
Radical promoted cationic RAFT polymerization by photo electron transfer reaction†
Shiliang
He
,
Xinrui
Yang
,
Yabing
Zhao
,
Yang
Liu
,
Bowen
Zhao
,
Xiangqiang
Pan
 ,
Jiajia
Li
*,
Jian
Zhu
* and
Na
Li
*
,
Jiajia
Li
*,
Jian
Zhu
* and
Na
Li
*
State and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, Jiangsu Key Laboratory of Advanced Functional Polymer Design and Application, Department of Polymer Science and Engineering, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: chemzhujian@suda.edu.cn; chemjjli@suda.edu.cn; chemlina@suda.edu.cn
First published on 18th September 2024
Abstract
The application of cationic RAFT polymerization in photocuring has enabled the fabrication of stimuli-responsive materials. However, these systems mainly rely on UV light, limiting their broader application. Herein, we developed a radical promoted cationic RAFT (RPC-RAFT) polymerization system utilizing a photoredox catalyst via a photo electron transfer reaction, extending the usable light wavelength to blue and red light. The well-defined polymerization of various vinyl ethers was achieved with controlled molecular weights and narrow molecular weight distributions. Moreover, we achieved successful photocuring using this system for the polymerization of a bifunctional vinyl ether monomer, offering a convenient approach for in-depth photocuring.
Introduction
Photoinduced cationic polymerization has been widely used for photocuring because of its advantages of inertness to oxygen, low energy requirement, low stress shrinkage and the ability to polymerize in the dark.1–5 However, conventional systems usually involve nonliving polymerization and lack control over the molecular weight and molecular weight distribution of the obtained polymers. This limitation hinders the production of tailor-made polymeric materials. Recently, photocontrolled cationic polymerization and photoinduced controlled cationic polymerization have attracted increasing attention.6–9 For instance, Nicewicz and You demonstrated the controlled cationic polymerization of para-methoxystyrene (p-MOS) by using a combination of pyrylium salts and methanol.10 In 2017, Yagci et al. reported a visible light-initiated controlled cationic polymerization system employing Mn2(CO)10 as a photoinitiator and benzyl bromide as an initiator.11 Zhou also developed a photocontrolled living cationic polymerization of p-MOS utilizing a triarylmethyl cation and a phosphate.12In 2015, cationic RAFT polymerization emerged as a powerful and versatile method for living cationic polymerization.13,14 The remaining thiocarbonyl thio chain ends of the polymers synthesized via this approach can be utilized not only for cationic chain extension but also for radical chain extension. This versatility allows for the combination of different polymerization mechanisms to prepare various block copolymers.15 Moreover, the photoinduced version of this technique has demonstrated excellent control over polymerization, along with superior temporal and spatial control. To date, various initiating systems have been developed. For example, Fors's group initially employed pyrylium salts as photocatalysts and later extended their work to iridium-based catalysts to achieve enhanced temporal control.16,17 Liao's group developed a series of organocatalytic cationic RAFT polymerization with strict temporal control utilizing various phosphonium salts.18–21 Additionally, Kamigaito's group reported the use of a series of acridinium salts as photoredox organocatalysts for photomediated cationic RAFT polymerization.22
Our group has also developed several photoinduced cationic RAFT polymerization by using metal carbonyl compounds as photoinitiators.23–25 Recently, we integrated free radical promoted cationic polymerization (FRPCP) with cationic RAFT polymerization to create a photoinduced 3D printing system capable of conveniently fabricating ‘living’ objects.26–29 FRPCP is a well-established method for achieving cationic polymerization under mild irradiation conditions, employing photosensitive dyes and onium salts.30 Upon excitation, these dyes activate onium salts by energy transfer or electron transfer, subsequently inducing cationic polymerization. Numerous photoinitiation systems based on organic and organometallic compounds have been reported, demonstrating excellent efficiency in photoinitiated cationic polymerization. The incorporation of a cationic RAFT agent into FRPCP provides excellent control over polymerization. Moreover, the diverse photoinitiating systems available for FRPCP offer various options for photoinduced cationic RAFT polymerization.
However, the range of photoinitiators available for this method remains limited (Fig. 1). Here, we demonstrate a RPC-RAFT system using a combination of photoredox catalysts and diphenyliodonium hexafluorophosphate (DPI) as initiating systems, offering alternative methods for RPC-RAFT polymerization under different light wavelengths and for photocuring applications.
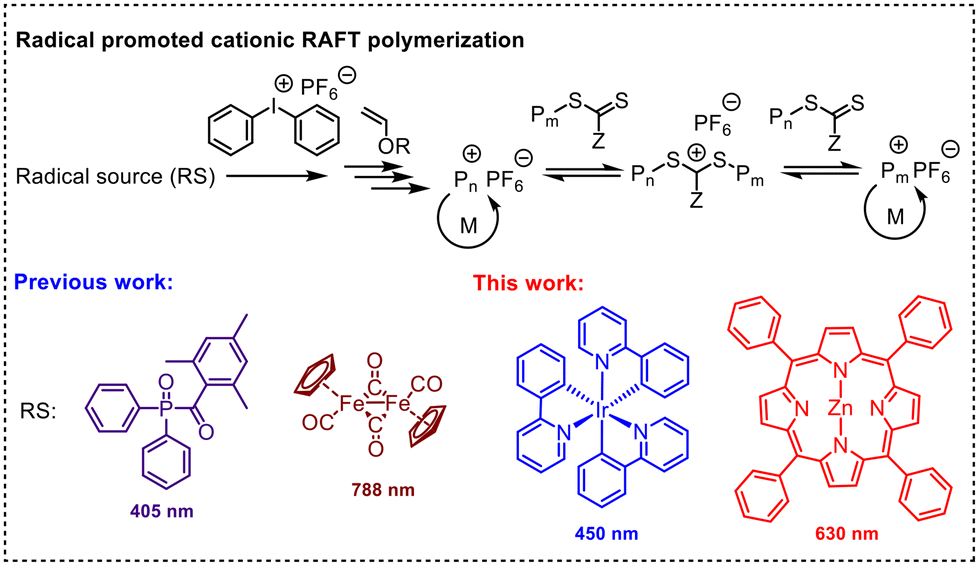 | ||
| Fig. 1 Radical sources used for radical promoted cationic RAFT polymerization. | ||
Results and discussion
Tris(2-phenylpyridine) iridium (Ir(ppy)3) was initially chosen as the photosensitizer due to its extensive use in reversible deactivation radical polymerization (RDRP).31,32S-1-Isobutoxyethyl N,N-diethyl dithiocarbamate (DTCB) was employed as the cationic RAFT agent because of its excellent controllability in cationic RAFT polymerization.14 We first screened the polymerization of isobutyl vinyl ether (IBVE) in various solvents under the conditions of [IBVE]0/[DTCB]0/[DPI]0/[Ir(ppy)3]0 = 100/1/0.1/0.0001. As shown in Table S1,† controlled molecular weight and narrow molecular weight distribution (MWD) of the obtained PIBVE were achieved in all the tested solvents, including dichloromethane (DCM), toluene, hexane and ethyl acetate (EA), although the polymerization rates varied. The polymerization in DCM proceeded faster than that in bulk or other solvents, likely due to the better solubility of DPI in polar solvents. Notably, significant heat release was observed during polymerization in DCM and the bulk (Table S1,† entries 1 and 2) due to the rapid polymerization rate. Therefore, toluene was selected as the solvent for further studies because of its moderate polymerization rate.As summarized in Table 1, we investigated the impact of each component on the polymerization process. The polymerization rate decreased with decreasing amounts of DPI, and no polymerization occurred in the absence of DPI, indicating that DPI is essential for generating initial initiating cations (Table 1, entries 1–4). Moreover, increasing the amount of Ir(ppy)3 accelerated the polymerization, likely by enhancing the generation of initiating cations (Table 1, entries 5 and 6). Remarkably, polymerization occurred even in the absence of Ir(ppy)3 (Table 1, entry 7), suggesting direct photolysis of DPI (Fig. S3†),5 since no polymerization was observed in the dark (Table 1, entry 8). Furthermore, the impact of the RAFT agent on the polymerization was examined (Table 1, entries 9–11). The molecular weight could be finely tuned by adjusting the molar ratio between IBVE and DTCB. The experimental molecular weight becomes much lower than theoretical value when targeting higher DP (Table 1, entry 11), which can be attributed to increased chain transfer reaction.33 Polymerization in the absence of DTCB resulted in PIBVE with a much greater molecular weight and broad MWD (Table 1, entry 12), demonstrating the essential role of the RAFT process in achieving controlled polymerization. The proposed polymerization mechanism is illustrated in Scheme 1. Upon visible light irradiation, the photocatalyst Ir(ppy)3 reaches its excited state and is then oxidized by DPI through a photoinduced electron/energy transfer (PET) reaction,34,35 reducing DPI and generating an aryl radical. This aryl radical adds to the monomer or RAFT agent to form a vinyl ether-type radical,3,36 which further reacts with DPI or Ir(IV) to generate the initiating cation for cationic RAFT polymerization.
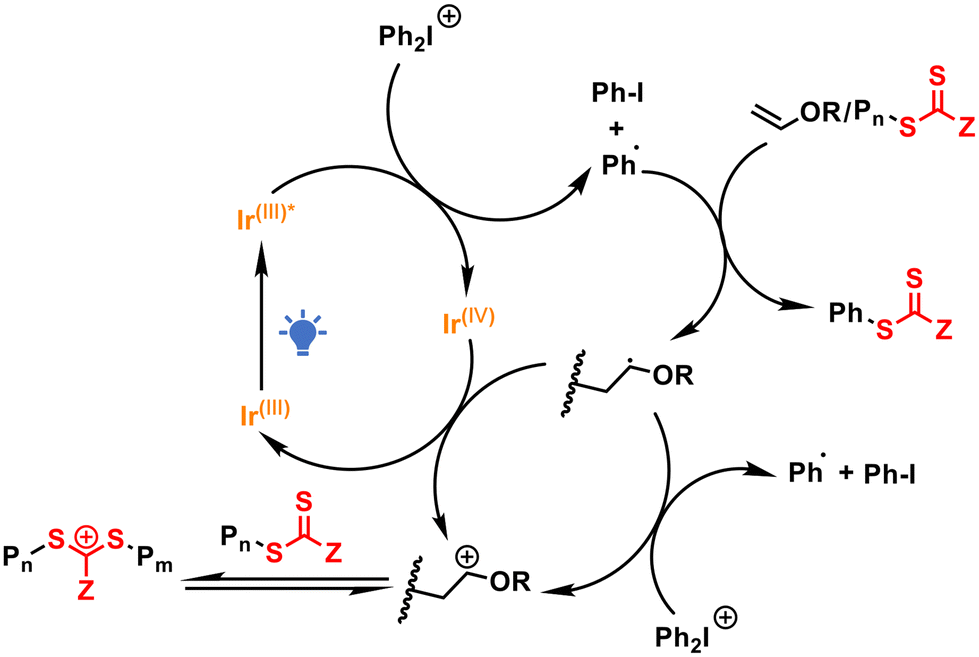 | ||
| Scheme 1 Proposed mechanism for RPC-RAFT polymerization by photo electron transfer reaction. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [IBVE]0/[DTCB]0/[DPI]0/[Ir(ppy)3]0 | Time | Conv. (%) |
M
n,th![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (g mol−1) a (g mol−1) |
M
n,SECb (g mol−1) |
Đ |
| a Calculated based on conversion (Mn,th = [M]0/[CTA]0 × MIBVE × conversion + MDTCB). b Determined by tetrahydrofuran (THF) SEC using polystyrene (PS) calibration. c Polymerization in the dark. | ||||||
| 1 | 100/1/0.2/0.0001 | 40 min | 96.3 | 9900 | 9900 | 1.15 |
| 2 | 100/1/0.1/0.0001 | 50 min | 98.7 | 10100 |
10400 |
1.12 |
| 3 | 100/1/0.01/0.0001 | 120 min | 95.4 | 9800 | 9600 | 1.13 |
| 4 | 100/1/0/0.0001 | 24 h | — | — | — | — |
| 5 | 100/1/0.1/0.01 | 5 min | 96.3 | 9900 | 9900 | 1.15 |
| 6 | 100/1/0.1/0.001 | 15 min | 95.4 | 9800 | 9600 | 1.13 |
| 7 | 100/1/0.1/0 | 120 min | 98.6 | 10100 |
11100 |
1.12 |
| 8c | 100/1/0.1/0.0001 | 24 h | — | — | — | — |
| 9 | 50/1/0.1/0.0001 | 50 min | 98.5 | 5200 | 5700 | 1.12 |
| 10 | 200/1/0.1/0.0001 | 50 min | 98.5 | 19900 |
18000 |
1.14 |
| 11 | 500/1/0.1/0.0001 | 230 min | 98.8 | 49700 |
31200 |
1.31 |
| 12 | 100/0/0.1/0.0001 | 80 min | 98.9 | 9900 | 48300 |
1.63 |
The polymerization kinetics were investigated under optimized conditions ([IBVE]0/[DTCB]0/[DPI]0/[Ir(ppy)3]0 = 100/1/0.01/0.0001) using a 450 nm LED at −30 °C in toluene and DCM (v/v = 9:1), which eliminates the solubility problem of DPI. As depicted in Fig. 2A, an induction period was observed in the semilogarithmic plot of monomer concentration versus polymerization time, likely due to the low concentration of Ir(ppy)3, which may result in insufficient decomposition of DPI. The polymerization rate can be improved by increasing the concentration of Ir(ppy)3 (Fig. S4†). Moreover, the polymerization rate dramatically decreased in the absence of Ir(ppy)3 (Fig. S5†), suggesting the important role of the photocatalyst. Fig. 2B shows that controlled molecular weights and narrow MWDs were achieved during polymerization, with the molecular weights aligning well with the theoretical values. In addition, the SEC traces shifted smoothly to higher molecular weight regions with increasing monomer conversion (Fig. 2C). Furthermore, an in situ chain extension experiment was performed to evaluate the living nature of the polymerization. As shown in Fig. 2D, the clear shift in the SEC trace after the addition of a second monomer demonstrates successful chain extension, consistent with the characteristics of a ‘living’ polymerization.
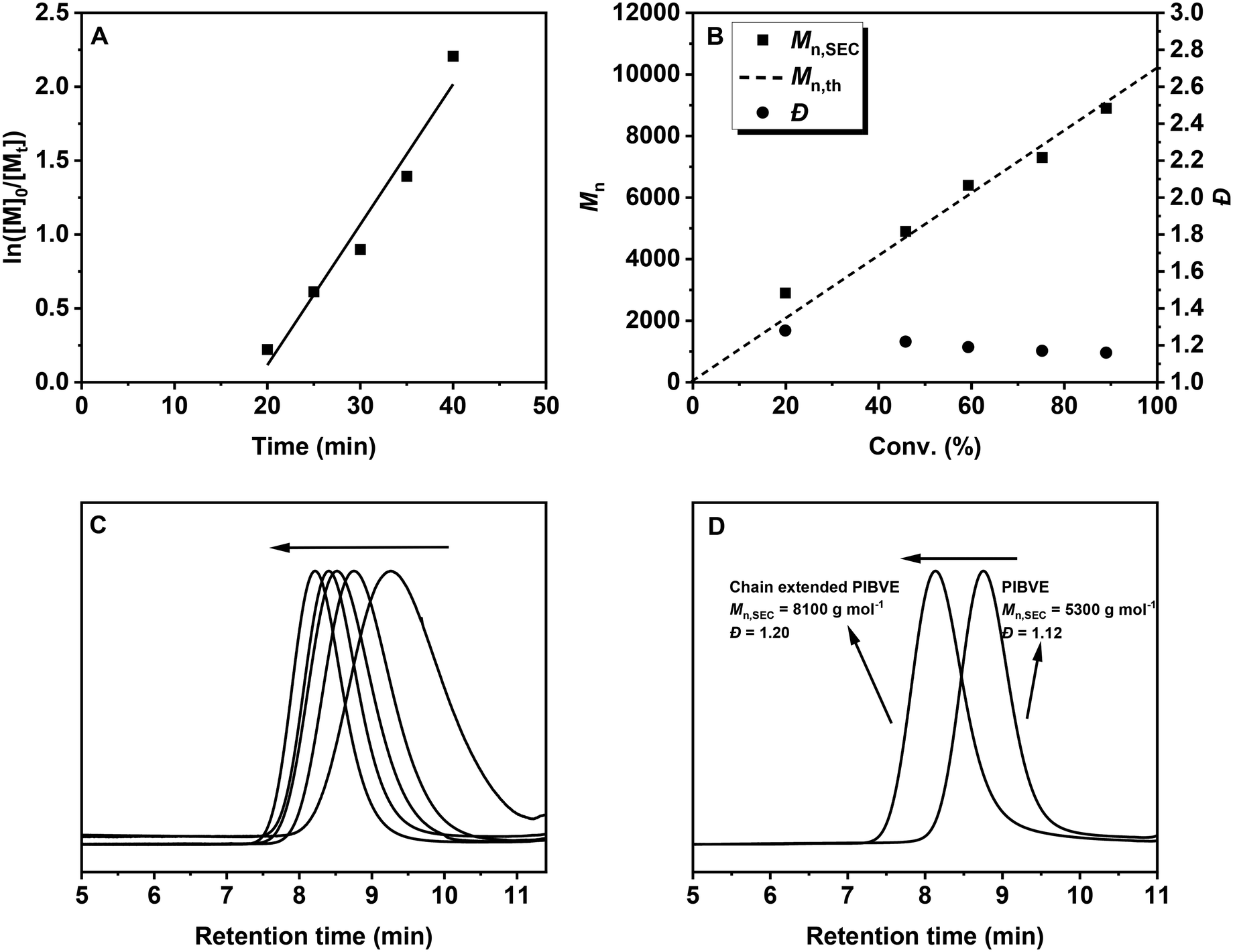 | ||
| Fig. 2 Polymerization of IBVE with [IBVE]0/[DTCB]0/[DPI]0/[Ir(ppy)3]0 = 100/1/0.01/0.0001 under a 450 nm blue LED at −30 °C in toluene and DCM (v/v = 9:1), VIBVE = 1 mL, Vsolvent = 1 mL. (A) ln([M]0/[M]t) versus time. (B) Molecular weight (Mn) and molecular weight distribution (Đ) versus monomer conversion; (C) SEC traces of the obtained PIBVE; (D) SEC traces of the PIBVE before and after chain extension. | ||
Then, proton nuclear magnetic resonance (1H NMR) and matrix-assisted laser desorption time-of-flight mass spectrometry (MALDI-TOF MS) were used to analyze the chain-end structure of the obtained PIBVE. The characteristic peak at 6 ppm in the 1H NMR spectrum, corresponding to the proton at the carbon connected to the sulfur in the dithiocarbamate-terminated chain end, confirmed the successful RAFT process (Fig. 3A). Moreover, the signal of aryl group was also found at around 7.2 ppm (peak j), further confirming the radical addition process between the aryl radical and monomer. The main series of peaks in the MALDI-TOF MS spectrum were attributed to PIBVE with a hydrogen-terminated chain end, likely resulting from the decomposition of the terminal RAFT agent during the MALDI-TOF MS process (Fig. 3B). These data confirm the relatively high chain-end fidelity of the PIBVE prepared by this method.
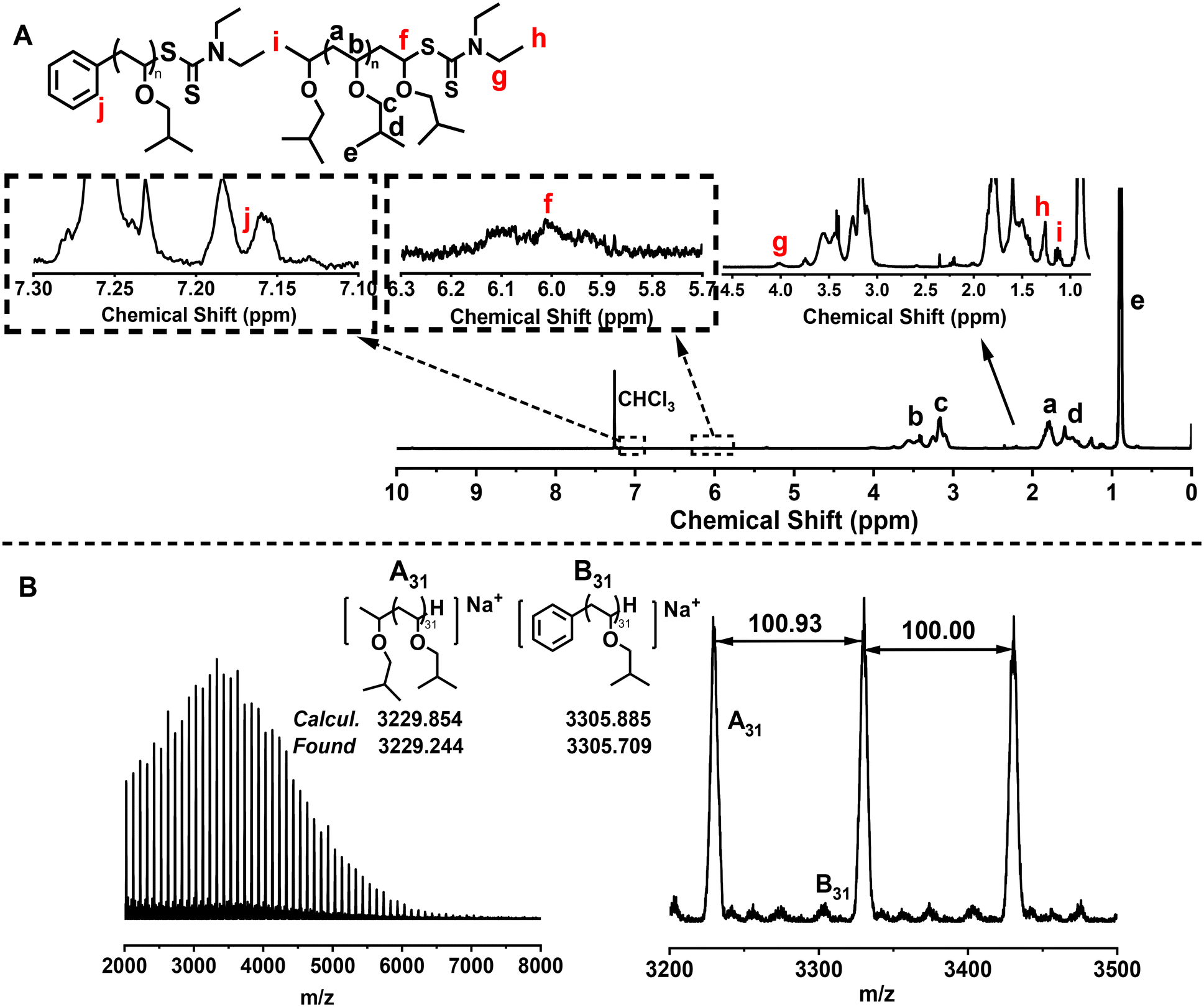 | ||
| Fig. 3 (A) 1H NMR spectrum of PIBVE (Mn = 5700 g mol−1, Đ = 1.07) and (B) MALDI-TOF MS spectrum of PIBVE (Mn = 5700 g mol−1, Đ = 1.07) obtained from the polymerization of IBVE with a molar ratio of [IBVE]0/[DTCB]0/[DPI]0/[Ir(ppy)3]0 = 100/1/0.1/0.0001 at 25 °C, VIBVE = 1 mL, Vtoluene = 1 mL. | ||
The monomer scope was then expanded using the optimized conditions. The results are summarized in Table 2. Controlled molecular weights and narrow MWDs were observed for the polymerization of various vinyl ethers, including butyl vinyl ether (BVE), ethyl vinyl ether (EVE), propoxy ethylene (PVE), 2-chloroethyl vinyl ether (Cl-EVE) and cyclohexyl vinyl ether (CyVE) (Table 2, entries 1–5), indicating the versatility of this method.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Monomer | Time (min) | Conv. (%) |
M
n,tha (g mol−1) |
M
n,SECb (g mol−1) |
Đ |
| a Calculated based on conversion (Mn,th = [M]0/[CTA]0 × Mmonomer × conversion + MDTCB). b Determined by tetrahydrofuran (THF) SEC using polystyrene (PS) calibration. | ||||||
| 1 | BVE | 40 | 96.4 | 9900 | 12600 |
1.13 |
| 2 | EVE | 20 | 94.2 | 7000 | 8100 | 1.20 |
| 3 | PVE | 20 | 98.7 | 8700 | 11300 |
1.10 |
| 4 | Cl-EVE | 70 | 90.9 | 10000 |
9900 | 1.24 |
| 5 | CyVE | 10 | 94.9 | 12200 |
10100 |
1.43 |
Zinc tetraphenylporphyrin (ZnTPP) is a red light-responsive photoredox catalyst, which has been widely used for PET-RAFT polymerization as an alternative substitute of Ir(ppy)3.37 Here we also examined the utilization of ZnTPP for RPC-RAFT polymerization to expand the light wavelength. As shown in Table 3, well-defined PIBVEs with controlled molecular weights and narrow MWDs were obtained under irradiation of red LED (λmax = 630 nm, 0.25 mW cm−2). Furthermore, no polymerization occurred after 24 h in the absence of ZnTPP (Table 3, entry 4), suggesting the photocatalyst plays an important role for initiating the polymerization.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X | Time | Conv. (%) |
M
n,tha (g mol−1) |
M
n,SECb (g mol−1) |
Đ |
| a Calculated based on conversion (Mn,th = [M]0/[CTA]0 × MIBVE × conversion + MDTCB). b Determined by tetrahydrofuran (THF) SEC using polystyrene (PS) calibration. | ||||||
| 1 | 50 | 120 min | 98.6 | 5200 | 5500 | 1.11 |
| 2 | 200 | 120 min | 97.3 | 19700 |
15900 |
1.15 |
| 3 | 500 | 300 min | 98.4 | 49400 |
29100 |
1.23 |
| 4 | 100 | 24 h | — | — | — | — |
Finally, we applied this polymerization method for photocuring. As depicted in Fig. 4, a photomask with the transparent slit word ‘RAFT’ was placed on a glass container filled with printing resins consisting of DDE, DTCB, DPI, and Ir(ppy)3 at a molar ratio of [DDE]0/[DTCB]0/[DPI]0/[Ir(ppy)3]0 = 100/1/0.1/0.01. Blue LED light (λmax = 450 nm, 1.5 mW cm−2) was then projected onto the photomask for 1 minute. After removing the photomask and unreacted solution, the clear 3D word ‘RAFT’ was observed (Fig. 4A). Similarly, red LED light (λmax = 630 nm, 1.5 mW cm−2) was used for the photocuring of resins with ZnTPP instead of Ir(ppy)3 (Fig. 4B). A longer curing time was required in this case, which could be due to the lower catalytic efficiency of ZnTPP. Overall, these results demonstrate successful photocuring of the RAFT resins under visible light.
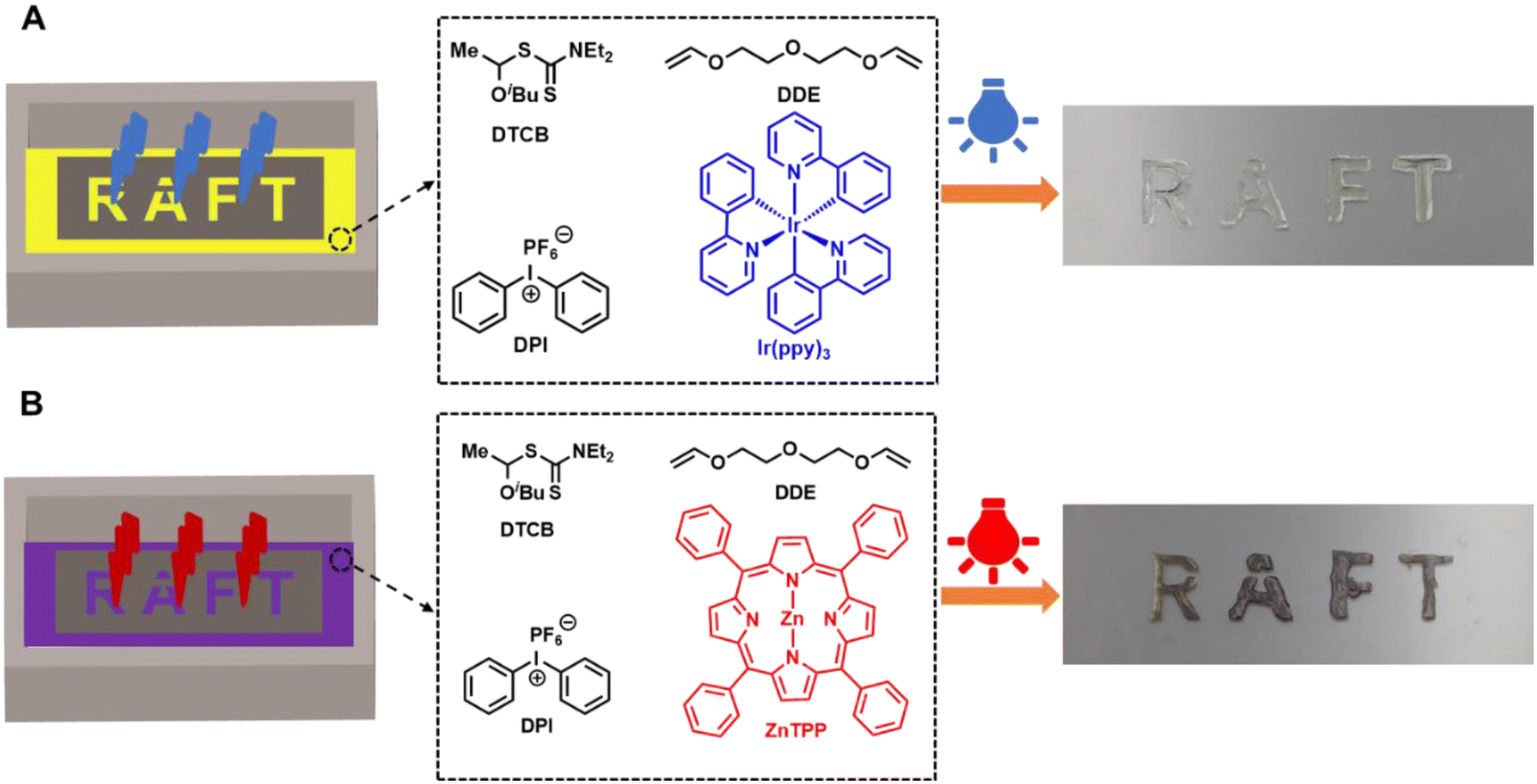 | ||
| Fig. 4 Photocuring using a photomask with an (A) blue LED at a molar ratio of [DDE]0:[DTCB]0:[DPI]0:[Ir(ypp)3]0 = 100:1:0.1:0.01 for 1 minute and (B) red LED [DDE]0/[DTCB]0/[DPI]0/[ZnTPP]0 = 100/1/0.2/0.1 for 5 minutes. | ||
Conclusion
In this study, we demonstrated successful RPC-RAFT polymerization using Ir(ppy)3 or ZnTPP as photoredox catalysts under visible light. The polymerization of various vinyl ethers under optimized conditions achieved controlled molecular weights and narrow MWDs, showcasing the versatility of this method. Our kinetic investigations revealed the importance of DPI as an initiator and the role of Ir(ppy)3 in accelerating the polymerization process. Furthermore, successful chain extension experiments confirmed the “living” nature of the polymerization. We also successfully applied this method to photocuring, creating well-defined 3D structures under both blue and red light. This highlights the potential of this technique for advanced manufacturing applications, such as 3D printing of “living” materials. Overall, our findings provide valuable insights into the development of efficient, visible-light-induced RAFT polymerization systems and their practical applications in polymer chemistry.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 22371199), Suzhou Cutting-edge Technology Research Project (SYG202350), the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions and the Program of Innovative Research Team of Soochow University.References
- K. Satoh, Z. Sun, M. Uchiyama, M. Kamigaito, J. Xu and C. Boyer, Polym. J., 2020, 52, 65–73 CrossRef CAS.
- Y. Zhou, J. Li, Y. Wu and Z. Luo, Chem. Rev., 2020, 5, 2950–3048 CrossRef PubMed.
- P. Xiao, J. Zhang, F. Dumur, A. Tehfe, F. Morlet-Savary, B. Graff, D. Gigmes, P. Fouassier and J. Lalevée, Prog. Polym. Sci., 2015, 41, 32–66 CrossRef CAS.
- P. Garra, C. Dietlin, F. Morlet-Savary, F. Dumur, D. Gigmes, J. P. Fouassier and J. Lalevée, Prog. Polym. Sci., 2019, 94, 33–56 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4241–4254 CrossRef CAS.
- Q. Michaudel, V. Kottisch and B. P. Fors, Angew. Chem., Int. Ed., 2017, 56, 9670–9679 CrossRef CAS PubMed.
- C. Aydogan, G. Yilmaz, A. Shegiwal, D. M. Haddleton and Y. Yagci, Angew. Chem., Int. Ed., 2022, 61, 2411–2425 CrossRef.
- K. Kaya, H. C. Kiliclar and Y. Yagci, Eur. Polym., 2023, 45, 1022–1336 Search PubMed.
- X. Feng, R. Liu, L. Liu, Y. Jin, Q. Shi, P. Yan and Y. Wu, J. Polym. Sci., 2023, 61, 2411–2425 CrossRef CAS.
- A. J. Perkowski, W. You and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 7580–7583 CrossRef CAS PubMed.
- M. Ciftci, Y. Yoshikawa and Y. Yagci, Angew. Chem., Int. Ed., 2017, 56, 519–523 CrossRef CAS PubMed.
- L. Wang, Y. Xu, Q. Zuo, H. Dai, L. Huang, M. Zhang, Y. Zheng, C. Yu, S. Zhang and Y. Zhou, Nat. Commun., 2022, 13, 3621 CrossRef CAS.
- M. Uchiyama, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2015, 54, 1924–1928 CrossRef CAS.
- M. Uchiyama, K. Satoh and M. Kamigaito, Giant, 2021, 5, 2666–5425 CrossRef.
- M. Kamigaito and M. Sawamoto, Macromolecules, 2020, 53, 6749–6753 CrossRef CAS.
- V. Kottisch, Q. Michaudel and B. P. Fors, J. Am. Chem. Soc., 2016, 138, 15535–15538 CrossRef CAS PubMed.
- V. Kottisch, M. J. Supej and B. P. Fors, Angew. Chem., Int. Ed., 2018, 57, 8260–8264 CrossRef CAS PubMed.
- Z. Yang, W. Xiao, X. Zhang and S. Liao, Polym. Chem., 2022, 13, 2776–2781 RSC.
- Z. Yang, J. Chen and S. H. Liao, ACS Macro Lett., 2022, 11, 1073–1078 CrossRef CAS.
- X. Zhang, Y. Jiang, Q. Ma, S. Hu and S. Liao, J. Am. Chem. Soc., 2021, 143, 6357–6362 CrossRef CAS PubMed.
- Z. Yang, X. Zhang, Y. Jiang, Q. Ma and S. Liao, Sci. China: Chem., 2021, 65, 304–308 CrossRef.
- M. Matsuda, M. Uchiyama, Y. Itabashi, K. Ohkubo and M. Kamigaito, Polym. Chem., 2022, 13, 1031–1039 RSC.
- J. Li, A. Kerr, S. Häkkinen, T. Floyd, M. Zhang, X. Pan, X. Zhu, S. Perrier and J. Zhu, Polym. Chem., 2020, 11, 2724–2731 RSC.
- J. Li, A. Kerr, Q. Song, J. Yang, S. Hakkinen, X. Pan, Z. Zhang, J. Zhu and S. Perrier, ACS Macro Lett., 2021, 10, 570–575 CrossRef CAS.
- J. Li, M. Chen, X. Lin, Q. Li, W. Zhang, G. Jin, X. Pan, J. Zhu and X. Zhu, ACS Macro Lett., 2020, 9, 1799–1805 CrossRef CAS PubMed.
- B. Zhao, J. Li, X. Pan, Z. Zhang, G. Jin and J. Zhu, ACS Macro Lett., 2021, 10, 1315–1320 CrossRef CAS PubMed.
- B. Zhao, J. Li, Z. Li, X. Lin, X. Pan, Z. Zhang and J. Zhu, Macromolecules, 2022, 55, 7181–7192 CrossRef CAS.
- B. Zhao, J. Li, G. Li, X. Yang, S. Lu, X. Pan and J. Zhu, Small, 2023, 19, 1613–5810 Search PubMed.
- B. Zhao, J. Li, X. Yang, S. He, X. Pan and J. Zhu, ACS Appl. Polym. Mater., 2024, 6, 1584–1591 CrossRef CAS.
- M. A. Tasdelen, J. Lalevée and Y. Yagci, Polym. Chem., 2020, 11, 1111–1121 RSC.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- M. D. Nothling, Q. Fu, A. Reyhani, S. Allison-Logan, K. Jung, J. Zhu, M. Kamigaito, C. Boyer and G. G. Qiao, Adv. Sci., 2020, 7, 2001656 CrossRef CAS.
- S. W. Spring, J. H. Hsu, R. J. Sifri, M. S. Yang, C. S. Cerione, H. Lambert, J. C. Ellison and B. P. Fors, J. Am. Chem. Soc., 2022, 144, 15727–15734 CrossRef CAS PubMed.
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS PubMed.
- M. L. Allegrezza and D. Konkolewicz, ACS Macro Lett., 2021, 10, 433–446 CrossRef CAS.
- J. Lalevee, N. Blanchard, M. A. Tehfe, M. Peter, F. Morlet-Savary and J. P. Fouassier, Macromol. Rapid Commun., 2011, 32, 917–920 CrossRef CAS PubMed.
- S. Shanmugam, J. T. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectrum and SEC of polymers. See DOI: https://doi.org/10.1039/d4py00774c |
| This journal is © The Royal Society of Chemistry 2024 |