
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicroneedle-assisted transdermal delivery of carvedilol nanosuspension for the treatment of hypertension
Anushri
Deshpande
*,
Vidhi
Mer
,
Darshana
Patel
and
Hetal
Thakkar

The Maharaja Sayajirao University Of Baroda, Vadodara, India. E-mail: hetal.thakkar-pharmacy@msubaroda.ac.in
First published on 24th May 2024
Abstract
Carvedilol nanosuspension loaded microneedles patch was formulated and characterized by particle size, zeta potential, solubility, Transmission Electron Microscopy, X-Ray Diffraction, in-vitro release and in-vivo pharmacokinetic studies A nanosuspension-loaded microneedle patch was successfully prepared and characterized by optical microscopy, scanning electron microscopy, axial fracture force, in vitro dissolution study, % drug content, in vitro drug-release study, ex vivo studies, an in vivo study, and stability studies. The particle size, PDI, and zeta potential of the carvedilol nanosuspension were found to be 179.6 ± 1.15 nm, 0.163 ± 0.01, and −14.2 ± 0.55 mV, respectively. There was a 9.21-fold increase in the saturation solubility of the carvedilol nanosuspension. Nanosuspension-loaded microneedles contained 98.78 ± 0.12% carvedilol. The relative bioavailability of the carvedilol from the microneedle patch was found to be 2.82-fold higher compared to the marketed formulation. The drug release from the microneedles followed zero-order kinetics, which is desirable in the case of transdermal delivery. The stability study indicated that the prepared formulation was stable under the storage conditions used. Thus, the developed transdermal microneedle patch containing the carvedilol nanosuspension seems to be a promising approach to foster greater patient compliance for the management of hypertension.
1. Introduction
Oral delivery is considered to be the most patient-compliant route for drug administration. However, certain limitations such as high first-pass metabolism, low oral bioavailability, and GI disturbance necessitate the exploration of alternative routes for drug delivery. The transdermal drug delivery system is associated with several advantages such as elimination of first-pass metabolism, the possibility of controlled release, and facile administration of a therapy.Carvedilol is a biopharmaceutics classification system (BCS) class II drug used in the management of hypertension, cardiac arrhythmias, angina pectoris, and symptomatic heart failure.1 Carvedilol is available in tablet form with strengths of 3.125 mg, 6.25 mg, 12.5 mg, and 25 mg. The plasma half-life of carvedilol is 6–10 h, which may necessitate a higher frequency of administration.2 Due to this limitation, controlled- and sustained-release formulations have been developed. Some of them are controlled-release tablets3 and pellets4 composed of hydroxypropyl methylcellulose, eudragit, or ethyl cellulose. Due to its low solubility and high first-pass metabolism, the oral bioavailability of carvedilol is low, at 25–30%. The oral bioavailability of a poorly water-soluble drug undergoing high first-pass metabolism can be enhanced by various approaches. Some of the approaches reported for carvedilol are solid lipid nanoparticles,5 niosomal carriers,6 solid self-nanoemulsifying drug delivery systems (S-SNEDDS),7 nanosuspension from an osmotic pump capsule,8 and solid dispersions.9
A nanosuspension is one of the approaches used for the enhancement of drug solubility.10 It has many advantages such as a simple method of preparation, less requirement of excipients, increased dissolution velocity and saturation solubility, improved adhesion, increased bioavailability leading to a decrease in the dose and fast-fed variability, and ease of large-scale manufacturing. The availability of the drug in nanosize leads to enhanced solubility due to a large increase in the surface area.11 Thus, it was expected that a nanosuspension of carvedilol would have greater solubility and consequently higher systemic absorption.
Transdermal drug delivery can overcome the limitations of oral and parenteral administration. However, this route is currently useful for only a small number of drugs, due to the impermeability of the outer layer of skin, the stratum corneum (SC).12 A penetration enhancement approach is usually required when pursuing transdermal drug delivery to achieve a desirable plasma concentration.13 Various penetration enhancement techniques have been reported, such as iontophoresis,14 sonoporation,15 microneedles,16 and chemical enhancers.17
MNs are minimally invasive devices that bypass the SC barrier of the skin, and thus, the dermal microcirculation and inferior layers of the skin can be accessed. When applied to the skin, they painlessly puncture the SC, creating microscopic channels through which drugs can diffuse. This provides a physical pathway for drugs to permeate the skin in a much higher concentration as compared to a normal topical formulation. Soluble MNs are composed of polymers that are biodegradable, biocompatible, and solubilize upon insertion into the skin. Thus, they reduce the risk of toxic or biohazardous waste.
Polymers such as poly(methyl vinyl ether-co-maleic acid),18,19 chitosan,20 and polyvinyl alcohol21 form a hydrogel when they come in contact with the interstitial fluid of the SC, and they are used for the preparation of microneedles. Once in the skin, they swell, thus creating a porous network through which drugs contained within a reservoir can diffuse, and subsequently move into the dermal microcirculation. Thus, they control the rate of drug release.
Amongst the different polymers available for microneedle preparation, poly(methyl vinyl ether-co-maleic anhydride) was selected due to its desirable properties such as biodegradable nature, capacity to form a hydrogel, bioadhesiveness, and ability to provide sustained release of a drug. The development of soluble microneedles for lipophilic drugs is challenging due to the uneven distribution of a drug in the microneedle matrix because of its insoluble nature. The non-uniform size of drug particles leads to the formation of imperfect microneedles. Formulation of a nanosuspension leads to uniformly sized particles and increased aqueous solubility, which allows for more rapid absorption into the systemic circulation. Thus, the principal objective of the present research was to formulate and evaluate carvedilol-loaded microneedles by in vitro, ex vivo, and in vivo studies and determine their ability to increase the bioavailability.
2. Materials and methods
2.1 Materials
Carvedilol was received as a gift from Zydus Pharmaceuticals Limited, Ahmedabad, India. Sodium deoxycholate (NaDC) and polymethyl vinyl ether-alt-maleic anhydride (PMVE/MA) were purchased from Sigma-Aldrich, India. Zirmil® (zirconium oxide) beads 0.4 mm in diameter were purchased from Magnum Apex, India, and Sylgard® 184: polydimethylsiloxane (PDMS) was procured from Dow Corning, USA. Potassium dihydrogen phosphate and sodium lauryl sulphate (SLS) were purchased from Fisher Scientific, India and sodium chloride was purchased from Sulab Chemicals, India. Disodium hydrogen phosphate and sodium hydroxide were purchased from Astron, India.2.2 Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio were added to the molding container and carefully mixed to prevent the formation of air bubbles. The air bubbles were removed by placing the mold in a vacuum desiccator for five minutes. After removal of the air bubbles, the MN derma stamp was vertically inserted and maintained overnight in this PDMS solution. The derma stamp was later removed from the molding container, and the container was maintained at 90 °C in a water bath for 5 hours. After completely curing, the mold was removed from the molding container.
:1 weight ratio were added to the molding container and carefully mixed to prevent the formation of air bubbles. The air bubbles were removed by placing the mold in a vacuum desiccator for five minutes. After removal of the air bubbles, the MN derma stamp was vertically inserted and maintained overnight in this PDMS solution. The derma stamp was later removed from the molding container, and the container was maintained at 90 °C in a water bath for 5 hours. After completely curing, the mold was removed from the molding container.
2.3 Characterization of the carvedilol nanosuspension
2.4 Characterization of nanosuspension-loaded microneedles
2.4.7.1 Drug permeation study. Freshly excised rat abdominal skin was hydrated in PBS (pH 7.4) for 30 min before the study. Subcutaneous fat was removed with a scalpel, and the skin was placed on a stack of filter papers pre-wetted with PBS (pH 7.4) to provide mechanical support. Simple patches (composition: water, glycerin as a plasticizer, carvedilol, and PVME/MA polymer), as well as microneedle patches containing 5 mg of carvedilol, were applied onto the skin using gentle thumb pressure. The skin was immediately mounted between the donor and receptor chambers of the diffusion cell with the patch on the SC facing towards the donor side. The receptor chamber of the cell was filled with 7 mL PBS (pH 7.4) containing 2% SLS as a diffusion medium, and then, the liquid in the receptor chamber was subjected to gentle stirring using a magnetic stirrer. Next, 1 mL samples were withdrawn from the receptor compartment at predetermined time points over 24 hours, and were replenished with 1 mL of fresh diffusion medium. Quantitative estimation of the drug in the samples was performed using UV-visible spectrophotometry at 242 nm. The percentage of drug that permeated through the skin was calculated. A graph of the cumulative amount of drug that permeated as a function of time was generated, and the steady flux (Jss, μg cm−2 h−1) was calculated from the slope of the linear portion of this graph. The permeation enhancement ratio (PER) compared to a plain patch (control) of PBS (pH 7.4) was calculated from the steady-state flux using the formula.27
2.4.7.2 Drug retained on the surface of the skin. At the end of 24 hours, the drug retained on the skin surface was estimated by washing the skin three times with PBS (pH 7.4) containing 2% SLS. These washings were collected, and the amount of drug present in them was estimated by UV-visible spectrophotometry at 242 nm. The drug deposited on the skin was calculated.24
2.4.7.3 Drug retained within the skin. After 24 hours, the washed skin was cut into small pieces using a chopper to estimate the drug deposition within the skin. The pieces were collected in diffusion media to extract the drug present in these pieces. The samples were then subject to homogenization (Remi homogenizer) for five minutes. This homogenized mixture was then subjected to a sonication in a bath sonicator for five 3 min cycles to extract the drug. The tissues were separated by centrifugation at 5000 rpm for 10 minutes. The supernatant was collected, and the drug present in it was analyzed by UV spectrophotometer at 242 nm after suitable dilution.24
2.4.7.4 Transdermal flux. The slope of the linear portion of the graph of the cumulative amount permeated as a function of time represents the steady-state flux (Jss, μg cm−2 h−1). The permeation enhancement ratio (PER) was calculated from the steady-state flux using the following equation.
where, Jss (test) = steady-state flux obtained via an optimized microneedles patch, and Jss (control) = steady-state flux obtained via a simple transdermal patch.24
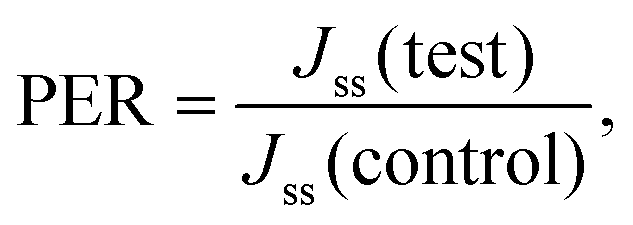
2.4.7.5 Histopathology study. The safety of breaching the SC with MNs was evaluated by performing a histopathology study. Rat abdominal skin was obtained by following protocol no: MSU/IAEC/2018-19/1806. Microneedles containing the drug or drug suspension were applied to freshly excised rat abdominal skin. A negative control and positive control were also prepared by treating rat skin with PBS (pH 7.4) and isopropyl alcohol (IPA), respectively, using the same method. After four hours, the skins were immersed in 10% buffered formalin (fixative), gradually dehydrated with increasing concentrations of ethanol, immersed in xylene, and finally embedded in paraffin. Then, 5-μm-thick sections of skin were cut from these paraffin blocks using a microtome, and the slices were placed on glass slides. The paraffin wax was removed by gently warming the slides and washing the molten wax with xylene. Sections were then washed with absolute alcohol and water and stained with hematoxylin and eosin to determine the histopathology. Commercial glycerol mounting fluid was used to finally mount the stained sections. The slides were analyzed at 10-fold magnification using an optical microscope.30
2.4.7.6 In vivo studies. The in vivo pharmacokinetic study was performed on male Wistar albino rats weighing 350–560 g. All animals were handled and housed according to CPCSEA guidelines, Department of Animal Welfare, government of India. The experimental protocol was approved by the Institutional Animal Ethics Committee (MSU/IAEC/2018-19/1824). Six sets of rats (3 animals in each set) were divided into 3 groups, with two sets of rats in each group. The oral suspension of carvedilol (0.5 mg kg−1) was administered to Group 1. Groups 2 and 3 received a carvedilol simple patch (0.5 mg kg−1) or a carvedilol MN patch, respectively. The marketed formulation of the carvedilol tablet was crushed and administered to rats fasted overnight as per the animal dose calculated (Group 1). The transdermal patch without microneedles and the microneedle patch were prepared according to animal dose and administered to Group 2 and Group 3, respectively.
The animal dose was calculated based on the following formula:
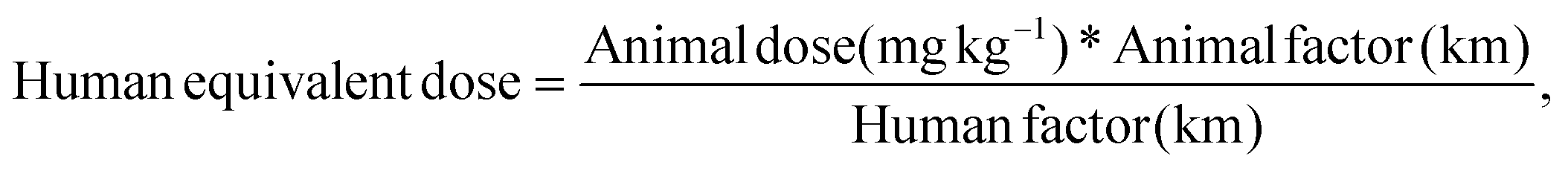
(1) At refrigeration temperature (2–8 °C).
(2) At room temperature (25–40 °C): the optimized MN patches were placed in an airtight plastic container with a bag containing silica as a desiccant. The containers were then tightly closed and sealed using Parafilm M® and finally with aluminum foil to avoid contact with moisture. One container was stored at room temperature (25–40 °C), and the other one was refrigerated (2–8 °C). The stability study was conducted by comparing the characterization parameters of the MN patches: AFF, % drug content, and % cumulative drug release of the MN patches under both conditions after 15 days and 30 days.24
3. Results and discussion
3.1 Nanosuspension analysis
 | ||
| Fig. 1 TEM of the carvedilol nanosuspension. | ||
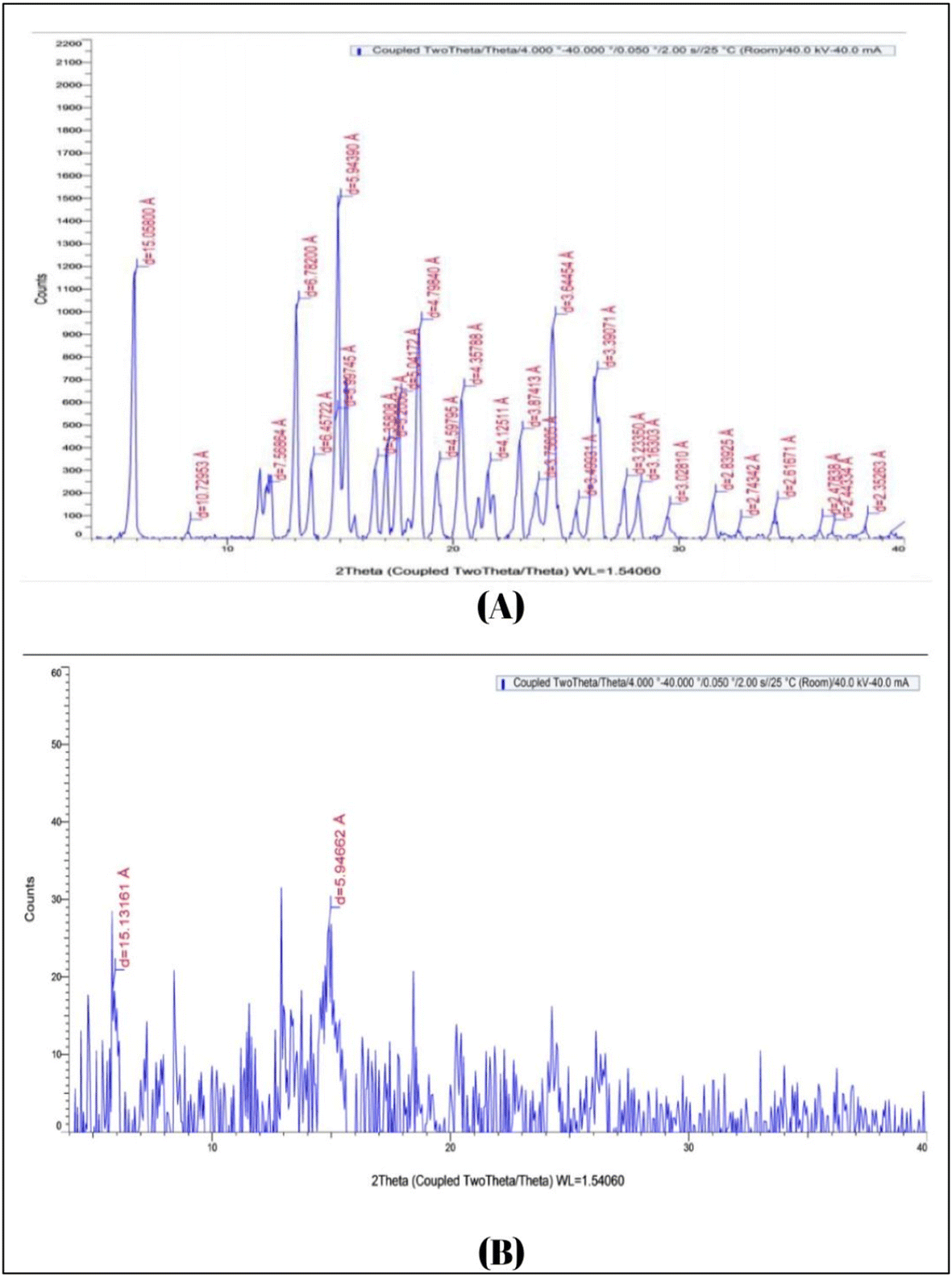 | ||
| Fig. 2 XRD of (A) pure carvedilol drug and (B) carvedilol nanosuspension. | ||
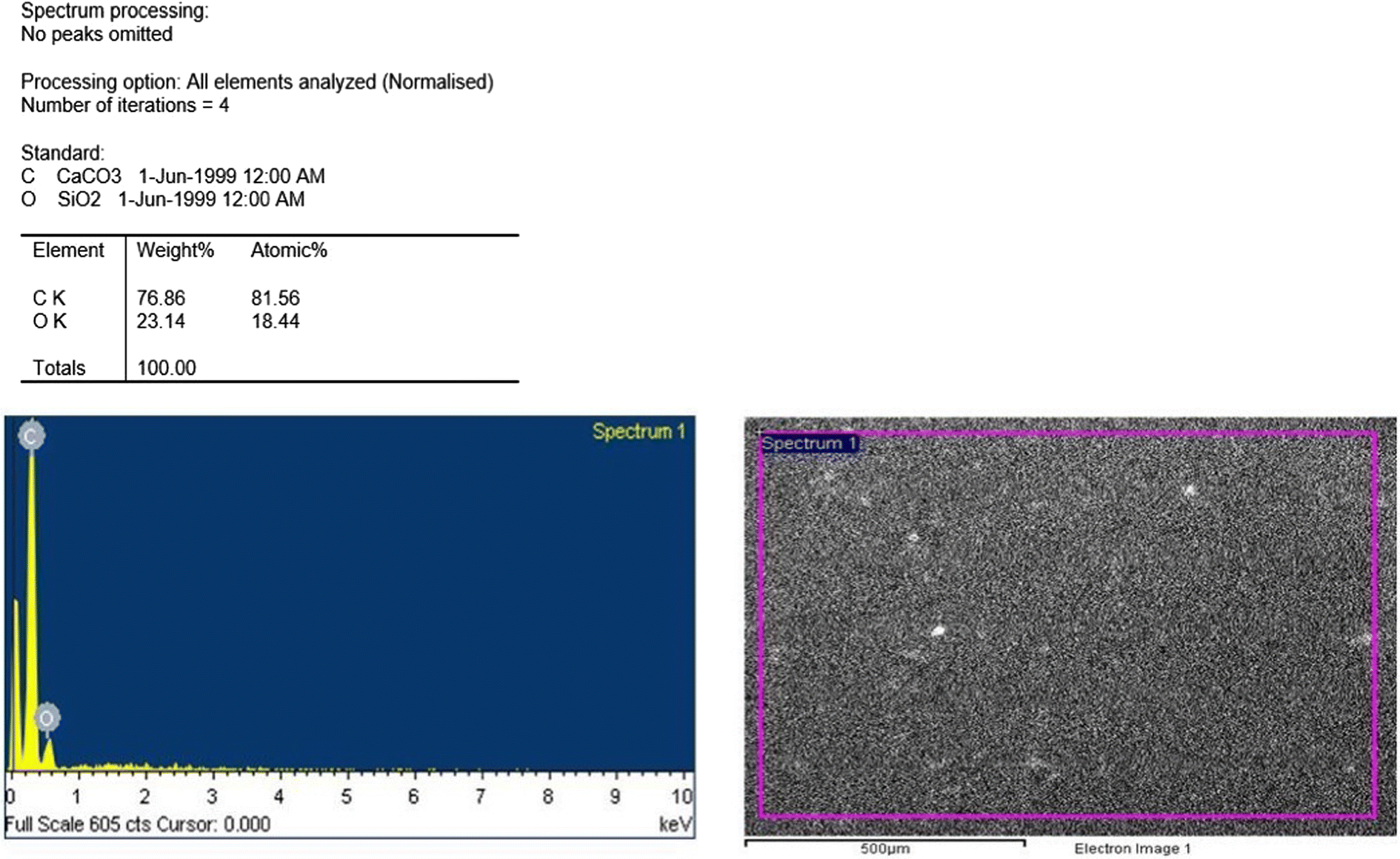 | ||
| Fig. 3 EDS report for the nanosuspension. | ||
 | ||
| Fig. 4 (A) SEM image of microneedles. Optical microscopy images at (B) 4× and (C) 10×. (D) SEM image of microneedles. | ||
3.2 Results and discussion of nanosuspension loaded microneedles
| Characterization | Observed value | Reference value |
|---|---|---|
| Length | 1.5–2 mm | 0.15–2.5 mm |
| Tip diameter | 5 μm | 1–25 μm |
| Base diameter | 260–400 μm | 50–250 μm |
| Distance between two needles | 581 μm | 500–800 μm |
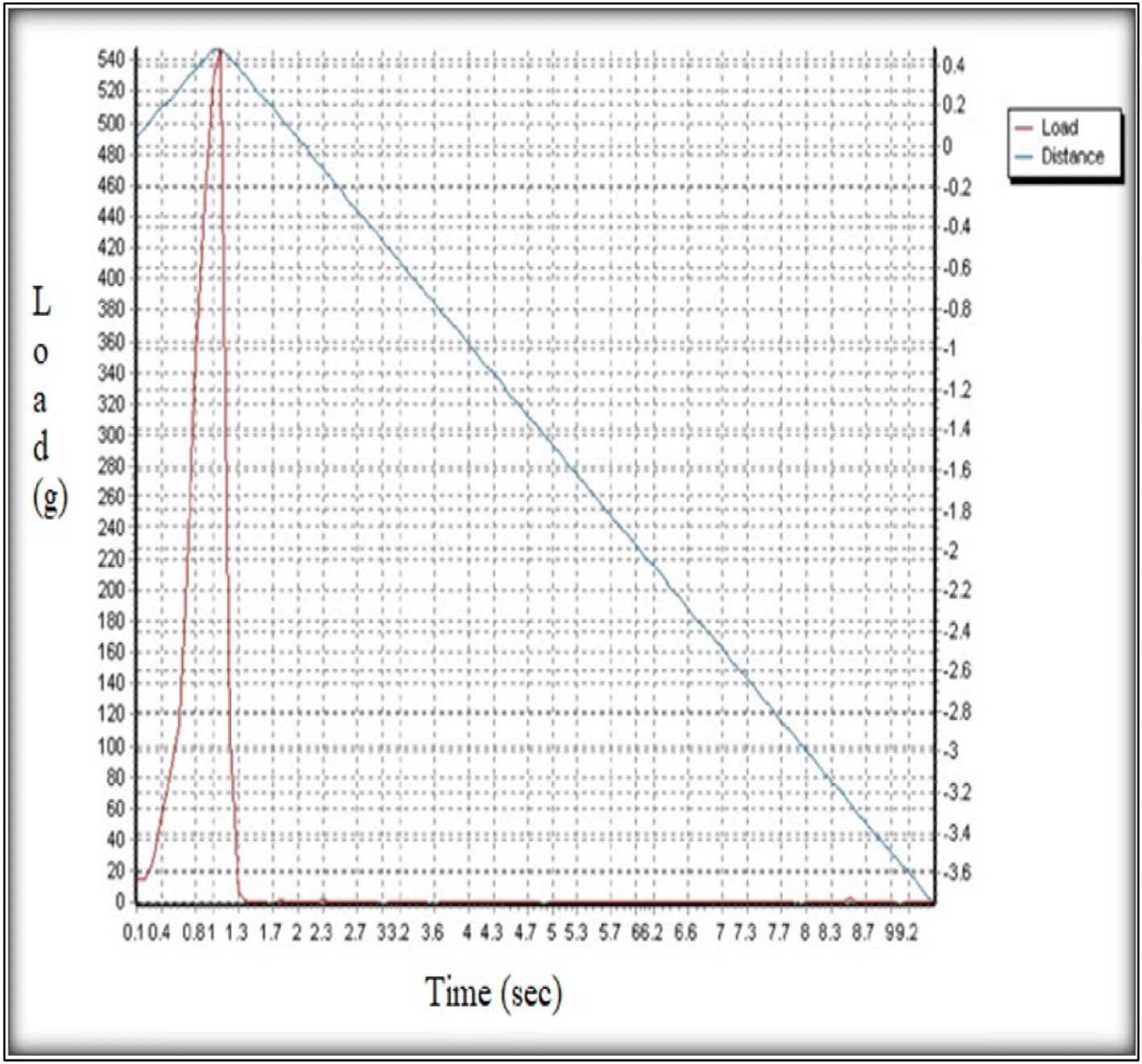 | ||
| Fig. 5 Graph of grams vs. time for the measurement of the axial fracture force. | ||
 | ||
| Fig. 6 Microscopic images of microneedles at different time points. At (A) 5 minutes, (B) 10 minutes, (C) 15 minutes, and (D) 20 minutes. | ||
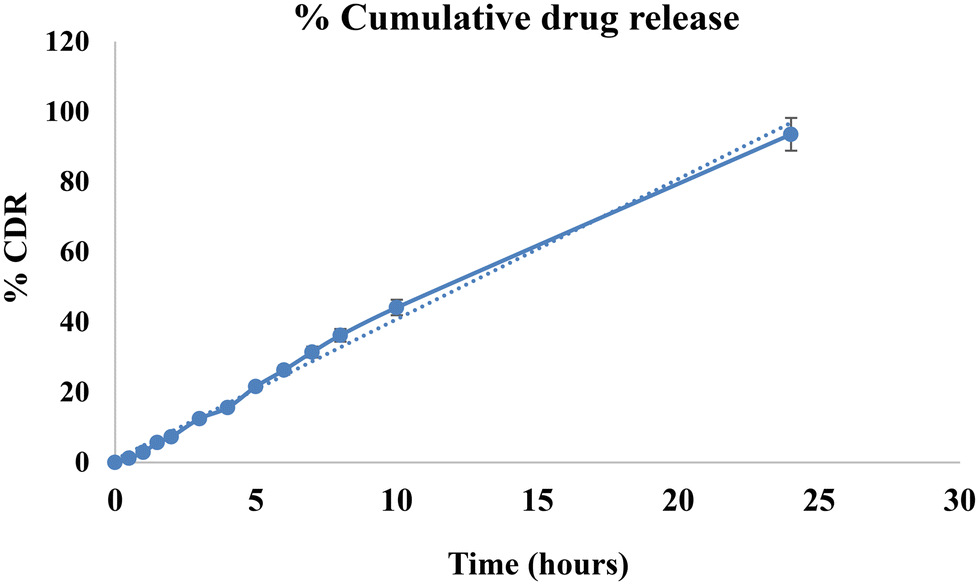 | ||
| Fig. 7 Cumulative drug release from microneedles. | ||
| Formulation | Regression coefficient (R2) | ||||
|---|---|---|---|---|---|
| Zero order | First order | Higuchi model | Hixson–Crowell model | Korsemeyer–Peppas model | |
| Microneedles | 0.9928 | 0.9468 | 0.916 | 0.9823 | 0.9823 |
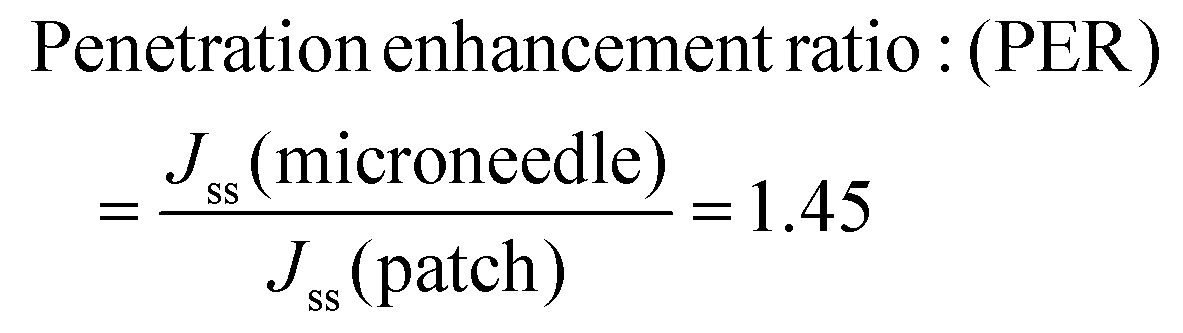
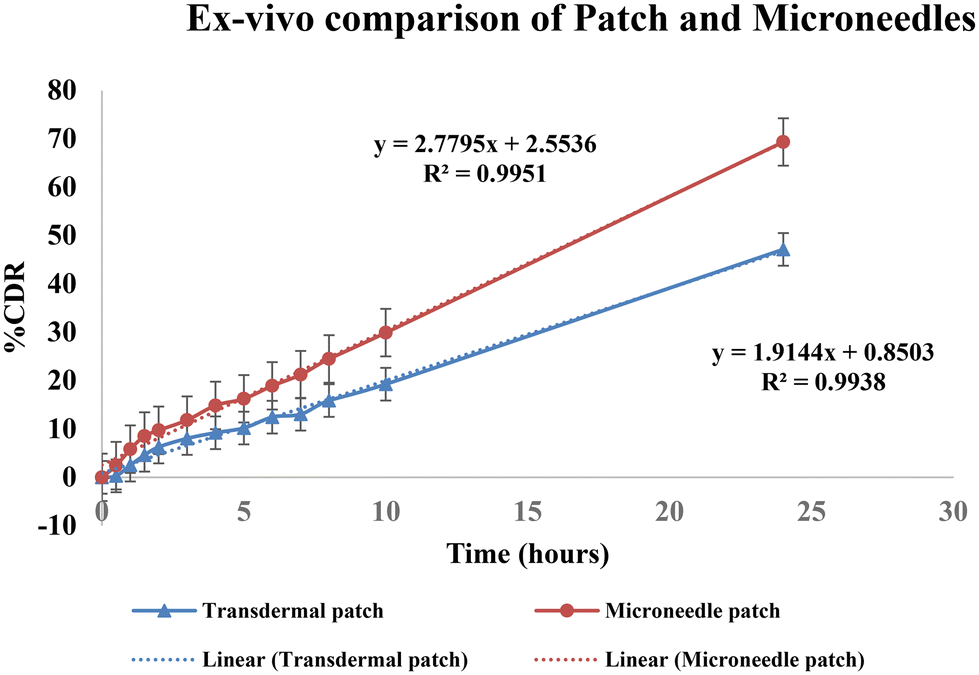 | ||
| Fig. 8 Comparison of % CDR of a simple patch and a microneedle patch. | ||
Thus, permeation was found to be increased by 1.45-fold by the microneedles patch. Image 11 shows a comparison of the drug deposition across the skin by the simple transdermal patch as well as the microneedles patch. The MN patch delivered 95.6 ± 0.77%, and in transdermal patches without MNs, it was found to be 66.29 ± 0.39% drug deposition across and within the skin. This indicates that greater drug permeation occurred from the MN patch due to the formation of microchannels in the skin. The amount of drug retained on the surface of the skin was significantly lower in the case of a microneedle patch, which indicates the ability of microneedles to increase the penetration of drug across the skin.
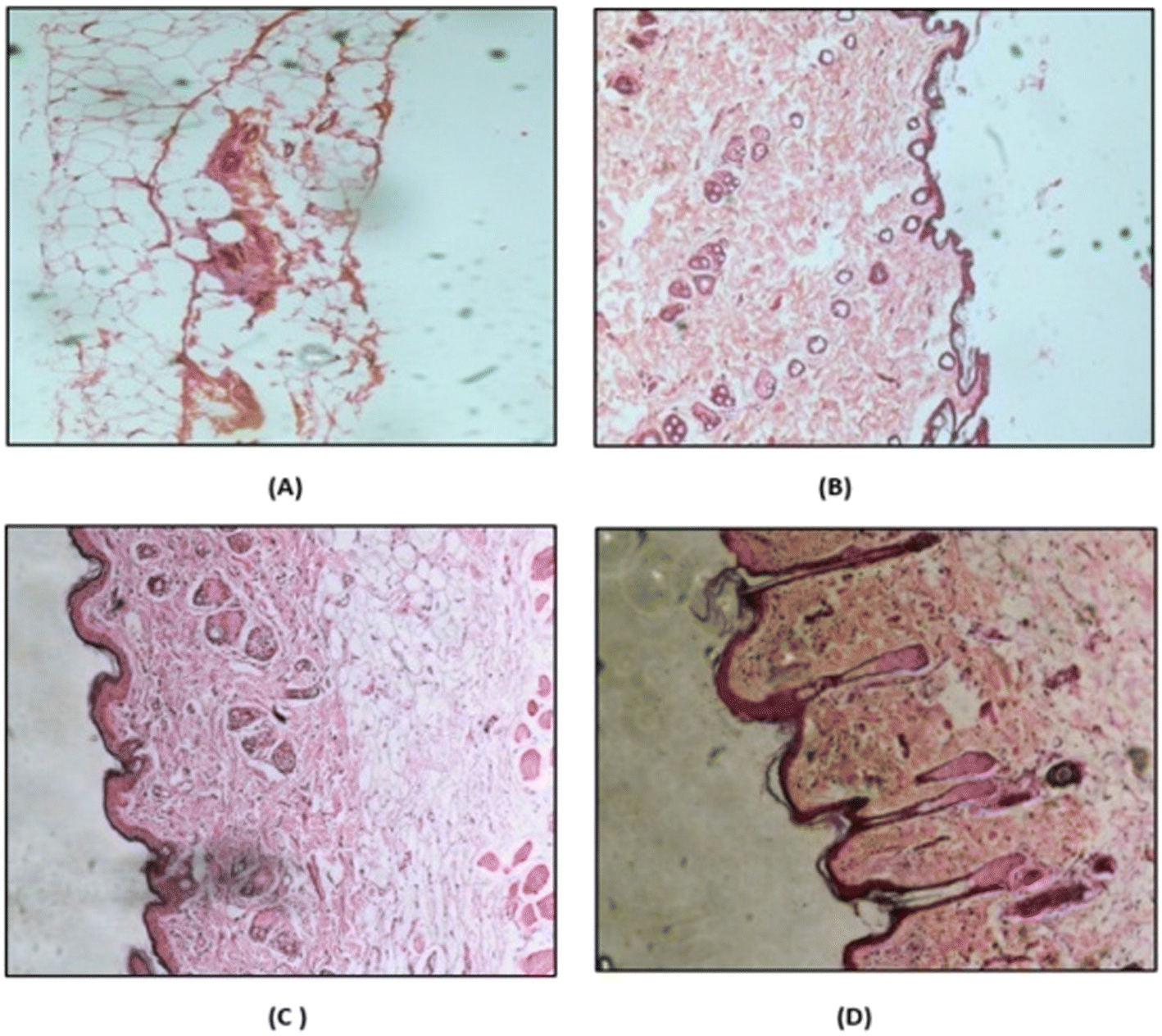 | ||
| Fig. 9 Histopathology images of (A) IPA (positive control), (B) PBS pH 7.4 (negative control), (C) drug suspension, and (D) microneedle patch. | ||
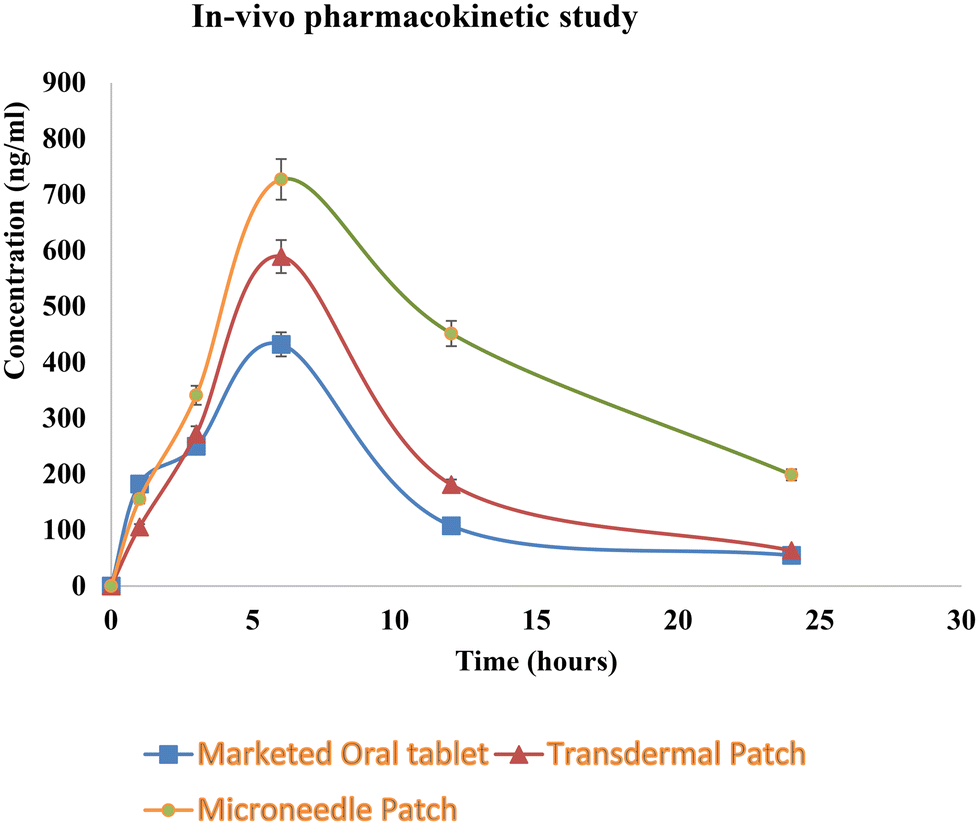 | ||
| Fig. 10 In vivo pharmacokinetic study. | ||
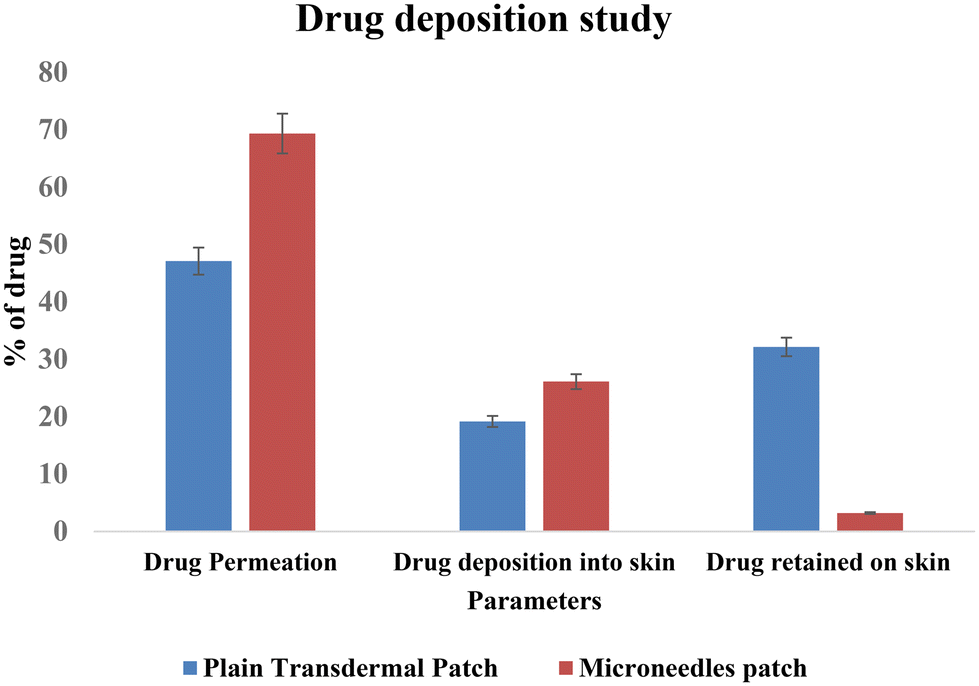 | ||
| Fig. 11 Skin permeation and deposition study. | ||
| Pharmacokinetic parameters | Marketed oral tablet | Simple transdermal patch | Microneedle patch |
|---|---|---|---|
| C max (ng ml−1) | 432.3 ± 20 | 589.3 ± 16 | 727.4 ± 18 |
| T max (h) | 6 | 6 | 6 |
| AUC (ng ml−1 h−1) | 4299 | 5686 | 12136 |
| t 1/2 (h) | 6.19 | 5.84 | 9.7 |
| MRT (h) | 11.10 | 11.47 | 17.24 |
| Relative bioavailability | — | 1.32 | 2.82 |
| AFF (N) | Drug content (%) | % CDR in 24 hours | |
|---|---|---|---|
| (25–40 °C) | |||
| Initial | 5.3 ± 0.06 | 97.92 ± 0.52 | 91.55% |
| After 15 days | 5.1 ± 0.03 | 96.24 ± 0.22 | 89.65% |
| After 30 days | 4.81 ± 0.09 | 95.89 ± 0.61 | 86.25% |
| (2–8 °C) | |||
| After 15 days | 5.34 ± 0.06 | 97.56 ± 0.32 | 91.25% |
| After 30 days | 5.08 ± 0.04 | 96.58 ± 0.12 | 89.15% |
| After 30 days | 4.88 ± 0.05 | 94.65 ± 0.57 | 86.57% |
| Ingredients | Concentration |
|---|---|
| Composition of nanosuspension | |
| Sodium deoxycholate (NaDC) | 0.5% |
| Distilled water | 5 mL |
| Carvedilol | 50 mg |
| Composition of nanosuspension-loaded microneedles | |
| Volume of nanosuspension in a single patch | 300 μL |
| Poly(methyl vinyl ether-alt-maleic anhydride) (PMVE/MA) | 20% |
| Glycerin | 100 μL |
| Sr. no. | Surfactant | Concentration of surfactant (%w/v) | Amount of drug (mg) | Size (nm) | PDI | Solubility (μg ml−1) |
|---|---|---|---|---|---|---|
| 1 | Poloxomer 407 | 0.5 | 50 | 450.9 | 0.235 | 30.56 |
| 2 | Poloxomer 188 | 0.5 | 50 | 401.9 | 0.2 | 25.8 |
| 3 | Polyvinyl alcohol (PVA) | 0.5 | 50 | 303.3 | 0.141 | 33.8 |
| 4 | Sodium taurocholate | 0.5 | 50 | 512.3 | 0.232 | 25.9 |
| 5 | Sodium cholate | 0.5 | 50 | 463.9 | 0.383 | 21.98 |
| 6 | Sodium deoxycholate (NaDC) | 0.5 | 50 | 189.5 | 0.129 | 51.47 |
4. Conclusion
A microneedle patch containing a carvedilol nanosuspension was developed and optimized using 32 full factorial designs. Carvedilol nanosuspension and carvedilol MN patches possess desirable characteristics that were confirmed by different evaluation tests. The microneedle patch led to significant enhancement of carvedilol bioavailability through the transdermal route. The drug release from the microneedles followed zero order kinetics, which is desirable in the case of transdermal delivery. The sustained release of the drug coupled with higher bioavailability leads to the possibility of reduction in dose and dosing frequency. Thus, the developed transdermal microneedle patch to administer carvedilol nanosuspension seems to be the promising and more patient-compliant approach for the management of hypertension.Author contributions
Anushri Deshpande: literature review for project selection conception, performing the laboratory experiments, and analyzing the results. Vidhi Mer: preparation of the draft manuscript. Darshana Patel: checking and revising the draft manuscript. Hetal Thakkar: providing overall guidance for project selection, manuscript preparation, final approval of the manuscript for communication, and final approval of the version to be communicated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Zydus Pharmaceuticals Limited, Ahmedabad, India for their kind gift of a sample of carvedilol and the All India Council for Technical Education for financial support.References
- R. R. Arun and J. Harindran, Enhancement of bioavailability of carvedilol using solvent deposition techniques, Int. J. Pharm. Sci. Res., 2017, 8(8), 3391–3401 CAS.
- P. C. Stafylas and P. A. Sarafidis, Carvedilol in hypertension treatment, Vasc. Health Risk Manage., 2008, 4(1), 23 CrossRef CAS PubMed.
- B. N. Nalluri, et al., Controlled release tablet formulations of carvedilol, J. Chem. Pharm. Res., 2012, 4, 4266–4274 CAS.
- A. M. Gupta and U. Shivhare, FORMULATION, AND Characterisation of Sustained Release Carvedilol Tablet from Reservoir Pellets, Int. J. Pharm. Sci. Res., 2016, 7(2), 626 CAS.
- V. K. Venishetty, et al., Design and evaluation of polymer coated carvedilol loaded solid lipid nanoparticles to improve the oral bioavailability: a novel strategy to avoid intraduodenal administration, Colloids Surf., B, 2012, 95, 1–9 CrossRef CAS PubMed.
- G. Arzani, et al., Niosomal carriers enhance oral bioavailability of carvedilol: effects of bile salt-enriched vesicles and carrier surface charge, Int. J. Nanomed., 2015, 10, 4797 CAS.
- B. Singh, et al., Optimized nanoemulsifying systems with enhanced bioavailability of carvedilol, Colloids Surf., B, 2013, 101, 465–474 CrossRef CAS PubMed.
- D. Liu, et al., Controlled delivery of carvedilol nanosuspension from osmotic pump capsule: in vitro and in vivo evaluation, Int. J. Pharm., 2014, 475(1–2), 496–503 CrossRef CAS PubMed.
- K. Yuvaraja and J. Khanam, Enhancement of carvedilol solubility by solid dispersion technique using cyclodextrins, water-soluble polymers, and hydroxyl acid, J. Pharm. Biomed. Anal., 2014, 96, 10–20 CrossRef CAS PubMed.
- H. P. Thakkar, B. V. Patel and S. P. Thakkar, Development and characterization of nanosuspensions of olmesartan medoxomil for bioavailability enhancement, J. Pharm. BioAllied Sci., 2011, 3(3), 426 CrossRef CAS PubMed.
- V. Patravale, A. A. Date and R. Kulkarni, Nanosuspensions: a promising drug delivery strategy, J. Pharm. Pharmacol., 2004, 56(7), 827–840 CrossRef CAS PubMed.
- M. G. Nava-Arzaluz, et al., Microneedles as transdermal delivery systems: combination with other enhancing strategies, Curr. Drug Delivery, 2012, 9(1), 57–73 CrossRef CAS PubMed.
- P. R. Nivrutti, P. A. Nath and J. H. Anantrao, Drug penetration enhancement techniques in transdermal drug delivery system: A review, J. Pharm. Res. Int., 2021, 33(19B), 46–61 Search PubMed.
- Y. Wang, et al., Influencing factors and drug application of iontophoresis in transdermal drug delivery: an overview of recent progress, Drug Delivery Transl. Res., 2022, 12(1), 15–26 CrossRef PubMed.
- B. C.-Q. Seah and B. M. Teo, Recent advances in ultrasound-based transdermal drug delivery, Int. J. Nanomed., 2018, 13, 7749 CrossRef CAS PubMed.
- N. Khare and P. Shende, Microneedle system: A modulated approach for penetration enhancement, Drug Dev. Ind. Pharm., 2021, 47(8), 1183–1192 CrossRef CAS PubMed.
- T. Haque and M. M. U. Talukder, Chemical enhancer: a simplistic way to modulate barrier function of the stratum corneum, Adv. Pharm. Bull., 2018, 8(2), 169 CrossRef CAS PubMed.
- M. M. D. C. Lobita, Development of dissolving microneedles for delivery of cancer cells membrane antigens, 2021 Search PubMed.
- V. Alimardani, et al., Recent advances on microneedle arrays-mediated technology in cancer diagnosis and therapy, Drug Delivery Transl. Res., 2021, 11(3), 788–816 CrossRef PubMed.
- H. Wei, et al., Hydrogel-based microneedles of chitosan derivatives for drug delivery, React. Funct. Polym., 2022, 172, 105200 CrossRef CAS.
- H. X. Nguyen, et al., Poly (vinyl alcohol) microneedles: Fabrication, characterization, and application for transdermal drug delivery of doxorubicin, Eur. J. Pharm. Biopharm., 2018, 129, 88–103 CrossRef CAS PubMed.
- K. D. Koradia, et al., Ziprasidone nanocrystals by wet media milling followed by spray drying and lyophilization: Formulation and process parameter optimization, J. Drug Delivery Sci. Technol., 2018, 43, 73–84 CrossRef CAS.
- Q. L. Wang, et al., A fabrication method of microneedle molds with controlled microstructures, Mater. Sci. Eng., C, 2016, 65, 135–142 CrossRef CAS PubMed.
- H. Thakkar, K. Pandya and B. Patel, Microneedle-mediated transdermal delivery of tizanidine hydrochloride, in Drug Delivery Systems, Springer, 2020, pp. 239–258 Search PubMed.
- S. K. Sukumaran and C. Venkatasubramaniyan, Design and Development of Chitosan Based Etravirine Nanosuspension, Nanomed. Res. J., 2022, 7(3), 270–287 Search PubMed.
- Q. Liu, et al., A wet-milling method for the preparation of cilnidipine nanosuspension with enhanced dissolution and oral bioavailability, J. Drug Delivery Sci. Technol., 2020, 55, 101371 CrossRef CAS.
- L. K. Vora, et al., Novel nanosuspension–based dissolving microneedle arrays for transdermal delivery of a hydrophobic drug, J. Interdiscip. Nanomed., 2018, 3(2), 89–101 CrossRef CAS PubMed.
- H. Kathuria, et al., Large size microneedle patch to deliver lidocaine through the skin, Pharm. Res., 2016, 33(11), 2653–2667 CrossRef CAS PubMed.
- A. Dąbrowska, et al., A water-responsive, gelatine-based human skin model, Tribol. Int., 2017, 113, 316–322 CrossRef.
- H. P. Thakkar, H. Savsani and P. Kumar, Ethosomal hydrogel of raloxifene HCl: statistical optimization & ex vivo permeability evaluation across microporated Pig ear skin, Curr. Drug Delivery, 2016, 13(7), 1111–1122 CrossRef CAS PubMed.
- S. Amodwala, P. Kumar and H. P. Thakkar, Statistically optimized fast dissolving microneedle transdermal patch of meloxicam: A patient-friendly approach to manage arthritis, Eur. J. Pharm. Sci., 2017, 104, 114–123 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |