
Comparison of the hydrophilicity of water-soluble poly(2-alkyl-2-oxazoline)s, poly(2-alkyl-2-oxazine)s and poly(2,4-dialkyl-2-oxazoline)s†
Kelly
Mint
ab,
Joshua P.
Morrow
b,
Nicole M.
Warne
b,
Xie
He
cd,
David
Pizzi
 b,
Shaffiq Zainal Osman
Shah
b,
Gregory K.
Pierens
cd,
Nicholas L.
Fletcher
cd,
Craig A.
Bell
cd,
Kristofer J.
Thurecht
*cde and
Kristian
Kempe
*ab
b,
Shaffiq Zainal Osman
Shah
b,
Gregory K.
Pierens
cd,
Nicholas L.
Fletcher
cd,
Craig A.
Bell
cd,
Kristofer J.
Thurecht
*cde and
Kristian
Kempe
*ab
aMaterials Science and Engineering, Monash University, Clayton, VIC 3800, Australia. E-mail: kristian.kempe@monash.edu
bDrug Delivery, Disposition, and Dynamics, Monash Institute of Pharmaceutical Science, Parkville, VIC 3052, Australia
cCentre for Advanced Imaging, The University of Queensland, St Lucia, QLD 4072, Australia
dAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, St Lucia, QLD 4072, Australia
eARC Training Centre for Innovation in Biomedical Imaging Technology, The University of Queensland, St Lucia, QLD 4072, Australia
First published on 6th June 2024
Abstract
Poly(cyclic imino ether)s (PCIEs) including poly(2-alkyl-2-oxazoline)s (POx), poly(2-alkyl-2-oxazine)s (POz) and poly(2,4-dialkyl-2-oxazoline)s (PdOx) are a rapidly emerging polymer class for use in biomedical and therapeutic applications due to the biocompatibility and “stealth-like” properties of their water-soluble homologues similar to poly(ethylene glycol) (PEG). The physico-chemical properties of PCIE can be easily “tuned” via appropriate monomer selection resulting for example in polymers ranging from water-soluble to water-insoluble. To date, studies focussing on the hydrophilicity of PCIEs have been limited to the well-known POx, with minimal comparison to POz and especially PdOx. In this study, the effect of degree of hydrophilicity for water-soluble POx, POz, and PdOx systems were assessed for the first time under one testing regime. Specifically, a library of 20 PCIEs was created, consisting of 10 different polymers each synthesised at two different degrees of polymerisation (DP = 20, 50). The hydrophilicity of each polymer was assessed by turbidimetry, high-performance liquid chromatography (HPLC), octanol–water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KOW), surface tension, and 1H NMR relaxometry. Additionally, logKOW was compared against in silico predictive techniques and hydrophilciity trends seen to correlate between the two techniques, though the predictive software utilised could not accurately predict logKOW for long polymer chains. This investigation lead to the elucidation of hydrophilicity trends stemming from molar mass, side chain length, backbone spacing, and additional backbone functionality for the case of PdOx. More specifically, hydrophilicity followed a POz > PdOx > POx trend when comparing between structural isomers, and a POx > POz > PdOx trend when comparing between polymers with the same 2-side chain. The knowledge resulting from this study can be utilised for the future design of smart, solubility-tailored PCIE systems for a range of biomedical applications.
KOW), surface tension, and 1H NMR relaxometry. Additionally, logKOW was compared against in silico predictive techniques and hydrophilciity trends seen to correlate between the two techniques, though the predictive software utilised could not accurately predict logKOW for long polymer chains. This investigation lead to the elucidation of hydrophilicity trends stemming from molar mass, side chain length, backbone spacing, and additional backbone functionality for the case of PdOx. More specifically, hydrophilicity followed a POz > PdOx > POx trend when comparing between structural isomers, and a POx > POz > PdOx trend when comparing between polymers with the same 2-side chain. The knowledge resulting from this study can be utilised for the future design of smart, solubility-tailored PCIE systems for a range of biomedical applications.
Introduction
Poly(ethylene glycol) (PEG) is widely regarded as the “gold standard” stealth polymer for use in biomedical applications. Indeed, “PEGylation” has been utilised in a plethora of bio- and nanomedical systems to increase their biocompatibility and blood circulation time.1–3 However, following the report of emerging immunogenicity as a result of anti-PEG antibodies in recent years, the need for PEG alternatives is becoming increasingly apparent.4–6 Of the many alternate polymer systems investigated, poly(cyclic imino ether)s (PCIE) are particularly promising.3,7 Perhaps the most notable groups of PCIE are polymers formed from the polymerisation of 2-substituted-2-oxazolines (poly(2-alkyl-2-oxazoline)s, POx) or their higher homologues, 2-substituted-2-oxazines (poly(2-alkyl-2-oxazine)s, POz), which contain an additional methylene unit in the polymer backbone. More recently, poly(2,4-dialkyl-2-oxazoline)s (PdOx), synthesised via the polymerisation of 2,4-substituted-2-oxazolines, have started to receive attention due to the increased versatility introduced through additional backbone functionalisation in the form of a 4-substituent group. Indeed, to date the vast majority of studies have been carried out on hydrophilic POx such as poly(2-methyl-2-oxazoline) (PMeOx) and poly(2-ethyl-2-oxazoline) (PEtOx), owing to their similar biocompatibility profile to PEG.7–12 Comparatively few studies have been undertaken for water-soluble POz systems,13–15 though interest in these systems is growing following reports that hydrophilic POz (poly(2-methyl-2-oxazine) (PMeOz) and poly(2-ethyl-2-oxazine) (PEtOz)) outperform both POx and PEG as antifouling coatings.16–19 PdOx systems are yet to be studied in similar applications, and to date have only been utilised as carrier materials in potential therapeutic polymer nanoparticles,20,21 though initial synthesis papers briefly compared the material properties of these systems to POx and POz isomers.22–24PCIE can be easily modified in a multitude of ways, ranging from simple changes in initiator, monomer, or termination agent choice, to the incorporation of reactive moieties to form complex architectures or allow for post-polymer modifications.25–29 This ease of modification leads to superior “tunability”; the ability to easily vary or “tune” polymer properties to specific applications. One such property that can be readily altered is the hydrophilicity of the polymer. Simple changes to side chain group and backbone spacing (e.g. via inclusion of an additional methylene unit such as in POz) provide access to diverse PCIE ranging from hydrophilic to hydrophobic.11,30–32 Incorporation of additional backbone functionality in the form of PdOx is also known to modulate the hydrophilicity of a polymer,33 though this has not yet been studied in direct comparison to POx or POz under unified parameters.
Hydrophilicity is an innate material property that is central to many PCIE applications, particularly those involving biological systems. Indeed, hydrophilicity modulates the solution behaviour of particles, affecting physical parameters such as chain conformation, end-to-end chain length and radius of gyration,34 alongside thermo-physical properties such as the display of lower solution critical temperature (LCST) behaviour. Hydrophilicity also underpins how polymers interact in a biological setting, including with drugs, cells, tissues, and surfaces, amongst others. For example, hydrophilicity has been linked to cellular interactions with nanoparticles (NPs), affecting both the cellular uptake of NPs35,36 and specific NP interaction with membranes,37–39 with more hydrophobic NPs being more readily internalised. Hydrophilicity of NP systems has also been linked to immune activity, where more hydrophobic NPs have been shown to elicit a stronger immune response in experiments conducted both in vitro and in vivo.40,41 Moreover, hydrophilicity influences low- and anti-fouling behaviour in aqueous environments, with hydrophilic polymer coatings often utilised to create a “hydration layer” on surfaces, preventing the attachment of proteins and microorganisms.42 Hydrophilicity of polymer systems is thus a highly important property to explore, considering how it underpins most behaviours utilised by researchers for drug delivery, biomedical, and antifouling applications. As such, understanding the factors affecting the hydrophilicity of PCIEs and how to precisely control this, will allow for the design of specific, tailored PCIE systems.
The hydrophilicity trend of PCIE as a family is broadly known, considering the insolubility and/or LCST behaviour of PCIEs with larger backbone spacing and longer side chain groups, allowing for a clear water-soluble/water-insoluble differentiation.18,31 However, the hydrophilicity of water-soluble PCIE, the most relevant for use in biological applications, is less well-defined. Polymers such as PMeOx and PMeOz, for example, are both known to be fully water-soluble up to 100 °C, thus would be considered equally hydrophilic via turbidimetry assessment.30,31 Assessment of the hydrophilicity of individual PCIEs has been largely limited in the literature to the measurement of cloud point temperature (Tcp),22,30,31,43 contact angle (both water and multi-solution)44–48 and surface energy,45,49 and, more recently, high-performance liquid chromatography (HPLC) retention time.10,50–53 While important properties to consider, some of these techniques have notable limitations. Turbidimetry, for example, is limited to the determination of Tcp for polymers that demonstrate phase separation in water at elevated temperatures; a property notably absent in the most hydrophilic PCIEs.18,31
As such, an in-depth understanding of the hydrophilicity of water-soluble PCIEs is yet to be achieved, limiting the current understanding of these systems’ behaviour and reducing our ability to design hydrophilicity-tailored water-soluble polymers.
As previously mentioned, to date there has been no study carried out which explicitly explores and compares the physicochemical properties and molecular behaviour of water-soluble POx, POz and PdOx using a consistent methodology. Because of the ever-growing interest in PCIE and ongoing discussions about their potential as PEG surrogates, a comprehensive comparative study of the different hydrophilic PCIE encompassing candidates from POx, POz and PdOx would provide a fundamental understanding of PCIE hydrophilicity as a function of monomer identity and physiochemical properties. This can in turn be used to design smart, application-tailored PCIE for use in bio- and nanomedicine in the future.
This study looks to use complementary characterisation methods such as log octanol–water partition coefficient (logKOW), 1H NMR relaxometry, and assessment of surface tension in solution, alongside more common techniques including turbidimetry and HPLC to demonstrate hydrophilicity trends and how these manifest in subsequent polymer properties across an extensive PCIE library. More specifically, we will elucidate in detail how variation in side chain length, backbone spacing, and inclusion of additional backbone functionality affect the hydrophilic character of PCIE, and in the process illustrate how understanding these structure–property relationships will assist in the design of tailored PCIE systems as PEG alternatives.
Results and discussion
Polymer synthesis and characterisation
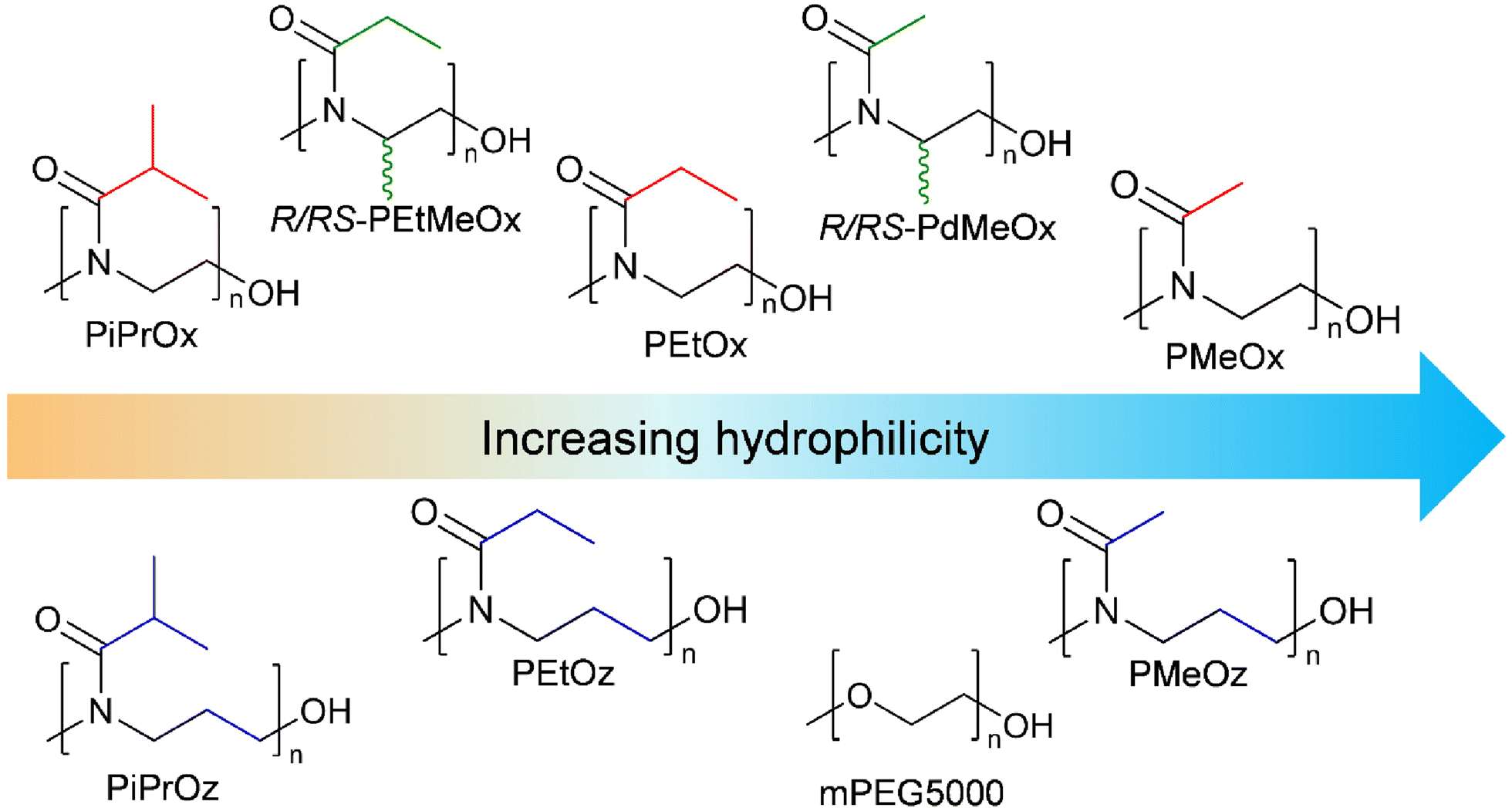
A key aim in this study was to establish a library of suitable materials to explore the effect of structure on function. Monomers and resulting polymers were selected to populate the library with a variety of chemistries in terms of the side chain (methyl-, ethyl-, or isopropyl-group), backbone spacing (with POz containing an additional methylene spacer compared to POx), molar mass (DP20 vs., DP50), and the presence/absence of additional backbone functionalisation via the inclusion of PdOx.The different POx, POz and PdOx included in this study are depicted in Scheme 1A. Structural isomers such as PEtOx and PMeOz were further grouped in terms of “additional” carbons compared to an unsubstituted poly(2-oxazoline) (Scheme 1B and Table S1†), to allow for comparison of the effect of atomic arrangement versus overall molar mass.
 | ||
| Scheme 1 (A) Polymer library synthesized. The library includes three poly(2-oxazolines): PMeOx, poly(2-ethyl-2-oxazoline) (PEtOx) and poly(2-isopropyl-2-oxazoline) (PiPrOx); three poly(2-oxazines): PMeOz, poly(2-ethyl-2-oxazoline) (PEtOz) and poly(2-isopropyl-2-oxazoline) (PiPrOz); and four poly(2,4-dialkyl-2-oxazolines): R-poly(2,4-dimethyl-2-oxazoline) (R-PdMeOx), RS-poly(2,4-dimethyl-2-oxazoline) (RS-PdMeOx), R-poly(2-ethyl-4-methyl-2-oxazoline) (R-PEtMeOx), and RS-poly(2-ethyl-4-methyl-2-oxazoline) (RS-PEtMeOx). Polymers were synthesized at both 50 and 20 DP. (B) Division of the polymer library into structural isomers based on groups of “additional” carbons comparative to an unsubstituted poly(2-oxazoline) (group 0, illustrated in black, not included in this study), to allow for easy graphical comparison between structural isomers. | ||
All polymers were initiated with methyl p-toluenesulfonate (MeOTs) and terminated with methanolic potassium hydroxide (KOH)54 or tetramethylammonium hydroxide (TMAH)55 in an effort to create polymers with comparable methyl α-end and ω-hydroxyl end groups. As reported by de la Rosa et al., termination with a strong base such as KOH can sometimes lead to compromised termination in the form of an amine ester group which is quickly hydrolysed, resulting in the formation of a small minority of alternatively end-capped polymers.55 To assess the effects of this on our PCIE library, a sub-set of DP20 polymers, expected to be more affected by end-group differences due to the greater relative contribution from end group properties, were additionally synthesised utilising TMAH as an alternate terminating agent. de la Rosa et al. reported successful termination for PEtOx20 with TMAH at one molar equivalent,55 and while this was not successful in our hands, three molar equivalents proved successful for PEtOx20 and R/RS-PdMeOx20 (Fig. S1 and 2†). However, it was observed that this termination strategy cannot be generalised for the entire PCIE family as other polymers, for example PEtOz20, could not be successfully terminated under these conditions (Fig. S1†). Overall, minimal differences were found between polymers terminated with TMAH or KOH, with both polymers displaying similar retention times and logKOW values, and KOH-terminated polymers occasionally showing a second low intensity peak in HPLC (Fig. S3†). Moreover, size exclusion chromatography (SEC) measurements revealed defined PCIE systems with dispersity (Đ) values <1.2 (Fig. 1 and Table S4†). Detailed synthesis procedures, calculated DP, structural analysis by 1H NMR spectroscopy, and DP20 SEC traces can be found in ESI (Fig. S4–S6 and Table S4†).
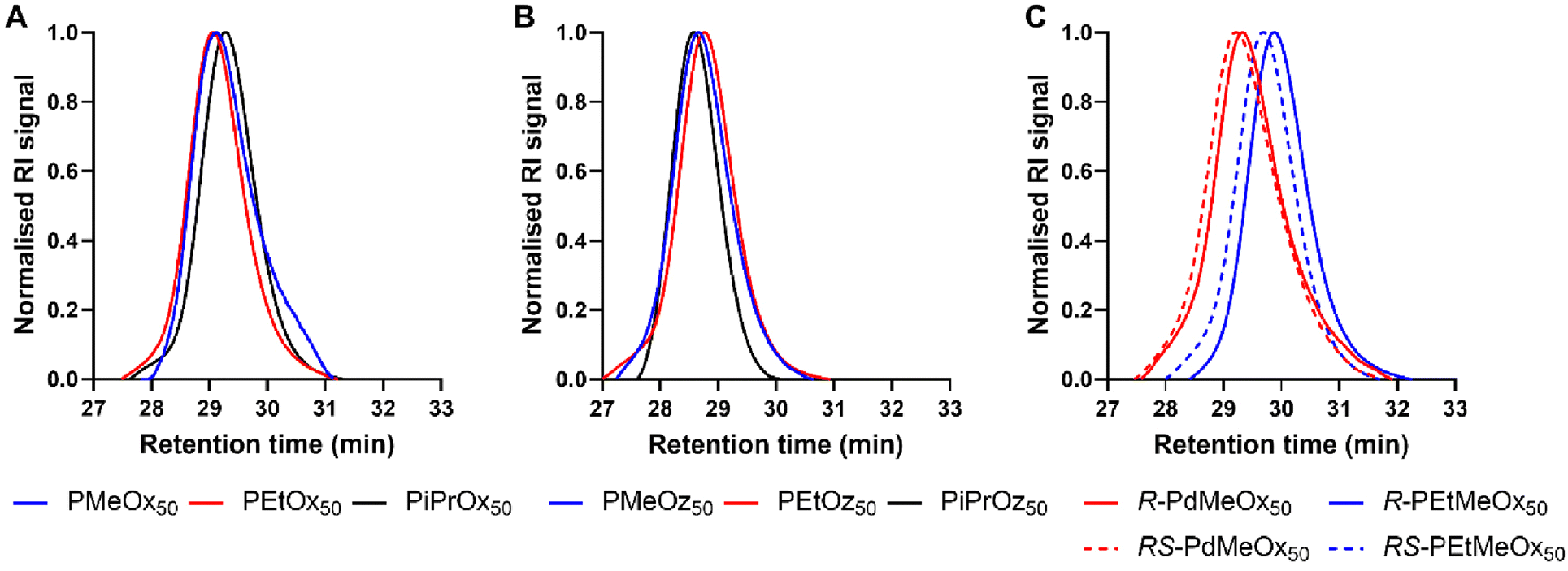 | ||
| Fig. 1 SEC traces (DMAc + 0.03% LiBr) for KOH terminated DP50 (A) POx; (B) POz and (C) PdOx. | ||
Alongside structural characterisation, the thermal properties of PCIEs were briefly assessed. Thermal properties are most commonly determined via techniques such as differential scanning calorimetry (DSC), resulting in measurements for Tg, the temperature at which the polymer transitions from glassy to amorphous behaviour, and Tm, the melting temperature for a crystalline or semicrystalline polymer. These values are needed to select appropriate polymers for a given application, as a shift from glass-like to rubber-like behaviour (or vice versa) during application can adversely affect behaviour. As such, thermal properties of the PCIE library were investigated, with Tg values determined from the second heating curve of a 0–150 °C (20 °C min−1) DSC run.
T g has been previously reported for all polymers in this library, but with somewhat differing conditions or methodology.19,31 Indeed, we found a good correlation with existing literature values for all polymers excluding PEtOz20 and PEtOz50, both of which were seen to be higher than the previously reported 8 °C for PEtOz200 (17.1 °C and 10.9 °C, respectively in this study (Fig. 2)).19 Similar to existing literature, PdOx are seen to have the highest Tg of any PCIE family (Fig. 2),22 with the lack of flexibility attributed to steric hindrance from the 4-methyl group. POz, comparatively, is seen to have the lowest Tg, due to the additional backbone spacing and resulting increased flexibility.19 As such, backbone spacing and additional backbone functionality is seen to exert a greater overall effect on polymer flexibility than side chain length. PiPrOx, the only semicrystalline polymer in the library, was observed to have both a Tg and Tm, similar to previous reports (Fig. S7†).56
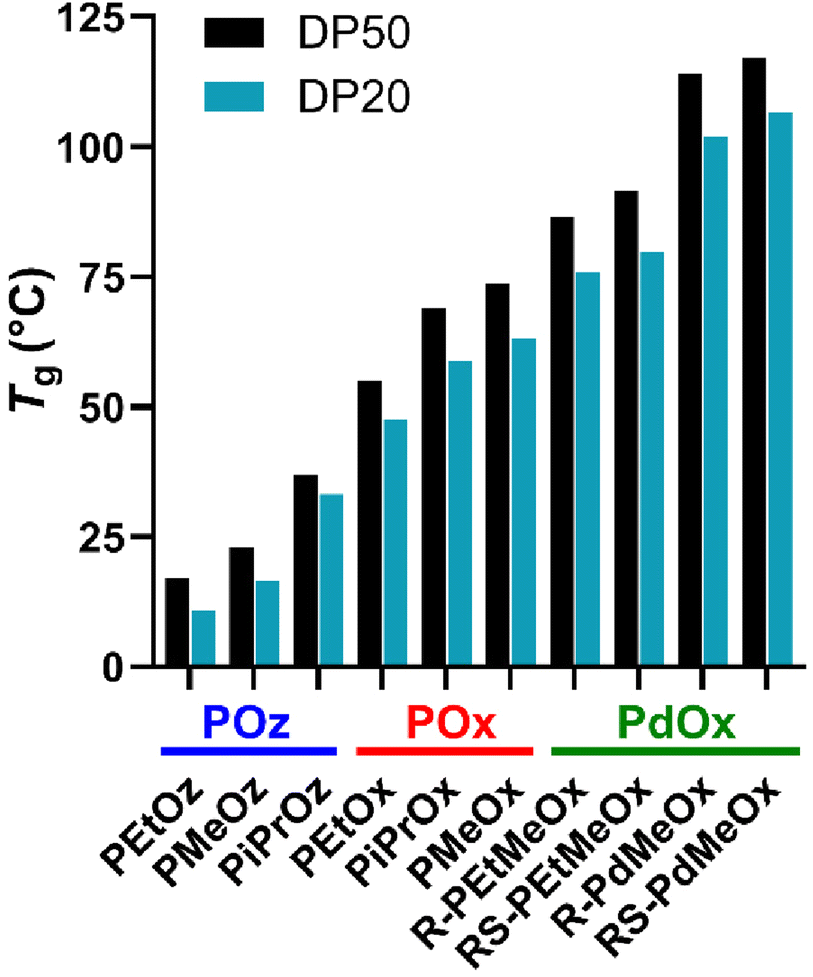 | ||
| Fig. 2 Glass transition temperatures (Tg) for PCIE library, measured from the second heating curve from a 0–150 °C (20 °C min−1) DSC run. DSC traces for individual polymers are included in ESI (Fig. S4†). Polymers are plotted in terms of increasing Tg, with PCIE families labelled in blue (POz), red (POx), or green (PdOx). | ||
Assessment of thermoresponsive behaviour of PCIE via turbidimetry
POx and POz bearing 2-ethyl or 2-(n/i/c)-propyl side chains are known to be thermoresponsive, demonstrating lower solution critical temperature (LCST) behaviour in water, wherein the polymer begins to phase separate at elevated temperatures. This behaviour has been instrumental to the growing interest in using these polymers as copolymer components for thermoresponsive systems.57–59T cp were observed for both DP50 and DP20 samples of PiPrOx, PiPrOz, R-PEtMeOx and RS-PEtMeOx (Fig. 3A and B), and match observations from previous studies.22,30,43 For both DPs, the trend followed PiPrOz < PiPrOx < R/RS-PEtMeOx, with DP20 polymers showing a higher Tcp, indicating a higher degree of hydrophilicity at shorter chain lengths. Indeed, PEtOz50 was observed to have a Tcp of 61.9 °C while no Tcp was observed for the DP20 equivalent (Fig. 3A and B), indicating a “critical” chain length between the two DPs where the polymer switches from fully water soluble to thermoresponsive. This is in opposition to Bloksma et al., who found similarly terminated PEtOz50 to show no Tcp.30 However, similarly to previously reported observations by Bloksma et al., POz demonstrated lower Tcp than POx with the same side group, indicating that the longer backbone may induce a behaviour more closely aligned to a hydrophobic polymer in solution.30 The same study also proposed that side chain identity has a greater effect on Tcp than the additional methylene group in the polymer backbone, since POx structural isomers of poly(n-propyl-2-oxazine) (PnPrOz) in the forms of poly(2-butyl-2-oxazoline) and poly(2-isobutyl-2-oxazoline) are known to be water insoluble, while PnPrOz was seen to be water-soluble. A similar trend was observed in this study (Fig. 3B and C), with PiPrOx50, the structural isomer to PEtOz50, demonstrating a lower Tcp, presumably due to the larger and more hydrophobic isopropyl side chain. R- and RS-PEtMeOx, also structural isomers of PiPrOx, were also observed to have a higher Tcp than PiPrOx at both DP, following the order POz > PdOx > POx (Fig. 3C and Fig. S8†). No differences were observed between stereoisomers, with both chiral R- and racemic RS-PEtMeOx having a similar Tcp, at both DPs. As such, we can see the hydrophilicity of structural isomers follows POz > PdOx > POx, in agreement with observations by Luxenhofer et al.,22 with the influence of 2-side chain within a polymer family following methyl > ethyl > isopropyl, similarly to observations for monosubstituted POx and POz.22,30,43
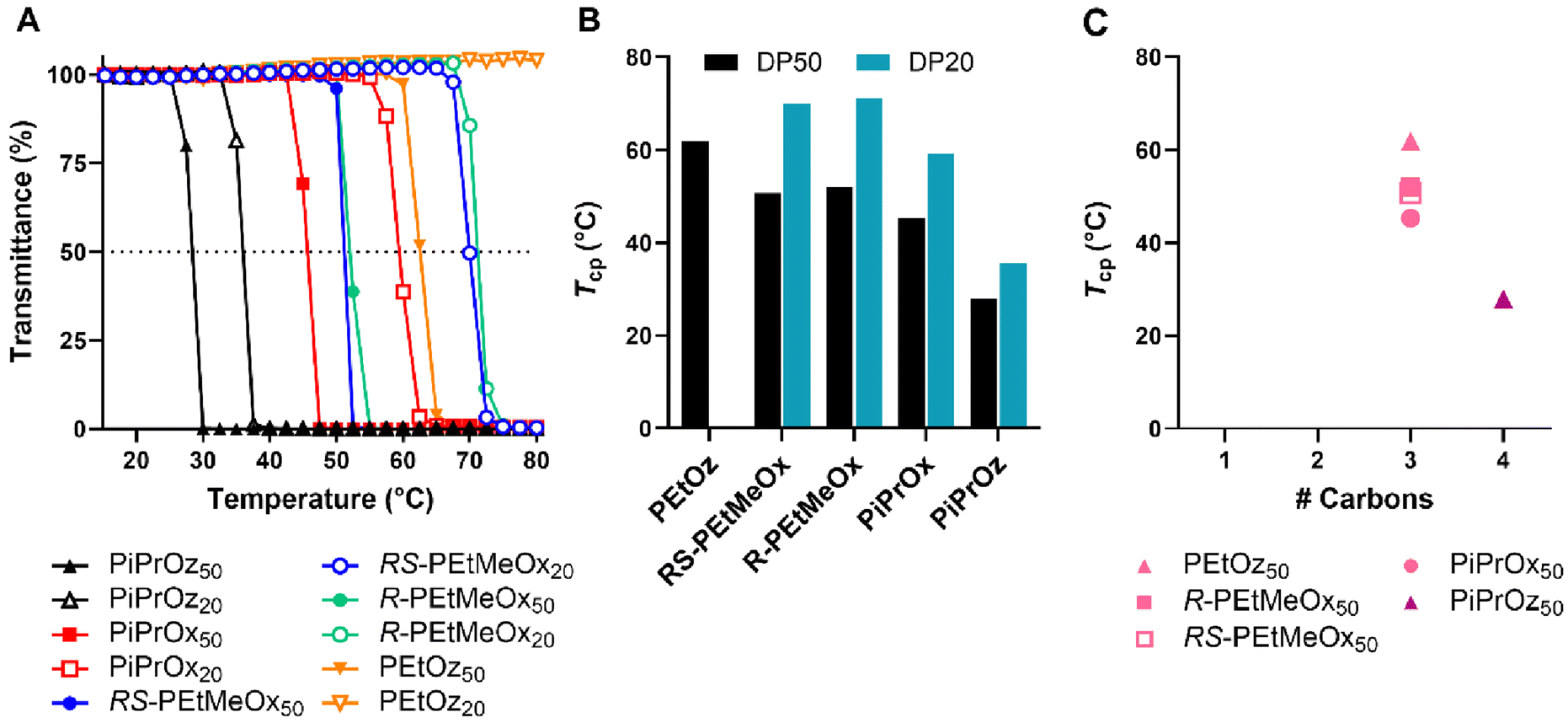 | ||
| Fig. 3 (A) Turbidimetry measurements for all polymers showing a Tcp under tested conditions in MilliQ water (5 mg mL−1 polymer solution heated from 15–80 °C at 1 °C min−1), alongside PEtOz20, which is included as a representative of the curve observed for polymers not demonstrating LCST behaviour under tested conditions. (B) Tcp values for polymers demonstrating LCST behaviour under tested conditions in MilliQ water (determined at 50% transmittance). Additional carbon groupings are denoted as underlines (pink = 3, purple = 4). (C) Tcp values in MilliQ water for DP50 thermoresponsive polymers according to the number of additional carbons as illustrated in Scheme 1B. | ||
To explore salt effects, especially those observed in biologically relevant solutions such as PBS, Tcp of polymers was also assessed in D-PBS. Broadly, Tcp of PCIEs decreased by 1–4 °C in D-PBS as compared to MilliQ water, indicating a “salting out” effect previously described for many PCIE systems (Fig. 4).43,60–63 In the case of D-PBS, the high Cl− concentration is thought to destabilise polymer–water hydrogen bonds, leading to lower Tcps.43 Nevertheless, overall hydrophilicity trends were maintained, with structural isomers following the same POz > PdOx > POx trend.
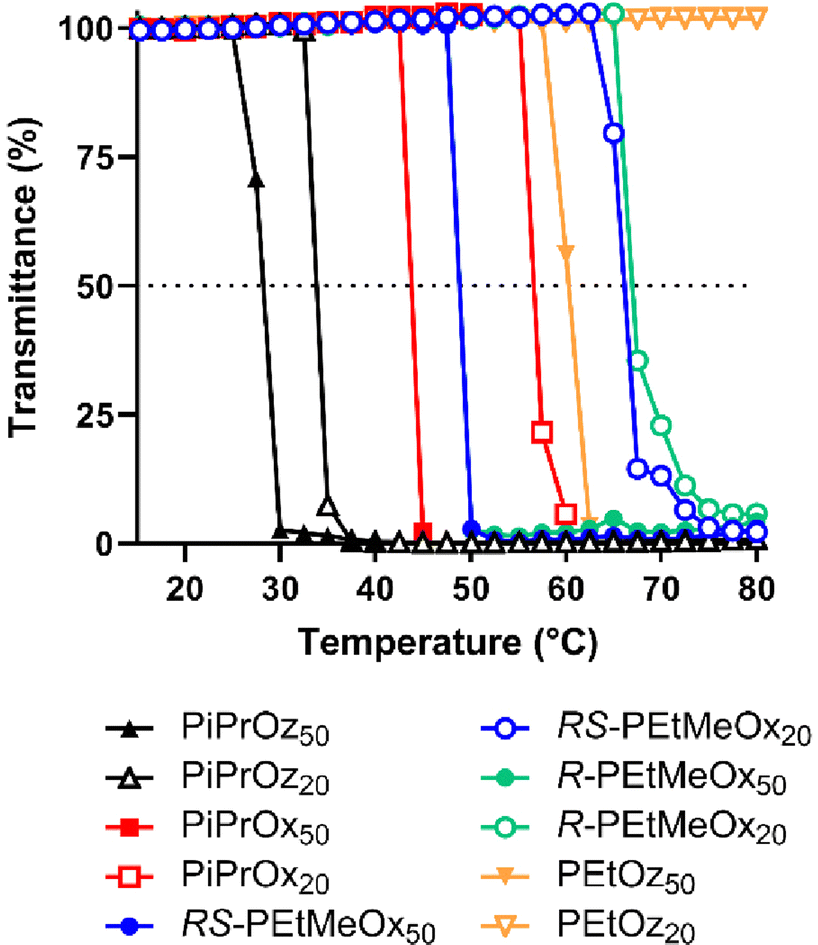 | ||
| Fig. 4 Turbidimetry measurements for all polymers showing a Tcp under tested conditions in D-PBS (5 mg mL−1 polymer solution heated from 15–80 °C at 1 °C min−1), alongside PEtOz20, which is included as a representative of the curve observed for polymers not demonstrating LCST behaviour under tested conditions. | ||
Octanol–water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) KOW)
KOW)
A common method for determination of hydrophilicity/hydrophobicity of small pharmaceutical compounds and, more recently, some polymer systems, is the water–octanol partition coefficient (logKOW). To determine logKOW, the compound or polymer of interest is added to pre-saturated water or octanol, and mixed with the opposing phase until equilibrium is reached, and the concentration in each phase is determined. The concentration of the compound in each phase determines the partitioning: a greater concentration in the aqueous phase indicates higher hydrophilicity and results in logKOW < 0, while a greater concentration in the organic phase indicates hydrophobicity with logKOW > 0. Here, a modified shake-flask technique based on a methodology published in relevant OECD guidelines was utilized to experimentally determine logKOW.64 Moreover, logKOW values were determined in silico. While in silico prediction models have been shown to reliably predict properties such as logKOW for small compounds such as drugs or monomers, this becomes less accurate for larger, more flexible polymer systems, though various derivations such as partition coefficient normalised to molecular or solvent-exposed surface area (logP/SA) have been developed in attempts to predict polymer hydrophilicity behaviour.65–67
To compare with in silico predictions, logP/SA was calculated for each polymer, since this is considered a more accurate prediction model for polymer chains than logP, which was observed to continuously increase/decrease with polymer DP, dependent on predicted hydrophilicity (Fig. 5A).65–67 First, logP/SA were predicted for PMeOx and PiPrOz as examples for a high and low hydrophilic polymer, respectively, up to a DP of 20 (Fig. 5B). DP 15 was selected as the DP at which a value plateau was reached for both polymers, and subsequently used for comparison between polymer systems (Fig. 5C).
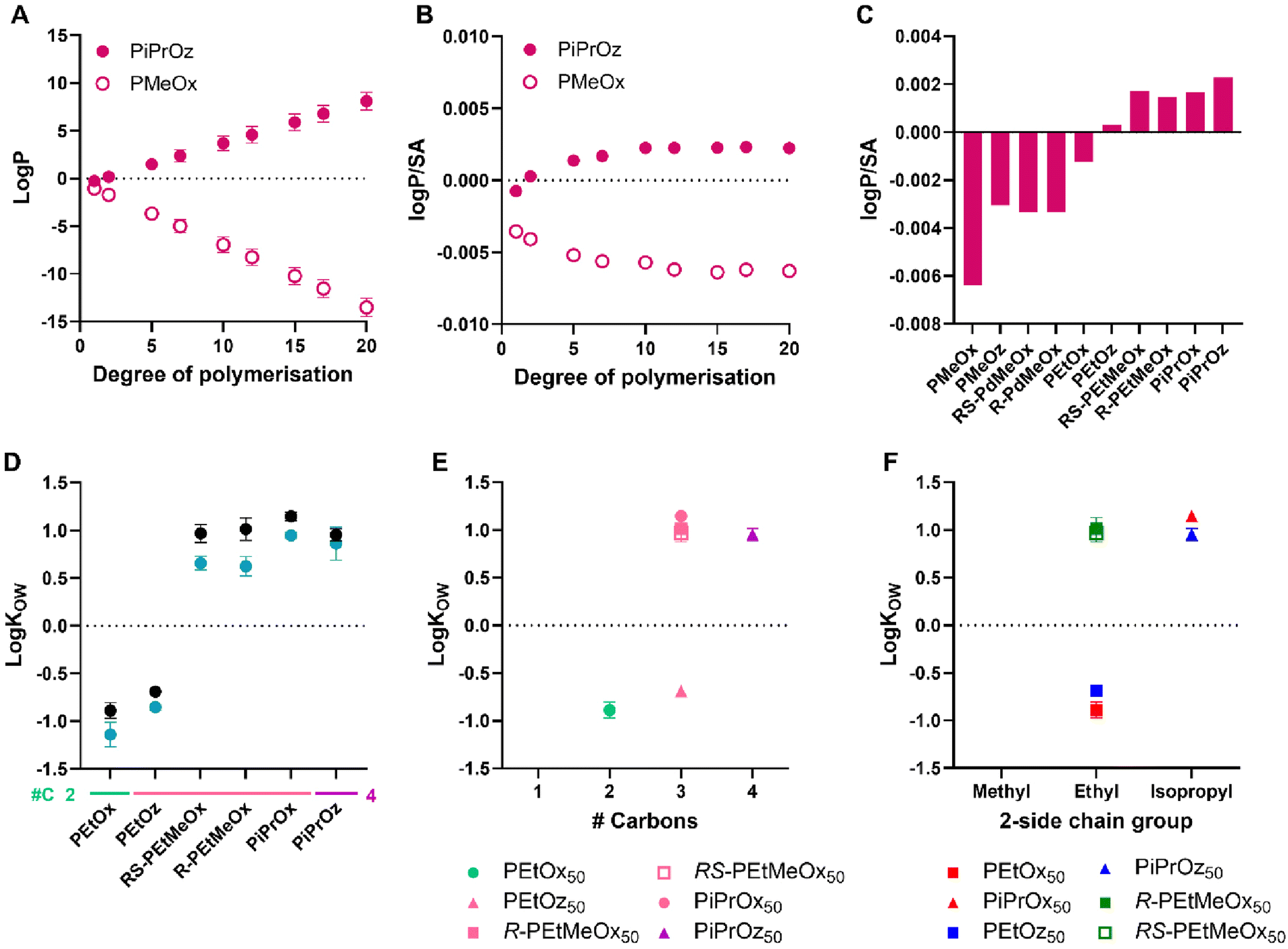 | ||
| Fig. 5 (A) LogP predictions for PMeOx and PiPrOz with increasing degree of polymerisation. (B) LogP/SA predictions for PMeOx and PiPrOz with increasing degree of polymerisation. (C) LogP/SA predictions for PCIEs at degree of polymerisation = 15. (D) LogKOW values for PCIE library at 20 °C. Polymers with no values plotted (PMeOx, PMeOz, R/RS-PdMeOx) showed no detectable partitioning into the 1-octanol phase and were observed only in the aqueous phase, thus logKOW could not be calculated for these polymers. Additional carbon groupings are denoted as underlines (light green = 2, pink = 3, purple = 4). (E) LogKOW values for DP50 polymers, plotted in terms of additional carbons as established in Scheme 1B. (F) LogKOW values for DP50 polymers plotted in terms of 2-side chain group and polymer family as established in Scheme 1A. Experimental error bars represent standard deviation (n = 3). | ||
While the values of experimental logKOW and logP/SA calculations cannot be compared, the trends observed therein can be, and indeed were seen to be similar across both in silico and experimental systems (Fig. 5C and D), with PiPrOx, PiPrOz, and R/RS-PEtMeOx having both higher logP/SA and higher logKOW, indicating lower hydrophilicity. Indeed, only these polymers obtained logKOW values greater than zero, indicating that they are significantly less hydrophilic than other water-soluble PCIEs. PEtOx and PEtOz were observed to have logKOW < 0 at both DPs, indicating stronger hydrophilicity (Fig. 5D). Notably, PEtOz has a positive logP/SA while having a negative logKOW, illustrating the difficulty with in silico prediction use. All polymers containing a methyl group in the 2-substituent position (PMeOx, PMeOz, R/RS-PdMeOx) show the lowest logP/SA, all <−0.002 (Fig. 5C), and also partitioned fully into the aqueous phase for both DPs under the conditions tested, thus logKOW could not be determined for these polymers. In this sense, both logP/SA and (lack of) logKOW show the high hydrophilicity of these polymers. Indeed, Sedlacek et al. reported a partition coefficient of approximately −2.5 for PMeOx80 in a PBS/octanol system,53 indicating high hydrophilicity. In this system, fluorescein-labelled polymers were used to calculate final polymer concentration in each phase, while we instead utilised the absorption of PCIEs at 200 nm to determine concentration, in order to mitigate any potential interactions based on the inclusion of additional moieties such as labelling groups. Sedlacek et al. also reported a similarly hydrophilic value for PEtOx80 (approximately −2), resembling trends found in this study, where PEtOx was seen to be the most hydrophilic polymer detectable using our experimental setup.
Molar mass is observed to affect logKOW values, with lower molar mass polymers being slightly more hydrophilic than higher molar mass polymers (Fig. 5D), potentially due to the higher relative contribution of the hydrophilic –OH end group for shorter polymer chains.
In more detail, POz demonstrated the lowest logKOW of structural isomer groups across both DPs (Fig. 5E and Fig. S9†). Exemplarily, PEtOz, PiPrOx, and R/RS-PEtMeOx are all structural isomers, yet PEtOz had logKOW < 0 while PiPrOx and R/RS-PEtMeOx had logKOW > 0, indicating that PEtOz is significantly more hydrophilic than other polymers with the same number of additional carbons. This is consistent with Tcp, showing again that hydrophilicity follows a POz > PdOx > POx trend for structural isomers. Similarly, if we look at a fixed side chain, such as all polymers with a 2-ethyl side chain (PEtOx, PEtOz, and R/RS-PEtMeOx), we can investigate which structural factors affect hydrophilicity (Fig. 5F and Fig. S9†). In this sense, we see that hydrophilicity follows POx > POz > PdOx, wherein both POz and PdOx have an additional carbon group compared to POx. Again, PdOx was seen to be less hydrophilic than POz, indicating that the inclusion of additional backbone functionality is more influential to the overall hydrophilicity of the polymer than increased backbone length; replicating the trend observed by Bloksma et al. and Luxenhofer et al.22,30
The influence of additional backbone functionality on hydrophilicity correlates with observations by Pizzi et al., where Cy5-labelled R-, S-, and RS-poly(oligo(2-ethyl-4-methyl-2-oxazoline)methacrylate) (R/S/RS-P(OEtMeOxMA)Cy5) bottle-brushes completely partitioned in the octanol phase.21
For 2-isopropyl containing polymers, the POx > POz trend was not observed, with logKOW of PiPrOz observed to be slightly lower than PiPrOx at both DPs. Across both DPs, structural isomer and 2-side chain trends remained the same (Fig. S9†). Overall, logKOW analysis resulted in the formation of three polymer groupings: very hydrophilic polymers which partitioned completely in the aqueous phase (PMeOx, PMeOz, R/RS-PdMeOx); hydrophilic polymers with 2–3 additional carbons and logKOW < 0 (PEtOx, PEtOz); and less hydrophilic polymers with 3–4 additional carbons and logKOW > 0 (PiPrOx, PiPrOz, R/RS-PEtMeOx).
Separation of PCIEs by HPLC
Similar to logKOW, separation via HPLC is a commonly used technique for the separation of hydrophilic and hydrophobic compounds in solution. Kim et al. have previously reported the successful separation of a family of hydroxyl-substituted POx via HPLC, showing that the inclusion of pendant hydroxyl groups increases the hydrophilicity of the polymer relative to PEtOx of the same DP.50 Sedlacek et al. similarly separated a library of POx with varying oligoether side chains, alongside PMeOx and PEtOx comparisons, and found poly(2-methoxymethyl-2-oxazoline) (PMeOMeOx) to be the most hydrophilic.53 More recently, Engel et al. separated a library of PMeOx and PEtOx polymers with various end-groups.51 Similarly to existing papers, a gradient methodology was utilised in this study, allowing the successful resolution of all 20 polymers under one method.
More hydrophilic polymers were seen to have retention times of <7 minutes across both DPs, and less hydrophilic polymers demonstrating retention times >7 minutes across both DPs (Fig. 6A and Fig. S10†). A visible jump in retention time is observed between PEtOz and RS-PEtMeOx in both DP50 and DP20 groups, indicating a clear divide between polymer groupings, similar to the hydrophilicity split observed for logKOW measurements. Retention time followed the same hydrophilicity pattern of POz > PdOx > POx for structural isomers (Fig. 6B and Fig. S11†), with the exception of R-PEtMeOx50 ∼ PiPrOx50. Interestingly, PiPrOx50 was again seen to be slightly less hydrophilic than PiPrOz50, while the reverse was seen for DP20 equivalents. Since PiPrOx and PiPrOz have both been the lest hydrophilic polymers in different tests thus far (Tcp: PiPrOz; logKOW: PiPrOx), we hypothesize that the two polymers have similar hydrophilicities, with test-specific factors such as solvent and, in the case of HPLC, molar mass, influencing which of the two polymers appear the least hydrophilic. Moreover, R-PEtMeOx50 is seen to have a marginally higher retention time than PiPrOx50, making it the least hydrophilic polymer assessed via HPLC. This is in contrast to RS-PEtMeOx50, which lies below both PiPrOx50 and PiPrOz50, indicating that R-PdOx may be less hydrophilic than RS-PdOx isomers. Indeed, the same difference was observed for PdMeOx50, and for both PdOx at lower DPs (Fig. S11†).
 | ||
| Fig. 6 (A) HPLC retention times for PCIE library (20–80% MQ-ACN gradient, C8 column). 0.5 mg mL−1 polymer in MQ-0.1% FA solutions were used with an injection volume of 10 μL. Additional carbon groupings are denoted as underlines (yellow = 1, light green = 2, pink = 3, purple = 4). (B) Retention times for DP50 polymers, plotted in terms of additional carbons as established in Scheme 1B. (C) Retention times for DP50 polymers, plotted in terms of 2-side chain group and polymer family as established in Scheme 1A. | ||
HPLC further allowed the determination of a hydrophilicity order for the fully water-soluble 2-methyl-containing polymers, which previously could not be differentiated via turbidimetry or logKOW measurements due to their high hydrophilicity. We see that, for both DPs, hydrophilicity follows PMeOx > PMeOz > R/RS-PdMeOx, establishing PMeOx as the most hydrophilic polymer in this library. While Kim et al. and Sedlacek et al. explored functionally-substituted-POx polymer libraries under different elution regimes,50,53 both utilised HPLC to measure retention time of PMeOx and PEtOx. Indeed, both observed similarly low retention times for PMeOx as in this study, reinforcing its high hydrophilicity. Similarly, both aforementioned papers and Rettler et al. investigated PMeOx and PEtOx retention times, with all finding PEtOx to have a higher retention time than PMeOx, and Rettler et al. finding PMeOx to have a lower retention time than a similarly sized PEG control.32,50,53 Nevertheless, to the best of the author's knowledge, no other study to date has assessed the retention time of non-POx PCIEs, e.g. the POz and PdOx included in this study, nor POx in relation to the full series of materials explored here.
Similar to logKOW trends, for polymers with the same 2-methyl- or ethyl-side chain, hydrophilicity followed a POx > POz > PdOx trend (Fig. 6C). Similar trends are observed for DP20 polymers, with the retention times for DP20 polymers seen to be lower than DP50, indicating increased hydrophilicity (Fig. 6A and Fig. S11†).
Surface tension of aqueous polymer solutions
The surface tension of a compound in solution is controlled by its interactions with the solvent and with the solvent–air interface.68 Thus, if assessed with water as a solvent, the surface tension of polymer solutions can provide an estimate of polymer hydrophilicity. Typically, the more hydrophobic regions of polymers will reorient themselves in solution such that hydrophobic components are aligned with the water–air interface rather than contained within the aqueous matrix, reducing the measured surface tension of the solution.69 This was observed for our PCIE library, with less hydrophilic polymers such as PiPrOx and PiPrOz demonstrating a lower surface tension than more hydrophilic PMeOx and PMeOz (Fig. 7A), indicating greater hydrophobicity. Further, since both surface tension is not dependent on UV-region absorbance detection in the same way that the logKOW and HPLC methodologies were, a PEG control was added, allowing us to assess PCIE properties against a widely used hydrophilic polymer control. mPEG5000 was selected for its similar molecular weight to DP50 PCIEs, and to maintain similar α- and ω-end groups to the synthesized PCIEs.
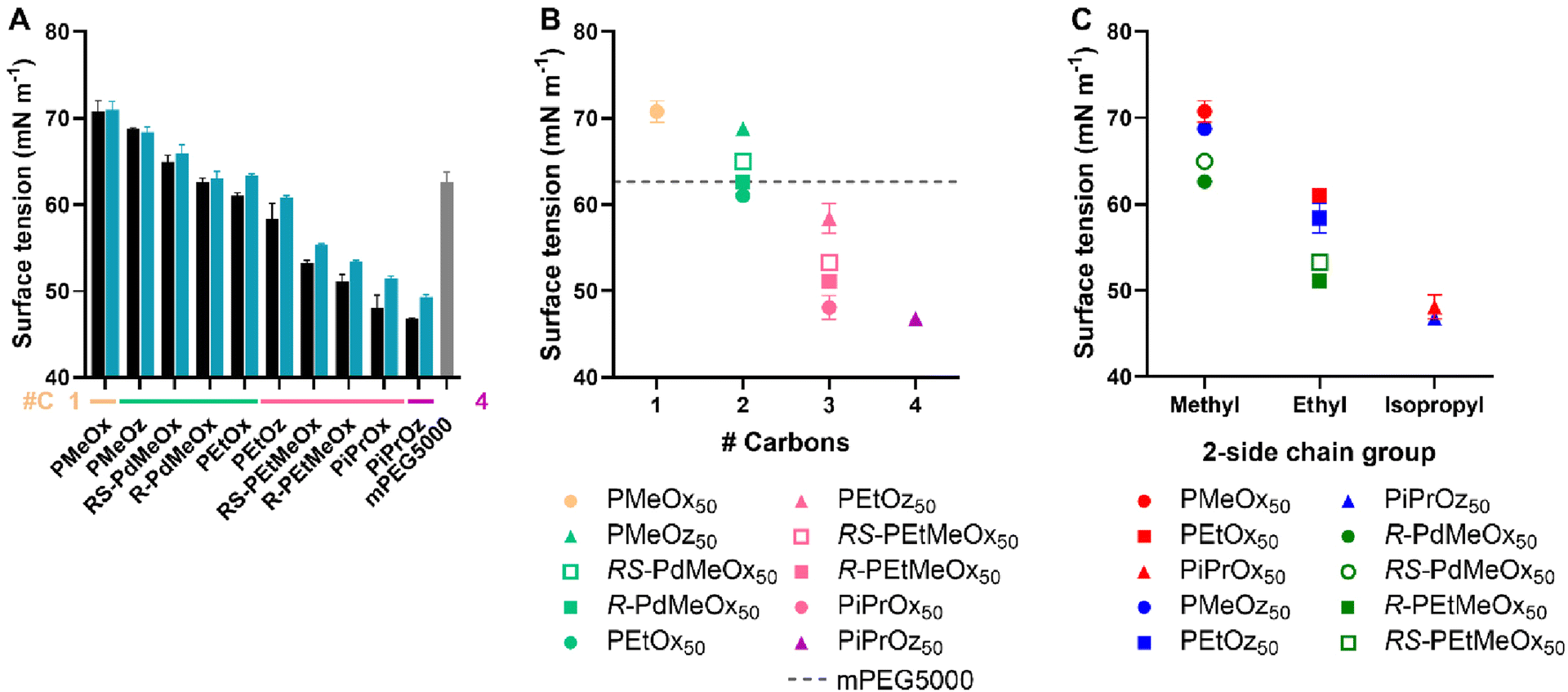 | ||
| Fig. 7 (A) Surface tension for 1 mg mL−1 polymer in MQ-0.1% FA solution. Additional carbon groupings are denoted as underlines (yellow = 1, light green = 2, pink = 3, purple = 4) (B) Surface tension for DP50 polymers, plotted in terms of additional carbons as established in Scheme 1B. (C) Surface tension for DP50 polymers, plotted in terms of 2-side chain and polymer family as established in Scheme 1A. Error bars represent standard deviation (n = 3). | ||
The trends observed in surface tension largely followed those established by HPLC and logKOW. Namely, polymers with a 2-methyl-group were the most hydrophilic, followed by polymers with a 2-ethyl-group, and finally polymers with a 2-isopropyl group. Within these groupings, the hydrophilicity trend followed POx > POz > PdOx, inclusive of isopropyl-containing polymers; opposing the variable isopropyl trend observed for logKOW and HPLC. Similarly, structural isomers followed the POz > PdOx > POx hydrophilicity trend established in previous tests (Fig. 7B). Molar mass trends broadly followed those observed for Tcp, logKOW and HPLC, with lower DP polymers generally having a higher surface tension and thus appearing more hydrophilic (Fig. 7A and Fig. S12†). Interestingly, minimal difference was observed between DPs for 2-methyl-containing polymers, indicating that the effects of molar mass on the surface tension of very hydrophilic PCIEs appear to be negligible. Similar to HPLC, R-PdOx were seen to have lower surface tension values and thus appear marginally less hydrophilic than the RS-PdOx stereoisomer, and the same POx > POz > PdOx hydrophilicity trend was observed for polymers with the same side chain group (Fig. 7C). mPEG5000 was seen to have a surface tension approximately equivalent to R-PdMeOx, making it similarly hydrophilic and showing it to lie on the more hydrophilic side of the polymer library (Fig. 7A and B). This places mPEG5000 as less hydrophilic than PMeOx50 and PMeOz50 but more hydrophilic than PEtOx50, which corroborates a similar trend observed by Viegas et al. via HPLC.10 This also presents PdMeOx as an interesting potential PEG alternative, though the slow polymerisation rate of PdOx compared to POx or POz may be considered a drawback.22
1H NMR relaxometry
1H NMR relaxometry was explored to assess the molecular mobility of polymer chains in water, with the aim of correlating this to predict the hydrophilicity of water-soluble polymers in D2O. Since all 2-methyl-containing polymers were previously assessed to be highly soluble in aqueous systems – indeed being inseparable via logKOW and Tcp measurements – this sub-group of polymers was selected for assessment to ensure that solubility of polymer chains would not confound data acquisition. 1H NMR relaxometry is a powerful tool by which the molecular mobility of polymer chains can be studied, with long-range interactions such as total polymer flexibility being described by spin–spin relaxation time (T2), and shorter-range interactions being described by spin–lattice relaxation time (T1).70T1 for all polymers was determined to be ≤ 1.04 seconds, indicating similar local mobility of polymer fragments (Table S4†). T2 was best fitted with a double exponential decay curve, indicating the presence of two relaxation regimes: T2(fast) and T2(slow) (Fig. S13†). T2(fast) describes the movement of more rigid segments of polymeric assemblies and was seen to be <0.1 s for all polymers, with R/RS-dMeOx50, in particular, showing T2(fast) of ∼0.015 s (Table 1), indicating a more rigid polymer structure in D2O comparative to PMeOx50 or PMeOz50. T2(slow) better describes the more flexible motion of polymer chains, and indeed was seen to differ greatly between polymers, with the more flexible PMeOz50 demonstrating the highest T2(slow), followed by PMeOx50, and finally R/RS-PdMeOx50.
| Polymer | T 2(slow) (s) | T 2(fast) (s) |
|---|---|---|
| PMeOz50 | 0.611 | 0.055 |
| PMeOx50 | 0.360 | 0.079 |
| R-PdMeOx50 | 0.131 | 0.016 |
| RS-PdMeOx50 | 0.151 | 0.017 |
Since a larger T2 value typically indicates greater mobility as a result of a more favourable interaction with the surrounding solvent, a larger T2 in D2O can be used to indicate greater hydrophilicity of the polymer. Irrespective of the component that was assessed from the double exponential decay in the measurement of the spin–spin relaxation time (T2(slow) or T2(fast)), the T2 of R/RS-PdMeOx50 is seen to be significantly shorter than either of the more flexible polymers, and thus can be concluded to be the least hydrophilic of the 2-methyl containing polymers, corresponding to trends observed in HPLC and surface tension measurements. Contrary to surface tension and HPLC trends, PMeOz was found to be more mobile and thus more hydrophilic than PMeOx. No major difference was seen between T2 values for R- and RS-PdMeOx50, suggesting minimal effect from secondary structure formation in D2O under the conditions tested, though further studies in this area are encouraged.
Comparison of techniques
As previously discussed, hydrophilicity affects a range of polymer behaviours across biomedical applications, including cellular uptake, drug loading capacity, antifouling and “stealth” behaviour, and biodistribution.35–41,71,72 As such, understanding hydrophilicity of biomedical polymers such as PCIEs and how to modulate it will allow for the design of precisely-engineered polymer therapeutic systems, capable of being tailored to specific applications. Techniques such as turbidimetry and logKOW as designed in this study, while useful indicators of relative hydrophilicity, were unable to differentiate the most water-soluble polymers in the library, hampering their use in the assessment of very hydrophilic systems. Similarly, in silico prediction of logP/SA was found to accurately predict trends observed in experimental logKOW measurements and as such is useful for predicting comparative differences in PCIE hydrophilicity, but could not reliably predict logP values for long polymer chains. HPLC and surface tension in aqueous solution were able to differentiate between all PCIEs in the library, with the note that PMeOx20, the most hydrophilic polymer, was on or close to the limit of detection for many of the tests. Relaxometry was used to study the hydrophilicity of the very hydrophilic PMeOx, PMeOz and R/RS-PdMeOx, and found R/RS-PdMeOx to be significantly less mobile and thus potentially less hydrophilic than the equivalent POx or POz systems. As such, techniques capable of evaluating the entire polymer library – namely HPLC and surface tension – are recommended methodologies by which to elucidate fine differences in the hydrophilicity of polymers, particularly when considering the ease of measurement for techniques such as surface tension.
Specific trends were elucidated for structural isomers and polymers with the same 2-side chain, allowing a “ranking” of polymers in terms of apparent hydrophilicity (Scheme 2). More specifically, structural isomers followed a POz > PdOx > POx hydrophilicity trend, while polymers with the same 2-side chain followed a POx > POz > PdOx trend. Differences in hydrophilicity between stereoisomers R- and RS-PdOx were established in some tests (HPLC, surface tension), but were not observed in most (Tcp, lowKOW, relaxometry), indicating that the effect of stereoisomerism on the hydrophilicity of PdOx is minimal. Across both structural isomer and side chain groupings, the inclusion of additional backbone functionality is seen to reduce the hydrophilicity of PCIE systems to a greater extent than the addition of a methylene unit in the polymer backbone. Thus, the incorporation of PdOx into PCIE copolymers presents an attractive method with which to modulate polymer hydrophilicity, with the added benefit of introducing an additional avenue of polymer functionalisation (e.g. by smart design of the 4-group).
 | ||
| Scheme 2 Relative hydrophilicity ranking of PCIE with mPEG5000 included as comparison. | ||
Haemocompatibility of PCIEs
Since nanomaterials are expected to circulate in the blood for hours to days, PCIE haemocompatibility is paramount. While the haemocompatibility of individual PCIE systems have been probed in various studies,7,13,43,73,74 none yet have encompassed all PCIEs included in this library. In this sense, to ensure the safety of these polymers in nanomedical applications, the hemocompatibility of polymers in the library was assessed using blood plasma from Balb/c nude mice. All PCIEs demonstrated <2% red blood cell (RBC) lysis in 0.5 mg ml−1 polymer solution, indicating good haemocompatibility (Fig. 8).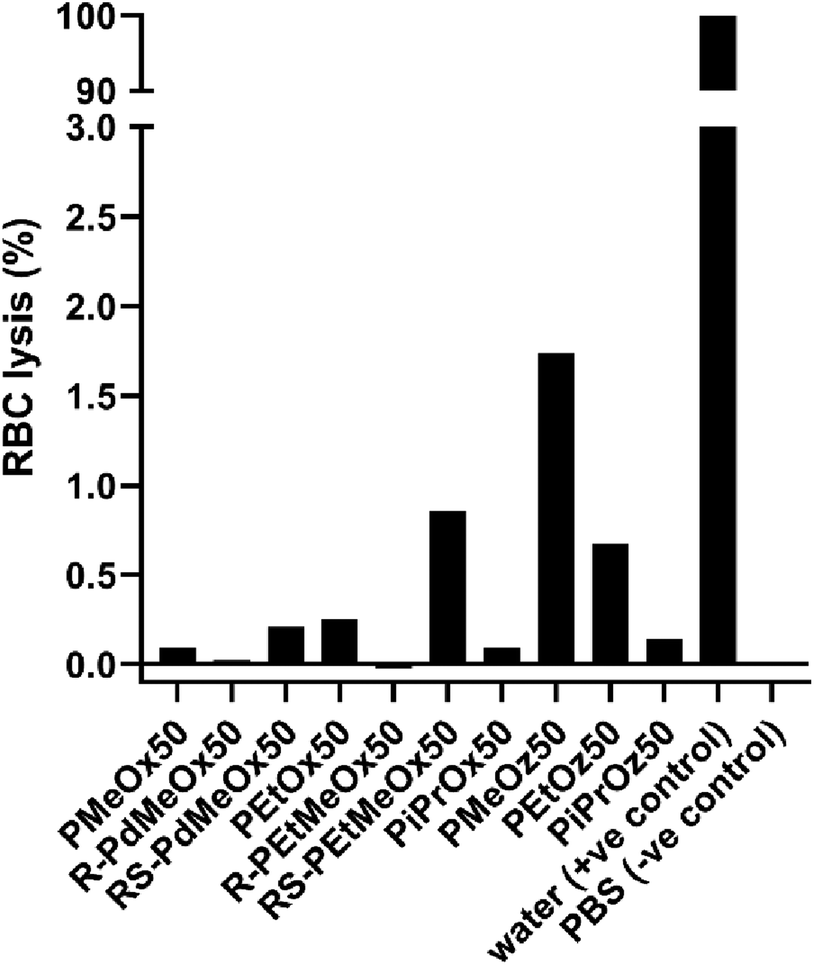 | ||
| Fig. 8 Red blood cell (RBC) lysis in the presence of 0.5 mg ml−1 polymer solution. A split y-axis is included for plotting of the positive control compared to the low RBC lysis seen for PCIE samples. | ||
Conclusions
A library of water-soluble PCIE was successfully synthesised and assessed on the basis of hydrophilicity through multiple tests in solution. Further, all PCIEs studies were confirmed to be non-haemolytic at 0.5 mg ml−1, illustrating their safety for use in nanomedicine. Broadly, polymers had varied degree of hydrophilicity, ranging from highly hydrophilic for PMeOx, PMeOz, and R/RS-PdMeOx, through hydrophilic for PEtOx and PEtOz, and down to the less hydrophilic PiPrOx, PiPrPOz, and R/RS-PEtMeOx. Trends predicted via in silico logP/SA calculations correlated well with experimental measurements such as logKOW, HPLC retention time, and surface tension, indicating a good ability to predict PCIE trends, but could not accurately predict logP values for long polymer chains. Trends among structural isomers and 2-side chain groups were elucidated as follows: structural isomers broadly followed a POz > PdOx > POx hydrophilicity trend, while polymers with the same 2-side chain followed a POx > POz > PdOx trend. Overall, fine differences in the hydrophilicity of a water-soluble PCIE library were established, and the knowledge presented in the elucidation of these trends will assist in the future design of hydrophilicity-tailored, application-specific PCIE systems for use in bio- and nanomedicine.
Materials and methods
Materials
Hypergrade acetonitrile (>99.9%), anhydrous acetonitrile (ACN) (99.8%), diethyl ether (>99.5%), dichloromethane (≥99.8%), methanol, 1-octanol (≥99%), chloroform (≥99.8%), petroleum benzine (60–80 °C b.p.), potassium hydroxide (KOH) (≥85%), tetramethylammonium hydroxide (TMAH) (25 wt% in MeOH), formic acid (95%), zinc acetate dihydrate (≥99%), propionitrile (99%), isobutyronitrile (99.6%), 3-amino-1-propanol (99%), 2-aminoethanol (≥99%), calcium hydride (95%), barium oxide (97%), and poly(ethylene glycol) methyl ether Mn = 5000 (mPEG5000) were purchased from Sigma-Aldrich and used as received. R-(−)-2-amino-1-propanol (98%) and RS-(±)-2-amino-1-propanol (97%) were purchased from AA BLOCKS and used as received. Deuterated chloroform (99.8%) was purchased from Cambridge Isotope Labs.2-Methyl-2-oxazoline (MeOx) (98%), 2-ethyl-2-oxazoline (EtOx) (>99%), and methyl p-toluenesulfonate (MeOTs) (98%) were purchased from Sigma-Aldrich, dried over barium oxide, and distilled to dryness before use.
Monomer synthesis
2-Isopropyl-2-oxazoline (iPrOx), 2-methyl-2-oxazine (MeOz), 2-ethyl-2-oxazine (EtOz), 2-isopropyl-2-oxazine (iPrOz), R/RS-2,4-dimethyl-2-oxazoline (R/RS-dMeOx) and R/RS-2-ethyl-4-methyl-2-oxazoline (R/RS-EtMeOx) were synthesised according to literature.22,30,43,75 Detailed procedures are included in the ESI.†Polymer synthesis
Polymers were synthesised via the cationic ring-opening polymerization (CROP) of POx, POz, or PdOx monomers as reported previously,22,30,43,49 with each polymer being synthesised at two [M/I] ratios: [M/I] = 20 (DP20) and [M/I] = 50 (DP50). Detailed polymer synthesis procedures are included in ESI.†Polymer characterisation
1H NMR spectra were obtained for each polymer using a Bruker AVANCE III HD 400 MHz spectrometer using CDCl3 as solvent. Molar mass and dispersity of polymers were determined using a Shimadzu modular system size exclusion chromatography (SEC) instrument comprising a DGU-12A degasser, an SIL-20 AD automatic injector, a 5.0 μm bead-size guard column (50 × 7.8 mm) followed by three KF-805 L columns (300 × 8 mm, bead size: 10 μm, pore size maximum: 5000 Å), an SPD-20A ultraviolet detector, and a RID-10A differential refractive index detector. N,N-Dimethylacetamide (DMAc) containing 0.03 wt% LiBr was used as the eluent, with all samples being filtered through 0.45 μm PTFE membranes before injection. Molar mass and dispersity of samples were determined comparative to polystyrene standards (0.5 to 2000 kg mol−1), with Shimadzu LabSolutions software being used to calculate values.DSC measurements
Thermal transitions of polymers were determined on a TA-2500 DSC (TA instruments) under nitrogen flow. For Tg, 10 mg of sample was placed in a pierced hermetically sealed pan and subjected to one heat/cool cycle from 0 °C to 150 °C at 20 K min−1 and a subsequent heat to 150 °C at the same rate. Tg values were determined from the second heating run. Since only PiPrOx has been reported to show any semicrystalline behaviour, Tm measurements were limited to PiPrOx20 and PiPrOx50. 10 mg of sample in a hermetically sealed pan was heated from 0–230 °C at 20 K min−1 following a preliminary heat to 150 °C (20 K min−1) to remove solvent effects. Tm was measured from the first heating curve.Cloud point temperature measurements
The cloud point temperature of samples was measured using a Shimadzu UV-3600 UV-Vis-NIR spectrophotometer. 5 mg mL−1 of each polymer was dissolved in MilliQ (MQ) water or D-PBS, filtered through a 0.45 μm PTFE filter and heated from 15–80 °C at 1 °C min−1. Absorbance was measured at 500 nm and Tcp was determined as the temperature corresponding to 50% transmittance.LogP and logP/SA predictions
LogP and surface area values were determined similar to a previously reported procedure,65 using the following software, ACD Labs and Chem3D 21.0.0. The chemical structures were composed and minimised with the MM2 force field. The solvent accessible surface area was calculated using a 1.4 Å probe.
Determination of logKOW
LogKOW of polymers was determined via a modified shake flask method. Briefly, MilliQ water and 1-octanol were mutually saturated for 24 hours, before separation into water-saturated 1-octanol (sat-oct) and 1-octanol-saturated MQ water (sat-MQ). 0.1 mg mL−1 solutions of each polymer were created in sat-MQ. 700 μL of each polymer solution was placed in an Eppendorf tube and 700 μL of sat-oct was added. Samples were shaken overnight at 175 rpm in a shaker incubator at room temperature, followed by centrifugation at 8000 rpm for 5 minutes to ensure separation of phases. Phases were then manually separated and a 100 μL aliquot of the aqueous phase sample was diluted 10-fold with fresh MQ water. 100 μL aliquots of the organic phase were dried in a vacuum oven for 24 hours at 25 °C to remove 1-octanol, then resuspended in a solvent mixture of 100 μL sat-MQ and 900 μL fresh MQ water, ensuring that diluted organic phase solutions matched the solvent composition of diluted aqueous phase solutions. The concentration of polymer in each phase was determined via the use of a standard calibration curve measured with a Shimadzu UV-3600 UV-Vis-NIR spectrophotometer, with absorbance measured at 200 nm. LogKOW was calculated from eqn (1) below, where [P]oct = polymer concentration in the 1-octanol phase and [P]aq = polymer concentration in the aqueous phase. Measurements were carried out in triplicate.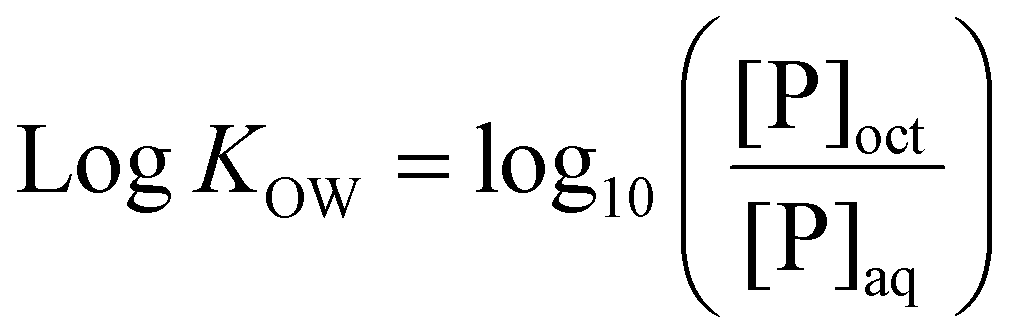 | (1) |
Separation of PCIEs via HPLC
0.5 mg mL−1 solutions of all polymers were created in MQ + 0.1% formic acid (FA) and filtered through a 0.45 μm PTFE membrane before injection. Samples (10 μL injection volume) were separated via gradient elution using MQ + 0.1% FA (Phase A) and hypergrade acetonitrile (Phase B) through a C8 column (Pinnacle DB C8 5 μm, 150 × 4.6 mm, Restek) at a flow rate of 1 mL min−1. The gradient program is as follows: 0–0.5 min: 20% phase B; 0.5–10.5 min: 20–80% phase B, 10.5–11.5 min: 80% phase B, 11.5–11.8 min: 80–20% phase B, 11.8–12 min: 20% phase B, totalling 12 minutes. A Shimadzu CBM-20A HPLC System Controller fitted with DGU-20A degassing units, a CTO-20AC oven, LC30AD pumps, and an SPD-M30A PDA detector was used. The column was held at 30 °C and absorbance was measured via a PDA UV-Vis detector at 203 nm (the maximum absorbance for most polymers in the chromatogram), with retention time determined using Shimadzu LabSolutions.Surface tension measurements
The same stock solutions as used in HPLC (1 mg mL−1 MQ + 0.1%FA) were subsequently used to determine the surface tension of polymer solutions via the pendant drop method. A 5 μL droplet was suspended and the surface tension was calculated using an Attension Theta Flex optical tensiometer (Biolin Scientific) and a Young-Laplace fit. Measurements were carried out in triplicate.1H NMR relaxometry
NMR relaxometry experiments were performed using a Bruker Avance III HD 700 MHz spectrometer at 298 K. Each polymer sample was dissolved in deuterium oxide (D2O) at 5 mg mL−1 and passed through a PTFE 0.22 μm filter. 500 μL of each sample was aliquoted into an NMR tube. The spin–lattice relaxation time (T1) was determined by the inversion recovery experiment., systematically incrementing the delay between the 180° pulse and the 90° pulse (t), and T1 relaxation times were obtained using the T1 fitting function in Dynamics Centre 2.8.3 (eqn (2)).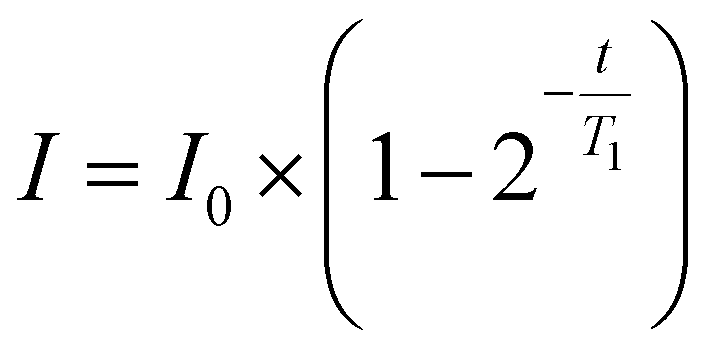 | (2) |
Spin–spin relaxation times (T2) were determined using the Carr–Purcell–Meiboom–Gill (CPMG) pulse sequence. T2 relaxation times were obtained by plotting peak integral vs. the length of the CPMG sequence, and values were determined through nonlinear regression curve fitting of a two-phase decay model using GraphPad Prism 9.
Details of t and CPMG pulse sequences can be found in ESI (Tables S2 and S3,† respectively).
Haemocompatibility testing of PCIEs
To measure the haemolysis percentage of the DP50 polymers, whole blood was collected from Balb/c nude mice as into EDTA coated tubes according to ethics application 2022/AE000135 as approved by UQ Animal Ethics Committee, and centrifuged at 1000g for 4 min. Plasma was then removed and the erythrocyte portion was washed with PBS (pH 7.4) twice at the above setting, and the supernatant was discarded. Finally, the blood cells were diluted to the initial volume by adding fresh PBS. To prepare the assay mixture, the polymer solutions (in PBS) and the blood cell suspension was mixed in equal volume (1:1, 0.5 mg mL−1 final polymer concentration). Blood cells mixed 1:1 with RO water was used as a positive control, while the PBS and whole blood mixture were taken as a negative control. All samples were incubated at 300 rpm in a thermomixer and maintained at 37 °C. After 1 h, the samples were centrifuged at 1000g for 4 min and 100 μL of the supernatant collected. These were then diluted 1:25 with PBS to achieve absorbance readings of the positive control of <1 and the OD value of the supernatant was measured at 540 nm using a Tecan plate reader.
Finally, the haemolysis percentage (HP) was calculated using the following formula (eqn (3)):
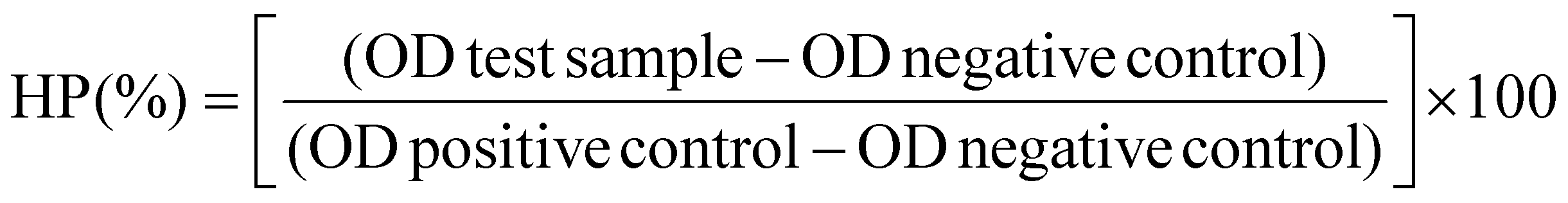 | (3) |
Author contributions
K. M.: methodology, investigation, formal analysis, validation, writing – original draft, and visualization; J. P. M.: methodology, investigation, formal analysis, and writing – review and editing; N. M. W.: methodology, investigation, formal analysis, and writing – review and editing; X. H.: methodology, investigation, formal analysis, visualization, and writing – review and editing; D. P.: methodology, investigation, and writing – review and editing; S. Z. O. S.: investigation, and writing – review and editing; G. K. P.: methodology, writing – review and editing; N. L. F.: writing – review and editing, and supervision; C. A. B.: methodology, writing – review and editing, and supervision; K. J. T.: methodology, writing – review and editing, and supervision; K. K.: methodology, conceptualization, writing – review and editing, supervision, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the support provided to this project by the HMSTrust Analytical Laboratory, based at the Monash Institute of Pharmaceutical Sciences. The authors also wish to acknowledge Dr Leonie van't Hag and Alice Tiong for the kind use of their equipment. K. K. gratefully acknowledges the award of an ARC Future Fellowship (FT190100572) from the Australian Research Council (ARC). KJT acknowledges the NHMRC (APP1148582) and ARC for support (IC170100035).References
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed
.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS PubMed
- M. Barz, R. Luxenhofer, R. Zentel and M. J. Vicent, Polym. Chem., 2011, 2, 1900–1918 RSC
- Y. Ju, W. S. Lee, E. H. Pilkington, H. G. Kelly, S. Li, K. J. Selva, K. M. Wragg, K. Subbarao, T. H. O. Nguyen, L. C. Rowntree, L. F. Allen, K. Bond, D. A. Williamson, N. P. Truong, M. Plebanski, K. Kedzierska, S. Mahanty, A. W. Chung, F. Caruso, A. K. Wheatley, J. A. Juno and S. J. Kent, ACS Nano, 2022, 16, 11769–11780 CrossRef CAS PubMed
- P. Zhang, F. Sun, S. Liu and S. Jiang, J. Controlled Release, 2016, 244, 184–193 CrossRef CAS PubMed
- G. T. Kozma, T. Shimizu, T. Ishida and J. Szebeni, Adv. Drug Delivery Rev., 2020, 154–155, 163–175 CrossRef CAS PubMed
- M. Bauer, C. Lautenschlaeger, K. Kempe, L. Tauhardt, U. S. Schubert and D. Fischer, Macromol. Biosci., 2012, 12, 986–998 CrossRef CAS PubMed
- J. Svoboda, O. Sedláček, T. Riedel, M. Hrubý and O. Pop-Georgievski, Biomacromolecules, 2019, 20, 3453–3463 CrossRef CAS PubMed
- R. Konradi, C. Acikgoz and M. Textor, Macromol. Rapid Commun., 2012, 33, 1663–1676 CrossRef CAS PubMed
- T. X. Viegas, M. D. Bentley, J. M. Harris, Z. Fang, K. Yoon, B. Dizman, R. Weimer, A. Mero, G. Pasut and F. M. Veronese, Bioconjugate Chem., 2011, 22, 976–986 CrossRef CAS PubMed
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed
- N. Adams and U. S. Schubert, Adv. Drug Delivery Rev., 2007, 59, 1504–1520 CrossRef CAS PubMed
- M. N. Leiske, M. Lai, T. Amarasena, T. P. Davis, K. J. Thurecht, S. J. Kent and K. Kempe, Biomaterials, 2021, 274, 120843 CrossRef CAS PubMed
- V. G. Deepagan, M. N. Leiske, N. L. Fletcher, D. Rudd, T. Tieu, N. Kirkwood, K. J. Thurecht, K. Kempe, N. H. Voelcker and A. Cifuentes-Rius, Nano Lett., 2021, 21, 476–484 CrossRef CAS PubMed
- N. M. Warne, A. Elbourne, M. P. Tran, J. R. Finnegan, O. M. Feeney and K. Kempe, Polym. Chem., 2023, 14, 2916–2929 RSC
- J. Svoboda, N. Lusiani, R. Sivkova, O. Pop-Georgievski and O. Sedlacek, Macromol. Rapid Commun., 2023, 44, 2300168 CrossRef CAS PubMed
- G. Morgese, B. Verbraeken, S. N. Ramakrishna, Y. Gombert, E. Cavalli, J. G. Rosenboom, M. Zenobi-Wong, N. D. Spencer, R. Hoogenboom and E. M. Benetti, Angew. Chem., Int. Ed., 2018, 57, 11667–11672 CrossRef CAS PubMed
- Z. Varanaraja, J. Kim and C. R. Becer, Eur. Polym. J., 2021, 147, 110299 CrossRef CAS
- M. M. Bloksma, U. S. Schubert and R. Hoogenboom, Macromol. Rapid Commun., 2011, 32, 1419–1441 CrossRef CAS PubMed
- M. Yang, M. S. Haider, S. Forster, C. Hu and R. Luxenhofer, Macromolecules, 2022, 55, 6176–6190 CrossRef CAS
- D. Pizzi, J. Humphries, J. P. Morrow, A. M. Mahmoud, N. L. Fletcher, S. E. Sonderegger, C. A. Bell, K. J. Thurecht and K. Kempe, Biomacromolecules, 2023, 24, 246–257 CrossRef CAS PubMed
- R. Luxenhofer, S. Huber, J. Hytry, J. Tong, A. V. Kabanov and R. Jordan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 732–738 CrossRef CAS
- M. M. Bloksma, U. S. Schubert and R. Hoogenboom, Polym. Chem., 2011, 2, 203–208 RSC
- M. M. Bloksma, S. Rogers, U. S. Schubert and R. Hoogenboom, Soft Matter, 2010, 6, 994–1003 RSC
- D. Pizzi, J. Humphries, J. P. Morrow, N. L. Fletcher, C. A. Bell, K. J. Thurecht and K. Kempe, Eur. Polym. J., 2019, 121, 109258 CrossRef
- R. Luxenhofer, Y. Han, A. Schulz, J. Tong, Z. He, A. V. Kabanov and R. Jordan, Macromol. Rapid Commun., 2012, 33, 1613–1631 CrossRef CAS PubMed
- M. W. M. Fijten, C. Haensch, B. M. van Lankvelt, R. Hoogenboom and U. S. Schubert, Macromol. Chem. Phys., 2008, 209, 1887–1895 CrossRef CAS
- Z. A. I. Mazrad, B. Schelle, J. A. Nicolazzo, M. N. Leiske and K. Kempe, Biomacromolecules, 2021, 22, 4618–4632 CrossRef CAS PubMed
- J. P. Morrow, Z. A. I. Mazrad, A. I. Bush and K. Kempe, J. Controlled Release, 2022, 350, 193–203 CrossRef CAS PubMed
- M. M. Bloksma, R. M. Paulus, H. P. C. van Kuringen, F. van der Woerdt, H. M. L. Lambermont-Thijs, U. S. Schubert and R. Hoogenboom, Macromol. Rapid Commun., 2012, 33, 92–96 CrossRef CAS PubMed
- M. Glassner, M. Vergaelen and R. Hoogenboom, Polym. Int., 2018, 67, 32–45 CrossRef CAS
- E. F. J. Rettler, J. M. Kranenburg, H. M. L. Lambermont-Thijs, R. Hoogenboom and U. S. Schubert, Macromol. Chem. Phys., 2010, 211, 2443–2448 CrossRef CAS
- M. M. Bloksma, S. Rogers, U. S. Schubert and R. Hoogenboom, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2790–2801 CrossRef CAS
- M. Mella and L. Izzo, Polymer, 2010, 51, 3582–3589 CrossRef CAS
- R. Luxenhofer, G. Sahay, A. Schulz, D. Alakhova, T. K. Bronich, R. Jordan and A. V. Kabanov, J. Controlled Release, 2011, 153, 73–82 CrossRef CAS PubMed
- S. Sun, Y. Huang, C. Zhou, S. Chen, M. Yu, J. Liu and J. Zheng, Bioconjugate Chem., 2018, 29, 1841–1846 CrossRef CAS PubMed
- W. H. Binder, R. Sachsenhofer, D. Farnik and D. Blaas, Phys. Chem. Chem. Phys., 2007, 9, 6435–6441 RSC
- Y. Li, X. Chen and N. Gu, J. Phys. Chem. B, 2008, 112, 16647–16653 CrossRef CAS PubMed
- C.-F. Su, H. Merlitz, H. Rabbel and J.-U. Sommer, J. Phys. Chem. Lett., 2017, 8, 4069–4076 CrossRef CAS PubMed
- D. F. Moyano, M. Goldsmith, D. J. Solfiell, D. Landesman-Milo, O. R. Miranda, D. Peer and V. M. Rotello, J. Am. Chem. Soc., 2012, 134, 3965–3967 CrossRef CAS PubMed
- F. Shima, T. Akagi, T. Uto and M. Akashi, Biomaterials, 2013, 34, 9709–9716 CrossRef CAS PubMed
- S. Chen, L. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283–5293 CrossRef CAS
- N. M. Warne, J. R. Finnegan, O. M. Feeney and K. Kempe, J. Polym. Sci., 2021, 59, 2783–2796 CrossRef CAS
- S. Wijnans, B.-J. de Gans, F. Wiesbrock, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2004, 25, 1958–1962 CrossRef CAS
- E. F. J. Rettler, J. M. Kranenburg, H. M. L. Lambermont-Thijs, R. Hoogenboom and U. S. Schubert, Macromol. Chem. Phys., 2010, 211, 2443–2448 CrossRef CAS
- J. Svoboda, N. Lusiani, R. Sivkova, O. Pop-Georgievski and O. Sedlacek, Macromol. Rapid Commun., 2023, 44, 2300168 CrossRef CAS PubMed
- Z. Varanaraja, N. Hollingsworth, R. Green and C. R. Becer, ACS Appl. Polym. Mater., 2023, 5, 5158–5168 CrossRef CAS
- J. F. R. Van Guyse, P. Cools, T. Egghe, M. Asadian, M. Vergaelen, P. Rigole, W. Yan, E. M. Benetti, V.-V. Jerca, H. Declercq, T. Coenye, R. Morent, R. Hoogenboom and N. De Geyter, ACS Appl. Mater. Interfaces, 2019, 11, 31356–31366 CrossRef CAS PubMed
- R. Hoogenboom, M. W. M. Fijten, H. M. L. Thijs, B. M. Van Lankvelt and U. S. Schubert, Des. Monomers Polym., 2005, 8, 659–671 CrossRef CAS
- J. Kim, V. Beyer and C. R. Becer, Macromolecules, 2022, 55, 10651–10661 CrossRef CAS
- N. Engel, M. Dirauf, J. A. Czaplewska, I. Nischang, M. Gottschaldt and U. S. Schubert, R. Soc. Open Sci., 2024, 11, 231008 CrossRef CAS PubMed
- M. Brunzel, M. Dirauf, M. Sahn, J. A. Czaplewska, N. Fritz, C. Weber, I. Nischang and U. S. Schubert, J. Chromatogr., A, 2021, 1653, 462364 CrossRef CAS PubMed
- O. Sedlacek, O. Janouskova, B. Verbraeken and R. Hoogenboom, Biomacromolecules, 2019, 20, 222–230 CrossRef CAS PubMed
- O. Nuyken, G. Maier, A. Groß and H. Fischer, Macromol. Chem. Phys., 1996, 197, 83–95 CrossRef CAS
- V. R. de la Rosa, S. Tempelaar, P. Dubois, R. Hoogenboom and L. Mespouille, Polym. Chem., 2016, 7, 1559–1568 RSC
- N. Oleszko-Torbus, A. Utrata-Wesołek, M. Bochenek, D. Lipowska-Kur, A. Dworak and W. Wałach, Polym. Chem., 2020, 11, 15–33 RSC
- A. Podevyn, S. Van Vlierberghe, P. Dubruel and R. Hoogenboom, Gels, 2022, 8, 64 CrossRef CAS PubMed
- M. Madau, G. Morandi, V. Lapinte, D. Le Cerf, V. Dulong and L. Picton, Polymer, 2022, 244, 124643 CrossRef CAS
- M. S. Haider, T. Ahmad, M. Yang, C. Hu, L. Hahn, P. Stahlhut, J. Groll and R. Luxenhofer, Gels, 2021, 7, 78 CrossRef CAS PubMed
- T. Klein, J. Parkin, P. A. J. M. de Jongh, L. Esser, T. Sepehrizadeh, G. Zheng, M. De Veer, K. Alt, C. E. Hagemeyer, D. M. Haddleton, T. P. Davis, M. Thelakkat and K. Kempe, Macromol. Rapid Commun., 2019, 40, 1800911 CrossRef PubMed
- M. M. Bloksma, D. J. Bakker, C. Weber, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 724–728 CrossRef CAS PubMed
- N. ten Brummelhuis, C. Secker and H. Schlaad, Macromol. Rapid Commun., 2012, 33, 1690–1694 CrossRef CAS PubMed
- M. A. Boerman, H. L. Van der Laan, J. C. M. E. Bender, R. Hoogenboom, J. A. Jansen, S. C. Leeuwenburgh and J. C. M. Van Hest, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1573–1582 CrossRef CAS
-
OECD, Test No. 107: Partition Coefficient (n-octanol/water): Shake Flask Method, 1995 Search PubMed
- A. J. D. Magenau, J. A. Richards, M. A. Pasquinelli, D. A. Savin and R. T. Mathers, Macromolecules, 2015, 48, 7230–7236 CrossRef CAS
- J. C. Foster, I. Akar, M. C. Grocott, A. K. Pearce, R. T. Mathers and R. K. O'Reilly, ACS Macro Lett., 2020, 9, 1700–1707 CrossRef CAS PubMed
- N. U. Dharmaratne, T. M. M. Jouaneh, M. K. Kiesewetter and R. T. Mathers, Macromolecules, 2018, 51, 8461–8468 CrossRef CAS
- J. E. Glass, J. Phys. Chem., 1968, 72, 4459–4467 CrossRef CAS
- D. J. F. Taylor, R. K. Thomas and J. Penfold, Adv. Colloid Interface Sci., 2007, 132, 69–110 CrossRef CAS PubMed
- D. Besghini, M. Mauri and R. Simonutti, Appl. Sci., 2019, 9, 1801 CrossRef CAS
- L. Wang, S.-Y. Li, W. Jiang, H. Liu, J.-X. Dou, X.-Q. Li and Y.-C. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 32312–32320 CrossRef CAS PubMed
- S. Bickerton, S. Jiwpanich and S. Thayumanavan, Mol. Pharm., 2012, 9, 3569–3578 CrossRef CAS PubMed
- Z. Kroneková, T. Lorson, J. Kronek and R. Luxenhofer, ChemRxiv, 2018, DOI:10.26434/chemrxiv.5793990.v1.
- M. Bauer, S. Schroeder, L. Tauhardt, K. Kempe, U. S. Schubert and D. Fischer, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1816–1821 CrossRef CAS
- K. Kempe, M. Lobert, R. Hoogenboom and U. S. Schubert, J. Comb. Chem., 2009, 11, 274–280 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00332b |
| This journal is © The Royal Society of Chemistry 2024 |