
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocurable acylhydrazone covalent adaptable networks with fast dynamic exchange and tunable viscoelastic properties†
Yunsheng
Xu
a,
Chonglin
Liu
a,
Yanying
Zhao
 b,
Xianming
Zhang
*a and
Minna
Hakkarainen
*c
b,
Xianming
Zhang
*a and
Minna
Hakkarainen
*c
aSchool of Materials Science and Engineering, Zhejiang Sci-Tech University, Hangzhou 310018, China. E-mail: xuyunsheng1019@zstu.edu.cn; 202130302137@mails.zstu.edu.cn; zhangxm@zstu.edu.cn
bSchool of Chemistry and Chemical Engineering, Zhejiang Sci-Tech University, Hangzhou 310018, China. E-mail: yyzhao@zstu.edu.cn
cDepartment of Fibre and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 58, 100 44 Stockholm, Sweden. E-mail: minna@kth.se
First published on 10th May 2024
Abstract
Acylhydrazone linkages are appealing for the design of covalent adaptable networks (CANs) due to their reversible nature and ease of formation from readily available starting materials. However, acylhydrazone CANs typically exhibit dynamic behavior within a limited temperature range due to their slow exchange rate. Here, vanillin was modified by a two-step reaction involving allylation and imination, followed by photo-curing with a four-armed thiol to fabricate acylhydrazone CANs. The dynamic behavior of these CANs could be controlled by adjusting the amount of p-anisidine (AD) organocatalyst. A model compound reaction revealed that the addition of a catalyst transforms the mechanism of acylhydrazone exchange from a 2 + 2 cycloaddition reaction to a nucleophilic addition reaction, effectively lowering the energy barrier of the intermediate state. CANs with higher amounts of AD catalyst exhibited shorter relaxation times and lower activation energies, thereby expanding the reprocessing temperature window and enhancing the recovery ratio of tensile stress after reprocessing. All the fabricated CANs demonstrated ready chemical recyclability in acidic or AD/DMSO solutions at 80 °C, following two different mechanisms depending on the reaction medium. These findings pave the way to photo-curable renewable acylhydrazone CANs with adjustable viscoelastic properties and mechanical and chemical recyclability enabled by rapid dynamic exchange reactions.
Introduction
Thermosets with superior mechanical properties, chemical resistance and dimensional stability have become indispensable in a wide range of areas. However, owing to their inherent crosslinked structure, they are typically not biodegradable and they cannot be thermally reprocessed like thermoplastics. Consequently, thermosetting materials, once they reach the end of their lifespan, are often subjected to incineration or landfilling, generating a series of environmental issues (e.g., carbon emissions and pollution).1–3 The building blocks of current commercial thermosets are also commonly derived from petroleum resources, which is not sustainable. Therefore, it is of great significance to develop recyclable thermosets from renewable resources.4,5The last decade has witnessed the success of imparting recyclability into thermosetting materials through the introduction of dynamic covalent bonds. These new categories of crosslinked materials are referred to as covalent adaptable networks (CANs).6–13 These crosslinked materials can be reprocessed by inducing network topology changes, providing CANs with the merits of both thermoplastics and thermosets. Currently, various types of dynamic covalent bonds have been successfully incorporated into CANs including esters,14–17 disulfides,18–21 vinylogous urethanes,22–27 acetals28–31 and imines.32–35 Among them, imines or Schiff bases have attractive dynamic covalent bonds for the preparation of CANs due to the wide range of biobased aldehydes and ketones, and often catalyst-free reactions with low activation energies. Furthermore, the susceptibility of imine bonds to enzymatic degradation was recently shown.36 Imines can both exchange with amine groups at room temperature or achieve dynamic exchange by imine metathesis.37–39 These multiple dynamic exchange mechanisms endow Schiff base CANs with the ability of rapid thermal reprocessing at lower temperatures and self-healing even at room temperature.40–42 However, the rapid dynamic nature of imines also gives rise to issues such as increased susceptibility to creep and limited acid resistance at operating temperatures.43 This limits their application in engineering environments requiring high dimensional stability and weathering resistance.44
Acylhydrazones, as imine derivatives, exhibit similar exchange reactions to Schiff bases. However, acylhydrazone compounds also exhibit a lower exchange rate and better acid resistance (pH > 4 stable) due to the mesomeric effect and hydrogen bonding caused by the amide bond adjacent to the imine bond.45–47 Therefore, acylhydrazones are promising for the production of CANs that are more creep and acid resistant compared to imine CANs.
The first report on acylhydrazone chemistry in the context of dynamic materials was published by Lehn and coworkers in 2004,48 followed by several studies focusing on fabricating self-healing dynamic gels.49–54 It is noticed that these properties were achieved by hydrolysis and reforming of the imine moiety with an assisted solvent.55,56 Recently, Roig and coworkers introduced acylhydrazone groups to epoxy resins.57 The achieved materials showed typical stress relaxation and thermal reprocessing properties. However, due to the inherently slow exchange rate of acylhydrazone bonds, these fabricated CANs exhibited relaxation times in the order of at least several hundreds of seconds, which hampers the thermal reprocessability.
Here, we present a new pathway for acylhydrazone CANs with fast dynamic exchange reactions catalyzed by p-anisidine. Previous literature studies have reported that primary amine compounds can act as nucleophilic catalysts, accelerating acyl imine exchange by forming Schiff base intermediates.58 Research by Andrea59 has demonstrated that aniline compounds, due to the electron-donating effect of their benzene ring, exhibit a much higher catalytic efficiency than aliphatic amine compounds. Considering the potential loss of the catalyst during the high-temperature reprocessing of CANs, a high-boiling-point aniline derivative i.e., p-anisidine was chosen as the catalyst for this study. Furthermore, the synthesis of photo-curable CANs starting from a renewable aromatic monomer i.e., vanillin has been demonstrated. The catalytic effect of p-anisidine on acylhydrazone exchange and the exchange mechanism were investigated via both experimental model compound reactions and computational calculations. The mechanical and viscoelastic properties as well as the mechanical and chemical recyclability of the synthesized acylhydrazone CANs were systematically investigated.
Experimental
Materials
Vanillin (VL, 99%), allyl bromide (AB, 98%), adipic acid dihydrazide (AAD, 99%), 1,6-hexamethylenediamine (HD, 99%), p-anisidine (AD, 99%), pentaerythritol tetrakis(mercaptoacetate) (SA, >96%), acetohydrazide (98%), benzaldehyde (99%), n-octanohydrazide (98%), sodium hydroxide (>96%), dimethyl sulfoxide (DMSO, 99%) and acetonitrile (99.9%) were supplied by Aladdin Reagent, China. Phenylbis(2,4,6-trimethylbenzoyl) phosphine oxide (BAPO, 98%), potassium carbonate (99%) and sodium sulfate (99%) were obtained from Shanghai Macklin Biochemical Co., Ltd. Ethyl acetate (EA, 99%), ethanol (EtOH, 99%), tetrahydrofuran (THF, 99%), N,N-dimethylformamide (DMF, 99%), acetic acid (99%), acetone (Ace, 99%), dichloromethane (DCM, 99%) and hydrochloric acid (HCl, 37%) were all purchased from Hangzhou High Crystal. All of the reagents were utilized as received.Synthesis of allylated vanillin (AVL)
Vanillin (45.65 g, 0.3 mol), allyl bromide (39.92 g, 0.33 mol) and potassium carbonate (45.61 g, 0.33 mol) were added to a round-bottom flask with a magnetic stirrer and dissolved in 300 ml ethanol. The mixture was heated to reflux and left to react for 12 h. After cooling to room temperature, the precipitate was filtered and ethanol was removed by rotatory evaporation to recover the reaction product. 150 ml of ethyl acetate was added to dissolve the reaction mixture. The obtained solution was washed 3 times with 100 ml of aqueous saturated sodium chloride, dried over sodium sulfate, and concentrated under reduced pressure to remove ethyl acetate residue, yielding AVL as a yellowish liquid (52.82 g, 91.6% yield). 1H NMR (400 MHz, CDCl3) δ: 9.83 (d, J = 2.9 Hz, 1H), 7.41 (dd, J = 8.1, 2.2 Hz, 2H), 6.96 (dd, J = 8.6, 2.8 Hz, 1H), 6.07 (dddd, J = 17.3, 10.7, 5.3, 3.1 Hz, 1H), 5.56–5.19 (m, 2H), 4.69 (tt, J = 3.2, 1.7 Hz, 2H), 3.92 (d, J = 2.9 Hz, 3H) ppm.Synthesis of vanillin-based resins containing an acylhydrazone bond (AVH)
AVL (48.0 g, 0.25 mol) and adipic acid dihydrazide (17.4 g, 0.1 mol) were added to a 500 ml round bottom flask attached with a magnetic mixer, and 200 ml of acetic acid/ethanol (volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution was added as a solvent and stirred at room temperature for 24 h. After the reaction, the formed precipitate was filtered and washed first with ethanol (3 × 100 ml) and then with hot deionized water (3 × 100 ml). The product was dried in a vacuum oven and AVH was obtained as a white powder (42.86 g, 82.1% yield): 1H NMR (400 MHz, DMSO-d6) δ: 11.14 (dd, J = 40.2, 3.1 Hz, 1H), 7.99 (dd, J = 76.5, 2.1 Hz, 1H), 7.45–6.71 (m, 3H), 6.42–5.65 (m, 1H), 5.55–5.10 (m, 2H), 4.70–4.43 (m, 2H), 4.02–3.56 (m, 3H), 2.77–2.56 (m, 1H), 2.35–2.02 (m, 1H), 1.82–1.42 (m, 2H) ppm.
:1) solution was added as a solvent and stirred at room temperature for 24 h. After the reaction, the formed precipitate was filtered and washed first with ethanol (3 × 100 ml) and then with hot deionized water (3 × 100 ml). The product was dried in a vacuum oven and AVH was obtained as a white powder (42.86 g, 82.1% yield): 1H NMR (400 MHz, DMSO-d6) δ: 11.14 (dd, J = 40.2, 3.1 Hz, 1H), 7.99 (dd, J = 76.5, 2.1 Hz, 1H), 7.45–6.71 (m, 3H), 6.42–5.65 (m, 1H), 5.55–5.10 (m, 2H), 4.70–4.43 (m, 2H), 4.02–3.56 (m, 3H), 2.77–2.56 (m, 1H), 2.35–2.02 (m, 1H), 1.82–1.42 (m, 2H) ppm.
Synthesis of vanillin-based resins containing a Schiff base bond (SVH)
AVL (48.0 g, 0.25 mol) and 1,6-hexamethylenediamine (11.6 g, 0.1 mol) were added to a 500 ml round bottom flask attached with a magnetic mixer, and 240 ml of water/ethanol (volume ratio 1:2) solution was added as a solvent and stirred at room temperature for 24 h. After the reaction, the formed precipitate was filtered and washed with a mixed solution of water/ethanol (1:2 v:v). The product was dried in a vacuum oven and SVH was obtained as a white powder (40.17 g, 80.3% yield): 1H NMR (400 MHz, DMSO-d6) δ 8.21 (s, 1H), 7.35 (d, J = 1.8 Hz, 1H), 7.18 (dd, J = 8.2, 1.9 Hz, 1H), 7.00 (d, J = 8.2 Hz, 1H), 6.05 (ddt, J = 17.3, 10.6, 5.3 Hz, 1H), 5.40 (dq, J = 17.3, 1.7 Hz, 1H), 5.26 (dq, J = 10.5, 1.5 Hz, 1H), 4.59 (dt, J = 5.4, 1.5 Hz, 2H), 3.79 (s, 3H), 3.55–3.48 (m, 2H), 1.61 (p, J = 6.7 Hz, 2H), 1.37–1.34 (m, 2H).
Preparation of acylhydrazone CANs
The AVH resin and pentaerythritol tetrakis(mercaptoacetate) (SA) were mixed according to the molar ratio of the vinyl bond to thiol as 1:1. The photoinitiator BAPO (2 wt%), different amounts of p-anisidine (ranging from 0, 0.05, 0.1, and 0.2 wt% of the total amount of resin) and a minimum amount of DMSO were added to form a homogeneous solution. The resin mixtures were cast into glass Petri dishes and photocured for 5 min under a 300 W UV curing lamp with a wavelength of 365 nm. The cured resins were further dried in an 80 °C vacuum oven for 48 h to remove any solvent residues. Finally, the photocured thermosets were hot pressed at 160 °C under 3 MPa pressure for 20 min to obtain dumbbell or rectangle-shaped specimens. The acylhydrazone CANs were denoted according to the feed ratio of the catalyst (e.g., X-AVSA10 represents a crosslinked resin containing 0.1 wt% of p-anisidine).
Model compounds to investigate the mechanism of the acylhydrazone exchange reaction
Compound a (N′-phenylmethylidene acetohydrazide) and b (N′-(4-hydroxy-3-methoxyphenyl)methylidene heptanehydrazide) were weighed at a molar ratio of 1:1 and added to a flask with DMSO as a solvent. The mixture was heated with stirring at 160 °C for 5 h under a nitrogen atmosphere. After heating, the solvent was dried, and the two groups of samples were dissolved in acetonitrile to be analyzed by gas chromatography-mass spectrometry (GC-MS). The same analysis was conducted without and with 0.2 wt% of p-anisidine as a catalyst for the acylhydrazone exchange reaction.
Computational calculations
Density functional theory (DFT) calculations were performed using Gaussian G09 software. All geometry optimizations and frequency calculations were performed using M062X/Def2-TZVP, and the reported energy was a single point calculation of M062X, with an implicit solvent model (SMD (DMSO)) for the gas-phase geometries. All optimized transition states (TSs) had only one hypothetical frequency along the reaction coordinates. The eigen reaction coordinates (ERC) were calculated to determine the adjacent intermediates connecting the relevant transition states. The energy reported in this study was 423.15 K free energy. CYLview20 was used to generate the 3D structures.Gel content and the swelling ratio test
Samples with a size of approximately 20 mm × 5 mm × 0.5 mm were immersed in 25 ml of DMSO for 48 h, after which they were dried at 120 °C for 24 h in a vacuum oven. The gel content was calculated from md/m0 × 100%, where m0 is the initial mass and md is the final mass after drying; the swelling ratio was calculated from (mw − m0)/m0 × 100%, where m0 is the initial mass and mw is the mass obtained immediately after immersion before drying.Thermal reprocessing of the thermosets
The thermosets were ground into powder using a freezing mill. The powders were transferred to a dumbbell mold (50 mm × 4.0 mm × 0.5 mm) and hot pressed at 160 °C under 3 MPa for 20 min.Chemical recycling of the thermosets
The samples with a size of 10 mm × 5 mm × 0.5 mm were immersed in 15 ml of 1 M HCl/DMSO (1:1.5 v:v) and AD/DMSO (2 wt%). The chemical recycling was conducted at room temperature and 80 °C. Visual changes in the materials were observed after 48 h, after which the solvent was removed and the dissolved products were further analyzed for their chemical structure.
Characterization
1H nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance AV (400 MHz) spectrometer, using dimethyl sulfoxide (DMSO)-d6 and deuterated chloroform (CDCl3) as solvents. An attenuated total reflection Fourier transform infrared spectrometer (ATR-FTIR, Nicolet IS50) was utilized to monitor the chemical change of AVH and SA before and after the curing process. All the spectra were scanned in the range of 4000–400 cm−1 with 16 scans. Differential scanning calorimetry (DSC) was performed under a nitrogen atmosphere using a Mettler Toledo DSC1 to determine the glass transition temperature (Tg) of cured resins. All the samples were heated from 25 °C to 250 °C, kept at 250 °C for 2 min, cooled to 25 °C and then heated to 250 °C, and the heating and cooling rates were set to 10 °C min−1. The glass transition temperature (Tg) was measured from the second heating curve of all samples. The thermal stability of cured resins was assessed by thermogravimetry (TGA) using a Mettler Toledo TGA1. All the samples were heated from 30 °C to 600 °C at a heating rate of 10 °C min−1 and a nitrogen flow rate of 50 ml min−1. Dynamic mechanical analysis (DMA) was performed in tension film mode on a Q800 DMA (TA Instruments) using rectangle film specimens (35 mm × 5 mm × 0.5 mm). All the measurements were conducted in the temperature range from −20 °C to 200 °C with a heating rate of 3 °C min−1. The strain was fixed at 0.5% and the oscillating frequency was set as 1 Hz. The stress relaxation test and dilatometry experiment were conducted on the Q800 DMA as well. For the stress relaxation test, the samples were subjected to 2% strain and varying temperatures (90 °C, 100 °C, 110 °C, and 120 °C), and the decrease in the stress relaxation modulus was recorded over time. For the dilatometry experiment, the sample was heated from 45 °C to 220 °C at a rate of 5 °C min−1 and the length of the samples was recorded. The force was fixed at 0.1 N. The frequency sweep measurements were performed on an Anton Paar MCR 702e. The experiments were conducted in parallel plate geometry using 0.5 mm sample disks and a normal force of 5 N with a strain of 0.1%. The frequency was varied from 100 to 0.1 rad s−1. The creep experiments were carried out on the Anton Paar MCR 702e. The sample was stretched at the set temperature (40, 50, 60, and 70 °C) with a constant force of 0.1 MPa for 5 min, followed by a recovery period of 20 min. The samples were also stretched at 100 °C with a constant force of 0.1 MPa for 30 min, followed by a recovery period of 90 min. Gas chromatography-mass spectrometry was carried out using a 5973I gas chromatograph-mass spectrometer (GC-MS, Agilent) with anhydrous acetonitrile as the solvent. The column used was an HP-5 ms Ultra Inert column with a maximum temperature of 280 °C. The shunt ratio and helium flow rate were set at 20:1 and 24.5 ml min−1, respectively; the heating rate was 10 °C min−1 from 50 °C to 280 °C. The samples were initially kept for 5 min isothermally at 50 °C and then for 5 min at 280 °C at the end of the program. The tensile test was performed using a universal tensile testing machine Instron 34TM-30. All the samples were conditioned at 22 °C and 40% relative humidity for 72 h before testing, and dumbbell shaped samples with sizes of 50 mm × 8.5 mm × 0.5 mm were tested at a tensile rate of 5 mm min−1. The tensile properties of each sample were calculated from the average of five repeated measurements.
Results and discussion
Investigation of the acylhydrazone exchange mechanism with the help of model compounds
As the primary purpose of this work was to develop acylhydrazone CANs exhibiting fast dynamic exchange, the underlying acylhydrazone exchange mechanism was first analyzed by investigating a model compound reaction. Four different acylhydrazone model compounds, denoted as a, b, c, and d, were synthesized through the reaction between two different aldehydes (vanillin and benzaldehyde) and two hydrazides (acetohydrazide and n-octanohydrazide) (Fig. 1a). The chemical structures of the synthesized compounds were verified from 1H NMR and mass spectra (Fig. S3–S6 and S9†). The model compounds a and b were mixed in DMSO without or with 0.2 wt% AD and maintained at 160 °C for 5 h to conduct the exchange reactions (Fig. 1b). The original model compounds and reaction mixtures after 5 h were investigated by GC-MS and the chromatograms are shown in Fig. 1c and Fig. S10–S12.† The results show that after mixing the two model compounds a and b, two new compounds emerged and could be identified as c and d. These compounds were identified in both reaction mixtures, showing successful exchange reactions between the acylhydrazones. Meanwhile, the catalyzed reaction produced higher amounts of compounds c and d, based on the peak areas, which indicates a clear catalytic effect induced by AD. By the quantitative analysis principle, the peak area of GC-MS is proportional to the concentration in the reaction mixture, and the ratio of the exchange rate between the catalyzed and non-catalyzed reaction can, thus, be calculated using the following equation:where m and mAD are the relative exchange yields of compound c in the reaction mixture with 0 wt% AD and 0.2 wt% AD, respectively. The m and mAD were obtained from the GC-MS analysis, while m0 and mAD0 are the theoretical yields of compound c in the reaction mixture with 0 wt% AD and 0.2 wt% AD, respectively. The GC-MS results (Table S1†) show that the addition of 0.2 wt% AD as a catalyst accelerated the dynamic exchange reaction by 2.1 times.
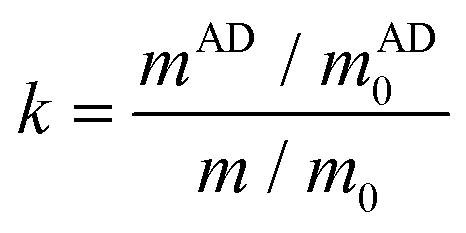
 | ||
| Fig. 1 The synthetic scheme for the model compounds (a), acylhydrazone exchange reaction scheme showing the exchange between a and b to form c and d and vice versa (b), GC chromatogram of a mixture of model compounds a and b (1), model compounds c and d, the expected reaction products from the acylhydrazone exchange reaction between a and b (2), and the reaction mixtures containing a, b, c, and d, after exchange reactions between a and b, without and with the AD catalyst (3) (c). | ||
Computational calculations were conducted to reveal the difference in the acylhydrazone exchange mechanism with or without AD as a catalyst. From Fig. 2a and b, it can be observed that for the non-catalyzed reaction, the imine metathesis involved a symmetry forbidden [2π + 2π] cycloaddition reaction and it further proceeded through a four-membered cyclic transition state, achieving the exchange of different acylhydrazones.60 This mechanism is also supported by the small molecule exchange kinetics between the model acylhydrazone compounds (Fig. S12 and S13†) investigated via GC-MS, as the uncatalyzed exchange of the acylhydrazone bond conformed to the pseudo first-order kinetics (Fig. S14 and S15†). In contrast, when AD is used as the catalyst, there will first be a nucleophilic attack on the acylhydrazone through transimination, and then the formed hydrazide would react with another acylhydrazone via two similar steps to form two new acylhydrazones. Fig. 2c and Table S3† show the Gibbs free energy and transition state geometry of acylhydrazone exchange obtained by DFT calculations (M062X/Def2-TZVP level). For the system with AD as the catalyst, the Gibbs free energy (11.55 kcal mol−1) is lower compared with the catalyst-free system (13.48 kcal mol−1), supporting the catalytic effect of AD on the acylhydrazone exchange reaction. The Cartesian coordinates of all calculated structures are shown in Table S4.†
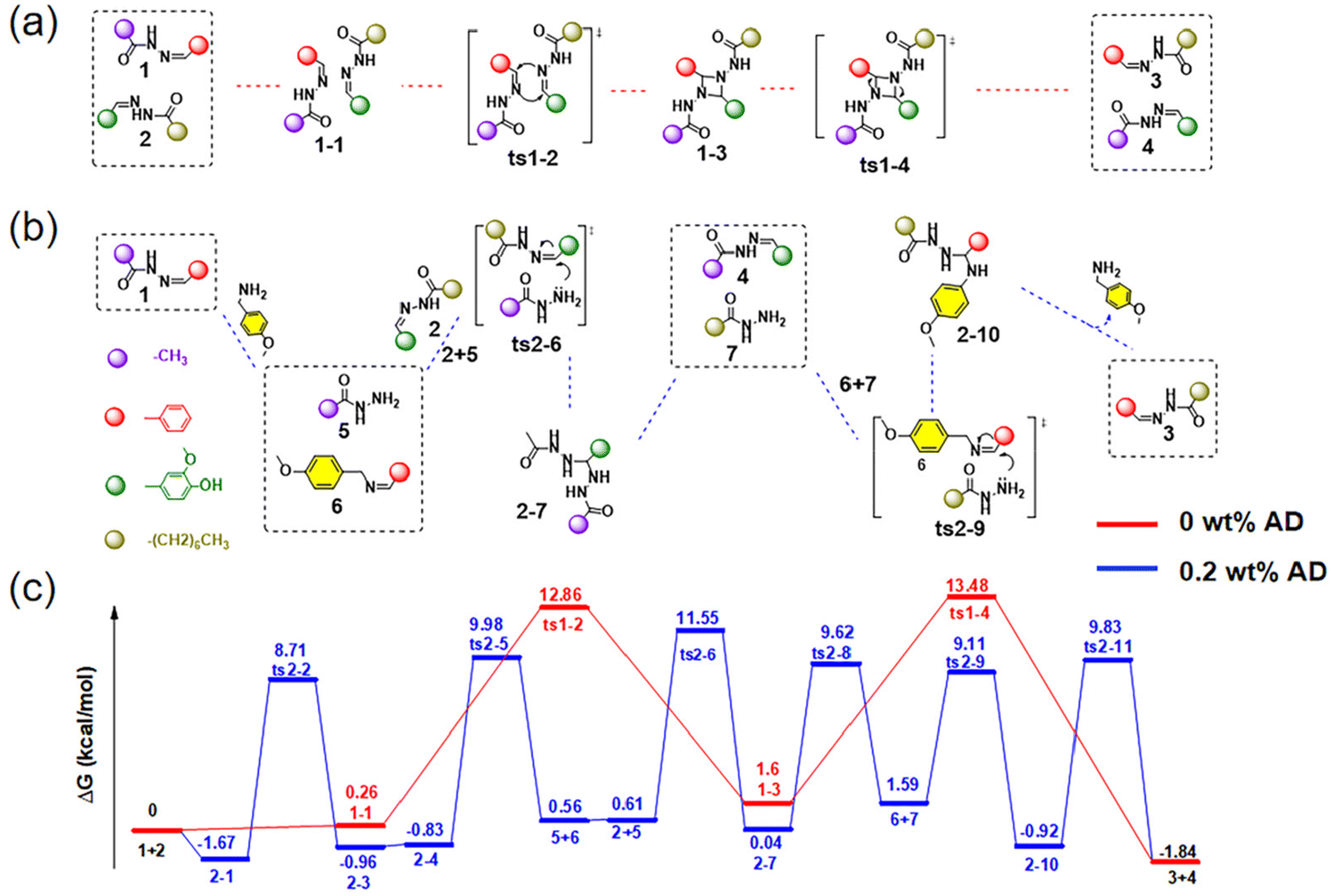 | ||
| Fig. 2 Exchange mechanism of the acylhydrazone without (a) and with AD (b), DFT-calculated energy surface for the exchange reactions of acylhydrazone compounds with or without AD (c). | ||
Synthesis of acylhydrazone CANs
To fabricate CANs containing acylhydrazone groups, vanillin as a starting monomer first underwent a two-step reaction, i.e., allylation followed by imination, to achieve acylhydrazones with vinyl ester groups. More specifically the phenol group in vanillin was first allylated to introduce a reactive vinyl group for further thiol–ene crosslinking. The chemical structure of allylated vanillin (AVL) was verified by 1H NMR (Fig. S1†), and it was consistent with the previous literature. The AVL was then reacted with dihydrazide adipate under mild conditions with acetic acid as the catalyst to produce the acylhydrazone monomer (AVH). The 1H NMR of AVH (Fig. S2†) showed the disappearance of the characteristic proton signals of the aldehyde (δ 9.83) and the newly formed acylhydrazone group (δ 11.14), indicating the completed imination reaction. Finally, the resins were formed by mixing AVH and a four-armed thiol (SA) with different loadings of the catalyst. The resins were photo-cured under 365 nm UV light to produce a crosslinked material with dynamic acylhydrazone groups (Fig. 3). To compare the acid and creep resistance between the acylhydrazone and imine CANs, an imine CAN (X-SVS) was synthesized according to the same route as the corresponding acylhydrazone, where only adipic acid dihydrazide was converted into hexamethylenediamine in step 2. The 1H NMR of the monomer SVH is shown in Fig. S7.† | ||
| Fig. 3 Synthesis scheme of acylhydrazone CANs. | ||
Chemical structure and properties of the thermosets
To confirm the successful photocuring of AVH/SA resins, ATR-FTIR spectra were recorded to monitor the changes in the chemical structure before and after curing (Fig. 4a). Compared with AVH and SA, the photocured resin (X-AVS) shows an obviously weakened intensity of SH at 2562 cm−1 and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– at 1560 cm−1, which illustrates a successful thiol–ene reaction. Moreover, the characteristic peaks of the acylhydrazone at 1667 cm−1 (–CO–NH–) and 1600 cm−1 (–CN–) remained in the spectra of the photocured thermosets, indicating that acylhydrazone-groups remained intact during the photocuring process. All the cured thermosets with different loadings of AD exhibited similar FTIR spectra (Fig. S16†), demonstrating no obvious differences in the chemical structure after the addition of AD as a catalyst for acylhydrazone CANs. In addition, the structure of the synthesized imine CAN was also confirmed from the ATR-FTIR spectrum (Fig. S17†).
C– at 1560 cm−1, which illustrates a successful thiol–ene reaction. Moreover, the characteristic peaks of the acylhydrazone at 1667 cm−1 (–CO–NH–) and 1600 cm−1 (–CN–) remained in the spectra of the photocured thermosets, indicating that acylhydrazone-groups remained intact during the photocuring process. All the cured thermosets with different loadings of AD exhibited similar FTIR spectra (Fig. S16†), demonstrating no obvious differences in the chemical structure after the addition of AD as a catalyst for acylhydrazone CANs. In addition, the structure of the synthesized imine CAN was also confirmed from the ATR-FTIR spectrum (Fig. S17†).
 | ||
| Fig. 4 ATR-FTIR spectra of vanillin-based resins before and after UV curing (a), the solvent resistance of acylhydrazone CANs in diverse solvents at room temperature after 3 days (b), photos of the acylhydrazone and imine CANs before and after immersion in 1 M HCl/DMSO at room temperature for 48 h (c), swelling ratio (d) and gel content (e) of the acylhydrazone CANs with different catalyst loadings. | ||
All the acylhydrazone CANs, X-AVS, X-AVS5, X-AVS10 and X-AVS20, exhibited a gel content of above 95% in DMSO as a solvent, indicating the robustness of the formed crosslinked networks. In order to further explore the chemical solvent resistance of the prepared acylhydrazone CANs, X-AVS and X-AVS20 were selected as benchmark samples and soaked in different solvents (EtOH, DCM, Ace, THF, DMF, DMSO and 1 M HCl) for 3 days at room temperature (Fig. 4b). For comparison the solvent resistance of the imine CAN (X-SVS) was also investigated. Both acylhydrazone thermosets remained visibly intact without obvious signs of degradation. Meanwhile, during soaking in 1 M HCl, the imine CAN (X-SVS) showed obvious swelling and only 39% of the weight remained intact (Fig. 4c): this comparison confirms the enhanced acid resistance of acylhydrazone CANs. Notably, the acylhydrazone CANs with a higher AD content exhibited slightly higher swelling ratios (Fig. 4d and e). This is likely explained by the presence of AD resulting in a slightly lower conversion of the thiol–ene reaction (96% for noncatalyzed and 95% in the presence of 0.2 wt% AD, as seen in Fig. S8†), leading to a lower crosslinking density for the acylhydrazone CANs with a higher AD content.
All the acylhydrazone CANs exhibited a Tg value of around 95 °C (Fig. 5a). The materials displayed the onset of degradation temperature (Td5) above 300 °C, which ensures stability for thermal processing and reprocessing (Fig. 5b). Fig. 5c and d show the temperature curves of storage modulus and tan delta as a function of temperature, which are similar to the DSC test results. The materials showed the same changing trend, and the peak values of tan delta of each material were close (Table 1). All the acylhydrazone CANs exhibited a broad Tα transition (from 75 to 85 °C), which indicates a non-homogeneous crosslinking. This may be due to the phase separation induced by the presence of a solvent during the photocuring process.61
| Sample | T ag (°C) | T bg (°C) | T d5 (°C) | Young's modulus (MPa) | Tensile strength (MPa) | Elongation at break (%) |
|---|---|---|---|---|---|---|
| Note: Tag is obtained from the peak of tan delta versus the temperature curve. Tbg is obtained from the DSC result. | ||||||
| X-AVS | 82 | 96 | 321 | 1620 ± 27 | 62.1 ± 2.1 | 21 ± 1.7 |
| X-AVS5 | 79 | 95 | 315 | 1850 ± 89 | 55.8 ± 1.6 | 24 ± 2.6 |
| X-AVS10 | 79 | 94 | 310 | 1620 ± 41 | 49.2 ± 1.9 | 26 ± 2.3 |
| X-AVS20 | 77 | 94 | 305 | 1560 ± 39 | 43.4 ± 1.7 | 28 ± 4.0 |
 | ||
| Fig. 5 DSC (a) and TGA (b) curve of the acylhydrazone CANs, storage modulus (c) and tan delta (d) of the acylhydrazone CANs as a function of temperature. | ||
Viscoelastic properties
A stress relaxation test was further performed within the linear viscoelastic regime to ascertain the CAN behavior. As shown in Fig. 6a–d, all the fabricated CANs exhibited typical stress relaxation curves in the temperature range of 80–120 °C and the viscoelastic behavior followed the Arrhenius equation:| τ*(T) = τo·eEa/RT |
 | ||
| Fig. 6 Double Maxwell model fit to the stress relaxation data of acylhydrazone CANs with different catalyst loadings from 90 °C to 120 °C (a–d), the relaxation time (τ) of acylhydrazone CANs as a function of 1000/T (e), and dilatometry experiments of all acylhydrazone CANs (f). | ||
The characteristic relaxation time (τ) was obtained from the fitting of the double Maxwell function62,63 (eqn (S1)†), and the activation energy (Ea) was calculated from the slope of the curve of ln(τ) vs. 1000/T (seen in Fig. 6e). The calculated activation energies of acylhydrazone CANs ranged from 81.2 to 45.4 kJ mol−1. Furthermore, a similar trend was found for the topology freezing transition temperatures (Tv). Both Ea and Tv are important parameters for determining the recyclability properties;64,65 lower Ea and Tv values represent a faster dynamic exchange rate at a certain reprocessing temperature. Therefore, these results demonstrate that the addition of AD as a catalyst may modulate the reprocessing temperature of acylhydrazone CANs.
Frequency sweep experiments were conducted to further investigate the influence of the catalyst on the rheological behavior of acylhydrazone CANs (Fig. 7). Overall, the shear storage modulus showed no significant decrease with temperature and frequency irrespective of the sample, indicating that acylhydrazone CANs follow an associative exchange mechanism. This also demonstrates that the impact of the catalyst-induced active end groups on the network density was negligible.66 Additionally, there was a varying degree of increase in the loss storage modulus for all the tested materials in the low-frequency region, suggesting a dissipation of mechanical energy due to the rearrangement of the dynamic network topology. Notably, the introduction of the AD catalyst allowed for a more obvious increase in the loss storage modulus at lower temperatures, confirming once again that AD, as a catalyst, can lower the activation energy barrier for dynamic exchange in acylhydrazone CANs.
 | ||
| Fig. 7 Frequency sweep measurements at temperatures ranging from 110 to 160 °C of acylhydrazone CANs with different catalyst loadings (a–d). | ||
Creep resistance of acylhydrazone CANs
Creep resistance of crosslinked materials is vital for maintaining structural integrity under prolonged exposure to high temperatures, ensuring dimensional stability in critical applications. In this study, we investigated the creep behavior of acylhydrazone-based CANs at temperatures ranging from 40 to 70 °C (Fig. 8a–d). All the acylhydrazone CANs exhibited a full recovery of strain below 40–60 °C, while they could also recover over 93% of the strain within 20 min at 70 °C. Specifically, at 70 °C, the acylhydrazone CANs with a higher catalyst loading show only slightly lower strain recovery (Fig. 8e), indicating that the addition of the catalyst did not significantly sacrifice their creep resistance performance. This could be explained by the inherent high Tg and Tv values (above 90 °C), contributing to highly restricted chain mobility and dynamic reactions at the tested temperatures. Meanwhile, the creep resistance of the material at 100 °C (above Tg) was tested (see Fig. S25 and S26†), and the ultimate recovery rates for X-AVS, heX-AVS5, X-AVS10 and X-AVS20 were 60%, 58%, 56% and 55%, respectively, indicating a slightly lower creep performance with increasing catalyst loading. Furthermore, we also performed creep tests on the imine CAN (X-SVS) at 30 °C and compared the results with those of the acylhydrazone CANs at 70 °C. From Fig. 8f, it is clear that the imine CANs, even when tested at a much lower temperature, demonstrate only 40% recovery of strain. It can therefore be concluded that acylhydrazone CANs possess superb creep resistance in comparison with the imine CANs, while the addition of AD did not significantly sacrifice their creep performance. | ||
| Fig. 8 The creep curves at different temperatures for the acylhydrazone CANs with different catalyst loadings (a–d), the creep curves of acylhydrazone CANs under different catalyst loadings at 70 °C (e), the strain recovery curves of acylhydrazone CANs under different catalyst loadings at 70 °C and the strain recovery curve of the Schiff base CAN at 30 °C (f). | ||
Reprocessability of acylhydrazone CANs
To demonstrate that the acylhydrazone CANs can be reprocessed while maintaining their mechanical properties, all the materials were hot pressed at 160 °C under 3 MPa for 20 min. The materials showed excellent remoldability after shredding, yielding fully transparent and homogeneous films (Fig. S18†) due to the exchange of acylhydrazone groups. Meanwhile, FTIR spectra of the materials after reprocessing did not show any additional signals or intensity changes in the spectra, indicating no significant changes in the chemical structure and functional groups (Fig. 9a). Compared with the pristine sample, all the recycled acylhydrazone CANs show a singular tan delta peak as a function of temperature (Fig. 9b), indicating a more homogeneous crosslinked system after reprocessing. Furthermore, the tan delta peaks of the reprocessed acylhydrazone CANs are positioned between the two peaks of pristine CANs. These peaks consistently decrease with an increase in catalyst loading, following the same trend of the pristine CANs. | ||
| Fig. 9 Comparison of the infrared spectra of original and reprocessed acylhydrazones (a), tan delta of the original and reprocessed acylhydrazone CANs as a function of temperature (b), mechanical properties of original and reprocessed acylhydrazone CANs (c), and photos showing a comparison of X-AVS, X-AVS20 and X-AVS (1:1.1) before and after hot pressing at 140 °C for 10 min (d). | ||
The original X-AVS exhibited a tensile strength of 62.1 MPa and an elongation at break of 21%. As a function of the increasing catalyst loading, the tensile strength slightly decreased and the elongation at break showed an inverse trend (Fig. 9c). After recycling, the mechanical properties of CANs showed different degrees of recovery (all the tensile stress curves are shown in Fig. S19 and S20†). It is noticed that the CANs with a higher content of catalyst exhibited higher recovery of the original mechanical properties. This could be explained by the Tv and Ea values, as previous results showed that acylhydrazone CANs with higher AD loading exhibited faster dynamic exchange rates. Therefore, under the same thermal processing conditions, the acylhydrazone with a higher AD content exhibited better reprocessability. This was also verified by the hot pressing of X-AVS and X-AVS20 at 140 °C for only 10 min (Fig. 9d). X-AVS20 could be fully recycled into a homogeneous film just after 10 min, while a major part of X-AVS remained as powder. Previous results reveal that the addition of a catalyst indeed promotes the dynamic exchange rate and reduce the Tv and Ea values. At the same time, the presence of a catalyst exerts the dangling end effect, resulting in a reduced crosslink density, which potentially contributes to the faster reprocessing rate. To further investigate this, we synthesized an acylhydrazone CAN via curing of vinyl bonds: sulfhydryl = 1:1.1 (X-AVS 1:1.1), with higher excess of thiol end groups in the materials. As expected, X-AVS (1:1.1) exhibited a lower Tg value (89 °C) and a higher swelling ratio (235%) compared with both X-AVS and X-AVS20 (Fig. S21 and S22†). However, the activation energy of X-AVS (1:1.1) (80.4 kJ mol−1) is close to that of X-AVS (81.2 kJ mol−1) and much higher than the activation energy of X-AVS 20 (45.4 kJ mol−1) (Fig. S23 and S24†). At the same time, after the reprocessing test at 140 °C, it was found that X-AVS (1:1.1) remained as powder, which is similar to the case of X-AVS. Based on the above results, we assume that the reprocessing efficiency of acylhydrazone CANs is governed by their dynamic exchange rate, which is modulated by AD as a catalyst.
Chemical recycling of acylhydrazone CANs
Acylhydrazone compounds are potentially hydrolyzed in an acidic aqueous solution or they could react with amines via transimination. Therefore, the chemical recycling of acylhydrazone CANs in acidic and amine solutions was explored. The previous solvent resistance test demonstrated that the fabricated acylhydrazone materials were stable in acidic solutions at room temperature; therefore, the chemical recycling temperature was raised to 80 °C to boost the reaction rate. As shown in Fig. 10a and b, all the materials could be fully dissolved to form homogeneous solutions within 2 days. The dissolved materials were further characterized by 1H NMR as shown in Fig. 10c. After the reaction in 1 M HCl/DMSO, the proton peaks of the acyl hydrazide (8.96 ppm) and aldehyde (9.84 ppm) appeared and that of the acylhydrazone (11.09 ppm) signal vanished, which demonstrated that the imine hydrolysis reaction occurred. On the other hand, a trace of the imine group was detected (8.53 ppm) after recycling in amine, indicating that the transimination reaction occurred. The mechanisms of both recycling processes are illustrated in Fig. 10d. | ||
| Fig. 10 Photos of acylhydrazone CANs before and after recycling in 1 M HCl/DMSO (a) and AD/DMSO (b) at 80 °C for 48 h, 1H NMR spectra of AVL, AVH and recycled products of X-AVS in HCl and AD (c), and the suggested chemical recycling mechanism of acylhydrazone CANs in HCl and AD (d). | ||
Conclusions
A new class of covalent adaptable networks was successfully fabricated based on the acylhydrazone motif. The viscoelastic behaviour and dynamic exchange reactions of the synthesized CANs could be modulated by the loading of AD as a catalyst. A model compound reaction revealed that AD, as a catalyst, lowers the energy barrier of the intermediate state in acylhydrazone metathesis reactions and increases the yield of the products. All the CANs exhibited good stability in common solvents, high thermal stability (Td5 is above 300 °C), and excellent mechanical properties (tensile stress above 40 MPa and elongation at break above 20%). Furthermore, the activation energy and the topology freezing transition temperature of CANs decreased with an increased concentration of the catalyst. As a result, the acylhydrazone CANs with a higher content of the catalyst exhibit a broader reprocessing temperature window and a higher recovery ratio of tensile stress after reprocessing. At the same time, the addition of catalyst did not sacrifice their creep performance, as all the acylhydrazone CANs exhibit a recovery of strain up to 93% at 40–70 °C, which is significantly higher compared to the imine CAN (40% at 30 °C). Finally, the synthesized CANs could be recycled in acidic and amine/DMSO solutions at 80 °C through two different mechanisms. In conclusion, we provide a facile synthetic route to photo-curable acylhydrazone CANs and a convenient way for modulating their dynamic behaviour.Author contributions
Yunsheng Xu conceptualized and wrote the original draft. Chonglin Liu performed the subject experiments and data curation. Yanying Zhao validated the studies and developed some methodologies. Xianming Zhang and Minna Hakkarainen conceptualized and supervised the work. Yunsheng Xu, Minna Hakkarainen, and Chonglin Liu reviewed and edited the final manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully appreciate the support from the National Natural Science Foundation of China (NSFC) (Grant No. 52303138), the Zhejiang Provincial Natural Science Foundation of China (Grant No. LQ22B040005) and the Science Foundation of Zhejiang Sci-Tech University (Grant No. 21212286-Y).References
- M. Hong and E. Y. X. Chen, Trends Chem., 2019, 1, 148–151 CrossRef CAS.
- L. T. J. Korley, T. H. Epps 3rd, B. A. Helms and A. J. Ryan, Science, 2021, 373, 66–69 CrossRef CAS PubMed.
- T. R. Walker and L. Fequet, TrAC, Trends Anal. Chem., 2023, 160, 116984 CrossRef CAS.
- C. R. Westerman, B. C. McGill and J. J. Wilker, Nature, 2023, 621, 306–311 CrossRef CAS PubMed.
- S. Zheng, Z. Wei, B. Wozniak, F. Kallmeier, E. Baráth, H. Jiao, S. Tin and J. G. de Vries, Nat. Sustainability, 2023, 6, 1436–1445 CrossRef.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- C. Taplan, M. Guerre, J. M. Winne and F. E. Du Prez, Mater. Horiz., 2020, 7, 104–110 RSC.
- Q. Li, S. Ma, P. Li, B. Wang, Z. Yu, H. Feng, Y. Liu and J. Zhu, Macromolecules, 2021, 54, 8423–8434 CrossRef CAS.
- S. Wang, S. Ma, J. Qiu, A. Tian, Q. Li, X. Xu, B. Wang, N. Lu, Y. Liu and J. Zhu, Green Chem., 2021, 23, 2931–2937 RSC.
- Y. Liu, B. Wang, S. Ma, X. Xu, J. Qiu, Q. Li, S. Wang, N. Lu, J. Ye and J. Zhu, Eur. Polym. J., 2021, 144, 110236 CrossRef CAS.
- Y. Zhang, L. Zhang, G. Yang, Y. Yao, X. Wei, T. Pan, J. Wu, M. Tian and P. Yin, J. Mater. Sci. Technol., 2021, 92, 75–87 CrossRef CAS.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- M. Podgorski, B. D. Fairbanks, B. E. Kirkpatrick, M. McBride, A. Martinez, A. Dobson, N. J. Bongiardina and C. N. Bowman, Adv. Mater., 2020, 32, e1906876 CrossRef PubMed.
- K. S. K. Reddy, W.-J. Gao, C.-H. Chen, T.-Y. Juang, M. M. Abu-Omar and C.-H. Lin, ACS Sustainable Chem. Eng., 2021, 9, 5304–5314 CrossRef CAS.
- J. Qiu, S. Ma, S. Wang, Z. Tang, Q. Li, A. Tian, X. Xu, B. Wang, N. Lu and J. Zhu, Macromolecules, 2021, 54, 703–712 CrossRef CAS.
- T. Liu, C. Hao, L. Wang, Y. Li, W. Liu, J. Xin and J. Zhang, Macromolecules, 2017, 50, 8588–8597 CrossRef CAS.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS PubMed.
- S. Xiang, L. Zhou, R. Chen, K. Zhang and M. Chen, Macromolecules, 2022, 55, 10276–10284 CrossRef CAS.
- N. J. Bongiardina, K. F. Long, M. Podgórski and C. N. Bowman, Macromolecules, 2021, 54, 8341–8351 CrossRef CAS.
- D. B. Tiz, F. A. Vicente, A. Kroflič and B. Likozar, ACS Sustainable Chem. Eng., 2023, 11, 13836–13867 CrossRef CAS.
- G. Li, H. S. Soo, J. Huang, T. Li, Y. Wang, S. Wang and W. Dong, Macromol. Rapid Commun., 2023, 44, 2300133 CrossRef CAS PubMed.
- J. O. Holloway, C. Taplan and F. E. Du Prez, Polym. Chem., 2022, 13, 2008–2018 RSC.
- Y. Ma, Z. Liu, S. Zhou, X. Jiang, Z. Shi and J. Yin, Macromol. Rapid Commun., 2021, 42, e2100394 CrossRef PubMed.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- S. Wang, L. Li and M. W. Urban, ACS Appl. Polym. Mater., 2022, 4, 9360–9367 CrossRef CAS.
- L. E. Diodati, S. Liu, C. M. Rinaldi-Ramos and B. S. Sumerlin, ACS Appl. Mater. Interfaces, 2023, 15, 32957–32966 CrossRef CAS PubMed.
- W. Denissen, M. Droesbeke, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef CAS PubMed.
- Q. Li, S. Ma, S. Wang, W. Yuan, X. Xu, B. Wang, K. Huang and J. Zhu, J. Mater. Chem. A, 2019, 7, 18039–18049 RSC.
- W. Yuan, S. Ma, S. Wang, Q. Li, B. Wang, X. Xu, K. Huang, J. Chen, S. You and J. Zhu, Eur. Polym. J., 2019, 117, 200–207 CrossRef CAS.
- S. Ma, J. Wei, Z. Jia, T. Yu, W. Yuan, Q. Li, S. Wang, S. You, R. Liu and J. Zhu, J. Mater. Chem. A, 2019, 7, 1233–1243 RSC.
- X. Xu, S. Ma, J. Wu, J. Yang, B. Wang, S. Wang, Q. Li, J. Feng, S. You and J. Zhu, J. Mater. Chem. A, 2019, 7, 15420–15431 RSC.
- H. Geng, Y. Wang, Q. Yu, S. Gu, Y. Zhou, W. Xu, X. Zhang and D. Ye, ACS Sustainable Chem. Eng., 2018, 6, 15463–15470 CrossRef CAS.
- S. Yu, G. Zhang, S. Wu, Z. Tang, B. Guo and L. Zhang, J. Mater. Chem. A, 2020, 8, 20503–20512 RSC.
- A. Liguori and M. Hakkarainen, Macromol. Rapid Commun., 2022, 43, e2100816 CrossRef PubMed.
- S. K. Schoustra, M. H. P. de Heer Kloots, J. Posthuma, D. van Doorn, J. A. Dijksman and M. M. J. Smulders, Macromolecules, 2022, 55, 10341–10355 CrossRef CAS PubMed.
- S. Subramaniyan, N. Najjarzadeh, S. R. Vanga, A. Liguori, P.-O. Syrén and M. Hakkarainen, ACS Sustainable Chem. Eng., 2023, 11, 3451–3465 CrossRef CAS.
- Z. Q. Lei, P. Xie, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2015, 3, 19662–19668 RSC.
- Y. Xu, K. Odelius and M. Hakkarainen, ACS Sustainable Chem. Eng., 2020, 8, 17272–17279 CrossRef CAS.
- A. Liguori, S. Subramaniyan, J. G. Yao and M. Hakkarainen, Eur. Polym. J., 2022, 178, 111489 CrossRef CAS.
- S.-H. Lee, S.-R. Shin and D.-S. Lee, Mater. Des., 2019, 172, 107774 CrossRef CAS.
- C. L. He, F. C. Liang, L. Veeramuthu, C. J. Cho, J. S. Benas, Y. R. Tzeng, Y. L. Tseng, W. C. Chen, A. Rwei and C. C. Kuo, Adv. Sci., 2021, 8, e2102275 CrossRef PubMed.
- D. Yu, X. Zhao, C. Zhou, C. Zhang and S. Zhao, Macromol. Chem. Phys., 2017, 218, 110236 Search PubMed.
- K. Liang, G. Zhang, J. Zhao, L. Shi, J. Cheng and J. Zhang, ACS Sustainable Chem. Eng., 2021, 9, 5673–5683 CrossRef CAS.
- S. K. Schoustra and M. M. J. Smulders, Macromol. Rapid Commun., 2023, 44, e2200790 CrossRef PubMed.
- G. Li, X. Zhang, J. Huang, T. Li, S. Yang, Y. Wang, J. Jiang, B. Xia, M. Chen and W. Dong, Chem. Eng. J., 2022, 435, 134766 CrossRef CAS.
- K. Caillaud and C. Ladavière, Macromol. Chem. Phys., 2022, 223, 2200064 CrossRef CAS.
- D. N. Barsoum, V. C. Kirinda, B. Kang and J. A. Kalow, J. Am. Chem. Soc., 2022, 144, 10168–10173 CrossRef CAS PubMed.
- W. G. Skene and J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8270–8275 CrossRef CAS PubMed.
- N. Kuhl, S. Bode, R. K. Bose, J. Vitz, A. Seifert, S. Hoeppener, S. J. Garcia, S. Spange, S. van der Zwaag, M. D. Hager and U. S. Schubert, Adv. Funct. Mater., 2015, 25, 3295–3301 CrossRef CAS.
- X. Jiang, X. Yang, B. Yang, L. Zhang and A. Lu, Carbohydr. Polym., 2021, 273, 118547 CrossRef CAS PubMed.
- D.-D. Zhang, Y.-B. Ruan, B.-Q. Zhang, X. Qiao, G. Deng, Y. Chen and C.-Y. Liu, Polymer, 2017, 120, 189–196 CrossRef CAS.
- Z. Guo, H. Gu, Y. He, Y. Zhang, W. Xu, J. Zhang, Y. Liu, L. Xiong, A. Chen and Y. Feng, Chem. Eng. J., 2020, 388, 124282 CrossRef CAS.
- G. Deng, F. Li, H. Yu, F. Liu, C. Liu, W. Sun, H. Jiang and Y. Chen, ACS Macro Lett., 2012, 1, 275–279 CrossRef CAS PubMed.
- R. Chang, X. Wang, X. Li, H. An and J. Qin, ACS Appl. Mater. Interfaces, 2016, 8, 25544–25551 CrossRef CAS PubMed.
- Y. Deng, Q. Zhang, D. H. Qu, H. Tian and B. L. Feringa, Angew. Chem., Int. Ed., 2022, 61, e202209100 CrossRef CAS PubMed.
- G. Li, J. Huang, X. Zhang, Y. Wang, J. Qiao, T. Liu, M. Du and W. Dong, Macromol. Mater. Eng., 2022, 308, 2200493 CrossRef.
- A. Roig, A. Petrauskaité, X. Ramis, S. De la Flor and À. Serra, Polym. Chem., 2022, 13, 1510–1519 RSC.
- A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS PubMed.
- M. Canal, Andrea, C. D. Navo, E. Sáez, D. Molero, G. Jiménez-Osés and R. Pérez-Fernández, Org. Biomol. Chem., 2021, 19, 7202–7210 RSC.
- M. Ciaccia and S. Di Stefano, Org. Biomol. Chem., 2015, 13, 646–654 RSC.
- C. R. Szczepanski, C. S. Pfeifer and J. W. Stansbury, Polymer, 2012, 53, 4694–4701 CrossRef CAS PubMed.
- F. Van Lijsebetten, K. De Bruycker, E. Van Ruymbeke, J. M. Winne and F. E. Du Prez, Chem. Sci., 2022, 13, 12865–12875 RSC.
- R. G. Ricarte and S. Shanbhag, Polym. Chem., 2024, 15, 815–846 RSC.
- Z. Yang, Q. Wang and T. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 21691–21699 CrossRef CAS PubMed.
- S. Kaiser, P. Novak, M. Giebler, M. Gschwandl, P. Novak, G. Pilz, M. Morak and S. Schlögl, Polymer, 2020, 204, 122804 CrossRef CAS.
- F. Van Lijsebetten, K. De Bruycker, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2022, 61, e202113872 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00230j |
| This journal is © The Royal Society of Chemistry 2024 |