropyloxycarbonyl, Boc tert-butyloxycarbonyl). | ||
To determine the torsion angle τmin (τ = C′C–OC–Cα–Cβ) at which there is maximum stabilization from carbonyl–carbonyl (n → π*) TBIs and intra-carbamate (n → σ*) TBIs in cisPro and transPro of 1–4, an energy screening at different torsion angles was performed as follows. τ was varied from −180° to +180° in steps of 1°–30° (as necessary) and the structure was allowed to optimize at each value of τ (Fig. 8a) using the B3LYP 6-311G(**)33 basis set in Gaussian 09W.34 Second order perturbation energies E(2) for the optimized structures were calculated for the cisPro and transPro isomers of 1–4 from these DFT-optimized structures.
These E(2) energies were normalized for comparison between homologues as follows. The n → σ* TBI energies in 1–4 contain two components: (a) one that is due to any n → σ* TBIs that are inherent in the carbamate (inherent n → σ*) in the absence of any n → π* TBIs and (b) another that is due to any changes induced in these n → σ* TBIs (induced n → σ*) by the presence of the n → π* TBIs. Hence, 1–4 contain the total energies due to three TBIs: (a) n → π*; (b) inherent n → σ*; and (c) induced n → σ*. Reference models 5–8 contain exclusively the inherent n → σ* TBIs. Note that these inherent TBI energies would vary with the homologation of the alcohol group in the carbamates. The energies of these inherent n → σ* TBIs in 5–8 (as a function of τ) were hence subtracted from the total (n → π* + inherent n → σ* + induced n → σ*) TBI energies of cisPro and transPro in 1–4 to give their normalized E(2) values (Fig. 8b).
In the plot of E(2) vs. τ, two sharp non-zero energy minima are observed, one each at positive and negative values of τ (termed τmin), for both the cisPro and transPro sets. This is expected because there are two gauche ranges of τ around the carbamate CO (τ = 0° to −60°, 0° to 60°) (Fig. 8a) where the σ* of the Cβ–Hβ is in a suitable orientation for n → σ* type TBIs (except in 1). In 1 there are no Cβ substituents and hence no such charge transfer can occur. The τmin values of 2 where E(2) is minimum are shown (Fig. 8b and c) and are representative of 3 and 4. The τmin values for cisPro are asymmetrically distributed about zero and have much lower E(2) values compared to transPro, whose E(2) values are symmetrical about zero and are shallower (Fig. 8b).
Closer examination showed that cisPro of 2 has three TBIs in the −ve and +ve τ values each, where NBO analyses show that D → A (donor → acceptor) orbital overlap (one np → σ* and two ns → σ*; σ* of Cβ–Hβ) interactions happen (Fig. 8d). Their corresponding O′C⋯Cβ distances are 2.86 ± 0.05 Å (Fig. 9) which are also well within the threshold range (3.22 Å)38,39 for O⋯C non-covalent interactions. These are consistently observed in cisPro of 3 and 4 as well (Fig. 9).
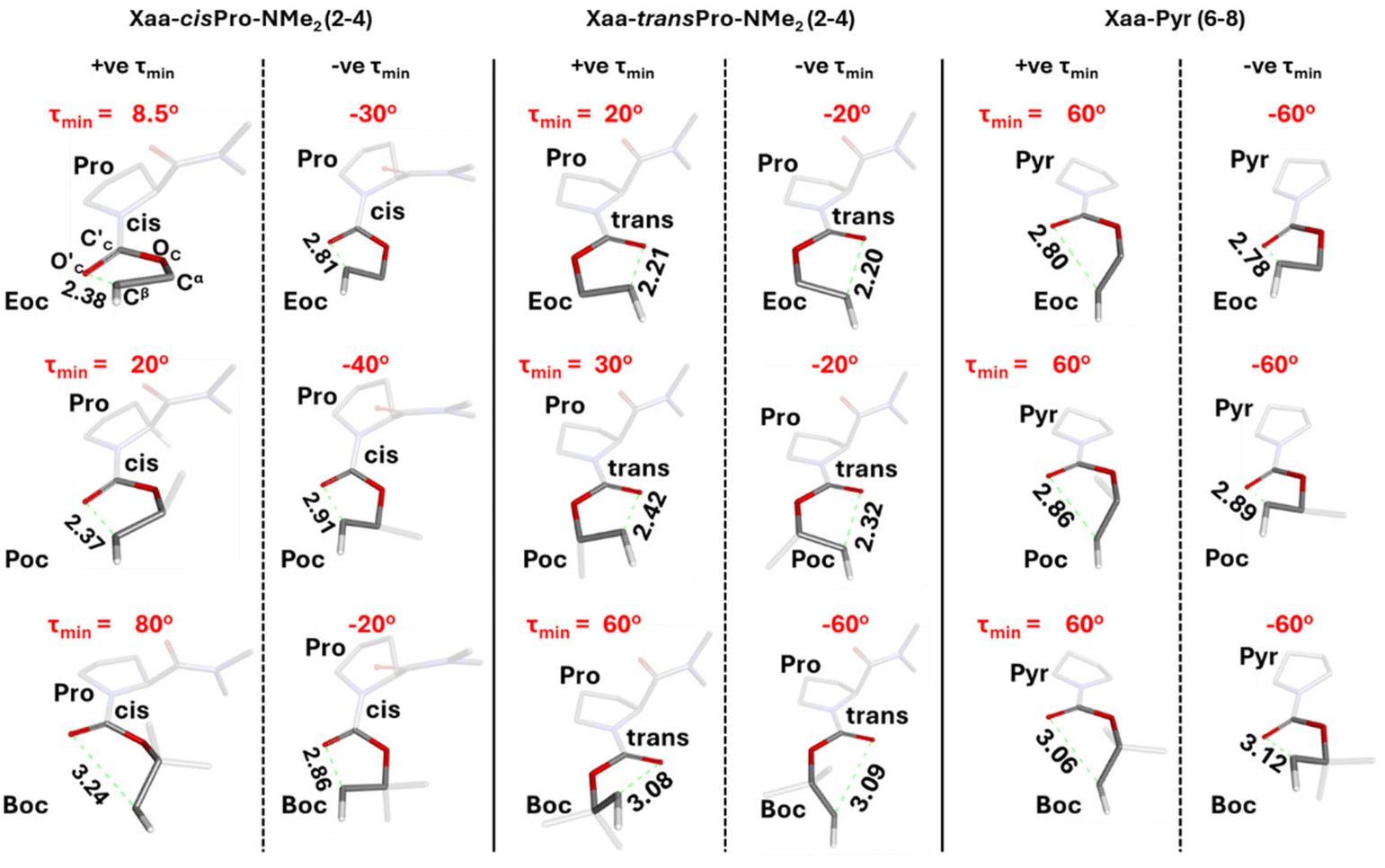 | ||
| Fig. 9 The pseudo-five-membered ring formed by the n → σ* type TBIs is shown for the cisPro and transPro isomers of all the homologues (2, 3, 4) and their corresponding pyrrolidine analogues (6, 7, 8). The green dotted lines depict the O′C⋯Cβ interactions. All structures are at their respective τmin (the dihedral angle C′C–OC–Cα–Cβ at which energy of n → σ* type TBI is minimum). | ||
On the other hand, in transPro of 1–4 although the O′C⋯Cβ (Å) and τ magnitudes (Fig. 9) are conducive for D → A interactions and there are two τmin values where O′C → Cβ interactions do occur (Fig. 8d), in both of them, there is only one np → σ* type TBI each. Notably the net NBO overlap energy in transPro is significantly lower than that in cisPro (Fig. 8d). Hence, the n → σ* type TBIs predominate in the cisPro conformers, and their energies improve relative to transPro with increasing number of σ* acceptors (of Cβ–Hβ). As a result, there is anomalous cisPro stabilization from Moc to Boc (1 to 4) despite the concomitant increase in steric bulk at R of the CO2R group in these carbamates.
In order to investigate the reason for the asymmetry of τmin (about 0) exclusively in cisPro of 2–4, the geometries of their carbamate groups in cisPro and transPro were compared. At τmin in cisPro of 2–4, the O′C⋯Cβ TBIs (depicted as green dotted lines in Fig. 9) result in the formation of a pseudo five-membered ring which adopts a half-chair conformation (Fig. 10)40 with O′C oriented exo and Cβ-endo, with respect to the proline substituent (Fig. 10) on the C′C–OC–Cα plane.41 The two Hαs on Cα in 2 are pseudo-equatorially (e′) and pseudo-axially (a′) oriented (Fig. 10). In 3, the CβH3 group that replaces one of the Hα occupies e′.42 In 4, both e′ and a′ are occupied by CβH3 groups. There is significant out of plane puckering of the torsion ρ (O′cC′C–OC–Cα, Fig. 10) by −40.0 ± 5.0° to accommodate the O′C → Cβ interactions in both the positive and negative τmin selectively in the cisPro isomers in 2–4.
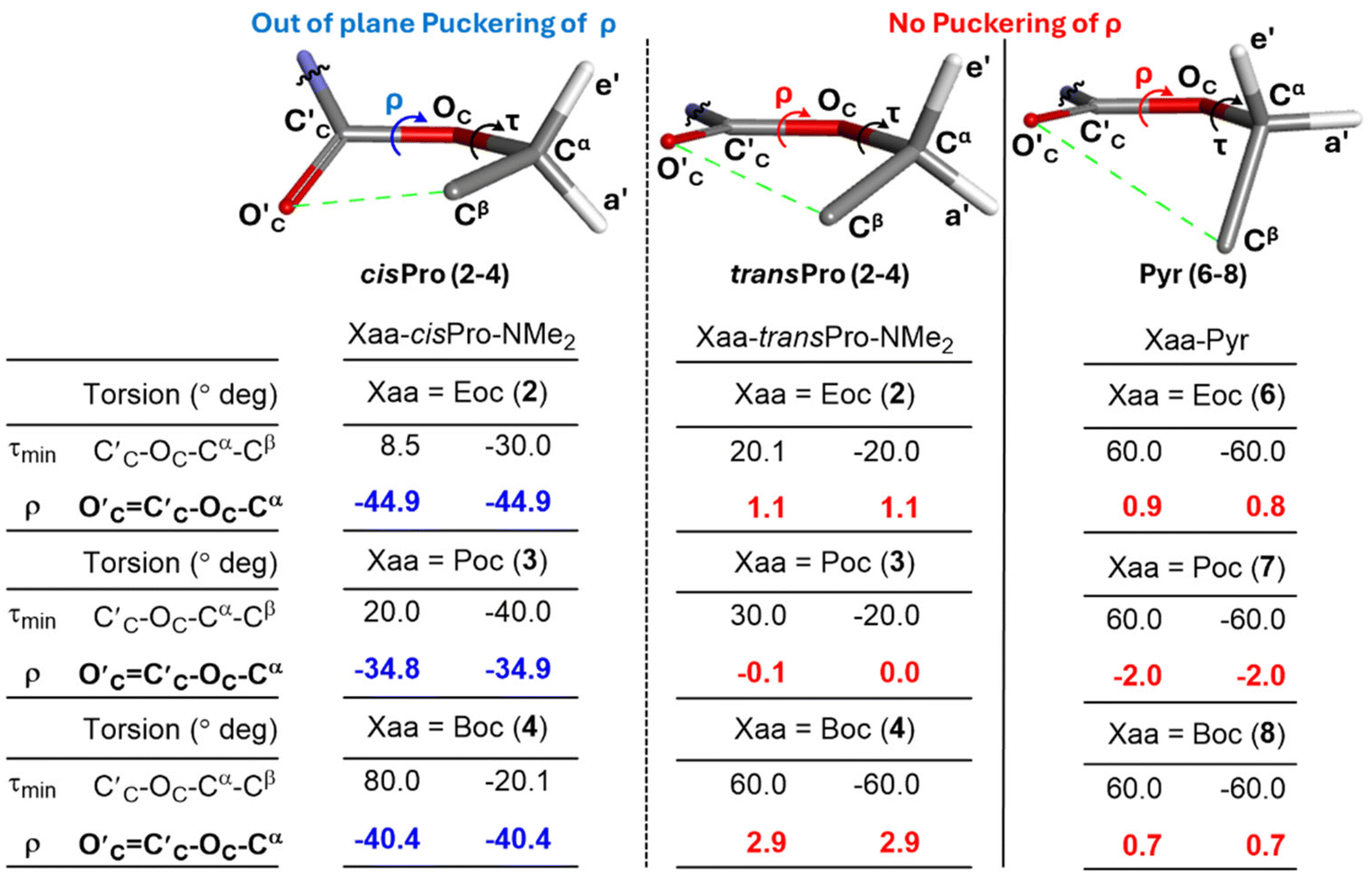 | ||
| Fig. 10 The pseudo-five-membered ring formed by the n → σ* type TBIs interactions is shown for the cisPro and transPro isomers of 2 along with the corresponding pyrrolidine analogue 6. The ρ torsions (O′cC′C–OC–Cα) are tabulated corresponding to the value at τmin (the dihedral angle C′C–OC–Cα–Cβ at which energy of n → σ* type TBI is minimum). | ||
Contrarily, in transPro of 2–4 as well as in the reference pyrrolidine models 6–8 – that lack the C′AOA charge donor source – there are no n → σ* type TBIs (O′C → Cβ) at τmin, as indicated by the absence of the ρ-puckering (Fig. 10). In both 2–4 and 6–8, the O′CC′C–OC–Cα group is largely in plane, indicating much less perturbation by any charge transfer unlike in cisPro. These selective puckering effects at the cisPro carbamate CO2R group are hence the steric consequences of the O′C⋯Cβ (n → σ*) TBIs which occur predominantly in the cisPro rotamers and cause the observed asymmetry of τmin.
Thus, a combination of two TBIs, one originating from the prolyl C-terminal amide oxygen (OA) to its N-terminal carbamate carbon (C′C) through carbonyl–carbonyl (OA → C′C) n → π* type interactions and the other from the carbamate oxygen (O′C) to the σ* of the Cβ–Hβ bond through intra-carbamate n → σ* type interactions, predominantly occurs in the cisPro conformers of prolyl carbamates and selectively stabilizes them compared to their transPro rotamers. Their interaction energies improve with increasing number of CβH3 substituents in the carbamate R group.
To observe the effect of solvent on these cisPro stabilizing TBIs, the Kc/t (Fig. 11a) and corresponding ΔG (kcal mol−1) (Fig. 11b) values (10 mM, 298 K, calculated using the Gibbs free energy equation ΔG = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnKc/t) of 1–4 were recorded in more polar solvents (DMSO-d6, D2O). The Kc/t and ΔG values of 1–4 improved as the solvent polarity increased (CDCl3 < DMSO-d6 < D2O). Notably, Kc/t of 4 shows a remarkable value of 2.60 (Fig. 10a) (ΔG = −0.56 kcal mol−1; >72% cisPro carbamate!) in aqueous medium (10% DMSO-d6 was added to D2O to improve solubility). To understand the source of such cisPro stabilization, studies were conducted to calculate the theoretical free energy difference ΔEc–t (corresponding to experimental ΔG) values of 1–4 with the PBE functional43 and def2-TZVPP basis sets using the TURBOMOLE V7.5 software.44
lnKc/t) of 1–4 were recorded in more polar solvents (DMSO-d6, D2O). The Kc/t and ΔG values of 1–4 improved as the solvent polarity increased (CDCl3 < DMSO-d6 < D2O). Notably, Kc/t of 4 shows a remarkable value of 2.60 (Fig. 10a) (ΔG = −0.56 kcal mol−1; >72% cisPro carbamate!) in aqueous medium (10% DMSO-d6 was added to D2O to improve solubility). To understand the source of such cisPro stabilization, studies were conducted to calculate the theoretical free energy difference ΔEc–t (corresponding to experimental ΔG) values of 1–4 with the PBE functional43 and def2-TZVPP basis sets using the TURBOMOLE V7.5 software.44
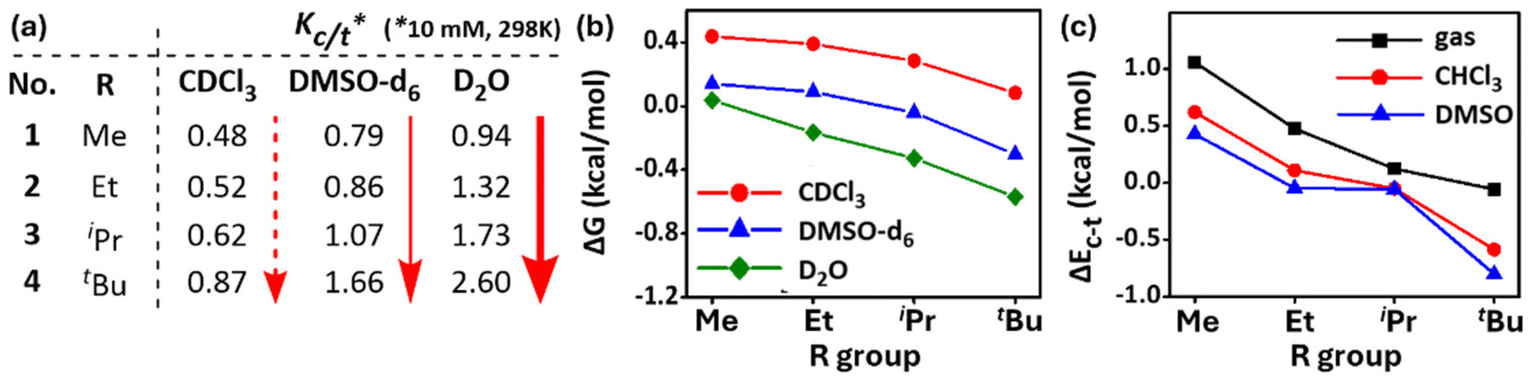 | ||
| Fig. 11 (a) Kc/t values of 1–4 in different solvents (10 mM, 298 K); plots of (b) experimental and (c) theoretically calculated conformational energy differences between transPro and cisPro isomers in 1–4 in different solvents (298 K). | ||
They showed similar trends for ΔEc–t (kcal mol−1) of 1–4 in the gas phase (ΔEc–t = EcisPro − EtransPro; more negative value for ΔEc–t implies greater stabilization of cisPro) (Fig. 10c). Importantly, when solvation effects were included via the COSMO solvation model,45 the theoretical ΔEc–t values also adopt more negative values with increasing solvent polarities (gas < CHCl3 < DMSO) (Fig. 10c). These data reveal better relative stabilization of cisPro containing the charge transfer TBIs in solvents of higher polarity. Hence, solvation effects also contribute significantly to cis/trans free energy differences in prolyl carbamates.
3 Conclusions
In this work, the conformations of cisPro and transPro rotamers of homologous prolyl carbamates and their corresponding pyrrolidine carbamates were investigated using NMR and theoretical (DFT, NBO) methods. These studies reveal an anomaly, where the relative stabilities of cisPro carbamates are found to increase with increasing steric bulk at R of the carbamate CO2R group. We observe the presence of a relay of two charge transfer tetrel bonding interactions (TBIs) predominantly in cisPro carbamates, which also stabilise them. First a charge transfer occurs from the amide oxygen to the carbamate carbon both of which flank the proline, through a carbonyl–carbonyl n → π* type TBI. This charge is further transferred from the carbamate oxygen to the σ* of the Cβ–Hβ bond (in C(O)O–Cα–Cβ–Hβ of the carbamate alcohol group) through an intra-carbamate n → σ* type TBI. Energies of the latter TBI improve with increase in the number of σ* acceptors (of Cβ–Hβ), despite the concomitant increase in steric bulk at the carbamate alcohol. This charge transfer relay of TBIs occurring in cisPro of prolyl carbamates is in the reverse direction (C-terminal to N-terminal across Pro) compared to that observed in transPro of prolyl amides (N-terminal to C-terminal across Pro) – both of which stabilize their corresponding conformers. A consequence of the stronger TBIs in cisPro carbamates is that there is significant puckering in the O′cC′C–OC–Cα (−40 ± 5°) torsion of the pseudo five-membered rings formed at the C(O)O–Cα–Cβ–Hβ group of carbamates. Increasing solvent polarities are observed to further improve the relative stabilities of cisPro carbamate conformers. This work provides first insights into the tetrel interactions that govern the cis–trans isomerism in carbamates. Given that carbamates appear in several biorelevant molecules46 and their cis–trans isomerism has been used to regulate ion flux in artificial ion channels,47 the current results are also important in showcasing the alcohol groups of carbamates as tools to regulate their cis–trans equilibria to suit various applications.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- D. N. Reddy and E. N. Prabhakaran, Steric and electronic interactions controlling the cis/trans isomer equilibrium at X-pro tertiary amide motifs in solution, Biopolymers, 2014, 101(1), 66–77 CrossRef CAS PubMed.
- G.-B. Liang, C. J. Rito and S. H. Gellman, Variations in the turn-forming characteristics of N-Acyl proline units, Biopolymers, 1992, 32(3), 293–301 CrossRef CAS PubMed.
- D. N. Reddy, G. George and E. N. Prabhakaran, Crystal-Structure Analysis of cis-X-Pro-Containing Peptidomimetics: Understanding the Steric Interactions at cis X-Pro Amide Bonds, Angew. Chem., Int. Ed., 2013, 52(14), 3935–3939 CrossRef CAS.
- S. Banerjee, S. K. Gupta, S. Pal and E. N. Prabhakaran, Crystal structures reveal that the sterically hindered pivaloyl–cisProlyl amide bond is energetically frustrated, Pept. Sci., 2024, 116(3), e24337 CrossRef CAS.
- C. Rao, P. Balaram and C. Rao, Infrared spectroscopic study of C7 intramolecular hydrogen bonds in peptides, Biopolymers, 1983, 22(9), 2091–2104 CrossRef CAS.
- B. N. Rao, A. Kumar, H. Balaram, A. Ravi and P. Balaram, Nuclear Overhauser effects and circular dichroism as probes of. beta.-turn conformations in acyclic and cyclic peptides with pro-X sequences, J. Am. Chem. Soc., 1983, 105(25), 7423–7428 CrossRef CAS.
- A. Choudhary, C. G. Fry, K. J. Kamer and R. T. Raines, An n→ π* interaction reduces the electrophilicity of the acceptor carbonyl group, Chem. Commun., 2013, 49(74), 8166–8168 RSC.
- N. J. Zondlo, Aromatic–Proline Interactions: Electronically Tunable CH/π Interactions, Acc. Chem. Res., 2013, 46(4), 1039–1049 CrossRef CAS.
- B. Dasgupta, P. Chakrabarti and G. Basu, Enhanced stability of cis Pro-Pro peptide bond in Pro-Pro-Phe sequence motif, FEBS Lett., 2007, 581(23), 4529–4532 CrossRef CAS.
- S. K. Gupta, S. Banerjee and E. N. Prabhakaran, van der Waals interactions to control amide cis–trans isomerism, New J. Chem., 2022, 46(26), 12470–12473 RSC.
- H. K. Ganguly, B. Majumder, S. Chattopadhyay, P. Chakrabarti and G. Basu, Direct Evidence for CH⋯π Interaction Mediated Stabilization of Pro-cisPro Bond in Peptides with Pro-Pro-Aromatic motifs, J. Am. Chem. Soc., 2012, 134(10), 4661–4669 CrossRef CAS PubMed.
- S. Banerjee, S. K. Gupta and E. N. Prabhakaran, Direct Evidence for Synchronicity between Rotation along Cα−C′ and Pyramidalization of C′ in Amides, ChemistrySelect, 2023, 8(17), e202301105 CrossRef CAS.
- S. Banerjee, S. K. Gupta, S. Pal and E. N. Prabhakaran, Crystal structures reveal that the sterically hindered pivaloyl–cisProlyl amide bond is energetically frustrated, Pept. Sci., 2024, e24337 CrossRef CAS.
- C. M. Lee and W. Kumler, The dipole moment and structure of the carbamate group, J. Am. Chem. Soc., 1961, 83(22), 4596–4600 CrossRef CAS.
- M. J. Deetz, C. C. Forbes, M. Jonas, J. P. Malerich, B. D. Smith and O. Wiest, Unusually low barrier to carbamate C–N rotation, J. Org. Chem., 2002, 67(11), 3949–3952 CrossRef CAS PubMed.
- B. Sahariah and B. K. Sarma, Spectroscopic evidence of n→ π* interactions involving carbonyl groups, Phys. Chem. Chem. Phys., 2020, 22(46), 26669–26681 RSC.
- E. N. Prabhakaran, S. Tumminakatti, K. Vats and S. Ghosh, Spectral evidence for generic charge→ acceptor interactions in carbamates and esters, RSC Adv., 2020, 10(20), 11871–11875 RSC.
- S. Scheiner, Origins and properties of the tetrel bond, Phys. Chem. Chem. Phys., 2021, 23(10), 5702–5717 RSC.
- V. L. Heywood, T. P. Alford, J. J. Roeleveld, S. J. L. Deprez, A. Verhoofstad, J. I. van der Vlugt, S. R. Domingos, M. Schnell, A. P. Davis and T. J. Mooibroek, Observations of tetrel bonding between sp 3-carbon and THF, Chem. Sci., 2020, 11(20), 5289–5293 RSC.
- T. Yoshimoto, K. Kawahara, F. Matsubara, K. Kado and D. Tsuru, Comparison of inhibitory effects of prolinal-containing peptide derivatives on prolyl endopeptidases from bovine brain and Flavobacterium, J. Biochem., 1985, 98(4), 975–979 CrossRef CAS PubMed.
- A. Dal Corso, V. Borlandelli, C. Corno, P. Perego, L. Belvisi, L. Pignataro and C. Gennari, Fast Cyclization of a Proline–Derived Self–Immolative Spacer Improves the Efficacy of Carbamate Prodrugs, Angew. Chem., 2020, 132(10), 4205–4210 CrossRef.
- J. Shi, T. Zheng, Y. Zhang, B. Guo and J. Xu, Cross-linked polyurethane with dynamic phenol-carbamate bonds: properties affected by the chemical structure of isocyanate, Polym. Chem., 2021, 12(16), 2421–2432 RSC.
- N. J. Baker, B. A. Bancroft and T. S. Garcia, A meta-analysis of the effects of pesticides and fertilizers on survival and growth of amphibians, Sci. Total Environ., 2013, 449, 150–156 CrossRef CAS PubMed.
- R. Karaman, Prodrugs Design Based on Inter–and Intramolecular Chemical Processes, Chem. Biol. Drug Des., 2013, 82(6), 643–668 CrossRef CAS.
- D. Chaturvedi, Perspectives on the synthesis of organic carbamates, Tetrahedron, 2012, 68(1), 15–45 CrossRef CAS.
- A. K. Ghosh and M. Brindisi, Organic carbamates in drug design and medicinal chemistry, J. Med. Chem., 2015, 58(7), 2895–2940 CrossRef CAS.
- A. Matošević and A. Bosak, Carbamate group as structural motif in drugs: A review of carbamate derivatives used as therapeutic agents, Arh. Hig. Rada Toksikol., 2020, 71(4), 285–299 Search PubMed.
- J. C. Verheijen, K. A. Wiig, S. Du, S. L. Connors, A. N. Martin, J. P. Ferreira, V. I. Slepnev and U. Kochendörfer, Novel carbamate cholinesterase inhibitors that release biologically active amines following enzyme inhibition, Bioorg. Med. Chem. Lett., 2009, 19(12), 3243–3246 CrossRef CAS.
- A. B. Smith III, A. K. Charnley and R. Hirschmann, Pyrrolinone-Based peptidomimetics.“Let the enzyme or receptor be the judge”, Acc. Chem. Res., 2011, 44(3), 180–193 CrossRef PubMed.
- S. K. Singh, K. K. Mishra, N. Sharma and A. Das, Direct spectroscopic evidence for an n→ π* interaction, Angew. Chem., Int. Ed., 2016, 55(27), 7801–7805 CrossRef CAS.
- S. K. Singh, P. Panwaria, K. K. Mishra and A. Das, Steric as well as n→ π* Interaction Controls the Conformational Preferences of Phenyl Acetate: Gas–phase Spectroscopy and Quantum Chemical Calculations, Chem. – Asian J., 2019, 14(24), 4705–4711 CrossRef CAS PubMed.
- G. J. Bartlett, A. Choudhary, R. T. Raines and D. N. Woolfson, n→π* interactions in proteins, Nat. Chem. Biol., 2010, 6(8), 615–620 CrossRef CAS PubMed.
- G. Purushothaman and V. Thiruvenkatam, Analysis of intermolecular interactions in 2, 3, 5 Trisubstituted Pyrazoles derivatives: insights into crystal structures, Gaussian B3LYP/6-311G (d, p), PIXELC and Hirshfeld surface, J. Chem. Crystallogr., 2016, 46, 371–386 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Google Scholar 2015, Gaussian Inc, Wallingford, CT, 2009 Search PubMed.
- H. Burgi, J. Dunitz and E. Shefter, Geometrical reaction coordinates. II. Nucleophilic addition to a carbonyl group, J. Am. Chem. Soc., 1973, 95(15), 5065–5067 CrossRef CAS.
- A. Lari, M. B. Pitak, S. J. Coles, E. Bresco, P. Belser, A. Beyeler, M. Pilkington and J. D. Wallis, The use of the triptycene framework for observing O⋯C=O molecular interactions, CrystEngComm, 2011, 13(23), 6978–6984 RSC.
- S. Scheiner and T. Kar, Spectroscopic and structural signature of the CH–O hydrogen bond, J. Phys. Chem. A, 2008, 112(46), 11854–11860 CrossRef CAS PubMed.
- A. v. Bondi, van der Waals Volumes and Radii, J. Phys. Chem., 1964, 68(3), 441–451 CrossRef CAS.
- V. R. Mundlapati, D. K. Sahoo, S. Bhaumik, S. Jena, A. Chandrakar and H. S. Biswal, Noncovalent Carbon–Bonding Interactions in Proteins, Angew. Chem., Int. Ed., 2018, 57(50), 16496–16500 CrossRef CAS.
- S. Saebø, F. R. Cordell and J. E. Boggs, Structures and conformations of cyclopentane, cyclopentene, and cyclopentadiene, J. Mol. Struct., 1983, 104(1–2), 221–232 CrossRef.
- C. t. Altona and M. Sundaralingam, Conformational analysis of the sugar ring in nucleosides and nucleotides. New description using the concept of pseudorotation, J. Am. Chem. Soc., 1972, 94(23), 8205–8212 CrossRef CAS PubMed.
- C. A. Stortz and A. M. Sarotti, Exhaustive exploration of the conformational landscape of mono-and disubstituted five-membered rings by DFT and MP2 calculations, RSC Adv., 2019, 9(42), 24134–24145 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- S. Balasubramani, G. Chen, S. Coriani, M. Diedenhofen, M. Frank, Y. Franzke, F. Furche, R. Grotjahn, M. Harding, C. Hättig, C. vanWüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed.
- A. Klamt, Conductor-like screening model for real solvents: a new approach to the quantitative calculation of solvation phenomena, J. Phys. Chem., 1995, 99(7), 2224–2235 CrossRef CAS.
- S. Šegan, I. Jevtić, T. Tosti, J. Penjišević, V. Šukalović, S. Kostić-Rajačić and D. Milojković-Opsenica, Determination of lipophilicity and ionization of fentanyl and its 3-substituted analogs by reversed-phase thin-layer chromatography, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2022, 1211, 123481 CrossRef.
- G. A. Woolley, A. S. Jaikaran, Z. Zhang and S. Peng, Design of regulated ion channels using measurements of cis-trans isomerization in single molecules, J. Am. Chem. Soc., 1995, 117(16), 4448–4454 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01539h |
| This journal is © The Royal Society of Chemistry 2024 |