
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The synthesis of polyisoxazoles incorporating fatty acids†
Andrew James
Clark
 *,
Nyle Owen Saul
Jones
and
Abdulrahman
Alhathir
*,
Nyle Owen Saul
Jones
and
Abdulrahman
Alhathir
Chemistry Department, The University of Warwick, Coventry, CV4 7AL, UK. E-mail: A.J.Clark@warwick.ac.uk
First published on 29th March 2024
Abstract
Polyisoxazoles derived from thermal and base-mediated nitrile-N-oxide cycloaddition to fatty amide derived alkynes with up to 44% biobased content are described and their structural and thermal properties reported. Glass transition temperatures, Tg, (ranged from −1.1 °C to 62.0 °C), and measured molecular weights were dependent upon the level of unsaturation in the fatty amide feedstock, the regiochemistry of the dinitrile-N-oxide monomer and whether polymerisation was thermally or base-mediated. For base-mediated processes the solvent played a critical role in controlling the molecular weights of the obtained polymers. MALDI-TOF-MS, infrared and 1H and 13C NMR analysis indicated polymerisation proceeds to give mixtures of linear, cyclic, branched or furoxan incorporated materials.
Introduction
Polytriazole synthesis via click reactions of suitably functionalised azides and alkynes is well documented.1,2 The analogous cycloaddition reactions between alkynes (A, Fig. 1) and suitably functionalized nitrile-N-oxides (B, Fig. 1) have received less attention.3 The uncatalyzed thermal cycloaddition of nitrile-N-oxides with alkynes normally proceeds with high regioselectivity for 3,5-isoxazoles.4 Early work on nitrile-N-oxide cycloaddition polymers was carried out in the 1960's and 1970's.5–7 More recently, Takata et al. have demonstrated a thermal, metal free, mild and high yielding approach to give both polyisoxazoles and polyisoxazolines from hydroximoyl chlorides 1a and 1b and conventional petrochemical based diyne monomers.8–10 Post polymerisation reductive modification of polyisoxazoles allows access to poly-β-aminoalcohols and poly-β-enaminoketones with interesting properties.9 However, the application of this approach toward biobased polymers is less developed. In 2023, a base-mediated thermal cycloaddition of vanillin derived nitrile-N-oxides to alkenes to give the related polyisoxazolines was described (71% biobased).11 In the study a range of biobased polyisoxazoline materials were prepared and used in encapsulating a thermoelectric generator with glass transition temperature ranging between 60–80 °C. The analysis and formation of polyisoxazoline derivatives however is complicated by the formation of diastereomers in each propagation event. Thermal self-dimerization of nitrile-N-oxides to give furoxans 2,12 can also complicate polymerisations and lead to lower molecular weights by removing desired reactive nitrile-N-oxide functional groups thus altering the stoichiometry of the reacting functional groups.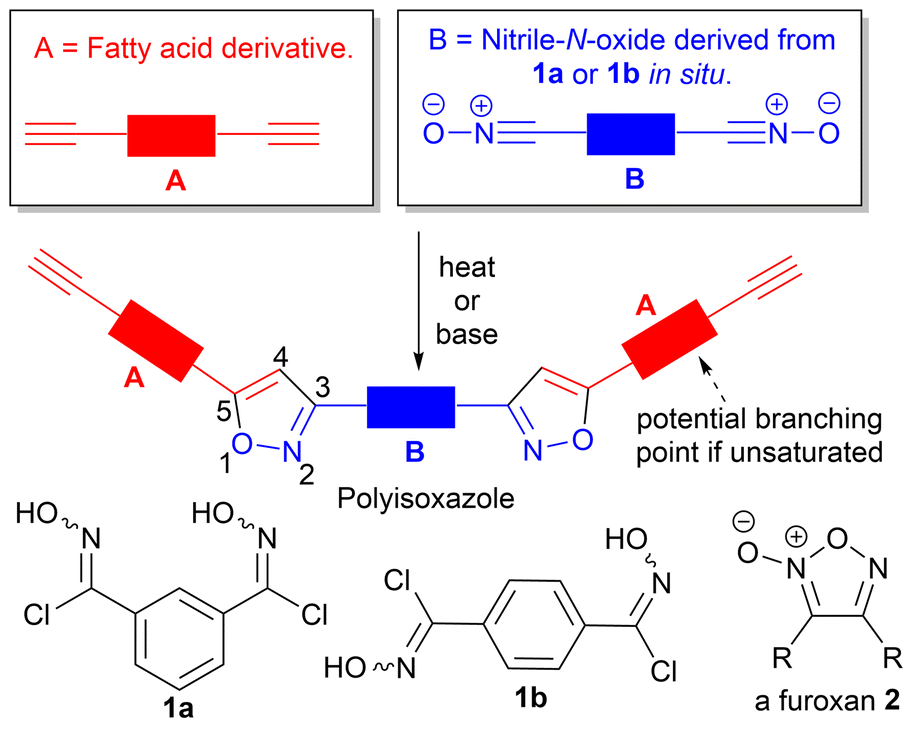 | ||
| Fig. 1 General approach to the synthesis of fatty acid derived polyisoxazoles. | ||
The use of vegetable oils and fatty acid derivatives to produce biobased materials is well established.13,14 While vegetable oil based polytriazoles, derived from azide–alkyne click reactions have been reported,15–17 the corresponding nitrile-N-oxide ‘click’ chemistry of fatty acid derivatives has not been explored. In principle, polyisoxazoles derived from fatty acid derivatives, should present with relatively low Tg's, complementing those prepared from vanillin.11 Incorporation of fatty acid derivatives derived from palmitic (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0), stearic (18:0), oleic (18:1), linoleic (18:2) and linolenic (18:3) acids should allow fine-tuning of polymer properties with unsaturated fatty acid derivatives potentially providing additional cross-linking opportunities via competing isoxazoline formation from their internal alkene groups, Fig. 1. To determine the value of such materials we undertook exploratory work to incorporate a range of different fatty acid derivatives into polyisoxazoles and report the effect on the molecular weight and thermal properties of the polymers produced.
:0), stearic (18:0), oleic (18:1), linoleic (18:2) and linolenic (18:3) acids should allow fine-tuning of polymer properties with unsaturated fatty acid derivatives potentially providing additional cross-linking opportunities via competing isoxazoline formation from their internal alkene groups, Fig. 1. To determine the value of such materials we undertook exploratory work to incorporate a range of different fatty acid derivatives into polyisoxazoles and report the effect on the molecular weight and thermal properties of the polymers produced.
In this study we show that the thermal properties of the fatty amide derived polyisoxazole materials are dependent upon the method of polymerisation, the regiochemistry of the nitrile-N-oxide precursor and the feedstock utilised, and that glass transition temperatures complement those reported for vanillin derived polymers. We use MALDI-TOF-MS to identify the types of species present in the new materials and show that depending upon the reaction conditions used, linear, cyclic, or furoxan incorporated materials can be detected. Attempts to limit competing furoxan formation by conducting the polymerisations at room temperature using base-mediated approaches in the renewable solvent EtOH are described. We also prepared a range of uniform oligomers to provide reference samples for GPC analysis and undertake variable temperature 1H NMR studies to understand barriers to rotation in the oligomers and by extrapolation the polymers.
Experimental
Material
All chemicals and solvents were purchased from Sigma Aldrich and used as received unless otherwise stated. Compounds 1a,81b,82 (R = Ph),18 and 4a–4f,19,20 were prepared according to literature procedures.Characterization
Monomer synthesis
:1 EtOAc:40–60° pet. ether) to give 3 as a pale-yellow liquid (1.4 g, 93%); νmax/cm−1: 3290 (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H), 3245 (C–H), 2978 (C–H), 2119 (CC), 1646 (C
C–H), 3245 (C–H), 2978 (C–H), 2119 (CC), 1646 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1413 (C–N); 1H NMR (500 MHz, CDCl3) δ 4.31 (d, J = 2.0 Hz, 2H, CH2), 4.18 (d, J = 2.0 Hz, 2H, CH2), 2.30 (t, J = 2.0 Hz, 1H, CH), 2.22 (t, J = 2.0 Hz, 1H, CH), 2.18 (s, 3H, CH3); 13C NMR (126 MHz, CDCl3) δ 170.06 (CO), 78.49 (C
O), 1413 (C–N); 1H NMR (500 MHz, CDCl3) δ 4.31 (d, J = 2.0 Hz, 2H, CH2), 4.18 (d, J = 2.0 Hz, 2H, CH2), 2.30 (t, J = 2.0 Hz, 1H, CH), 2.22 (t, J = 2.0 Hz, 1H, CH), 2.18 (s, 3H, CH3); 13C NMR (126 MHz, CDCl3) δ 170.06 (CO), 78.49 (C![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H), 77.79 (CH), 73.09 (CH), 72.40 (CH), 37.08 (CH2), 33.90 (CH2), 21.58 (CH3); m/z (ES+) calcd (C8H9NO + Na+): 158.0576, found: 158.0577 (M + Na+).
H), 77.79 (CH), 73.09 (CH), 72.40 (CH), 37.08 (CH2), 33.90 (CH2), 21.58 (CH3); m/z (ES+) calcd (C8H9NO + Na+): 158.0576, found: 158.0577 (M + Na+).
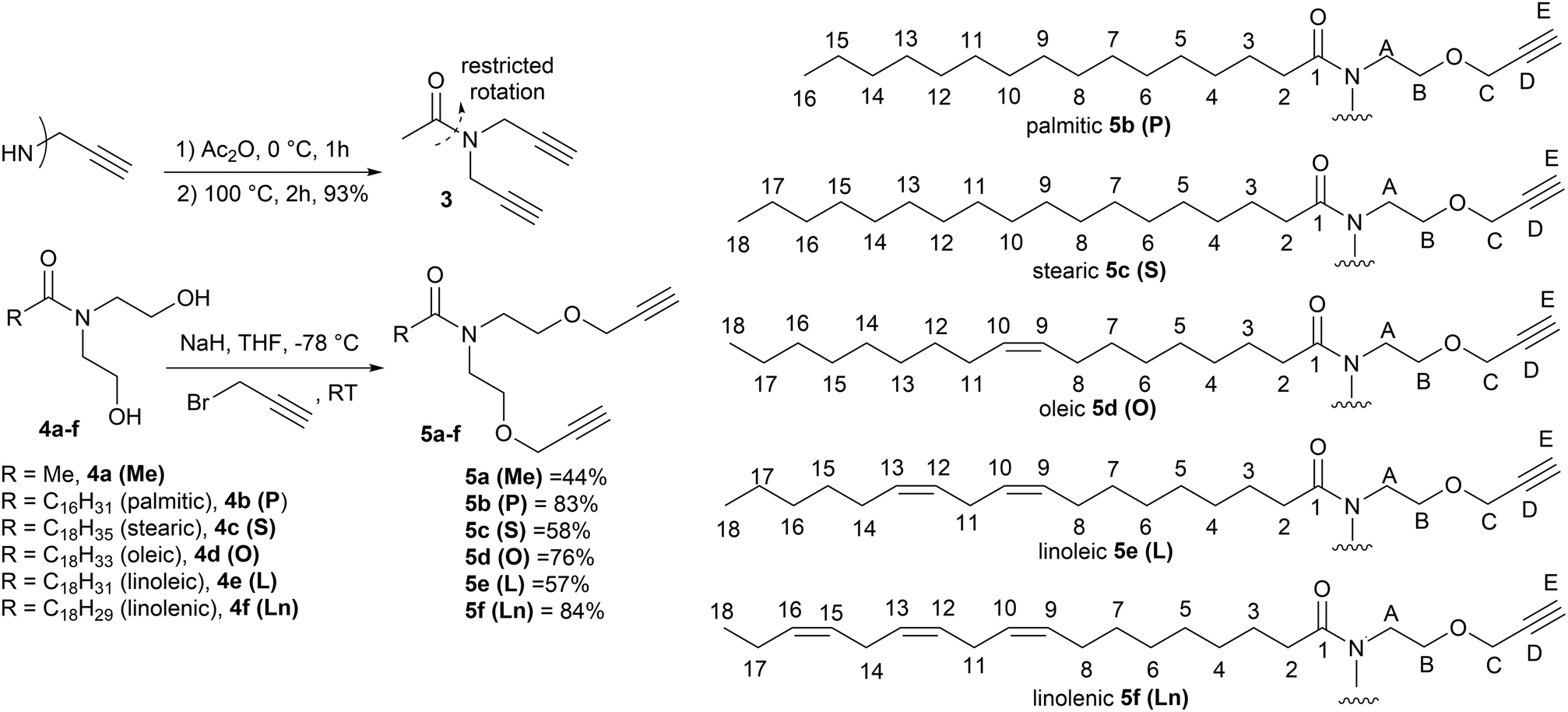 | ||
| Fig. 2 Synthetic route to the monomers used in this study, (3 and 5a–f) and the numbering system used in the NMR assignments in the Experimental section. | ||
The organic layer washed with water (2 × 50 mL) and brine (50 mL), before drying over MgSO4. The solvent was removed in vacuo to leave a crude product. Unsaturated amides 4d–f were purified by column chromatography through a silica plug (EtOAc, Rf = 0.2). Saturated amides 4b–c were purified by recrystallisation from Et2O. Part 2: The amides 4b–f from part 1 and 4a18 (1 eq., 0.5 M in dry THF) were added to NaH (60% in mineral oil, 2.2 eq.) under N2. [For unsaturated compounds the reactions were carried out at −78 °C, saturated compounds were reacted at 0 °C.] After 30 min propargyl bromide (80 wt% in toluene, 2.2 eq.) was added dropwise and the reaction mixture was left to warm to room temperature overnight. Water (10 mL) was added dropwise followed by 2 M HCl (10 mL). The solvent was removed in vacuo and the residue was taken into Et2O (150 mL) and washed with water (2 × 50 mL) and brine (50 mL) before drying over MgSO4. After removal of the solvent in vacuo the products 5a–f were purified through a silica plug (1:4 EtOAc:40–60° pet. ether).
:1 EtOAc:40–60° pet. ether; νmax/cm−1: 3283 (CC–H), 2941 (C–H), 2869 (C–H), 2114 (CC), 1625 (CO), 1419 (C–N), 1098 (C–O); 1H NMR (500 MHz, CDCl3) δ 4.10 (d, J = 2.5 Hz, 2H, HC), 4.08 (d, J = 2.5 Hz, 2H, HC), 3.66–3.58 (m, 4H, HB), 3.58–3.47 (m, 4H, HA), 2.42 (t, J = 2.5 Hz, 1H, HE), 2.39 (t, J = 2.5 Hz, 1H, HE), 2.08 (s, 3H, CH3); 13C NMR (126 MHz, CDCl3) δ 171.33 (CO), 79.63 (CD), 79.32 (CD), 74.88 (CE), 74.56 (CE), 68.65 (CB), 67.88 (CB), 58.49 (CC), 58.29 (CC), 49.70 (CA), 46.16 (CA), 21.79 (CH3); m/z: (ES+) calcd (C12H17NO3 + Na+): 246.1101, found: 246.1102 (M + Na+).
:4 EtOAc:40–60° pet. ether; m.p. 38–40 °C; νmax/cm−1 3306 (C–H), 2916 (C–H), 2849 (C–H), 2114 (CC), 1611 (CO); 1466 (C–H), 1412 (C–N), 1098 (C–O), 1037 (C–O); 1H NMR (500 MHz, CDCl3) δ 4.14 (d, J = 2.0 Hz, 2H, HC), 4.12 (d, J = 2.0 Hz, 2H, HC), 3.70–3.62 (m, 4H, HB), 3.62–3.54 (m, 4H, HA), 2.43 (t, J = 2.0 Hz, 1H, HE), 2.41 (t, J = 2.0 Hz, 1H, HE), 2.38–2.33 (t, J = 7.5 Hz, 2H, H2), 1.70–1.57 (m, 2H, H3), 1.36–1.21 (m, 24H, H4–15), 0.87 (t, J = 7.0 Hz, 3H, H16); 13C NMR (126 MHz, CDCl3) δ 173.94 (CO), 79.80 (CD), 79.45 (CD), 74.90 (CE), 74.54 (CE), 68.87 (CB), 68.17 (CB), 58.63 (CC), 58.41 (CC), 48.91 (CA), 46.52 (CA), 33.25 (C2), 32.06 (C14), 29.91–29.40 (Cchain), 25.47 (C3), 22.83 (C15), 14.27 (C16); m/z: (ES+) calcd (C26H45NO3 + Na+): 442.3292, found: 442.3283 (M + Na+).
:4 EtOAc:40–60° pet. ether; m.p. 41–43 °C; νmax/cm−1 3307 (C–H), 2915 (C–H), 2848 (C–H), 2114 (CC), 1612 (CO), 1466 (C–H), 1414 (C–N), 1099 (C–O), 1037 (C–O); 1H NMR (500 MHz, CDCl3) δ 4.15 (d, J = 2.0 Hz, 2H, HC), 4.12 (d, J = 2.0 Hz, 2H, HC), 3.67 (t, J = 5.0 Hz, 2H, HB), 3.64 (t, J = 5.0 Hz, 2H, HB), 3.61–3.57 (m, 4H, HA), 2.43 (t, J = 2.0 Hz, 1H, HE), 2.41 (t, J = 2.0 Hz, 1H, HE), 2.36 (t, J = 7.5 Hz 2H, H2), 1.65–1.59 (m, 2H, H3), 1.32–1.24 (m, 28H, H4–17), 0.88 (t, J = 7.0 Hz, 3H, H18); 13C NMR (126 MHz, CDCl3) δ 173.93 (CO), 79.81 (CD), 79.45 (CD), 74.90 (CE), 74.54 (CE), 68.88 (CB), 68.18 (CB), 58.63 (CC), 58.41 (CC), 48.91 (CA), 46.52 (CA), 33.25 (C2), 32.07 (C16), 29.93–29.40 (Cchain), 25.47 (C3), 22.84 (C17), 14.27 (C18); m/z: (ES+) calcd (C28H49NO3 + Na+): 470.3605, found: 470.3606 (M + Na+).
:4 EtOAc:40–60° pet. ether νmax/cm−1 3308 (C–H), 3003 (C–H), 2922 (C–H), 2852 (C–H), 2115 (CC), 1639 (CO), 1463 (C–H), 1442 (C–N), 1102 (C–O), 1031 (C–O), 755 (C–H); 1H NMR (500 MHz, CDCl3) δ 5.47–5.27 (m, 2H, H9–10), 4.14 (d, J = 2.0 Hz, 2H, HC), 4.12 (d, J = 2.0 Hz, 2H, HC), 3.72–3.63 (m, 4H, HB), 3.63–3.53 (m, 4H, HA), 2.45 (t, J = 2.0 Hz, 1H, HE), 2.42 (t, J = 2.0 Hz, 1H, HE), 2.36 (t, J = 7.5 Hz 2H, H2), 2.02–1.99 (m, 4H, H8, H11), 1.67–1.58 (m, 2H, H3), 1.31–1.27 (m, 20H, H4–7, H12–17), 0.88 (t, J = 7.0 Hz, 3H, H18); 13C NMR (126 MHz, CDCl3) δ 173.74 (CO), 129.95 (CC), 129.82 (CC), 79.67 (CD), 79.32 (CD), 74.83 (CE), 74.47 (CE), 68.72 (CB), 68.04 (CB), 58.49 (CC), 58.27 (CC), 48.79 (CA), 46.39 (CA), 33.09 (C2), 31.92 (C16), 29.86–29.02 (Cchain), 27.23 (C8/11), 25.29 (C3), 22.70 (C17), 14.14 (C18); m/z: (ES+) calcd (C28H47NO3 + Na+): 468.3448, found: 468.3454 (M + Na+).
:4 EtOAc:40–60° pet. ether; νmax/cm−1 3305 (C–H), 3008 (C–H), 2924 (C–H), 2853 (C–H), 1638 (CO), 1464 (C–H), 1444 (C–N), 1102 (C–O), 1031 (C–O), 755 (C–H); 1H NMR (500 MHz, CDCl3) δ 5.37–5.21 (m, 4H, H9–10, H12–13), 4.09 (d, J = 2.0 Hz, 2H, HC), 4.06 (d, J = 2.0 Hz, 2H, HC), 3.65–3.57 (m, 4H, HB), 3.55–3.51 (m, 4H, HA), 2.71 (t, J = 7.0 Hz, 2H, H11), 2.40 (t, J = 2.0 Hz, 1H, HE), 2.37 (t, J = 2.0 Hz, 1H, HE), 2.31 (t, J = 7.5 Hz, 2H, H2), 2.01–1.97 (m, 4H, H8, H14), 1.60–1.54 (m, 2H, H3), 1.34–1.18 (m, 14H, H4–7, H15–17), 0.83 (t, J = 7.0 Hz, 3H, H18); 13C NMR (126 MHz, CDCl3) δ 173.71 (CO), 130.15 (C13), 130.05 (C12), 127.97 (C10), 127.90 (C19), 79.64 (CD), 79.29 (CD), 74.81 (CE), 74.46 (CE), 68.66 (CB), 67.99 (CB), 58.44 (CC), 58.22 (CC), 48.75 (CA), 46.35 (CA), 33.05 (C2), 31.51 (C16), 29.64 (C7), 29.40 (C15/4), 29.33 (C5), 29.19 (C6), 27.22 (C14), 27.19 (C8), 25.62 (C11), 25.28 (C3), 22.57 (C17), 14.08 (C18); m/z: (ES+) calcd (C28H45NO3 + Na+): 466.3292, found: 466.3299 (M + Na+).
:4 EtOAc:40–60° pet. ether; νmax/cm−1 3296 (C–H), 3009 (C–H), 2926 (C–H), 2854 (C–H), 2115 (CC), 1639 (CO), 1463 (C–H), 1443 (C–N), 1100 (C–O), 1031 (C–O), 666 (C–H); 1H NMR (500 MHz, CDCl3) δ 5.47–5.26 (m, 6H, H9–10, H12–13, H15–16), 4.13 (d, J = 2.0 Hz, 2H, HC), 4.11 (d, J = 2.0 Hz, 2H, HC), 3.67 (t, J = 5.5 Hz, 2H, HB), 3.63 (t, J = 5.5 Hz, 2H, HB), 3.60–3.56 (m, 4H, HA), 2.87–2.74 (m, 4H, H11, H14), 2.43 (t, J = 2.0 Hz, 1H, HE), 2.40 (t, J = 2.0 Hz, 1H, HE), 2.36 (t, J = 7.5 Hz, 2H, H2), 2.08–2.02 (m, 4H, H8, H17), 1.76–1.45 (m, 2H, H3), 1.39–1.22 (m, 8H, H4–7), 0.96 (t, J = 7.5 Hz, 3H, H18); 13C NMR (101 MHz, CDCl3) δ 175.85 (CO), 132.07 (CC), 130.44 (CC), 128.39 (CC), 128.38 (CC), 127.80 (CC), 127.24 (CC), 79.77 (CD), 79.42 (CD), 74.89 (CE), 74.53 (CE), 68.83 (CB), 68.13 (CB), 58.60 (CC), 58.38 (CC), 48.88 (CA), 46.48 (CA), 33.20 (C2), 29.75 (C7), 29.53 (C4/5), 29.32 (C6), 27.35 (C8), 25.73 (C11), 25.64 (C14), 25.41 (C3), 20.66 (C17), 14.40 (C18); m/z: (ES+) calcd (C28H43NO3 + Na+): 464.3135, found: 464.3145 (M + Na+).
Polymer synthesis
 | ||
| Fig. 3 Numbering system used in the NMR assignments in the Experimental section. | ||
C–H), 2961 (C–H), 1651 (CO), 1603 (CO), 1406 (C–N), 901 (C–H); 1H NMR (500 MHz, CDCl3) δ 8.04 (brs, H6′), 7.77 (brs, H7′), 7.50 (brs, H8′), 6.67–6.39 (brm, H3′), 4.91–4.70 (brm, H1′), 4.34 (brs, HA-min), 4.18 (brs, HA-maj), 2.36 (brs, HB-maj), 2.27 (brs, HB-min), 2.25 (s, CH3); 13C NMR (126 MHz, CDCl3) δ 170.89 (CO), 168.98 (C2′), 168.06 (C2′), 162.11 (C4′), 129.78 (C8′), 129.48 (C5′), 129.17 (C5′), 128.67 (C7′), 128.38 (C7′), 125.25 (C6′), 125.21 (C6′), 101.67 (C3′), 101.46 (C3′), 101.18 (C3′), 100.93 (C3′), 73.74 (CC), 73.26 (CC), 44.89 (C1′), 43.39 (C1′), 41.15 (C1′), 40.83 (C1′), 38.85 (CA), 38.81 (CA), 34.66 (CA), 21.69 (CMe), 21.59 (CMe). (Mn = 4.4k, Mw = 6.5k, Đ = 1.5.)
O), 1613 (CO), 1433 (C–N), 1099 (C–O), 908 (C–H); 1H NMR (500 MHz, CDCl3) δ 8.48 (s, minor signal for 3,4-isoxazole), 8.18 (brs, H7′), 7.84 (brs, H9′), 7.45 (brs, H10′), 6.60 (brs, H5′), 4.62 (brs, H3′), 4.13 (d, J = 2.0 Hz, HC), 4.11 (d, J = 2.0 Hz, HC), 3.72 (brs, H2′), 3.61 (brs, H1′/A/B), 2.43 (t, J = 2.0 Hz, HE), 2.41 (t, J = 2.0 Hz, HE), 2.15 (brs, CH3); 13C NMR (126 MHz, CDCl3) δ 171.97–171.45 (CO), 169.89 (C4′), 169.82 (C4′), 161.96 (C6′), 161.91 (C6′), 129.80 (C9′), 129.69 (C7′), 128.45 (C8′), 125.30 (C10′), 101.45 (C5′), 101.35 (C5′), 75.05 (CE), 74.72 (CE), 69.88 (C2′), 69.46 (C2′), 68.85 (CB), 75.05 (CE), 74.72 (CE), 69.88 (C2′), 69.46 (C2′), 68.85 (CB), 68.05 (CB), 64.21 (C3′), 63.92 (C3′), 58.62 (CC), 58.44 (CC), 50.34–49.82 (CA/1′), 46.62 (CA/1′), 46.43 (CA/1′), 21.97 (CMe). (Mn = 5.9k, Mw = 8.7k, Đ = 1.5.)
O), 1464 (C–H), 1437 (C–N), 1157 (C–O), 1102 (C–O), 912 (C–H); 1H NMR (500 MHz, CDCl3) δ 8.20 (brs, H8′), 7.85 (brs, H9′), 7.53–7.50 (m, H10′), 6.65–6.54 (brm, H5′), 4.76–4.52 (brm, H3′), 4.13 (d, J = 2.0 Hz, HC), 4.11 (d, J = 2.0 Hz, HC), 3.78–3.54 (m, H1′, H2′, HA, HB), 2.43 (t, J = 2.0, HE), 2.40 (t, J = 2.0, HE), 2.39–2.30 (m, H2), 1.65–1.51 (m, H3), 1.33–1.13 (brm, H4–15), 0.86 (brt, J = 6.5 Hz, H16); 13C NMR (126 MHz, CDCl3) δ 173.97 (C1), 169.93 (C4′), 169.81 (C4′), 161.93 (C6′), 161.87 (C6′), 129.82 (C7′), 129.80–129.73 (C10′), 129.71 (C7′), 128.43 (C9′), 125.32–125.18 (C8′), 101.39–101.23 (C5′), 79.74 (CD), 79.38 (CD), 74.99 (CE), 74.62 (CE), 70.04–69.83 (C2′), 69.69–69.54 (C2′), 68.91 (CB), 68.21 (CB), 64.35–64.14 (C3′), 64.04–63.81 (C3′), 58.60 (CC), 58.41 (CC), 49.23–48.81 (CA/1′), 46.92–46.50 (CA/1′), 33.44–33.19 (C2), 32.03 (C14), 29.99–29.17 (C4–13), 25.42 (C3), 22.80 (C15), 14.24 (C16). (Mn = 4.8k, Mw = 11.9k, Đ = 2.5.)
O), 1464 (C–H), 1438 (C–N), 1157 (C–O), 1102 (C–O), 913 (C–H); 1H NMR (500 MHz, CDCl3) δ 8.21 (brs, H8′), 7.85 (brs, H9′), 7.58–7.43 (m, H10′), 6.66–6.54 (brm, H5′), 4.67–4.56 (brm, H3′), 4.13 (d, J = 2.0 Hz, HC), 4.11 (d, J = 2.0 Hz, HC), 3.77–3.56 (m, H1′, H2′, HA, HB), 2.43 (t, J = 2.0, HE), 2.40 (t, J = 2.0, HE), 2.39–2.32 (m, H2), 1.66–1.54 (m, H3), 1.36–1.08 (brm, H4–17), 0.86 (brt, J = 6.5 Hz, H18); 13C NMR (126 MHz, CDCl3) δ 173.98 (C1), 169.94 (C4′), 169.81 (C4′), 161.93 (C6′), 161.87 (C6′), 129.82 (C7′), 129.81–129.73 (C10′), 129.72 (C7′), 128.43 (C9′), 125.34–125.19 (C8′), 101.43–101.20 (C5′), 79.74 (CD), 79.39 (CD), 74.99 (CE), 74.62 (CE), 70.04–69.86 (C2′), 69.71–69.52 (C2′), 68.92 (CB), 68.21 (CB), 64.32–64.14 (C3′), 64.02–63.81 (C3′), 58.61 (CC), 58.41 (CC), 49.19–48.90 (CA/1′), 46.88–46.55 (CA/1′), 33.42–33.18 (C2), 32.04 (C16), 29.92–29.32 (C4–15), 25.42 (C3), 22.81 (C17), 14.25 (C18). (Mn = 48k, Mw = 12.7k, Đ = 2.7.)
C–H), 2923 (C–H), 2853 (C–H), 1637 (CO), 1462 (C–H), 1438 (C–N), 1159 (C–O), 1102 (C–O), 912 (C–H); 1H NMR (500 MHz, CDCl3) δ 8.21 (brs, H8′), 7.85 (brs, H9′), 7.58–7.44 (brm, H10′), 6.65–6.55 (brm, H5′), 5.30 (brs, H9/10), 4.71–4.52 (brm, H3′), 4.49–4.44 (m, HCH2-min/crosslinked), 4.14 (brs, HC), 4.10 (brs, HC), 3.78–3.54 (m, H1′, H2′, HA, HB), 2.43 (brs, HE), 2.40 (br, HE), 2.39–2.30 (m, H2), 2.03–1.90 (brm, H9/11), 1.60 (brs, H3), 1.25 (brs, H4–7/12–17), 0.86 (brs, H18); 13C NMR (126 MHz, CDCl3) δ 173.97 (C1), 169.94 (C4′), 169.81 (C4′), 161.94 (C6′), 161.88 (C6′), 130.05 (C10), 129.90 (C9), 129.72 (C7′), 129.83–129.74 (C10′), 129.72 (C7′), 128.45 (C9′), 125.35–125.16 (C8′), 101.35 (C5′), 75.02 (CE), 74.66 (CE), 70.06–69.81 (C2′), 69.71–69.48 (C2′), 68.91 (CB), 68.20 (CB), 64.32–64.08 (C3′), 63.98–63.81 (C3′), 58.61 (CC), 58.42 (CC), 49.19–48.87 (CA/1′), 46.88–46.55 (CA/1′), 33.37–33.19 (C2), 32.02 (C16), 29.96–29.12 (C4–7/12–15), 27.39–27.27 (C11), 25.41 (C3), 22.80 (C17), 14.25 (C18). (Mn = 5.6k, Mw = 19.4k, Đ = 3.4.)
C), 3007 (C–H), 2925 (C–H), 2853 (C–H), 1636 (CO), 1463 (C–H), 1437 (C–N), 1158 (C–O), 1101 (C–O), 912 (C–H); 1H NMR (500 MHz, CDCl3) δ 8.21 (brs, H8′), 7.85 (brs, H9′), 7.59–7.41 (brm, H10′), 6.64–6.51 (brm, H5′), 5.40–5.20 (brm, H9/10/12/13), 4.68–4.56 (brm, H3′), 4.49–4.44 (m, HCH2-min/crosslinked), 4.13 (brs, HC), 4.11 (brs, HC), 3.78–3.53 (m, H1′, H2′, HA, HB), 2.79–2.69 (brm, H11), 2.51 (brs, Hcross-linked), 2.43 (brs, HE), 2.40 (br, HE), 2.39–2.29 (m, H2), 2.07–1.95 (brm, H8/14), 1.60 (brs, H3), 1.27 (brs, H4–7/15–17), 0.87 (brs, H18); 13C NMR (126 MHz, CDCl3) δ 173.93 (C1), 170.37–169.69 (C4′), 162.04–161.84 (C6′), 130.33 (C13), 130.20 (C9), 129.84–129.74 (C10′), 129.71 (C7′), 128.44 (C9′), 128.09 (C10), 128.02 (C12), 125.32–125.18 (C8′), 101.33 (C5′), 85.47 (Ccross-linked), 79.73 (CD), 79.37 (CD), 74.99 (CE), 74.63 (CE), 70.04–69.78 (C2′), 69.73–69.49 (C2′), 68.90 (CB), 68.19 (CB), 64.35–64.10 (C3′), 64.02–63.80 (C3′), 58.60 (CC), 58.40 (CC), 49.15–48.82 (CA/1′), 46.86–46.48 (CA/1′), 33.37–33.09 (C2), 31.61 (C16), 29.88–29.18 (C4–7/15), 27.35–27.27 (C8/14), 25.73 (C11), 25.38 (C3), 22.68 (C17), 14.19 (C18). (Mn = 6.3k, Mw = 33.6k, Đ = 5.3.)
C), 3008 (C–H), 2925 (C–H), 2854 (C–H), 1635 (CO), 1461 (C–H), 1438 (C–N), 1159 (C–O), 1101 (C–O), 912 (C–H); 1H NMR (500 MHz, CDCl3) δ 8.21 (brs, H8′), 7.85 (brs, H9′), 7.58–7.42 (brm, H10′), 6.67–6.53 (brm, H5′), 5.43–5.23 (brm, H9/10/12/13/15/16), 4.73–4.50 (brm, H3′), 4.50–4.45 (m, HCH2-min/cross-linked), 4.13 (brs, HC), 4.11 (brs, HC), 3.72–3.52 (brm, H1′, H2′, HA, HB), 2.78 (brs, H11/14), 2.57 (brs, Hcross-linked), 2.43 (brs, HE), 2.40 (br, HE), 2.39–2.25 (m, H2), 2.08–1.95 (brm, H8/17), 1.60 (brs, H3), 1.37–1.16 (brm, H4–7), 0.96 (brs, H18); 13C NMR (126 MHz, CDCl3) δ 173.95 (C1), 170.27–169.70 (C4′), 162.09–161.76 (C6′), 132.09 (C9/10/12/13/15/16), 130.44 (C9/10/12/13/15/16), 129.85–129.75 (C10′), 128.55–128.43 (C9′), 128.41 (C9/10/12/13/15/16), 128.39 (C9/10/12/13/15/16), 127.83–127.75 (C9/10/12/13/15/16), 127.24 (C9/10/12/13/15/16), 125.33–125.17 (C8′), 101.34 (C5′), 85.39 (Ccross-linked), 79.74 (CD), 79.39 (CD), 75.04 (CE), 74.66 (CE), 70.04–69.79 (C2′), 69.66–69.47 (C2′), 68.91 (CB), 68.21 (CB), 64.34–64.13 (C3′), 64.02–63.80 (C3′), 58.62 (CC), 58.42 (CC), 49.18–48.83 (CA/1′), 46.90–46.54 (CA/1′), 33.38–33.17 (C2), 29.87–29.20 (C4–7), 27.35 (C8), 25.74 (C11), 25.65 (C14), 25.40 (C3), 20.68 (C17), 14.42 (C18). (Mn = 4.3k, Mw = 16.5k, Đ = 3.9.)
O), 1592 (CN), 1573 (CN), 1101 (C–O); 1H NMR (500 MHz, CDCl3) δ 7.85 (s, H8′), 6.56–6.52 (m, H5′), 4.63 (d, J = 2.0 Hz, H3′), 4.12 (d, J = 2.0 Hz, HC), 3.67 (m, H1′, H2′, HA, HB), 2.42–2.40 (m, HE), 2.36–2.30 (m, H2), 1.63–1.52 (m, H3), 1.34–1.03 (m, H4–17), 4.12 (brt, J = 6.8 Hz, H18); 13C NMR (126 MHz, CDCl3) δ 173.86 (C1), 169.79 (C4′), 169.69 (C4′), 161.71 (C1), 161.65 (C6′), 130.79 (C7′), 127.34 (C8′), 101.17 (C5′), 101.08 (C5′), 79.67 (CE), 79.27 (CE), 74.86 (CD), 74.49 (CD), 69.81 (C2′), 69.51 (C2′), 68.72 (CB), 68.04 (CB), 64.10 (C3′), 63.77 (C3′), 58.49 (CC), 58.27 (CC), 48.94 (C1′/A), 48.77 (C1′/A), 46.62 (C1′/A), 46.37 (C1′/A), 33.19 (C2), 31.92 (C16), 29.71 (C4–15), 29.67 (C4–15), 29.57 (C4–15), 29.51 (C4–15), 29.37 (C4–15), 25.31 (C3), 22.69 (C17), 14.13 (C18); (Mn = 4.9k, Mw = 25.3k, Đ = 5.9).
Results and discussion
Isoxazole polymers are normally prepared by the reaction of ester or ether derived alkyne monomers and aromatic derived nitrile-N-oxides, (e.g., derived from thermal elimination of HCl from 1a–b).8 Few examples of reactions with amide derived alkyne monomers have been reported. Due to restricted rotation around the amide bond the analysis, structure determination and conformational space of the products is often not straightforward. A further issue is that conventional polystyrene or PMMA standards in GPC analysis can lead to inaccurate measures of molecular weights. Consequently, our initial investigations focused on the polymerization of the non-bioderived model propargyl amide 3 and propargyl ether 5a where the potential fatty acid chain was replaced with a methyl group to facilitate analysis. Monomer 3 was prepared in 93% yield from the acetylation of dipropargyl amine with acetic anhydride at 100 °C, while the propargyl ether 5a was prepared in 44% yield by NaH deprotonation of the known diol214a at −78 °C followed by reaction with two equivalents of propargyl bromide at room temperature, Fig. 2. As expected, both compounds exhibited two sets of sharp signals for the two inequivalent alkyne groups in both the 1H 500 MHz and 13C 126 MHz NMR. Variable temperature analysis (1H) of 5a indicated a barrier to rotation around the amide bond, of ΔG = 75.7 ± 1.3 kJ mol−1 which is identical to that reported for 4a (75.7 kJ mol−1).21Polyisoxazole formation has mainly been undertaken using thermal polymerisation between 80–100 °C and so thermal analysis (TGA) of both monomers (heating at 10 °C min−1 from 25–600 °C under N2) was undertaken to determine if they exhibited suitable stability for further reactions (3, T5% = 133 °C, T20% = 170 °C, 5a, T5% = 240 °C, T20% = 285 °C where T5% and T20% are the temperature for 5% and 20% mass loss respectively), see ESI.† Although the less flexible monomer 3 showed lower stability, both were suitable for further study. Reacting an appropriate excess of either 3 or 5a (dipolarophile, monomer A) with nitrile-N-oxide precursor 1a (monomer B) using the procedure of Takata8 (80 °C, DMF, 4 Å MS, 24 h) produced authentic small oligomer samples of (AB)1A type (e.g. O13 = (3–1a–3), O15a = (5a–1a–5a)) and (AB)2A type (O23 = (3–1a–3–1a–3), O25a = (5a–1a–5a–1a–5a)) with alkyne end groups for 1H NMR and GPC analysis, Fig. 4a. In addition, a 1:1 stoichiometry of 3 or 5a with 1a under the same reaction conditions was used to prepare the polymers p(1a-co-3) and p(1a-co-5a) respectively. After precipitation from MeOH the crude polymers were further washed with MeOH to remove the lower molecular weight materials. Initial analysis of the 1H NMR of the reactions of the propargyl ether 5a indicates that the major mode of cycloaddition proceeds to give the expected 3,5-isoxazole product, Fig. 4b, although traces (<5%) of the alternative 3,4-regioisomer (8.49 ppm and 4.50 ppm) were detected, Fig. 4d. Relative integration of the H3′ signals adjacent to the isoxazole moiety with the HC signals of the end group allowed information on the degree of polymerisation to be determined, (p(1a-co-3) DP = 16, p(1a-co-5a) DP = 10). For the oligomer O15a at room temperature, the 1H NMR spectrum showed two singlets for the acetyl group (0.55:0.45), while four distinct singlet chemical environments were observed for the isoxazole H5′ proton, indicating a relatively complicated conformational analysis for such a simple molecule, Fig. 4c.
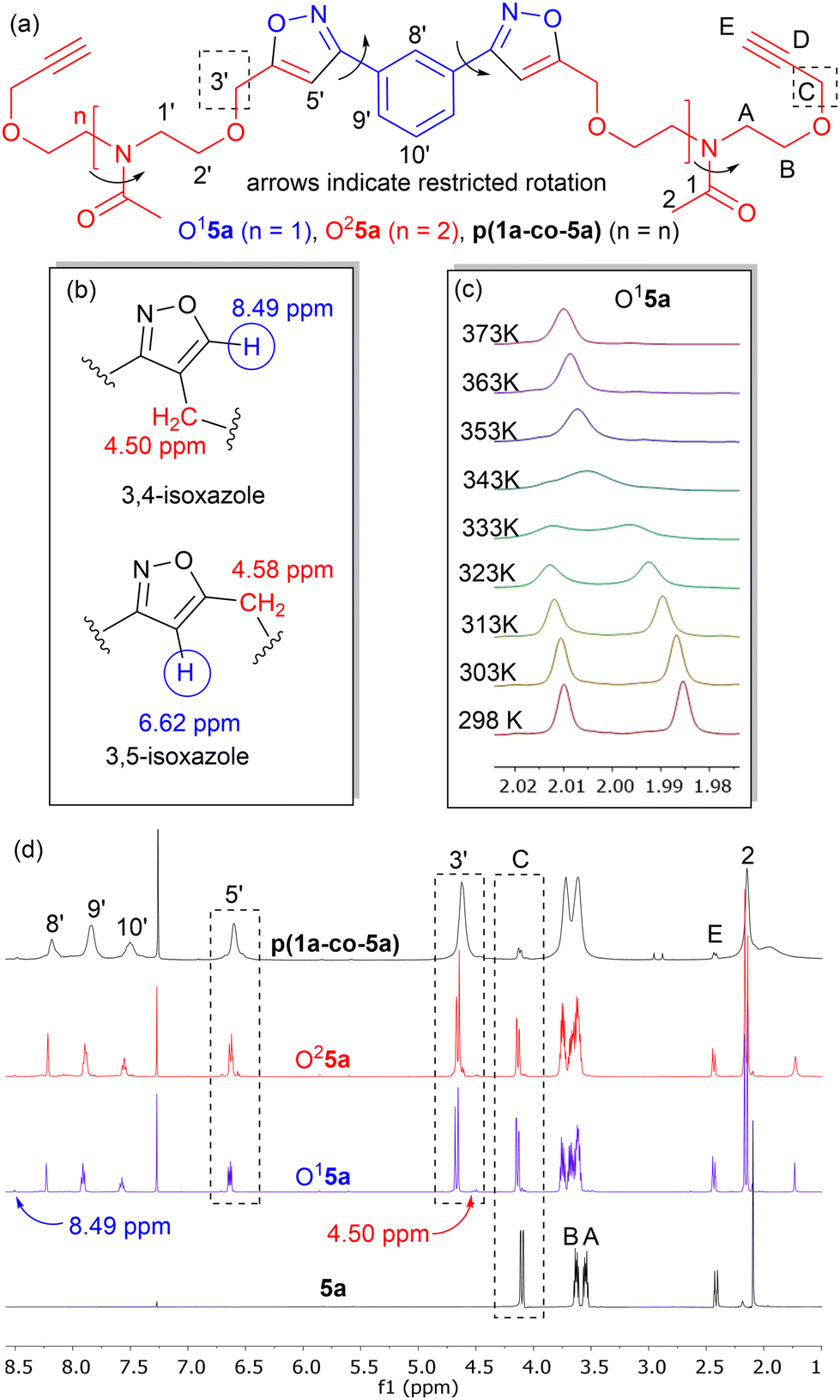 | ||
| Fig. 4 (a) Structures of oligomers, O15a and O25a, and polymer p(1a-co-5a) with the numbering system used in this paper; (b) the 500 MHz 1H NMR values for 3,4- and 3.5-isoxazoles in O15a; (c) the 500 MHz 1H variable temperature NMR of oligomer O15a between 298–373 K showing Tc = 343 K; (d) stacked plot of 500 MHz 1H NMR spectrum of monomer 5a, the two oligomers O15a and O25a, and the polymer p(1a-co-5a) showing (i) minor amounts of 3,4-isoxazole at 8.49 ppm and 4.50 ppm, (ii) the position of signals representative of polymer chains (3′) and end groups (C) with the relative integration providing the degree of polymerisation, (iii) the positions and assignments of key signals. | ||
Restricted rotation around the two amides, as well as the C–C aryl-isoxazole bonds of the meta substituted aromatic ring needs to be considered, Fig. 4a. Variable temperature 1H NMR of O15a in d6-DMSO provided an estimated ΔG barrier to rotation around the amide functionality of 75.1 ± 1.3 kJ mol−1, (Tc = 343 K), which is similar to that observed in the monomer. The four environments for the isoxazole protons reduced to two at elevated temperature but coalescence was not observed at 373 K. Interestingly, the more compact oligomer O13 (based upon the reaction of 2 equivalents of 3 with 1a) exhibited a lower barrier to rotation of ΔG = 72.1 ± 1.3 kJ mol−1.
The GPC results for the reactions of propargyl ether 5a are shown in Fig. 5a. Authentic samples of the monomer 5a and the two (AB)nA type oligomers O15a (ABA) and O25a ((AB)2A) were used as references to confirm polymerisation. GPC data for both polymers confirmed that washing the crude polymers with MeOH (p′(1a-co-5a)) facilitated removal of lower molecular weight oligomers and unreacted starting materials, Fig. 5a.
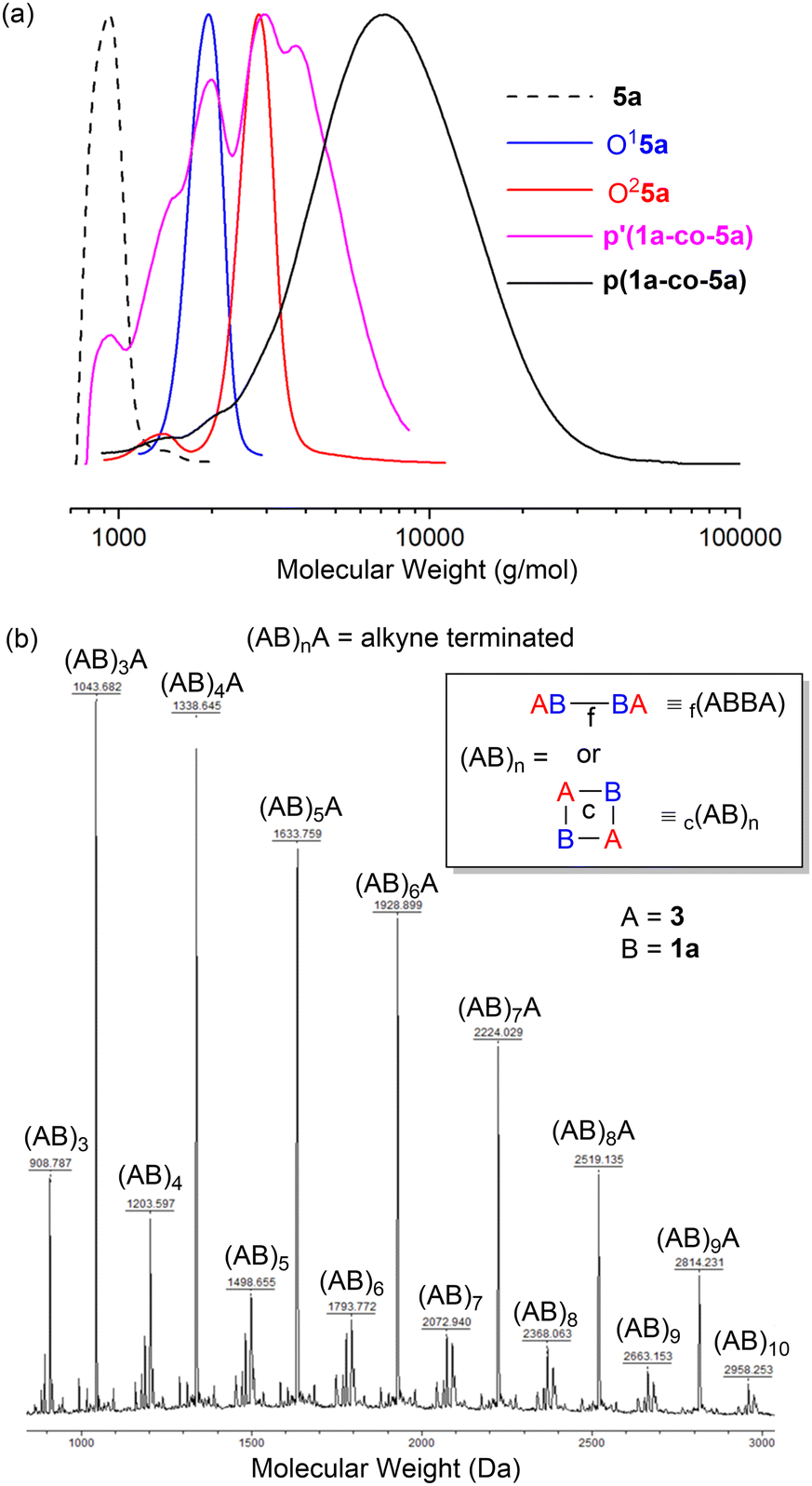 | ||
| Fig. 5 (a) GPC in DMF of propargyl ether monomer 5a, oligomers O15a and O25a, MeOH wash of p(1a-co-5a) and polymer p(1a-co-5a); (b) MALDT-TOF-MS of P6 between 900–3000 Da using DCTB as matrix showing a repeat unit of 295 Da for two species (AB)nA and (AB)n type. | ||
Trace amounts of lower molecular weight species were detected in the GPC of the polymers themselves. p(1a-co-5a), Mn = 5.9k, Mw = 8.7k, Đ = 1.5, repeat unit = 383 Da, p(1a-co-3), Mn = 4.4k, Mw = 6.5k, Đ = 1.5, repeat unit = 295 Da.
Insight into the nature of the linkages present in the polymers was investigated using MALDI-TOF-MS which indicated several species with the correct repeating unit for both polymers. The MALDI-TOF-MS for p(1a-co-3) is shown as an example and shows the expected linear alkyne terminated (AB)nA series (monomer A = alkyne 3 and monomer B = nitrile-N-oxide derived from 1a, the linear polymer is terminated with an alkyne end group). No termination of the polymer with an oximoyl chloride (CN(OH)Cl) end group was detected, indicating HCl elimination must be complete under the reaction conditions. One series was assigned as (AB)n type polymers. This is unlikely to be an ABAB type oligomer terminated with one end group of an alkyne (monomer A) and the other a nitrile-N-oxide (monomer B). While this is the norm for step-growth polymerisation, the nitrile-N-oxide itself is not normally a stable functional group at elevated temperature instead undergoing dimerization to give either 1,4,2,5-dioxadiazines or furoxans.22 A few poorly stable nitrile-N-oxides are known at room temperature, but they are restricted to inherently sterically encumbered cases (e.g. 4-substituted-2,6-dimethylbenzonitrile oxide).23,24 It is very unlikely that the polymers contain nitrile-N-oxide end groups due to the high temperature of the reaction and the lack of an intense peak in the IR spectrum at around 2200–2300 cm−1. Consequently, the (AB)n type polymers may be tentatively assigned as either (a) a cyclic species, c(AB)n, with no end groups, or (b) a species where any growing chain nitrile-N-oxide undergoes dimerization to give a furoxan of type 2, f(ABBA)n, with alkyne end groups, Fig. 5b. No characteristic signals of aryl furoxans were detected in the infrared spectrum of either p(1a-co-3) or p(1a-co-5a), (namely the characteristic peaks at 1590 cm−1, 1572 cm−1 and 1503 cm−1), suggestive that the (AB)n species are cyclic in nature, for infrared spectra of polymers see ESI.†
Both polymers p(1a-co-3) and p(1a-co-5a) exhibited similar thermal stability: p(1a-co-3) (T5% = 251 °C, T20% = 349 °C), and p(1a-co-5a) (T5% = 249 °C, T20% = 339 °C). These values were significantly higher than those of their respective monomers: p(1a-co-3) (T5% = +116 °C, T20% = +179 °C), and p(1a-co-5a) (T5% = +9 °C, T20% = +54 °C). As expected, the more flexible polymer p(1a-co-5a) exhibited a lower glass transition temperature (Tg = 51.0 °C) compared to p(1a-co-3) (Tg = 112.0 °C). Given that the primary focus of this work was to prepare biobased polyisoxazoles with lower Tg's than vanillin derived materials11 (ranging from 60–80 °C), our next step concentrated on incorporating fatty acid derivatives using the more flexible ethanolamine derived chains.
Biobased polymers from fatty amides
Polymerisation of the saturated monomers derived from palmitic 5b(P) and stearic 5c(S) acids were expected to behave similarly to 5a. However, the unsaturated analogues based upon oleic 5d(O), linoleic 5e(L) and linolenic 5f(Ln) acids have the potential to undergo cross-linking/branching to form isoxazolines via addition to the differing levels of unsaturation present in their chains, Fig. 6. However, it was expected that the competing reaction to give the isoxazolines would be relatively slow due to the internal nature of the alkene groups compared to the terminal alkynes.25 To quantify how likely this was and to obtain spectroscopic information to help determine the level of cross-linking/branching in subsequent soluble polymers, we reacted methyl oleate 6 with phenyloximoyl chloride 7 under the same polymerisation conditions as the model studies. The reaction produced four compounds 8a-d, two regioisomers in equal amounts, with each regioisomer consisting of two diastereomers in a roughly 1:1 ratio (based upon the 1H NMR) in very low yield (8%) after 24 hours. While the formation of two regioisomers was expected due to the limited regiospecific driving force between the nitrile-N-oxide and the internal alkene26 the formation of diastereomers was not expected due to the stereospecific nature of the cycloaddition. The low yield, after an extended reaction time at elevated temperature, indicated that while isoxazoline formation was possible under these conditions it was not particularly fast. It is also possible that the extended reaction time allowed the generated HCl to mediate an epimerisation of the acidic proton adjacent to the imine group in 8a–d or that reversible HCl addition to the unreacted methyl oleate alkene in 6 allowed equilibration of the dienophile stereochemistry.
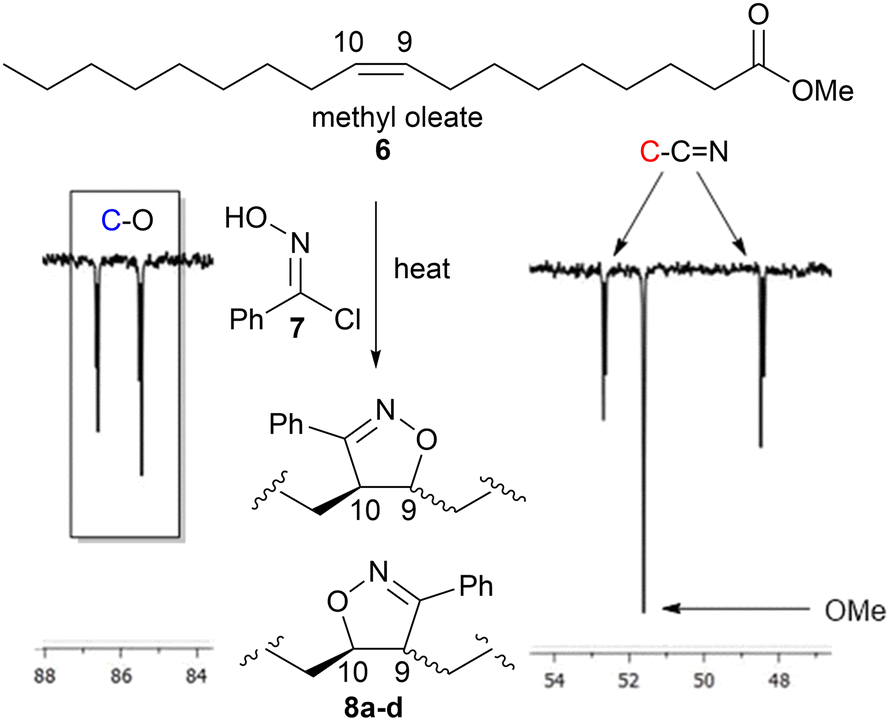 | ||
| Fig. 6 Portion of the 126 MHz 13C spectrum of 8a–d on CDCl3 showing the characteristic isoxazoline signals at 86.6, 85.4, 52.6 and 48.4 ppm. | ||
The position of the characteristic 1H NMR signals for the CH protons next to the oxygen atom and one diastereomer next to the nitrogen atom of the isoxazoline group 8a–d resonated in areas that would make them inappropriate as reference handles due to overlap with polymer resonances. On the other hand, the positions of the C9 and C10 carbon signals in the 13C NMR spectrum for each compound were characteristic (86.6, 85.4, 52.6, 48.4 ppm) and appeared in spectral regions expected to be clear in the linear polymers (based upon the model studies p(1a-co-3) and p(1a-co-5a)), Fig. 6.
Reaction of each of the fatty acid derived monomers 5b–f with 1.0 equivalent of 1a at 80 °C in DMF for 24 h with 4 Å molecular sieves produced the corresponding polymeric materials, Table 1. Interestingly, as the level of unsaturation in the fatty acid side chains increased, the degree of polymerisation (as determined by 1H NMR spectra) of the materials reduced, Fig. 7. Tentative traces of branching were confirmed through the 13C spectrum of the soluble materials, presumably due to the slower rate of addition to the disubstituted alkene group compared to the terminal alkyne groups. The yields of the materials also dropped with the level of unsaturation, due to increased levels of unreacted monomers but not significantly.
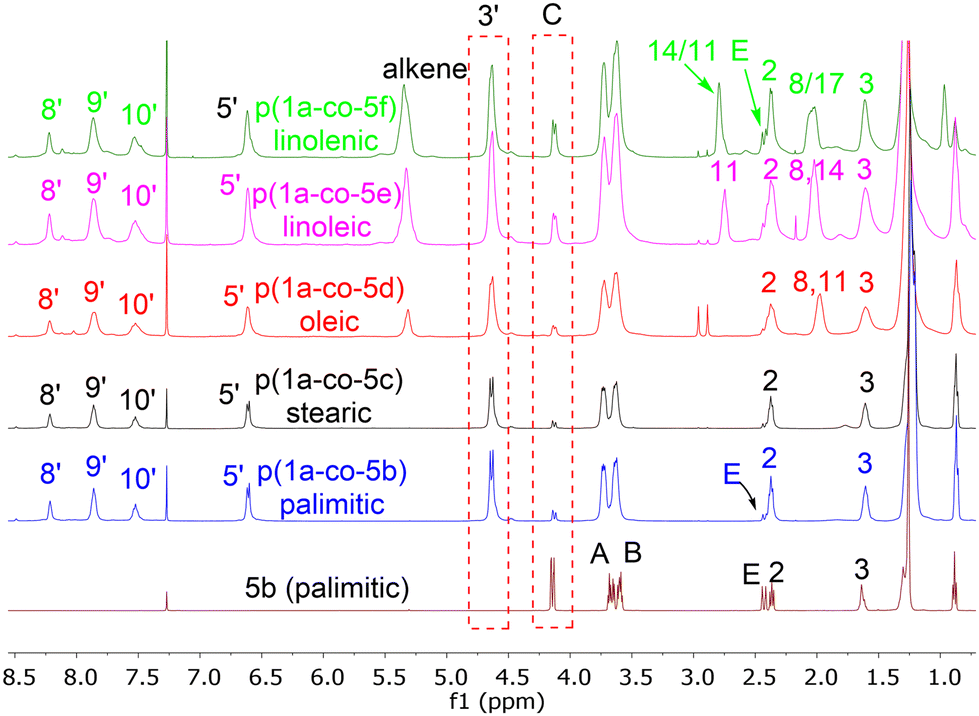 | ||
| Fig. 7 Stacked plot of 500 MHz 1H NMR of fatty acid derived polymers p(1a-co-(5a–f)) and palmitic monomer 5b(P) showing increasing levels of unreacted alkyne end groups as evidenced by peaks ‘C’ and ‘E’ relative to polymer chains. | ||
| Polymer | Yield (%) | DPa |
T
gb (°C) |
T
5%c (°C) |
M
nd (kDa) |
M
wd (kDa) |
Đ |
|---|---|---|---|---|---|---|---|
| a Determined from 1H NMR by integration of the peaks at 4.63 ppm (peak 3′, in the polymer) and 4.12 ppm (peak C, CH2 of the end group in polymer), see Fig. 7. b Determined from DSC, heat rate 10 °C min−1. c Temperature of 5% decomposition from TGA, heat rate 10 °C min−1. d Determined from GPC, eluent CHCl3. | |||||||
| 1a-co-3 | 64 | 16 | 112 | 249 | 4.4 | 6.5 | 1.5 |
| 1a-co-5a | 78 | 10 | 51.0 | 251 | 5.9 | 8.7 | 1.5 |
| 1a-co-5b(P) palmitic | 87 | 9 | 5.7 | 272 | 4.8 | 11.9 | 2.5 |
| 1a-co-5c(S) stearic | 84 | 10 | 4.8 | 279 | 4.8 | 12.7 | 2.7 |
| 1a-co-5d(O) oleic | 80 | 6 | −1.1 | 255 | 5.6 | 19.4 | 3.4 |
| 1a-co-5e(L) linoleic | 76 | 5 | 3.2 | 253 | 6.3 | 33.6 | 5.3 |
| 1a-co-5f(Ln) linolenic | 71 | 3 | 6.1 | 247 | 4.3 | 16.5 | 3.9 |
It was possible to isolate a pure sample of the stearate ABA oligomer O15c(S) to serve as a reference for GPC and for VT 1H NMR analysis, (ΔGrotation = 75.2 ± 1.3 kJ mol−1), Fig. 8a. The normalised GPC data shows a relatively wide molar mass dispersion (Đ) for the unsaturated polymers: oleic p(1a-co-5d(O))Đ = 3.4, linoleic p(1a-co-5e(L))Đ = 5.3 and linolenic p(1a-co-5f(Ln))Đ = 3.9. These values are higher than expected for a typical step growth polymerisation and are most likely attributed to branching due to competing isoxazoline formation by reaction with the internal alkene groups, although more complex processes such as competing cyclic formation or furoxan formation, or a mixture of all three cannot be ruled out.
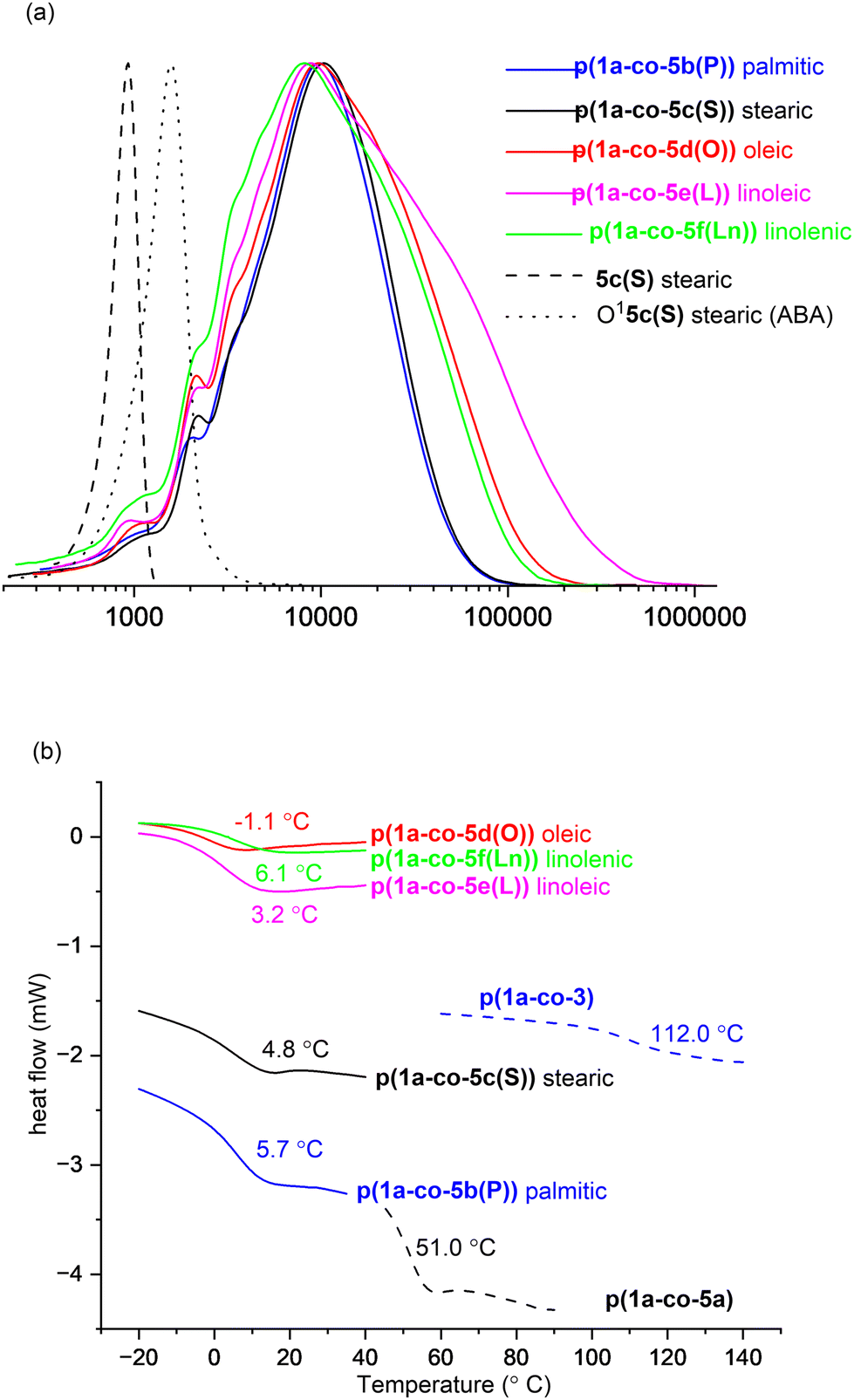 | ||
| Fig. 8 (a) Normalised GPC data for the stearate monomer 5c and ABA type oligomer O15c and fatty acid derived polymers p(1a-co-(5b–f)) showing wider molar mass dispersity for unsaturated oleic p(1a-co-5d(O)), linoleic p(1a-co-5e(L)), and linolenic p(1a-co-5f(Ln)) derivatives; (b) Tg values for p(1a-co-3) and fatty acid derived polymers p(1a-co-(5b–f)) from differential scanning calorimetry (DSC), heat rate 10 °C min−1. | ||
Thermal analysis of the fatty acid derived polymers p(1a-co-(5b–f)) indicated significantly lower Tg values (−1.1 °C to 6.1 °C) compared to p(1a-co-5a) as expected, (Fig. 8b), due to the incorporation of the flexible C16–C18 chains. Assuming no branching occurred during the polymerisation the expected order of Tg values should be linolenic p(1a-co-5f(Ln)) < linoleic p(1a-co-5e(L)) < oleic p(1a-co-5d(O)) < stearic p(1a-co-5c(S)) < palmitic p(1a-co-5b(P)) < p(1a-co-5a) < p(1a-co-3). The fact that the linolenic p(1a-co-5f(Ln)) and linoleic p(1a-co-5e(L)) materials have marginally higher Tg values than the oleic p(1a-co-5d(O)) material suggests that branching is occurring in these polyunsaturated derivatives. Additionally, all the biobased polymers show degraded thermal stability compared to the stearate ABA type oligomer O15c(S). While the polyunsaturated linolenic p(1a-co-5f(Ln)) and linoleic p(1a-co-5e(L)) materials exhibit initially lower thermal stability (T5%) below 300 °C and slightly higher stability above 300 °C than the saturated analogues, the inherent complication of branching discouraged us from any further investigation, Fig. 9.
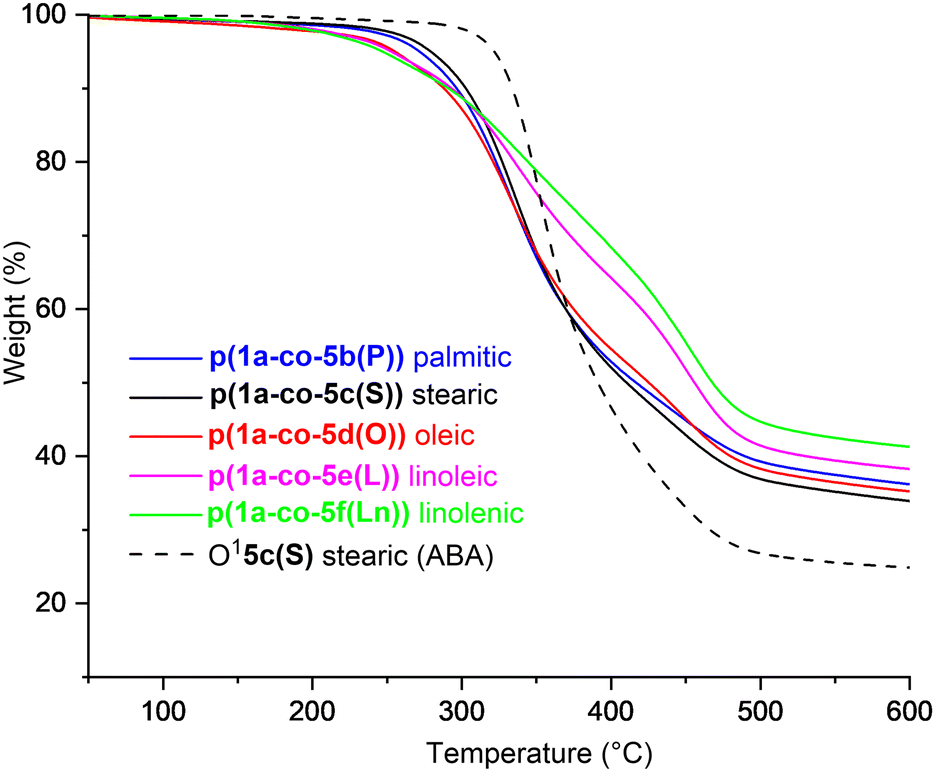 | ||
| Fig. 9 Thermal gravimetric analysis plots from 50–600 °C for fatty acid derived polymers p(1a-co-(5b–f)), compared to the stearate ABA type oligomer O15c, heat rate 10 °C min−1. | ||
MALDI-TOF-MS of the stearate polymer p(1a-co-5c(S)), Fig. 10, indicated the presence of two repeating species with a repeating unit of 607 Da. These species represent linear (AB)nA type polymers terminated by alkyne end groups (1684, 2291, 2896, 3505 Da respectively) and additionally (AB)2–4 type species tentatively assigned as cyclic c(AB)n materials. The alternative linear structure (AB)n terminated with one alkyne and one nitrile oxide end group was discounted due to the instability of the nitrile oxide under the reaction conditions and its propensity to dimerise via furoxan formation although it can't be completely ruled out. The alternative linear furoxan (f) linked material f(AB-BA)n with two alkyne end groups was also discounted due to the absence of evidence of furoxan formation in the infrared spectrum. Although the infrared spectrum of the fatty acid derived polymers p(1a-co-(5b-f)) did not indicate furoxan formation, it would be more energy efficient to be able to undertake the polymerisation at ambient temperatures. This approach could help suppress any competing side reactions that might affect the efficiency of the polymerisation and consequently lower molecular weights. We briefly explored differing base-mediated polymerisation conditions using the stearate derived monomer 5c(S) as a test substrate. While organic bases such as triethylamine have been used to facilitate nitrile oxide formation from oximoyl chloride 4a at elevated temperatures,27 we found that inorganic bases, specifically K2CO3, were the most efficient for mediating the reaction at room temperature in our experiments. A summary of the different conditions investigated, including added copper salts and different bases, NaHCO3, Na2CO3, and K2CO3 in various solvents (DCM, DMF, acetone, THF, diethyl ether, ethanol) can be found in the ESI.† The best conditions involved utilising K2CO3 in the renewable solvent ethanol, which furnished a material with the highest observed molecular weight (Mn = 19.0k, Mw = 36.2k, Đ = 1.9), [5c and 1a, K2CO3, EtOH], Fig. 11b, but with a bimodal molar mass distribution. Scaling up of the test reactions from 150 mg to 6.0 g of stearate monomer 5c(S) utilising 1 equivalent of either 1a or 1b with K2CO3 in ethanol over 24 hours resulted in the formation of the base mediated meta linked stearate derived polymer pbase(1a-co-5c(S)) as a sticky pale solid and the corresponding para derived material pbase(1b-co-5c(S)) as a white solid in yields of 88% and 90% respectively. Both materials completely dissolved in CHCl3 and THF and both exhibited large dispersity in their GPC traces in these solvents indicative of secondary processes to the desired step growth polymerisation occurring. Indeed, the low molar mass tailing in both GPC traces could be indicative of an inactive species, tentatively assigned as the cyclic (AB)n species which can no longer propagate [pbase(1b-co-5c(S)) (Mn = 4.9k, Mw = 25.3k, Đ = 5.9), pbase(1a-co-5c(S)) (Mn = 11.4k, Mw = 88.4k, Đ = 25.8), Fig. 11b]. The meta derived material pbase(1a-co-5c(S)) did not exhibit any observable signals in the 1H NMR at 4.12 ppm for the end group –CH2– protons of the propargyl group, confirming high molecular weights, while trace evidence of alkyne terminated end groups in the 1H NMR of pbase(1b-co-5c(S)) was observed indicating a DP of 21, Fig. 11a.
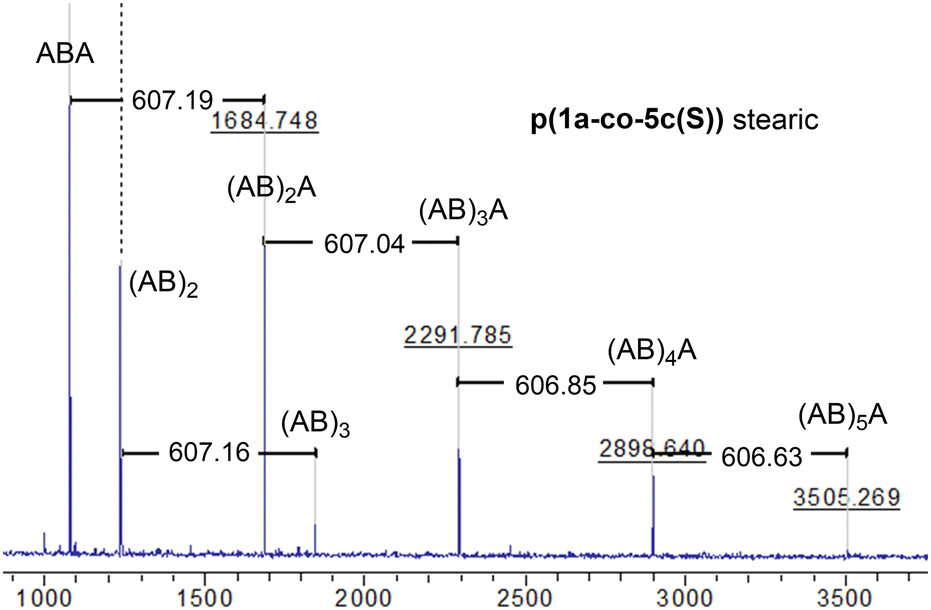 | ||
| Fig. 10 MALDI-TOF-MS of stearate p(1a-co-5c(S)) showing two polymeric species (the (AB)nA type and the (AB)n type, both with a repeat unit of 607 Da). | ||
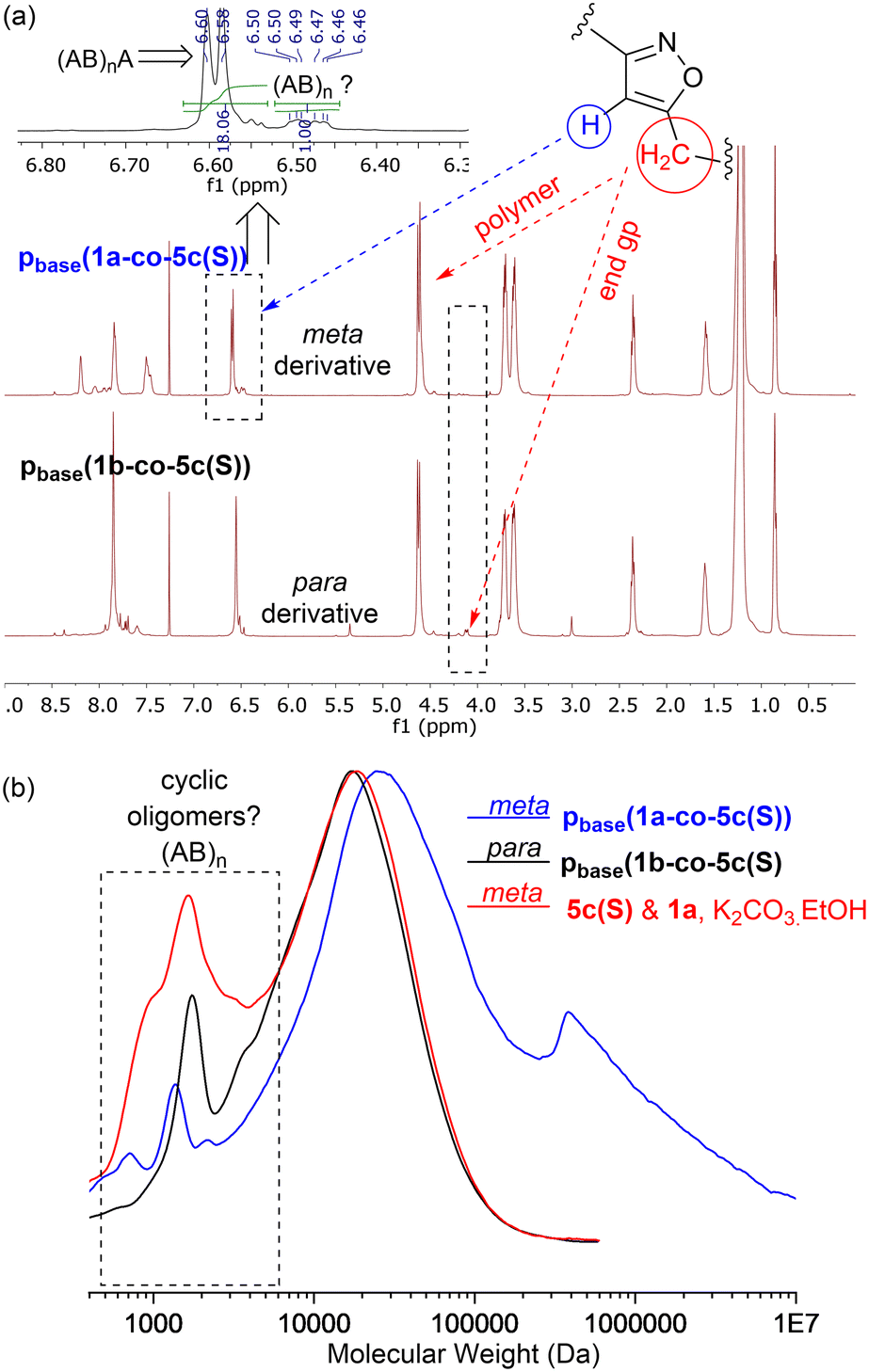 | ||
| Fig. 11 (a) 500 MHz 1H NMR spectra of stearate derived pbase(1a-co-5c(S)) and pbase(1b-co-5c(S)) showing high number of repeat units and tentative assignment of cyclic oligomers; (b) GPC in CHCl3 of pbase(1a-co-5c(S)) and pbase(1b-co-5c(S)) showing area tentatively assigned as cyclic oligomers. | ||
Further evidence that the low molar mass species observed from the GPC data were cyclic in nature came from the MALDI-TOF-MS, Fig. 12b. Both materials showed two peaks at 1237 Da and 1845 Da for stearate derived (AB)2 and (AB)3 oligomers. No higher homologues were detected in the MS range (0–5000 Da) suggesting these species were inactive cyclic homologues which cannot propagate further, Fig. 12a. If these low molar mass (AB)n type oligomers were not cyclic in nature, but instead were the alternative linear furoxan linked materials of the type f(AB–BA) with alkyne end groups, these linear chains would continue to grow and would have reacted further to give higher homologues which were not detected. This allows us to tentatively speculate that the minor peaks in the range of 6.50–6.46 ppm in the 1H NMR of pbase(1a-co-5c(S)), can be assigned to H5′ isoxazole proton in these cyclic materials.
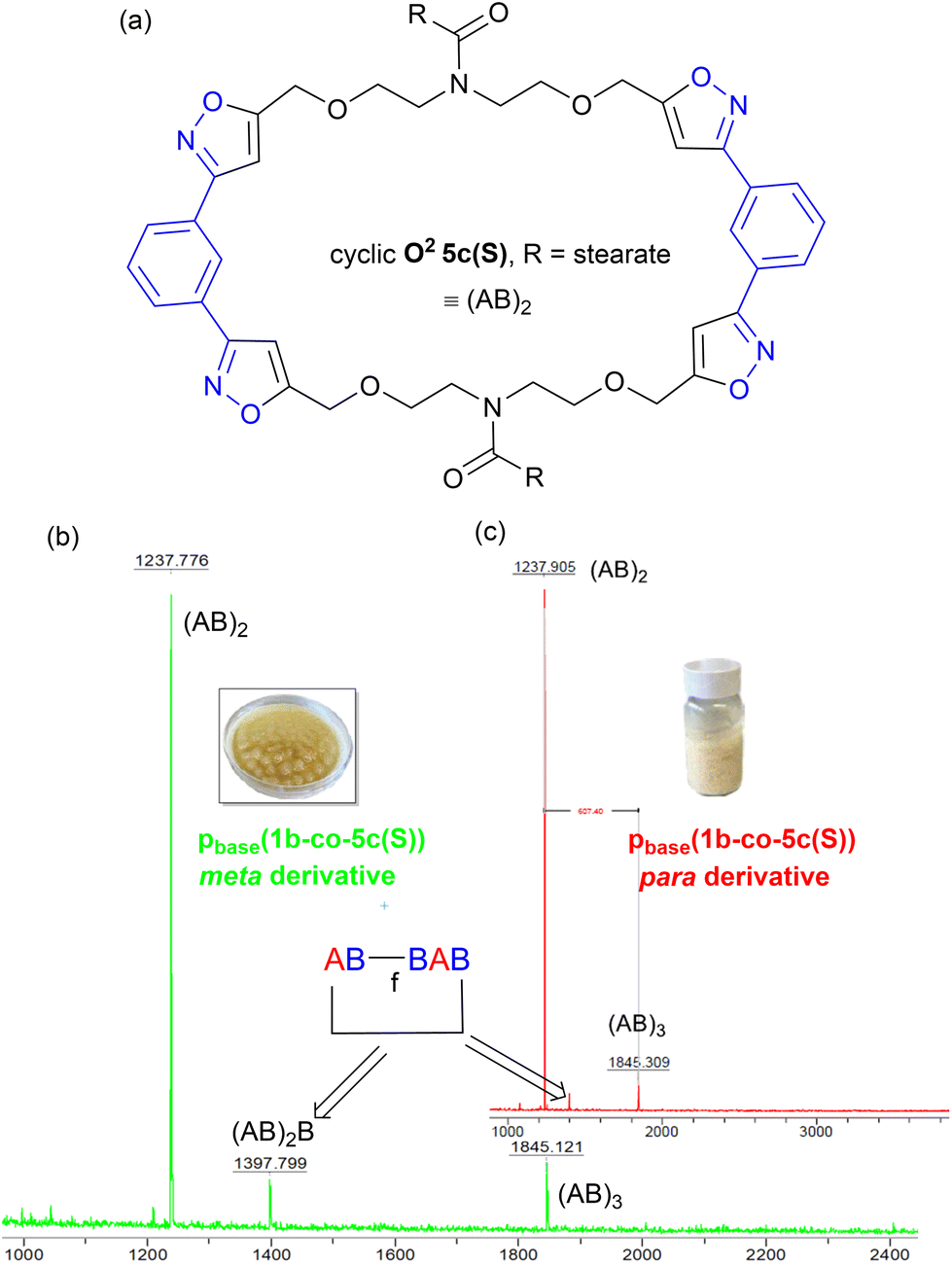 | ||
| Fig. 12 (a) Postulated structure of the stearate derived (AB)2 species O25c, (b) the MALDI-TOF-MS spectrum of metapbase(1a-co-5c(S)), and (c) the MALDI-TOF-MS spectrum of parapbase(1b-co-5c(S)). | ||
In both polymers, traces of a species at 1397 Da were observed which must contain at least one furoxan linkage, most likely in the cyclic material of type c(ABBAB). Indeed, analysis of the infrared spectrum of the stearate derived pbase(1a-co-5c(S)) indicated a shoulder on the main CN stretch peaks at 1592 cm−1 indicative of furoxan formation. If furoxan formation is detected within the cyclic oligomer it is also statistically likely that it is incorporated into the growing linear polymer chains in a second polymerisation process. This undesired secondary polymerisation process could be the origin of the broader molar mass distribution (1 × 105–1 × 107 Da) observed for pbase(1a-co-5c(S)), Fig. 11b. If K2CO3 mediated polymerisation does in fact produces a polyisoxazole with random incorporation of furoxan linkages it might be expected to have different thermal properties. Thermal analysis indicated the polymers produced via the room temperature base-mediated approach were significantly more thermally stable than those derived from heating in DMF alone. This is presumably due to the significantly larger molecular weights and/or furoxan incorporation, [stearate derived p(1a-co-5c(S))T5% = 279 °C vs.pbase(1a-co-5c(S))T5% = 339 °C (+60 °C), pbase(1a-co-5c(S))T5% = 328 °C (+49 °C)]. In addition, both polymers exhibited higher Tg values than the smaller molecular weight polymer p(1a-co-5c(S)) prepared thermally. The DSC of both polymers indicated melting points (Tm = 31 °C for pbase(1a-co-5c(S))), and (Tm = 48 °C for pbase(1b-co-5c(S))), and DMA analysis provided Tg's of 31 °C and 62 °C respectively. The higher value for para derived pbase(1b-co-5c(S)) is presumably due to the linear nature of the linking nitrile oxide influencing packing and chain mobility of the polymer ends, with the linear chains packing more efficiently than those derived from the metapbase(1a-co-5c(S)). Although nitrile-N-oxides are known to react with nucleophiles to give hydroxamic acids,28,29 no evidence of this reaction manifold incorporating ethoxide (from K2CO3 and EtOH) was detected.
Conclusions
Biobased polyisoxazoles derived from thermal polymerisation of both saturated and unsaturated C16–C18 fatty acid derived alkynes 5b–f with 1a exhibit lower Tg's, but similar thermal stability compared to those derived from acetate materials 3 and 5a. While the degree of polymerisation determined by 1H NMR drops as the level of unsaturation in the fatty chains increases: palmitic derivative p(1a-co-5b(P)) ∼ stearic derivative p(1a-co-5c(S)) > oleic derivative p(1a-co-5d(O)) > linoleic derivative p(1a-co-5e(L)) > linolenic derivative p(1a-co-5f(Ln)), the Mn, Mw and dispersity increases for the unsaturated derivatives. This increase is due to competitive isoxazoline formation resulting from the competing reaction with the internal alkene groups with 1a in the unsaturated derivatives, which (a) introduces branching within the growing polymer chains and (b) changes the stoichiometry of the desired reacting functional groups. Although evidence of branching could not be confirmed in the 1H NMR spectra, very weak signals around 86 ppm and 48 ppm in the 13C spectra of the oleic p(1a-co-5b(O)), linolenic p(1a-co-5b(L)), and linoleic p(1a-co-5b(Ln)) derived polymers confirmed this process, as did GPC data. Normally Tg would decrease as the level of unsaturation in the side chains increased, as they are more kinked, reducing the ability to pack, this is observed in the series: palmitic derivative p(1a-co-5b(P)) > stearic derivative p(1a-co-5c(S)) > oleic derivative p(1a-co-5d(O)). However, the Tg increases marginally from the oleic derivative p(1a-co-5d(O)) < the linoleic derivative p(1a-co-5e(L)) < the linolenic derivative p(1a-co-5f(Ln)), presumably caused by the greater opportunities for branching.Conducting the polymerisation of stearate monomer 5c(S) with either the meta1a or para1b nitrile-N-oxide precursors under basic conditions (K2CO3, EtOH) led to polymers with higher molecular weights and Đ's and higher Tg values, although GPC indicated a small oligomeric distribution followed by the main polymeric distribution. MALDI-TOF-MS of all the polymers indicated the expected repeat units for linear (AB)nA type polymers (with terminal alkyne end groups) as well as a (AB)n species which could be cyclic in nature (lacking end groups) or incorporate a furoxan linkage. However, while there was little evidence of competing furoxan formation for polymers produced thermally as observed in both the MALDI-TOF-MS or the infrared spectra, (p(1a-co-3) and p(1a-co-(5a–f))), there was some evidence for furoxan incorporation in polymers pbase(1a-co-5c(S)) and pbase(1b-co-5c(S)) prepared using the base-mediated process.
In general, the Tg values from the biobased materials in this study (−1.1 °C to 62.0 °C) complement those derived from biobased vanillin derivatives (60–80 °C).11 This suggests a broad spectrum of thermal properties can be achieved with biobased materials, making them versatile for various potential applications.
Author contributions
AJC was the principal investigator responsible for the conceptualisation, project administration, supervision and original draft writing. NOSJ prepared polymers p(1a-co-3) and p(1a-co-(5a–f)) using thermal conditions and undertook the investigation, helped develop the methodology, undertook formal analysis and reviewed the writing, AA undertook polymerisation tests at room temperature and prepared polymers pbase(1a-co-5c(S)) and pbase(1b-co-5c(S)) and undertook the investigation, helped develop the methodology, undertook formal analysis and reviewed and edited the writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AA's thanks SASO (the Saudi Standards, Metrology, and Quality Organization) for PhD scholarship and research support. NOSJ's PhD scholarship was supported by the University of Warwick.Notes and references
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS PubMed.
- Z. Geng, J. J. Shin, Y. Xi and C. J. Hawker, J. Polym. Sci., 2021, 59, 963 CrossRef CAS.
- G. Delaittre, N. K. Guimard and C. Barner-Kowollik, Acc. Chem. Res., 2015, 48, 1296 CrossRef CAS PubMed.
- E. Coutouli-Argyropoulou, P. Lianis, M. Mitakou, A. Giannoulis and J. Nowak, Tetrahedron, 2006, 62, 1494 CrossRef CAS.
- C. G. Overberger and S. Fujimoto, J. Polym. Sci., Part B: Polym. Lett., 1965, 3, 735 CrossRef CAS.
- Y. Iwakura, S. Shiraishi, M. Akiyama and M. Yuyama, Bull. Chem. Soc. Jpn., 1968, 41, 1648 CrossRef CAS.
- Y. Iwakura, K. Uno, S.-J. Hong and H. A. Tatsuhiko, Polym. J., 1971, 2, 36 CrossRef CAS.
- Y. Koyama, M. Yonekawa and T. Takata, Chem. Lett., 2008, 37, 918 CrossRef CAS.
- Y.-G. Lee, Y. Koyama, M. Yonekawa and T. Takata, Macromolecules, 2009, 42, 7709 CrossRef CAS.
- Y.-G. Lee, M. Yonekawa, Y. Koyama and T. Takata, Chem. Lett., 2010, 39, 420 CrossRef CAS.
- J. J. Q. Mah, C.-G. Wang, N. Surat'man, S. F. D. Solco, A. Suwardi, S. Wang, X. J. Loh and L. Zibiao, ACS Appl. Polym. Mater., 2023, 5, 6757 Search PubMed.
- T. Pasinszki, B. Hajgató, B. Havasi and N. P. C. Westwood, Phys. Chem. Chem. Phys., 2009, 11, 5263 RSC.
- U. Biermann, U. T. Bornscheuer, I. Feussner, M. A. R. Meier and J. O. Metzger, Angew. Chem., Int. Ed., 2021, 60, 20144 CrossRef CAS PubMed.
- U. Biermann, U. T. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854 CrossRef CAS PubMed.
- J. Hong, Q. Luo and B. K. Shah, Biomacromolecules, 2010, 11, 2960 CrossRef CAS PubMed.
- J. Hong, Q. Luo, X. Wan, Z. S. Petrović and B. K. Shah, Biomacromolecules, 2012, 13, 261 CrossRef CAS PubMed.
- M. C. Floros, A. L. Leão and S. S. Narine, BioMed Res. Int., 2014, 2014 DOI:10.1155/2014/792901.
- D. R. Kelly, S. C. Baker, D. S. King, D. S. deSilva, G. Lord and J. P. Taylor, Org. Biomol. Chem., 2008, 6, 877 Search PubMed.
- L. P. Guan, X. Sui, X. Q. Deng, D. H. Zhao, Y. L. Qu and Z. S. Quan, Med. Chem. Res., 2011, 20, 601 CrossRef CAS.
- S. M. Sagnella, C. E. Conn, I. Krodkiewska and C. J. Drummond, Phys. Chem. Chem. Phys., 2011, 13, 13370 RSC.
- R. A. Aitken, M. H. Smith and H. S. Wilson, J. Mol. Struct., 2016, 1113, 151 CrossRef.
- F. DeSarlo, J. Chem. Soc., Perkin Trans. 1, 1974, 1951 RSC.
- M. Shiro, M. Yamakawa, T. Kubota and H. Koyama, Chem. Commun., 1968, 1409 RSC.
- M. M. Krayushkin, L. G. Vorontsova, M. G. Kurella and M. A. Kalik, Russ. Chem. Bull., 1993, 42, 689 CrossRef.
- J. Plumet, ChemPlusChem, 2020, 85, 2252 CrossRef CAS PubMed.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 633 CrossRef.
- P. Carmella, A. Corsaro, A. Compagnini and F. Marinone Albini, Tetrahedron Lett., 1983, 24, 4377 CrossRef.
- N. Masina, Y. E. Choonara, P. Kumar, L. C. du Toit, M. Govender, S. Indermun and V. A. Pillay, Carbohydr. Polym., 2017, 157, 1226 CrossRef CAS PubMed.
- M. Volanti, D. Cespi, F. Passarini, E. Neri, F. Cavani, P. Mizsey and D. Fozer, Green Chem., 2019, 21, 885 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional 1H, 13C, COSY, HMBC and HMQC spectra and variable temperature NMR spectra, infrared spectra, MALDI-TOF-MS, SEC traces and TGA, DSC and DMA thermogram data. See DOI: https://doi.org/10.1039/d4py00102h |
| This journal is © The Royal Society of Chemistry 2024 |