
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Block copolymer micelles as colloidal catalysts for photocatalytic NAD+ reduction†
Jonas
Eichhorn
 ab,
Alexander K.
Mengele
c,
Christof
Neumann
bde,
Johannes
Biskupek
f,
Andrey
Turchanin
bde,
Ute
Kaiser
f,
Sven
Rau
c and
Felix H.
Schacher
*abe
ab,
Alexander K.
Mengele
c,
Christof
Neumann
bde,
Johannes
Biskupek
f,
Andrey
Turchanin
bde,
Ute
Kaiser
f,
Sven
Rau
c and
Felix H.
Schacher
*abe
aInstitute of Organic Chemistry and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstraße 10, 07743 Jena, Germany. E-mail: felix.schacher@uni-jena.de
bJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany
cInstitute of Inorganic Chemistry I, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany
dInstitute of Physical Chemistry and Abbe Center of Photonics, Friedrich Schiller University Jena, Lessingstraße 10, 07743 Jena, Germany
eCenter for Energy and Environmental Chemistry Jena (CEEC Jena), Philosophenweg 7a, 07743 Jena, Germany
fElectron Microscopy of Materials Science, Central Facility for Electron Microscopy, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany
First published on 16th September 2024
Abstract
We herein report the preparation, characterization and (photo)catalytic investigation of block copolymer micelles based on amphiphilic, pH-responsive block copolymers featuring pendant bipyridyl rhodium complexes as NAD+ reduction catalyst and ruthenium polypyridyl complexes as photosensitizers. A well-defined polystyrene-block-poly((acrylic acid)-co-(2-(4-(4′-methyl-2,2′-bipyridyl))ethylacrylate)) (PS-b-P(AA-co-bpyEA)) block copolymer was synthesized via nitroxide-mediated polymerization (NMP). In post-polymerization functionalization reactions utilizing the pendant bpy moieties rhodium and ruthenium centers were covalently incorporated into the pH-responsive segment of the block copolymer. Proof of successful metal attachment was provided by nuclear magnetic resonance spectroscopy (NMR) and X-ray photoelectron spectroscopy (XPS) as well as size-exclusion chromatography (SEC). Morphological and structural investigations of the obtained block copolymer micelles in aqueous solutions using dynamic light scattering (DLS), transmission electron microscopy (TEM), scanning transmission electron microscopy in combination with energy dispersive X-ray spectroscopy (STEM-EDX) were performed. The respective model complexes and the metal-functionalized block copolymer micelles were finally tested as soft matter-based photocatalytic systems for NADH formation.
Introduction
Many advances in catalyst design, activity and selectivity have made homogeneous catalysis an attractive method for performing organic transformations in research as well as in industry.1–4 However, homogeneous catalysis encounters several difficulties in recovering the catalyst, preventing degradation processes and keeping products free of catalyst residues. In contrast, for heterogeneous systems the recovery/reactivation of deactivated catalysts from inorganic supports is a standard procedure in industry, albeit energy-intensive (smelting, refining, electrolysis).5–7 Furthermore, the activity of catalysts in heterogeneous regimes can be limited by diffusion effects and the effective available surface area of the catalyst. At this point a nanostructured, polymeric support (micelle, vesicle, bilayer, membrane) in solution may offer a promising and sustainable alternative. Such systems may provide significant synergism by combining high selectivity and activity based on molecular catalysts with ease of recovery and possibility of reactivation from heterogeneous catalysis. Hence, soft matter integration of catalytically active centers is very appealing for enabling the possibility of importing advantages from heterogeneous catalysis, such as simplified recovery, recyclability, and reuse. Further on, specifically generating and controlling local environments upon matrix incorporation allows a defined adjustment of reaction surroundings. Thus, upon covalently incorporating catalyst systems into such polymer-based nanostructures would enable the possibility to specifically target the prevention of degradation pathways such as leaking and provide long-term stability of active centers.8 Acquiring an in-depth understanding on how structure design of soft matter matrices can lead to improvements beyond just mechanical support is crucial. Therefore, the implementation of well-studied catalytic reactions into such matrices provide a profound basis for obtaining detailed insights in how structure influences the reaction and hence is the primary objective of this work.In our previous work we provided pH-responsive polystyrene-block-poly((acrylic acid)-co-(2-(4-(4′-methyl-2,2′-bipyridyl))-ethylacrylate)) (PS-b-P(AA-co-bpyEA)) block copolymer micelles as tunable matrix and potential nanoreactor for light-driven catalysis.9 In the latter case we demonstrated the suitability of the pendant bipyridine units for complexing platinum centers. In another work, we demonstrated the successful covalent embedding of molecular ruthenium(II) photosensitizers within block copolymer micelles, facilitating outstanding light-driven catalysis for the hydrogen evolution reaction (HER) using molecularly dissolved thiomolybdate clusters as catalysts.10 The resulting colloidal nanoreactors served beyond mechanical support and appeared to actively engage in the light-driven catalytic reaction, prompting the need for comprehensive investigations to elucidate the involvement of polymer-assisted transport and substrate diffusion processes. Following up on this, we herein use pH-sensitive micelles as soft matter matrix for both covalently attached rhodium catalysts and ruthenium photosensitizers to drive nicotinamide reduction. This would open the intriguing possibility to generate polymeric scaffolds where either the Rhodium-based catalyst or the Ruthenium-based photosensitizer would be immobilized and the complementary unit could be added as molecular entity. This would aid in understanding the effect of restrained diffusion imposed by the polymer backbone on the overall photocatalysis. Therefore, a polystyrene-block-poly((tert-butyl acrylate)-co-(2-(4-(4′-methyl-2,2′-bipyridyl))-ethylacrylate)) (PS-b-P(tBuA-co-bpyEA)) diblock terpolymer acts as soft matter matrix for immobilizing both rhodium and ruthenium centers. Subsequently, tert-butyl ester functionalities are deprotected to acrylic acid. The resulting amphiphilic block copolymers containing either rhodium, ruthenium or both metals retain the capacity to form micelles in aqueous solution and are tested in the following as soft matter-based colloidal catalyst, photosensitizer and photocatalyst for light-driven nicotinamide reduction.
Results and discussion
Preparation of [Rh]-functionalized block copolymers
A thoroughly investigated catalyst for nicotinamide reduction is [Rh(bpy)(Cp*)Cl]Cl and its various derivatives (bpy = 2,2′-bipyridine, Cp* = pentamethylcyclopentadienyl). These complexes have been used for a variety of catalytic applications such as the reduction of protons,11–13 CO2,14,15 carbonyl compounds16,17 as well as nicotinamides18–20 and the molecular mechanism of these reactions has been worked out in detail.21–24 Reductive activation of the Rh complexes is either possible electrochemically,14,19 using formate,20 or by suitable photosensitizers in the presence of appropriate electron donors.17,25 Resulting from the ubiquity of NAD(P)H in biological processes, it is possible to interlock the Rh-catalyzed formation of the reduced cofactors with enzymatic reactions giving rise to a broad range of possible applications such as stereoselective synthesis of chiral alcohols26 or the production of solar fuels.27,28The block copolymers for functionalization were synthesized by block extension of a polystyrene (PS) macroinitiator with tert-butyl acrylate (tBuA) and 2-(4-(4′-methyl-2,2′-bipyridyl))-ethylacrylate (bpyEA) utilizing nitroxide-mediated polymerization (NMP) (Scheme S1†). The functionalization of the obtained block copolymer PS277-b-P(tBuA99-co-bpyEA9) (BCP1, ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n = 44
n = 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1, Đ = 1.16) and PS277-b-P(tBuA262-co-bpyEA28) (BCP2, n = 70400 g mol−1, Đ = 1.28) was carried out upon cleavage of bridging chlorides in the [Rh(Cp*)Cl2]2 dimer while providing the block copolymer attached bpyEA units as ligands. In the following procedure an excess TFA was applied to deprotect tBuA units yielding amphiphilic, pH-responsive and [Rh]-functionalized diblock terpolymers [Rh]@BCP3 and [Rh]@BCP4 (Fig. 1).
300 g mol−1, Đ = 1.16) and PS277-b-P(tBuA262-co-bpyEA28) (BCP2, n = 70400 g mol−1, Đ = 1.28) was carried out upon cleavage of bridging chlorides in the [Rh(Cp*)Cl2]2 dimer while providing the block copolymer attached bpyEA units as ligands. In the following procedure an excess TFA was applied to deprotect tBuA units yielding amphiphilic, pH-responsive and [Rh]-functionalized diblock terpolymers [Rh]@BCP3 and [Rh]@BCP4 (Fig. 1).
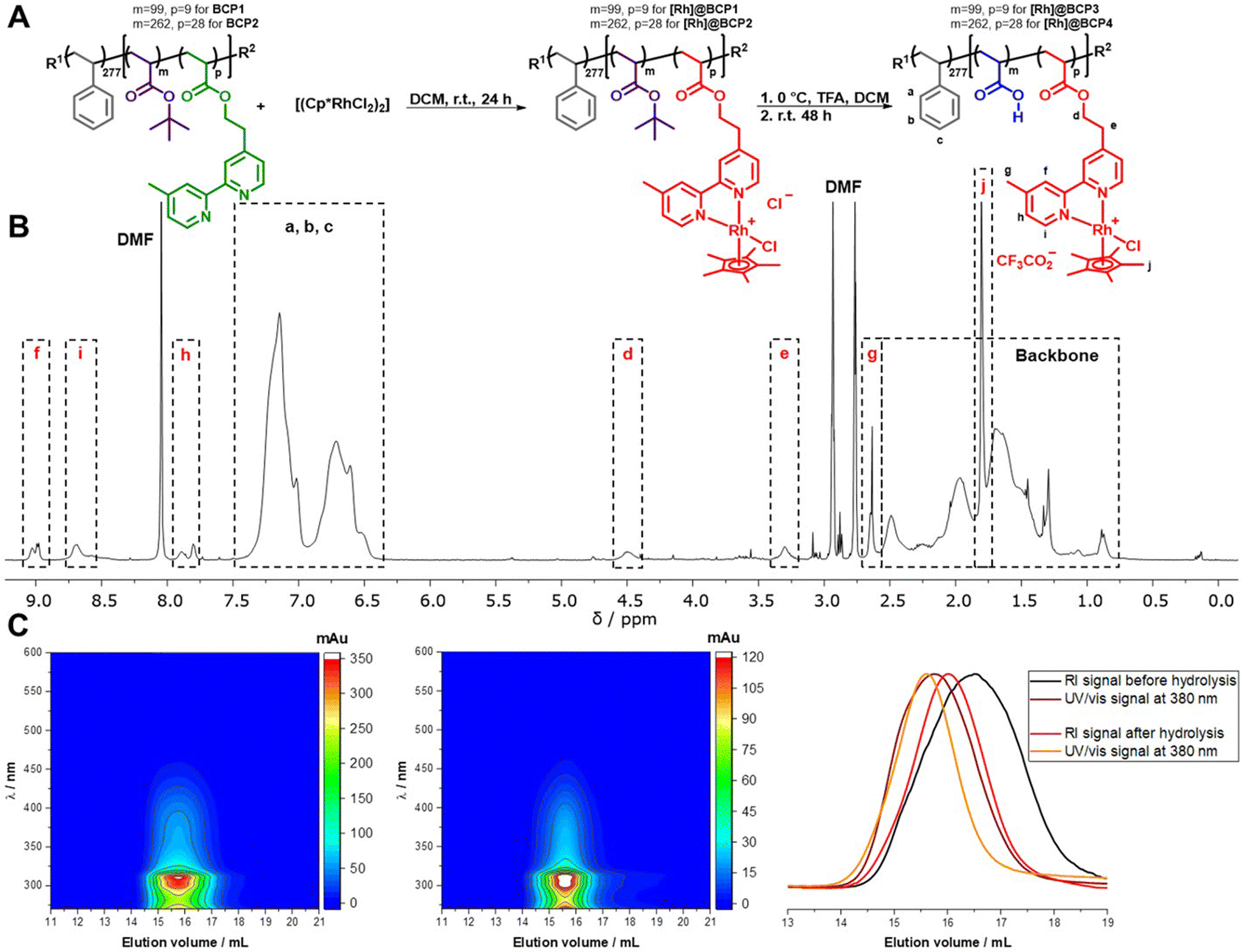 | ||
| Fig. 1 (A) Reaction scheme depicting the post-polymerization functionalization of PS277-b-P(tBuAm-co-bpyEAp) (the subscripts denote the corresponding degree of polymerization) with [Rh(Cp*)Cl2]2 and subsequent acidic deprotection of tBuA ester moieties, m = 99, 262, p = 9, 28, R1 = 1,1-dimethyl-2-carboxyethyl, R2 = N-tert-butyl-N-(1-(diethoxyphosphoryl)-2,2-dimethylpropyl)-N-oxyl nitroxide (SG1). (B) 1H-NMR spectrum of PS277-b-P(AA99-co-([Rh(bpyEA)(Cp*)Cl]CF3CO2)9) ([Rh]@BCP3) in DMF-d7 + TFA and signal assignment. (C) SEC-coupled in-line diode array spectra for PS277-b-P(tBuA99-co-([Rh(bpyEA)(Cp*)Cl]Cl)9) ([Rh]@BCP1) (left) and [Rh]@BCP3 (middle); RI elution traces and UV/vis absorption traces before and after deprotection of tBuA units (right). | ||
As reported for small molecular reference complexes,11 the covalent attachment of the [Rh] complex to the block copolymer is approximated to be quantitative (degree of functionalization (DoF) for bpyEA units >99%) on the basis of 1H-NMR analysis (Fig. S4 and S5†). Characteristic low-field shifts for bipyridine proton signals f, g, h, i as well as the presence of pentamethylcyclopentadienyl proton signal j in 1H-NMR spectra before and after deprotection with TFA provide further evidence for successful functionalization (Fig. 1B and S1–S7†). Furthermore, the block copolymer was purified by preparative SEC removing low-molecular weight components (such as unreacted [Rh(Cp*)Cl2]2), and thus, the pentamethylcyclopentadienyl proton signal j can be attributed to the attached [Rh]-complex. 1H-NMR spectra after deprotection imply almost complete cleavage of tBuA groups to AA upon comparing the NMR spectra before (Fig. S4 and S5†) and after treatment with TFA (Fig. 1B, S6 and S7†). The measurement of the above shown 1H-NMR spectra (Fig. 1B) was conducted using small amounts of TFA to shift the water signal to low-field and prevent overlap with protons d and e. The PAA content renders the block copolymers hygroscopic, and thus water could not be quantitatively removed from the sample. In case of PS277-b-P(AA262-co-([Rh(bpyEA)(Cp*)Cl]CF3CO2)28) ([Rh]@BCP4) the addition of TFA to perform 1H-NMR led to aggregate formation. Instead, measurements were performed using water signal suppression and as a result, the signal for protons e vanished (Fig. S6 and S7†).
From SEC-coupled in-line diode array spectra of the block copolymers before and after deprotection a successful attachment of the rhodium complex to the polymer structure can be qualitatively verified (Fig. 1C left and middle, S14†). First, we observe matching UV/vis spectra extracted from SEC analysis and separate UV/vis measurements of the respective compounds (Fig. S15 and S16†). Second, the characteristic UV/vis spectrum is recorded at elution volumes distinctive for polymeric structures with molar masses above 10000 g mol−1. Additionally, RI traces accompany the UV/vis signal at 380 nm (Fig. 1C, right). A general shift towards lower elution volumes observed for the UV/vis signal presumably occurs due to a device specificity.
The obtained increased dispersities and asymmetries in RI elution traces from SEC analysis for [Rh]-functionalized samples arise from potential polymer–column interactions of the polyionic, second segment alongside with altered solution structures after [Rh]-attachment and tBuA deprotection. The successful attachment of the Rh-complex to the polymer is confirmed via XPS. As seen in Fig. 2, a rhodium signal can be detected at a binding energy (BE) of 310.0 eV (Rh 3d5/2) at the same BE detected for the model complex [Rh(dmbpy)(Cp*)Cl]Cl (1, dmbpy = 4,4′-dimethyl-2,2′-bipyridine). In parallel the position of the N 1s signal is found at a BE of 400.4 eV matching to those of the bare Rh-complex 1 as well. This BE is higher in comparison to characteristic positions for pyridinic nitrogen (398.7 eV) and confirms the presence of the coordinated metal ions.29 Furthermore, a chlorine signal according to the Rh–Cl bonds was found (Fig. S19†). For [Rh]@BCP3, a F 1s peak was observed to be associated with CF3COO− as well as the respective C–F3 component in the C 1s spectrum. This supports the hypothesis of exchanging non-coordinating chloride in [Rh]-complexes with CF3COO−, introduced by TFA.
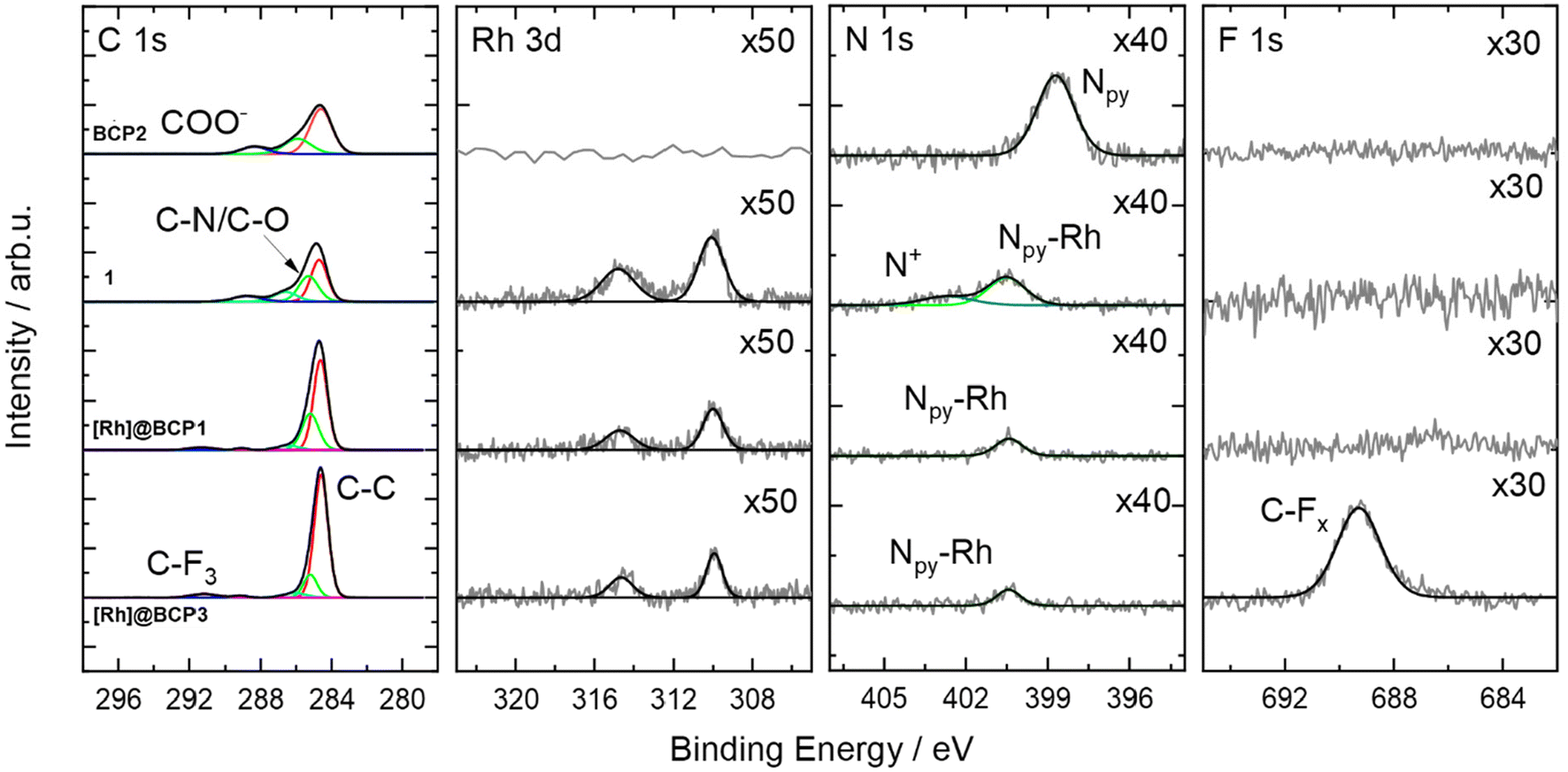 | ||
| Fig. 2 High resolution C 1s, Rh 3d, N 1s and F 1s (left to right) XP spectra of BCP2, [Rh(dmbpy)(Cp*)Cl]Cl (1), [Rh]@BCP1 and [Rh]@BCP3. For better representation, intensities of the Rh 3d, N 1s and F 1s spectra are multiplied by a factor given in the figure. | ||
Preparation of [Ru]-functionalized block copolymers
Generally, well-established systems for light-driven solar fuel generation often utilize ruthenium polypyridyl complexes as robust and efficient photosensitizers.30–37 While the vast majority of ruthenium-based photosensitizers ([Ru]) are applied in homogeneous systems, reports investigating its usage as covalently attached photosensitizer in non-conjugated soft matter matrices are rather limited.38–41 In principle, two different strategies to incorporate [Ru] into polymeric structures are available.42–45 Either a polymerizable ruthenium complex is used as comonomer39,46–56 or [Ru] is anchored via post-polymerization functionalization.38,40,56–70 Focusing on post-polymerization modifications, often pendant bipyridyl moieties act as attachment sites. Typically, procedures for grafting ruthenium polypyridyl complexes to existing polymer structures rely on [Ru(bpy)2Cl2] as readily available precursor. In this manner accessible asymmetric complexes commonly require aqueous mixtures with methanol or ethanol in combination with elevated temperatures,54,63,71–76 ethanol/methanol,52,53,56,66,77,78 DMF,79,80 acetone64,81–83 or dimethoxyethane65 under reflux. With respect to amphiphilic block copolymers obtained with nitroxide-mediated radical polymerization (NMP) as polymer matrix for ruthenium incorporation, solubility and integrity of the polymer chain cannot be ensured upon the latter instances. While polar solvents, such as aqueous mixtures of alcohols or acetone are not suitable to dissolve the hydrophobic compartment of amphiphilic block copolymers, a non-selective solvent should be chosen to dissolve the entire block copolymer for ensuring sufficient accessibility of the anchoring units in post-polymerization approaches. The above-mentioned reaction pathways for synthesizing asymmetric ruthenium bipyridyl complexes require elevated temperatures to cleave nitroxide-bearing chain ends and therefore, facilitate degradation of the polymer chain. For milder approaches in most cases [Ru(bpy)2Cl2] is necessary to be activated with silver salts,64,65,78,79,81–83 however silver colloid residues resulting from polynuclear anionic argentate side products may cause problems during subsequent catalysis.84 In addition for post-polymerization approaches utilizing copper salts in click chemistry67–70 a later potential interference of copper residues in (light-driven) catalysis cannot be excluded. A report from Brennan et al. provides a desirable approach in DMF with temperatures below 100 °C, thus avoiding polymer degradation and ensuring good solubility.85 Still, the activation of the chloro ligands in [Ru(bpy)2Cl2] is desirable to further increase reactivity. Isolating the respective activated complex, where the chloro ligands are exchanged with coordinating solvent molecules, allows the removal of potential silver colloids for subsequent application. In fact, a number of literature protocols utilized isolated [Ru(bpy)2(solvent)2] species as activated complex.63,65,86 In this work we select [Ru(bpy)2(ACN)2](PF6)2 as activated complex for its good thermal stability86 to covalently attach a ruthenium photosensitizer complex via post-polymerization functionalization within the later hydrophilic and pH-responsive segment of an amphiphilic block copolymer.Therefore, BCP2 was treated in a ligand exchange reaction with [Ru(bpy)2(ACN)2](PF6)2, where two acetonitrile ligands of the precursor are substituted with one pendant bpyEA unit at the block copolymer. In the next step the obtained ruthenium functionalized block copolymer was treated with an excess of TFA to deprotect tBuA units under acidic conditions yielding an amphiphilic, pH-responsive and [Ru]-functionalized diblock quaterpolymer (Fig. 3).
 | ||
| Fig. 3 (A) Reaction scheme depicting the post-polymerization functionalization of BCP2 with [Ru(bpy)2(ACN)2](PF6)2 and subsequent acidic deprotection of tBuA ester moieties, R1 = 1,1-dimethyl-2-carboxyethyl, R2 = SG1. (B) 1H-NMR spectrum of PS277-b-P(AA262-co-bpyEA12-co-([Ru(bpyEA)(bpy)2](CF3CO2)2)16) ([Ru]@BCP4) in DMF-d7 and signal assignment. (C) SEC-coupled in-line diode array spectra for PS277-b-P(tBuA262-co-bpyEA12-co-([Ru(bpyEA)(bpy)2](PF6)2)16) ([Ru]@BCP2) (left) and [Ru]@BCP4 (middle); RI elution traces and UV/vis absorption traces before and after deprotection of tBuA units (right). | ||
Through 1H-NMR analysis the degree of functionalization of bpyEA units with [Ru] was determined to 57% resulting in 16 out of 28 available bpyEA units per polymer chain to act as ligands for the covalent attachment of the ruthenium precursor (Fig. S9†). In analogy to [Rh]-containing block copolymers an approximated quantitative deprotection of tBuA can be assumed. The 1H-NMR spectrum for [Ru]@BCP4 was measured using water- and solvent signal suppression, thus the protons e and e′ are not present (Fig. 3B). The high PAA content causes water entry, analogously to the earlier presented [Rh] containing block copolymers [Rh]@BCP3 and [Rh]@BCP4. As a result, bpyEA side chain signals as well as protons j and i′ are superimposed by the water and DMF solvent signal (Fig. S10†). SEC-coupled in-line diode array spectra of the block copolymers before and after deprotection verify a successful attachment of the ruthenium chromophore to the polymer structure in a qualitative manner (Fig. 3C and S17†). Dispersity and shape of traces determined by the used SEC are not fully representative, since strong column interactions are a general issue for [Ru]-functionalized polymers, causing falsified results or even preventing analysis.54,64,65 In this case, polymer characteristics such as a narrow molecular weight distribution and monomodality determined before the attachment are still valid as we assume no change in the degree of polymerization throughout the performed reactions (Table 1).
| Block copolymer | Composition |
n
/g mol−1 |
Đ | Block ratioc | Metal contentd/wt% |
|---|---|---|---|---|---|
| a NMR analysis, based on the degree of polymerization (DP) of tBuA and presumed quantitative deprotection to AA. b SEC-analysis, PS standard for calibration. c Molecular weight of second block/molecular weight of first block. d NMR analysis, based on DP of [Rh] or [Ru]. | |||||
| BCP1 | PS277-b-P(tBuA99-co-bpyEA9) | 44300 |
1.16 | 0.52 | — |
| BCP2 | PS277-b-P(tBuA262-co-bpyEA28) | 70400 |
1.28 | 1.43 | — |
| [Rh]@BCP1 | PS277-b-P(tBuA99-co-([Rh(bpyEA)(Cp*)Cl]Cl)9) | 47100 |
1.33 | 0.62 | 1.97 Rh |
| [Rh]@BCP2 | PS277-b-P(tBuA262-co-([Rh(bpyEA)(Cp*)Cl]Cl)28) | 79000 |
1.48 | 1.72 | 3.65 Rh |
| [Rh]@BCP3 | PS277-b-P(AA99-co-([Rh(bpyEA)(Cp*)Cl]CF3CO2)9) | 41600 |
1.26 | 0.43 | 2.23 Rh |
| [Rh]@BCP4 | PS277-b-P(AA262-co-([Rh(bpyEA)(Cp*)Cl]CF3CO2)28) | 64300 |
1.54 | 1.21 | 4.48 Rh |
| [Ru]@BCP2 | PS277-b-P(tBuA262-co-bpyEA12-co-([Ru(bpyEA)(bpy)2](PF6)2)16) | 81600 |
1.54 | 1.81 | 1.98 Ru |
| [Ru]@BCP4 | PS277-b-P(AA262-co-bpyEA12-co-([Ru(bpyEA)(bpy)2](CF3CO2)2)16) | 66900 |
1.15 | 1.30 | 2.42 Ru |
However, SEC analysis shows reduced absolute absorption for the block copolymer after deprotection, that may arise from polymer–column interactions or the change in solubility by further charge introduction through PAA. It cannot be excluded that a significant amount of sample is removed by the pre-column. Thus, a shift to higher elution volumes can result, which also is caused by a reduction of the molar mass through cleavage of the tBuA groups. Nevertheless, sufficiently good qualitative analyzability with this SEC system is given for the presented [Ru]-containing polymer samples.
The presence of non-attached complex can be excluded since after preparation the block copolymers were passed over preparative SEC columns to remove on the one hand unreacted precursor and on the other hand potential low-molecular weight substances either as reagents or side-products. When comparing NMR spectra before and after deprotection with TFA, a reduction of [Ru(bpyEA)(bpy)2]2+ content is not observed. Exemplary, integrals of signals for protons f′, m, f, i and l remain constant relative to those for protons a, b, and c (Fig. S9 and S10†). However, the treatment of [Ru]@BCP2 with TFA also resulted in an exchange of PF6− ions with trifluoroacetate (CF3COO−) to a large extend. XPS analysis (see Fig. 4) of the block copolymer after deprotection with TFA shows a shift of the F 1s signal to higher BEs, characteristic for CFx binding modes, that can be associated with CF3COO−. Furthermore, a rather weak signal corresponding to remaining PF6− is observed, which is confirmed by a small P 2p signal (see Fig. S21†). Nevertheless, Ru 3d and N 1s BEs from [Ru(bpy)2(dmbpy)](PF6)2 (model complex) are analogously found in the block copolymer spectra after functionalization with [Ru] and after treatment with TFA. A successful attachment of [Ru] is further verified upon comparing the N 1s signal of the block copolymer before functionalization with those of the model complex. Similar to the observation after attachment of the Rh-complex, for BCP2 the N 1s binding mode is shifted to higher BEs after attachment of [Ru], matching those of the model complex (400.0 eV). Additionally, a second N 1s binding mode for [Ru]@BCP2 can be found related to non-functionalized bpyEA units (398.7 eV, light green). Please note, the different intensities of ruthenium and nitrogen in the XP spectra of the model complex and after attachment of [Ru] to the block copolymer are related to the sample fabrication via drop casting (see ESI† for details).
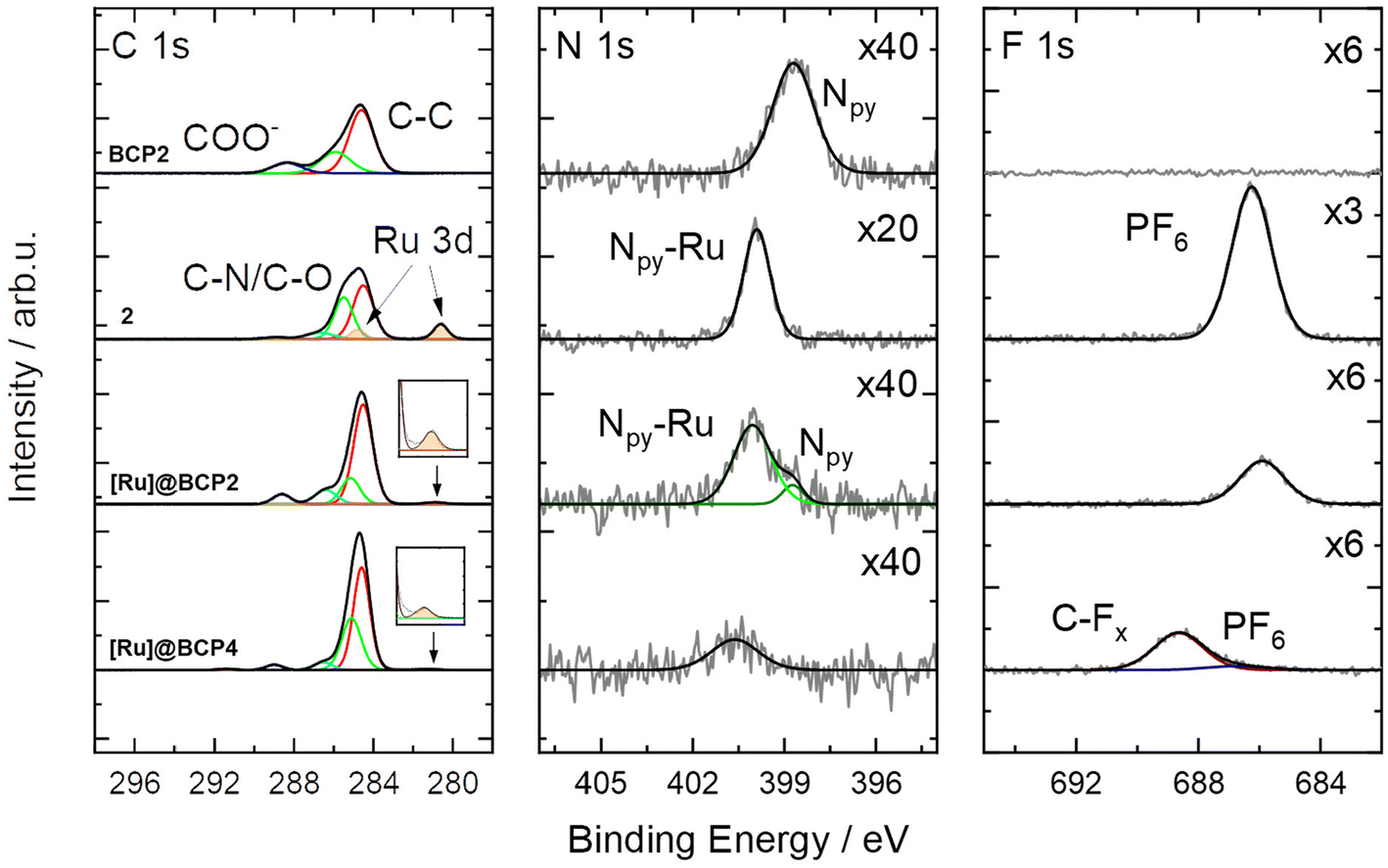 | ||
| Fig. 4 High resolution C 1s (left), N 1s (middle) and F 1s (right) XP spectra of BCP2, [Ru(bpy)2(dmbpy)](PF6)2 (2), [Ru]@BCP2 and [Ru]@BCP4. For better representation, intensities of the N 1s and F 1s spectra are multiplied by a factor given in the figure. | ||
Preparation of [Ru]- and [Rh]-functionalized block copolymer
With [Ru]@BCP2 on hand, remaining unfunctionalized bpyEA units are available for the attachment of [Rh] to form a block copolymer containing both photosensitizer and catalyst. In analogy to the [Rh]-functionalized block copolymer the functionalization of the [Ru] centers bearing block copolymer and subsequent acidic deprotection was performed (Fig. 5).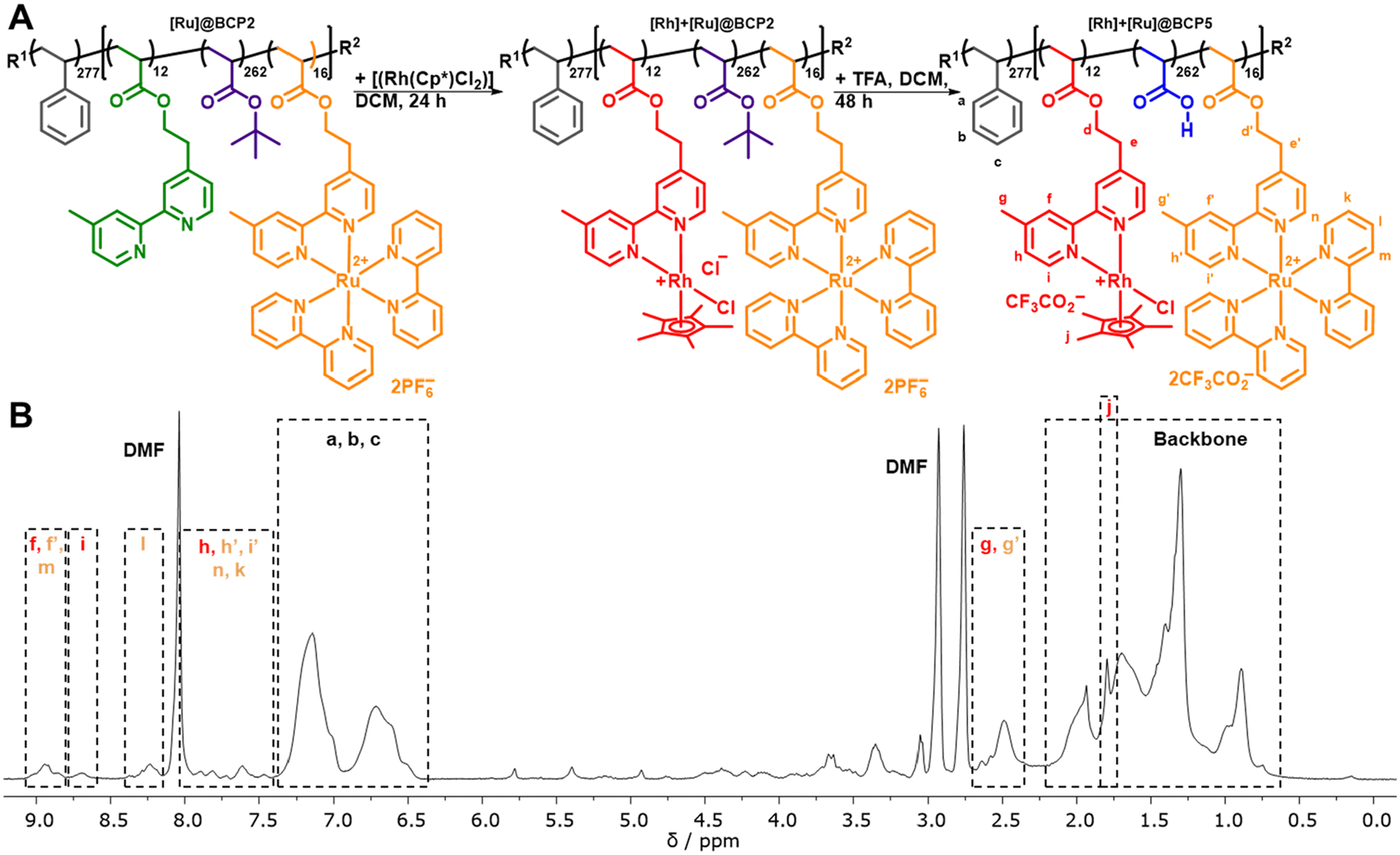 | ||
| Fig. 5 (A) Reaction scheme depicting the functionalization of [Ru]@BCP2 and subsequent acidic deprotection of tBuA ester moieties, R1 = 1,1-dimethyl-2-carboxyethyl, R2 = SG1. (B) 1H-NMR spectrum of PS277-b-P(AA262-co-([Rh(bpyEA)(Cp*)Cl]CF3CO2)12-co-([Ru(bpyEA)(bpy)2] (CF3CO2)2)16) ([Rh] + [Ru]@BCP5) in DMF-d7 + TFA and signal assignment. | ||
Characteristic signals for both [Ru] and [Rh] can be found in the 1H-NMR spectrum of [Rh] + [Ru]@BCP5 confirming approximated no residual unfunctionalized bpyEA units per polymer chain (Fig. 5B and Fig. S12†). The high efficiency of attaching [Rh] to pendant bipyridine units was additionally proven in this approach. Most pronounced evidence for successful [Rh] incorporation is given by signals attributed to protons i and j. Especially the presence of proton i allows the determination of the DoF for [Rh] to 12 units per polymer chain upon comparing to signals caused by the PS block a, b and c. As during purification, the block copolymer was passed over preparative SEC, collecting the high molecular weight fraction only, the signal of proton j can be assigned exclusively to immobilized [Rh] complex. SEC-coupled in-line diode array spectra further provide evidence for the presence of [Rh] in the copolymer architecture (Fig. 6). Both features of [Ru] and [Rh] in the UV/vis spectrum at elution volumes of 14.9 mL (before deprotection) and 15.1 mL (after deprotection) show characteristic bands for both complexes. Despite the stronger absorption of [Ru] bands (289 nm and 456 nm) the shoulder at 311 nm reveals the distinctive absorption of [Rh] (compare to Fig. S17A†). Reduced absolute absorption of the block copolymer after deprotection is explained by strong polymer–column interactions, also noted previously for the [Ru]-containing block copolymer. The shoulder for lower elution volumes (sample after deprotection) is most likely due to partial aggregate formation of the block copolymer in the solvent for SEC analysis (DMAc + 0.05 wt% NH4PF6, Fig. 6D).
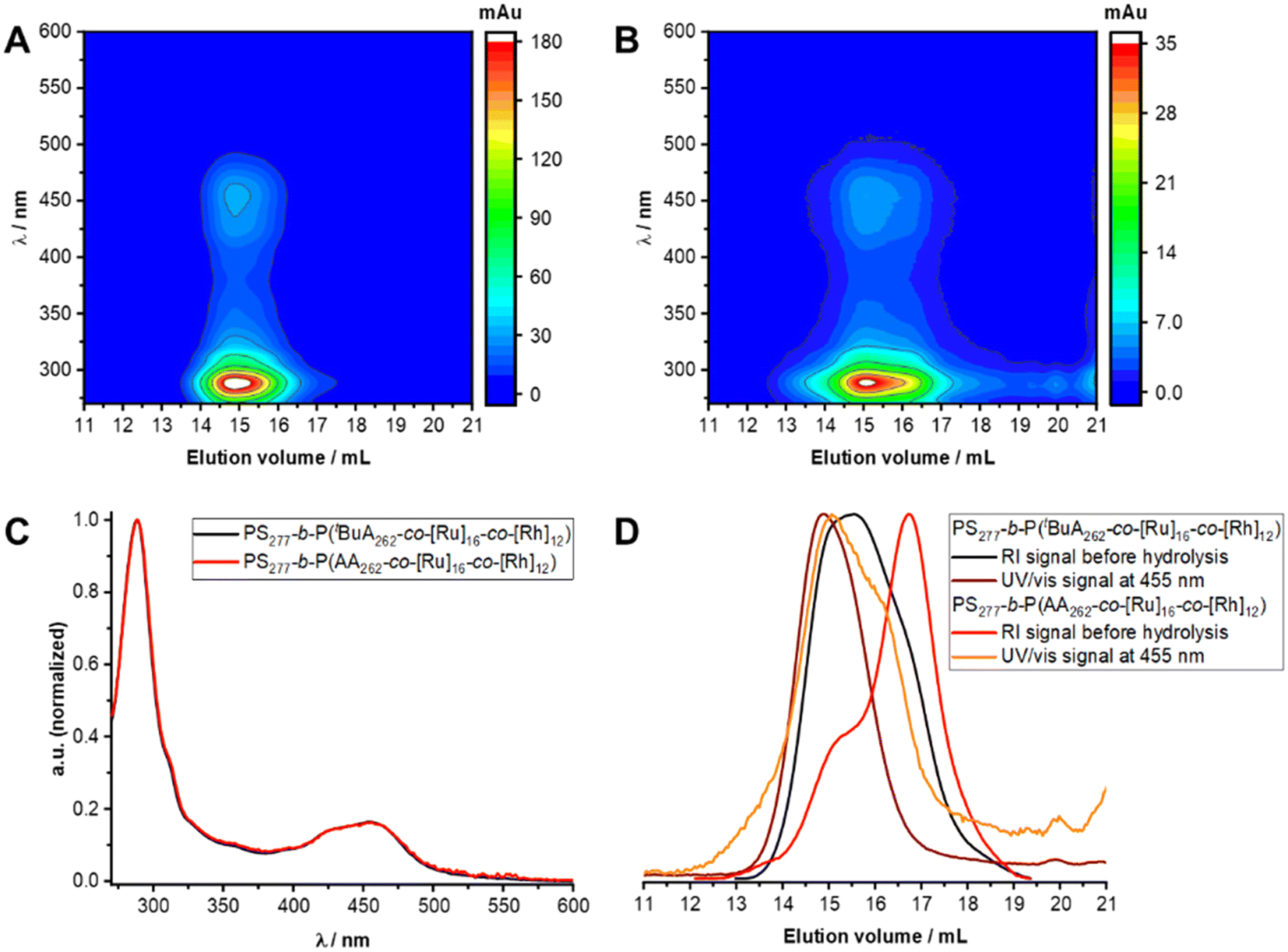 | ||
| Fig. 6 (A) SEC-coupled in-line diode array spectra for [Rh] + [Ru]@BCP2. (B) SEC-coupled in-line diode array spectra for [Rh] + [Ru]@BCP5. (C) Black: diode array spectrum of [Rh] + [Ru]@BCP2 from in-line SEC analysis, elution volume = 14.9 mL, arbitrary unit (a.u.) normalized; red: diode array spectrum of [Rh] + [Ru]@BCP5 from in-line SEC analysis, elution volume = 15.1 mL, normalized. (D) RI elution traces and UV/vis absorption traces before and after deprotection of tBuA units. | ||
Micellization of amphiphilic block copolymers in water
Micellization of metal-functionalized block copolymers in water was carried out similarly to previously published results with small changes (Section S3†).9 Rhodium- and ruthenium-functionalized block copolymers form spherical micelles in water, which were investigated by dynamic light scattering (DLS) and transmission electron microscopy (TEM) (Fig. S22–S25†). Apparent hydrodynamic radii (〈Rh〉) of the formed micelles were determined by DLS analysis showing a clear relation of DP in the second, hydrophilic segment and measured 〈Rh〉 for [Rh]-micelles. The TEM images (Fig. S23–S25†) show isolated micelles as well as aggregated constructs most probably as the result of drying processes on the TEM grid. A clear trend of influence from electrostatic repulsion in the corona is observed for [Rh]-micelles resulting in lower values for average aggregation number![[N with combining macron]](https://www.rsc.org/images/entities/i_char_004e_0304.gif) agg and chain density
agg and chain density 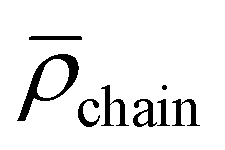 (see Section S1† for details) with increasing DP in the second segment. Compared to [Rh]@BCP4 a decrease in 〈Rh〉 of 8 nm is observed for using [Ru]@BCP4 that can be explained by decreased electrostatic repulsion of the micellar corona introduced by permanently, twice positively charged [Ru] units (Table 2). Although the degree of functionalization with [Rh] is higher, the average number of positive charges in the second segment of the ruthenium functionalized block copolymer is higher. Consistent with results obtained from [Rh]-containing micelles a trend in further reducing electrostatic repulsion causing higher agg and
(see Section S1† for details) with increasing DP in the second segment. Compared to [Rh]@BCP4 a decrease in 〈Rh〉 of 8 nm is observed for using [Ru]@BCP4 that can be explained by decreased electrostatic repulsion of the micellar corona introduced by permanently, twice positively charged [Ru] units (Table 2). Although the degree of functionalization with [Rh] is higher, the average number of positive charges in the second segment of the ruthenium functionalized block copolymer is higher. Consistent with results obtained from [Rh]-containing micelles a trend in further reducing electrostatic repulsion causing higher agg and 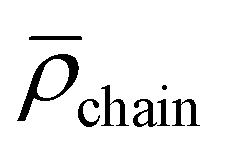 is observed. Obtained results for [Rh]- and [Ru]-containing micelles are rather similar to previously reported PS-b-PAA and PS-b-P(AA-co-bpyEA) micelles, however secondary aggregation is less pronounced for [Ru]@BCP4 micelles (second mode between 60–200 nm in Fig. S22†).9 A powerful method to verify the presence of ruthenium in the micellar structure is achieved by scanning transmission electron microscopy (STEM) accompanied by energy dispersive X-ray (EDX) analysis which allows spatially resolving elemental composition of nanostructures. Although disintegrating most of the carbon of the micelles upon this process, the presence of ruthenium in the micellar fragments was proven. In addition, remaining carbon and oxygen residuals spatially correlate with ruthenium (Fig. S26–S28†). STEM-EDX analysis of [Rh]-containing micelles ([Rh]@BCP4) led to a rather scattered deposition of the polymer, presumably due disintegration of the micelles under plasma treatment. Thus, a spatial correlation of, for instance, carbon, oxygen and rhodium for distinctive micellar fragments was not possible (Fig. S29†). Furthermore, the strong Cl–K line overlaps with Rh–L lines and strong K and Ca–L lines with Rh–M lines, respectively (Fig. S30†). Additionally, the Rh–K lines (at 20.2 keV) are outside the standard range of 0–20 keV. Upon using the extended keV-range the presence of Rh could be verified (see the Ru–K line in Fig. S30†). However, the local mapping showed a rather homogenous Rh distribution (Fig. S29†). Micellization of [Rh] + [Ru]@BCP5 yielded spherical micelles and analysis was carried out via DLS and TEM (Fig. 7).
is observed. Obtained results for [Rh]- and [Ru]-containing micelles are rather similar to previously reported PS-b-PAA and PS-b-P(AA-co-bpyEA) micelles, however secondary aggregation is less pronounced for [Ru]@BCP4 micelles (second mode between 60–200 nm in Fig. S22†).9 A powerful method to verify the presence of ruthenium in the micellar structure is achieved by scanning transmission electron microscopy (STEM) accompanied by energy dispersive X-ray (EDX) analysis which allows spatially resolving elemental composition of nanostructures. Although disintegrating most of the carbon of the micelles upon this process, the presence of ruthenium in the micellar fragments was proven. In addition, remaining carbon and oxygen residuals spatially correlate with ruthenium (Fig. S26–S28†). STEM-EDX analysis of [Rh]-containing micelles ([Rh]@BCP4) led to a rather scattered deposition of the polymer, presumably due disintegration of the micelles under plasma treatment. Thus, a spatial correlation of, for instance, carbon, oxygen and rhodium for distinctive micellar fragments was not possible (Fig. S29†). Furthermore, the strong Cl–K line overlaps with Rh–L lines and strong K and Ca–L lines with Rh–M lines, respectively (Fig. S30†). Additionally, the Rh–K lines (at 20.2 keV) are outside the standard range of 0–20 keV. Upon using the extended keV-range the presence of Rh could be verified (see the Ru–K line in Fig. S30†). However, the local mapping showed a rather homogenous Rh distribution (Fig. S29†). Micellization of [Rh] + [Ru]@BCP5 yielded spherical micelles and analysis was carried out via DLS and TEM (Fig. 7).
 | ||
| Fig. 7 (A) DLS-CONTIN plots from three individual measurements for [Rh] + [Ru]@BCP5 based micelles in deionized water, c = 0.5 g L−1. (B) TEM image of [Rh] + [Ru]@BCP5 based micelles in deionized water, c = 0.5 g L−1 and schematic representation of the anticipated micellar structure. | ||
| Block copolymer | Composition | Block ratioa | 〈Rh〉b/nm |
![[r with combining macron]](https://www.rsc.org/images/entities/i_char_0072_0304.gif) core
/nm
core
/nm |
agg
|
|
|---|---|---|---|---|---|---|
| a Molecular weight of second block/molecular weight of first block. b DLS analysis. c TEM analysis. | ||||||
| [Rh]@BCP3 | PS277-b-P(AA99-co-([Rh(bpyEA)(Cp*)Cl]CF3CO2)9) | 0.43 | 24 | 14.0 ± 2.0 | 237 | 0.097 ± 0.028 |
| [Rh]@BCP4 | PS277-b-P(AA262-co-([Rh(bpyEA)(Cp*)Cl]CF3CO2)28) | 1.21 | 33 | 12.5 ± 1.9 | 171 | 0.087 ± 0.026 |
| [Ru]@BCP4 | PS277-b-P(AA262-co-bpyEA12-co-([Ru(bpyEA)(bpy)2](CF3CO2)2)16) | 1.30 | 25 | 15.9 ± 1.7 | 348 | 0.110 ± 0.024 |
| [Rh] + [Ru]@BCP5 | PS277-b-P(AA262-co-([Rh(bpyEA)(Cp*)Cl]CF3CO2)12-co-([Ru(bpyEA)(bpy)2] (CF3CO2)2)16) | 1.43 | 20 | 12.7 ± 1.3 | 179 | 0.088 ± 0.019 |
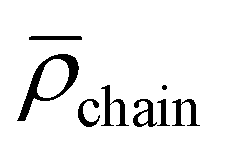
DLS analysis reveals micelles exhibiting 〈Rh〉 of 20 nm with a second mode notable between 50–120 nm. Compared to the ruthenium-functionalized micelles 〈Rh〉 is reduced by 5 nm due to further decreased electrostatic repulsion of the micellar corona introduced by permanently positively charged [Rh] units into an already [Ru] functionalized matrix (Table 2). As expected, utilizing the block copolymer containing both [Ru] and [Rh] centers in the PAA segment, the lowest value of 〈Rh〉 for all metal functionalized block copolymers was obtained. From TEM analysis core could be determined to 12.7 ± 1.3 nm resulting in an approximated agg = 179. Therefore, 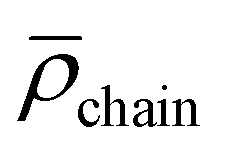 can be calculated to 0.088 ± 0.019 nm−2. Counterintuitively, core and agg show decreased values, although increased attractive electrostatic forces are expected in the corona driving micellization towards larger core sizes. This observation may be explained by the altered micellization process compared to [Rh]- or [Ru]-functionalized samples.
can be calculated to 0.088 ± 0.019 nm−2. Counterintuitively, core and agg show decreased values, although increased attractive electrostatic forces are expected in the corona driving micellization towards larger core sizes. This observation may be explained by the altered micellization process compared to [Rh]- or [Ru]-functionalized samples.
Thermal and photocatalytic NAD+ reduction studies
In a final step the [Ru]- and [Rh]-containing micelles were analyzed with respect to their contribution to purely thermal as well as light-driven catalytic NADH formation. The presence of [Rh(bpy)(Cp*)Cl]+-like centers allowed to probe the principal catalytic activity of all [Rh] embedded materials in a light-independent fashion by incubation with sodium formate as reductant (all experimental details can be found in the ESI, section S4†). Using this method, it was initially tested whether the different molecular compositions, more specifically, the degree of polymerization in the second block of [Rh]@BCP3 and [Rh]@BCP4 had any effect on the efficiency of the NADH-forming catalytic process. At a temperature of 45 °C and a concentration of 5 μM micelle-anchored [Rh] centers, both [Rh]-functionalized polymeric systems behaved comparable (Table S3,† entries 1 and 2). As can be seen in Fig. S31,† the emission band centered at 462 nm, being characteristic for NADH, increased in a linear fashion and reached for both micelles a TON of ca. 1 after 90 min (TON = turnover number). As for analogous small molecular Rh complexes the β-hydride elimination process has been reported to represent the rate determining step of the overall NADH-forming process,20 both [Rh]-containing micelles were also investigated at higher temperatures, i.e. 60 °C, to accelerate this process or other activity-limiting steps such as substrate diffusion towards the micelle-embedded [Rh] centers. As shown in Fig. 8A the linearly increasing amount of NADH is again clearly observed by the rising emission band at 462 nm as well as the rising intensity of the absorption band at 340 nm (see Fig. S32†). After 90 min a TON of ca. 4 was observed for both nearly identically behaving [Rh]-containing micelles (see Fig. 8A, Fig. S32 and Table S3,† entries 3 and 4). As neither at 45 °C nor at 60 °C any notable difference in catalytic activity was observed between the two different [Rh] containing micelles, it is concluded that the rate of formate-driven NADH formation is independent of the micellar corona size for the two herein presented [Rh] micelles.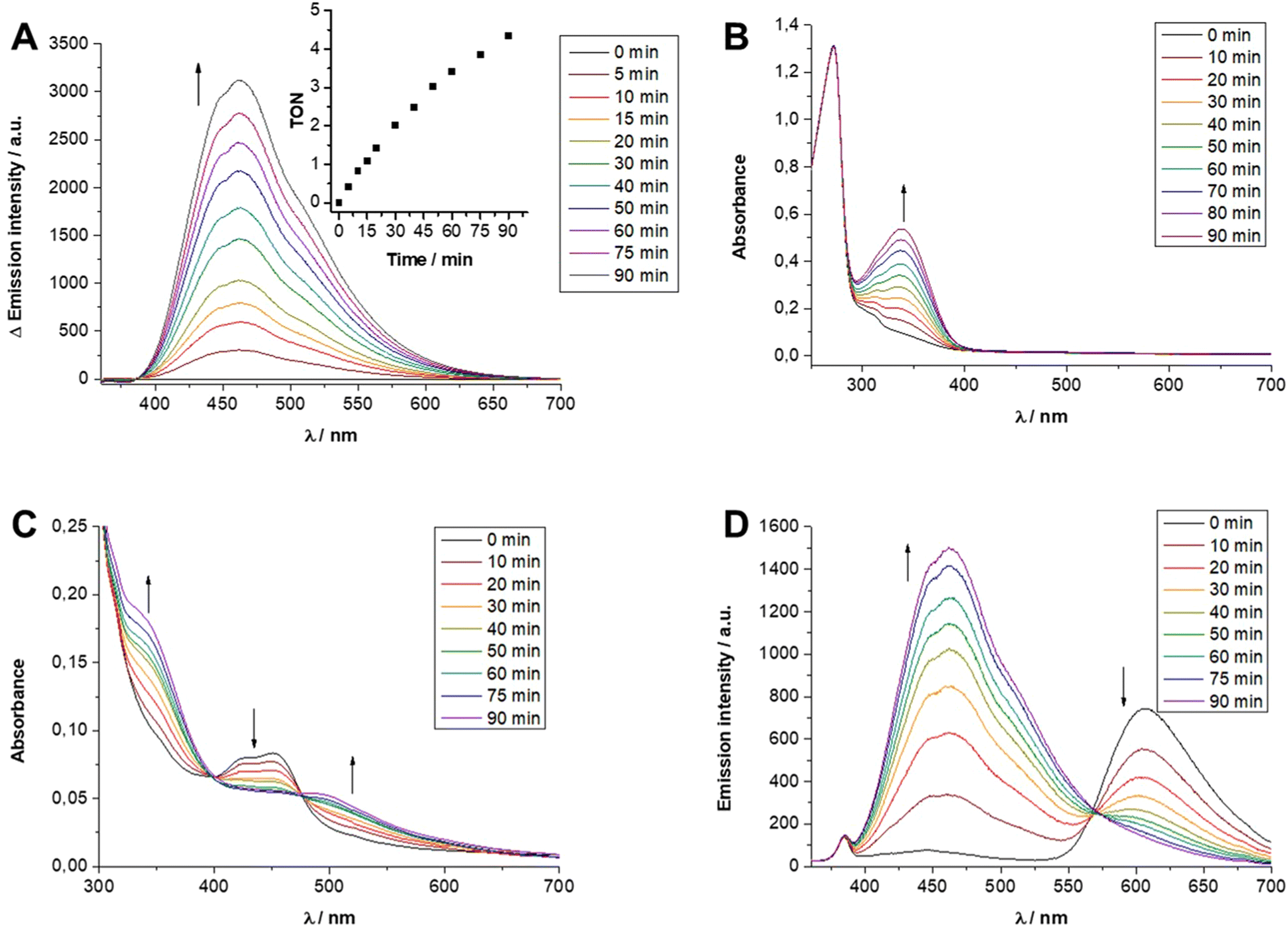 | ||
| Fig. 8 (A) Emission spectroscopic changes and corresponding TON plot during formate-driven NAD+ reduction at 60 °C using 5 μM [Rh]@[Rh]@BCP3. (B) UV/vis spectroscopic changes during formate-driven NAD+ reduction at r.t. with 5 μM 1. (C and D) UV/vis and emission spectroscopic changes during photocatalytic NAD+ reduction using 5 μM [Ru(bpy)3]Cl2 and 5 μM [Rh]@BCP3. | ||
In contrast to the elevated temperatures described above, no NAD+ reduction was observed at room temperature for both [Rh]-containing micelles (Fig. S33 and Table S3,† entries 5 and 6). This is also true for the bimetallic [Ru] and [Rh] moieties containing [RuRh] micelles based on [Rh] + [Ru]@BCP5 which otherwise yielded a TON for the thermal NADH formation of 1.8 after 90 min at 45 °C (Fig. S33 and Table S3,† entries 7 and 8). However, when comparing the polymeric systems with [Rh(dmbpy)(Cp*)Cl]Cl (Rh complex 1) as small molecular reference catalyst, substantially higher formate-driven NADH formation activity was observed for 1. Contrary to the micelles, this reference catalyst was active at room temperature and showed a TON of 15 after 90 min (see Fig. 8B and Table S3,† entry 9). At 45 °C complex 2 even converts ca. 80% of NAD+ into NADH within only 20 min, i.e. a TON of 40 after 20 min is observed, accompanied by a maximum TOF of 150 h−1 (TOF = turnover frequency, see Fig. S34 and Table S3,† entry 10). The superior activity of complex 1 compared to the [Rh] functionalized micelles might be explained by either a very slow β-hydride elimination process inside the micelles, a slow diffusion rate of substrates (HCO2− or NAD+) towards the micelle-embedded [Rh] centers or a blocking of the Rh binding site by the high local concentration of polymer integrated acid functionalities preventing the necessary coordination of formate to the Rh center.
After elucidating the capability of all [Rh]-containing micelles for NADH formation in the presence of a suitable reductant, their activity for cofactor reduction under irradiation with visible light (λ = 465 nm) was investigated (for experimental details see ESI, section S4†). During the photocatalytic process in presence of triethylamine (TEA) as sacrificial electron donor, the [Rh] centers (i.e.1 as well as micelle-incorporated [Rh] centers) were reduced by Ru polypyridine moieties, i.e. either by molecular [Ru(bpy)3]Cl2 or polymeric [Ru]@BCP4 as well as pendant [Ru] sites in the bifunctional micelle [Rh] + [Ru]@BCP5.
Based on the results from the light-independent catalysis results described above as well as prior results on heterodinuclear RuRh photocatalysts showing improved (photo)catalytic activity at elevated temperatures,87 light-driven catalysis was initially investigated at 45 °C. As for the formate-driven catalytic process, both only [Rh] centers containing micelles exhibited identical photocatalytic activity in the presence of 5 μM [Ru(bpy)3]Cl2 showing a TON of 2 after 90 min (see Fig. S35 and Table S3,† entries 11 and 12). This indicated that the varying ratio of repeating units in the polymeric backbones also induced no reactivity differences in the photocatalytic processes.
However, the analysis of the absorption and emission spectroscopic changes during this process in combination with the course of the respective TOF values suggested that higher TONs were likely prevented by the parallel occurring photodegradation of [Ru(bpy)3]Cl2 which was concluded from the vanishing 3MLCT band absorbance at around 450 nm and the decreasing intensity of the 3MLCT emission band with a maximum at around 605 nm (see Fig. 8C and D). Thus, photocatalytic runs with an increased concentration of 25 μM [Ru(bpy)3]Cl2 were performed. Although this did not lead to an increase in longevity of the overall photocatalytic system, as a consequence of an initially faster photocatalysis still an improved TON of ca. 4.5 after 120 min at 45 °C was observed (Fig. S36†). Interestingly, when keeping 25 μM [Ru(bpy)3]Cl2 but varying the temperature from room temperature (r.t.) to 45 °C and to 60 °C, the initial TOF for NADH formation as well as the rate of the photodegradation increased (see Fig. S36†). These opposed effects lead to TONs of ca. 3.5, 4.5 and 3.0 at r.t., 45 °C and 60 °C after 120 min, respectively (Table S2,† entries 13–15).
However – in contrast to the formate-driven process – under photocatalytic conditions NAD+ can also be directly reduced by Ru polypyridine complexes, leading to the formation of NAD-dimers (NAD2)88 as non-luminescent89 side product. Thus, the selectivity for NADH at different [Ru(bpy)3]Cl2 concentrations and temperatures was determined as well (see ESI section S4† for the details on selectivity determination). With 5 μM [Ru(bpy)3]Cl2 at r.t. a selectivity for NADH formation of 85% was obtained using both [Rh]-functionalized micelles. At 25 μM [Ru(bpy)3]Cl2 the selectivity drops to 58% at r.t. and to 47% and 35% at 45 °C and 60 °C, respectively. Control experiments where either only the [Rh] functionalized micelles or only [Ru(bpy)3]Cl2 were irradiated in the presence of NAD+ indicated that (i) irrespective of the applied temperature (r.t. or 60 °C) a very limited NADH-forming background activity was observed for the [Rh] micelles (Fig. S37†) but (ii) significant photocatalytic activity with low selectivity (<50% for NADH) was detected for [Ru(bpy)3]Cl2 (Fig. S38†) at elevated temperatures. In the absence of NAD+ no Ru-induced emission spectroscopic changes at 462 nm were observed (Fig. S39†). As from these experiments it became clear that a highly selective NADH forming photocatalytic process involving [Rh]-containing micelles is only achieved at low, i.e. 5 μM [Ru(bpy)3]Cl2 as well as at r.t., all further experiments were performed under these conditions unless otherwise noted (see Fig. S40†).
In contrast to the formate-driven process where complex 1 outperformed the [Rh] functionalized micelles by at least one order of magnitude, the light-dependent reference experiment involving [Rh(dmbpy)(Cp*)Cl]Cl instead of the [Rh]-modified micelles resulted in very similar NADH production. At r.t. and within 90 min a TON of 2 (93% selectivity) and at 45 °C a TON of 4 (77% selectivity) for complex 2 was obtained (see Fig. S41 and Table S3,† entries 16 and 17; at 5 μM [Ru(bpy)3]Cl2 the [Rh]-functionalized micelles gave TONs of ca. 2, irrespective whether r.t. (85% selectivity) or 45 °C (82% selectivity) was applied, see Fig. S35 and S42 as well as Table S3,† entries 18–21) which highlights the photochemical reduction of [Rh] centers as activity limiting factor.
As discussed above, the use of rather photolabile [Ru(bpy)3]Cl2 might additionally limit the photocatalytic system to reach higher cofactor conversion. Therefore the [Ru]-containing micelles were analyzed as alternative photosensitizers. In fact, the irradiation time-dependent luminescence loss for [Ru]@BCP4 within the solution used for all photocatalytic experiments (0.12 M TEA and 0.1 M NaH2PO4 in degassed H2O) was only 19% within 120 min at r.t. compared to 47% for 5 μM [Ru(bpy)3]Cl2 (Fig. S43†). However, when 5 μM Ru polypyridine moieties via[Ru]@BCP4 were combined with either 5 μM 1 or 5 μM [Rh]@[Rh]@BCP4, only TONs of 0.8 and 0.4 were obtained at r.t., respectively (Fig. S44 and Table S3,† entries 22 and 23). This suggested that despite the improved photostability of the [Ru] moieties when being embedded into the micellar environment, only very limited photocatalytic activity was observed. While indeed photocatalytic activity was observed for the system with [Rh] micelles and molecular [Ru], in the opposite case a fundamental difference in mass and/or energy/electron transfer seems to hinder any activity. It can be discussed whether molecular or micelle-embedded [Rh] centers are prevented from entering the corona of [Ru] micelles, or whether altered electronic properties of [Ru] upon embedding changed reactivity. Considering the additional observation that in absence of any [Rh] moiety the [Ru]@BCP4 micelles also produced ca. 0.4 molecules of NADH per [Ru] center (Fig. S45†), close to no independent activity of the [Rh] micelles in combination with [Ru]@BCP4 was observed. Inter-micellar corona penetration is unlikely to happen, due to electrostatic repulsive forces between individual micelles. Thus, a close interaction of [Ru] and [Rh] centers inside the micellar corona is prevented.
To overcome this low efficiency of inter-micellar activity, the bifunctional micelle [Rh] + [Ru]@BCP5 was prepared. However, also in this case TONs of only 0.5 were observed (Fig. S46 and Table S3,† entry 24) despite a very similar photostability (20% luminescence loss within 120 min) as for [Ru]@BCP4. Analysis of the luminescence quenching within the [RuRh] micelles revealed that in comparison to [Ru]@BCP4 almost no luminescence loss upon introduction of [Rh] moieties was obtained (Fig. S47†). Thus, in addition to substrate diffusion towards the metal centers, the low intermetallic charge transfer efficiency within the [RuRh] micelles might be another important factor limiting photocatalytic activity. It seems that proximity between [Ru] and [Rh] sites is insufficient to enable electron transfer. Since the photocatalytic experiments were conducted in basic pH regime induced by TEA as essential electron donor for the photocatalytic process, a rather stretched conformation of individual chains can be expected, increasing distance between the functional units.
Conclusions
In the herein presented work, block copolymers featuring pendant bipyridyl rhodium complexes and ruthenium polypyridyl complexes were successfully prepared. For this, well-defined polystyrene-block-poly((acrylic acid)-co-(2-(4-(4′-methyl-2,2′-bipyridyl))ethylacrylate)) (PS-b-P(AA-co-bpyEA)) block copolymers were prepared via nitroxide-mediated polymerization (NMP). In post-polymerization functionalization approaches utilizing the pendant bpy moieties rhodium centers as potential NAD+ reduction catalysts and ruthenium centers as potential photosensitizers were covalently incorporated, leading to block copolymers with [Rh] or [Ru] only, or even containing both metals. A successful metal attachment and integrity of embedded complexes was investigated by NMR, SEC and XPS analysis. The obtained block copolymers self-assembled in water generating spherical micelles, bearing the metal sites inside the PAA segment forming the corona. Micellar size and morphology were characterized using a combination of dynamic light scattering (DLS), transmission electron microscopy (TEM), and in case of [Ru] only containing micelles scanning transmission electron microscopy in combination with energy dispersive X-ray spectroscopy (STEM-EDX). Differences in micellar size could be explained by the nature and amount of incorporated metal complexes inside the corona as well as the preparation strategy.The analysis of the (photo)catalytic activity of the [Ru], [Rh] and [RuRh] modified micelles showed, that under formate-driven NAD+ reduction conditions model complex 2 outperformed the polymeric architectures by at least one order of magnitude at all investigated temperatures. However, under light-dependent conditions using TEA as electron donor, the difference in catalytic activity became less pronounced, likely due to a slow photochemical [Rh] catalyst reduction also in the case of complex 2. When the [Rh] containing micelles were activated by [Ru(bpy)3]Cl2 TONs for the cofactor reduction of up to 4.5 per [Rh] moiety could be observed. Although the chemical integrity of the [Ru] centers in an intermicellar photocatalysis approach or by the use of a bifunctional [RuRh] micelle was improved, the amount of generated NADH was lower compared to the experiments when [Ru(bpy)3]Cl2 served as molecular, freely diffusing photosensitizer. This likely indicates that, substrate diffusion and charge transfer in these promising materials is an important factor determining overall catalytic activity which will be analyzed in future studies.
Author contributions
J. E. performed the synthesis and characterization (NMR, SEC, UV/vis absorption spectroscopy) of block copolymers, the preparation of block copolymer micelles and analysis (DLS, TEM). A. M. performed all catalytic investigations. C. N. performed XPS measurements and analysis, and J. B. performed STEM-EDX analysis. J. E., A. M., C. N., J. B., S. R. and F. H. S. wrote the manuscript. All authors read and commented on the manuscript.Data availability
All of the experimental data have been included in the ESI.†All raw data is listed according to figures (main text and ESI†) and is available at Zenodo as public repository under the following DOI: https://doi.org/10.5281/zenodo.12409998.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the Deutsche Forschungsgemeinschaft (Project A01, B03, B05, B07 and Z02 within the Sonderforschungsbereich SFB/TRR 234 “Catalight”, project ID: 364549901). The TEM/cryo-TEM facilities of the Jena Center for Soft Matter (JCSM) were established with a grant from the DFG (German Research Foundation) and the EFRE (European Fonds for Regional Development). C.N. and A. T. acknowledge the financial support of the DFG through research infrastructure grant INST 275/357-1 FUGG-Projektnummer 313713174. We thank Alexander Kleine, Michael Jäger and Grit Festag for analytical and technical support regarding SEC analysis. We are grateful for the support from NMR department at FSU Jena.References
-
P. W. N. M. van Leeuwen, Homogeneous Catalysis, Springer Netherlands, Dordrecht, 2004 Search PubMed
.
- Y. G. Shelke, A. Yashmeen, A. V. A. Gholap, S. J. Gharpure and A. R. Kapdi, Chem. – Asian J., 2018, 13, 2991 CrossRef PubMed
- G. W. Parshall, J. Mol. Catal., 1978, 4, 243 CrossRef
-
S. Bhaduri and D. Mukesh, Homogeneous Catalysis. Mechanisms and Industrial Applications, Wiley, Hoboken, 2014 Search PubMed
-
G. Ertl, H. Knözinger and J. Weitkamp, Handbook of Heterogeneous Catalysis, Wiley, 1997 Search PubMed
- C. H. Bartholomew and J. B. Butt, Catalyst deactivation, 1991. Proceedings of the 5th international symposium, Evanston IL, June 24–26, 1991, Elsevier, Amsterdam, New York, 1991.
- M. Argyle and C. Bartholomew, Catalysts, 2015, 5, 145 CrossRef
- R. Lambert, A.-L. Wirotius, J. Vignolle and D. Taton, Polym. Chem., 2019, 10, 460 RSC
- J. Eichhorn, Y. D. Gordievskaya, E. Y. Kramarenko, A. R. Khokhlov and F. H. Schacher, Macromolecules, 2021, 54, 1976 CrossRef
- J. Eichhorn, P. Hofmann, B. Bagemihl, C. Streb, S. Rau and F. H. Schacher, J. Mater. Chem. A, 2023, 11, 11334 RSC
- U. Kölle and M. Grützel, Angew. Chem., Int. Ed. Engl., 1987, 26, 567 CrossRef
- S. Fukuzumi, T. Kobayashi and T. Suenobu, Angew. Chem., Int. Ed., 2011, 50, 728 CrossRef CAS PubMed
- D. Ghosh, D. C. Fabry, D. Saito and O. Ishitani, Energy Fuels, 2021, 35, 19069 CrossRef CAS
- C. Caix, S. Chardon-Noblat and A. Deronzier, J. Electroanal. Chem., 1997, 434, 163 CrossRef CAS
- F. M. Wisser, M. Duguet, Q. Perrinet, A. C. Ghosh, M. Alves-Favaro, Y. Mohr, C. Lorentz, E. A. Quadrelli, R. Palkovits, D. Farrusseng, C. Mellot-Draznieks, V. de Waele and J. Canivet, Angew. Chem., Int. Ed., 2020, 59, 5116 CrossRef CAS PubMed
- C. Leiva, H. Christine Lo and R. H. Fish, J. Organomet. Chem., 2010, 695, 145 CrossRef CAS
- T. Ghosh, T. Slanina and B. König, Chem. Sci., 2015, 6, 2027 RSC
- K. T. Oppelt, J. Gasiorowski, D. A. M. Egbe, J. P. Kollender, M. Himmelsbach, A. W. Hassel, N. S. Sariciftci and G. Knör, J. Am. Chem. Soc., 2014, 136, 12721 CrossRef CAS PubMed
- E. Steckhan, S. Herrmann, R. Ruppert, E. Dietz, M. Frede and E. Spika, Organometallics, 1991, 10, 1568 CrossRef CAS
- H. C. Lo, C. Leiva, O. Buriez, J. B. Kerr, M. M. Olmstead and R. H. Fish, Inorg. Chem., 2001, 40, 6705 CrossRef CAS
- L. M. A. Quintana, S. I. Johnson, S. L. Corona, W. Villatoro, W. A. Goddard, M. K. Takase, D. G. VanderVelde, J. R. Winkler, H. B. Gray and J. D. Blakemore, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6409 CrossRef PubMed
- A. Marrone and R. H. Fish, J. Organomet. Chem., 2021, 943, 121810 CrossRef CAS
- T. K. Todorova, T. N. Huan, X. Wang, H. Agarwala and M. Fontecave, Inorg. Chem., 2019, 58, 6893 CrossRef CAS PubMed
- C. L. Pitman, O. N. L. Finster and A. J. M. Miller, Chem. Commun., 2016, 52, 9105 RSC
- S. H. Lee, D. H. Nam and C. B. Park, Adv. Synth. Catal., 2009, 351, 2589 CrossRef CAS
- S. Choudhury, J.-O. Baeg, N.-J. Park and R. K. Yadav, Angew. Chem., Int. Ed., 2012, 51, 11624 CrossRef CAS
- R. K. Yadav, J.-O. Baeg, A. Kumar, K. Kong, G. H. Oh and N.-J. Park, J. Mater. Chem. A, 2014, 2, 5068 RSC
- R. K. Yadav, J.-O. Baeg, G. H. Oh, N.-J. Park, K. Kong, J. Kim, D. W. Hwang and S. K. Biswas, J. Am. Chem. Soc., 2012, 134, 11455 CrossRef CAS
- Z. Tang, E. Chulanova, M. Küllmer, A. Winter, J. Picker, C. Neumann, K. Schreyer, F. Herrmann-Westendorf, A. Arnlind, B. Dietzek, U. S. Schubert and A. Turchanin, Nanoscale, 2021, 13, 20583 RSC
- Y.-J. Yuan, Z.-T. Yu, D.-Q. Chen and Z.-G. Zou, Chem. Soc. Rev., 2017, 46, 603 RSC
- S. Losse, J. G. Vos and S. Rau, Coord. Chem. Rev., 2010, 254, 2492 CrossRef
- S. Fukuzumi, Y.-M. Lee and W. Nam, Coord. Chem. Rev., 2018, 355, 54 CrossRef
- T. Stoll, C. E. Castillo, M. Kayanuma, M. Sandroni, C. Daniel, F. Odobel, J. Fortage and M.-N. Collomb, Coord. Chem. Rev., 2015, 304–305, 20 CrossRef
- V. Balzani, P. Ceroni, A. Credi and M. Venturi, Coord. Chem. Rev., 2021, 433, 213758 CrossRef
-
S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, in Topics in Current Chemistry, ed. V. Balzani and S. Campagna, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 117–214 Search PubMed
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS
- J.-F. Yin, M. Velayudham, D. Bhattacharya, H.-C. Lin and K.-L. Lu, Coord. Chem. Rev., 2012, 256, 3008 CrossRef CAS
- A. Chettri, J.-H. Kruse, K. Kumar Jha, L. Dröge, I. Romanenko, C. Neumann, S. Kupfer, A. Turchanin, S. Rau, F. H. Schacher and B. Dietzek, Chemistry, 2021, 27, 17049–17058 CrossRef CAS
- B. Happ, J. Kübel, M. G. Pfeffer, A. Winter, M. D. Hager, B. Dietzek, S. Rau and U. S. Schubert, Macromol. Rapid Commun., 2015, 36, 671 CrossRef CAS PubMed
- H. Shimakoshi, M. Nishi, A. Tanaka, K. Chikama and Y. Hisaeda, Chem. Commun., 2011, 47, 6548 RSC
- M. Suzuki, O. Bartels, R. Gerdes, G. Schneider, D. Wöhrle, G. Schulz-Ekloff, M. Kimura, K. Hanabusa and H. Shirai, Phys. Chem. Chem. Phys., 2000, 2, 109 RSC
- V. Marin, E. Holder, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 618 RSC
- T. Mede, M. Jäger and U. S. Schubert, Chem. Soc. Rev., 2018, 47, 7577 RSC
- N. Zabarska, A. Stumper and S. Rau, Dalton Trans., 2016, 45, 2338 RSC
- U. S. Schubert and C. Eschbaumer, Angew. Chem., Int. Ed., 2002, 41, 2892 CrossRef CAS
- P. Giannopoulos, A. K. Andreopoulou, C. Anastasopoulos, D. Raptis, G. Sfyri, J. K. Kallitsis and P. Lianos, RSC Adv., 2016, 6, 8256 RSC
- M. Heller and U. S. Schubert, Macromol. Rapid Commun., 2002, 23, 411 CrossRef CAS
- E. Holder, M. A. R. Meier, V. Marin and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3954 CrossRef CAS
- B. S. Kim, C. Basavaraja, E. A. Jo, D. G. Kim and D. S. Huh, Polymer, 2010, 51, 3365 CrossRef CAS
- E. K. Pefkianakis, N. P. Tzanetos and J. K. Kallitsis, Chem. Mater., 2008, 20, 6254 CrossRef CAS
- F. Pinaud, R. Millereux, P. Vialar-Trarieux, B. Catargi, S. Pinet, I. Gosse, N. Sojic and V. Ravaine, J. Phys. Chem. B, 2015, 119, 12954 CrossRef CAS PubMed
- B. Happ, C. Friebe, A. Winter, M. D. Hager and U. S. Schubert, Eur. Polym. J., 2009, 45, 3433 CrossRef CAS
- B. Happ, J. Schäfer, R. Menzel, M. D. Hager, A. Winter, J. Popp, R. Beckert, B. Dietzek and U. S. Schubert, Macromolecules, 2011, 44, 6277 CrossRef CAS
- J.-K. Lee, D. Yoo and M. F. Rubner, Chem. Mater., 1997, 9, 1710 CrossRef CAS
- Y. Lu, Photochem. Photobiol. Sci., 2010, 9, 392 CrossRef CAS
- X. Schultze, J. Serin, A. Adronov and J. M. J. Fréchet, Chem. Commun., 2001, 1160 RSC
- Z. Fang, A. Ito, S. Keinan, Z. Chen, Z. Watson, J. Rochette, Y. Kanai, D. Taylor, K. S. Schanze and T. J. Meyer, Inorg. Chem., 2013, 52, 8511 CrossRef CAS PubMed
- Z. Fang, A. Ito, H. Luo, D. L. Ashford, J. J. Concepcion, L. Alibabaei and T. J. Meyer, Dalton Trans., 2015, 44, 8640 RSC
- J.-S. Moon, C. Kim, W.-G. Kim, I. Kim, K. Kyhm, J.-W. Oh and N. Kim, J. Photochem. Photobiol., A, 2015, 310, 141 CrossRef CAS
- K. Peter and M. Thelakkat, Macromolecules, 2003, 36, 1779 CrossRef CAS
- T. Schlotthauer, R. Schroot, S. Glover, L. Hammarström, M. Jäger and U. S. Schubert, Phys. Chem. Chem. Phys., 2017, 19, 28572 RSC
- R. Schroot, T. Schlotthauer, B. Dietzek, M. Jäger and U. S. Schubert, Chemistry, 2017, 23, 16484 CrossRef
- J. R. Carlise and M. Weck, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2973 CrossRef
- V. Marin, E. Holder and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 374 CrossRef
- J. Serin, X. Schultze, A. Adronov and J. M. J. Fréchet, Macromolecules, 2002, 35, 5396 CrossRef
- K. Yoshinaga, I. Toyofuku, K. Yamashita, H. Kanehara and K. Ohkubo, Colloid Polym. Sci., 2000, 278, 481 CrossRef
- G. Leem, S. Keinan, J. Jiang, Z. Chen, T. Pho, Z. A. Morseth, Z. Hu, E. Puodziukynaite, Z. Fang, J. M. Papanikolas, J. R. Reynolds and K. S. Schanze, Polym. Chem., 2015, 6, 8184 RSC
- G. Leem, Z. A. Morseth, E. Puodziukynaite, J. Jiang, Z. Fang, A. T. Gilligan, J. R. Reynolds, J. M. Papanikolas and K. S. Schanze, J. Phys. Chem. C, 2014, 118, 28535 CrossRef
- G. Leem, Z. A. Morseth, K.-R. Wee, J. Jiang, M. K. Brennaman, J. M. Papanikolas and K. S. Schanze, Chem. – Asian J., 2016, 11, 1257 CrossRef PubMed
- Y. Sun, Z. Chen, E. Puodziukynaite, D. M. Jenkins, J. R. Reynolds and K. S. Schanze, Macromolecules, 2012, 45, 2632 CrossRef
- D. L. Ashford, M. K. Brennaman, R. J. Brown, S. Keinan, J. J. Concepcion, J. M. Papanikolas, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2015, 54, 460 CrossRef PubMed
- A. Ito, N. Kobayashi and Y. Teki, Inorg. Chem., 2017, 56, 3794 CrossRef CAS PubMed
- K. Peuntinger, T. D. Pilz, R. Staehle, M. Schaub, S. Kaufhold, L. Petermann, M. Wunderlin, H. Görls, F. W. Heinemann, J. Li, T. Drewello, J. G. Vos, D. M. Guldi and S. Rau, Dalton Trans., 2014, 43, 13683 RSC
- R. P. Thummel and F. Lefoulon, Inorg. Chem., 1987, 26, 675 CrossRef CAS
- M. N. Jackson, S. Oh, C. J. Kaminsky, S. B. Chu, G. Zhang, J. T. Miller and Y. Surendranath, J. Am. Chem. Soc., 2018, 140, 1004 CrossRef CAS PubMed
- H. Shalan, A. Colbert, T. T. Nguyen, M. Kato and L. Cheruzel, Inorg. Chem., 2017, 56, 6558 CrossRef CAS
- N. H. Damrauer, T. R. Boussie, M. Devenney and J. K. McCusker, J. Am. Chem. Soc., 1997, 119, 8253 CrossRef CAS
- M. Kakoti, A. K. Deb and S. Goswami, Inorg. Chem., 1992, 31, 1302 CrossRef CAS
- N. Armaroli, G. Accorsi, D. Felder and J.-F. Nierengarten, Chem. – Eur. J., 2002, 8, 2314 CrossRef CAS PubMed
- J. G. Gaudiello, P. R. Sharp and A. J. Bard, J. Am. Chem. Soc., 1982, 104, 6373 CrossRef CAS
- E. Holder, G. Schoetz, V. Schurig and E. Lindner, Tetrahedron: Asymmetry, 2001, 12, 2289 CrossRef CAS
- E. Holder, O. Trapp, G. Trapp, V. Marin, R. Hoogenboom and U. S. Schubert, Chirality, 2004, 16, 363 CrossRef CAS PubMed
- V. Marin, E. Holder, R. Hoogenboom, E. Tekin and U. S. Schubert, Dalton Trans., 2006, 1636 RSC
- S. Kaufhold, L. Petermann, D. Sorsche and S. Rau, Chemistry, 2017, 23, 2271 CrossRef CAS PubMed
- B. J. Brennan, A. E. Keirstead, P. A. Liddell, S. A. Vail, T. A. Moore, A. L. Moore and D. Gust, Nanotechnology, 2009, 20, 505203 CrossRef
- M. Hirahara and Y. Umemura, Inorg. Chem., 2021, 60, 13193 CrossRef CAS PubMed
- L. Zedler, P. Wintergerst, A. K. Mengele, C. Müller, C. Li, B. Dietzek-Ivanšić and S. Rau, Nat. Commun., 2022, 13, 2538 CrossRef CAS PubMed
- R. Wienkamp and E. Steckhan, Angew. Chem., Int. Ed. Engl., 1983, 22, 497 CrossRef
- E. J. Land and A. J. Swallow, Biochim. Biophys. Acta, Bioenerg., 1971, 234, 34 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00693c |
| This journal is © The Royal Society of Chemistry 2024 |