
Targeting delivery of CRISPR systems into tumours to edit glutamine metabolism for cancer therapy by DPA-Zn-modified nanoparticles†
Qi
Shao
a,
Chang-Hui
Wang
*b,
Han
Gu
c,
Xiao-Hong
Zhou
c,
Xuan
Nie
c,
Wei-Qiang
Huang
 c,
Fan
Gao
c,
Fei
Wang
*d and
Ye-Zi
You
*ac
c,
Fan
Gao
c,
Fei
Wang
*d and
Ye-Zi
You
*ac
aDepartment of Pharmacy, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: yzyou@ustc.edu.cn
bDepartment of Cardiology, The First Affiliated Hospital of Anhui Medical University, Hefei, Anhui 230022, China. E-mail: wangchanghui@ahmu.edu.cn
cDepartment of Polymer Science and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, China
dDepartment of Neurosurgical, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: neurosurgeonwf1@ustc.edu.cn
First published on 26th August 2024
Abstract
Tumour cells exhibit distinct metabolism to sustain their proliferation and survival, making targeting metabolic pathways an appealing option for tumour therapy. Glutamine metabolism plays a crucial role in fuelling tumour growth and modulating the tumour microenvironment. However, the clinical translation of glutamine metabolism-targeting therapies faces poor efficiency and systemic toxic effects. Here, we constructed biocompatible and functional polymer nanoparticles to deliver CRISPR-Cas9 into tumours for efficient and simultaneous gene editing, which can cut off two genes that express glutaminase (GLS1) and phosphoribosyl pyrophosphate amidotransferase (PPAT) to manipulate glutamine metabolism. The results demonstrated that genetic manipulation of glutamine metabolism significantly inhibited tumour development and metastasis while also favourably altering the tumour microenvironment. Importantly, this method improved antitumour immunity and promoted long-term immunological memory. This work highlights the potential of simultaneously targeting multiple glutamine metabolic pathways through gene editing, providing a promising strategy for cancer therapy.
Introduction
Compared with their normal counterparts, tumour cells exhibit heightened metabolic activity and energy demands to fuel their aberrant requirements for growth, proliferation, and metastasis.1–4 Cancer metabolism displays significant heterogeneity, and certain features, such as the upregulation of aerobic glycolysis,2 enhanced glutamine metabolism,5–7 and increased lipid and amino acid synthesis capabilities.8 These features are intrinsic to cancer cells, representing fundamental biochemical necessities widely utilized in cancer diagnosis,9 monitoring,10 and treatment.11,12 Generally, tumour cells also heavily consume glutamine, which is converted to glutamate by glutaminase (GLS) and then metabolized to α-ketoglutarate (α-KG) to enter the TCA cycle.5,6,13 This procedure provides resources and energy for tumour proliferation. Glutamine not only serves as a crucial “carbon source” but also as a major nitrogen contributor that is vital for the cellular synthesis of various compounds, including purines, pyrimidines, nucleotides, and some non-essential amino acids.14 Given the pivotal role of glutamine in cellular metabolism, its metabolic pathways have emerged as potential targets for antitumour therapy.Recently, in-depth investigations have revealed that glutamine also plays an essential role in immune1,15,16 cell proliferation and immunological functions. Glutamine, a critical amino acid, serves as a metabolic intermediate required for promoting the proliferation of T cells and the production of cytokines. However, the extensive consumption of glutamine by tumour cells leads to severe glutamine deficiency in the tumour microenvironment, causing dysregulation of the antitumour immune response of T cells.17–19 Simultaneously, glycolytic metabolism in tumour cells generates a large amount of lactate, promoting the polarization of tumour-associated macrophages (TAMs) toward the M2 phenotype, which inhibits the activity of CD8+ T cells, and this process facilitates immune evasion and exacerbates immune suppression within the tumour microenvironment.15,20–22 Therefore, blocking tumour glutamine metabolism has great promise for cancer treatment. Previous investigations have revealed that selectively suppressing GLS has a significant impact on the differentiation of CD4+ and CD8+ immune cell sub-populations.23 Broad-spectrum glutamine antagonism alters the nutrient composition of the tumour microenvironment (TME),24 reducing the production of immunosuppressive metabolites such as kynurenine in the TME, preventing the generation and recruitment of myeloid-derived suppressor cells (MDSCs), and alleviating immune checkpoint blockades while restoring antitumour immunity,25 thus inhibiting tumour growth. Common glutamine inhibitors, including glutamine antimetabolites and their prodrugs (such as L-DON and JHU083)16,26 and allosteric glutaminase inhibitors (such as BPTES and 968),23 generally exhibit hydrophobicity, poor delivery, and low specificity, which limits their clinical application. To maximize the efficacy of glutamine inhibition, it is essential to evaluate the detrimental impact of glutamine metabolism inhibition on antitumour therapy as well as metabolic compensation and immunological resistance. Current research focuses on the combination of glutamine metabolism inhibition with various tumour-treatment strategies, such as the codelivery of glycolysis and glutamine metabolism inhibitors27 (such as GOD and BPTES), the combination of CB-839 with photothermal therapy,28 and synergistic therapy with anti-PD-L1 and DON.29 These strategies have demonstrated outstanding therapeutic effects in the treatment of various malignancies, underscoring the immense potential and broad prospects of glutamine inhibition in the field of tumour therapy.
The enzyme glutaminase30 converts glutamine into glutamate and free ammonia. Glutamate is further converted into α-KG, which then participates in the tricarboxylic acid cycle (TCA), providing energy and reactants for cancer cells. In addition to functioning as a carbon source glutamine also acts as a nitrogen donor, which is essential for tumour cell proliferation.7 Phosphoribosyl pyrophosphate amidotransferase14 (PPAT) primarily catalyses the transfer reaction between phosphoribosyl and phosphoribosylamine (an ATP precursor), transferring the γ-nitrogen of glutamine to 5-phosphoribosyl pyrophosphate (PRPP). PRPP is a key intermediary in the purine biosynthesis pathway and is used for the synthesis of DNA and RNA. Phosphoribosylamine is an ATP precursor; hence, PPAT activity is directly tied to cellular energy metabolism. Therefore, simultaneous blockade of the carbon and nitrogen metabolic pathways of glutamine will provide an innovative therapeutic strategy for tumours. CRISPR gene editing allows precise and specific modifications to practically any genomic region within cells.31,32 By selecting appropriate combinations of sgRNAs, multiple gene edits can be accomplished.33,34 Herein, we created a CRISPR Cas9 plasmid (Cas9-sgPPAT + sgGLS) that targets both glutaminase (GLS1) and phosphoribosyl pyrophosphate amidotransferase (PPAT). DPA-Zn-modified nanoparticles were used as vehicles for the targeted delivery of Cas9-sgPPAT + sgGLS into tumours to express the Cas9 protein, sgPPAT and sgGLS, which could effectively inhibit the enzyme expression of PPAT and GLS in tumour cells (Scheme 1). The targeted nature of the carrier ensures the precision of genetic deletion of GLS and PPAT in tumour cells. The results demonstrated that glutamine metabolism is highly impacted, leading to insufficient cellular energy supply and material production in tumours, significantly inhibiting tumour growth and metastasis. In addition, the infiltration of endogenous CD8+ T cells into tumour tissues, along with the levels of the proinflammatory cytokines TNF-α, IFN-γ, and IL-12, is improved, markedly ameliorating the immunosuppressive tumour microenvironment.
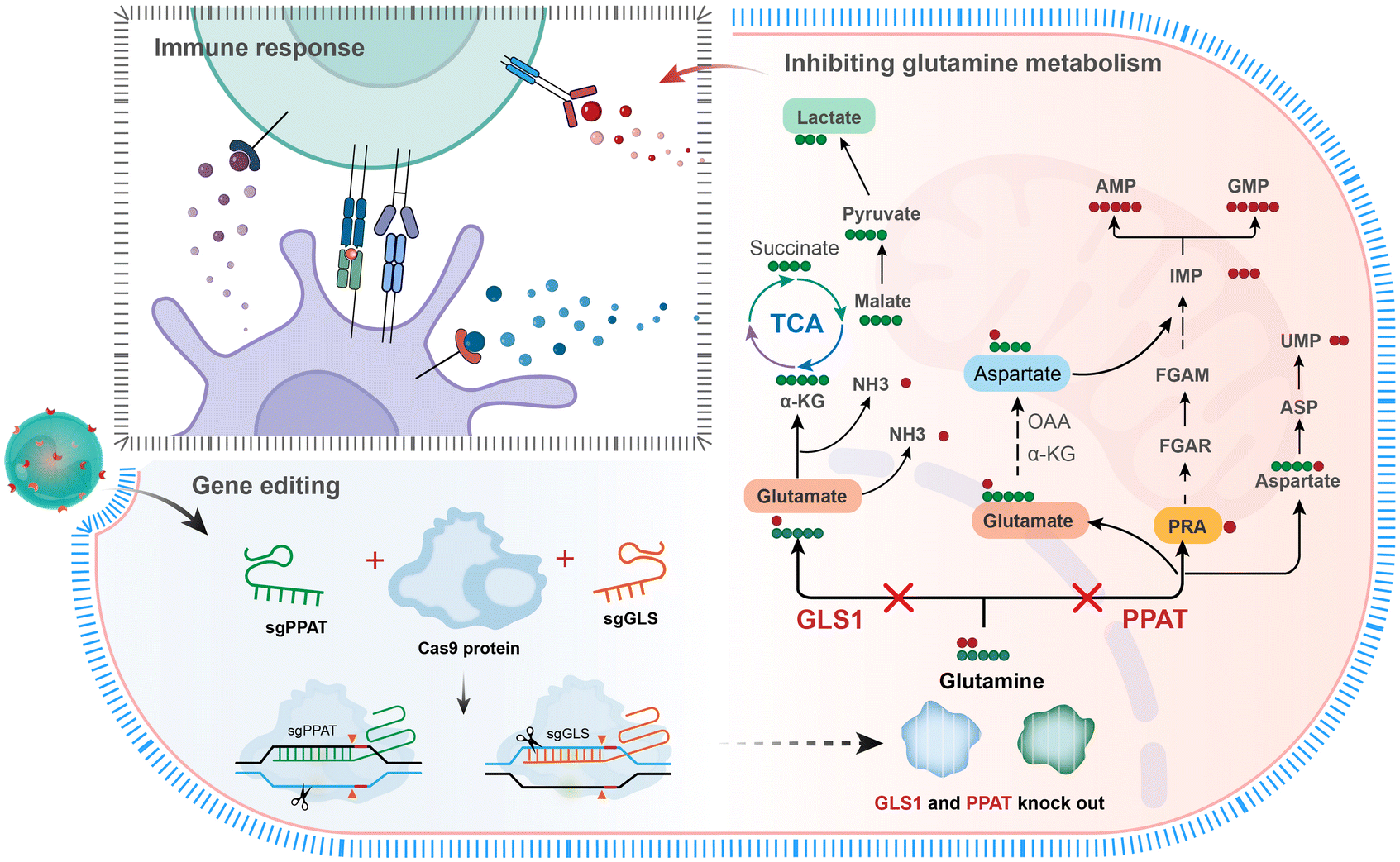 | ||
| Scheme 1 Schematic illustration of the targeted delivery of CRISPR-Cas9 by DPA-functionalized polymer nanoparticles to disrupt glutamine metabolism. | ||
Results and discussion
Through the Michael addition reaction of N,N′-hexamethylene bisacrylamide, N,N′-dimethyl-1,3-propanediamine, and N,N′-cystamine bisacrylamide, hyperbranched polyamide amine (PAA) containing disulfide bonds (PAA, molecular weight 10.8 kDa) was obtained.35 The reaction of perfluorooctanoyl chloride with PAA results in the formation of fluorocarbon chain-modified PAA. Subsequently, amphiphilic small molecules with fluorocarbon chains and hydrophilic heads (C7F15-DPA-Zn) were prepared according to previously reported methods.36 The DPA-Zn-CFPNs are prepared from a polymer (PAA-nC7F15) and a small molecule (C7F15-DPA-Zn). Because they have the same hydrophobic part (C7F15), the small molecule and polycation could be assembled into stable nanoparticles. Additionally, the DPA-Zn group located on the surface of the nanoparticles is used to enhance the interaction of nanoparticles with tumour-cell membranes, thereby improving the efficiency of targeted gene editing. Then, the fluorocarbon chain-modified PAA was co-assembled with the prepared amphiphilic small molecules, forming DPA-Zn-modified cationic polymer nanoparticles (DPA-Zn-FCPN), which were prepared using a thin-film dispersion method, with zeta potential of +32.6 mV and size of 176 ± 21 nm (Fig. s2†). Polyethyleneimine (PEI), which has a high charge density, is widely regarded as the gold standard for gene delivery.37,38 Its CE50 (described as the “charge ratio” required to induce a 50% drop in fluorescence in the exclusion experiment with ethidium bromide) is approximately 1.32. But, DPA-Zn-FCPN had a lower CE50 of 0.37, indicating that DPA-Zn-FCPN has greater DNA binding capacity (Fig. s3†). Furthermore, the surface of the nanoparticles formed by the DPA-Zn-FCPN and DNA contained DPA-Zn structures, which can highly recognize phosphatidylserine (PS) on the surface of cancer cells, facilitating DNA delivery to tumour tissues (Fig. s5†). Once inside the cell, the complexes can disassemble under intracellular GSH, releasing the loaded plasmids.By targeting the intracellular enzymes of glutaminase (GLS1) and phosphoribosyl pyrophosphate amidotransferase (PPAT), we designed sgRNAs aimed at these two enzymes. The sgRNA strands were integrated into single plasmids (Cas9-sgGLS + sgPPAT) (Fig. s1†), which simultaneously express Cas9, sgRNAGLS and sgRNAPPAT (Fig. 1a). DPA-Zn-FCPN condensed Cas9-sgGLS + sgPPAT plasmids into DPA-Zn-functionalized polyplexes, which can effectively deliver Cas9-sgPPAT + sgGLS plasmids into 4T1 and CT26 cells and tumour tissue, as shown in Scheme 1 and Fig. s4.† The protein knockout efficiency of each group was evaluated through western blot experiments, and grayscale analysis using ImageJ. The results revealed significant reductions in PPAT and GLS1 protein expression within the Cas9-sgPPAT + sgGLS group of 4T1 cells by 65% and 37%, while the Cas9-sgPPAT and Cas9-sgGLS groups exhibited reductions of 30% and 15% in PPAT and GLS expression, respectively (Fig. 1b). This indicates that the DPA-Zn-functionalized polyplex-mediated delivery of the dual-targeting plasmid successfully diminished the expression of both enzymes within the cells, with greater efficiency observed for the combined knockout than for the individual knockouts. Similar results were observed in CT26 cells (Fig. 1c). It is conceivable that concurrently impeding the pathway of glutamine as an energy metabolite and synthetic precursor may have a more pronounced effect on cell growth and protein synthesis, whereas singularly affecting the energy pathway (GLS1) or synthetic pathway (PPAT) may prompt compensatory metabolic pathways.
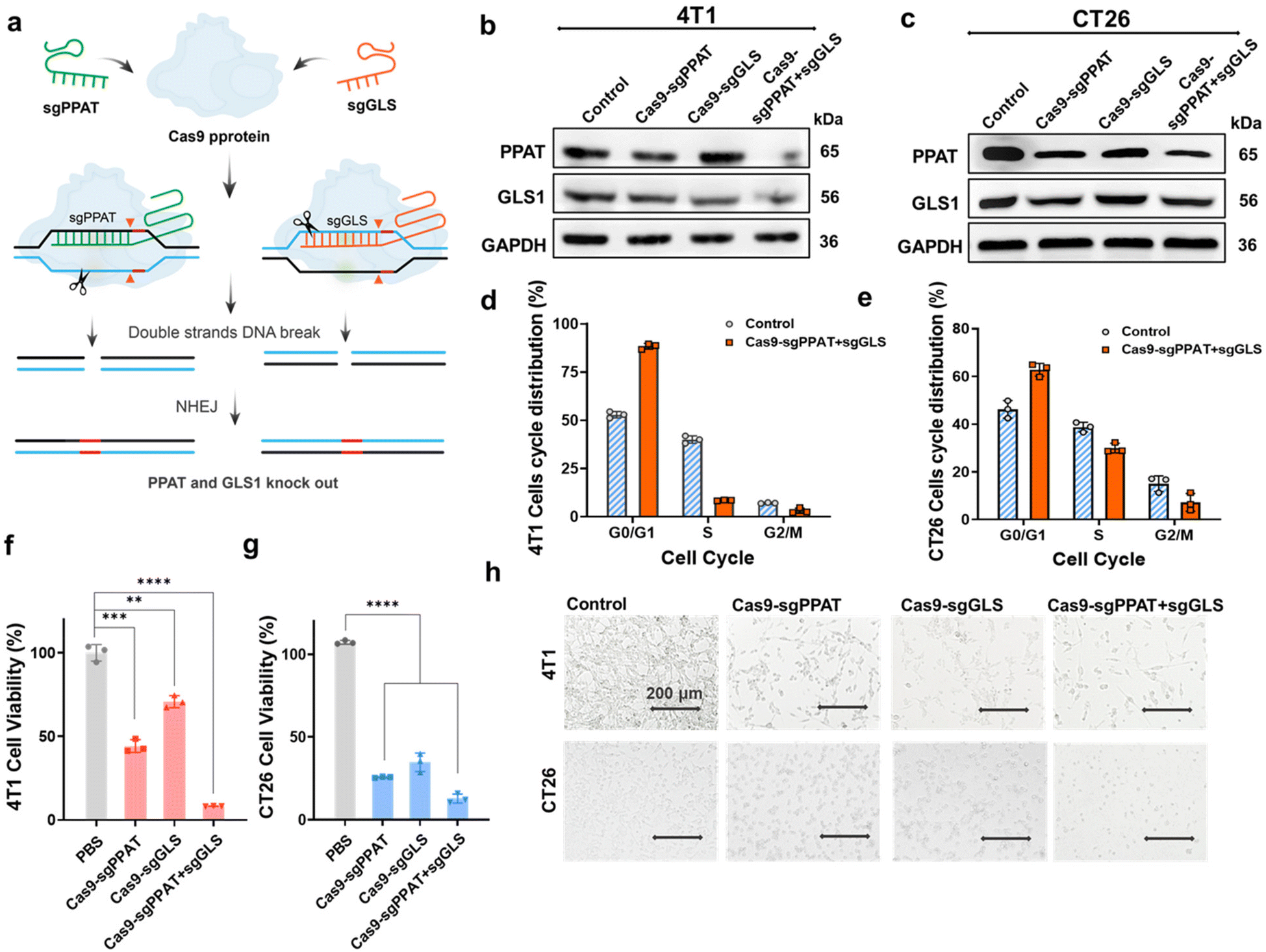 | ||
| Fig. 1 In vitro gene editing efficiency, cell apoptosis and cell-cycle arrest evaluation. (a) Schematic diagram of double gene knockout mediated by the Cas9-sgPPAT + sgGLS1 plasmid. (b) and (c) Western blot analysis of PPAT and GLS expression in 4T1 cells and CT26 cells after treatment with Cas9-sgPPAT NPs, Cas9-sgGLS NPs or Cas9-sgPPAT + sgGLS NPs. GAPDH was used as a loading control. (d) and (e) Cell-cycle analysis of 4T1 cells and CT26 cells 72 hours after gene editing. (f) and (g) Cell viability of 4T1 cells and CT26 cells 72 hours after treatment with different Cas9 sgRNA NPs. Cell viability was assessed using the MTT assay. The data in the panels are presented as the mean ± SDs (n = 3). P values were determined by Student's t test (NS: not significant, *: P < 0.05, **: P < 0.01, ***: P < 0.001, ****: P < 0.0001). (h) Images of 4T1 and CT26 cells post-treatment with Cas9-sgPPAT NPs, Cas9-sgGLS NPs, and Cas9-sgPPAT + sgGLS NPs. Scale bar, 200 μm. | ||
Glutamine metabolism plays a pivotal role in the cellular energy supply and substance synthesis and is intricately linked with the progression of the cell cycle.14,30 We conducted cell-cycle analysis on genetically edited 4T1 and CT26 cells. As depicted, the population of cells in the G0/G1 phase significantly increased following treatment with Cas9-sgPPAT + sgGLS, while the number of cells in the S phase markedly decreased, which demonstrates that after treatment, most cells are either in interphase or have halted division due to reduced energy supply or insufficient synthetic substrates, indicating a distinct phenomenon of cell-cycle arrest.39 Subsequent MTT assays were conducted to assess cell viability 72 hours post-transfection, the results revealing that knocking out a single enzyme has a limited effects on cell activity. However, when both enzymes were simultaneously knocked out, the cell survival rate decreased to less than 15% (Fig. 1f, g and Fig. s6, s7†), accompanied by noticeable changes in cell morphology (Fig. 1h), indicating a pronounced growth inhibition effect. In addition, knocking out PPAT alone had a more pronounced inhibitory effect on cell growth than knocking out GLS1. These findings underscore the significant role of glutamine as a source of energy and biosynthetic material in various cellular growth and proliferation processes, particularly in cancer cells.
We then measured the levels of glutamine and glucose in the cells after gene editing. Normalizing the intracellular glucose and glutamine levels against those of the control group, as shown in Fig. 2a and b, the results revealed a fivefold increase in intracellular glutamine levels following simultaneous knockout of the two enzymes involved in glutamine metabolism. Moreover, despite no direct intervention in the glucose metabolic pathway, there was a noticeable increase in intracellular glucose levels following the alteration in glutamine metabolism, elucidating the intricate interplay between these metabolic processes. To further elucidate the energy supply blockade mediated by glutamine metabolism inhibition, we employed a Seahorse extracellular flux analyser to examine the changes in the two primary metabolic pathways of tumour cells—aerobic glycolysis (AGlyc) and oxidative phosphorylation (OxPhos)—following gene editing. The experiment involved the sequential addition of the following targeted drugs for the mitochondrial electron transport chain (ETC): oligomycin, FCCP, and rotenone/antimycin A to measure the oxygen consumption rate (OCR) of cells and obtain key parameters reflecting mitochondrial function.27 We investigated the effect of glutamine metabolism pathway inhibition on cellular oxidative phosphorylation by measuring the oxygen consumption rate (OCR) of 4T1 and CT26 cells 72 h after gene editing. As depicted in Fig. 2c and e, cells treated with Cas9-sgPPAT + GLS exhibited more pronounced respiratory inhibition than cells in the other groups, with a respiratory rate less than 2% of the maximum respiration rate induced by FCCP,40 which indicates that dual gene editing not only exerts an acute effect on oxidative phosphorylation but also exerts a significant and persistent influence on mitochondrial respiration, effectively impeding the cellular energy supply.
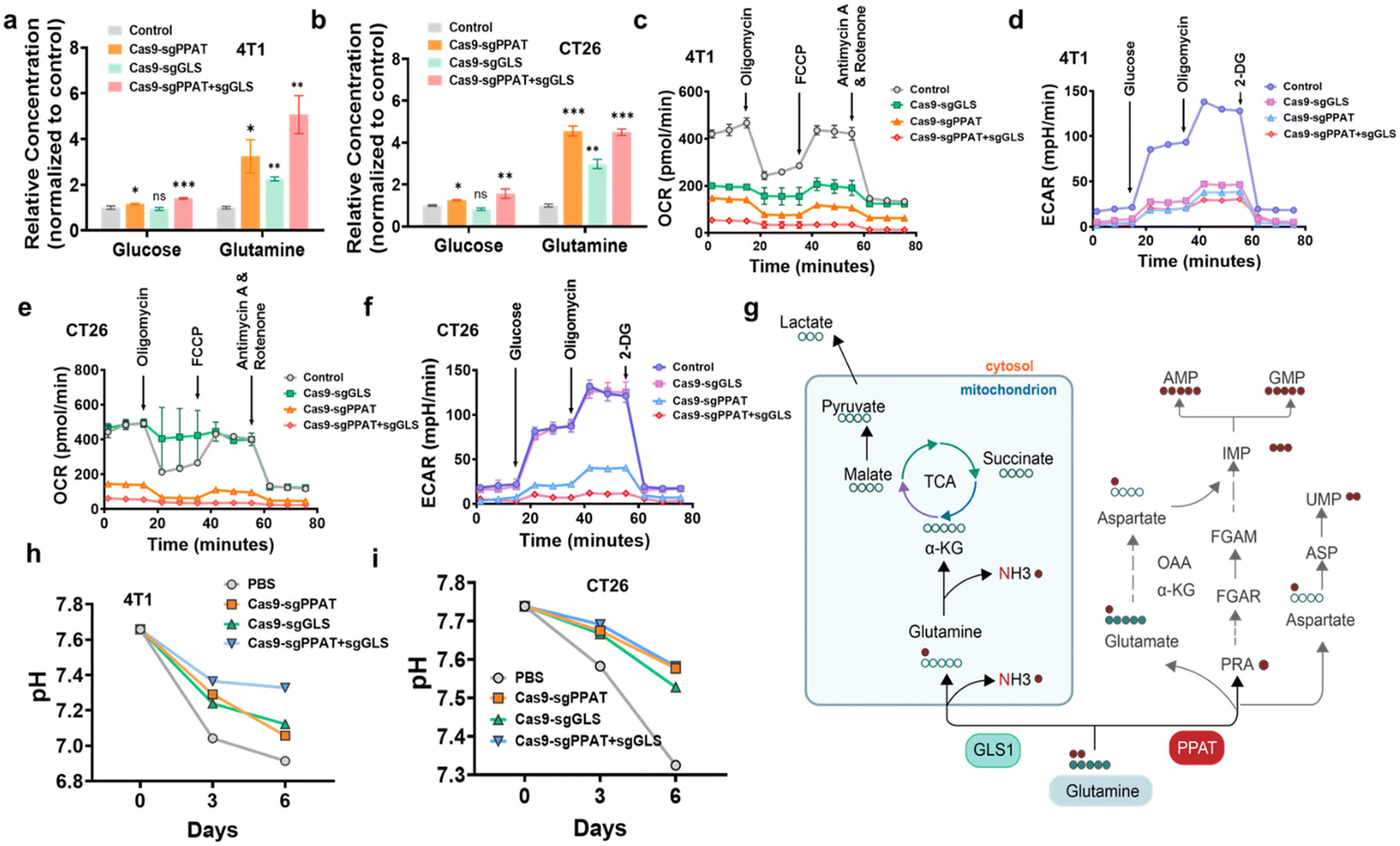 | ||
| Fig. 2 Intracellular glutamine content and metabolic analysis. (a) and (b) Relative contents of glucose and glutamine in 4T1 and CT26 cells after treatment with Cas9-sgPPAT NPs, Cas9-sgGLS NPs or Cas9-sgPPAT + sgGLS NPs (μmol mg−1 protein, normalized to the vehicle group). The data in the panels are presented as the mean ± SDs (n = 3). P values were determined by Student's t test (NS: not significant, *: P < 0.05, **: P < 0.01, ***: P < 0.001, ****: P < 0.0001). (c) and (e) Effect of different treatments on the OCRs of 4T1 cells and CT26 cells 72 hours after gene editing. (d) and (f) Effect of different treatments on the ECARs of 4T1 cells and CT26 cells 72 hours after gene editing. (h) and (i) Changes in the pH of the 4T1 and CT26 cell culture media following treatment with various gene editing agents for three and six days. (g) Schematic illustration of glutamine metabolism. Glutamine synthesizes nucleotides through de novo nucleotide biosynthesis pathways while concurrently catabolizing glutamine to support the replenishment reactions of the TCA cycle. Red circles represent nitrogen (N), while green circles represent carbon (C). | ||
In aerobic glycolysis, when glucose is converted into acidic molecules, including pyruvate and lactate, it releases protons into the extracellular environment, leading to acidification. Similarly, glutamine, under the action of glutaminase, is converted into glutamate, which subsequently participates in the TCA cycle as α-KG, thereby contributing to the aforementioned process2,13 (Fig. 2g). Successive additions of glucose, oligomycin, and 2-DG in the experiment allowed for the assessment of cellular glycolytic function by monitoring changes in the extracellular acidification rate (ECAR).41 As depicted in Fig. 2d and f, compared with those of the control group, all groups of cells subjected to gene editing exhibited a downward shift in their curves, indicating a reduction in all key indicators of glycolysis, thus confirming its inhibition. Furthermore, the Cas9-sgPPAT + GLS group demonstrated the most potent inhibition of glycolysis, attributed to the inhibition of multiple metabolic pathways, rendering tumour cells unable to replenish energy through alternative compensatory pathways. These results suggest that simultaneous knockout of the enzymes PPAT and GLS can effectively induce cellular starvation by inhibiting the glycolytic pathway, thereby suppressing tumour growth. We then measured the pH of the cell culture after transfection with Cas9-sgPPAT, Cas9-sgGLS, and Cas9-sgPPAT + sgGLS. As depicted in Fig. 2h and i, the pH of the untreated control group noticeably decreased, from an initial value of 7.6 to 6.8 by the sixth day. Upon intervention in glutamine metabolism, the magnitude of pH reduction in the environment decreased, with the most pronounced attenuation observed in the multiple gene editing group. This further underscores that the restriction of glutamine metabolism not only influences cell growth but also affects the surrounding environment through changes in metabolic byproducts.8,16,42
Tumour cells undergo metabolic reprogramming, altering both glucose and glutamine metabolism, to meet their heightened metabolic and proliferative demands.22 This metabolic competition within the tumour microenvironment deprives immune cells of accessible metabolic substrates, leading to immune evasion and malignant growth of tumour cells. Therefore, inhibiting glutamine metabolism and glycolysis in tumour cells is crucial for restraining tumour growth, ameliorating the immunosuppressive tumour microenvironment, and facilitating the antitumour immune response.18,43 To explore the correlation between Cas9-sgPPAT + sgGLS-mediated suppression of tumour cell glutamine metabolism and immune activation, we established a 4T1 tumour model (Fig. 3a). Through immunofluorescence and flow cytometry analyses, we investigated alterations in the immunosuppressive microenvironment, as well as changes in lymphocyte infiltration, proliferation, and functionality. As depicted in Fig. 3b and g, following Cas9-sgPPAT + sgGLS treatment, the proportion of M1 macrophages increased from 7.8% to 45.7%, while that of M2 macrophages, which are representative of immunosuppression, decreased from 50.5% to 13.3%. The M1/M2 ratio reached 3.4, which was 23 times greater than that of the PBS-treated group. Although the metabolic suppression of tumour glutamine was limited in the Cas9-sgPPAT and Cas9-sgGLS groups, similar results were observed, with the M1/M2 ratio increasing by 10-fold and 6-fold, respectively, compared with that in the PBS group. This finding implies that the acidic microenvironment within the TME is ameliorated, facilitating the transition of macrophages toward a proinflammatory phenotype.
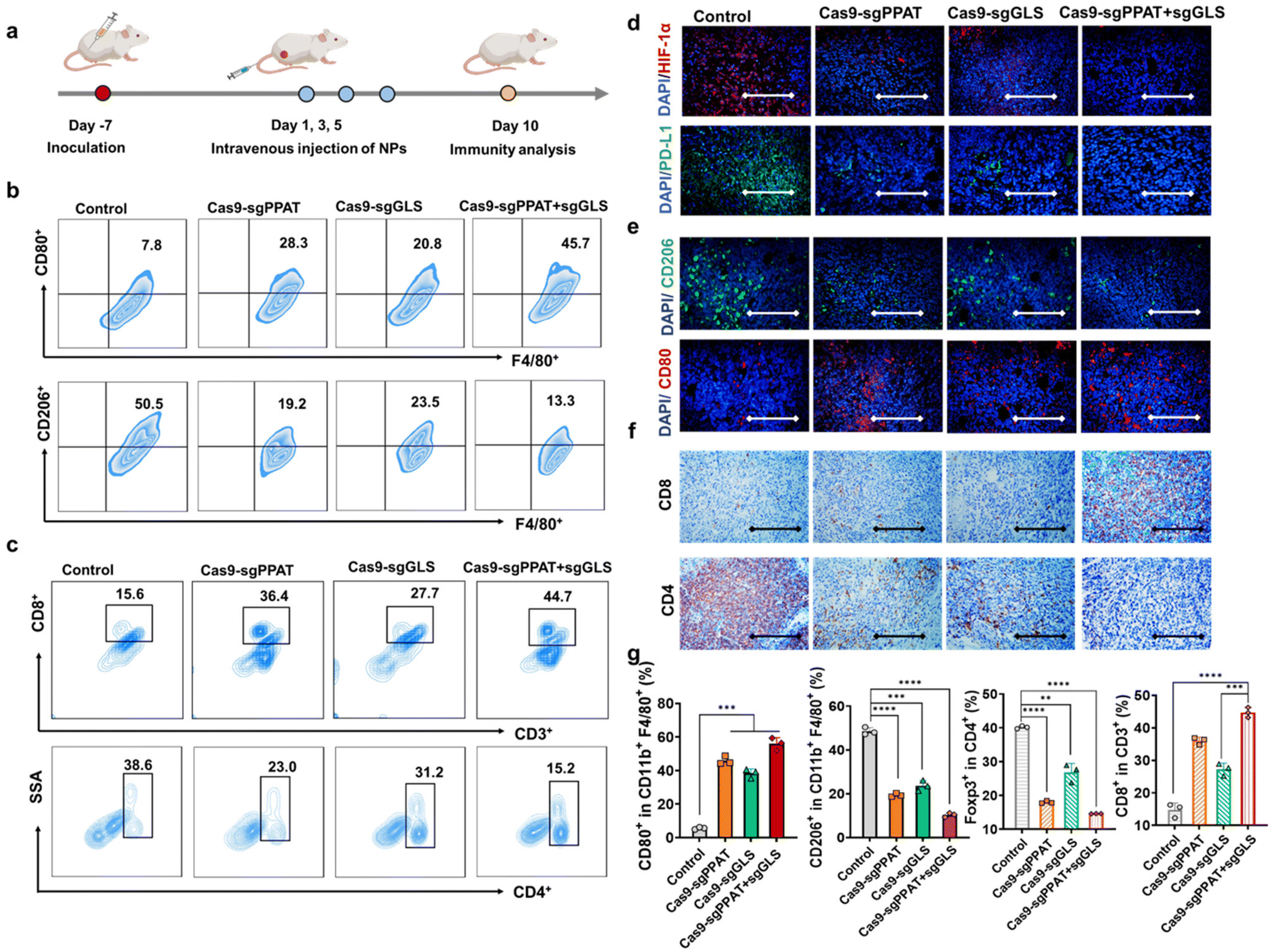 | ||
| Fig. 3 Inhibition of glutamine metabolism improves the tumour microenvironment in vivo. (a) Schematic illustration for construction of the 4T1 tumour model where the 4T1 tumour-bearing mice were treated with 0.3 mg kg−1 Cas9 plasmids three times. (b) Flow cytometric plots of the M1-type (CD80) and M2-type (CD206) macrophages in tumours from mice sacrificed 10 days post-treatment. (c) Representative flow cytometry data and quantification of CD8+ T cells and CD4+ T cells in tumours. (d) Immunofluorescence staining of HIF-1α and PD-L1 at tumour sites. Scale bar: 100 μm. (e) Immunofluorescence staining of CD80 and CD206 at tumour sites. Scale bar: 100 μm. (f) IHC analyses of CD4 and CD8 at tumour sites. Scale bar: 100 μm. (g) Quantitative analysis of the percentages of M1-type (CD80+, CD11b+, F4/80+) and M2-type (CD206+, CD11b+, F4/80+) macrophages at tumour sites and CD8+ T cells and CD4+ T cells in the tumour after various treatments (n = 3). Data are shown as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. | ||
HIF-1α serves as a crucial transcriptional regulator of cell survival under hypoxic conditions, and its expression levels reflect the hypoxic status and malignancy of tumours.44 PD-L1, an essential immune checkpoint molecule, is expressed on the surface of tumour cells. By binding to the programmed death receptor PD-1 on T cells, PD-1 suppresses T-cell function and causes T-cell apoptosis, assisting tumour cells in immune evasion.29,45 Initially, we measured HIF-1α and PD-L1 levels through immunofluorescence staining. The PBS group exhibited prominent fluorescence signals in both HIF-1α and PD-L1 images, whereas the Cas9-sgPPAT + sgGLS group showed negligible fluorescence signals (Fig. 3d), indicating an amelioration of the immunosuppressive microenvironment within the tumour area and laying the groundwork for eliciting antitumour immune responses in vivo. Subsequently, the efficiency of antitumour immune responses was investigated by measuring the infiltration, proliferation, and differentiation of immune cells. As expected, substantial infiltration of T lymphocytes into the TME was observed in the Cas9-sgPPAT + sgGLS group, with the proportion of CD3+CD8+ T cells reaching 44.7%, whereas that in the PBS group reached only 15.6% (Fig. 3c). Immunofluorescence analysis of DCs and immunohistochemical analysis of tumour-infiltrating T cells also revealed similar trends (Fig. 3e). These findings suggest that disrupting tumour-cell metabolism can reduce the production of immunosuppressive metabolic byproducts, effectively inducing TAM differentiation into a proinflammatory immune phenotype while also increasing T-cell infiltration.
Next, through flow cytometry and immunofluorescence, we further analysed the proliferation and functionality of tumour-infiltrating lymphocytes, as well as the phenotype of macrophages and the abundance of T cells in the lymph nodes. As depicted in Fig. 4a, the number of CD8+ T cells in the lymph nodes of mice treated with Cas9-sgPPAT + sgGLS was significantly greater than that in the lymph nodes of the other groups and was doubled compared to that number in the control group, while the percentage of CD4+ T cells decreased by 34%. In addition, flow cytometry analysis of macrophage phenotypes within the lymph nodes revealed a 41.3% increase in proinflammatory M1 macrophages compared to those in the PBS group, accompanied by a 37.9% reduction in immunosuppressive M2 macrophages, resulting in an 11.1-fold increase in the M1/M2 ratio (Fig. 4c). Similar results were observed in the Cas9-sgPPAT and Cas9-sgGLS groups, with M1/M2 ratios increasing by 8-fold and 3.7-fold, respectively, consistent with the quantitative analysis results mentioned above (Fig. s8†). Furthermore, immunofluorescence staining was utilized to assess the tumour infiltration of other immune cells in the Cas9-sgPPAT + sgGLS group. Compared with those in the PBS group, there was a significant reduction in tumour infiltration by myeloid-derived suppressor cells (MDSCs, marked by Gr-1), considered a major contributor to immunosuppression, and a decrease in the percentage of regulatory T cells (Tregs, marked by Foxp3), which maintain immune tolerance and inhibit immune responses.46
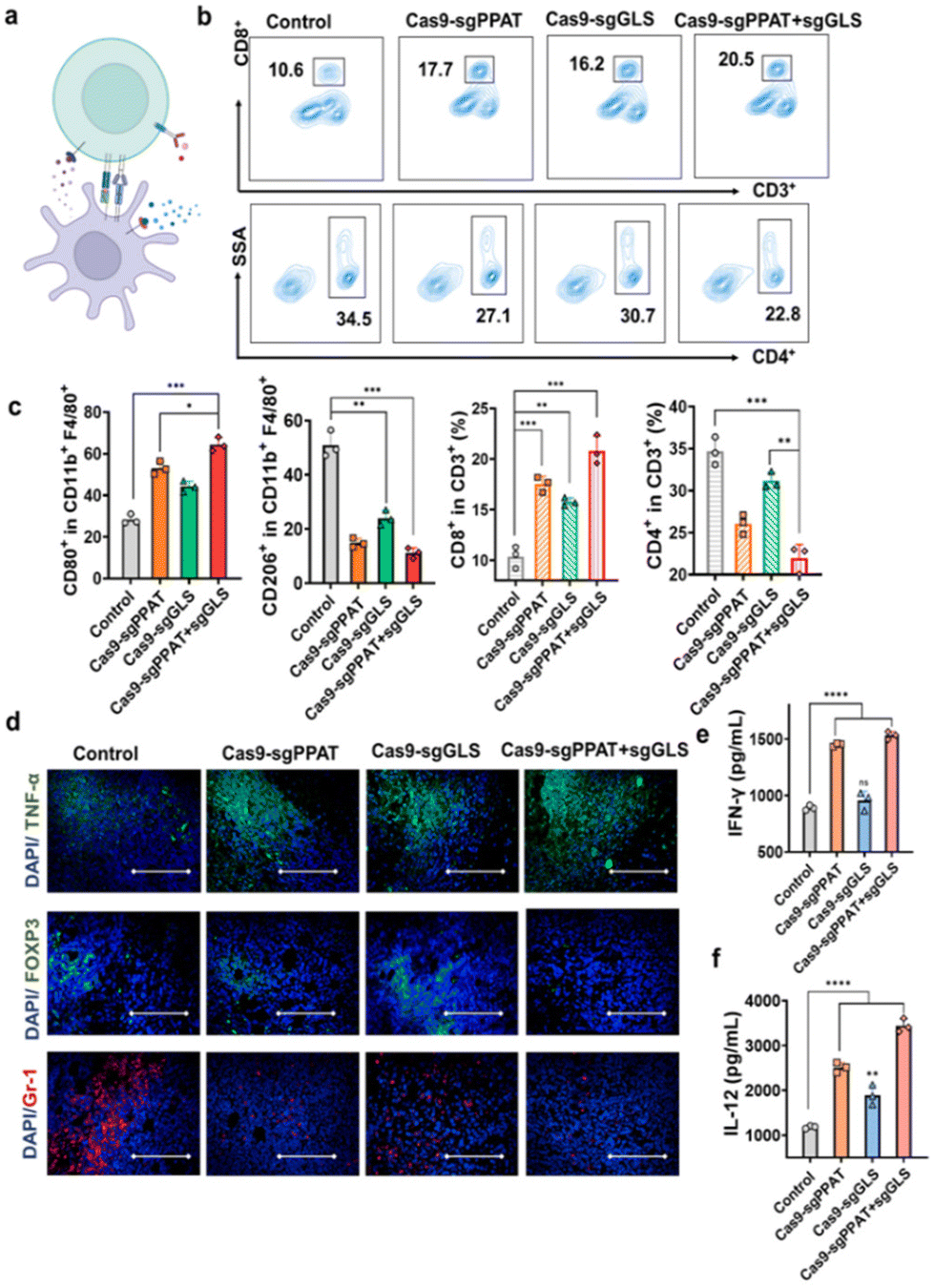 | ||
| Fig. 4 Immune activation effect of Cas9-sgPPAT + sgGLS1 on 4T1 tumour-bearing mice. (a) Schematic diagram of immune cell activation in 4T1 tumour-bearing mice. (b) Flow cytometry analysis of the M1-type and M2-type phenotypes in lymph nodes after various treatments. (c) Quantitative analysis of the percentages of M1-type (CD80+, CD11b+, F4/80+) and M2-type (CD206+, CD11b+, F4/80+) macrophages in the lymph nodes and of CD8+ T cells and CD4+ T cells (gated on CD3+ T cells) in the lymph nodes after various treatments (n = 3). (d) Immunofluorescence staining of TNF-α, Foxp3 and Gr-1 at tumour sites. Scale bar: 100 μm. (e) ELISA analysis of interferon-γ (IFN-γ) and (f) interleukin-12 (IL-12) in serum from mice after various treatments. The data are shown as the mean ± SDs. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. | ||
Cytokines play a pivotal role in regulating and influencing the interactions, proliferation, differentiation, and functions of immune cells,47 exerting a significant influence on tumour immunity.48,49 Through ELISA, we assessed the secretion of proinflammatory cytokines within the tumour milieu, including tumour necrosis factor-alpha (TNF-α), tumour-associated interferon-gamma (IFN-γ), interleukin-12 (IL-12), and the anti-inflammatory cytokine interleukin-10 (IL-10), to evaluate the functionality of T cells and macrophages. Previous studies revealed that the Cas9-sgPPAT + sgGLS group exhibited the most robust expression of CD8+ T cells, thus demonstrating the highest secretion of intratumoural IFN-γ. Furthermore, the levels of TNF-α and IL-12 significantly increased, while the level of IL-10 markedly decreased (Fig. 4e, f and Fig. s9†). In essence, these findings underscore that the metabolic blockade of intracellular glutamine mediated by Cas9-sgPPAT + sgGLS can markedly reverse the immunosuppressive tumour microenvironment and further enhance antitumour immune responses through intratumoural metabolic reprogramming.
To further explore the potential biological mechanisms of glutamine metabolism blockade in tumour therapy, metabolic profiling analysis50 was conducted on 4T1 tumour cells subjected to different treatments. Partial least squares discriminant analysis (PLS-DA) was employed to segregate the samples, establishing a relationship model between metabolite expression and sample categories to ascertain sample validity. The PLS-DA results in Fig. 5a indicate minimal differences between samples within the Cas9-sgPPAT + sgGLS and PBS groups, but significant differences exist between the groups. Using liquid chromatography-mass spectrometry (LC-MS) to assess the levels of metabolites in the two groups, compared with the control group, cells treated with Cas9-sgPPAT + sgGLS exhibited 534 differentially abundant metabolites, with 24 significantly different metabolites, 20 of which showed noticeable decreases and 4 of which showed significant increases (FC ≥ 1.2||FC ≤ 1/1.2, OPLS-DA_VIP ≥ 1, P value ≤ 0.05). In comparison with those in the control group, the majority of metabolites exhibited decreased levels, with a minority associated with cellular oxidative stress and DNA damage showing increased levels, signifying significant disparities in the levels of various metabolites (Fig. 5b). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis51,52 revealed the predominant enrichment of metabolites involved in amino acid metabolism, carbohydrate metabolism, and nucleotide metabolism pathways (Fig. 5d). Specifically, following Cas9-sgPPAT + sgGLS treatment, the expression of NADH, 2-deoxyglucose-6-phosphate, and citric acid significantly decreased, indicating a notable impact on energy generation within the tumour,45 with both glucose metabolism pathways and the TCA cycle being suppressed. Adenine, GDP, and cGMP also exhibited significant decreases, indicating a dramatic decrease in the generation of nucleotide precursors essential for cellular DNA and RNA synthesis.53 Furthermore, the expression of Cys-Gly, N-acetylsphingosine, spermine, and acetoacetate notably declined, indicating a significant reduction in the nutrients required for the metabolism of essential substances such as proteins and lipids,54 which influenced multiple critical processes, including tumour growth, proliferation,22,30 and cellular transformation (Fig. 5c). Metabolic pathway enrichment analysis55 of differentially abundant metabolites in the KEGG pathway further demonstrated that the most significant changes induced by Cas9-sgPPAT + sgGLS occurred in energy metabolism pathways, nucleotide metabolism, cofactor biosynthesis, and secondary metabolite biosynthesis. The decreased levels of metabolites associated with the Rap1 signalling pathway, Ras signalling pathway, and HIF-1 signalling pathway (Fig. 5d) impact tumour cell adhesion, growth, reproduction, and response to hypoxic environments, consistent with previous conclusions regarding tumour therapy and antitumour immunity.
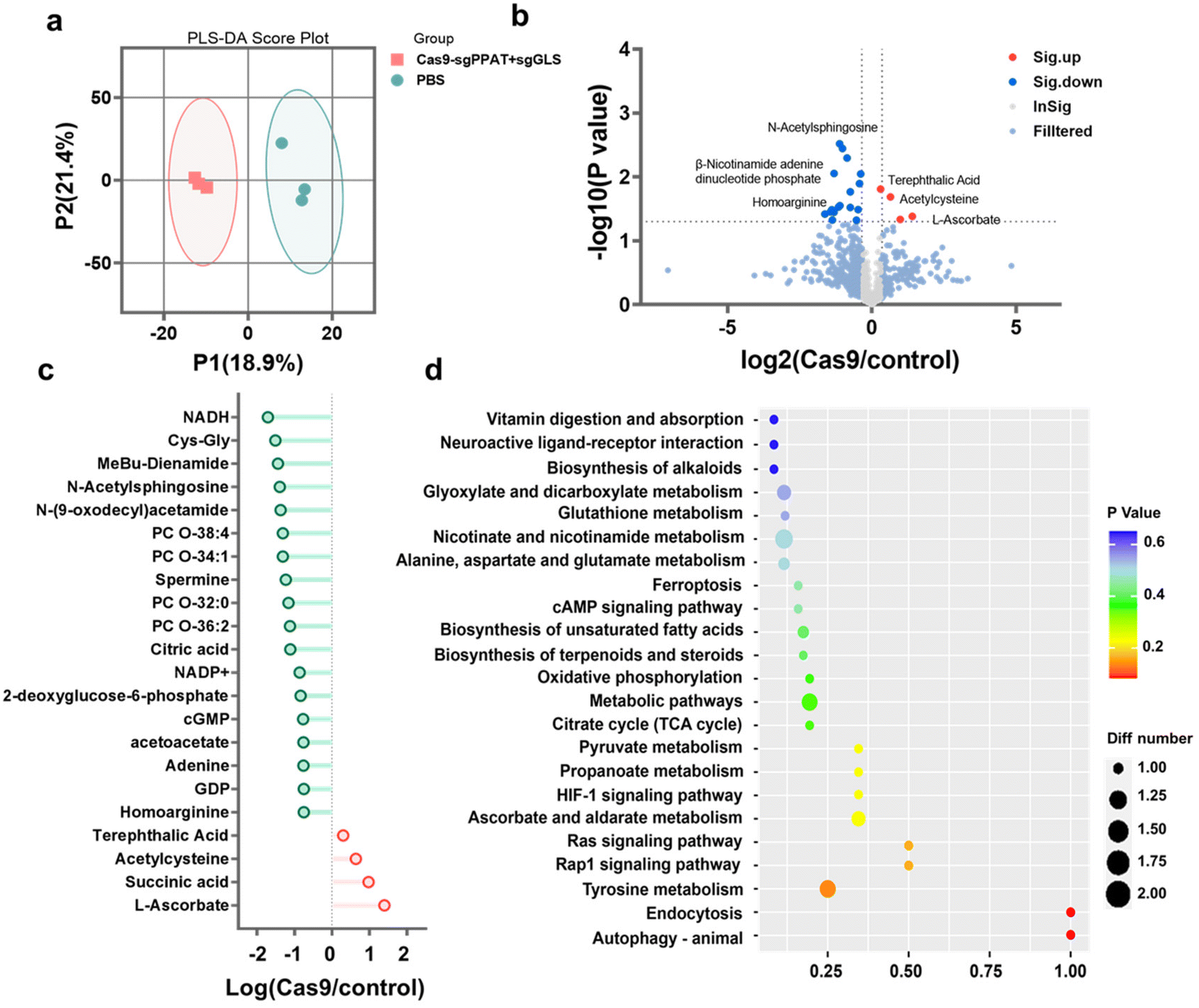 | ||
| Fig. 5 Metabolomic analysis reveals that inhibition of glutamine reshapes the metabolic pathways of tumour cells. (a) Partial least squares discriminant analysis (PLS-DA) was performed based on differentially expressed metabolites from the Cas9-sgPPAT + sgGLS1 and PBS groups. Each point represents one sample. (b) The volcano plot highlights the differentially expressed metabolites compared with the control group. (c) The analysis of upregulated and downregulated metabolites associated with metabolic pathways after Cas9-sgPPAT + sgGLS1 treatment. (d) KEGG enrichment analysis of cellular metabolic pathways after Cas9-sgPPAT + sgGLS1 treatment. | ||
To assess the role of gene editing-mediated glutamine metabolism blockade in antitumour therapy in vivo, we established a 4T1 tumour model. BALB/c mice were subcutaneously injected with 100 μL of 4T1 cell suspension (approximately 1 × 108 cells per mL). One week later, when the average tumour volume reached approximately 100 mm3, the 4T1 tumour-bearing mice were randomly divided into four groups (PBS, Cas9-sgPPAT, Cas9-sgGLS, and Cas9-sgPPAT + sgGLS). The mice were treated every other day, in total on seven occasions, and the tumour volume and mouse weight were continuously measured to determine the efficacy of the treatments (Fig. 6a and b). The untreated PBS group demonstrated rapid tumour growth, with an average size of 1258 mm3 on day 16. However, inhibition of tumour glutamine metabolism significantly improved the therapeutic outcome, with tumour sizes of 321 mm3 and 456 mm3 in the PPAT and GLS groups, respectively, by day 16 (Fig. 6b and c). Notably, the combined knockout group showed an average tumour size of only 76 mm3, with the tumour volume gradually decreasing over time, indicating controlled tumour development (Fig. 6g). Mice were euthanized, dissected, weighed, and photographed 16 days after treatment (Fig. 6h). The Cas9-sgPPAT + sgGLS group demonstrated superior antitumour activity, with an average tumour mass of 69 mg, 7% of that of the PBS group. Similar results were observed in H&E staining and TUNEL immunofluorescence images of tumour sections (Fig. 6h). H&E staining revealed that the Cas9-sgPPAT + sgGLS treatment group had fewer tumour cells. Moreover, distinct green fluorescence areas in the TUNEL images indicated tumour cell apoptosis. Further investigation of the biosafety of gene editing for the blockade of glutamine metabolism involved H&E staining of major organs, including the heart, liver, spleen, lungs, and kidneys (Fig. s10†). Compared with those observations in the PBS group, no substantial atrophy, lesions, or necrosis was detected.11,56 Furthermore, there were no significant fluctuations in mouse weight (Fig. s11†) during the treatment period, demonstrating reliable biosafety.
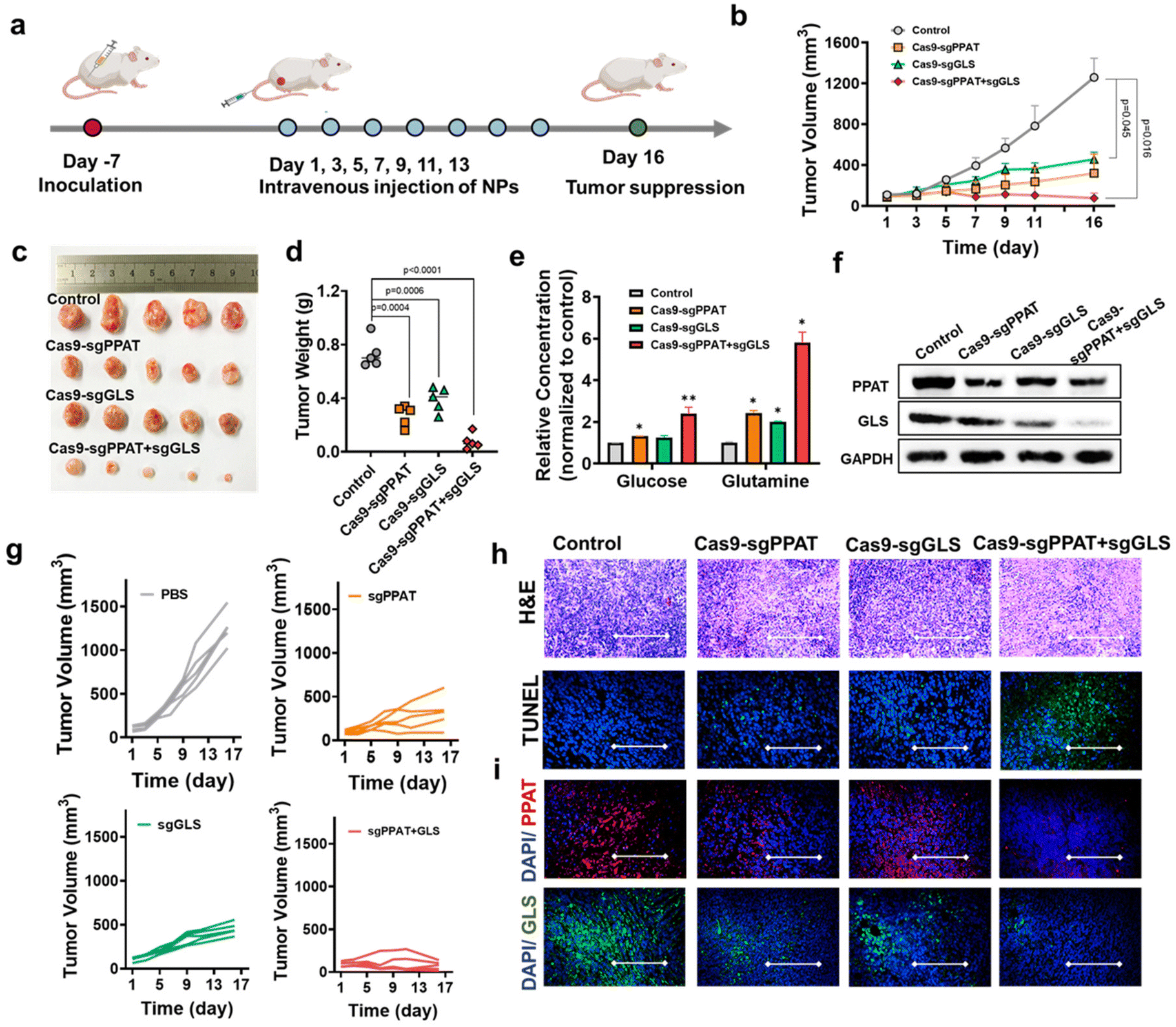 | ||
| Fig. 6 Antitumour effect of glutamine metabolic pathway blockade on the 4T1 tumour model. (a) Schematic illustration of the administration design. 4T1 tumour-bearing mice were treated with DPA-Zn-functionalized polyplexes with CRISPR Cas9 plasmids (Cas9-sgPPAT, Cas9-sgGLS and Cas9-sgPPAT + sgGLS; dosage: 0.3 mg kg−1) 7 times. (b) Growth curves of the 4T1 tumours during treatment. (c) Photographs and (d) weights of the excised tumours from treated mice. (e) Relative levels of glucose and glutamine in 4T1 tumours after treatment with Cas9-sgPPAT NPs, Cas9-sgGLS NPs or Cas9-sgPPAT + sgGLS NPs (μmol mg−1, normalized to the vehicle group). The data in the panels are presented as the mean ± SDs (n = 3). P values were determined by Student's t test (NS: not significant, *: P < 0.05, **: P < 0.01, ***: P < 0.001, ****: P < 0.0001). (f) Western blot analyses of the expression of PPAT and GLS1 in each treatment group. (g) Tumour volume change curves of the mice in the PBS, Cas9-sgPPAT NP, Cas9-sgGLS NP and Cas9-sgPPAT + sgGLS NP treatment groups. (h) H&E and TUNEL staining of tumour sites. In TUNEL staining, normal cell nuclei are stained blue, and apoptotic cell nuclei are stained green. Scale bar: 100 μm. (i) Immunofluorescence staining of PPAT and GLS1 at tumour sites. Scale bar: 100 μm. | ||
Additional investigation of PPAT and GLS levels inside tumour tissues was performed using western blotting (Fig. 6f) and immunofluorescence (Fig. 6h). The results revealed decreases of 47% for PPAT and 54% for GLS in the Cas9 sgPPAT and Cas9 sgGLS groups, while in the Cas9-sgPPAT + sgGLS group, the decreases were 65% for PPAT and 70% for GLS. Immunofluorescence tests confirmed the findings by directly observing variations in red fluorescence (PPAT) and green fluorescence (GLS) within tumour tissues. Following treatment cessation, the glucose and glutamine levels in fresh tumour tissues from various groups were assessed. Fig. 6e shows that inhibiting tumour glutamine metabolism significantly increased the overall glucose and glutamine levels, which were normalized to the levels in the PBS group by 2.4-fold and 5.8-fold, respectively. These findings suggest that, in addition to reducing the activity of a single metabolic enzyme, blocking multiple glutamine metabolic pathways can markedly inhibit tumour growth, improve the tumour microenvironment, provide metabolic substrates for the proliferation and activation of immune cells, and further enhance the function and efficacy of tumour therapy.
Experimental
Materials
The Seahorse XF Glycolytic Rate Assay Kit (Cat. 103020-100) and Seahorse XF Cell Mito Stress Test Kit (Cat. 103015-100) were purchased from Agilent (USA). A glutamine (Gln) content assay kit and a glucose content assay kit were purchased from Solarbio (Beijing, China). mCherry-labelled Cas9 and sgRNA “all-in-one” plasmids (pCas9-sgPPAT, pCas9-sgGLS, pCas9-sgPPAT + GLS) were constructed with V-solid (Beijing, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT, 98%), DMSO, RIPA cell lysis buffer, protease inhibitor, and an enhanced BCA protein assay kit were procured from Beyotime Biotechnology (Shanghai, China). The anti-PPAT antibody (Cat. 15401-1-AP), GLS antibody (Cat. 12855-1-AP) and GAPDH antibody (Cat. 60004-1-Ig) were obtained from Proteintech (USA). HRP-labelled secondary antibodies were purchased from ZSGB-Bio (Beijing, China). The antibodies used in the immunological assays included APC-conjugated anti-mouse/human CD11b (BioLegend, Cat. 101211), PerCP/Cyanine5.5 anti-mouse F4/80 (BioLegend, Cat. 123127), FITC-conjugated anti-mouse CD80 (BioLegend, Cat. 104705), PE-conjugated anti-mouse CD86 (BioLegend, Cat. 104705), APC-conjugated anti-mouse CD86 (BioLegend, Cat. 105011), FITC-conjugated anti-mouse CD206 (BioLegend, Cat. 141703), FITC-conjugated anti-mouse CD3 (BioLegend, Cat. 141703), APC-conjugated anti-mouse CD4 (BioLegend, Cat. 100515), PerCP/Cyanine5.5-conjugated anti-mouse CD8a (BioLegend, Cat. 155013), PE-conjugated anti-mouse/rat/human FOXP3 (BioLegend, Cat. 320007), and APC-conjugated anti-mouse IFN-γ (BioLegend, Cat. 505809).Cell culture
The cell lines utilized in this study included human cervical cancer cells (HeLa), murine colon cancer cells (CT26), murine breast cancer cells (4T1), and human breast cancer cells (MCF-7), all of which were procured from the American Type Culture Collection (ATCC). HeLa and MCF-7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (HyClone), penicillin (100 U mL−1), and streptomycin (100 μg mL−1) (Gibco); CT26 and 4T1 cells were cultured in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum (Gibco), penicillin (100 U mL−1), and streptomycin (100 μg mL−1). All cells were maintained in a cell culture incubator at 37 °C with 5% CO2.Preparation of the DPA-Zn-FCPN
The nanomicelles were prepared by film dispersion. Precalculated amounts of PA and DPA-Zn (Zn ions![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N atoms in PA = 1:3) were dissolved in methanol. Subsequently, the methanol was removed using a rotary evaporator to form a thin film on the surface of the container. Next, ultrapure water (final concentration of DPA-Zn-coated polyplexes = 1.07 mg mL−1) was added to the container to form composite micelles.
:N atoms in PA = 1:3) were dissolved in methanol. Subsequently, the methanol was removed using a rotary evaporator to form a thin film on the surface of the container. Next, ultrapure water (final concentration of DPA-Zn-coated polyplexes = 1.07 mg mL−1) was added to the container to form composite micelles.
Preparation of DPA-Zn-FCPN and pCas9-sgRNA polyplexes
The pCas9-sgRNA plasmid is capable of simultaneously expressing the Cas9 protein and sgRNA. Following DNA gel electrophoresis and transfection experiments, the optimal complex ratio of DPA-Zn-FCPN to the pCas9-sgRNA plasmid was validated as N/P = 11, which was subsequently utilized in both transfection and in vivo experiments. An appropriate volume of DPA-Zn-FCPN was added to pCas9-sgRNA (0.1 μg μL−1 in pure water), and the mixture was gently vortexed and then incubated at room temperature for 30 minutes to ensure the formation of a stable complex system.Cell viability assays
The MTT method was used to assess the impact of individually and concurrently knocking out two glutamine metabolic enzymes on the viability of different tumour cells. HeLa, MCF-7, CT26, and 4T1 cells were seeded at a density of 8 × 103 cells per well in a 96-well plate and cultured overnight. Once the cell density reached the transfection threshold, PBS and the prepared DPA-Zn-FCPN/pCas9-sgGLS NPs, DPA-Zn-FCPN/pCas9-sgPPAT NPs, or DPA-Zn-FCPN/pCas9-sgGLS + sgPPAT NPs were added to each well at a dose of 0.5 μg per well. After 6 hours, the culture medium was replaced with fresh medium, and the cells were incubated for another 48 hours. Subsequently, 100 μL of MTT solution (5 mg mL−1, 20 μL) in DMEM was added to each well and incubated at 37 °C for 4 hours, followed by the addition of 150 μL of DMSO to fully dissolve the formed formazan crystals. Finally, the absorbance at 490 nm was measured using a microplate reader (Thermo Fisher, USA). Each experimental group was tested in triplicate.Western blotting assay
4T1 cells (or CT26 cells) were seeded in 6-well plates at a density of 3 × 105 cells per well and cultured overnight. Once the cell density reached the transfection threshold, the subsequent experiments commenced. DPA-Zn-FCPN/pCas9-sgGLS NPs, DPA-Zn-FCPN/pCas9-sgPPAT NPs, and DPA-Zn-FCPN/pCas9-sgGLS + sgPPAT NPs were prepared at a dose of 5 μg per well for the CRISPR Cas9 plasmids. The prepared DNA complexes of each group were dispersed in serum-free medium and cocultured with cells for 6 hours, followed by replacement with fresh medium containing 10% FBS and culture continued for 72 hours. After culturing, the cells were washed with PBS and lysed on ice with 300 μL of pre-cooled cell lysis buffer (containing 10 μL of PMSF per mL) for 20 minutes. The supernatant was collected after centrifugation at 12000 rpm and 4 °C for 10 minutes, and the protein concentration was quantified using the BCA method. Subsequently, 8 μg of total protein was separated by 10% SDS-PAGE and transferred onto PVDF membranes (Beyotime). The membranes were blocked at room temperature for 1 hour, followed by overnight incubation at 4 °C with primary antibodies (GLS antibody, diluted 1:30000, Proteintech; PPAT antibody, diluted 1:2000, Proteintech). After washing, the membranes were incubated at room temperature for 1 hour with HRP-conjugated secondary antibodies (goat anti-rabbit IgG, diluted 1:2000, ZSGB-Bio; goat anti-mouse IgG, diluted 1:2000, ZSGB-Bio). An anti-GAPDH antibody (diluted 1:200000, Proteintech) served as an internal control. Finally, the membranes were visualized on an LAS4000 (GE) molecular imager, and the images were analysed for grayscale using ImageJ.
Intracellular glutamine content detection
4T1 and CT26 cells were seeded and transfected with CRISPR/Cas9 plasmids, followed by continued incubation for 72 hours. After incubation, the cells were processed according to the instructions provided by the glutamine detection kit (Solarbio) to determine the intracellular glutamine content. The cells in the wells were washed, and then the cells were collected into centrifuge tubes and centrifuged to remove the supernatant. The extraction volume was determined at a ratio of 500:1 based on the cell number (104 cells):extraction solution volume (mL), resulting in the addition of 0.5 mL of extraction solution to approximately 2.5 million cells. The cells were then sonicated and centrifuged at 12000g for 5 min at 4 °C to collect the supernatant. Thirty microliters of the supernatant from each group were taken for subsequent protein concentration determination. The remaining supernatant was mixed with 250 μL of extraction solution 2, vigorously shaken for 5 min, and then centrifuged at 12000g for 5 min at 4 °C. The upper clear liquid layer was collected and placed on ice for testing.
The enzyme reaction reagent was added to the supernatant, and the mixture was incubated at 37 °C for 1 hour for the enzyme reaction, followed by the addition of the remaining chromogenic reagent and further incubation at 37 °C for 1 hour. After centrifugation at 12000g for 5 min at room temperature, 200 μL of the supernatant was aspirated, and the absorbance at 450 nm was measured in a 96-well plate; the results were recorded as Atest, Acontrol, Astandard, and Ablank. The ΔAtest was calculated as Atest − Acontrol, and the ΔAstandard was calculated as Astandard − Ablank. The protein concentration of each group was determined by the BCA method and is denoted as Cpr.
The glutamine content was calculated based on the sample protein concentration:
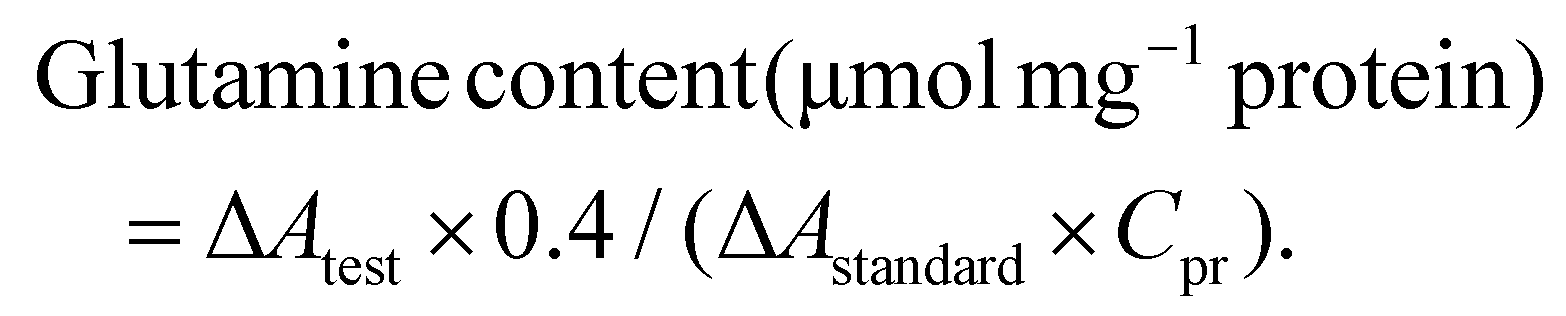
Intracellular glucose content measurement
After cultivation, the cells were processed following the protocol provided by the Glucose Assay Kit (Solarbio) for intracellular glucose content measurement. The cells were collected into centrifuge tubes and centrifuged, and the supernatant was discarded. At a 500:1 ratio of cell number (104 cells) to distilled water volume (mL), the cells were sonicated, boiled in a water bath for 10 minutes, cooled, and then centrifuged at 8000g and 25 °C for 10 minutes, after which the supernatant was collected for analysis.
Standard and blank tubes were set up, and the reaction reagent was added according to the instructions. After vortexing, the tubes were placed in a 37 °C constant temperature incubator for 15 minutes. The absorbance at 505 nm was then measured for the blank, standard, and test tubes and recorded as Ablank, Astandard, and Atest, respectively, with ΔAtest = Atest − Ablank and ΔAstandard = Astandard − Ablank. The protein concentration for each group was determined by the BCA method and is denoted as Cpr.
The glucose content was calculated based on the sample protein concentration:
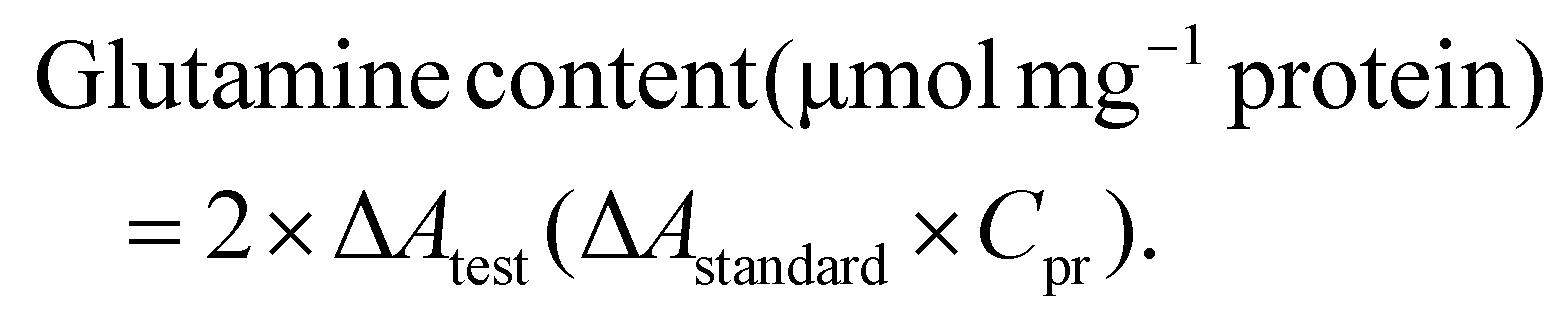
Assessing cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR)
The cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using an XF96 extracellular flux analyser from Seahorse Bioscience.20,27 The cells were seeded at a density of 8 × 103 cells per well in XF 96 polystyrene cell culture plates and cultured overnight. After the transfection density was reached, PBS and DPA-Zn-FCPN/pCas9-sgGLS NPs, DPA-Zn-FCPN/pCas9-sgPPAT NPs, or DPA-Zn-FCPN/pCas9-sgGLS + sgPPAT NPs (with a plasmid dosage of 0.5 μg per well) were added to the cells, which were then co-incubated for 6 hours before they were replaced with fresh culture medium, after which the cells were incubated for 48 hours. One day prior to the OCR and ECAR measurements, the sensor probe plate was hydrated overnight at 37 °C in a CO2-free incubator using Seahorse XF calibration solution. For the assay preparation, in the OCR experiment, the required oligomycin, FCCP, and rotenone/antimycin A were diluted in assay medium to the final concentrations and added to the assay plate. After aspirating the culture medium from the cell plate, each well was washed twice with assay medium before adding 180 μL of assay medium per well. Following the setup of the assay program, the measurements were conducted using the Seahorse XF analyser, and the baseline OCR report was generated by Wave Desktop software (Agilent Technologies).In the ECAR experiment, the required saturated glucose, oligomycin, and 2-DG were diluted in the assay medium to final concentrations and added to the assay plate. Similarly, the culture medium from the cell plate was aspirated, and each well was washed twice with assay medium before 180 μL of assay medium was added to each well. Following the setup of the assay program, the measurements were conducted using the Seahorse XF analyser, and the baseline ECAR report was generated by Wave Desktop software (Agilent Technologies).
4T1 murine tumour model
Cells were collected by centrifugation to ensure that the optimal conditions were met. The cell suspension was then adjusted with PBS, and at a density of 5 × 106 cells per mouse, the cells were inoculated subcutaneously into the right hind limb of female BALB/c mice aged 4 to 6 weeks, thereby constructing the 4T1 subcutaneous tumour model. Following inoculation, continuous observation of the tumour volume at the inoculation site ensued. The dimensions of the tumour, including its length (a) and width (b), were recorded. The formula for calculating tumour volume (V) was V = 0.5a × b2. The subsequent experiments commenced when the tumour volume reached approximately 100 mm3. The BALB/c mice were procured from Shanghai Slac Laboratory Animal Co., Ltd. They were housed under constant temperature (25 ± 3 °C) and relative humidity (40%–70%) conditions with a 12-hour light–dark cycle. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the University of Science and Technology of China and approved by the Animal Ethics Committee of the University of Science and Technology of China (no: 2022-N(A)-078).Preparation of single-cell suspensions from tumour and lymphoid tissues
The aforementioned tumour-bearing mice were randomly divided into four groups, each comprising ten individuals: PBS, DPA-Zn-FCPN/pCas9-sgGLS NPs, DPA-Zn-FCPN/pCas9-sgPPAT NPs, and DPA-Zn-FCPN/pCas9-sgGLS + sgPPAT NPs (the dosage of Cas9-sgRNA was 0.3 mg kg−1). Medication was administered via tail vein injection every other day, with simultaneous monitoring of mouse body weight and tumour volume changes. After three doses were administered, blood was collected from the orbital cavity, and the mice were euthanized. Peripheral blood was collected by centrifugation at 3000 rpm for 10 minutes and stored at −80 °C for subsequent cytokine analysis.Tumour tissues (or lymph nodes adjacent to the tumour site) were retrieved, washed with prechilled PBS, and then minced on ice. The tissue fragments were rapidly and gently ground on a 200 μm sieve while being rinsed with prechilled PBS. The resulting suspension was collected in 1.5 mL EP tubes and kept on ice. After all the tumour tissues from each group were ground, the suspension was passed through a sieve to remove incompletely dissociated tissues and cell clusters. The suspension was then centrifuged at 1500 rpm and 4 °C for 5 minutes, after which the supernatant was discarded. An appropriate volume of red blood cell lysis buffer was added to ice for erythrocyte lysis, followed by excess PBS to terminate the lysis process.57 The cell pellet was collected by centrifugation, washed with PBS, and centrifuged again. Finally, an appropriate volume of PBS was added to resuspend the cells, resulting in a single-cell suspension of tumour tissues, which was stored on ice for subsequent experiments.
Phenotypic analysis of DCs in tumour tissues
First, prepared single-cell suspensions from tumours were diluted according to the requirements for antibody usage and grouped accordingly. First, add the fluorescent dyes PerCP/Cyanine5.5-anti-F4/80, APC-anti-CD11b, FITC-anti-CD80, and FITC-anti-CD206 to the prepared experimental cell tubes as single-staining tubes to delineate the positive cell positions. The experimental groups were supplemented with combinations of antibodies against CD11b + F4/80 + CD80+ or CD11b + F4/80 + CD206+ to detect different phenotypes of DCs. After the membranes were incubated with antibodies at 4 °C for 30 minutes, flow cytometry was performed.Ratio of CD4+ T cells to CD8+ T cells in tumour tissues
Following the same procedures as above, prepared single-cell suspensions from tumours were diluted according to the requirements for antibody usage and grouped accordingly to prepare individual staining tubes. Fluorescent dyes such as FITC-conjugated anti-CD3, APC-conjugated anti-CD4, and PerCP/cyanine5.5-conjugated anti-CD8a were added to the cell tubes of the experimental groups. Combinations of CD3+ and CD4+ or CD3+ and CD8+ antibodies were added to the experimental groups to detect different ratios of T cells.18 After the membranes were incubated with antibodies at 4 °C for 30 minutes, flow cytometry was performed.Immunofluorescence detection of the expression of CD4, CD8, Tregs, and MDSCs in tumour tissues
Mice from the respective groups were dissected to obtain fresh tumour tissues, which were fixed in 4% paraformaldehyde. Tissue sections were prepared by embedding in paraffin wax, followed by incubation with antibodies against CD4, CD8, FoxP3 (Treg marker), and Gr-1 (MDSC marker) and subsequent incubation with secondary antibodies labelled with different fluorochromes.26 Finally, the nuclei were stained with DAPI and stored in the dark. Observation and imaging were performed using a fluorescence microscope.Tumour therapeutic experiment
BALB/c mice were implanted with 4T1 tumours and randomly divided into four groups, each comprising six individuals: PBS, DPA-Zn-FCPN/pCas9-sgGLS NPs, DPA-Zn-FCPN/pCas9-sgPPAT NPs, and DPA-Zn-FCPN/pCas9-sgGLS + sgPPAT NPs. The dosage of Cas9-sgRNA in each group was 0.3 mg kg−1. Treatment commenced when the average tumour volume reached 100 mm3, and intravenous injections were administered every other day. Simultaneously, the body weights and tumour volumes of the mice were recorded. After six consecutive administrations, the treatment was halted, and the tumour growth in each mouse was monitored continuously. The experiment was concluded when the tumour volume reached 1500 mm3 in the PBS control group. After adhering to ethical standards for animal welfare, the mice were euthanized, and major organs and tumours were dissected for subsequent analysis.Statistical analysis
Statistical data are reported as the mean ± SD. The significance of differences between two groups was analysed using two-tailed Student's t tests. For multiple comparisons, one-way analysis of variance (ANOVA) was used. A P value of less than 0.05 was considered significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. All the statistical analyses were performed using GraphPad Prism 9, Excel 2016 and Origin. Flow cytometry data were processed using FlowJo.Conclusions
In conclusion, glutamine metabolism in tumour cells plays a key role in fuelling tumour growth and modulating the tumour microenvironment, presenting compelling targets for innovative cancer therapies. Here, DPA-Zn-modified polymer nanoparticles were used as vehicles for the targeted delivery of Cas9-sgPPAT + sgGLS into tumours to express the Cas9 protein, sgPPAT and sgGLS, which can effectively inhibit the enzyme expression of PPAT and GLS within tumour cells to effectively block glutamine metabolism, leading to insufficient cellular energy supply and material production in tumours, significantly inhibiting tumour growth and metastasis. In addition, the immunosuppressive tumour microenvironment is markedly ameliorated by improving the infiltration of endogenous CD8+ T cells and the levels of the proinflammatory cytokines TNF-α, IFN-γ, and IL-12 in tumour tissues. Targeting the delivery of CRISPR systems into tumours to edit glutamine metabolism not only suppresses tumour progression but also potentiates antitumour immune responses, offering a promising avenue for cancer treatment.Author contributions
Q. S. carried out the laboratory research and wrote the draft of the manuscript. Y. Z. Y., F. W., F. G., C. H. W., X. N., and H. L. W. guided the whole project. W. Q. H. and H. G. performed the data analysis. Y. Z. Y. contributed to the writing of the theory section of the manuscript. All the authors discussed the results and contributed to the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52021002, 52131305, 52073269, 51873202, U22A20154, 22131010, 52203196, 22101275, 31870993), and USTC Research Funds of the Double First-Class Initiative (YD2060002016, WK9110000005).References
- E. N. Arner and J. C. Rathmell, Cancer Cell, 2023, 41, 421–433 CrossRef CAS PubMed.
- C. Chelakkot, V. S. Chelakkot, Y. Shin and K. Song, Int. J. Mol. Sci., 2023, 24, 2606–2640 CrossRef CAS PubMed.
- B. Faubert, A. Solmonson and R. J. DeBerardinis, Science, 2020, 368, 152–153 CrossRef PubMed.
- Z. E. Stine, Z. T. Schug, J. M. Salvino and C. V. Dang, Nat. Rev. Drug Discovery, 2022, 21, 141–162 CrossRef CAS PubMed.
- B. J. Altman, Z. E. Stine and C. V. Dang, Nat. Rev. Cancer, 2016, 16, 619–634 CrossRef CAS PubMed.
- A. A. Cluntun, M. J. Lukey, R. A. Cerione and J. W. Locasale, Trends Cancer, 2017, 3, 169–180 CrossRef CAS PubMed.
- J. Jin, J.-K. Byun, Y.-K. Choi and K.-G. Park, Exp. Mol. Med., 2023, 55, 706–715 CrossRef CAS PubMed.
- B. I. Reinfeld, M. Z. Madden, M. M. Wolf, A. Chytil, J. E. Bader, A. R. Patterson, A. Sugiura, A. S. Cohen, A. Ali, B. T. Do, A. Muir, C. A. Lewis, R. A. Hongo, K. L. Young, R. E. Brown, V. M. Todd, T. Huffstater, A. Abraham, R. T. O’Neil, M. H. Wilson, F. Xin, M. N. Tantawy, W. D. Merryman, R. W. Johnson, C. S. Williams, E. F. Mason, F. M. Mason, K. E. Beckermann, M. G. Vander Heiden, H. C. Manning, J. C. Rathmell and W. K. Rathmell, Nature, 2021, 593, 282–288 CrossRef CAS PubMed.
- Q. Yan, D. Yang, N. Xue, Y. Zhu and Y. Yang, Curr. Precis. Chem., 2023, 1, 468–479 CrossRef CAS.
- E. Sanz-Garcia, E. Zhao, S. V. Bratman and L. L. Siu, Sci. Adv., 2022, 8, 8618–8632 CrossRef PubMed.
- S. Gao, X. Yang, J. Xu, N. Qiu and G. Zhai, ACS Nano, 2021, 15, 12567–12603 CrossRef CAS PubMed.
- L. Yang, A. Achreja, T.-L. Yeung, L. S. Mangala, D. Jiang, C. Han, J. Baddour, J. C. Marini, J. Ni, R. Nakahara, S. Wahlig, L. Chiba, S. H. Kim, J. Morse, S. Pradeep, A. S. Nagaraja, M. Haemmerle, N. Kyunghee, M. Derichsweiler, T. Plackemeier, I. Mercado-Uribe, G. Lopez-Berestein, T. Moss, P. T. Ram, J. Liu, X. Lu, S. C. Mok, A. K. Sood and D. Nagrath, Cell Metab., 2016, 24, 685–700 CrossRef CAS PubMed.
- G.-F. Zhang, M. V. Jensen, S. M. Gray, K. El, Y. Wang, D. Lu, T. C. Becker, J. E. Campbell and C. B. Newgard, Cell Metab., 2021, 33, 804–817 CrossRef CAS PubMed.
- M. Kodama, K. Oshikawa, H. Shimizu, S. Yoshioka, M. Takahashi, Y. Izumi, T. Bamba, C. Tateishi, T. Tomonaga, M. Matsumoto and K. I. Nakayama, Nat. Commun., 2020, 11, 1320–1336 CrossRef CAS PubMed.
- K. DePeaux and G. M. Delgoffe, Nat. Rev. Immunol., 2021, 21, 785–797 CrossRef CAS PubMed.
- R. D. Leone, L. Zhao, J. M. Englert, I.-M. Sun, M.-H. Oh, I.-H. Sun, M. L. Arwood, I. A. Bettencourt, C. H. Patel, J. Wen, A. Tam, R. L. Blosser, E. Prchalova, J. Alt, R. Rais, B. S. Slusher and J. D. Powell, Science, 2019, 366, 1013–1021 CrossRef CAS PubMed.
- M. Corrado and E. L. Pearce, J. Clin. Invest., 2022, 132, 148546 CrossRef PubMed.
- J. Park, P. C. Hsueh, Z. Li and P.-C. Ho, Immunity, 2023, 56, 32–42 CrossRef CAS PubMed.
- Y. Wang, F. Wang, L. Wang, S. Qiu, Y. Yao, C. Yan, X. Xiong, X. Chen, Q. Ji, J. Cao, G. Gao, D. Li, L. Zhang, Z. Guo, R. Wang, H. Wang and G. Fan, Cell Rep., 2021, 36, 109516–109525 CrossRef CAS PubMed.
- M. J. Allegrezza and J. R. Conejo-Garcia, Trends Cancer, 2017, 3, 19–27 CrossRef CAS PubMed.
- J. E. Bader, K. Voss and J. C. Rathmell, Mol. Cell, 2020, 78, 1019–1033 CrossRef CAS PubMed.
- I. Martinez-Reyes and N. S. Chandel, Nat. Rev. Cancer, 2021, 21, 669–680 CrossRef CAS PubMed.
- W.-H. Yang, Y. Qiu, O. Stamatatos, T. Janowitz and M. J. Lukey, Trends Cancer, 2021, 7, 790–804 CrossRef CAS PubMed.
- L. Yang, Z. Chu, M. Liu, Q. Zou, J. Li, Q. Liu, Y. Wang, T. Wang, J. Xiang and B. Wang, J. Hematol. Oncol., 2023, 16, 59 CrossRef CAS PubMed.
- G. Morad, B. A. Helmink, P. Sharma and J. A. Wargo, Cell, 2021, 184, 5309–5337 CrossRef CAS PubMed.
- C. Guo, Z. You, H. Shi, Y. Sun, X. Du, G. Palacios, C. Guy, S. Yuan, N. M. M. Chapman, S. A. Lim, X. Sun, J. Saravia, S. Rankin, Y. Dhungana and H. Chi, Nature, 2023, 620, 200–208 CrossRef CAS PubMed.
- H. Du, S. Meng, M. Geng, P. Zhao, L. Gong, X. Zheng, X. Li, Z. Yuan, H. Yang, Y. Zhao and L. Dai, Small, 2023, 19, 2303253 CrossRef CAS PubMed.
- Z. Li, X. Li, Y. Lu, X. Zhu, W. Zheng, K. Chen, S. Liu, J. Wu and W. Guan, Small, 2024, 20, 2305174 CrossRef CAS PubMed.
- X. K. Jin, S. M. Zhang, J. L. Liang, S. K. Zhang, Y. T. Qin, Q. X. Huang, C. J. Liu and X. Z. Zhang, Adv. Mater., 2023, 36, 2309094 CrossRef PubMed.
- Y. Xiang, Z. E. Stine, J. Xia, Y. Lu, R. S. O'Connor, B. J. Altman, A. L. Hsieh, A. M. Gouw, A. G. Thomas, P. Gao, L. Sun, L. Song, B. Yan, B. S. Slusher, J. Zhuo, L. L. Ooi, C. G. L. Lee, A. Mancuso, A. S. McCallion, A. Le, M. C. Milone, S. Rayport, D. W. Felsher and C. V. Dang, J. Clin. Invest., 2015, 125, 2293–2306 CrossRef PubMed.
- N. S. McCarty, A. E. Graham, L. Studena and R. Ledesma-Amaro, Nat. Commun., 2020, 11, 1281 CrossRef CAS PubMed.
- C. Yang, D. Lee, M. S. Zhang, A. P. W. Tse, L. Wei, M. H. R. Bao, B. P. Y. Wong, C. Y. K. Chan, V. W. H. Yuen, Y. Chen and C. C. L. Wong, Adv. Sci., 2022, 9, 2202104 CrossRef CAS PubMed.
- Q. Liu, J. Cai, Y. Zheng, Y. Tan, Y. Wang, Z. Zhang, C. Zheng, Y. Zhao, C. Liu, Y. An, C. Jiang, L. Shi, C. Kang and Y. Liu, Nano Lett., 2019, 19, 7662–7672 CrossRef CAS PubMed.
- P. L. H. Vo, C. Ronda, S. E. Klompe, E. E. Chen, C. Acree, H. H. Wang and S. H. Sternberg, Nat. Biotechnol., 2021, 39, 480–489 CrossRef CAS PubMed.
- X. Nie, W. You, Z. Zhang, F. Gao, X. H. Zhou, H. L. Wang, L. H. Wang, G. Chen, C. H. Wang, C. Y. Hong, Q. Shao, F. Wang, L. Xia, Y. Li and Y. Z. You, Adv. Healthcare Mater., 2023, 12, 2203252 CrossRef CAS PubMed.
- S. Liu, D. Zhou, J. Yang, H. Zhou, J. Chen and T. Guo, J. Am. Chem. Soc., 2017, 139, 5102–5109 CrossRef CAS PubMed.
- J. Yang, P. Zhang, L. Tang, P. Sun, W. Liu, P. Sun, A. Zuo and D. Liang, Biomaterials, 2010, 31, 144–155 CrossRef CAS PubMed.
- M.-M. Xun, Y.-H. Liu, Q. Guo, J. Zhang, Q.-F. Zhang, W.-X. Wu and X.-Q. Yu, Eur. J. Med. Chem., 2014, 78, 118–125 CrossRef CAS PubMed.
- H. K. Matthews, C. Bertoli and R. A. M. de Bruin, Nat. Rev. Mol. Cell Biol., 2022, 23, 74–88 CrossRef CAS PubMed.
- M. Huang, D. Xiong, J. Pan, Q. Zhang, S. Sei, R. H. Shoemaker, R. A. Lubet, L. M. Montuenga, Y. Wang, B. S. Slusher and M. You, Adv. Sci., 2022, 9, 2105885 CrossRef CAS PubMed.
- J. Jin, J.-K. Byun, Y.-K. Choi and K.-G. Park, Exp. Mol. Med., 2023, 55, 706–715 CrossRef CAS PubMed.
- M. Reina-Campos, N. E. Scharping and A. W. Goldrath, Nat. Rev. Immunol., 2021, 21, 718–738 CrossRef CAS PubMed.
- D. J. Kloosterman and L. Akkari, Cell, 2023, 186, 1627–1651 CrossRef CAS PubMed.
- C. Liao, X. Liu, C. Zhang and Q. Zhang, Semin. Cancer Biol., 2023, 88, 172–186 CrossRef CAS PubMed.
- Y. Gao, H. Zhang, L. Tang, F. Li, L. Yang, H. Xiao, J. Karges, W. Huang, W. Zhang and C. Liu, Adv. Sci., 2024, 11, 2300806 CrossRef CAS PubMed.
- D. N. Edwards, V. M. Ngwa, A. L. Raybuck, S. Wang, Y. Hwang, L. C. Kim, S. H. Cho, Y. Paik, Q. Wang, S. Zhang, H. C. Manning, J. C. Rathmell, R. S. Cook, M. R. Boothby and J. Chen, J. Clin. Invest., 2021, 131, 148546 CrossRef PubMed.
- A. Iwasaki and R. Medzhitov, Nat. Immunol., 2015, 16, 343–353 CrossRef CAS PubMed.
- P. G. Holder, S. A. Lim, C. S. Huang, P. Sharma, Y. S. Dagdas, B. Bulutoglu and J. T. Sockolosky, Adv. Drug Delivery Rev., 2022, 182, 114112 CrossRef CAS PubMed.
- F. Zipp, S. Bittner and D. P. Schafer, Immunity, 2023, 56, 914–925 CrossRef CAS PubMed.
- Y. Wang, B. Zhang, Q. Xi, C. Chen, Z. Wang, F. Li, S. Wang, W. Yang, X.-J. Liang, Z. Yu and M. Yu, Nano Today, 2023, 53, 102009 CrossRef CAS.
- E. G. Armitage and C. Barbas, J. Pharm. Biomed. Anal., 2014, 87, 1–11 CrossRef CAS PubMed.
- F. Danzi, R. Pacchiana, A. Mafficini, M. T. Scupoli, A. Scarpa, M. Donadelli and A. Fiore, Signal Transduction Targeted Ther., 2023, 8, 137 CrossRef PubMed.
- D. Huang, Y. Wang, J. W. Thompson, T. Yin, P. B. Alexander, D. Qin, P. Mudgal, H. Wu, Y. Liang, L. Tan, C. Pan, L. Yuan, Y. Wan, Q.-J. Li and X.-F. Wang, Nat. Cell Biol., 2022, 24, 230–241 CrossRef CAS PubMed.
- Y.-J. Li, C. Zhang, A. Martincuks, A. Herrmann and H. Yu, Nat. Rev. Cancer, 2023, 23, 115–134 CrossRef CAS PubMed.
- D. R. Schmidt, R. Patel, D. G. Kirsch, C. A. Lewis, M. G. Vander Heiden and J. W. Locasale, Ca-Cancer J. Clin., 2021, 71, 333–358 CrossRef PubMed.
- D. Rosenblum, A. Gutkin, R. Kedmi, S. Ramishetti, N. Veiga, A. M. Jacobi, M. S. Schubert, D. Friedmann-Morvinski, Z. R. Cohen, M. A. Behlke, J. Lieberman and D. Peer, Sci. Adv., 2020, 6, 9450 CrossRef PubMed.
- S. Chen, Y. Zhong, W. Fan, J. Xiang, G. Wang, Q. Zhou, J. Wang, Y. Geng, R. Sun, Z. Zhang, Y. Piao, J. Wang, J. Zhuo, H. Cong, H. Jiang, J. Ling, Z. Li, D. Yang, X. Yao, X. Xu, Z. Zhou, J. Tang and Y. Shen, Nat. Biomed. Eng., 2021, 5, 1019–1037 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00591k |
| This journal is © The Royal Society of Chemistry 2024 |