
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphorus acid: an asset for flame-retardant sustainable vitrimers†
Florian
Cuminet
ab,
Nathan
Vanachte
a,
Chloé
Farina
 a,
Maxinne
Denis
a,
Claire
Negrell
a,
Sylvain
Caillol
*a,
Éric
Dantras
b,
Éric
Leclerc
a and
Vincent
Ladmiral
*a
a,
Maxinne
Denis
a,
Claire
Negrell
a,
Sylvain
Caillol
*a,
Éric
Dantras
b,
Éric
Leclerc
a and
Vincent
Ladmiral
*a
aICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: sylvain.caillol@enscm.fr; vincent.ladmiral@enscm.fr
bCIRIMAT, Université Toulouse 3 – Paul Sabatier, Physique des Polymères, 118 Route de Narbonne, 31062 Toulouse, France
First published on 20th February 2024
Abstract
Three biobased epoxy resins from vanillic alcohol, linseed oil and cardanol were crosslinked with phosphoric acid to prepare vitrimers. The reactivities of the different types of epoxy towards phosphoric acid were assessed by 1H and 31P NMR spectroscopy on model molecules. The resulting phosphate esters constitute the nodes of the network, which can exchange with free hydroxy groups upon heating, thus conferring reshaping capability to the materials. Additionally, the phosphate groups endow flame retardant properties to the materials. When exposed to fire a char layer is formed, which insulates the inner part of the material and slows down its combustion. Finally, mixtures of the three epoxy resins led to dynamic materials with both fire retardancy properties and low Tg.
Introduction
Vitrimers are polymeric materials at the crossroads of thermosets and thermoplastics, able to be mechanically reprocessed and endowed with high chemical resistance.1 They are made of a tridimensional macromolecular network whose covalent bonds are dynamic thanks to bond or functional group exchanges.2 This peculiar property makes them able to flow and change shape upon heating.Since the first vitrimer, based on catalyzed transesterification,3 several strategies have been developed to obtain fast exchanges in such materials. Indeed, sufficiently fast exchange at moderately high temperature is desirable to attain acceptable reprocessing times and avoid degradation. In particular, numerous different exchange reactions were investigated4,5 such as vinylogous urethane transamination,6 imine metathesis7 or olefins metathesis8 to name a few. Alternatively, the rate of a slow exchange reaction can be enhanced by the use of catalysts. This approach is typical for transesterification vitrimers for instance.3,5 Another strategy consists in exploiting well-chosen and judiciously located activating groups in the network to accelerate the exchange reaction.9–14 Surprisingly, in this quest for fast exchanges, limited interest was given to phosphate esters despite their known easy exchange with alcohols.15 Phosphates were used as adhesion promoters in polyimine vitrimers.16 The transesterification of carboxylate esters in vitrimers was also catalyzed by phosphates and phosphonates.17–22 Nonetheless, few articles reported the use of phosphate esters as the exchange sites in vitrimers. Exchanges can happen between phosphate esters and carboxylate esters,23 and even via a dissociative pathway involving a cyclization to make a dynamic network.24 However, the associative exchange of a phosphate ester with a hydroxyl moiety is the most common. For example, a vitrimer was prepared by anionic polymerization of bisphenol A diglycidyl ether (BADGE) in ionic liquid containing phosphates. The resulting phosphate esters were the crosslinking nodes of the network.25 Feng and Li also reported vitrimers based on BADGE crosslinked by phosphate diesters.26,27 The dynamic behaviour of phosphate ester based vitrimers was much faster compared to their carboxylic ester counterparts. The exchange can even be accelerated by hydroxyl groups in close vicinity to phosphate ester bonds.28 Furthermore, phosphates added fire retardancy properties to the materials.29 Another article reported epoxidized rubber crosslinked by D-fructose 1,6-bisphosphoric acid as an example of biobased vitrimer elastomer.30 Phytic acid, a naturally occurring polyphosphoric acid, was used to crosslink epoxidized itaconic acid to obtain fully biobased fire retardant vitrimers.31 Finally, Sijbesma et al. described a network with phosphate triesters as crosslinking nodes.32 The material was a vitrimer, resulting from the reaction between a polyol and triphenyl phosphate in the presence of sodium hydride. The phosphate ester vitrimers described so far were based on difunctional phosphoric acid monoesters. However, the use of phosphoric acid H3PO4 has not been reported yet in such materials. Phosphoric acid bears three acid functions able to react with epoxides to produce beta-hydroxyl phosphoester functional groups susceptible to fast transesterification.
In this work, the reactivities of different epoxy functions towards phosphoric acid were assessed via1H NMR spectroscopy studies on model molecules. Then, three biobased epoxy resins with different structures were polymerized with phosphoric acid H3PO4 and the thermal and mechanical behaviors of the materials obtained were compared, in particular their dynamic and flame-retardant properties. Binary and ternary mixtures of the epoxy monomers could also be used to tune the properties of the final materials.
Results and discussion
Epoxy resins
Diglycidyl ether of vanillic alcohol (DGEVA) is derived from vanillin,33 a compound found in vanilla which can be produced industrially from lignocellulosic materials.34 Vanillin is first reduced to vanillic alcohol prior to glycidylation by epichlorohydrin, which can be biobased,35 to finally reach DGEVA (Scheme 1). DGEVA is based on a phenolic structure and could replace DGEBA, the diepoxy found in more than 90% of commercial epoxy formulas and derived from bisphenol A, a known endocrine disruptor.36 | ||
| Scheme 1 Synthesis of DGEVA from vanillin. | ||
Polyfunctional epoxy resins can also be obtained from vegetable oils, by oxydation of unsaturated triglycerides (triesters of fatty acid with glycerol, Scheme 2).36 Epoxidized linseed oil (ELO) was used in this study to cover a broad spectrum of mechanical properties for the final materials.
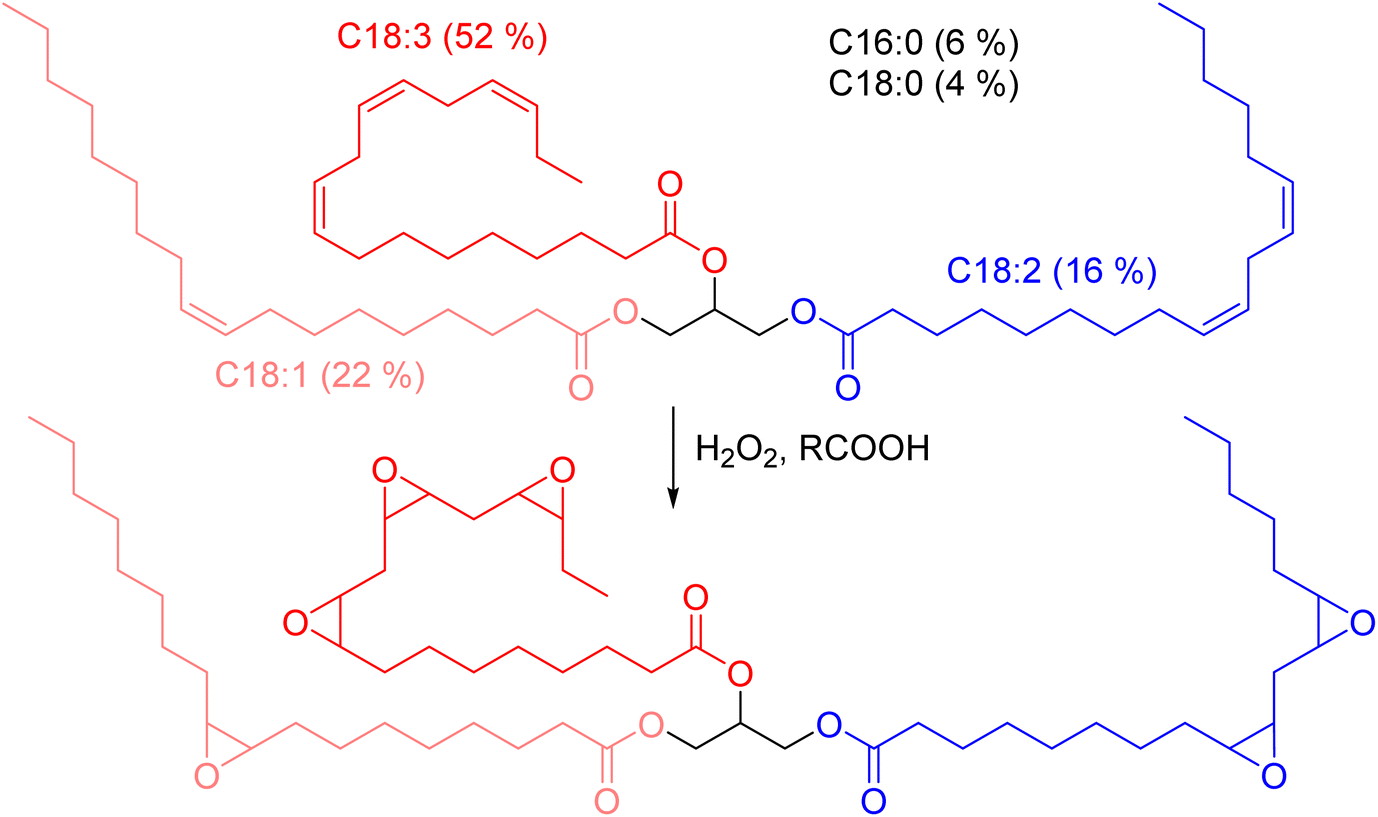 | ||
| Scheme 2 Epoxidation of vegetable oils unsaturations. Example of the typical triglyceride composition of linseed oil.37 | ||
Finally, a commercial cardanol-based difunctional epoxy (Cardolite NC-514) was also used. Cardanol is extracted from cashew nut shell liquid (CNSL), a waste from the production of cashew nuts. Cardanol is composed of a phenol with a C15 aliphatic side chain (Scheme 3).38–40 Cardanol can be modified to produce an epoxy resin with an average functionality of 2.41 These three monomers were endowed with different types of epoxy function, internal, terminal and glycidyl, and thus with different reactivities, which were evaluated with model compounds.
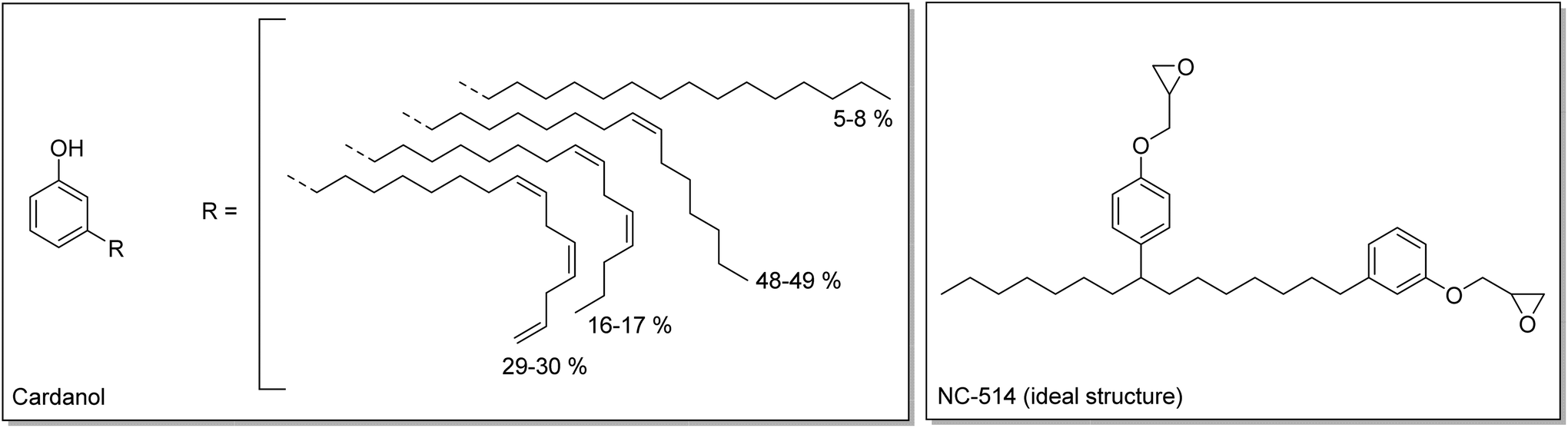 | ||
| Scheme 3 Structures of cardanol42 and of NC-514 (ideal structure expected). | ||
Reactivity study
The reactivity of phosphoric acid towards epoxy moieties was investigated by 1H NMR spectroscopy. Three model molecules were chosen to compare the reactivity of terminal or internal epoxides and of phenolic glycidyl ethers. The monophosphoric acid dibutyl phosphate was chosen to simplify the interpretation of the spectra, and was used in a 10-fold molar excess compared to epoxy groups to conduct the kinetics experiments under pseudo-first order conditions. 9,10-Epoxyoctadecane, 1,2-epoxydodecane and phenyl glycidyl ether were used as epoxy substrates and the disappearance of cyclic ether signals were monitored (Fig. 1).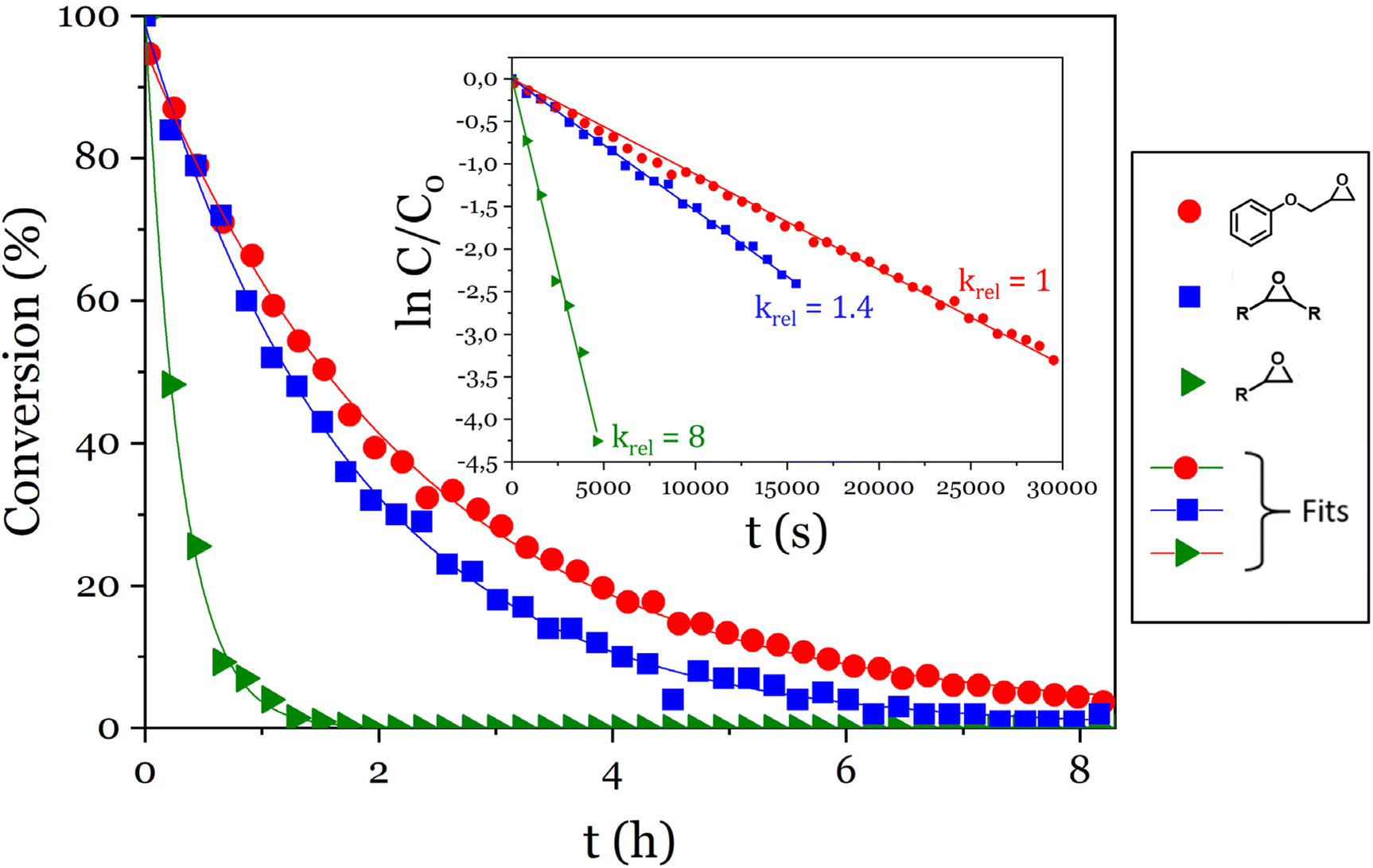 | ||
| Fig. 1 Evolution of the conversion of the epoxy model molecules versus time and pseudo-first order plots (inset). | ||
A 90% conversion was reached in 5 h 50 min for the glycidyl ether, 4 h for the internal epoxide (9,10-epoxyoctadecane) and only 40 min for the terminal epoxide (1,2-epoxydodecane). For each epoxy, ln(C/C0) was plotted against time and kobs were determined as the slopes of the linear fits (Fig. 1 inset). The kobs measured this way were 1.120 ± 0.006 10−4 s−1, 1.55 ± 0.01 10−4 s−1 and 9.0 ± 0.3 10−4 s−1 for the glycidyl ether, the internal epoxide and the terminal epoxide, respectively (Fig. 1 inset).
The aliphatic terminal epoxides were converted 8 times faster than the glycidyl ether. Such a difference in reaction rates between the opening of the phenoxyepoxide and of the alkoxyepoxide was unexpected. One possible explanation may be that the phenoxy substituent, slightly less electron donor than its alkoxy counterpart, provides a lower stabilization of the positive charge on the protonated epoxide intermediate. The slight difference in regioselectivity (Scheme 4) is in agreement with this explanation since the phenoxyepoxide led to a greater proportion of terminal phosphate than its alkoxy analog, likely due to the lower stabilization of the secondary (internal) phenoxyepoxide–derived carbocation. However, this explanation remains speculative and was not fully demonstrated. The ring opening reaction was also much faster for terminal epoxy than for internal epoxy, probably due to steric effects.
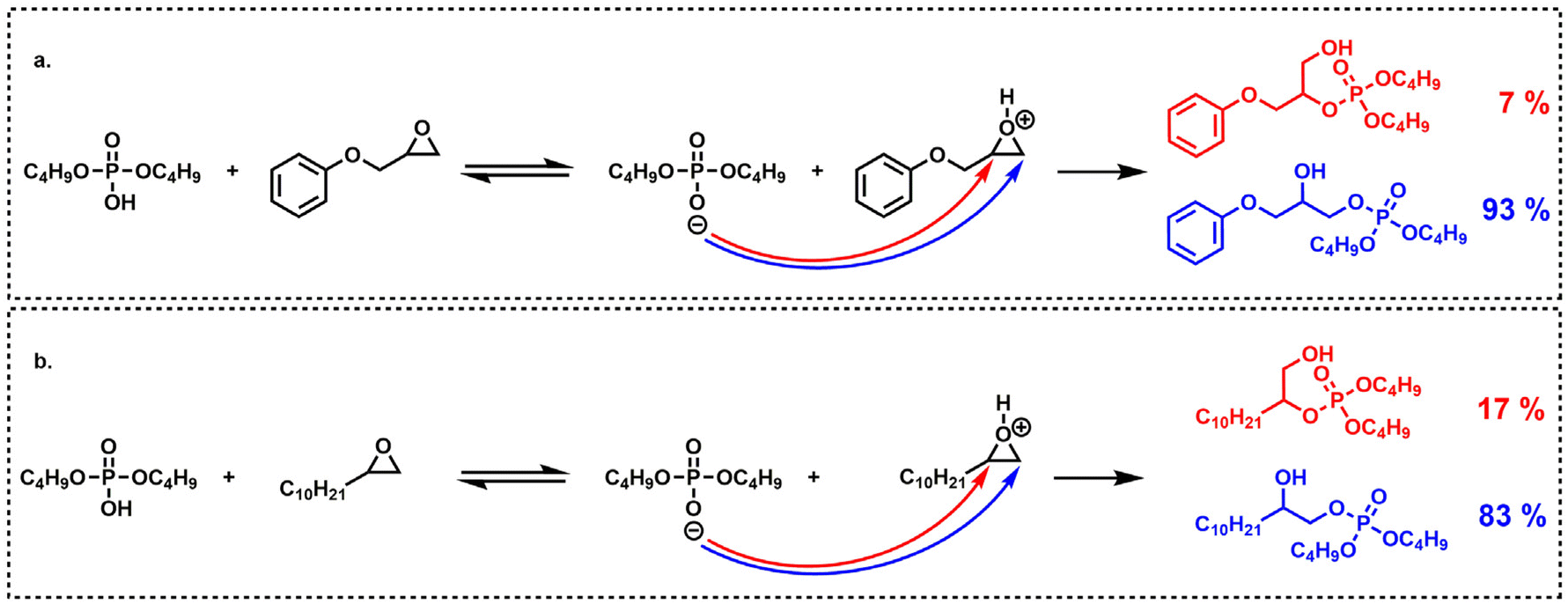 | ||
| Scheme 4 Regioselectivity (mol%) of oxirane opening by dibutylphosphoric acid on: (a) phenylglycidyl ether and, (b) 1,2-epoxydodecane. | ||
Although the methodology adopted reveals the consumption of the epoxide functions in presence of phosphoric acid, it does not give access to information about the reactions at stakes and the products formed. Considering the reactions were carried out at room temperature and with a large excess of acid (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 POH/epoxy), the self-condensation or homopolymerization of epoxides to produce ether bonds is very unlikely.
:1 POH/epoxy), the self-condensation or homopolymerization of epoxides to produce ether bonds is very unlikely.
Once the reactivity of the different types of epoxy towards a monophosphoric acid group was verified, reversely the reactivity of the three acid functions of H3PO4 towards an epoxy function was also assessed, to ensure the formation of a trisubstituted phosphate ester by this reaction. Thus, the evolution of a binary mixture of 1 equivalent of phosphoric acid and 28 equivalents of phenyl glycidyl ether in acetone was monitored by 31P NMR spectroscopy (≈10:1 epoxy/acid functions). In this case, the epoxy order was degenerate and the reactions were run under pseudo-first order for the phosphoric acid. The kinetic equations to model this system are given in ESI.†
The 31P NMR spectra showed that the substitution of the three acid functions happened simultaneously (Fig. 2 and S15–S17†). To simplify the fitting, T was determined as T = 1 − (A + M + D), thus only A, M and D were fitted altogether with the model equations (eqn (S5)–(S8)†), and the values obtained for k1, k2 and k3 were reported in Table 1.
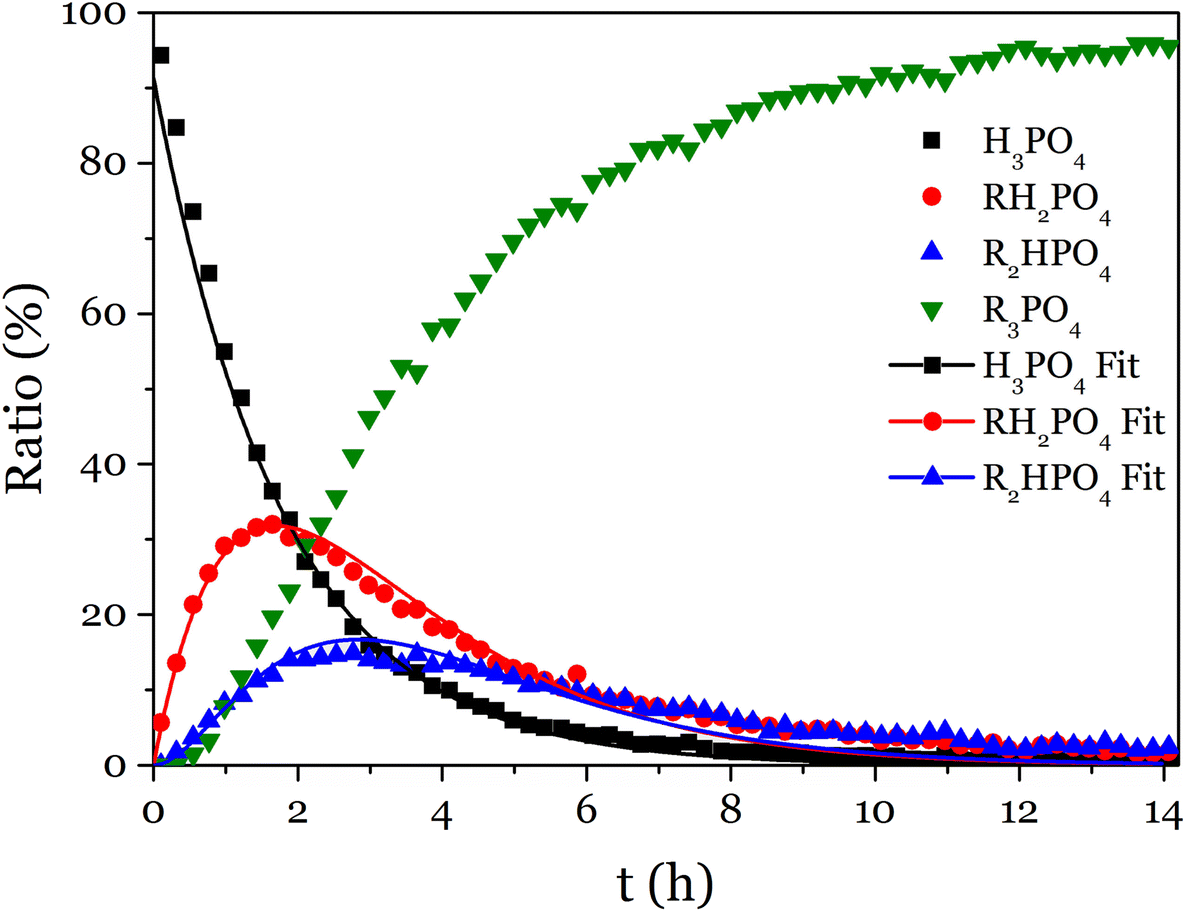 | ||
| Fig. 2 Evolution with time of the proportion of P-containing species during the reaction of phosphoric acid with phenyl glycidyl epoxy. | ||
| k 1 (h−1) | k 2 (h−1) | k 3 (h−1) | R 2 |
|---|---|---|---|
| N.B.: the kinetics parameters were determined by a single fit to optimize the three curves at a time. | |||
| 0.56 ± 0.01 | 0.62 ± 0.02 | 1.01 ± 0.03 | 0.98373 |
According to these rate constants values (ki, i = 1–3), the disubstituted product is more reactive than the monosubstituted product, itself slightly more reactive than the phosphoric acid toward the addition to an epoxy. This result proves that the formation of trisubstituted phosphate esters by reaction of epoxy and phosphoric acid is favored, and could lead to crosslinked networks.
Vitrimers synthesis
Vitrimers were synthesized from commercially available biobased epoxy resins and phosphoric acid using a 1:1 epoxy:acid equivalent ratio. The ring opening polymerization of polyfunctional epoxy by phosphoric acid leads to β-hydroxy phosphate esters. Phosphoric acid is trifunctional, it thus played the roles of both hardener and crosslinker (Scheme 5).
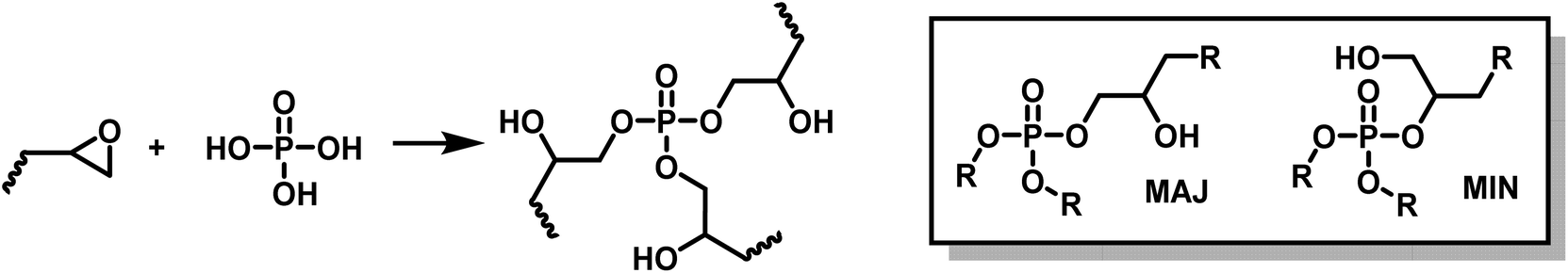 | ||
| Scheme 5 Synthesis of phosphate triesters by ring opening polymerization (ROP) of epoxides by phosphoric acid. | ||
DGEVA, ELO and NC-514 were used as epoxy precursors, and the influence of their structures on the materials properties was assessed. The aromatic structure of DGEVA was expected to confer rigidity and thermal resistance to the materials. The aliphatic structure of ELO was expected to lower the Tg and confer flexibility to the final materials. NC-514 was expected to yield materials with intermediate properties compared to DGEVA-based and ELO-based materials.
Due to the strong acidity (pKa) of phosphoric acid, the reaction with the epoxy ring is fast and very exothermic. The high viscosities of the epoxy resins do not allow an efficient heat dissipation, and the direct mixing of concentrated phosphoric acid and epoxy resin on a 10-grams scale can lead to a thermal runaway. Such runaway reaction effectively happened in the course of this study and led to a small fire which caused limited damage to a lab instrument. To prevent this issue, a solution is to dilute the reaction medium by using a solvent with a suitable boiling point, sufficiently high not to evaporate during the polymerization, but not too high to prevent removal under reduced pressure after synthesis. ELO and DGEVA epoxy resins were thus first diluted with 50 wt% and 20 wt% of acetone, respectively. The formulation for NC-514 was solvent-free owing to the lower reactivity of the glycidyl ether moieties towards phosphoric acid. Then, phosphoric acid was added as a concentrated aqueous solution (Table S1†). After one night at room temperature, the DGEVA, ELO and NC-514 formulations led to a rigid clear yellow material (DGEVAPE, Fig. 3a), a flexible brown material (ELOPE, Fig. 3b) and a flexible dark brown material (CEPE, Fig. 3c), respectively.
 | ||
| Fig. 3 Photos of (a) DGEVAPE, (b) ELOPE and (c) CEPE. | ||
Curing
The materials were dried 15 h in an oven at 60 °C and grinded into powders. The powders underwent a curing step at 130 °C under vacuum to ensure complete drying and curing. The dry powders were then pressed at 150 °C for 1.5 h to obtain the final materials.FTIR revealed that the oxirane C–O–C stretching band at 915 cm−1 for NC-514, 918 cm−1 for DGEVA (glycidyl ethers) and at 830 cm−1 for ELO (aliphatic oxirane) were not visible on the final materials, suggesting that all the epoxy function had reacted (Fig. S1–S3†).
No residual exothermic phenomenon was visible on DSC thermograms and the Tg of the materials were stable at 115 °C, 12 °C and 25 °C for DGEVAPE, ELOPE and CEPE, respectively (Table 2 and Fig. S4–S6†). Thus, the materials were considered fully cured. As expected, DGEVAPE exhibited a high Tg due to the rigidity and high aromatic carbon content of DGEVA and the high crosslinking density, whereas ELOPE had a relatively low Tg due to the flexibility of the fatty chains of ELO. NC-514 which contains both aromatic cycles and aliphatic chains afforded a material (CEPE) with an intermediate Tg. The materials thus prepared cover a large range of Tg and may be suitable to various applications.
| Material | Epoxy EEW (g eq.−1) | T g (°C) | T α (°C) | Max tan δ (°C) | E′Gb (GPa) | E′Rc (MPa) | Gel contentd (%) | Swelling indexd (%) |
|---|---|---|---|---|---|---|---|---|
| a Determined as the maximum of E′′. b At −50 °C. c At 175 °C. d In THF. | ||||||||
| DGEVAPE | 269 | 115 | 83 | 121 | 4.4 | 6.5 | 97 ± 2 | 76 ± 2 |
| ELOPE | 191 | 20 | 39 | 60 | 1.2 | 1.2 | 89 ± 1 | 186 ± 10 |
| CEPE | 448 | 25 | 22 | 41 | 1.6 | 1.9 | 71 ± 1 | 219 ± 14 |
The gel contents of the materials were determined in THF. DGEVAPE, ELOPE and CEPE exhibited gel contents values of 97, 89, and 71%, respectively (Table 2). These results confirmed that these materials were indeed crosslinked 3D networks. The swelling indices were 76, 186, and 219% for DGEVAPE, ELOPE and CEPE respectively.
Mechanical characterizations
The thermomechanical properties of the materials were determined by DMA (Table 2 and Fig. S7–S9†). The Tα was determined as the maximum of E′′, at 83, 39 and 22 °C for DGEVAPE, ELOPE and CEPE, respectively. Two subvitreous modes (β and γ mechanical relaxations) are visible below Tα for DGEVAPE. A deconvolution of the E′′ curve was performed with a sum of three Gaussian curves to determine Tα. The value for DGEVAPE is far from the Tg determined by DSC. This is likely due to the breadth of the transition from the glassy to the rubbery states (Fig. S7†). Similarly, the relatively broad transition for ELOPE can explain the difference between Tg and Tα. For CEPE, the values of Tg and Tα are consistent with each other. Taking into account the broad transition observed for DGEVAPE, the storage modulus values on the glassy plateau and rubbery plateau were taken at −50 °C and +175 °C, respectively, to compare the three materials. The moduli on the glassy plateaus were 4.4, 1.2 and 1.6 GPa for DGEVAPE, ELOPE and CEPE, respectively. This trend was expected as it follows the aromatic carbon content. The moduli on the rubbery plateaus were 6.6, 1.2 and 1.9 MPa, respectively. For a similar matrix, the value of the modulus on the rubbery plateau evolves with the crosslinking density. Here, the polymers have very different structures, thus the values are a combination of these factors and no clear trend can be observed.The materials are networks of β-hydroxy phosphate triesters. Hydroxyl groups can engage into transesterification with the phosphate esters inducing exchanges in the network. This exchange happens upon heating and leads to the reshaping capability of the materials (Fig. 4). Stress relaxation experiments were performed to characterize the dynamic behavior of the materials. The 3 materials DGEVAPE, ELOPE and CEPE fully relaxed the stress applied above their Tg (Fig. 5). The stress-relaxation curves obtained at different temperatures were fitted with a Kohlrausch–Williams–Watts equation. The relaxation times at 130 °C were 894, 242, and 238 s for DGEVAPE, ELOPE and CEPE, respectively. The long relaxation time of DGEVAPE is not surprising as the transition to the rubbery state for this material was very broad, and extended up to 170 °C. Thus, at 130 °C the stress relaxation is hindered by the alpha relaxation mobility. The relaxation times τKWW obtained were plotted in an Arrhenius diagram and the flow activation energies, Ea, were calculated from the slope for each material (Table S2†).
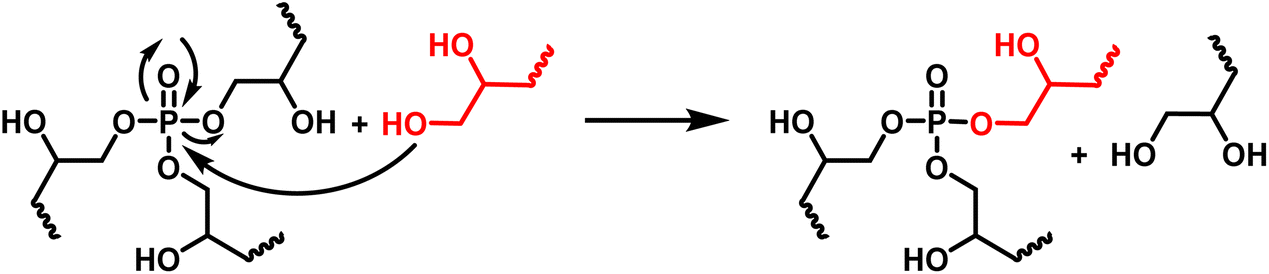 | ||
| Fig. 4 Exchange in the polymer network by phosphate transesterification. | ||
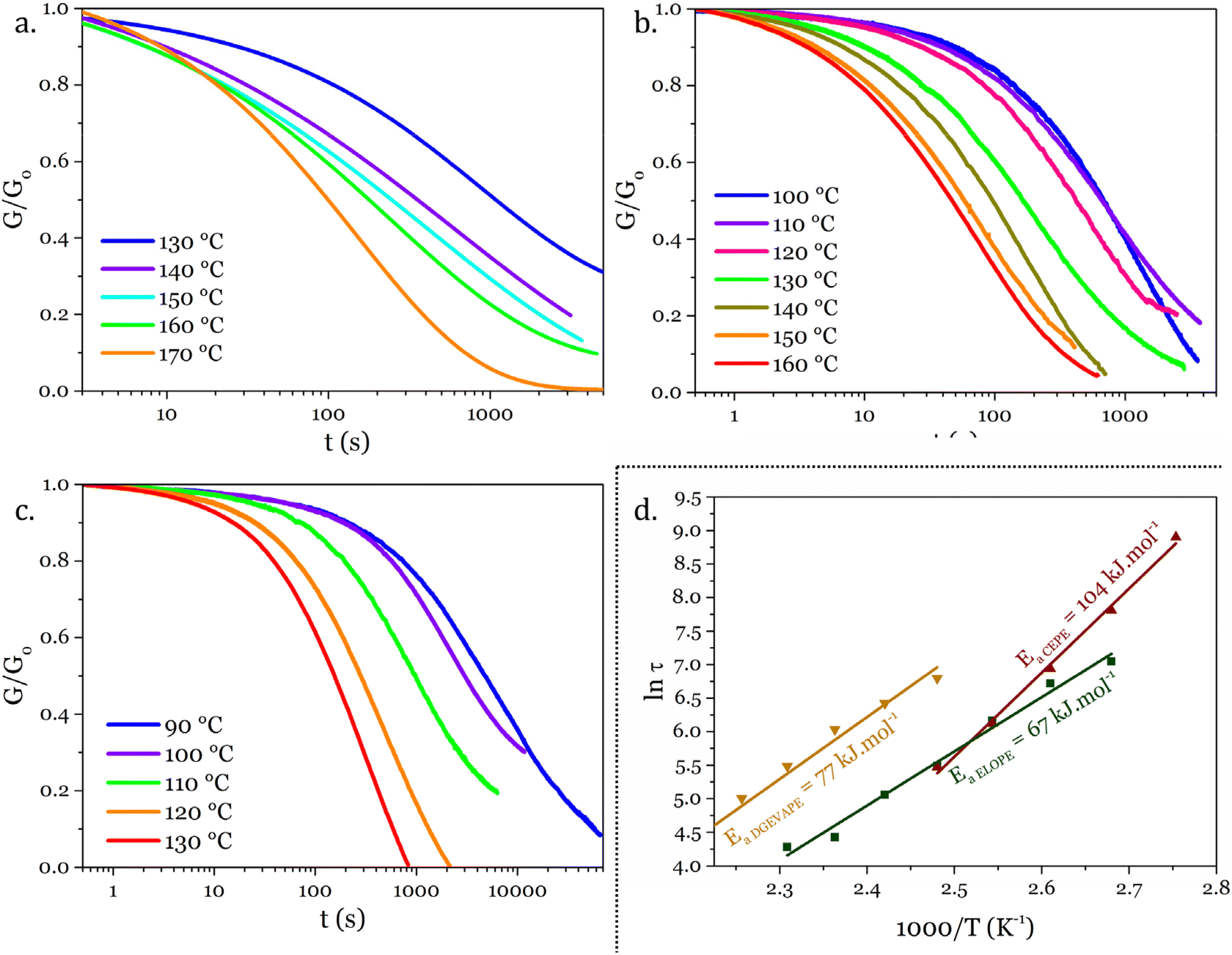 | ||
| Fig. 5 Stress-relaxation experiments results on (a) DGEVAPE, (b) ELOPE and (c) CEPE. (d) Arrhenius diagram of the τ determined from Kohlrausch–Williams–Watt fits for each material, and determination of the flow activation energies. | ||
These Ea values were 77, 67, and 104 kJ mol−1 for DGEVAPE, ELOPE and CEPE respectively. Activation energies depend on the exchange reaction,5 which is the same in the materials reported here, on the polymer matrix43 and on the mobility at the local scale.44–46 The network of CEPE is looser due to the high EEW, so one would expect the activation energy to be lower.47 The high activation energy might thus be due to the relatively low density of exchangeable bond compared to the other networks presented here.47 The dangling fatty chains might also play a role as a plasticizer48 and keep the exchangeable bonds apart. This hypothesis could also explain the slight difference between the activation energies of DGEVAPE and ELOPE.
Thermal and flame retardant properties
Phosphorus-free counterpart materials of DGEVAPE, ELOPE and CEPE, (called DGEVACE, ELOCE and CECE, respectively) but based on citric acid were prepared and examined to highlight the influence of phosphorus in terms of flame retardant (FR) properties. Citric acid was chosen because it is a short trifunctional carboxylic acid, and should thus lead to crosslinking density relatively close to that imparted by phosphoric acid (Table S1†). The thermal properties of the materials were first assessed by TGA under nitrogen (Table 3 and Fig. S13†). The temperatures at 5% weight loss were measured at 234, 219, and 298 °C for DGEVAPE, ELOPE and CEPE, and 277, 239 and 305 °C for DGEVACE, ELOCE and CECE, respectively. A single step of decomposition was observed for phosphorus-free counterpart materials and for DGEVAPE contrary to CEPE and ELOPE materials. The temperatures at the maxima of the TG derivatives (Td max) were 403, 413 and 452 °C for DGEVACE, ELOCE and CECE. DGEVAPE exhibited a single Td max at 317 °C, while for ELOPE and CEPE two maxima were visible at 280 and 454 °C, and at 351 and 483 °C, respectively. The decrease of Td5% for the phosphorus-containing materials compared to phosphorus-free ones is due to the lower stability of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P–O bond compared to C–C bond. The phosphate groups first decomposed and during this step, they increased the crosslinking carbonization that prevented further decomposition of the epoxy resin. For CEPE and ELOPE, the second degradation step is the aliphatic chain decomposition (Fig. S13†).49 Thus, the residues at 700 °C represented 49 wt% for DGEVAPE, 26 and 23 wt% for ELOPE and CEPE, when for DGEVACE, ELOCE and CECE the residues were 16, 2 and 3 wt%, respectively. The first difference of residue yields between the two families (phosphorus-containing and -free materials) is explained by the phosphorus action. The second main difference in residues between aliphatic and aromatic compounds is explained by the interaction of phosphorus and aromatic groups, resulting in polyaromatic char.50 The better residue yield for ELOPE compared to CEPE is the result of the high phosphorus ratio in this material (9.2 wt% vs. 4.3 wt%). Such increase of the residue on phosphorus-containing materials clearly points out its FR efficiency in condensed phase, and the high performance of both materials made with DGEVA is likely due to their high aromatic carbon content, and the high cross-linking density.51 Indeed, the aromatic content in CECE is not sufficient to favor the char formation, as the crosslinking density is lower than in DGEVACE.
P–O bond compared to C–C bond. The phosphate groups first decomposed and during this step, they increased the crosslinking carbonization that prevented further decomposition of the epoxy resin. For CEPE and ELOPE, the second degradation step is the aliphatic chain decomposition (Fig. S13†).49 Thus, the residues at 700 °C represented 49 wt% for DGEVAPE, 26 and 23 wt% for ELOPE and CEPE, when for DGEVACE, ELOCE and CECE the residues were 16, 2 and 3 wt%, respectively. The first difference of residue yields between the two families (phosphorus-containing and -free materials) is explained by the phosphorus action. The second main difference in residues between aliphatic and aromatic compounds is explained by the interaction of phosphorus and aromatic groups, resulting in polyaromatic char.50 The better residue yield for ELOPE compared to CEPE is the result of the high phosphorus ratio in this material (9.2 wt% vs. 4.3 wt%). Such increase of the residue on phosphorus-containing materials clearly points out its FR efficiency in condensed phase, and the high performance of both materials made with DGEVA is likely due to their high aromatic carbon content, and the high cross-linking density.51 Indeed, the aromatic content in CECE is not sufficient to favor the char formation, as the crosslinking density is lower than in DGEVACE.
| Material | P ratioa (wt%) |
T
d 5%b (°C) |
T
d maxb (°C) |
Residue (char)b,c (wt%) |
|---|---|---|---|---|
a Determined as:  b Under nitrogen atmosphere – Tdmax read on the TG derivatives.
c At 700 °C.
b Under nitrogen atmosphere – Tdmax read on the TG derivatives.
c At 700 °C.
|
||||
| DGEVACE | 0 | 277 | 403 | 16 |
| DGEVAPE | 6.8 | 234 | 317 | 49 |
| ELOCE | 0 | 239 | 413 | 2 |
| ELOPE | 9.2 | 219 | 280 and 454 | 26 |
| CECE | 0 | 305 | 452 | 3 |
| CEPE | 4.3 | 298 | 351 and 483 | 23 |
Flame retardancy were investigated to evaluate the effect of the phosphate esters crosslinking nodes on this property. Table 4 summarizes the data obtained from PCFC analyses and the heat release rate (HRR) vs. temperature curves are presented in Fig. 6.
 | ||
| Fig. 6 PCFC curves of the phosphate ester vitrimers DGEVAPE, ELOPE, CEPE and their carboxylic ester counterparts DGEVACE, ELOCE and CECE. | ||
| Samples | pHRR (W g−1) | T at pHRR (°C) | THR (kJ g−1) | Residue content (wt%) | EHC (kJ g−1) |
|---|---|---|---|---|---|
| DGEVACE | 364.3 ± 2 | 397 ± 3 | 16.6 ± 0.2 | 9.1 ± 0.6 | 18.3 ± 0.2 |
| DGEVAPE | 107.9 ± 5 | 317 ± 1 | 4.4 ± 0.2 | 49.4 ± 3 | 8.7 ± 0.2 |
| ELOCE | 268.3 ± 10 | 407 ± 8 | 23.5 ± 0.7 | 1.4 ± 0.6 | 23.8 ± 0.6 |
| ELOPE | 125.2 ± 9 | 282 ± 2 | 18.5 ± 0.3 | 20.6 ± 3 | 23.3 ± 0.5 |
| 114.5 ± 8 | 445 ± 3 | ||||
| CECE | 331.3 ± 7 | 451 ± 6 | 28.2 ± 0.8 | 0.0 ± 0.5 | 28.2 ± 0.8 |
| CEPE | 109.3 ± 10 | 346 ± 5 | 24.8 ± 1 | 12.5 ± 3 | 28.3 ± 0.2 |
| 209.6 ± 10 | 479 ± 5 |
The curves (Fig. 6) showed a strong influence of the phosphate on the FR properties. The peak of heat release rate (pHRR), the total heat release (THR), the energy of combustion (EHC) as well as the residue are important parameters to determine the ability of a material to provide flame retardant properties. First, a fuel release at lower temperature was observed for phosphorus-containing materials compared to phosphorus-free ones. It comes from the early stage of decomposition for phosphorus groups which leads to fuel emission at lower temperature. This decomposition released phosphoric species resulting in high level of charring. The pHRR of the DGEVAPE material was 107.9 W g−1, 70% lower than DGEVACE (364.3 W g−1), with a decrease of the associated temperature, highlighting the early decomposition of phosphate species and the charring effect. Moreover, the ELOPE and the CEPE materials exhibited two pHRR. Those results were consistent with the TGA analysis: one or two peaks were observed corresponding to the decomposition steps. Moreover, the first pHRR could be attributed to the phosphoric acid parts decomposition. That is why the first peak of ELOPE exhibited a value of 125.2 W g−1 (slightly higher value due to the phosphorus ratio in the material) and the first peak of CEPE showed a value of 109.3 W g−1 which was in the same range as the DGEVAPE peak. Then, the second pHRR could be mainly ascribed to the decomposition of the aliphatic chains of the network not able to create efficient charring. The effective heat of combustion (EHC) values such as the total heat release (THR) decreased when phosphoric acid was used as crosslinker for the material. The best result was obtained with DGEVAPE because of the absence of aliphatic chains. In fact, these chains are unfavorable to char promotion and contain a high amount of carbon, contributing to heat release. Additionally, the residue contents were consistent with the char content measured by TGA.
Cone calorimetry (Fig. 7) provides important information on the fire behavior of a material.52 The results of cone calorimeter tests are summarized in Table 5 and Fig. 8 exhibits the associated curves. Time to ignition (TTI) measures the time to achieve sustained flaming combustion at a particular external heat flux. All the TTI were higher for the phosphorus-containing materials. Indeed, DGEVACE exhibited a TTI of 77 s whereas DGEVAPE showed a 44% longer TTI of 117 s. Moreover, the TTI of ELOPE and CEPE were 43% and 37% longer than those of ELOCE and CECE, respectively. Despite a lower degradation temperature measured in TGA, phosphate materials exhibit a higher TTI. This may be ascribed to the lower heat of combustion (EHC) of gases released by the decomposition. Indeed, ignition occurs when a critical density of energy is reached, and this density depends not only on the gases concentration but also on their heat of combustion.53 The rate of heat release (HRR) and the total heat release (THR) are important parameters to characterize the fire performance of materials. The introduction of phosphoric acid, a phosphate compound, has a strong influence on the THR due to its ability to act in condensed phase. Indeed, the THR of DGEVAPE, ELOPE and CEPE were 63%, 27% and 50% lower than those of DGEVACE, ELOCE and CECE, respectively. From these differences, it is clear that both phosphorus and aromatics are active in improving properties.
 | ||
| Fig. 7 DGEVACE (reference) (a) before and (b) after cone calorimeter analysis, and DGEVAPE (vitrimer) (c) before and (d) after cone calorimeter analysis. | ||
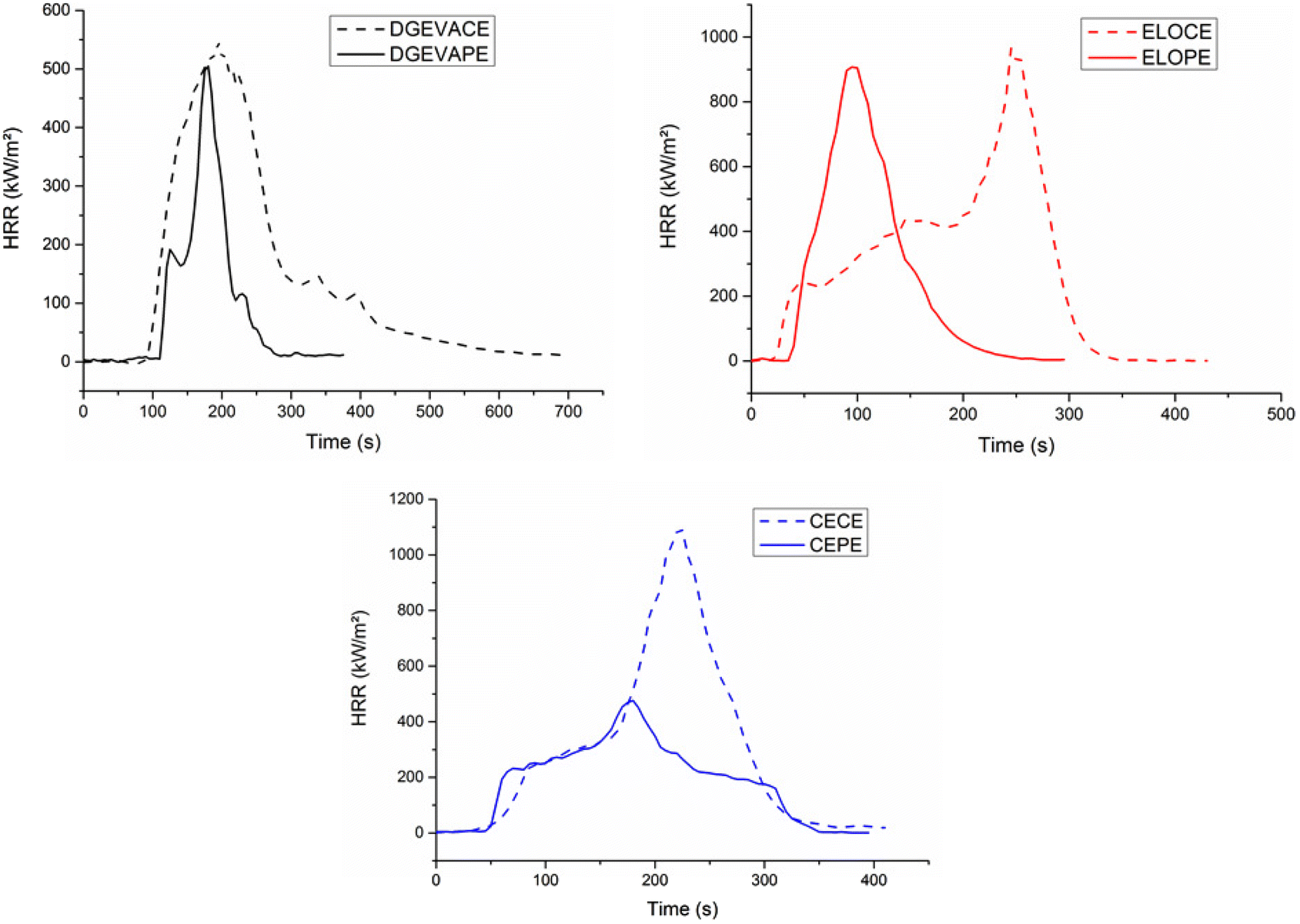 | ||
| Fig. 8 Cone calorimeter curves of the phosphate ester vitrimers DGEVAPE, ELOPE, CEPE and their carboxylic ester counterparts DGEVACE, ELOCE, CECE. | ||
| Samples | TTI (s) | pHRR (kW m−2) | THR (MJ g−1) | Residue content (wt%) | EHC (MJ kg−1) |
|---|---|---|---|---|---|
| DGEVACE | 77 ± 3 | 550 ± 9 | 18.2 ± 0.4 | 2.6 ± 0.3 | 18.7 ± 0.4 |
| DGEVAPE | 117 ± 4 | 503 ± 9 | 6.8 ± 0.2 | 44.0 ± 0.9 | 12.1 ± 0.2 |
| ELOCE | 19 ± 3 | 955 ± 10 | 23.6 ± 0.3 | 0.4 ± 0.2 | 23.7 ± 0.3 |
| ELOPE | 36 ± 3 | 907 ± 10 | 17.3 ± 0.3 | 21.8 ± 0.5 | 22.1 ± 0.4 |
| CECE | 31 ± 3 | 1090 ± 12 | 15.5 ± 0.2 | 0.1 ± 0.1 | 15.5 ± 0.2 |
| CEPE | 49 ± 4 | 477 ± 9 | 7.7 ± 0.4 | 22.6 ± 0.6 | 9.9 ± 0.5 |
The effective heat of combustion (EHC) was also lower for the phosphoric acid-based CANs compared to the citric acid analogs. As expected, the residue contents were higher for the phosphoric acid-containing materials than for the citric acid ones (Fig. 7). This is particularly remarkable in the case of DGEVAPE which residue content reached 44% whereas it was only 2.6% for DGEVACE. The residue content values were consistent with the ones measured by TGA. The decomposition of phosphorus species takes place in the first stage of degradation with the generation of radicals able to initiate effective char entailed insulating effect. Indeed, phosphate functions are known to provide a continuous protective carbon layer due to rearrangement and Diels–Alder reactions.54 This carbon layer prevent heat and gas transfer between the gas and condensed phases.55,56
The pHRR of DGEVAPE and ELOPE was close respectively to their carboxylic ester counterparts DGEVACE and ELOCE. However, the pHRR of CEPE was half lower compared to CECE. An hypothesis which may explain this result relies on the fact that cardanol possesses conjugated double bonds on the alkyl chain, close to an aromatic cycle. In the presence of phosphoric acid, these double bonds favor Diels–Alder reaction responsible for the charring is favored. This charring would protect more efficiently the material against the combustion, which could explain the lower pHRR in the case of cardanol-based materials.
With all the parameters taken into account, the best fire retardancy results were obtained with DGEVAPE and caused by the joint action of the high content of aromatic rings, the high cross-linking ratio, and the presence of phosphate.
Materials from epoxy mixtures
Crosslinked materials prepared from epoxy mixtures were investigated. Four materials were prepared by reacting phosphoric acid with epoxy resin mixtures (Fig. 9, and Table 6). Three materials were prepared from binary mixtures of epoxy: CardVan from NC-514 and DGEVA; VanELO from DGEVA and ELO, and CardELO from NC-514 and ELO. The fourth material, CardVanELO was prepared from an equimolar ternary mixture of NC514, DGEVA and ELO. | ||
| Fig. 9 Pictures of vitrimers from epoxy mixtures. (a) CardELO, (b) CardVAn, (c) VanELO, (d) CardVanELO. | ||
All 4 materials exhibited gel contents over 86% (up to 97% for Van ELO), which confirms that crosslinked materials were obtained. The swelling indices of CardVan and VanELO (189% and 85% respectively) were lower than those of CEPE (219%) and ELOPE (186%), confirming that DGEVA densifies the polymer networks.
All materials exhibited a single Tg, indicating homogeneity and the absence of phase segregation (Fig. S14†). The Tg of CardVan is 29 °C, the presence of DGEVA in the formula does not have a significant effect compared to CEPE (Tg = 25 °C). In VanELO this effect is also observed, as DGEVA is the main component, but the Tg remains low at 28 °C, and closer to ELOPE (20 °C). CardELO has a Tg of 38 °C, comparable to the value for CEPE (25 °C), which was expected as NC-514 is the main component and the difference between the Tg of CEPE and ELOPE is low (25 °C and 12 °C). Finally, the Tg of CardVanELO is 24 °C, which can also be expected as 71% of the formula is composed of NC-514 and ELO giving a low Tg compared to DGEVA (Table 7).
| Sample | T d 5% (°C) | T d 50% (°C) | Residue (char)a,b (wt%) | Swelling indexc (%) | Gel contentc (%) | T g (°C) |
|---|---|---|---|---|---|---|
| a Under nitrogen atmosphere. b At 700 °C. c In THF. | ||||||
| CardVan | 292 | 464 | 41 | 189 ± 18 | 92 ± 2 | 29 |
| VanELO | 262 | 459 | 44 | 85 ± 11 | 97 ± 1 | 28 |
| CardELO | 282 | 458 | 23 | 204 ± 20 | 86 ± 2 | 38 |
| CardVanELO | 281 | 445 | 31 | 141 ± 16 | 88 ± 2 | 24 |
TGA under nitrogen of CardVan revealed that the temperature of 5% weight loss at 292 °C is close to the value of CEPE (298 °C) which is the main component, and higher than the value for DGEVAPE (234 °C). VanELO has a higher Td5% at 262 °C than the two corresponding single-component materials. Interestingly, the residue at 700 °C for CardVan and VanELO are 41 and 44%, close to the value of DGEVAPE (49%) and much higher than the values for CEPE and ELOPE (23% and 26%). Comparatively, the value for CardELO is close to both CEPE and ELOPE, at 23%. As expected, the mixture of DGEVA to other epoxy seems to increase significantly the char content of the materials obtained. CardVanELO exhibits mean properties between the three monoepoxy materials with a Td5% of 281 °C and a residue of 31%.
Stress-relaxation experiments were performed at 150 °C on each of these 4 materials. They all relaxed the stress applied, as their three monoepoxy-derived counterparts (Fig. 10).
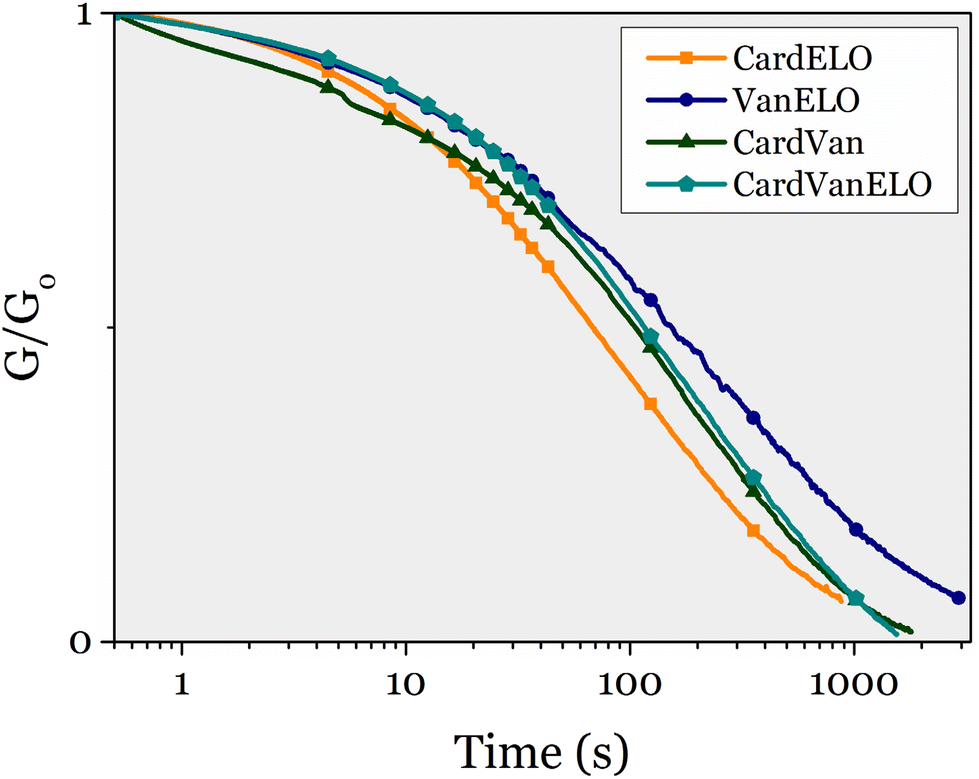 | ||
| Fig. 10 Stress-relaxation experiments on epoxy mixtures vitrimers at 150 °C. | ||
Conclusion
Biobased vitrimers were synthesized from three biobased epoxy monomers derived from vanillin, linseed oil and cardanol. Phosphoric acid was used as the hardener to prepare networks of trifunctional phosphate esters. The reactivity of phosphoric acid towards epoxy was studied on model molecules by 1H and 31P NMR. The kinetics of the different types of epoxy functions were also compared and showed that internal and terminal aliphatic epoxides are respectively 1.4 and 8 times more reactive compared to glycidyl ether epoxides. The material derived from vanillin exhibited a high Tg due to its high aromatic carbon content, contrary to the aliphatic material made from linseed oil and the material prepared from cardanol containing both aromatic cycles and aliphatic chains. Thanks to the transesterification reaction on phosphate esters and the presence of hydroxy groups in the material, the materials exhibited dynamic properties and could be reprocessed by compression molding without any catalyst. However, ELOPE exhibited a sensitivity to moisture and hydrolysis and flowed after several weeks, which could hinder its use for durable applications. DGEVAPE and CEPE appeared much more resistant to moisture, as they did not flow after over a year without any storage precaution. The high phosphate content, but also a high ratio of aromatic groups for the vanillin-based vitrimer, conferred strong flame retardancy properties to the materials, due to the formation of an insulating char layer during the combustion, limiting the heat flow in the materials. The synthesis of such phosphate vitrimers is straightforward and readily scalable, and the DGEVA could be mixed with ELO and NC-514 to improve the flame retardancy properties, while keeping a relatively low Tg. Such materials could find applications in flexible halogen-free fire-retardant polymers.Experimental section
Materials
9,10-Epoxyoctadecane (purity not specified), 1,2-epoxydodecane (≥90%) and phenyl glycidyl ether (≥99%) were purchased from Sigma-Aldrich. DGEVA was obtained from Sherwin-Williams (Eposir VANDGE batch RE1086B) and kindly supplied by DIAM Bouchage SAS. Epoxydized cardanol NC-514 was supplied by Cardolite. Epoxydized linseed oil Merginat ELO was supplied by Hobum Oleochemicals GmbH. Phosphoric acid (85% in H2O), citric acid (99%) and deuterated chloroform (99.8%) were supplied by Sigma-Aldrich. Acetone (≥99.8%) was supplied by Carlo Erba. Deuterated acetone (99.8%) was supplied by Eurisotop.Synthetic procedures
:1 epoxy/OH ratio. The mixtures were stirred manually until they were homogeneous (clear) and then were cast in silicone molds. The mixtures were left overnight under a fume hood to slowly evaporate the acetone and allow the polymerization. The materials were then left 16 h at 60 °C under vacuum in an oven, and then 16 h at 130 °C to evaporate acetone and ensure complete polymerization.
:1 epoxy/OH ratio and the same curing process as the previous materials was applied, one night at 60 °C and one night at 130 °C under vacuum.
Instrumentation
Kinetics study of the epoxy reactivity: 30 to 35 mg of epoxy were dissolved in 0.25 mL of CDCl3. 10 equivalents of dibutyl phosphate were mixed in 0.25 mL of CDCl3. The epoxy and the phosphate solutions were quickly mixed together in a test tube with a vortex, transferred into an NMR tube, which was then placed into the NMR spectrometer as fast as possible, and the NMR data acquisition was started. The disappearance of the peaks associated to the epoxy function was followed with time by 1H NMR.
Kinetics study of the phosphoric acid reactivity: 30 mg of phosphoric acid (85% aqueous solution, 25.5 mg H3PO4, 0.26 mmol, 1 eq.) was mixed with 0.25 mL of deuterated acetone. 1 mL of phenyl glycidyl ether (1.109 g, 7.38 mmol, 28 eq.) was mixed with 0.25 mL of deuterated acetone. The two solutions were quickly mixed together in a test tube with a vortex, transferred into an NMR tube, which was then placed into the NMR spectrometer as fast as possible, and the NMR data acquisition was started. The reaction was followed by 31P NMR.
The swelling index (SI) was calculated using eqn (1), where m2 is the mass of the swollen material and m1 is the initial mass. The reported SI are average values of three samples.
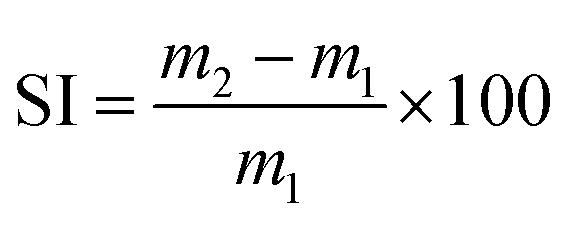 | (1) |
The gel content (GC) was calculated using eqn (2), where m3 is the mass of the dried material and m1 is the initial mass. Reported GC are average values of three samples.
 | (2) |
Pyrolysis combustion flow calorimeter analysis
Fire behavior of vitrimers was analyzed using a pyrolysis combustion flow calorimeter (PCFC). About 3–4 mg were placed in the pyrolyzer, undergoing an increase of temperature from 20 °C to 750 °C at a rate of 1 °C s−1 under a nitrogen flow. Pyrolytic gases were sent to a combustor heated at 900 °C under air flow (N2/O2 = 80/20). At this temperature and with 20% of oxygen, combustion was considered to be complete. HRR was determined according to oxygen depletion (Huggett's relation) as cone calorimeter test. PCFC analyses correspond to anaerobic pyrolysis followed by high temperature oxidation of decomposition products (complete combustion).59 All samples were tested in triplicate. The EHC (effective heat of combustion) is the heat released in relation to the quantity of volatile products between the TTI and the extinction time, and mathematically represents the total heat released divided by the loss of mass in the same time.Cone calorimeter test
Flammability was studied using a cone calorimeter in order to investigate the fire behavior of the samples. The samples dimensions were 100 × 100 × 3 mm3. All the samples exhibited a weight of 45 g ± 2 g. The samples were placed at 2.5 cm below a conic heater and isolated by rock wool. The samples were exposed to a 35 kW m−2 heat flux in well-ventilated conditions (air rate 24 L s−1) in the presence of a spark igniter to force the ignition. Heat release rate (HRR) was determined by oxygen depletion according to Huggett principle (1 kg of consumed oxygen corresponds to 13.1 MJ of heat released).58 Peak of Heat Release Rate (pHRR) is the maximal value of the heat release rate. The total heat released (THR) was obtained by integration of HRR curves. All samples were tested in triplicate.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by the Institut Carnot Chimie Balard CIRIMAT (16CARN000801) and the French National Research Agency ANR (AFCAN project – ANR-19-CE06-0014). FC would like to thank Professor Rinaldo Poli for his help on the mathematical treatment of the H3PO4-epoxy reactivity kinetics study. SC would like to thank Diam Bouchage for the supply of DGEVA. MD and CN would like to thank Dr Rodolphe Sonnier and Loic Dumazert for their help with the cone calorimetry and PCFC analyses.References
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- J. Zheng, Z. M. Png, S. H. Ng, G. X. Tham, E. Ye, S. S. Goh, X. J. Loh and Z. Li, Mater. Today, 2021, 51, 586–625 CrossRef CAS.
- F. Cuminet, S. Caillol, É. Dantras, É. Leclerc and V. Ladmiral, Macromolecules, 2021, 54, 3927–3961 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- X. Liu, L. Liang, M. Lu, X. Song, H. Liu and G. Chen, Polymer, 2020, 210, 123030 CrossRef CAS.
- Y.-X. X. Lu, F. Tournilhac, L. Leibler and Z. Guan, J. Am. Chem. Soc., 2012, 134, 8424–8427 CrossRef CAS PubMed.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- F. Cuminet, D. Berne, S. Lemouzy, E. Dantras, C. Joly-Duhamel, S. Caillol, E. Leclerc and V. Ladmiral, Polym. Chem., 2022, 8, 5255–5446 Search PubMed.
- D. Berne, F. Cuminet, S. Lemouzy, C. Joly-Duhamel, R. Poli, S. Caillol, E. Leclerc and V. Ladmiral, Macromolecules, 2022, 55, 1669–1679 CrossRef CAS.
- M. Delahaye, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2019, 141, 15277–15287 CrossRef CAS PubMed.
- H. Zhang, S. Majumdar, R. A. T. M. van Benthem, R. P. Sijbesma and J. P. A. Heuts, ACS Macro Lett., 2020, 9, 272–277 CrossRef CAS PubMed.
- S. Penczek, J. B. Pretula, K. Kaluzynski and G. Lapienis, Isr. J. Chem., 2012, 52, 306–319 CrossRef CAS.
- Z. Zhou, X. Su, J. Liu and R. Liu, ACS Appl. Polym. Mater., 2020, 2, 5716–5725 CrossRef CAS.
- E. Rossegger, R. Höller, D. Reisinger, M. Fleisch, J. Strasser, V. Wieser, T. Griesser and S. Schlögl, Polymer, 2021, 221, 123631 CrossRef CAS.
- K. Moazzen, E. Rossegger, W. Alabiso, U. Shaukat and S. Schlögl, Macromol. Chem. Phys., 2021, 222, 2100072 CrossRef CAS.
- F. Chen, Q. Cheng, F. Gao, J. Zhong, L. Shen, C. Lin and Y. Lin, Eur. Polym. J., 2021, 147, 110304 CrossRef CAS.
- C. Dertnig, G. Guedes de la Cruz, D. Neshchadin, S. Schlögl and T. Griesser, Angew. Chem., Int. Ed., 2023, 62, e202215525 CrossRef CAS PubMed.
- M. Bergoglio, D. Reisinger, S. Schlögl, T. Griesser and M. Sangermano, Polymers, 2023, 15, 1024 CrossRef CAS PubMed.
- U. Shaukat, E. Rossegger and S. Schlögl, Polymer, 2021, 231, 124110 CrossRef CAS.
- L. Cheng, S. Liu and W. Yu, Polymer, 2021, 222, 123662 CrossRef CAS.
- S. Majumdar, B. Mezari, H. Zhang, J. Van Aart, R. A. T. M. Van Benthem, J. P. A. Heuts and R. P. Sijbesma, Macromolecules, 2021, 54, 7955–7962 CrossRef CAS PubMed.
- J.-H. Lu, Z. Li, J.-H. Chen, S.-L. Li, J.-H. He, S. Gu, B.-W. Liu, L. Chen and Y.-Z. Wang, Research, 2022, 2022, 9846940, DOI:10.34133/2022/9846940.
- X. Feng and G. Li, Chem. Eng. J., 2021, 417, 129132 CrossRef CAS.
- X. Feng and G. Li, ACS Appl. Mater. Interfaces, 2020, 12, 57486–57496 CrossRef CAS PubMed.
- R. Wink, S. Majumdar, R. A. T. M. van Benthem, J. P. A. Heuts and R. P. Sijbesma, Polym. Chem., 2023, 14, 4294–4302 RSC.
- Y. Chen, Y. Zeng, Y. Wu, T. Chen, R. Qiu and W. Liu, ACS Appl. Mater. Interfaces, 2023, 15, 5963–5973 CrossRef CAS PubMed.
- L. Huang, Y. Yang, Z. Niu, R. Wu, W. Fan, Q. Dai, J. He and C. Bai, Macromol. Rapid Commun., 2021, 42, 2100432 CrossRef CAS PubMed.
- Y. Liu, B. Wang, S. Ma, X. Xu, J. Qiu, Q. Li, S. Wang, N. Lu, J. Ye and J. Zhu, Eur. Polym. J., 2021, 144, 110236 CrossRef CAS.
- S. Majumdar, H. Zhang, M. Soleimani, R. A. T. M. van Benthem, J. P. A. Heuts and R. P. Sijbesma, ACS Macro Lett., 2020, 9, 1753–1758 CrossRef CAS PubMed.
- M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol and B. Boutevin, Green Chem., 2014, 16, 1987–1998 RSC.
- E. A. B. da Silva, M. Zabkova, J. D. Araújo, C. A. Cateto, M. F. Barreiro, M. N. Belgacem and A. E. Rodrigues, Chem. Eng. Res. Des., 2009, 87, 1276–1292 CrossRef.
- A. Almena and M. Martín, Ind. Eng. Chem. Res., 2016, 55, 3226–3238 CrossRef CAS.
- R. Auvergne, S. Caillol, G. David, B. Boutevin and J.-P. Pascault, Chem. Rev., 2014, 114, 1082–1115 CrossRef CAS PubMed.
- H. B. W. Patterson, Hydrogenation of Fats and Oils: Theory and Practice, 2nd edn, 2011, pp. 189–278 Search PubMed.
- M. Ionescu, X. Wan, N. Bilić and Z. S. Petrović, J. Polym. Environ., 2012, 20, 647–658 CrossRef CAS.
- W. S. J. Li, F. Cuminet, V. Ladmiral, P. Lacroix-Desmazes, S. Caillol and C. Negrell, Prog. Org. Coat., 2021, 153, 106093 CrossRef CAS.
- C. K. S. Pillai, V. S. Prasad, J. D. Sudha, S. C. Bera and A. R. R. Menon, J. Appl. Polym. Sci., 1990, 41, 2487–2501 CrossRef CAS.
- F. Jaillet, E. Darroman, B. Boutevin and S. Caillol, OCL, 2016, 23, D511 CrossRef.
- S. Caillol, Curr. Opin. Green Sustain. Chem., 2018, 14, 26–32 CrossRef.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- F. Meng, M. O. Saed and E. M. Terentjev, Nat. Commun., 2022, 13, 5753 CrossRef CAS PubMed.
- B. R. Elling and W. R. Dichtel, ACS Cent. Sci., 2020, 14, 53 Search PubMed.
- A. Breuillac, A. Kassalias and R. Nicolaÿ, Macromolecules, 2019, 52, 7102–7113 CrossRef CAS.
- Y. Spiesschaert, C. Taplan, L. Stricker, M. Guerre, J. M. Winne and F. E. Du Prez, Polym. Chem., 2020, 11, 5377–5385 RSC.
- C. Li, S. Zhang and B. Ju, Ind. Crops Prod., 2023, 191, 115941 CrossRef CAS.
- Q. Wang and W. Shi, Polym. Degrad. Stab., 2006, 91, 1289–1294 CrossRef CAS.
- U. Braun, A. I. Balabanovich, B. Schartel, U. Knoll, J. Artner, M. Ciesielski, M. Döring, R. Perez, J. K. W. Sandler, V. Altstädt, T. Hoffmann and D. Pospiech, Polymer, 2006, 47, 8495–8508 CrossRef CAS.
- R. Ménard, C. Negrell, M. Fache, L. Ferry, R. Sonnier and G. David, RSC Adv., 2015, 5, 70856–70867 RSC.
- T. McNally, P. Pötschke, P. Halley, M. Murphy, D. Martin, S. E. J. Bell, G. P. Brennan, D. Bein, P. Lemoine and J. P. Quinn, Polymer, 2005, 46, 8222–8232 CrossRef CAS.
- R. E. Lyon and J. G. Quintiere, Combust. Flame, 2007, 151, 551–559 CrossRef CAS.
- J. Green, J. Fire Sci., 1996, 14, 353–366 CrossRef CAS.
- J. Qi, Q. Wen and W. Zhu, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 392, 032007 Search PubMed.
- S. Hamdani, C. Longuet, D. Perrin, J. M. Lopez-Cuesta and F. Ganachaud, Polym. Degrad. Stab., 2009, 94, 465–495 CrossRef CAS.
- J. G. Dorsey, G. F. Dorsey, A. C. Rutenberg and L. A. Green, Anal. Chem., 1977, 49, 1144–1145 CrossRef CAS.
- F. I. Altuna, V. Pettarin and R. J. J. Williams, Green Chem., 2013, 15, 3360–3366 RSC.
- C. Huggett, Fire Mater., 1980, 4, 61–65 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01328f |
| This journal is © The Royal Society of Chemistry 2024 |