
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acid-labile and non-degradable cross-linked star polymer model networks by aqueous polymerization for in situ encapsulation and release†
Gavin
Irvine
 a,
Stuart
Herron
a,
Daniel W.
Lester
b and
Efrosyni
Themistou
*a
a,
Stuart
Herron
a,
Daniel W.
Lester
b and
Efrosyni
Themistou
*a
aSchool of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Belfast, BT9 5AG, UK. E-mail: e.themistou@qub.ac.uk
bPolymer Characterisation Research Technology Platform, University of Warwick, Library Road, Coventry, CV4 7AL, UK
First published on 14th October 2024
Abstract
Biocompatible, acid-labile cross-linked star polymer model networks (CSPMNs) have great potential for use in drug delivery. However, a primary complication of this research stems from the prevalence of their synthesis to take place in organic solvents. Herein, to minimize CSPMN potential cytotoxicity, aqueous reversible addition–fragmentation chain transfer polymerization is employed for their synthesis. Initially, “arm-first” star polymers were synthesized in water using a poly[oligo(ethylene glycol) methyl ether methacrylate] (POEGMA) homopolymer and a non-degradable ethylene glycol dimethacrylate or acid-labile diacetal-based bis[(2-methacryloyloxy)ethoxymethyl] ether cross-linker. Subsequently, OEGMA addition resulted in the preparation of “in–out” star polymers (with higher molecular weights) followed by cross-linker addition to form CSPMNs. Rhodamine B dye encapsulation was performed during CSPMN synthesis and its release was observed under biologically relevant conditions. Having shown the effective breakdown of the diacetal-based CSPMNs, their potential for use in drug delivery in low pH environments (i.e. cancerous tumors) is expected to be high.
Introduction
Biocompatible synthetic polymer hydrogels1 (water-swellable polymer networks) are attracting increasing attention as drug delivery systems, tissue engineering matrices and biosensors, due to their ability to absorb large quantities of water or biological fluids.2 A class of these materials, with great structural control, is polymer model networks,3 prepared by “living” polymerization techniques.4 These polymer networks have well-defined structures consisting of precise lengths of (elastic) chains between their cross-links and therefore, it is expected that they can give more consistent and predictable results when used multiple times in a specific application.A relatively new type of model network developed by Patrickios and coworkers in 20015 is that of CSPMNs6 comprising interconnected “in–out” star polymers. These model networks are based on cross-linked star polymers bearing both elastic and dangling chains and two different types of cores, the primary and the secondary. The primary cores have both elastic and dangling chains connected to them, whereas the secondary cores bear only elastic chains. Various types of CSPMNs, such as homopolymer hydrophilic,5 amphiphilic,7–10 double-hydrophilic,11,12 acid-labile13–15 and double network amphiphilic,16–20 were synthesized by the Patrickios group using various monomers and two different polymerization techniques, group transfer polymerization (GTP)21,22 at ambient temperature5,8–16,23 and reversible addition–fragmentation chain transfer (RAFT) polymerization24,25 at 6519–70 °C.18 However, the research surrounding CSPMNs has focused on their formation only in tetrahydrofuran (THF)5,8–16,23 and 1,4-dioxane.18,19 According to Prat et al.,26 these organic solvents are considered to be ‘problematic’ or ‘hazardous’ and they should be avoided, especially in cases where the synthesized polymers are intended to be used in biomedical applications, since they can affect their toxicity. It is worth mentioning here that there are only two reports in the literature where CSPMNs have been used for biomedical applications, and more specifically, for DNA adsorption,10,12 whereas these well-defined polymeric nanostructures were not used before for drug encapsulation and delivery.
Star polymers,27–31 which are the precursors of CSPMNs, consist of linear polymer chains (star polymer “arms”) connected together at a central point called the “core”. These branched polymeric nanostructures have been widely utilized in the biomedical field for various applications,32 such as gene33–42 and drug delivery.43–45 Although in most synthetic procedures in the literature star polymers were prepared using organic solvents,35,46–49 contrary to the CSPMNs, in some recent studies by our group38 and other researchers, heterogeneous RAFT emulsion and dispersion polymerization in water or water/alcohol mixtures50–57 was used to synthesize star polymers. This is a biologically friendly synthetic methodology, since only water and alcohols (reported as ‘recommended’ solvents that are good alternatives to organic solvents for the preparation of polymers26) are used. Another advantage of this aqueous synthetic procedure is that it is more efficient than RAFT polymerization in organic solvents since it can result in low dispersity (Đ) values and high yields with high solids content. Therefore, this synthetic methodology appears to be promising for the development of both biocompatible CSPMNs and their star polymer precursors.
The synthesis of CSPMNs using biologically friendly conditions, such as an aqueous solution at physiological temperature (37 °C), can immensely increase their potential biological applications. Although RAFT polymerization has been an ever more frequently used “living” controlled polymerization technique since its conception by researchers in CSIRO, Australia,24,25 there are currently no reports in the literature on the synthesis of CSPMNs or “in–out” star polymers by aqueous RAFT polymerization. In our previous work, we investigated the formation of biocompatible cationic “arm-first” star polymers via aqueous RAFT polymerization.38 In this work, we use a similar methodology for the preparation of more advanced nanostructures: “in–out” star polymers and their CSPMN derivatives.
Herein, in order to make sure that the resulting CSPMNs will have increased biocompatibility for use in biomedical applications and especially in drug delivery, both the neutral polyethylene glycol (PEG)-based monomer, OEGMA,58 and the acid-labile diacetal-based bis[(2-methacryloyloxy)ethoxymethyl] ether (MOEME)15,38,59 cross-linker were used. PEG is extensively used in biomedical applications60 due to its high biocompatibility and OEGMA was used before for the preparation of CSPMNs11 for use in biomedical applications. The use of MOEME produces CSPMNs with degradable cores15 that can break down in acidic environments (such as tumors). Their low molecular weight (MW) degradation products can potentially be easily removed from the body through normal metabolic pathways. In more detail, the preparation of novel biocompatible PEG-based CSPMNs (star-based gels) with acid-labile diacetal MOEME-based and non-degradable ethylene glycol dimethacrylate (EGDMA)-based cores by a facile RAFT polymerization methodology under biologically friendly conditions, i.e. with water as polymerization solvent and physiological temperature (37 °C), is described here (see Scheme 1). The synthetic procedure involved four steps: synthesis of a linear polymer in ethanol by the addition of an OEGMA monomer to a mixture of solvent and polymerization initiator/agent (Scheme 1a), followed by the preparation of two different types of star polymers, “arm-first” and “in–out”61 (Scheme 1b and c, respectively), and finally, the synthesis of the CSPMN (Scheme 1d)5 in water at 37 °C. In more detail, the first star polymer precursors were synthesized using the “arm-first” approach by the addition of a cross-linker to a linear POEGMA polymer (Scheme 1b). Subsequently, a second type of star polymer precursor was prepared using the “in–out” approach following the addition of more monomer to the “arm-first” star polymers (Scheme 1c). Then, the CSPMNs were formed by the addition of cross-linkers to the “in–out” star polymers (Scheme 1d). The polymerization conditions were optimized for the formation of the CSPMNs.
 | ||
| Scheme 1 Reaction scheme for the synthesis of the (a) macro-CTA by RAFT solution polymerization in ethanol, synthesis of (b) “arm-first” star polymers, (c) “in–out” star polymers and (d) CSPMNs by aqueous RAFT polymerization, and degradation trial for (e) EGDMA-based and (f) MOEME-based CSPMNs under acidic conditions. | ||
This preparation method of CSPMNs, which is based on “in–out” star polymer precursors,16,19 was chosen here since it is considered to be better than the “core-first” type star polymer precursor method.18 The “in–out” method can give less star–star coupling and better star polymer size homogeneity due to steric hindrance conferred by the star polymer arms prepared in the first polymerization step. Furthermore, multi-functional initiators or RAFT agents normally require more multi-step procedures as they are not usually commercially available. It is also possible to add different functionalities to “in–out” star polymers by using different monomers for the second arm formation and, depending on the desired application, this characteristic may be desirable. This can be exploited to add further functionality, such as the ability to conjugate to peptides for biomedical applications.28
The use of the acid-labile diacetal-based MOEME cross-linker (prepared using the procedure described by Themistou et al.62) for both the preparation of the “arm-first” (Scheme 1b) and the preparation of the CSPMN (Scheme 1d) resulted in the formation of a CSPMN that, contrary to the non-degradable EGDMA-based CSPMN (Scheme 1e) prepared by the commercially available EGDMA cross-linker, can degrade to a linear polymer under acidic conditions similar to the ones found in cancerous tumors (Scheme 1f). Finally, rhodamine B dye (used as a surrogate drug entity) encapsulation in situ during the synthesis of the MOEME-based CSPMN was performed followed by its release monitoring from the acid-labile CSPMN. To our knowledge, this is the first report on CSPMN and “in–out” star polymer synthesis in water and in physiological temperature. This is also the first encapsulation (during polymerization) and release study using a CSPMN.
Results and discussion
Preparation of the hydrophilic POEGMA macro-CTAs
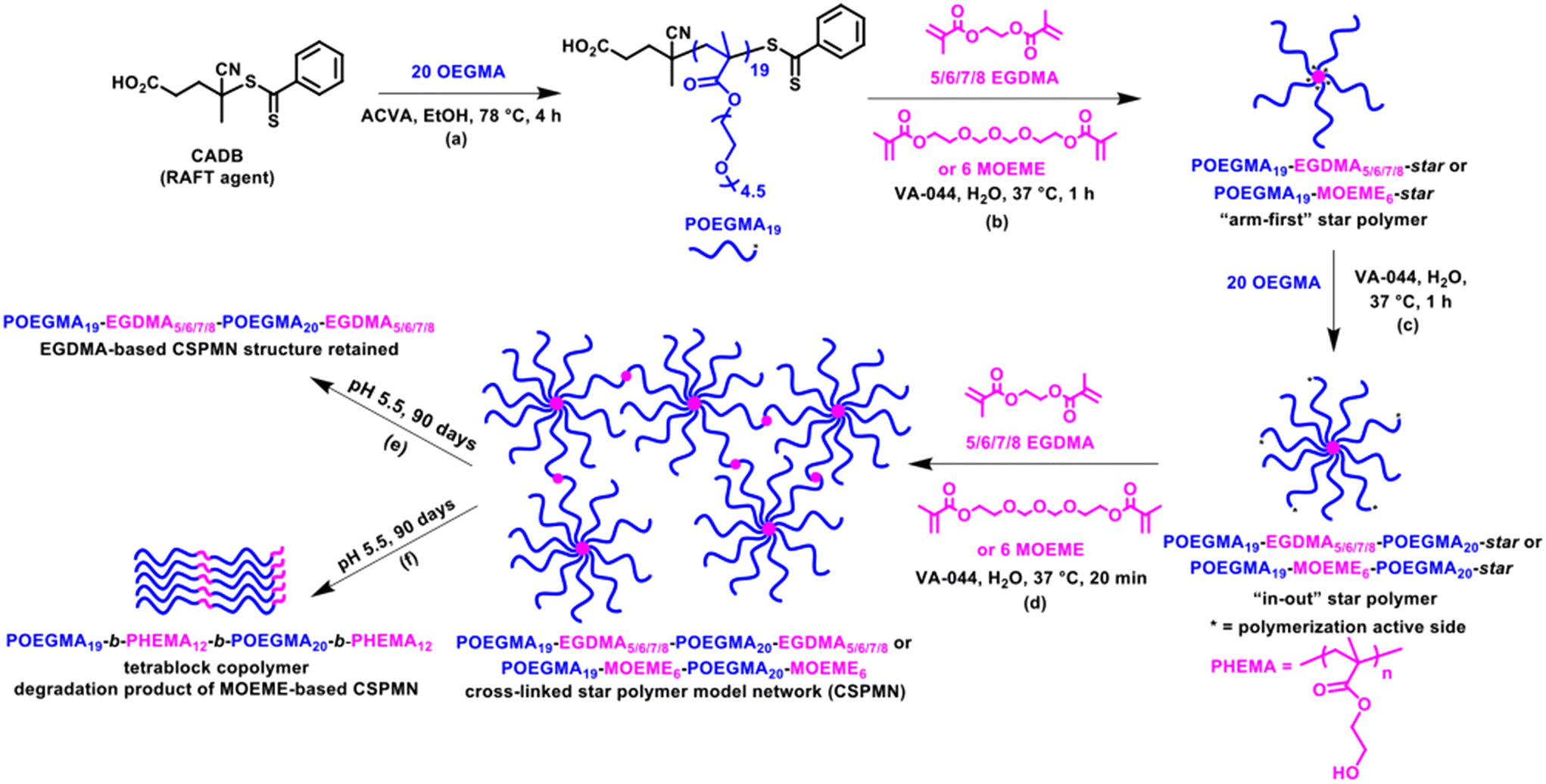
The hydrophilic POEGMA19 macro-chain transfer agent (macro-CTA) was prepared in ethanol at 78 °C for 4 h (Scheme 1a). This was performed using an OEGMA monomer with a short PEG chain (4–5 PEG units), as opposed to longer PEG chained OEGMA monomers, to minimize any steric hindrance issues in the incorporation of the POEGMA linear polymer arms to the star polymer core during the “arm-first” star polymer formation (Scheme 1b). The successful preparation of the macro-CTA was confirmed by proton nuclear magnetic resonance spectroscopy (1H NMR) spectroscopy in deuterated chloroform (CDCl3) and size exclusion chromatography (SEC) in THF. More specifically, a high monomer conversion of 97% was confirmed by 1H NMR spectroscopy by comparing the area under the methacrylic peaks at 5.55 and 6.11 ppm with the area under the polymer/monomer peak at 4.07 ppm. This corresponds to a POEGMA macro-CTA with a mean degree of polymerization (DP) of 19 and a MW of 5979 g mol−1. The purified POEGMA19 macro-CTA (after dialysis using methanol), was characterized by 1H NMR spectroscopy and its 1H NMR characteristic peaks are shown in Fig. S1a.† The obtained SEC (THF) chromatogram of the macro-CTA (Fig. 1) showed a relatively narrow molecular weight distribution (MWD) with a Đ value of 1.27. A number-average MW (Mn) value of 5110 g mol−1 and a peak molecular weight (weight at the peak maximum, Mp) value of 6780 g mol−1 were determined. The Mn value was in relatively good agreement with the 1H NMR MW value. The slightly high Đ value obtained is attributed to traces of EGDMA cross-linker impurity present in the commercially available OEGMA monomer.38,63,64 This can cause some chain–chain coupling reactions during the formation of POEGMA, which were not found to significantly affect the subsequent “arm-first” polymer formation.38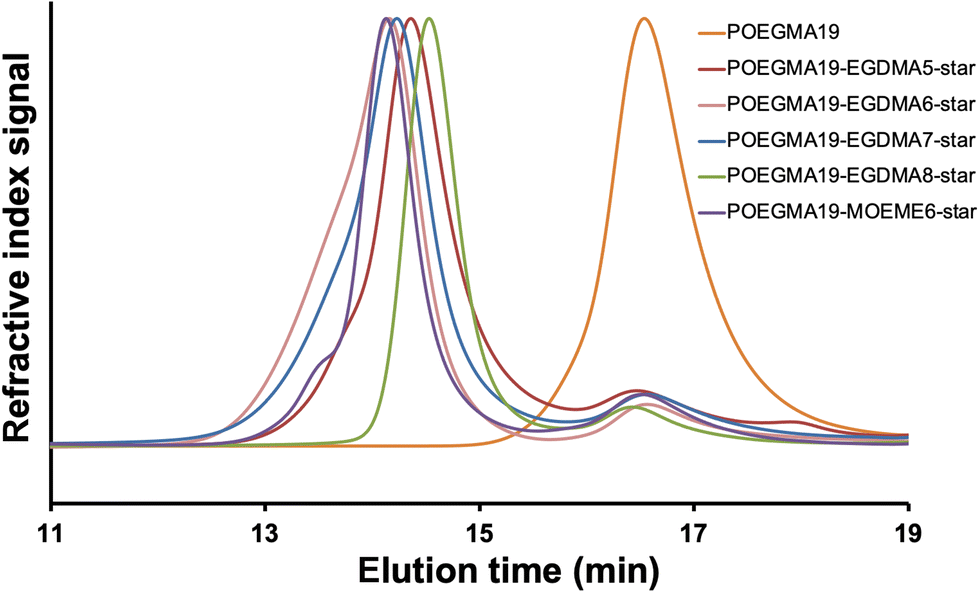 | ||
Fig. 1 SEC (THF) chromatograms of all POEGMA-EGDMA-star (EGDMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :macro-CTA molar ratios = 5:1, 6:1, 7:1, 8:1) and POEGMA-MOEME-star (MOEME:macro-CTA molar ratio = 6:1) “arm-first” star polymers and their linear POEGMA macro-CTA precursors. :macro-CTA molar ratios = 5:1, 6:1, 7:1, 8:1) and POEGMA-MOEME-star (MOEME:macro-CTA molar ratio = 6:1) “arm-first” star polymers and their linear POEGMA macro-CTA precursors. | ||
Preparation of “arm-first” star polymers
In order to investigate the likelihood and efficacy of CSPMN formation, initially the POEGMA macro-CTA was reacted with either the degradable acid-labile MOEME or the non-degradable EGDMA cross-linker, to form star polymers (CSPMN precursors) using an “arm-first” approach61,62,65 (Scheme 1b). These star polymers were synthesized by aqueous RAFT polymerization with a 25% w/w solids content at 37 °C over 1 h, the time required to reach full cross-linker conversion and maximum conversion of the arm into the star polymer. A series of non-degradable “arm-first” star polymers were prepared with varying EGDMA cross-linker:macro-CTA (5:1, 6:1, 7:1 and 8:1) molar ratios to optimize the amount of cross-linker used for the synthesis of CSPMNs and their “arm-first” and “in–out” star polymer precursors. The results of their characterization by SEC and static light scattering (SLS) are presented in Table 1. Dynamic light scattering (DLS) data are presented in Table 1 and Fig. S2† and 1H NMR (CDCl3) data are shown in Fig. S7–S10.†
| Star polymer structure | M n [g mol−1] | Đ valuea | % Linear polymera | Hydrodynamic diameterb [nm] | Number of armsc | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Data obtained by THF SEC versus PMMA standards. b Data obtained by DLS in water (intensity-average diameter distribution). c Data obtained by SLS. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POEGMA19-EGDMA5-star | 49400 |
1.31 | 14.1 | 14.1 | 33 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POEGMA19-EGDMA6-star | 76700 |
1.32 | 9.6 | 18.8 | 79 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POEGMA19-EGDMA7-star | 65200 |
1.45 | 13.6 | 18.7 | 39 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POEGMA19-EGDMA8-star | 115200 |
1.12 | 8.4 | 21.5 | 78 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POEGMA19-MOEME6-star | 70800 |
1.23 | 17.0 | 12.3 | 27 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The SEC (THF) chromatograms of the “arm-first” star polymers, overlaid in Fig. 1 together with their POEGMA macro-CTA linear polymer precursor, showed that a shift to lower elution times was observed when compared to their respective macro-CTA, indicative of an increase in MW due to the interconnection of the linear polymer arms to the cross-linker core and successful formation of the star-shaped polymers. The SEC data presented in Table 1, also shows that the “arm-first” star polymers have much higher Mn values (49400–115200 g mol−1) than the macro-CTA (5110 g mol−1). Their light scattering Mw values were used to give an indication of the approximate number of arms in each star polymer (Table 1). The number of arms was calculated as the star polymer light scattering Mw value after subtracting the cross-linker contribution (number of cross-linker units multiplied by the cross-linker MW) divided by the light scattering Mw of the POEGMA macro-CTA. All light scattering Mw values are provided in Table S1.† A small amount (8–17%) of unreacted linear polymer precursors is still present in all reaction products after the synthesis of the “arm-first” star polymer, which is common in “arm-first” RAFT polymerization.38,55,66 This is also evident in the SEC chromatograms of all the star polymers (Fig. 1), where the MWD appeared to be bimodal. The higher peak that appears at the lower elution time corresponds to the star polymer, while the small peak appearing at around 16 to 17 min elution time is due to the remaining linear polymer that was not attached to the star polymer core. The areas under the two peaks in the chromatogram were used to calculate the % unreacted linear polymer (8–17%, Table 1). These values were relatively low compared to others in the literature and therefore purification of the star polymers was deemed unnecessary and was avoided to minimize product loss. The Đ values of the “arm-first” star polymers were found to range from 1.12 to 1.45 for the EGDMA-based star polymers. The MOEME-based star polymer had a Đ value of 1.23. These relatively low values for “arm-first” star polymer formation compared to previous studies59,62,67,68 suggest that well-defined star polymers were formed, which was also evident by the hydrodynamic diameter values obtained for all the “arm-first” star polymers, ranging from 12 to 22 nm (Fig. 2 and Table 1). The POEGMA19-EGDMA8-star appeared to have the highest hydrodynamic diameter (22 nm) while POEGMA19-MOEME6-star had the lowest hydrodynamic diameter (12 nm).
 | ||
| Fig. 2 DLS volume weighted diameter distribution for all “arm-first” POEGMA19-EGDMAx-star and POEGMA19-MOEMEy-star star polymers (1 mg mL−1 in deionized water). | ||
For the EGDMA-containing “arm-first” star polymers, it was found that the cross-linker:macro-CTA molar ratio of 8:1 was the most efficient for star polymer formation as it yielded the lowest % linear polymer (8.4%), the lowest Đ value (1.12) and the highest Mn value (115200 g mol−1) when compared to the other molar ratios used (5:1, 6:1 and 7:1). On the other hand, the 5:1 molar ratio gave the highest % linear polymer (14.1%) for the EGDMA-based star polymers, which meant that POEGMA19-EGDMA5-star was excluded from the experiments that followed for the synthesis of “in–out” star polymers and CSPMNs. Additionally, POEGMA19-EGDMA5-star and POEGMA19-EGDMA7-star had a larger % linear polymer in their samples, which led to them having fewer attached arms than POEGMA19-EGDMA6-star and POEGMA19-EGDMA8-star that exhibited a smaller % linear polymer through SEC. The EGDMA:macro-CTA molar ratio of 7:1 also appeared to give a high % linear polymer (13.6%) and the highest Đ value (1.45) of all the EGDMA “arm-first” star polymers prepared, with a number of arms of 39. A general trend was observed whereby the number of arms increased as the EGDMA:macro-CTA molar ratio increased, with POEGMA19-EGDMA6-star having the most arms (79) although there is a shoulder peak evident in its SEC chromatogram (Fig. 1), which may contribute to this high value and large light scattering MWs (Table S1†). All EGDMA star polymers had a larger number of arms than POEGMA19-MOEME6-star (27). However, in subsequent experiments, the 8:1 molar ratio resulted in aggregation in the “in–out” star polymer synthesis (aggregates of around 230 nm were observed based on DLS measurements, Table 2), while the formation of CSPMNs (in 10% w/w or 25% w/w solids content) was not successful. Therefore, an EGDMA cross-linker:macro-CTA molar ratio of 6:1, with a relatively good % linear polymer of 9.6% and a Đ value of 1.32, was considered to be the optimum for the synthesis of a non-degradable EGDMA-based CSPMN. This cross-linker:macro-CTA molar ratio was also used for the preparation of the acid-labile CSPMN using the acid-labile MOEME cross-linker.
| Star polymer structure | % Solids content (w/w) | OEGMA % conversion | M n [g mol−1] | Đ valuea | % Linear polymera | Hydrodynamic diameterb [nm] | Number of armsc | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Data obtained by THF SEC versus PMMA standards. b Data obtained by DLS in water (intensity-average diameter distribution). c Data obtained by SLS. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POEGMA19-EGDMA6-POEGMA20-star | 10 | 97.6 | 122400 |
1.96 | 6.2 | 41.2 | 69 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POEGMA19-EGDMA7-POEGMA20-star | 25 | ∼100.0 | 149900 |
2.77 | 13.9 | 48.6 | 107 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POEGMA19-EGDMA7-POEGMA20-star | 10 | 97.7 | 106900 |
2.32 | 6.5 | 55.8 | 63 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POEGMA19-EGDMA8-POEGMA20-star | 10 | 99.3 | 315800 |
2.01 | 4.4 | 230.1 | 89 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POEGMA19-MOEME6-POEGMA20-star | 25 | 98.1 | 104700 |
1.77 | 8.3 | 23.9 | 42 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POEGMA19-MOEME6-POEGMA20-star | 10 | ∼100.0 | 75700 |
1.42 | 6.4 | 25.1 | 30 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Similarly, a core degradable “arm-first” POEGMA19-MOEME6-star star polymer was synthesized to be used as a precursor for the synthesis of a core degradable “in–out” star polymer and a core degradable CSPMN. The successful preparation of this “arm-first” star polymer is evidenced by the presence of the diacetal protons of the MOEME cross-linker at 4.81 ppm in its 1H NMR (CDCl3) spectrum (Fig. S1b and S11†). The two “arm-first” star polymers prepared using a molar ratio of 6:1 and the two different cross-linkers (EGDMA and MOEME) appeared to have similar SEC Mn values (76700 and 70800 g mol−1, respectively). However, POEGMA19-EGDMA6-star yielded a higher number of arms (79) than POEGMA19-MOEME6-star (27). As also shown in our previous work,38 the bulkier, degradable MOEME cross-linker is not as efficient as the non-degradable EGDMA cross-linker in forming “arm-first” star polymers, with the % unreacted linear polymer for POEGMA19-MOEME6-star of 17.0% compared to 9.6% for its corresponding POEGMA19-EGDMA6-star. However, although the EGDMA cross-linker appeared to be better in incorporating linear polymer chains to the star polymer core, the MOEME cross-linker gave a more uniform star polymer with a lower Đ value (1.23 compared to 1.32 for EGDMA). Furthermore, in the subsequent “in–out” star polymer synthesis, the MOEME star polymer was found to be more efficient and successful in two concentrations (10% and 25% w/w) compared with one for the EGDMA star polymer (10% w/w).
Further investigation of the DLS data (Table 1) of the EGDMA-based “arm-first” star polymers shows a general trend of an increase in the hydrodynamic diameter with a general increase in the amount of cross-linker used in the star polymer synthesis and/or an increase in the number of arms incorporated in the star polymer (from 14 nm for the star polymer with a 5:1 cross-linker:macro-CTA molar ratio with only 33 arms to 22 nm for the one with an 8:1 molar ratio with 78 arms). The other two “arm-first” star polymers (6:1 and 7:1 ratios) have intermediate hydrodynamic diameter values. More specifically, POEGMA19-EGDMA6-star with a 6:1 ratio and 79 arms appeared to have a similar hydrodynamic diameter (18.8 nm) to the POEGMA19-EGDMA7-star with a 7:1 ratio and 39 arms (18.7 nm). These results suggest that an increase in the amount of cross-linker can give a higher hydrodynamic diameter for the “arm-first” star polymer, due to a larger core and/or a higher number of arms attached to the core resulting in the chains being more extended.
Since the POEGMA19-MOEME6-star and POEGMA19-EGDMA6-star appeared to have a similar increase in MW with comparatively good Đ values (1.23 and 1.32, respectively), they were selected as the best candidates for “in–out” star polymer and CSPMN syntheses.
Preparation of “in–out” star polymers
After optimizing the preparation of “arm-first” star polymers, an addition of more OEGMA monomers resulted in the formation of new arms growing from the center (core) of the star polymers and therefore, an increase in the MW of the star polymers, resulting in what is known as “in–out” star polymers (Scheme 1c).61,69,70 The “living” character of the polymer chain facilitates this step as the polymerization active sites are on the core of the “arm-first” star polymers. Therefore, the newly formed POEGMA linear chains emanate from the core.Initially, two kinetic studies were conducted for the synthesis of POEGMA19-EGDMA7-POEGMA20-star at 37 °C using two different % solids content values (25% w/w and 10% w/w). Samples were taken at different time points (every hour) over a course of 4 hours to study the effect of reaction time on “in–out” star polymer formation. From these experiments, it was found that for both % solids content values, a reaction time of 1 h was sufficient for the formation of the “in–out” star polymer, since lower Đ values were obtained (Table S2†) in 1 h, while the molecular weight was not changed significantly by increasing the reaction time. By comparing the two different % solids content values, it was concluded that 10% w/w was more favorable for the formation of the EGDMA-based “in–out” POEGMA19-EGDMA7-POEGMA20-star due to the lower Đ value (2.32 compared to 2.77 for the 25% w/w) obtained for the 1 hour reaction time.
Following the kinetic studies, a series of non-degradable “in–out” POEGMA19-EGDMAx-POEGMA20-star polymers (where x = 6, 7, and 8) and the acid-labile “in–out” POEGMA19-MOEME6-POEGMA20-star star polymers were prepared using their corresponding synthesized “arm-first” star polymer precursors (1H NMR (CDCl3) spectra shown in Fig. S12–S17†). All EGDMA-based “in–out” star polymers were prepared using 10% w/w solids content, as this was found to give more uniform star polymers based on the results of the kinetic studies and the formation of a POEGMA19-EGDMA6-POEGMA20-star chemical gel at 25% w/w concentration. For the MOEME-based POEGMA19-MOEME6-POEGMA20-star both 10% w/w and 25% w/w solids contents were used. As an example, the 1H NMR characteristic peaks for the “in–out” POEGMA19-MOEME6-POEGMA20-star, a precursor of the MOEME-based CSPMN, are shown in Fig. S1c†, and they indicate the successful incorporation of the POEGMA arms in the “arm-first” star polymer core to form the “in–out” star polymer. Table 2 shows the % solids contents, % OEGMA monomer conversions, MWs, Đ values and % unattached linear polymer values obtained by SEC (THF), hydrodynamic diameters determined by DLS (Fig. S3†) and the total number of arms by SLS for these “in–out” star polymers.
The % OEGMA monomer conversions were very high (97.6–100%) for all “in–out” star polymers, indicating that the reaction time used (1 h) was sufficient to polymerize almost all of the OEGMA monomer. This corresponds to a DP value of 20 for the new POEGMA20 chains that grew out from the “arm-first” star polymer core in this polymerization step. To calculate how many new POEGMA chains were created in this polymerization step, the light scattering Mw value of their corresponding “arm-first” star polymer precursors was subtracted from the light scattering Mw value of the “in–out” star polymer and the obtained value was divided by the light scattering Mw value of the POEGMA19 macro-CTA. Then to find the total number of arms in the “in–out” star polymer (Table 2) the new number of arms found by this calculation was added to the number of arms of the “arm-first” star polymer (Table 1). The SEC (THF) data for all of these “in–out” star polymers showed a lower elution time compared to both their linear and “arm-first” star polymer precursors, as observed in Fig. 3, indicating the successful formation of “in–out” star polymers. As expected, the Mn values of the “in–out” star polymers (75700–149900 g mol−1) and their Đ values (1.42–2.77) were higher than the ones of their corresponding “arm-first” star polymer precursors (70800–115200 g mol−1 and 1.12–1.45, respectively), while the calculated % linear polymer values (4.4–13.9) are lower than the calculated “arm-first” star polymer values (8.4–17.0). It is interesting to mention here that both MOEME-based “in–out” star polymers yielded much lower Đ values (1.42–1.77) than their EGDMA-based equivalents (1.96–2.77). When EGDMA was used as a cross-linker a number of issues arose with “in–out” star polymer formation. POEGMA19-EGDMA6-POEGMA20-star yielded fewer arms from SLS (69) when compared to its POEGMA19-EGDMA6-star precursor (79) and this can be attributed to a large number of “arm-first” star polymers present in this sample. This can be observed as a shoulder peak around 14.5 min in its SEC chromatogram (Fig. 3(a)) and suggests that the formation of POEGMA19-EGDMA6-POEGMA20-star is inefficient. Similarly, the formation of POEGMA19-EGDMA7-POEGMA20-star in 25% w/w solids concentration yielded 107 arms, which is more than double the number of arms of its star polymer precursor (39) and suggests an inefficient star preparation (possibly due to star–star coupling) as the number of arms in the “in–out” star polymer cannot be more than double that of its “arm-first” star polymer precursor. The large light scattering MW values observed (Table S1†) are likely a result of the wide MWD observed by SEC (Fig. 3(b)). When POEGMA19-EGDMA7-POEGMA20-star was synthesized in 10% w/w solids concentration the number of arms observed was 63, further adding credence to our observation that 10% w/w is a more suitable concentration for the synthesis of EGDMA “in–out” star polymers. This difference is also seen in their SEC chromatograms where POEGMA19-EGDMA7-POEGMA20-star has a narrower MWD in 10% w/w compared to 25% w/w. The synthesis of POEGMA19-EGDMA8-POEGMA20-star yielded a small increase in the number of arms (from 78 to 89) although its synthesis was shown to result in aggregation as determined by DLS. When MOEME was used as a cross-linker, as also found with the EGDMA-based star polymers, the more dilute reaction mixture (10% w/w solids content) gives more uniform POEGMA19-MOEME6-POEGMA20-star star polymers with lower Đ values: 1.42 for the 10% w/w compared to 1.77 for the 25% w/w solids content. However, the reaction for the synthesis of the POEGMA19-MOEME6-POEGMA20-star using 10% w/w solids content did not appear to work well, since the Mn value (75700 g mol−1) of the resulting “in–out” star polymer (Table 2) was only slightly higher than its “arm-first” star polymer precursor (70800 g mol−1), as shown in Table 1. Light scattering data also indicated that only 3 extra arms were attached to POEGMA19-MOEME6-POEGMA20-star (30) compared to POEGMA19-MOEME6-star (27). On the other hand, when 25% w/w solids content was used for the POEGMA19-MOEME6-POEGMA20-star synthesis, a higher Mn value (104700 g mol−1) was determined and more arms were observed (42), showing that this higher solids content is more efficient in this synthetic step. These results suggest that MOEME is a better cross-linker to use for preparing these star polymers as it yields more desirable values from SEC and SLS.
 | ||
| Fig. 3 SEC (THF) chromatograms showing the synthesis (macro-CTA, “arm-first” star polymers, and “in–out” star polymers) of (a) POEGMA19-EGDMA6-POEGMA20-star, (b) POEGMA19-EGDMA7-POEGMA20-star, (c) POEGMA19-EGDMA8-POEGMA20-star and (d) POEGMA19-MOEME6-POEGMA20-star “in–out” star polymers. | ||
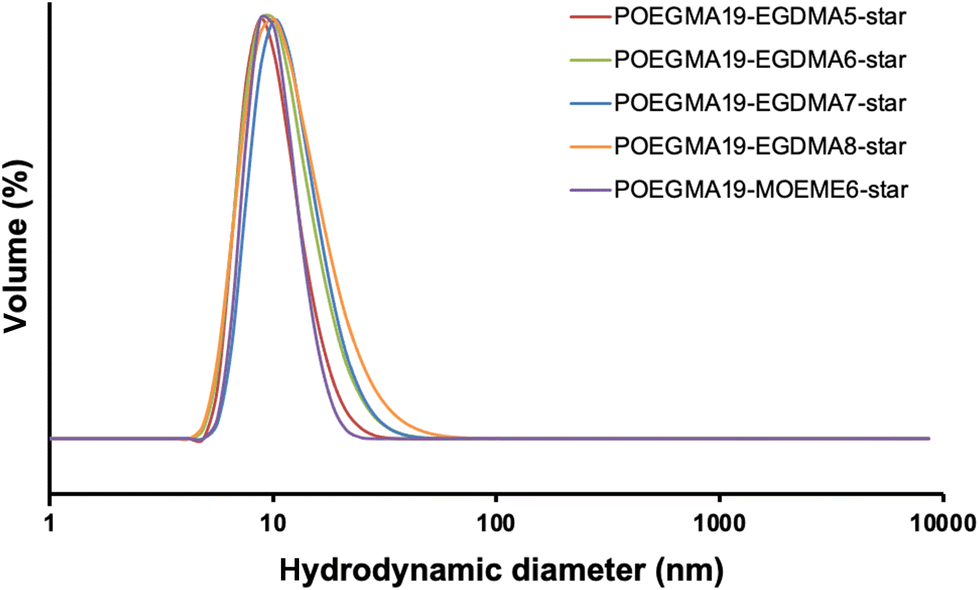
The DLS volume data for all the synthesized “in–out” star polymers are presented in Fig. 4 and their hydrodynamic diameters (based on intensity) are shown in Table 2. All “in–out” star polymers appeared to have hydrodynamic diameters in the range of 23.9–55.8 nm except for the POEGMA19-EGDMA8-POEGMA20-star (10% w/w), which has a noticeably higher hydrodynamic diameter, possibly due to some star–star polymer coupling or formation of star polymer aggregates due to the high amount of cross-linker used for its preparation. These hydrodynamic diameters were higher than the ones determined for the “arm-first” star polymers (12.3–21.5 nm). The % solids content used in the synthesis of the “in–out” star polymers does not seem to significantly affect their hydrodynamic diameter. More specifically, POEGMA19-MOEME6-POEGMA20-star and POEGMA19-EGDMA7-POEGMA20-star were found to have similar hydrodynamic diameters at both 10% and 25% w/w solids content. POEGMA19-MOEME6-POEGMA20-star had a hydrodynamic diameter of 24 nm at 10% w/w compared to 25 nm at 25% w/w, while POEGMA19-EGDMA7-POEGMA20-star appeared to have higher hydrodynamic diameters: 56 nm at 10% w/w and 49 nm at 25% w/w. As shown in Table 2, the degradable MOEME cross-linker resulted in the formation of “in–out” star polymers with smaller (24–25 nm) hydrodynamic diameters than those of EGDMA (41–230 nm), consistent with smaller MWs. For the EGDMA-based “in–out” star polymers, the higher amount of cross-linker used during the synthesis, the higher was the observed star polymer hydrodynamic diameter.
 | ||
| Fig. 4 DLS volume weighted diameter distribution for all “in–out” POEGMA19-EGDMAx-POEGMA20-star and POEGMA19-MOEMEy-POEGMA20-star star polymers (1 mg mL−1 in deionized water). | ||
Preparation of CSPMNs
After the synthesis of the “in–out” star polymers, where the polymerization active sites are on the ends of the newly prepared polymer arms, at the periphery of the star polymers, attempts were made to prepare cross-linked star polymer gels (CSPMNs5). The synthesis of the CSPMNs was investigated by adding more cross-linker to connect the “in–out” star polymers together, as can be seen in Scheme 1d, and observing if gelation occurred. Based on the results presented above for the synthesis of the star polymers, the best “in–out” star polymers for the synthesis of core degradable and non-degradable CSPMNs are the ones with a cross-linker:macro-CTA molar ratio of 6:1, namely the non-degradable POEGMA19-EGDMA6-POEGMA20-star and the core degradable POEGMA19-MOEME6-POEGMA20-star. The synthesized CSPMNs were hydrophilic as they were prepared using the PEG-based methacrylic monomer OEGMA, so that they can be used in biomedical applications.11 Gelation, which indicates successful formation of the CSPMN, was obtained for the non-degradable POEGMA19-EGDMA6-POEGMA20-EGDMA6 at 10% w/w solids content and the core degradable POEGMA19-MOEME6-POEGMA20-MOEME6 at 25% w/w solids content (Fig. S4†).
The SEC data of the CSPMN star polymer precursors (Fig. 1 and 3, Tables 1 and 2) indicate that only a small amount of unattached linear polymer is present at the end of their synthesis. More specifically, the non-degradable “arm-first” POEGMA19-EGDMA6-star and the core degradable POEGMA19-MOEME6-star star polymers had 9.6% and 17.0% unattached linear polymer (Table 1), respectively. The non-degradable “in–out” POEGMA19-EGDMA6-POEGMA20-star (10% w/w solids content) and the core degradable “in–out” POEGMA19-MOEME6-POEGMA20-star (25% w/w solids content) appeared to have only 6.2% and 8.3% unattached linear polymer (Table 2), respectively. Therefore, purification was deemed unnecessary and was not performed in order to simplify the synthetic procedure and to avoid product loss. Also, a very small amount of OEGMA monomer is present at the end of the “in–out” star polymer reaction, 2.4% (97.6% conversion, Table 2) for the non-degradable “in–out” POEGMA19-EGDMA6-POEGMA20-star (10% w/w solids content) and 1.9% (98.1% conversion, Table 2) for the core degradable “in–out” POEGMA19-MOEME6-POEGMA20-star (25% w/w solids content). Both the small amounts of POEGMA linear polymer and OEGMA monomer present in the “in–out” star polymer precursors did not appear to impede the formation of the CSPMNs.
The formation of the non-degradable POEGMA19-EGDMA6-EGDMA20-MOEME6 CSPMN at 10% w/w solids content was quick as gelation time was only 20 min at 37 °C. However, when CSPMN formation for POEGMA19-MOEME6-POEGMA20-MOEME6 was attempted with 10% w/w solids content, gelation did not occur even after 48 h. The solution was possibly too dilute for the MOEME CSPMN to form, while also it is possible that the formation of the “in–out” POEGMA19-MOEME6-POEGMA20-star star polymer in the previous step was incomplete (as discussed above). Therefore, CSPMN formation was carried out for POEGMA19-MOEME6-POEGMA20-MOEME6 using the 25% w/w “in–out” POEGMA19-MOEME6-POEGMA20-star precursor. This yielded a CSPMN with a gelation time of only 20 min at 37 °C. The extractables from the CSPMN POEGMA19-MOEME6-POEGMA20-MOEME6 were collected and analyzed by SEC (THF) (Fig. S5†). The Mn, Mp and Đ values of the two extractables’ peaks determined by SEC (THF) were 29630 g mol−1, 28170 g mol−1 and 1.38, respectively, for the peak at the lower elution time in the chromatogram. The same values were 6450 g mol−1, 3230 g mol−1 and 1.68, respectively, for the peak at the higher elution time. The peak at the higher elution time is likely unreacted POEGMA19, due to their similar Mp value. The peak at the lower elution time is theorized to be the result of an extension of the unreacted linear polymer present in the sample. The unreacted POEGMA19 linear polymer will polymerize further with the excess OEGMA monomer added in the “in–out” star polymer synthetic step yielding linear polymers with a higher DP and larger MWs than the POEGMA19 macro-CTA.
Degradation of CSPMNs under acidic conditions
The POEGMA19-EGDMA6-POEGMA20-EGDMA6 CSPMN is not degradable since it contains non-degradable EGDMA cross-links; therefore, it retains its structure under acidic conditions (Scheme 1e). On the contrary, the POEGMA19-MOEME6-POEGMA20-MOEME6 CSPMN has both primary (formed during the “arm-first” synthesis, Scheme 1b) and secondary (formed during the CSPMN synthesis, Scheme 1d) degradable diacetal-based MOEME cores. The acetals in the CSPMN cores can break down under acidic conditions15,38,59,62,71–74 similar to the ones found in tumors (i.e. pH 5.5),75 transforming the CSPMN structure to linear polymers,15 with each unit of MOEME cross-linker giving two 2-hydroxyethyl methacrylate (HEMA) units. The degradation of the cross-linker, and therefore the CSPMN cores, eliminates the interconnection between the linear chains that are holding the CSPMN structure together and this results in the collapse of the network. As presented schematically in Scheme 1f, after the full degradation (90 days) of the POEGMA19-MOEME6-POEGMA20-MOEME6 CSPMN, its degradation product should be a POEGMA19-b-PHEMA12-b-POEGMA20-b-PHEMA12 tetrablock copolymer. The 1H NMR (CDCl3) spectrum of the degradation product is presented in Fig. S1d,† together with the linear POEGMA19 (Fig. S1a†), “arm-first” POEGMA19-MOEME6-star (Fig. S1b†) and “in–out” POEGMA19-MOEME6-POEGMA20-star (Fig. S1c†) CSPMN precursors. The HEMA oxymethylene proton peaks appear in the 1H NMR (CDCl3) spectrum of the degradation product at ∼4.2 ppm (Fig. S1d†). The Mn, Mp and Đ values of one of the CSPMN degradation products determined by SEC (THF) were 6720 g mol−1, 7050 g mol−1 and 1.51, respectively, with the SEC trace shown in Fig. 5. These MW values are slightly higher than those of the linear POEGMA19 (Mn = 5110 g mol−1 and Mp = 6780 g mol−1), indicating that the degradation was successful since a linear polymer, which is longer than the initial linear POEGMA precursor, was obtained from the collapse of the CSPMN structure. There is a shoulder peak, present at a lower elution time, in the SEC trace of the degradation product with Mn, Mp and Đ values of 20120 g mol−1, 10720 g mol−1 and 1.41, respectively.
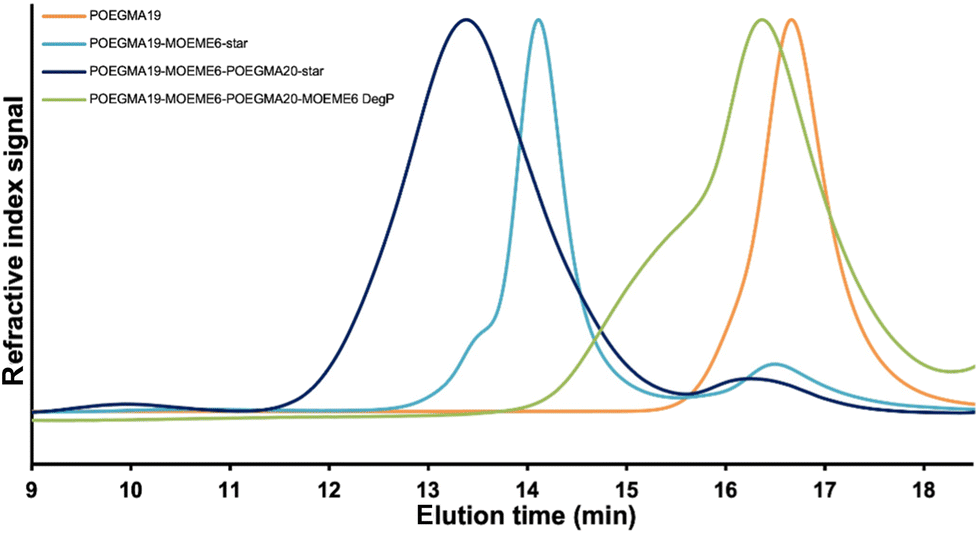 | ||
| Fig. 5 SEC (THF) trace for the degradation product (DegP) of the POEGMA19-MOEME6-POEGMA20-MOEME6 CSPMN. Also included for comparison are the SEC (THF) traces of the precursors to POEGMA19-MOEME6-POEGMA20-MOEME6. | ||
Encapsulation of rhodamine B dye within an acid-labile MOEME-based CSPMN
Once the most favorable conditions for the formation of an acid labile CSPMN were optimized (25% w/w, 37 °C), encapsulation of a drug model was performed in situ during the synthesis of the CSPMN. Since the polymerization reaction takes place under physiological conditions (aqueous solution, 37 °C), developing a methodology to encapsulate a biologically important molecule in situ during polymerization is very important for biomedical applications, and especially for drug delivery. This was tested here during the synthesis of the POEGMA19-MOEME6-POEGMA20-MOEME6 CSPMN using rhodamine B as a surrogate drug. The addition of the dye in the polymerization mixture for the CSPMN synthesis did not seem to affect the polymerization, since a gel was formed and the gelation time was the same as with the polymerization reaction without the dye (20 min). At the end of the polymerization, the dye was encapsulated (entrapped) in the CSPMN nanostructure, giving an intense pink color to the formed gel.Release kinetics of rhodamine B from MOEME-based CSPMN
In order to mimic the conditions inside the body for drug release, two different dye release experiments were set up. One dye release study of the rhodamine B loaded POEGMA19-MOEME6-POEGMA20-MOEME6 CSPMN was conducted in a pH 7.4 buffer in order to mimic the physiological conditions while the other was conducted in a pH 5.5 buffer to imitate the pH of a cancerous late stage tumor.75 The drug release was monitored over a period of a few days and samples were taken at various timepoints. The absorbance of the samples was measured using UV-vis spectroscopy (wavelength range 510–590 nm) and calibration curves were prepared for rhodamine B release in both pH 5.5 and pH 7.4 solutions (Fig. S6†). At around 72 h the fluorescence signal reached its maximum value in both pH values, therefore almost all of the amount of the rhodamine B dye that could be released from the CSPMN was transferred to its surrounding environment (7.80 μg for pH 7.4 and 2.99 μg for pH 5.5). Numerous studies have been carried out on how lower pH environments affect the structure and make-up of rhodamine B. It has been shown that not only do acidic environments protonate and alter the maximum UV wavelength absorbance of the dye but they also degrade rhodamine through protonation.76–78 This goes someway to explaining the difference in the calculated amount of rhodamine released between the two pH environments (probably underestimated at pH 5.5). The color of each solution upon completion of the study is shown in Fig. S4.† A % cumulative release profile of rhodamine B for POEGMA19-MOEME6-POEGMA20-MOEME6 CSPMN in both pH 5.5 and pH 7.4 was constructed to observe the release of rhodamine B over time (with fluorescence maximum signal at 100%) and it is presented in Fig. 6. The release profiles of the rhodamine B encapsulated MOEME-based CSPMN show that the release rate decreased gradually during the period of 72 h at both pH values. At pH 5.5, a higher release rate was observed consistently over this period, which is attributed to some degradation of the CSPMN cores under these acidic conditions.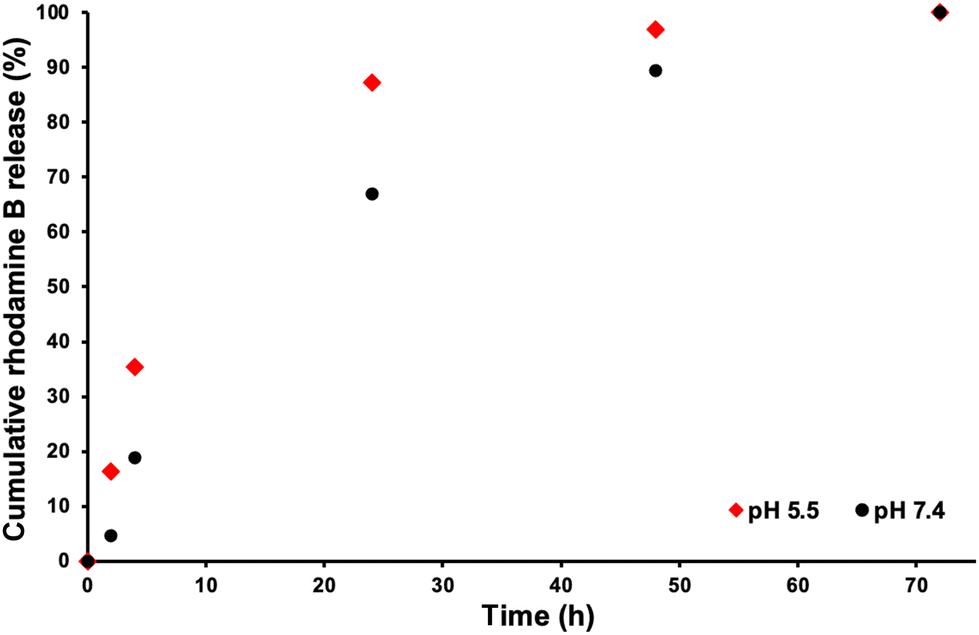 | ||
| Fig. 6 % Cumulative rhodamine B release profile for the POEGMA19-MOEME6-POEGMA20-MOEME6 CSPMN in physiological pH (7.4) and acidic pH (5.5) at 37 °C over a period of 72 h. | ||
Conclusions
The preparation of well-defined polymeric networks under biologically friendly conditions has great potential in the biomedical field and especially in drug encapsulation and delivery. In this work, PEG-based CSPMNs with both acid-labile diacetal MOEME cores and non-degradable EGDMA cores were synthesized by aqueous RAFT polymerization at 37 °C from their linear, “arm-first” and “in–out” star polymer precursors. The successful formation of the CSPMNs was confirmed by facile gelation (in only 20 min). Both the core degradable and non-degradable CSPMNs, and their star polymer precursors, were of high quality, comparable with those synthesized previously in organic solvents. In situ encapsulation of rhodamine B in the acid-labile MOEME-based CSPMN was successful and dye release was observed by UV-vis. In conclusion, CSPMN formation using a biologically friendly aqueous RAFT polymerization approach was successful, and it presents a facile method of forming these acid-labile well-defined networks in a biologically friendly medium, which is desirable for drug delivery and other biomedical applications.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Department for the Economy (DfE) for funding the Ph.D. studentship of G.I.References
- T. Tanaka, Sci. Am., 1981, 244, 124–138 CrossRef CAS PubMed.
- Q. Chai, Y. Jiao and X. Yu, Gels, 2017, 3, 6 CrossRef PubMed.
- G. Hild, Prog. Polym. Sci., 1998, 23, 1019–1149 CrossRef CAS.
- O. W. Webster, Science, 1991, 4996, 887–893 CrossRef PubMed.
- M. Vamvakaki, S. C. Hadjiyannakou, E. Loizidou, C. S. Patrickios, S. P. Armes and N. C. Billingham, Chem. Mater., 2001, 13, 4738–4744 CrossRef CAS.
- A. I. Triftaridou, D. Kafouris, M. Vamvakaki, T. K. Georgiou, T. C. Krasia, E. Themistou, N. Hadjiantoniou and C. S. Patrickios, Polym. Bull., 2007, 58, 185–190 CrossRef CAS.
- M. Vamvakaki, C. S. Patrickios, P. Lindner and M. Gradzielski, Langmuir, 2007, 23, 10433–10437 CrossRef CAS PubMed.
- X. Zhang, K. Kyriakos, M. Rikkou-Kalourkoti, E. N. Kitiri, C. S. Patrickios and C. M. Papadakis, Colloid Polym. Sci., 2016, 294, 1027–1036 CrossRef CAS.
- M. Vamvakaki and C. S. Patrickios, Chem. Mater., 2002, 14, 1630–1638 CrossRef CAS.
- D. S. Achilleos, T. K. Georgiou and C. S. Patrickios, Biomacromolecules, 2006, 7, 3396–3405 CrossRef CAS PubMed.
- S. N. Georgiades, M. Vamvakaki and C. S. Patrickios, Macromolecules, 2002, 35, 4903–4911 CrossRef CAS.
- T. K. Georgiou and C. S. Patrickios, Biomacromolecules, 2008, 9, 574–582 CrossRef CAS PubMed.
- E. Themistou and C. S. Patrickios, Macromolecules, 2004, 37, 6734–6743 CrossRef CAS.
- D. Kafouris, E. Themistou and C. S. Patrickios, Chem. Mater., 2006, 18, 85–93 CrossRef CAS.
- E. Themistou and C. S. Patrickios, Macromol. Chem. Phys., 2008, 209, 1021–1028 CrossRef CAS.
- M. Rikkou-Kalourkoti, E. N. Kitiri, C. S. Patrickios, E. Leontidis, M. Constantinou, G. Constantinides, X. Zhang and C. M. Papadakis, Macromolecules, 2016, 49, 1731–1742 CrossRef CAS.
- X. Zhang, K. Kyriakos, M. Rikkou-Kalourkoti, E. N. Kitiri, C. S. Patrickios and C. M. Papadakis, Colloid Polym. Sci., 2016, 294, 1027–1036 CrossRef CAS.
- E. N. Kitiri, C. S. Patrickios, C. Voutouri, T. Stylianopoulos, I. Hoffmann, R. Schweins and M. Gradzielski, Polym. Chem., 2017, 8, 245–259 RSC.
- E. N. Kitiri, C. K. Varnava, C. S. Patrickios, C. Voutouri, T. Stylianopoulos, M. Gradzielski and I. Hoffmann, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2161–2174 CrossRef CAS.
- N. Ghasdian, E. Church, A. P. Cottam, K. Hornsby, M. Y. Leung and T. K. Georgiou, RSC Adv., 2013, 3, 19070–19080 RSC.
- O. W. Webster, W. R. Hertler, D. Y. Sogah, W. B. Farnham and T. V. RajanBabu, J. Am. Chem. Soc., 1983, 105, 5706–5708 CrossRef CAS.
- O. W. Webster, Adv. Polym. Sci., 2004, 167, 1–34 CAS.
- T. K. Georgiou and C. S. Patrickios, Macromolecules, 2006, 39, 1560–1568 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 857–871 CrossRef CAS.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS PubMed.
- H. Gao, M. C. Jones, J. Chen, R. E. Prud'homme and J. C. Leroux, Chem. Mater., 2008, 20, 3063–3067 CrossRef CAS.
- S. S. Nagarkar, M. Tsujimoto, S. Kitagawa, N. Hosono and S. Horike, Chem. Mater., 2018, 30, 8555–8561 CrossRef CAS.
- W. Xu, A. A. Steinschulte, F. A. Plamper, V. F. Korolovych and V. V. Tsukruk, Chem. Mater., 2016, 28(3), 975–985 CrossRef CAS.
- W. Wu, W. Wang and J. Li, Prog. Polym. Sci., 2015, 46, 55–85 CrossRef CAS.
- T. K. Georgiou, M. Vamvakaki, L. A. Phylactou and C. S. Patrickios, Biomacromolecules, 2005, 6, 2990–2997 CrossRef CAS PubMed.
- T. K. Georgiou, L. A. Phylactou and C. S. Patrickios, Biomacromolecules, 2006, 7, 3505–3512 CrossRef CAS PubMed.
- T. K. Georgiou, Polym. Int., 2014, 63, 1130–1133 CrossRef CAS.
- X. Liao, G. Walden, N. D. Falcon, S. Donell, M. J. Raxworthy, M. Wormstone, G. P. Riley and A. Saeed, Eur. Polym. J., 2017, 87, 458–467 CrossRef CAS.
- F. Dai, P. Sun, Y. Liu and W. Liu, Biomaterials, 2010, 31, 559–569 CrossRef CAS PubMed.
- T. J. Gibson, P. Smyth, M. Semsarilar, A. P. McCann, W. J. McDaid, M. C. Johnston, C. J. Scott and E. Themistou, Polym. Chem., 2020, 11, 344–357 RSC.
- K. S. Pafiti, C. S. Patrickios, T. K. Georgiou, E. N. Yamasaki, N. P. Mastroyiannopoulos and L. A. Phylactou, Eur. Polym. J., 2012, 48, 1422–1430 CrossRef CAS.
- M. Ahmed and R. Narain, Prog. Polym. Sci., 2013, 38, 767–790 CrossRef CAS.
- F. J. Xu, Z. X. Zhang, Y. Ping, J. Li, E. T. Kang and K. G. Neon, Biomacromolecules, 2009, 10, 285–293 CrossRef CAS PubMed.
- T. K. Georgiou, M. Vamvakaki, C. S. Patrickios, E. N. Yamasaki and L. A. Phylactou, Biomacromolecules, 2004, 5, 2221–2229 CrossRef CAS PubMed.
- D. P. Yang, M. N. N. L. Oo, G. R. Deen, Z. Li and X. J. Loh, Macromol. Rapid Commun., 2017, 38, 1700410 CrossRef PubMed.
- J. Liu, H. Duong, M. R. Whittaker, T. P. Davis and C. Boyer, Macromol. Rapid Commun., 2012, 33, 760–766 CrossRef CAS PubMed.
- B. Jeong, Y. K. Choi, Y. H. Bae, G. Zentner and S. W. Kim, J. Controlled Release, 1999, 62, 109–114 CrossRef CAS PubMed.
- J. Ferreira, J. Syrett, M. Whittaker, D. Haddleton, T. P. Davis and C. Boyer, Polym. Chem., 2011, 2, 1671–1677 RSC.
- D. H. Rein, P. Rempp and P. J. Lutz, Macromol. Chem. Phys., 1998, 199, 569–574 CAS.
- F. Dai, P. Sun, Y. Liu and W. Liu, Biomaterials, 2010, 31, 559–569 CrossRef CAS PubMed.
- B. Mendrek, L. Sieroń, M. Libera, M. Smet, B. Trzebicka, A. L. Sieroń, A. Dworak and A. Kowalczuk, Polymer, 2014, 55, 4551–4562 CrossRef CAS.
- Q. Qiu, G. Liu and Z. An, Chem. Commun., 2011, 47, 12685–12687 RSC.
- X. Cao, C. Zhang, S. Wu and Z. An, Polym. Chem., 2014, 5, 4277–4284 RSC.
- X. Shi, M. Miao and Z. An, Polym. Chem., 2013, 4, 1950–1959 RSC.
- Q. Chen, X. Cao, H. Liu, W. Zhou, L. Qin and Z. An, Polym. Chem., 2013, 4, 4092–4102 RSC.
- X. Shi, W. Zhou, Q. Qiu and Z. An, Chem. Commun., 2012, 48, 7389–7391 RSC.
- K. Wang, H. Peng, K. J. Thurecht, S. Puttick and A. K. Whittaker, Polym. Chem., 2014, 5, 1760–1771 RSC.
- B. S. Tucker, S. G. Getchell, M. R. Hill and B. S. Sumerlin, Polym. Chem., 2015, 6, 4258–4263 RSC.
- Q. Chen, F. Han, C. Lin, X. Wen and P. Zhao, Polymer, 2018, 146, 378–385 CrossRef CAS.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- E. Themistou and C. S. Patrickios, Macromolecules, 2007, 40, 5231–5234 CrossRef CAS.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- V. Lotocki and A. Kakkar, Pharmaceutics, 2020, 12, 827 CrossRef CAS PubMed.
- E. Themistou and C. S. Patrickios, Macromolecules, 2006, 39, 73–80 CrossRef CAS.
- T. J. Gibson, P. Smyth, W. J. McDaid, D. Lavery, J. Thom, G. Cotton, C. J. Scott and E. Themistou, ACS Macro Lett., 2018, 7, 1010–1015 CrossRef CAS PubMed.
- P. Smyth, T. J. Gibson, G. Irvine, G. Black, D. Lavery, M. Semsarilar, C. J. Scott and E. Themistou, Eur. Polym. J., 2020, 124, 109471 CrossRef CAS.
- A. Blencowe, J. F. Tan, T. K. Goh and G. G. Qiao, Polymer, 2009, 50, 5–32 CrossRef CAS.
- X. Wei, G. Moad, B. W. Muir, E. Rizzardo, J. Rosselgong, W. Yang and S. H. Thang, Macromol. Rapid Commun., 2014, 35, 840–845 CrossRef CAS PubMed.
- J. T. Wiltshire and G. G. Qiao, Macromolecules, 2006, 39, 9018–9027 CrossRef CAS.
- P. Lang, W. Burchard, M. S. Wolfe, H. J. Spinelli and L. Page, Macromolecules, 1991, 24, 1306–1314 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 7216–7223 CrossRef CAS.
- H. Gao, N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 5995–6004 CrossRef CAS.
- J. Hu, J. He, M. Zhang and P. Ni, Polym. Chem., 2015, 6, 1553–1566 RSC.
- R. Banerjee, S. Parida, C. Maiti, M. Mandal and D. Dhara, RSC Adv., 2015, 5, 83565–83575 RSC.
- M. Kawamura, A. Kanazawa, S. Kanaoka and S. Aoshima, Polym. Chem., 2015, 6, 4102–4108 RSC.
- E. Themistou and C. S. Patrickios, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5853–5870 CrossRef CAS.
- D. Delli Castelli, G. Ferrauto, J. C. Cutrin, E. Terreno and S. Aime, Magn. Reson. Med., 2014, 71, 326–332 CrossRef CAS PubMed.
- R. W. Ramette and E. B. Sandell, J. Am. Chem. Soc., 1956, 78, 4872–4878 CrossRef CAS.
- C. J. Miller, H. Yu and T. D. Waite, Colloids Surf., A, 2013, 435, 147–153 CrossRef CAS.
- N. Pourreza, S. Rastegarzadeh and A. Larki, Talanta, 2008, 77, 733–736 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section; additional 1H NMR spectra; DLS, SEC, SLS and UV-vis data. See DOI: https://doi.org/10.1039/d3py00677h |
| This journal is © The Royal Society of Chemistry 2024 |