
Strategies to prepare polymers with cleavable linkages releasing active agents in acidic media
Nachnicha
Kongkatigumjorn
and
Daniel
Crespy
 *
*
Department of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), Rayong 21210, Thailand. E-mail: daniel.crespy@vistec.ac.th
First published on 1st October 2024
Abstract
Incorporating labile bonds in the chemical structure of polymers yields interesting materials that are responsive to changes in environmental stimuli, such as pH. Active agents can be conjugated to various polymers through acid-labile linkages, conferring upon them spatiotemporal control over the cleavage and subsequent release of drugs, antibacterial agents or corrosion inhibitors. One key factor influencing the release of active agents is the hydrolytic stability of the linkages. We compared the hydrolytic kinetics of acid-cleavable linkages and discuss the effect of structural fine tuning of molecules on the hydrolysis rate. Synthetic routes towards various acid-sensitive linkages are discussed in relation with the functional unit available on the active substances to be released.
1. Introduction
Materials can be engineered to confer spatiotemporal control over the change in their physicochemical properties, leading to a subsequent release of active agents, such as drugs,1–3 antibacterial agents,4–6 or corrosion inhibitors,7,8 while preserving the structural integrity of the materials.9,10Active agents can be encapsulated via physical entrapment in a large variety of carriers, such as liposomes, particles, hydrogels, or fibers.11 However, encapsulation via physical entrapment is typically associated with a burst release of active agents and an undesirable leakage of payloads from carriers.12 Thus, acid-cleavable linkages are valuable building blocks because they allow for the selective release of functional compounds from parent molecules.13
Local pH near anodic areas of steel substrates during corrosion in 0.05 M NaCl is typically acidic (pH ≈ 3.5).7 This finding has inspired research on the preparation of polymer-corrosion-inhibitor conjugates as coatings, whereby the active moiety, i.e. the corrosion inhibitor, is linked to the polymer via an acid-cleavable bond.8 The decrease in the local pH value, as a consequence of corrosion, acts as the trigger to release the corrosion inhibitor, which is adsorbed on the metal's surface to form a protective surface as a barrier to water and oxygen. Another acidic medium is the extracellular environment of tumors (pH ≈ 6.5) owing to the hypoxic microenvironment produced by irregular angiogenesis in fast-growing tumors.3 The reduction in pH values is also found from pH ≈ 7.4 in the bloodstream to pH ≈ 6.5 in the early endosomal compartment and below pH ≈ 5 in the lysosomal compartments of normal cells.14 Besides, an increase in lactic acid production due to a high rate of glycolysis induces a decrease in the pH value from ≈7.4 to ≈4 in the inflammatory phase of infected areas of wounds,15 which has led to the development of acid-responsive materials for wound healing.16 Besides, sweat is also slightly acidic, and therefore, polymers have been designed to release fragrances through the cleavage of acid-labile linkages.17
There are several reviews discussing pH-cleavable linkages, including the hydrolysis of acid-cleavable linkages.18–23 Herein, we provide a systematic review on the structural factors influencing the sensitivity of pH-cleavable linkages to acidic conditions and the reactivity of possible reagents for synthesizing the linkages. Moreover, we extend the discussion, which is typically focused only on the release of drugs, to other active substances, such as fragrances, self-healing agents, and corrosion inhibitors. We compare the hydrolytic kinetics of acid-cleavable linkages and discuss the effect of structural fine tuning of the molecules on the hydrolysis rate. An overview of various linkages is proposed, whose selection depends on the functional unit available on the active substance to be released.
2. Substituents effects on the dissociation of pH-sensitive linkages
The hydrolysis kinetics of acid-cleavable linkages was assessed by high-performance liquid chromatography (HPLC) with an ultraviolet (UV) detector in buffer solution at pH 5.5 and pH 7.4 at 25 °C.1,2 Acid-cleavage linkages (acetals/ketals, imines, hydrazones, orthoesters, and thioether esters) were conjugated with UV traceable molecules. Then, the linkages were ranked according to their hydrolytic rate and their selectivity in aqueous solutions (see Table 1). To quantify the relative rates of hydrolysis, the half-life (t1/2) of the acid-cleavable groups was calculated from the HPLC results. When the half-life at pH 5.5 was lower than 1 h, the hydrolysis was considered by the authors to be “fast”. The selectivity of hydrolysis regarding the pH of the solution was evaluated by comparing the ratio of the half-life at pH 7.4 over the half-life at pH 5.5. When the ratio was more than 15, the cleavage was considered by the authors to be highly selective. For ratios lower than 2, it was considered non-selective. For ratios between 2 and 15, the cleavage was interpreted as selective. Because there was no justification for the choice of these numbers, the results were highly empirical; however, they did allow for a comparison of the reactivities.| Acid-cleavage linkages | Chemical structures | t 1/2 (pH 5.5) |
|
|---|---|---|---|
| Acetal/ketal |
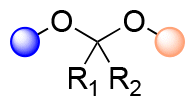
|
<1 h | <2 |
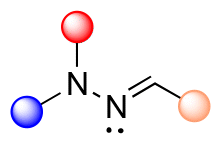
| Hydrazone |
|
<1 h | <2 |
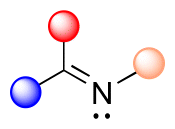
| Imine |
|
<1 h | <2 |
| Orthoester |
|
<1 h | >15 |
| Thioether ester |
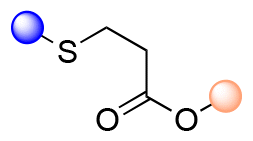
|
>1 h | <2 |
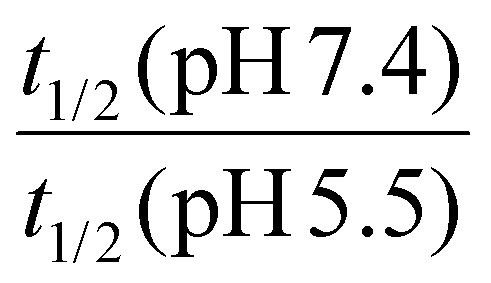
The hydrolytic rate can also change dramatically, depending on the chemical structures of the parents and the released molecules. Herein, we discuss the various approaches for tuning the hydrolytic rates of acid-cleavage linkages in aqueous media.
2.1 Hydrolysis of acetal/ketal linkages
Acetals and ketals groups are suitable acid-sensitive linkages for releasing molecules containing primary, secondary, or tertiary alcohols.25 Both linkages are stable under basic conditions due to the presence of lone electron pairs on the oxygen atoms. They act as Lewis bases and are fairly labile in the presence of Lewis or protic acids. Moreover, acetals and ketals can be readily hydrolyzed to the corresponding carbonyl compound (aldehyde or ketone) through a carboxonium ion intermediate upon acid cleavage. In the hydrolysis process, the formation of the resonance-stabilized carboxonium ions intermediate (Fig. 1) is the rate-determining step. When the intermediate carboxonium ion is more stabilized, the hydrolysis rate is faster. Acetals and ketals can be formed using primary, secondary, and tertiary alcohols and syn-1,2 and syn-1,3 diols.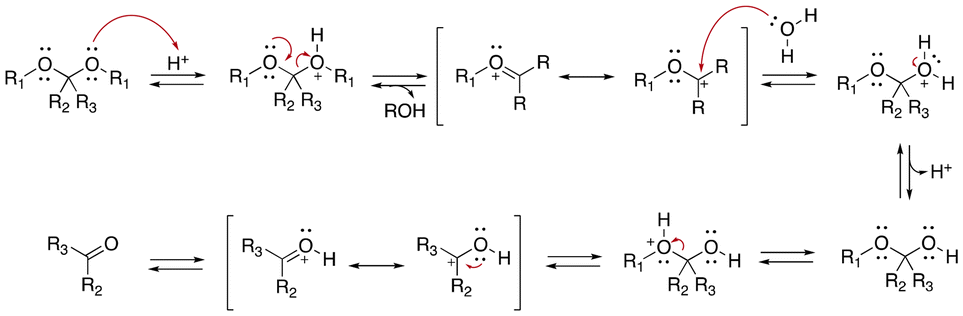 | ||
| Fig. 1 Scheme of the acidic hydrolysis of acetal groups.25 | ||
The substituent effects in the acidic hydrolysis of acetal groups was investigated by quantitative 1H NMR spectroscopy (Table 2).26 It was found that acyclic acetals were hydrolyzed faster than cyclic acetals. Moreover, the presence of carbocation-stabilizing substituents, such as aromatic substituents, in acetal groups significantly accelerated the hydrolysis rate.
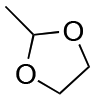
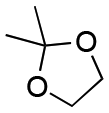
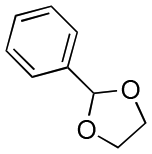
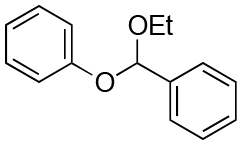
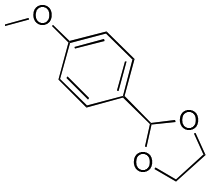
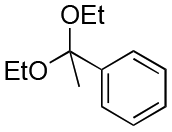
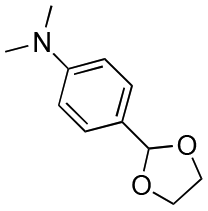
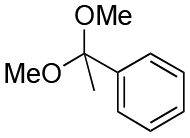
| Entry | Acetal | k H+ (L mol−1 S−1) | Entry | Acetal | k H+ (L mol−1 S−1) |
|---|---|---|---|---|---|
| 1 |
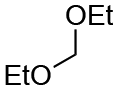
|
1.5 × 10−4 | 8 |
|
0.013 |
| 2 |
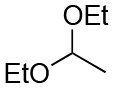
|
1.6 | 9 |
|
0.13 |
| 3 |
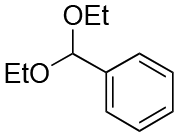
|
160 | 10 |
|
0.5 |
| 4 |
|
41 | 11 |
|
130 |
| 5 |
|
6000 | 12 |
|
3000 |
| 6 |
|
1600 | 13 |
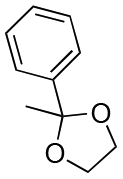
|
1.2 |
| 7 |

|
2.6 × 10−6 | 14 |
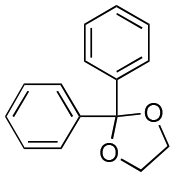
|
0.05 |
The control of the pH-sensitivity of acetal- and ketal-based linkers by structural variations was discussed recently.27 Three important effects of the substituents in acetal/ketal linkages were reported to be the distance between the R1 substituent (Fig. 1) and the ketal moiety, the presence of alkyl and aryl substituents at R2 and R3 positions, and the effect of electron-withdrawing/donating substituents in benzylidene acetals. The hydrolysis kinetics was measured by 1H NMR spectroscopy at 25 °C. To quantify the relative rates of hydrolysis, the half-life of these molecules was calculated from first-order kinetics fitting curves (see Fig. 2).
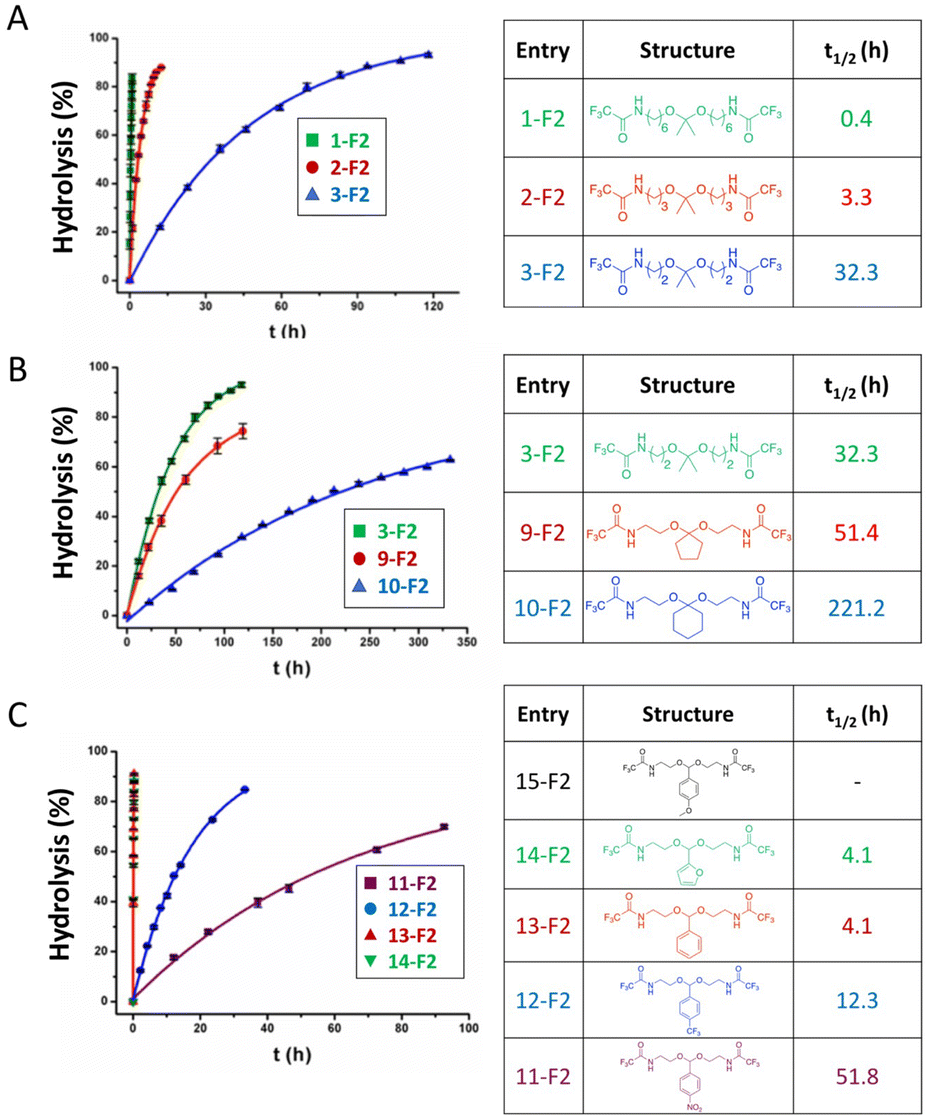 | ||
| Fig. 2 Relative hydrolysis rates of acetals/ketals depending on the substituents at the R1, R2 and R3 positions, with varying the (A) distance between the electron-withdrawing amide moiety and ketal moiety, (B) substituents effects of acrylic and cyclic ketones, (C) effects of electron-withdrawing and electron-donating substituents. The solid lines are fitting curves.27 (Copyright 2017 American Chemical Society). | ||
In this study, the amide group was used as an inductive electron-withdrawing moiety. The effect of the number of carbon atoms between the amide and the carboxonium ion intermediate is shown in Fig. 2A. When the six-carbon linker 1-F2 was changed to a two-carbon-linker (3-F2), the half-life of the ketals was increased from 0.4 h to 32.3 h. The longer half-lives were induced by the higher stability of the carboxonium ions. An inductive effect-based destabilization of the carboxonium ion intermediate was observed in the molecule 3-F2 (Fig. 2B), which raised the energy of the transition state. Moreover, the hydrolysis rates of the cyclopentyl 9-F2 and cyclohexanone ketals 10-F2 were found to be about two times and seven times slower, respectively, compared with the alkyl analogues (Fig. 2B). The difference in hydrolysis rates between the five-membered ring 9-F2 and the six-membered ring 10-F2 was attributed to the difference in torsional energy strain. The lower torsional energy strain in 10-F2 induced a slower hydrolytic rate of ketals.
The half-lives of 15-F2 were not provided because the hydrolysis rate of 15-F2 was too high to be measured. Indeed, incorporating an electron-donating p-methoxy moiety on the acetal bond of 15-F2 significantly accelerated the hydrolytic rate of the acetal bond. Interestingly, the presence of a resonance-based electron-withdrawing group in 13-F2, 14-F2, 15-F2 reduced the half-life, whereas incorporating an electron-donating p-methoxy moiety in 11-F2, 12-F2 significantly enhanced the hydrolysis rate (Fig. 2C).
2.2 Hydrolysis of imine and hydrazone linkages
Imine linkages can be easily obtained from the reactions of primary amines with carbonyl groups of aldehydes or ketones.28 Imines are usually stable in neutral and basic conditions, but they are hydrolyzed in aqueous acidic media (see Fig. 3).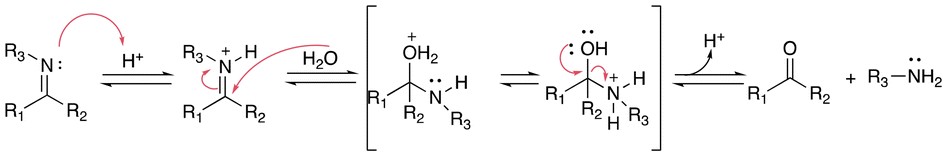 | ||
| Fig. 3 Scheme of the hydrolysis of imine bonds in acidic aqueous media. | ||
The hydrolytic stability of imine bonds depends on the pH value of the medium, the basicity of the imine nitrogen, and the substituents effects surrounding the imine bonds. Imines with higher basicity are more easily protonated, and hence hydrolyzed more rapidly.
Cordes et al. investigated aryl and alkyl substituents effects on the R1, R2, R3 positions in imine linkages.29 Imines with aryl groups were found to be more stable towards hydrolysis than alkyl imines groups due to electron delocalization with π-bond conjugations between the aromatic ring and the imine bond. Electron-withdrawing substitutions enhanced the hydrolysis rate of imines in acidic media. Moreover, iminium salts (R1R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+R3R4) in the intermediate state were found to be very sensitive to the presence of water and can easily hydrolyze at any pH value in aqueous media.30
N+R3R4) in the intermediate state were found to be very sensitive to the presence of water and can easily hydrolyze at any pH value in aqueous media.30
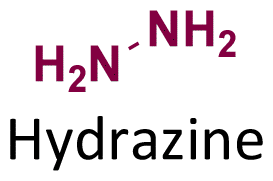
Hydrazone (R1–C1N1–N2H–R3) linkages are one of the most common linkages for providing a pH-responsive release because they are easy to synthesize and display a moderate sensitivity. Hydrazones are created via a formation of a stable imine from the reaction of aldehydes and ketones with α-nucleophiles, such as hydrazines and aminooxy groups.31 At pH 7.4, the hydrazone bond is relatively stable and hydrolyzes very slowly, while the rate of hydrolysis increases in mild acidic environments (pH 5–6). Due to electron delocalization in hydrazone structures (Fig. 4), hydrazones are more stable towards acid hydrolysis than imines, while charge delocalization in alkylhydrazones II-F4 and acylhydrazones IV-F4 enhances the density of negative charge on the carbon, resulting in a decrease in its electrophilicity. An alternative explanation for the decreasing electrophilicity is that delocalization causes a decrease in π-electron repulsion between the electron lone pairs of nitrogen.
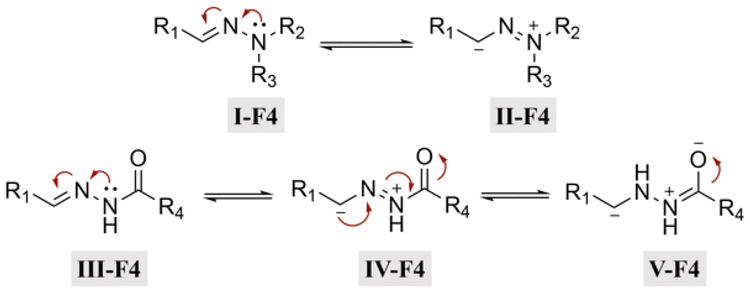 | ||
| Fig. 4 Scheme showing the electron delocalization in alkyl and acyl hydrazones. | ||
The hydrolytic cleavage of hydrazones groups is reversible. The hydrolytic stability of different structural hydrazone linkages was determined by NMR spectroscopy of the conjugates at different pH values.32 The hydrolytic stability of hydrazone linkages with a positive charge on the nitrogen (trimethylhydrazoniumion 6-T3) was higher than that for other hydrazone linkages at pH 7.0 (Table 3). Moreover, the hydrolysis of trimethylhydrazonium ions at pH 5.0–9.0 was very slow, but was not due to electron delocalization and repulsion between lone pairs of electrons in nitrogen atoms. The protonation of the nitrogen atom was unfavourable, due to the presence of an adjacent quaternary ammonium group yielding potentially positive–positive charge repulsion, which explains why the trimethylhydrazonium showed a very high hydrolytic stability. To quantify the hydrolytic rate, the half-lives of hydrazone linkages at pH 5.0–9.0 were calculated by fitting release curves with the first-order kinetics (Table 3). The hydrolytic rate of the tertiary amine 2-T3 on the hydrazone linkage was higher than that for the secondary amine 1-T3. The presence of the highly electronegative substituent 4-T3 on the hydrazone linkage induced a slower hydrolytic rate than with substituents with lower electronegativity.
| Entry | Chemical structure | pH | ||||
|---|---|---|---|---|---|---|
| 5.0 | 6.0 | 7.0 | 8.0 | 9.0 | ||
| 1 |
|
9 min | 24.5 min | 1 h | 4.2 h | 19.5 h |
| 2 |
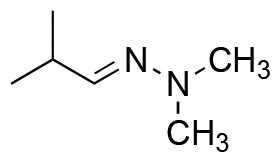
|
7.4 min | 11.3 min | 32 min | 2.0 h | 11.7 h |
| 3 |
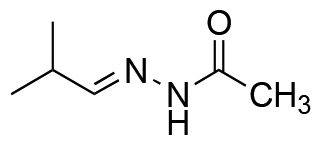
|
2.4 min | 21.4 min | 2 h | 10.17 h | 4.2 h |
| 4 |
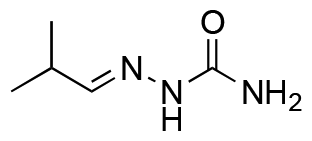
|
8.5 min | 36.0 min | 3.8 h | 12.3 h | 2.9 h |
| 5 |
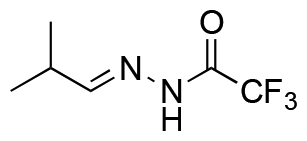
|
7.5 min | 12.4 min | 14 min | 23 min | 1.0 h |
| 6 |
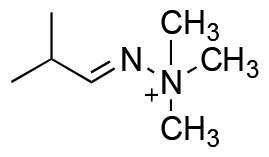
|
10.3% hydrolysis in 17 d | ND | no hydrolysis in 22 d | ND | ND |
The hydrolytic stability of hydrazone linkages can be controlled by changing the different substitutions around the hydrazone linkages.33 Zhang et al. investigated the substituent effects around hydrazone linkages of pH-sensitive poly(ethylene glycol)hydrazone-farnesylthiosalicylate conjugates by measuring the amount of cleaved farnesylthiosalicylate by HPLC with an UV detector.34 Farnesylthiosalicylate is a hydrophobic compound and an antagonist for RAS (renin–angiotensin system, an endocrine system), which can decrease tumor development and progression. Hydrophilic poly(ethylene glycol) was conjugated to hydrophobic farnesylthiosalicylate-hydrazide to form a polymer with different substituents on the hydrazone linkers. The increase in alkyl chain length led to a decrease in the hydrolytic rate at pH 5.5 relative to PHF-2 (Fig. 5). This phenomenon was attributed to the decrease in π-electron repulsion between the electron lone pairs of nitrogen atoms when the distance between them was increased (linker X in Fig. 5A). Besides, the presence of aryl substituents on hydrazone linkages led to a further decrease in the hydrolysis rate at pH 5.5. This behaviour was attributed to electron delocalization of the π-bonds in the –CN– group of the hydrazone and the aryl substituents, which contributed to hydrolytic stability.
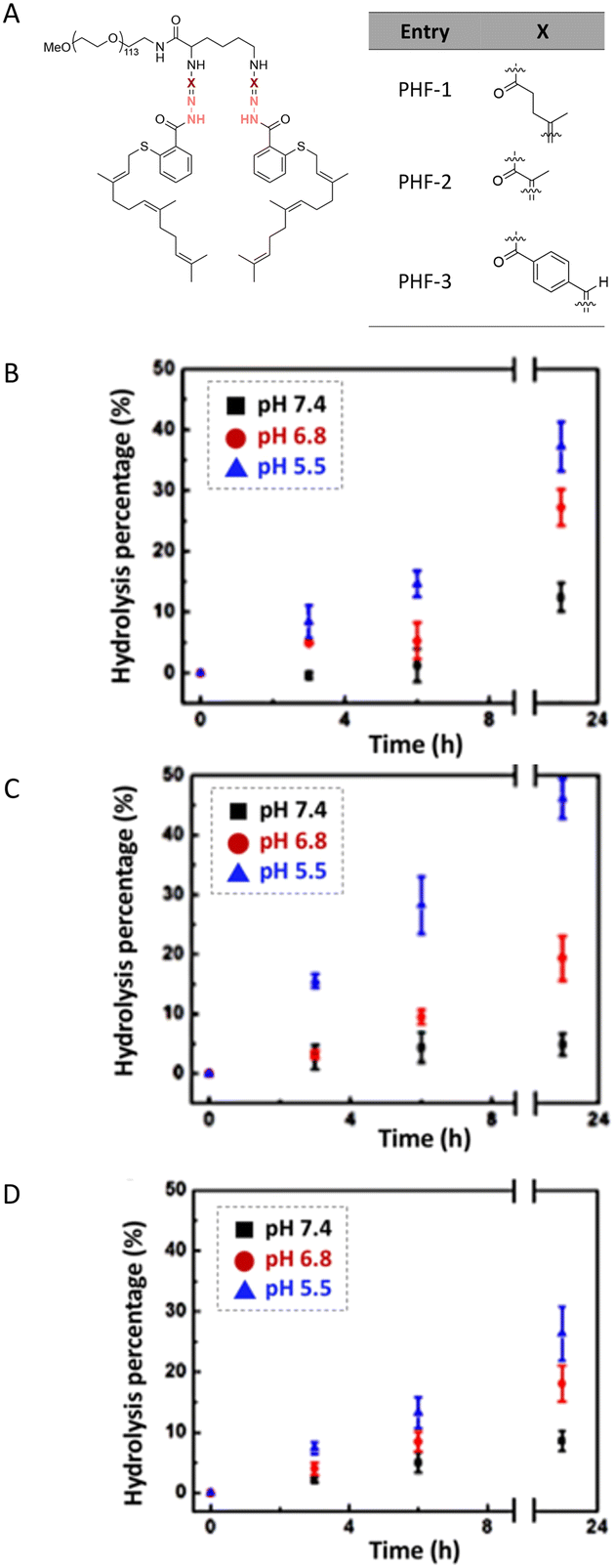 | ||
| Fig. 5 Structure of poly(ethylene glycol)-hydrazone-farnesylthiosalicylate (PHF) (A). Temporal evolution of the amount of hydrolyzed hydrazone linkers at different pH values for PHF-1 (B), PHF-2 (C), and PHF-3 (D).34 (Copyright 2015 American Chemical Society). | ||
2.3 Hydrolysis of orthoester linkages
Orthoesters were noted to hydrolyze faster in response to a mild acidic environment than ketal and acetal groups with similar structures due to the formation of a stable dialkoxy carbon cation intermediate.34 The hydrolysis of alkyl orthoesters (Fig. 6) implies the cleavage of three alkoxy groups.35 The first and second alkoxy groups were rapidly hydrolyzed in acidic conditions. Then, the cleavage of the third alkoxy group was slower compared with the cleavage of the first and second alkoxy groups. The cyclic orthoesters can be in principle protonated at two oxygen positions: the endocyclic atom O-1 (or O-2) or the exocyclic atom O-3 (Fig. 6).36 Protonation at one of the endocyclic atom induces a ring opening to afford a cation. Water then attacks the cation to yield an ester or a diol. Protonation at the O-3 position yields methanol and a cyclic cation. Then, the cyclic cation is attacked with water to generate an ester.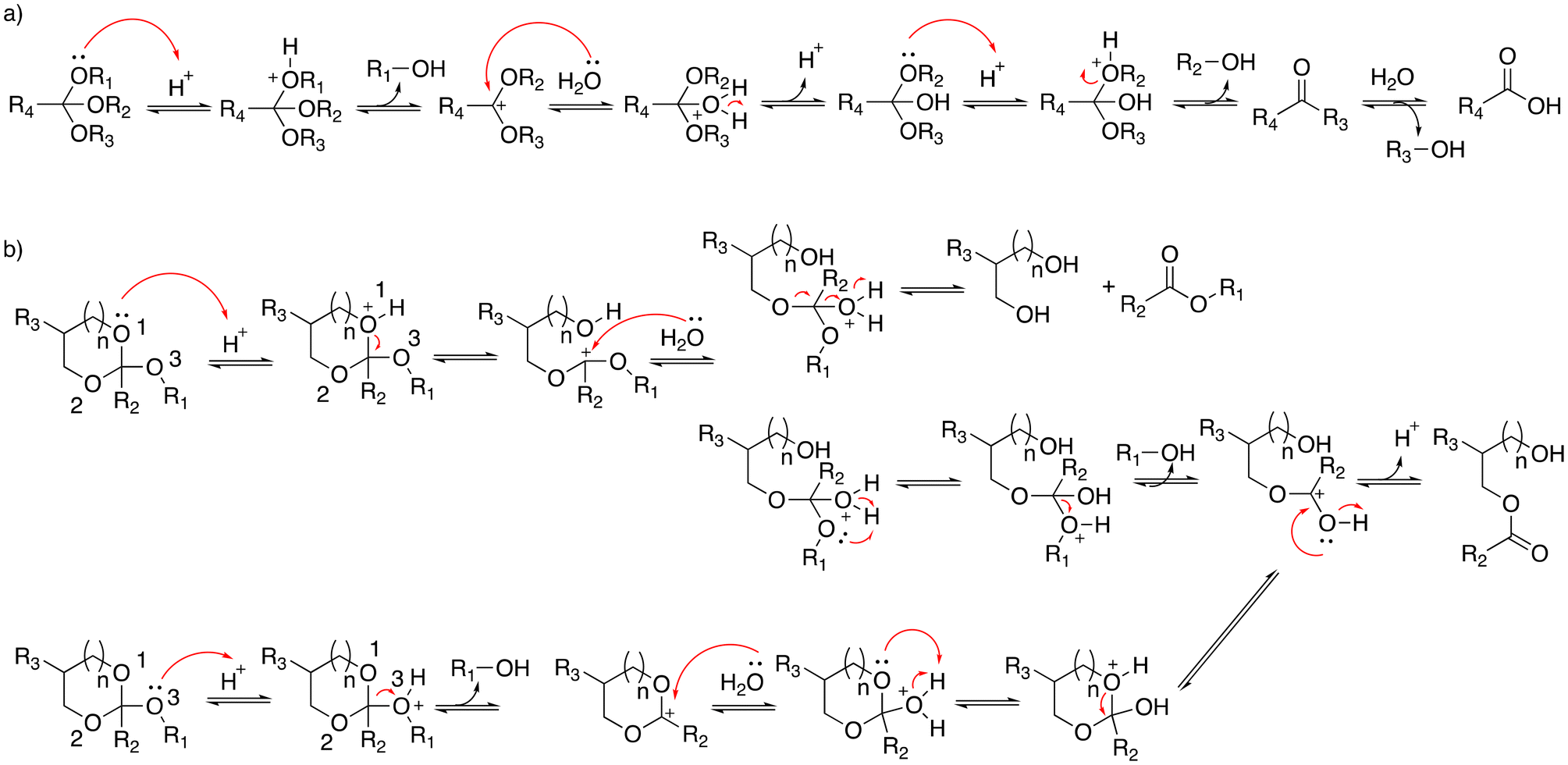 | ||
| Fig. 6 Scheme of the hydrolysis of (a) alkyl orthoesters and (b) cyclic orthoesters.35 | ||
Cheng et al. reported the synthesis of a family of acid-labile polymers with pendent cyclic orthoesters.37 The hydrolytic kinetics of six cyclic orthoesters were monitored by 1H NMR spectroscopy at pH 5.6, 6.5, 7.5, and 8.5 (Table 4). For identical exocyclic alkyloxy groups, the hydrolysis rate decrease could be ranked as 1a > 2a > 3a and 1b > 2b > 3b. The faster hydrolysis rate of the six-membered cyclic orthoesters (1a or 2b) compared with the five-membered cyclic ortho esters (2a and 2b) can be explained by anomeric effects. The ideal chair conformation of the six-membered ring induced a higher basicity of the exocyclic oxygen atoms. Thus, the protonated exocyclic alkoxy groups on the six-membered ring orthoesters were hydrolyzed faster than the five-membered ring orthoesters.
| Entry | Orthoester | Temperature (°C) | pH | |||
|---|---|---|---|---|---|---|
| 5.4 | 6.5 | 7.5 | 8.5 | |||
| The absence of t1/2 was due to the hydrolytic reaction being too fast to be measured. | ||||||
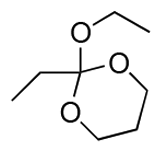
| 1a |
|
15 | ∼1 | ∼2 | 14 | 50 |
| 2a |
|
15 | ∼2 | ∼3 | 20 | 120 |
| 3a |
|
15 | 36 | ∼1900 | ||
| 3a |
|
23 | 15 | ∼720 | ||
| 1b |
|
23 | ∼8 | 28 | ||
| 2b |
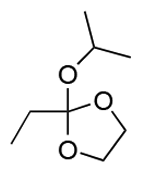
|
23 | 12 | 80 | ||
| 3b |
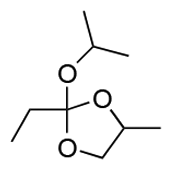
|
23 | 17 | ∼560 | ||
However, another study reported that the hydrolytic rate of five-membered ring orthoesters was faster than for six-membered rings due to amide substituents on the ring.17 Amide substituents affected the conformation of cyclic orthoesters via intramolecular hydrogen bonding, which influenced the hydrolysis.36,37 Löw and coworkers explored substituents effects on the hydrolytic rate of alkyl orthoesters at the R4 position (Fig. 6a).38 The hydrolysis reaction was monitored by 1H NMR spectroscopy. The half-lives (t1/2) and kinetic rates (kobs) were calculated by fitting release curves with pseudo-first-order kinetics (Table 5). The electron-rich orthoesters (1-T5) were observed to hydrolyze readily even at pH 7 with a hydrolytic rate of 1.3 × 10−3 s−1. However, the electron-deficient orthoesters (5-T5, 6-T5) were remarkably stable at pH 1 and their half-lives were more than 16 h. The orthoformates and chloromethyl-substituted orthoesters (2-T5, 3-T5) were stable at pH 7, whereas they were readily hydrolyzed at pH 5, with t1/2 values of 20 min and 121 min.
| Entry | R4 | pH 8 | pH 7 | pH 6 | pH 5 | pH 4 | pH 3 | pH 1 | |
|---|---|---|---|---|---|---|---|---|---|
| a 1-Benzyl-4-(trimethoxymethyl)-1H-1,2,3-triazole. b 1-Benzyl-3-methyl-4-(trimethoxymethyl)-1H-1,2,3-triazol-3-ium. | |||||||||
| 1-T5 | –CH3 | t 1/2 [min] | 60 | 10 | 2 | <1 | |||
| k obs [s−1] | 1.8 × 10−4 | 1.3 × 10−3 | 6.1 × 10−3 | ||||||
| 2-T5 | –H | t 1/2 [min] | >1000 | 510 | 70 | 20 | 7 | <1 | |
| k obs [s−1] | 2.5 × 10−5 | 1.6 × 10−4 | 8.8 × 10−4 | 2.0 × 10−3 | |||||
| 3-T5 | –CH2Cl | t 1/2 [min] | >1000 | 660 | 120 | 30 | <1 | ||
| k obs [s−1] | 1.7 × 10−5 | 8.6 × 10−5 | 2.9 × 10−4 | ||||||
| 4-T5 | –Triazolea | t 1/2 [min] | <1 | ||||||
| 5-T5 | –Triazoliumb | t 1/2 [min] | Inert | >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
|||||
| k obs [s−1] | 1.6 × 10−6 | ||||||||
| 6-T5 | –CCl3 | t 1/2 [min] | >1000 | ||||||
2.4 Hydrolysis of thioether esters
β-Thiopropionate esters (RCOOCH2CH2SR′) are specific thioether esters that are very sensitive to the presence of acids.13 The hydrolysis of β-thiopropionate esters is faster than the hydrolysis of ester bonds (RCOOR′) in acid conditions due to the inductive effect and the protonation of the neighbouring sulfur atom. The presence of electron-withdrawing groups in close proximity to ester bonds reduced the electron density at the carbonyl groups. Consequently, the carbonyl group was easily protonated and an electron lone pair of water rapidly attacked the electrophilic carbons, resulting in an accelerated hydrolysis. Moreover, the protonation of the sulfur atom and subsequent formation of a six-membered ring by intramolecular hydrogen bonding (Fig. 7) caused a rapid hydrolysis of the β-thiopropionate esters under mild acidic conditions in aqueous media.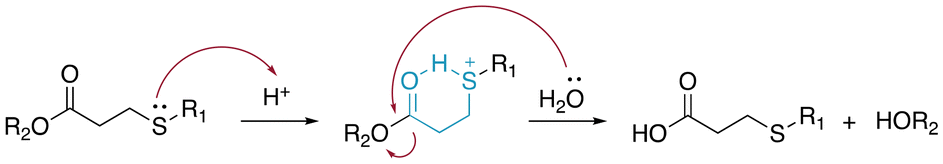 | ||
| Fig. 7 Mechanism of the hydrolysis of β-thiopropionate groups in aqueous acidic media. | ||
Bowman et al. investigated the influence of the number of carbons between the sulfide and ester bonds on ester hydrolysis using quantitative 1H NMR spectroscopy.39 The trithiol molecules displayed either one (1, trimethylolpropane tris(2-mercaptoacetate)) or two (2, trimethylolpropane tris(3-mercaptopropionate)) carbons between the ester bond and the sulfur atom. The hydrolysis of the thiol-based esters was performed at pH 8. The rate constants of for the ester hydrolysis of 2 and 1 were 0.022 ± 0.001 and 0.08 ± 0.015 day−1, respectively. The effects of the number of methylene units between the carbonyl and sulfur was also studied by Schoenmakers and coworkers.40 The drug paclitaxel (PTX) was coupled using a thioether ester linker to poly(ethylene glycol) via a reaction between a thiol of mercaptoacid and an acrylamide group in PEG-diacrylamide (Fig. 8A). The measured release profiles of PTX at pH 7.4 could be well described by first-order kinetics (Fig. 8B). The half-lives for the hydrolysis of 3-sulfanylpropionyl and 4-sulfanylbutyryl were 4.2 ± 0.1 and 14.0 ± 0.2 days, respectively.
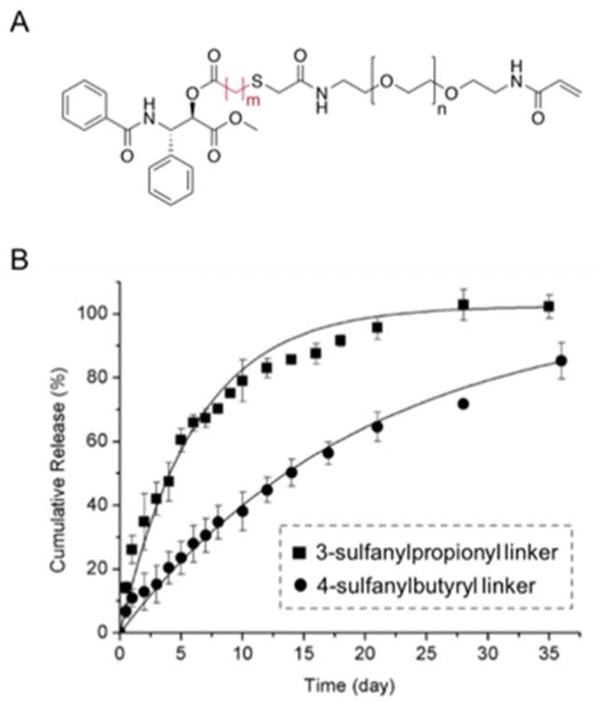 | ||
| Fig. 8 (A) Chemical structure of paclitaxel conjugated with poly(ethylene glycol) via a β-thiopropionate linkage (A) (m = 2: 3-sulfanylpropionyl linker, m = 3: 4-sulfanylbutyryl linker). (B) Release kinetics of paclitaxel from PEG conjugates at pH 7.4 (■: 3-sulfanylpropionyl linker; ●: 4-sulfanylbutyryl linker).40 (Copyright 2004 Elsevier). | ||
The hydrolytic rate of thioether esters can be accelerated by increasing the degree of oxidation on the sulfur atom.41 Hydroxyethylmethacrylate was condensed with dexamethasone to form a polymerizable dexamethasone (DMSL) with different thioether ester linkers (Fig. 9A) containing a sulfide (DMSL1), a sulfoxide (DMSL2), or a sulfone (DMSL3) group. Oxidation of the sulfide group induced a faster hydrolysis rate due to the stronger electron-withdrawing properties of the oxidized sulfur. The hydrolysis kinetics was measured by 1H NMR spectroscopy at 37 °C and at pH 7.4 and 9.4. To quantify the hydrolytic kinetics, the half-lives of these molecules were calculated from first-order kinetics (Fig. 9B). The hydrolysis of DMSL3 (t1/2 = 0.89 ± 0.1 days) was faster than that of DMSL2 (t1/2 = 18.4 ± 0.4 days) at pH 7.4. After 7 days and at pH 7.4, <5% dexamethasone was released from DMSL1 whereas >15% was released from DMSL2 and DMSL3. Dexamethasone was released much faster at pH 9.4 than at pH 7.4 for all three compounds. Also, the sulfone DSML3 was hydrolyzed faster (t1/2 = 3.2 ± 0.3 h) than the sulfoxide DMSL2 (t1/2 = 7.1 ± 0.4 h), which was hydrolyzed faster than the sulfite DSML1 (t1/2 = 41.3 ± 1.5 h) (Fig. 9B.).
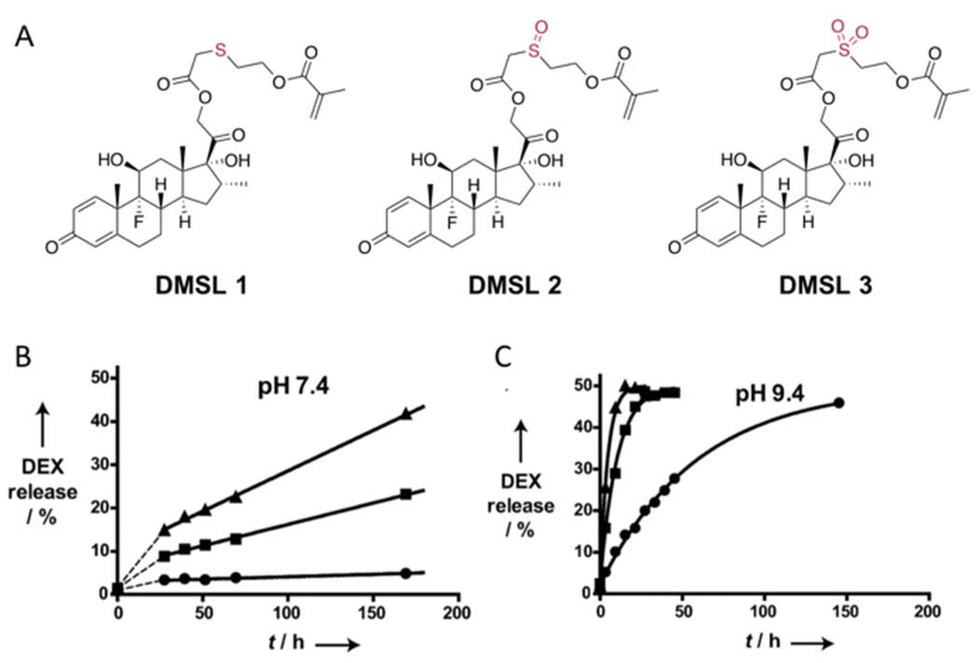 | ||
| Fig. 9 (A) Chemical structure of polymerizable dexamethasone (DMSL) derivatives with sulfide (DMSL 1), sulfone (DMSL 2), and sulfoxide (DMSL 3) ester linkages. Release kinetics of DEX from polymeric micelles with crosslinked cores loaded with different thioether esters (●, DMSL1), a sulfoxide ester (■, DMSL2), or a sulfone ester (▲, DMSL3) at 37 °C at pH 7.4 (B) or (C) pH 9.4.41 (Copyright © 2012 WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
Recently, a series of 8-hydroxyquinoline derivatives was designed with structural fine tuning of the thioether ester linkages.42 Their hydrolytic rate and hydrolysis selectivity under acidic, neural, and basic conditions were measured and compared (Fig. 10). The hydrolytic stability of the 8HQ-derivatives was studied in a mixture of 600 μL of DMSO-d6 and 150 μL of D2O (neutral condition) or 150 μL of 0.5 M DCl solution in D2O (acidic condition), or 150 μL of 5.0 M DCl solution in D2O (conc. acidic condition). It was found that steric hindrance effects induced a significant decrease in the hydrolysis rate, whereas the hydrolysis rate was accelerated by oxidation of the sulfur atom on the thioether ester linkages. The presence of steric hindrance and stronger van de Waals repulsion induced a higher selectivity for hydrolysis of the thioether ester linkages.
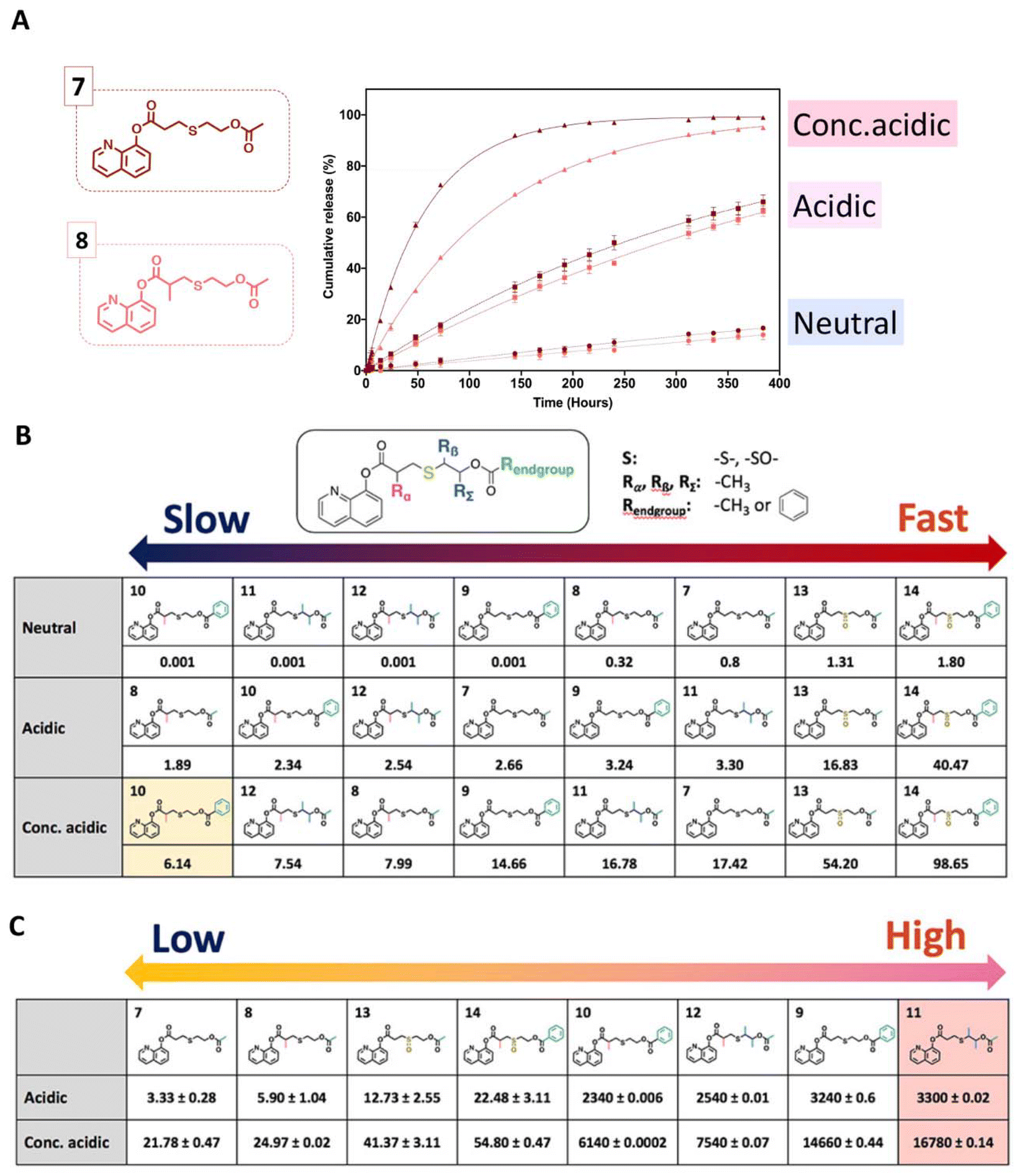 | ||
| Fig. 10 Temporal evolution of the release of 8HQ from 7–8 (A) in a mixture of D2O/DMSO-d6 (neutral condition) = (1:4 v/v), a mixture of 0.5 M DCl in D2O/DMSO-d6 (acidic condition) = (1:4 v/v), a mixture of 5.0 M DCl in D2O/DMSO-d6 (conc. acidic condition) = (1:4 v/v), or a mixture of 5.0 M DCl in D2O/DMSO-d6 = (1:4 v/v). Kinetic constants of the hydrolysis (10−3 h−1) (B) and selectivity of the hydrolysis of thioether esters (C) by calculating the ratio of k values under neutral conditions to k values under acid conditions.42 (Copyright 2022 American Chemical Society). | ||
3. Selection of acid-labile linkages
The hydrolytic rate of pH-cleavable linkages and the type of functional groups of the active agents to be released are two crucial points to take into account when designing polymeric materials or substrates with pH-cleavable linkages (Fig. 11).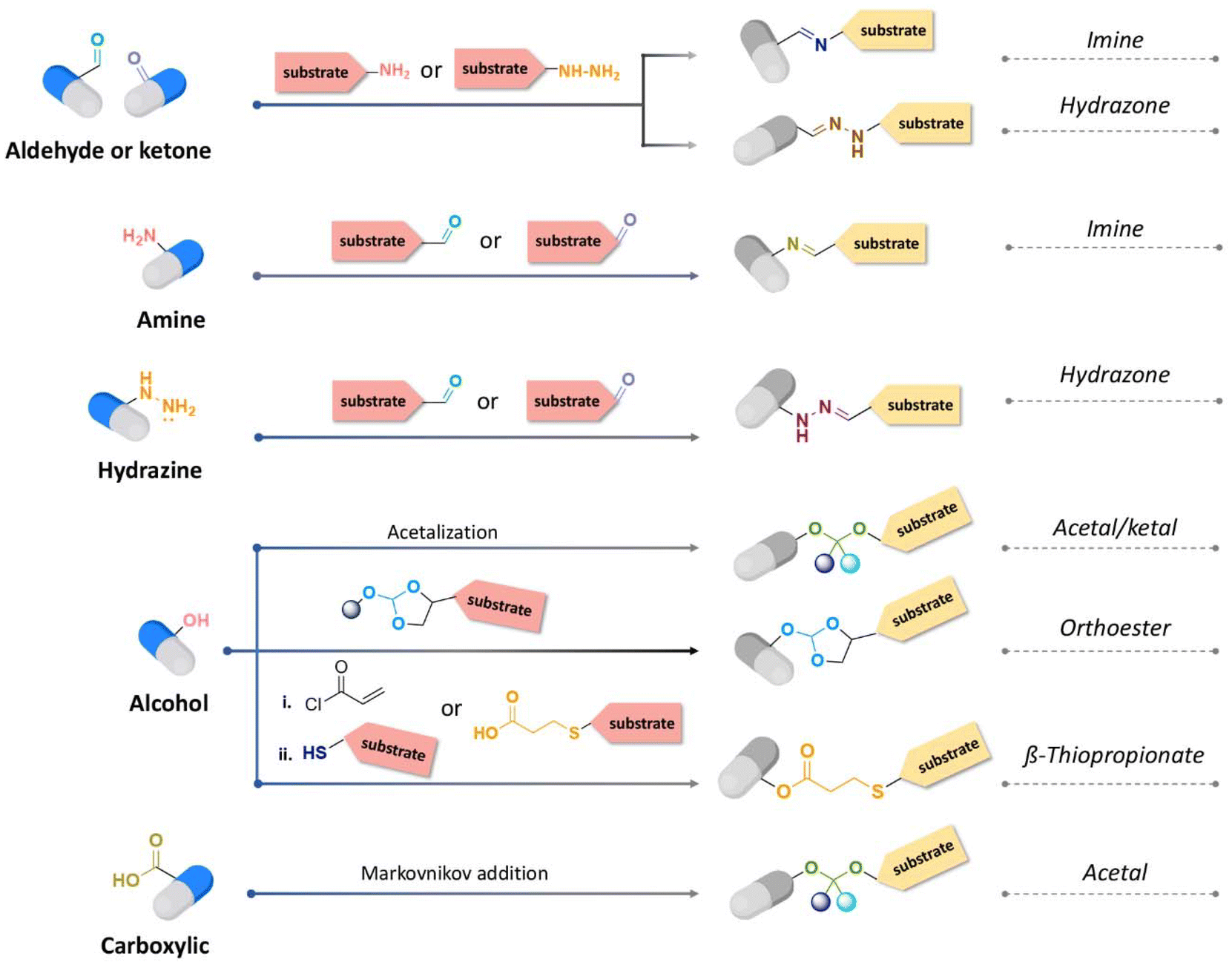 | ||
| Fig. 11 Synthetic route for acid-cleavable linkages depending on the chemical functions presented by the active substance to be released. | ||
3.1 Active agents containing aldehyde or ketone groups
The nucleophilic and addition reaction of primary amines to carbonyl groups of ketones or aldehydes is a reversible process forming imine linkages (Fig. 12).43 A proton transfer forms carbinolamine, which is eliminated as water to produce an iminium ion. The nitrogen atom of the iminium ion is then deprotonated to provide imine linkages. Removal of the water is required to drive the equilibrium towards the formation of the imine.44 Some examples of active agents bearing aldehyde or ketone groups and their derivatives, as well as the reagents used for preparing imine and hydrazone linkages, are listed in Table 6.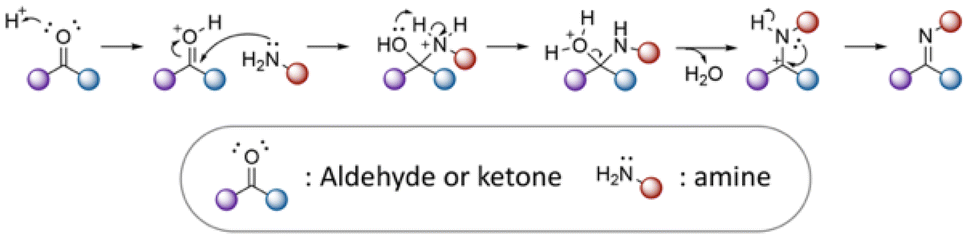 | ||
| Fig. 12 Mechanism of imine formation. | ||
| Active agents | Chemical structure of the active agent | Derivative | Active functional group | Linkages | Reagents for linkage formation | Ref. |
|---|---|---|---|---|---|---|
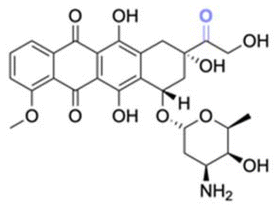
| Doxorubicin (DOX) |
|
Ketone | Hydrazone |
|
69–85 | |
|
86–88 | |||||
|
89–99 | |||||
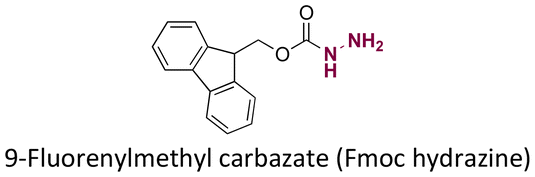
|
100 | |||||
|
101 and 102 | |||||
|
103 | |||||
|
104–111 | |||||
| Imine | 112 | |||||
| Dexamethasone (DEX) |
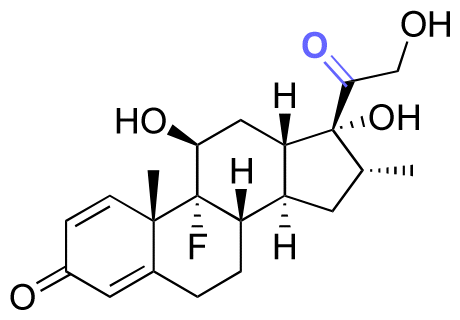
|
Ketone | Hydrazone |
|
113 | |
|
114 and 115 | |||||
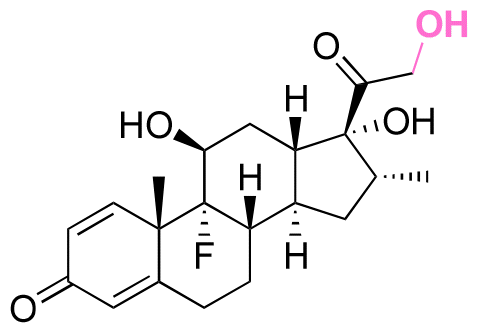
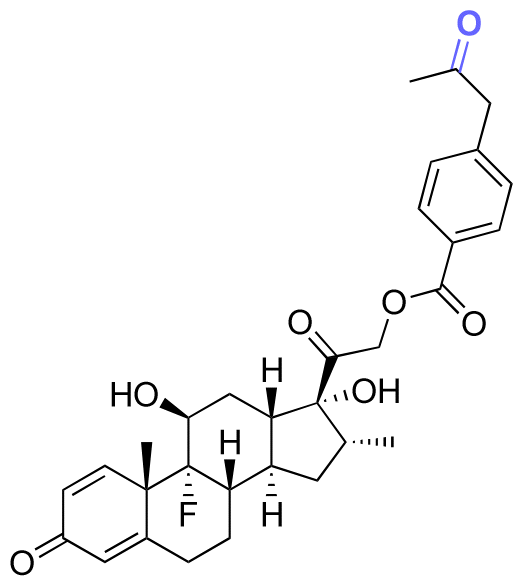
| Dexamethasone (DEX) |
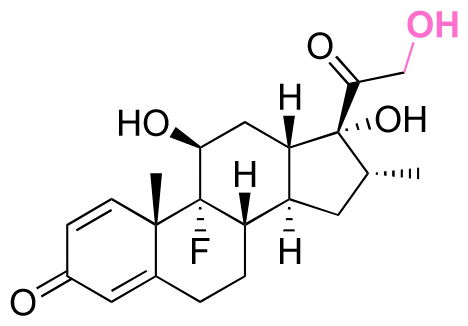
|
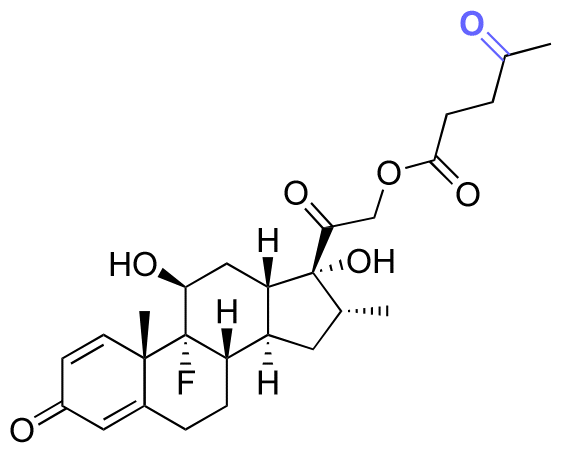
|
Ketone | Hydrazone |
|
114 and 115 |
|
|
|||||
| Docetaxel (DTX) |
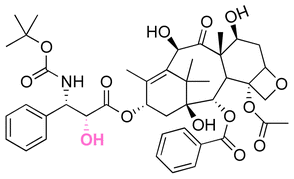
|
|
Ketone | Hydrazone |
|
116 |
| Paclitaxel (PTX) |
|
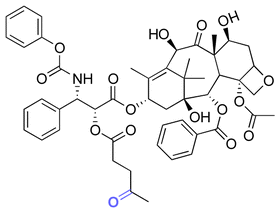
|
Ketone | Hydrazone |
|
116 |
| Pirarubicin |
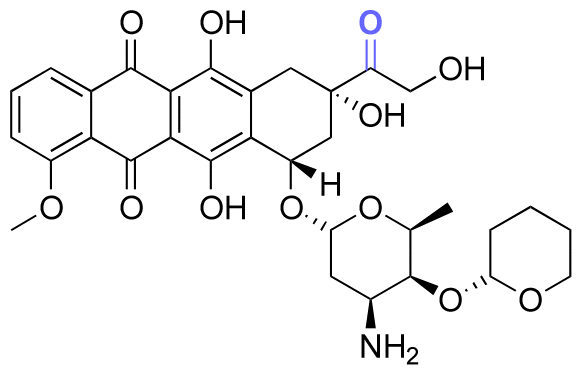
|
Ketone | Hydrazone |
|
117 | |
|
118 | |||||
| Curcumin |
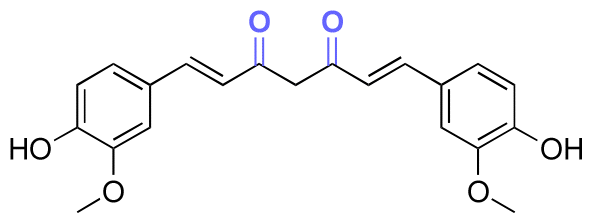
|
Ketone | Hydrazone |
|
119 | |
| Curcumin |
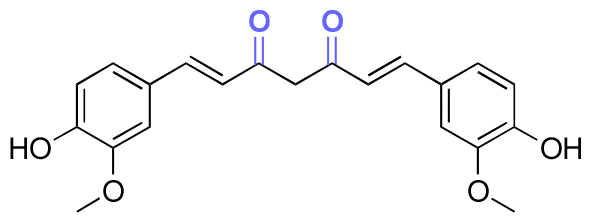
|
Ketone | Hydrazone |
|
119 | |
| β-Ionone |
|
Ketone | Hydrazone | 63 | ||
| 3-(9-Acidinylamino)-5-hydroxymethylaniline (AHMA) |
|
|
Ketone | Hydrazone |
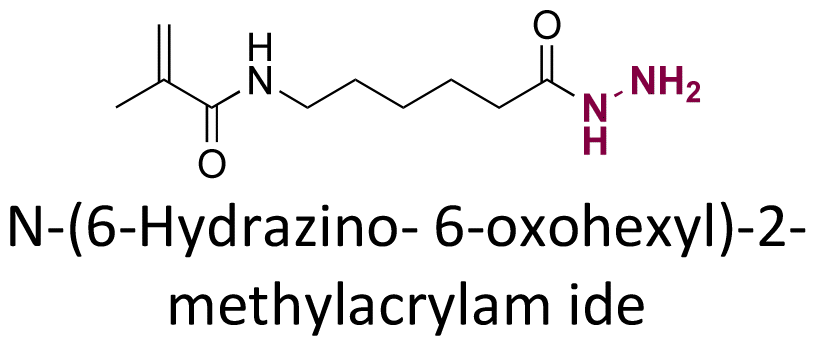
|
120 |
| Cyclamen aldehyde |
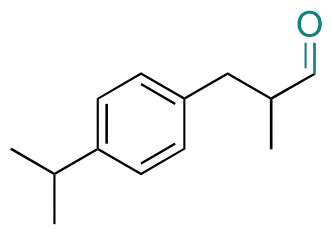
|
Aldehyde | Hydrazone/imine | 61 and 63 | ||
| Cinnamaldehyde |
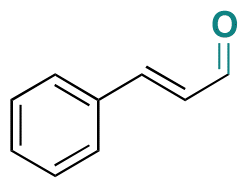
|
Aldehyde | Hydrazone/imine | 63 and 121 | ||
| Helional |
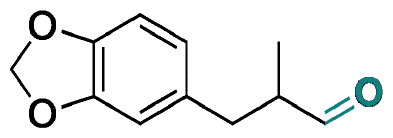
|
Aldehyde | Hydrazone/imine | 61 and 63 | ||
| S-hyNic linker terephthaldehydic acid | Amine/aldehyde | Hydrazone | 122 | |||
| p-Anisaldehyde |
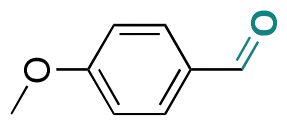
|
Aldehyde | Imine |
|
123 | |
| Streptomycin |
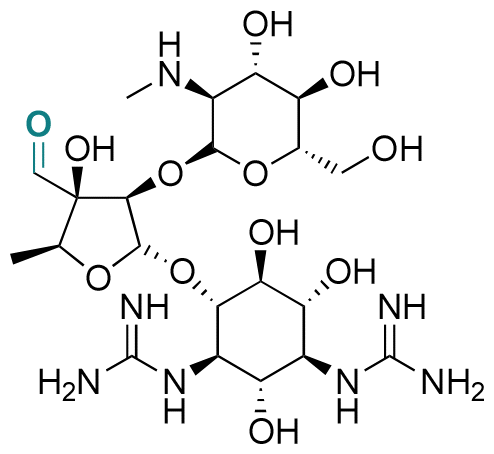
|
Aldehyde | Imine | 124 | ||
| Chromone-3-carboxaldehyde |
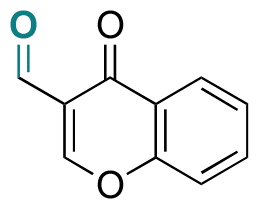
|
Aldehyde | Imine | 125 | ||
| Hydroxycitronellal |
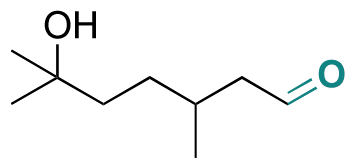
|
Aldehyde | Imine | 61 | ||
| Triplal |
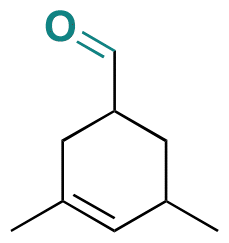
|
Aldehyde | Imine | 61 | ||
| Hexylcinnamic aldehyde |
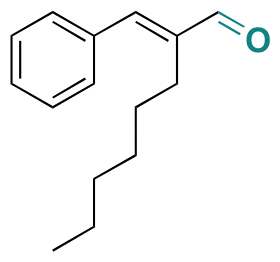
|
Aldehyde | Imine | 126 | ||
| (Z)-4-Dodecenal |
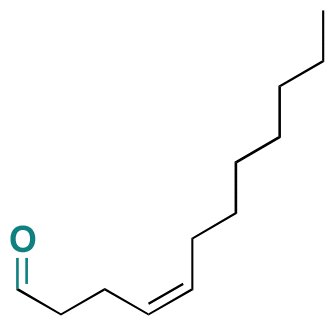
|
Aldehyde | Imine | 126 |
The rate and yield of imine formation depends on the temperature, pH, and concentration of active agents and the substrate.45 Moreover, steric hindrance and substituent effects at both the carbonyl and amine compounds can affect the kinetics of imine formation. A slower rate of imine formation by active agents containing ketones compared to aldehydes has been observed, attributed to the larger steric hindrance in the former molecules.46 Imine formation can be accelerated in the presence of organic base catalysts as a result of a reduction of carbinolamine conversion.47 Sayer and coworkers conjugated streptomycin, bearing a bulky aldehyde, with amine groups of a cationic polymer in the presence of diethylenetriamine as a catalyst.47 The cationic polymer was prepared by the copolymerization of cysteine dibenzyl and hexamethylene diisocyanate, which was further reacted with 4-aminobenzylamine. After the reaction of streptomycin with the dissolved cationic polymer, the polymer contained ∼0.15 streptomycin per polymer repeating unit, corresponding to a 10% drug-loading content.
Mechanochemical conjugation is an alternative synthesis method to overcome the limitation of linkage formation due to incompatibility of the solubility between the active agents and substrates.48 Ethoxyethyl glycidyl ether (EEGE) and azidohexyl glycidyl ether (AHGE) were polymerized via an anionic ring-opening polymerization to obtain poly(EEGE-block-AHGE). Subsequently, azide moieties on the PAHGE block were converted to primary amines by the Staudinger reduction reaction, which were then reacted with cinnamaldehyde.
Fragrances are active substances that are exploited in cosmetics, food, and healthcare products; however, they are susceptible to fast evaporation due to their volatility. Thus, volatile active agents containing aldehyde or ketone were conjugated via various hydrolyzable labile moieties, such as imine,49–52 thioether,53,54 ester, and acetals,55–59 on polymers to prolong their long-term storage and allow their controlled release. Thus, fragrances containing aldehyde groups, such as vanillin, cinnamaldehyde, citral, citronellal, cyclamen aldehyde, β-ionone, helional, and p-anisaldehyde, were reacted with substrates containing amine groups, such as polymers or surfactants, to form Schiff bases.60,61 Up to 80% fragrance was released within 2 weeks in acidic aqueous conditions, while 40% was released in neutral conditions.
Hydrazone is another type of linkage produced by Schiff base reaction and possesses a higher hydrolytic stability than imine linkages, which can allow a reduction in the unwanted release of the active agents from substrates in neutral conditions.18,33,62 In one study, the reversible properties of hydrazine bonds allowed the controlled release of highly aldehyde- or ketone-bearing volatile fragrances due to a dynamic equilibrium between the carbonyl compounds and hydrazine substrates.63 The mechanism of hydrazone formation is provided in Fig. 13.64,65 Generally, hydrazone linkages obtained from ketones have higher stability than from aldehydes.66 The fastest rate of hydrazone formation was observed in acidic conditions (pH ∼ 4.5) in the presence of acid catalysts, where carbonyl carbon was protonated but hydrazine was not yet protonated,67 whereas the rate of hydrazone formation was found to be limited in neural conditions (pH ∼ 7.4).68
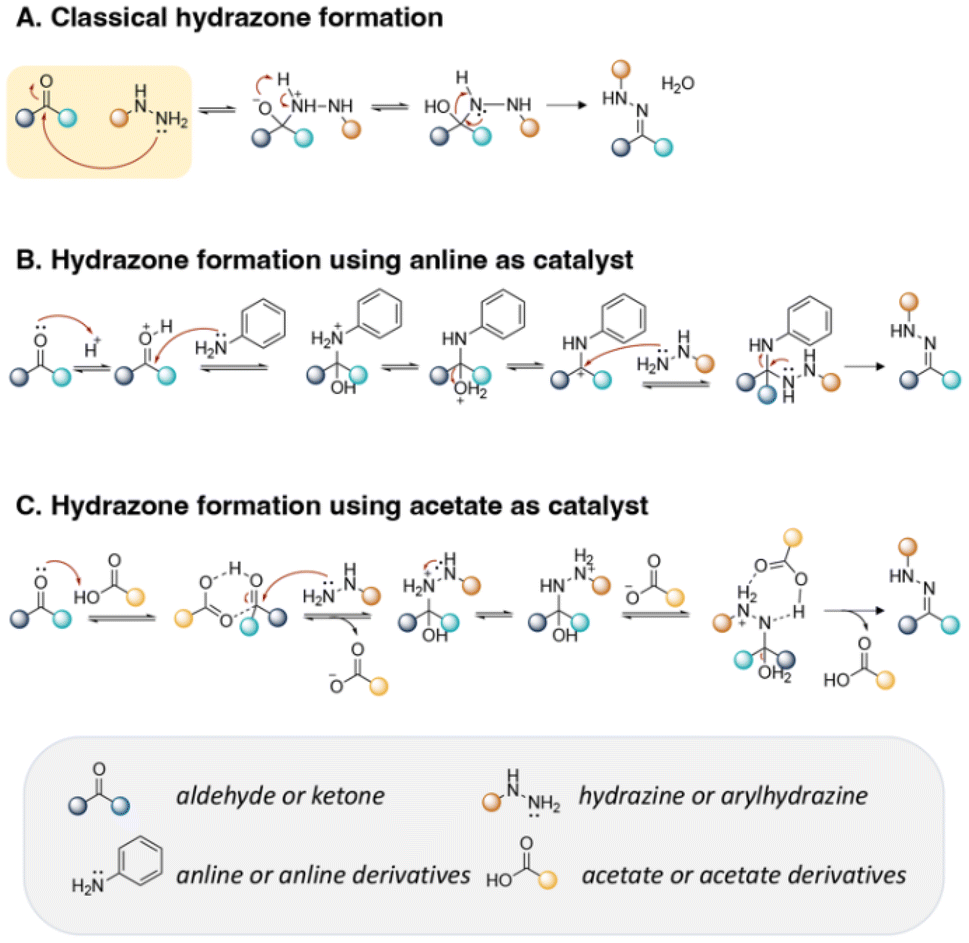 | ||
| Fig. 13 Mechanism of hydrazone formation (A) in the presence of aniline derivatives (B) or acetate derivatives (C). | ||
Recently, aldehyde and ketone fragrances were conjugated to polymers in coatings for releasing the fragrances in the presence of human sweat.17 Benzaldehyde and heptanal were reacted with methacryloyl hydrazide to form pH-responsive fragrance monomers, namely (E)-N′-benzylidene methacrylohydrazine (BMH) and (E)-N′-heptylidenemethacrylohydrazide (HMH). A photocurable urethane acrylate (UA) resin was copolymerized with BMH or/and HMH to produce P(UA-co-BMH), P(UA-co-HMH), or P(UA-co-BMH-HMH). The release of benzaldehyde was detected at 863 ppm in artificial sweat at pH 4.5 and at 600 ppm in buffer solution at pH 7.4 after 7 weeks, indicating the acidity of artificial sweat accelerated the hydrolysis of the hydrazone linkages.
Kool and coworkers investigated the kinetics of hydrazone formation by reacting 50 structurally different aryl- and alkyl-aldehydes and ketones with phenyl-hydrazines, (see Fig. 14).127 Moreover, it was found that butyraldehyde (1) formed a hydrazone bond 65 times more rapidly than benzaldehyde. The faster reaction of alkyl-carbonyls compared to bulky- or aryl-carbonyls was attributed to the lower steric hindrance effects for forming the tetrahedral intermediate. Besides, the lesser reactivity of ketones over aldehydes was also due to the presence of larger steric hindrance provided by there being more alkyl groups nearby the carbonyl groups of ketones than aldehydes. Indeed, the reactivity rate of butyraldehyde with hydrazine was 44 times larger than the reactivity rate of 2-butanone with the same hydrazine. Also, the reactivity of 2-butanone for hydrazone formation was lower than the reactivity of di-isopropyl ketone and di-tert-butyl ketone, which was attributed to steric hindrance effects.
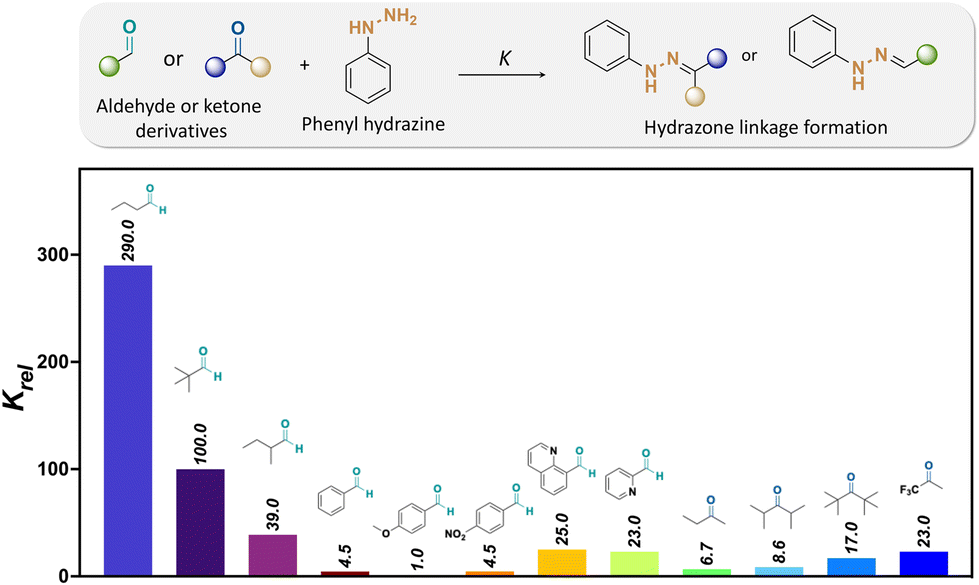 | ||
| Fig. 14 Relative formation rates (Krel) of hydrazones from a selected set of aldehydes and ketones at pH 7.127 The relative reaction rates refer to the hydrazone formation between either aldehyde or ketones and phenylhydrazine [Krel(4-methoxybenzaldehyde) = 1].67 | ||
The influence of the aldehyde or ketone containing electron-deficient groups on hydrazone formation was also investigated. The reaction rate of 4-nitrobenzaldehyde with hydrazine was 4.5 times faster than with 4-methoxybezaldehye. Moreover, the rate of hydrazone formation from trifluoracetone as a reagent was 3.4 times larger than the rate of hydrazone formation from 2-butaone, which was attributed to the increased electron-drawing effects at the carbonyl group of aldehydes and ketones. Besides, the rate of hydrazone formation by aldehydes containing basic groups, such as 2-formylpyridine and 8-formylquinoline, was increased up to 8 times compared to that with aldehydes without basic groups as a result of internal acid–base catalysis at the reaction transition state.
Trifluoracetic acid (TFA) is a suitable catalyst to form hydrazone bonds because H-bonding between the aldehyde/ketone of active agents and carboxylic residue of TFA allow an increase in the nucleophilic character at the carbonyl group of the aldehyde/ketone and removal of the nitrogen and oxygen protecting groups of the hydrazine reagents, inducing an accelerated rate of hydrazone formation.128 Effective bifunctional catalysts consisting of a nucleophilic amine and an anthranilic acid in their structures were also employed as catalysts to accelerate the conjugation reaction of active agents containing an aldehyde or ketone at pH 5–9 (Fig. 13B and C).129,130,131–133
Cellular alkyl-aldehydes acting as carcinogens in cardiovascular and lung diseases134–136 were detected through their conjugation with the fluorescein dye (7-(diethylamino)courmarin) via hydrazone linkages in the presence of series of arylamine catalysts. The rate of linkages formation in the presence of 1,4-phenylenediamine as a catalyst was increased up to 5 times compared with the rate of linkages formation in the presence of aniline as a catalyst.137 The conjugation of 7-(diethylamino)courmarin with alkyl-aldehydes present in mammalian cells (acetaldehyde, acrolein, formaldehyde, and glycolaldehyde) via hydrazone linkages led to a fluorescent signal at 477 nm, which allowed direct monitoring of the relative levels of intracellular aldehydes. The amounts of ketone/aldehyde molecules conjugated with substrates containing hydrazine though hydrazone linkages could be quantified by reacting fluorescent dyes with the remaining hydrazine groups of the substrates after their conjugation. The hydrazine-precursor was synthesized by reacting the amine group of 8-arm poly(ethylene glycol)-amine (PEG-NH2) with the carboxylic groups of tri-tert-butyl N-(carboxymethyl)hydrazine tricarboxylate.114 The tert-butyl carbazate (boc) groups were then removed in the presence of TFA in DCM to produce poly(ethylene glycol)-hydrazine (PEG-HY). The ketone group of dexamethasone (DEX), an anti-inflammatory corticosteroid and osteogenic inducer, was covalently linked to a hydrazine group of PEG-HY though hydrazone linkages to form PEG-DEX. Then, the PEG-DEX was crosslinked with oxidized pullulan to obtain a DEX-conjugated hydrogel via hydrazone linkages. 2,4,6-Trinitrobenzene sulfonic acid (TNBS) was complexed with free hydrazine groups in PEG-DEX and PEG-HY to investigate the amount of conjugated DEX in the hydrogel. Specifically, the amount of conjugated DEX on PEG-NH2via hydrazone linkages was determined by measuring the reduction of fluorescence intensity of PEG-NH2 using a 2,4,6-trinitrobenzene sulfonic acid assay, which indicated the presence of ∼14 wt% of DEX in the PEG-DEX conjugates. The release rate of DEX from the hydrogel at pH 6.5 was larger than the release rate of DEX from the hydrogel at pH 7.4. Overall, the released DEX from the hydrogel induced osteoblastic differentiation, leading to an increase in cell viability and proliferation, resulting in bone regeneration.
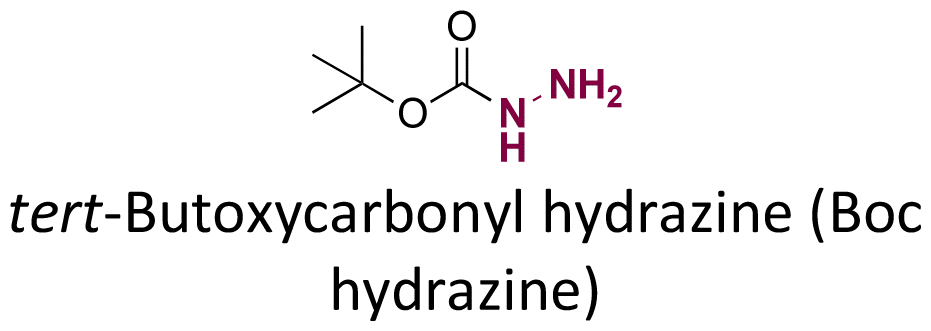
Doxorubicin (DOX), an anticancer agent, can induce cardiotoxicity at treatment dosages >400–700 mg m−2. To address this problem, hydrazone-based conjugation chemistry was used for inserting acid-labile linkages by the reaction between the ketone group of DOX in the C-13 position with the hydrazine groups of selected molecules and polymers (Fig. 15). Methotrexate (MTX), a hydrophilic folic acid antagonist, was reacted with tert-butoxycarbonyl hydrazine with a carbonyl group of MTX to form MTX-hydrazine. Then, DOX was attached to the MTX-hydrazine via a hydrazone linkage to form an amphiphilic prodrug assembly in micelles. It was found that 87% of the DOX was released from MTX-DOX nanoparticles after incubating the nanoparticles for 72 h at pH 5.0.138
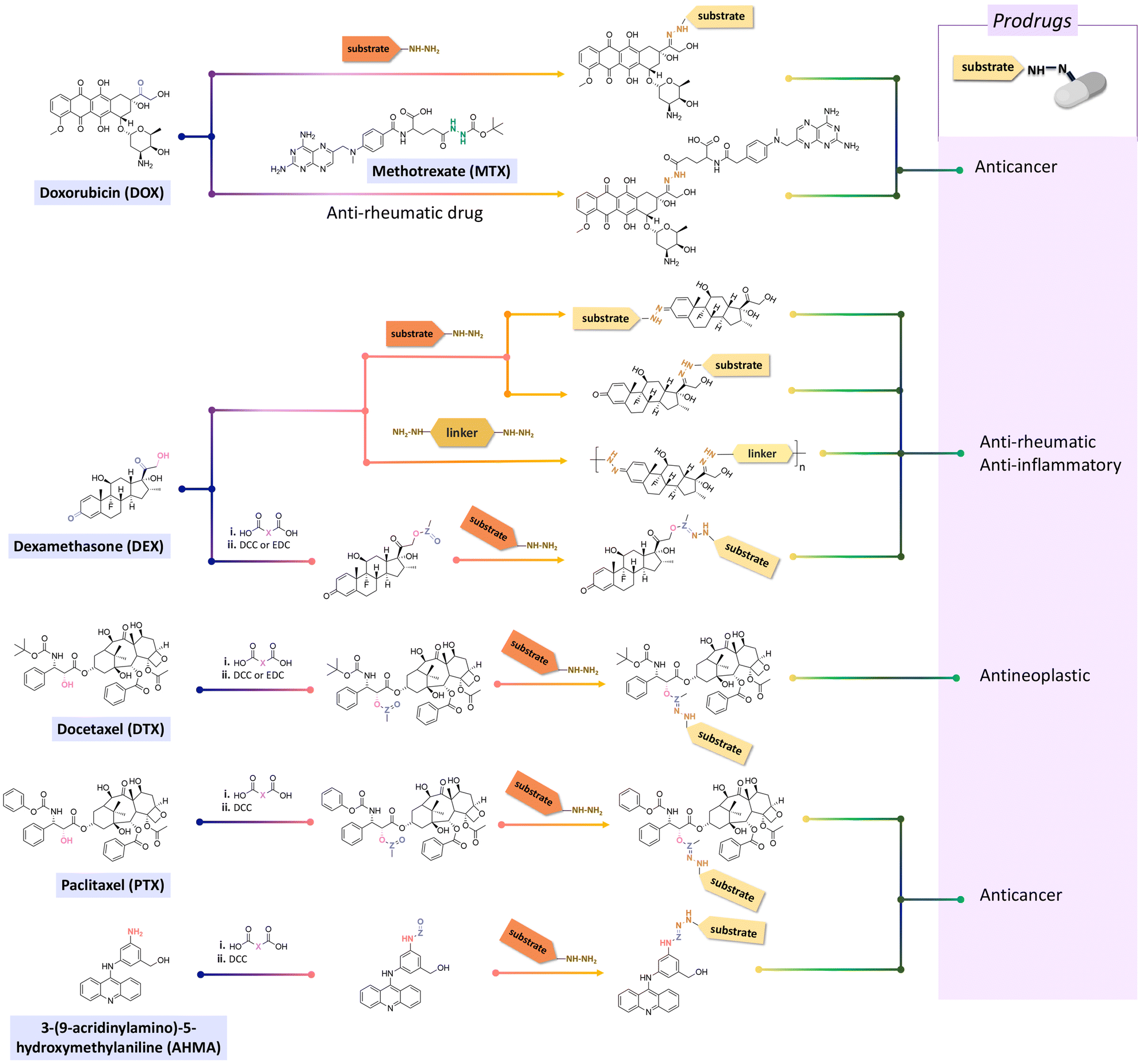 | ||
| Fig. 15 Chemical structure and scheme for modifying active agents bearing ketone groups to produce hydrazone derivatives. Anticancer agents (doxorubicin, methotrexate, paclitaxel and 3-(9-acridinylamino)-5-hydroxymethylaniline), anti-rheumatic and anti-inflammatory agents (dexamethasone), or antineoplastic agents (docetaxel) can be directly reacted with substrates containing hydrazine groups or via aldehyde-based spacers. | ||
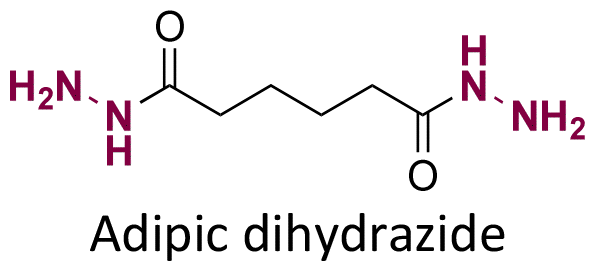
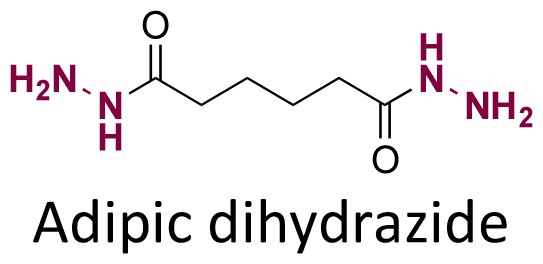
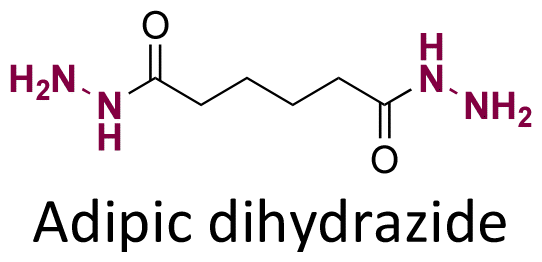
Both ketone groups of DEX were reacted with adipic acid dihydrazine to prepare a polyprodrug (PDEX) containing pH-cleavable hydrazone bonds in the polymer backbone.115 PDEX with number average molecular weights ranging from 2kDa to 133 kDa were synthesized by varying the reaction time, temperature, and concentration of catalyst in the reaction. The PDEXs were then encapsulated in mesoporous silica nanocapsules, which could then be used to reduce the inflammatory effect in chronic diseases.
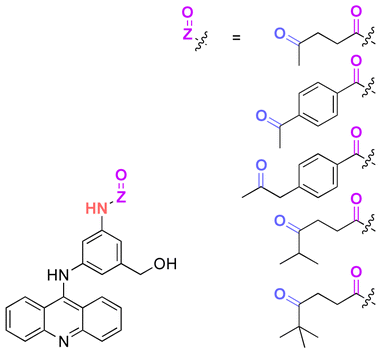
Some active agents containing an aldehyde or ketone cannot be directly conjugated with substrates via imine or hydrazone linkages due to their limited reactivity. Thus, some additional spacers are sometimes required to obtain derivatives of active agents that are then conjugated with substrates. Thus, oxo-acids (e.g. levulinic acid, 4-(2-oxopropyl)benzoic acid and 4-oxopentanoic acid) were selected as spacers to react with the hydroxy groups of DEX, paclitaxel (PTX), docetaxel (DTX), or hexyl aminolevulinate (HAL) by an esterification reaction using N,N′-dicyclohexylcarbodiimide (DCC) or 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and as coupling agents for forming drug derivatives containing ketone groups.116,139,140 Then, the ketone groups of the derivatives were reacted with the side chains of polymers containing hydrazide groups. The amine groups of 3-(9-acridinylamino)-5-hydroxymethylaniline, an anticancer drug, were reacted with thiazolidine-2-thione-activated carboxylic acids in an acylation reaction to introduce carbonyl groups, which were conjugated to polymers containing hydrazine side-chains.120
3.2 Active agents containing amines and hydrazine groups
The influence of the amine structure on imine formation was quantified by reacting amines with 2-formaylbenzesulfonic acid (Fig. 16) by 1H NMR spectroscopy in neutral and alkaline media. The equilibrium constant (K) of the reaction increased with increasing the basicity of the amines.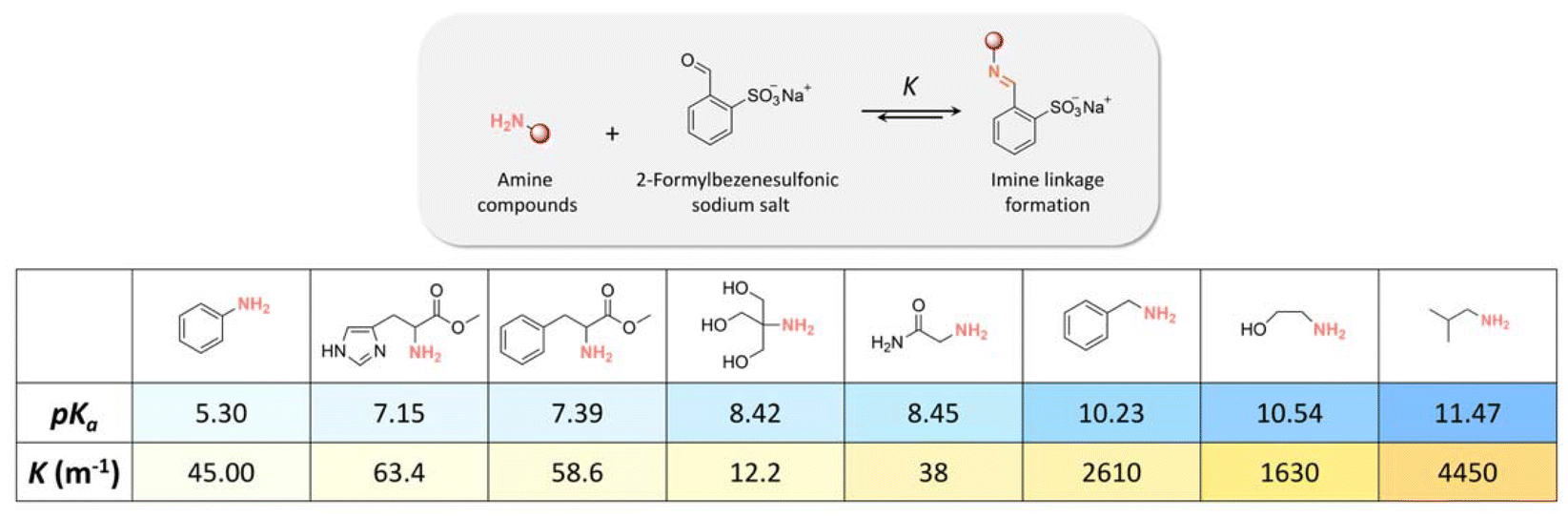 | ||
| Fig. 16 pKa of amine compounds and equilibrium constants (K, m−1) of imine linkage formation via the condensation reaction of amine compounds with 2-formylbenzenesulfonate. | ||
Twenty different hydrazine derivatives were reacted with 2-formylpyridine as a chromophore for quantitative measurement of the hydrazone formation rate by UV–spectroscopy (Fig. 17).141 The slowest relative rates (Krel) of hydrazone formation were observed for the reactions involving fluorophenylhydrazine or diphenylhydrazine due to the influences of their electron-deficient groups. Notably, the larger Krel of hydrazone formation with ortho-carboxyphenylhydrazine and 2-(dimethylamino) ethylhydrazine were attributed to the acid/base groups on the hydrazines, which catalyzed the reaction up to 23 or 13 times faster in comparison with hydrazone formation with fluorophehylhydrazine. Also, compounds with methoxy- or methyl substituted on arylhydrazines exhibited a faster rate of hydrazone formation than with fluorophenylhydrazine or diphenylhydrazine. Electron-poor arylhydrazines reacted slower than electron-rich arylhydrazines due to their lower nucleophilicity. The lower Krel for hydrazone formation was due to the lower inductive effect from the electron-withdrawing substituent groups on the hydrazine reagents.
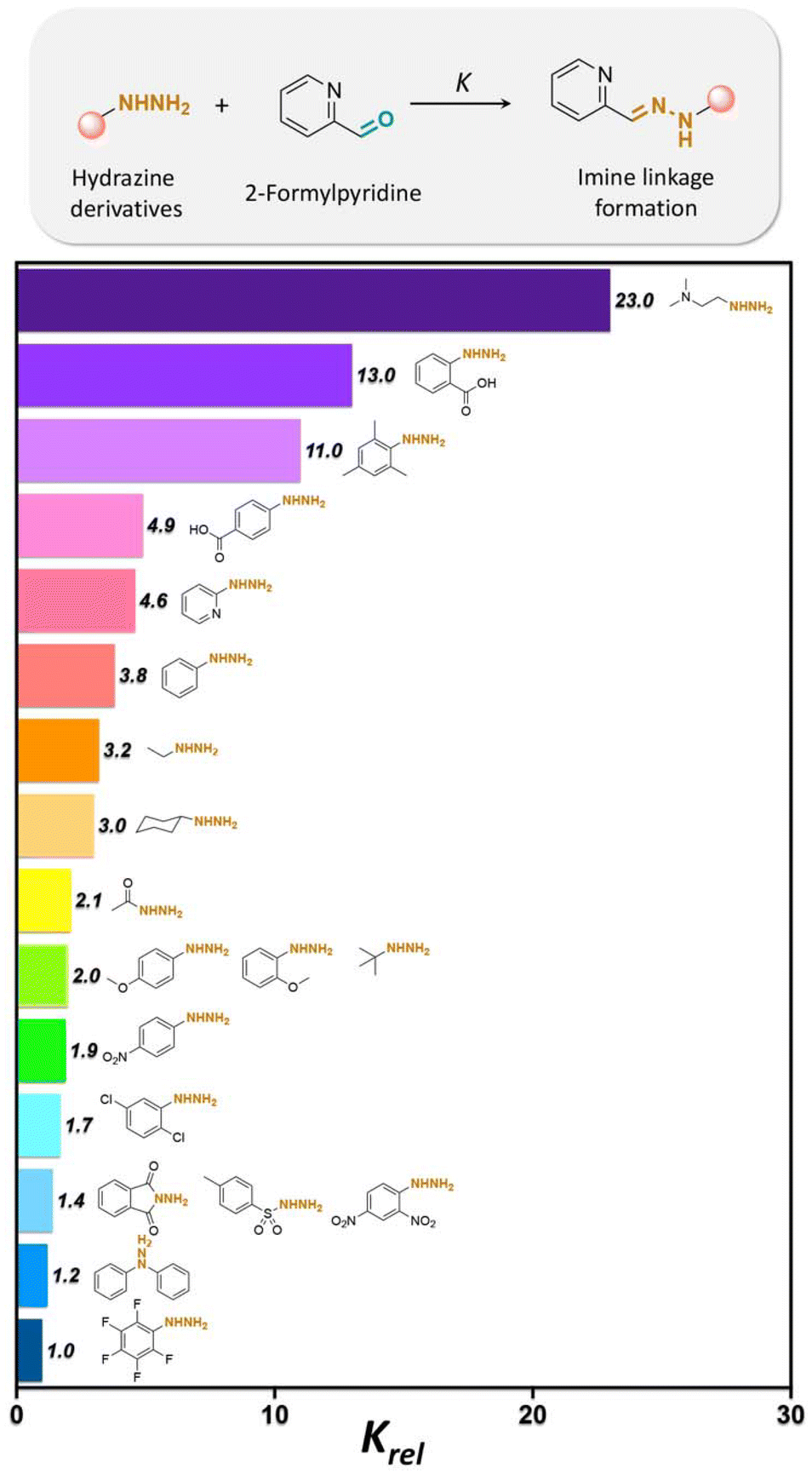 | ||
| Fig. 17 Relative formation rates (Krel) of hydrazones from a selected set of hydrazines at pH 7.4. Krel is defined as the ratio between the rate of hydrazone formation of hydrazines with 2-formylpyridine over the rate of hydrazone formation of fluorophenylhydrazine with 2-formylpyridine.141 | ||
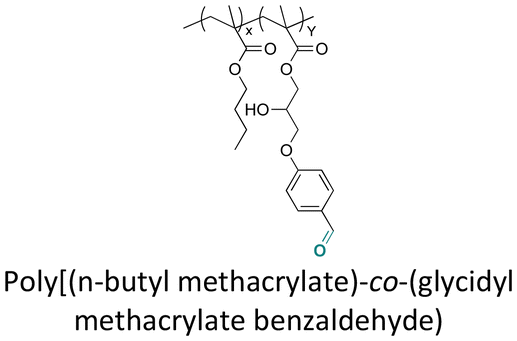
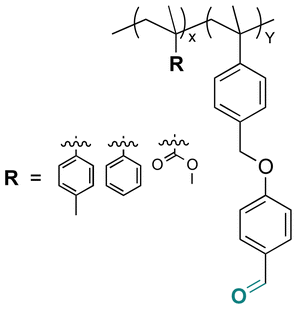
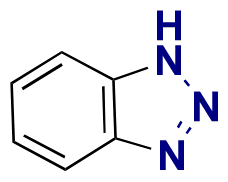
Some examples of active agents bearing amine and hydrazine groups, and their derivatives, as well as the reagents used for preparing imine and hydrazone linkages are listed in Table 7. Tryptamine, benzotriazole-5-carbonydrazide (BTA-hydrazine), and 5-aminomethyl-8-hydroxyquinoline (AM8HQ), which are corrosion inhibitors containing –NH2 groups, were recently conjugated with copolymers bearing pendent aldehyde units via imine or hydrazone linkages (Fig. 18A and B).142,143 Polymer-inhibitors containing 13–19 wt% conjugated inhibitors were further processed as nanoparticles via a mini-emulsion–solvent evaporation method.10,144 It was found that 100% BTA-hydrazine was released at pH 3.5, while 55% was released at pH 7.0 after 30 min.143 Besides, the faster release rate of tryptamine compared with AM8HQ from similar linkages was attributed to the larger basicity of tryptamine (pKa ∼ 10.2) than of AM8HQ (pKa ∼ 9.0).
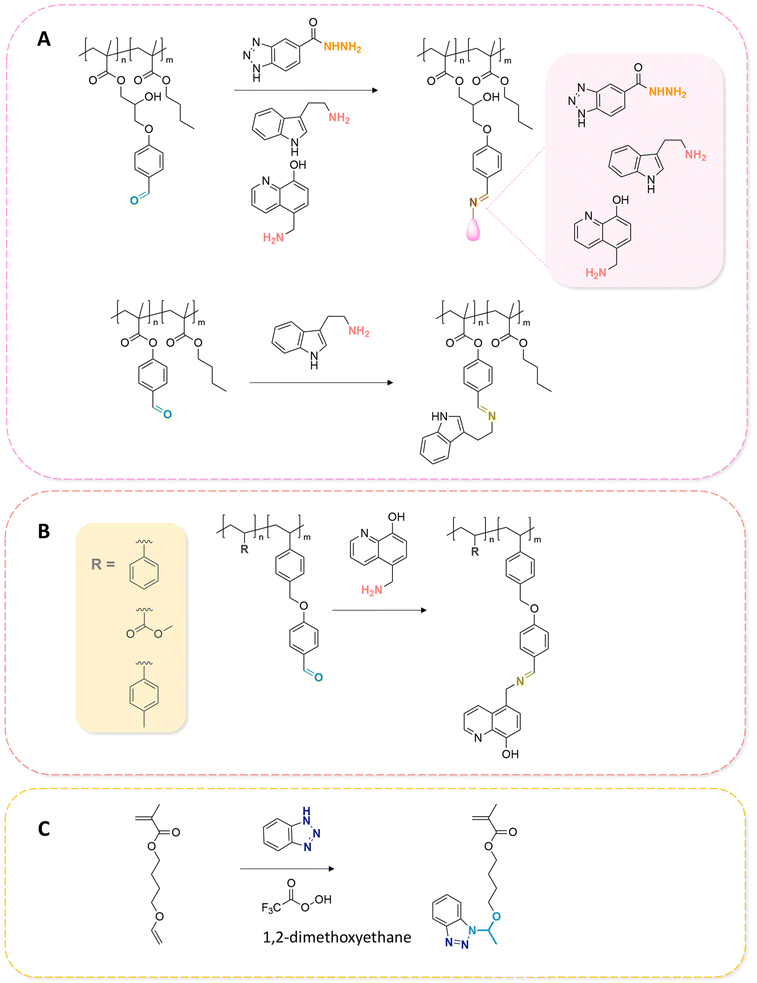 | ||
| Fig. 18 Schematic description of the synthesis of (A) aldehyde-containing polymethacrylates conjugated with primary amine- or hydrazine-containing corrosion inhibitors;143 (B) 4-((4-formylphenyl)oxymethyl)styrene and its copolymerization with styrene, 4-methylstyrene or methyl methacrylate and conjugation of the corresponding polymers with 5-(aminomethyl)-8-hydroxyquinoline;142 (C) preparation of 4-(1-(benzotriazole-N-yl)ethyoxy)butyl methacrylate, a monomer with a hemiaminal ether linkage.145 | ||
| Active agents | Chemical structure of the active agent | Derivative | Active functional group | Linkages | Reagents for linkage formation | Ref. |
|---|---|---|---|---|---|---|
| Vancomycin |
|
|
Hydrazine | Hydrazone |

|
146 |
| Tryptamine |
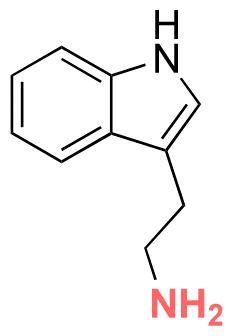
|
Amine | Imine |
|
143 | |
| 8-Hydroxyquinoline (8HQ) |
|
|
Amine | Imine |
|
143 and 147 |
|
142 | |||||
| Benzotriazole |
|
Triazole | Hemiaminal ether |
|
145 and 147 | |
| Cy5-hydrazine |
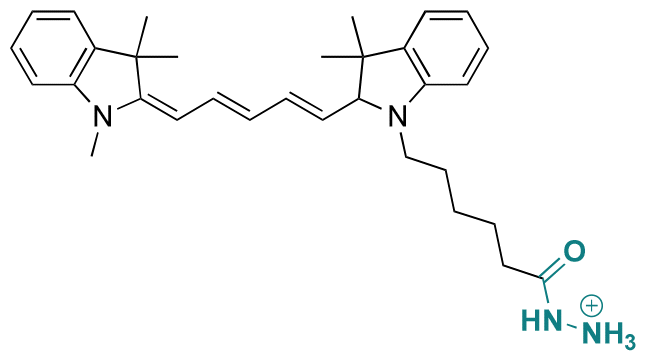
|
Hydrazine | Hydrazone | 148 |
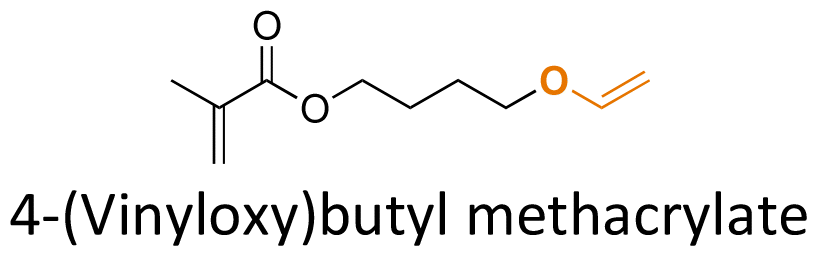
Hemiaminal ether linkages have been investigated as new mild acid-sensitive linkages for conjugating drugs or other active agents containing secondary amine groups,149,150 and drug derivatives of imidazoles,151,152 tetrazoles,145,153 and indoles154 to polymer chains. The synthesis mechanism of hemiaminal ether is provided in Fig. 19. Polymerizable benzotriazole, 4-(1-(benzotriazole-N-yl)ethyoxy)butyl methacrylate (BEBMA) was prepared by the addition of benzotriazole (BTA) to 4-(vinyloxy)butyl methacrylate in the presence of trifluoracetic acid (TFA) as a catalyst (Fig. 13C).145 The amounts of BTA released from nanoparticles of the copolymers were 20%–30% at pH 3.5 and 7%–9% at pH 5.0 after 11 days. On the contrary, no BTA was released from the nanoparticles after 3 weeks at pH 7.0.
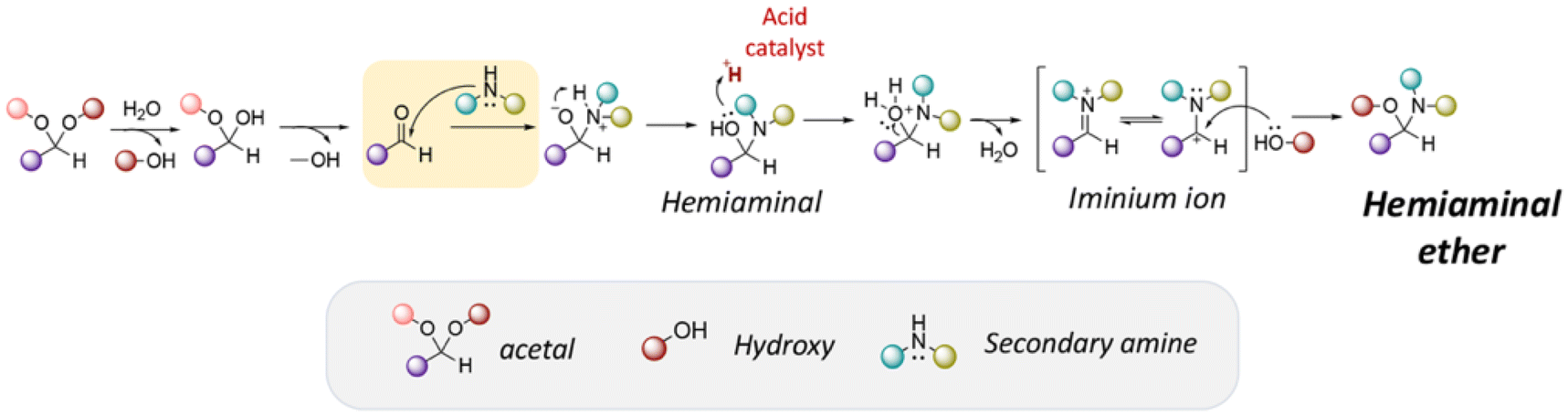 | ||
| Fig. 19 Mechanism of hemiaminal ether formation. | ||
3.3 Active agents containing hydroxyl groups
Active agents bearing hydroxy groups have been widely conjugated to substrates via acetal,155–159 orthoester,160,161 and thiopropionate linkages,162–165 allowing a controlled release of the active agent at desirable pH value at targeting areas. The general mechanism of acetal bond formation is provided in Fig. 20. Some examples of active agents bearing hydroxyl groups, and their derivatives, as well as the reagents used for preparing acetal, orthoester, and β-thiopropionate linkages are listed in Table 8.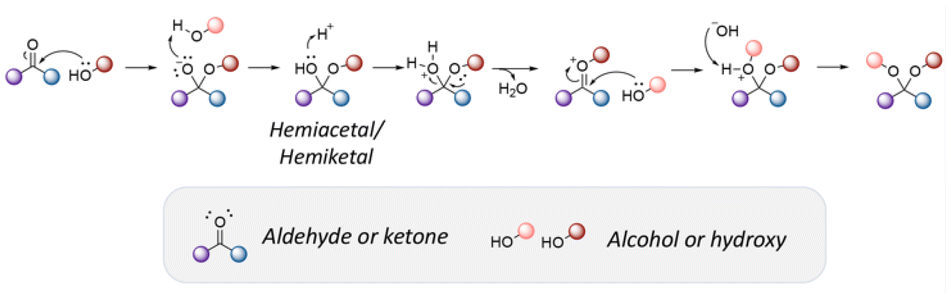 | ||
| Fig. 20 Mechanism of acetal formation via the nucleophilic addition of alcohols to aldehyde. | ||
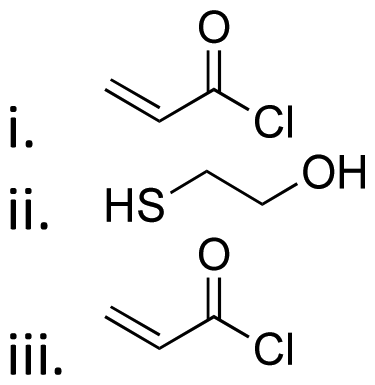
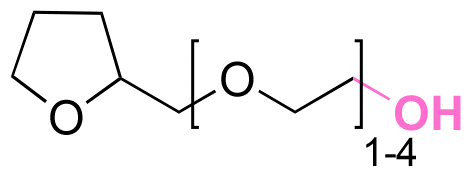
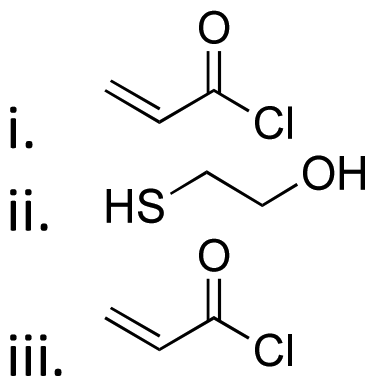
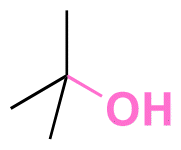
| Active agents | Chemical structure of the active agent | Derivative | Active functional group | Linkages | Reagents for linkage formation | Ref. |
|---|---|---|---|---|---|---|
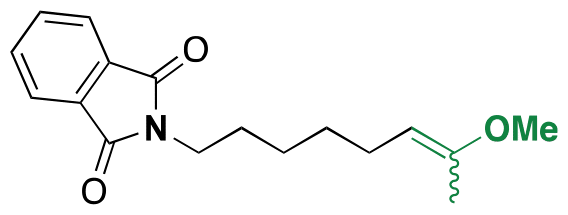
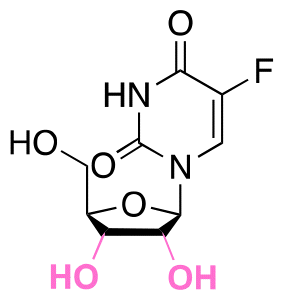
| 5-Fluro-2′-dexoyuridine |
|
Hydroxyl | Acetal |
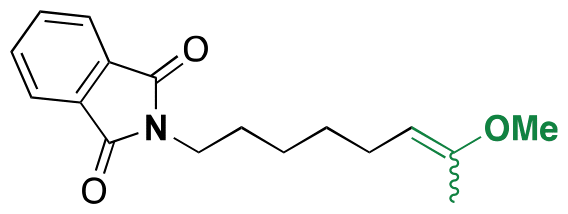
|
155 | |
| Estradiol |
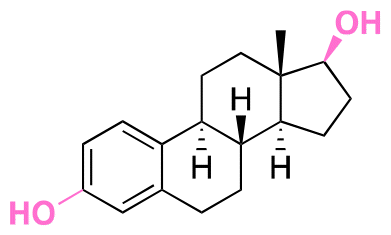
|
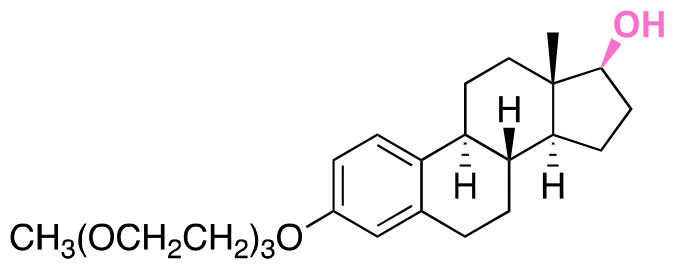
|
Hydroxyl | Acetal |
|
155 |
| 5-Fluorouridine |
|
Hydroxyl | Acetal |
|
155 | |
| Lumefantrine |
|
Hydroxyl | Acetal |
|
159 | |
| SN38 |
|
|
Hydroxyl | Acetal |
|
157 |
| Combretastatin A-4 |
|
Hydroxyl | Aromatic orthoester |
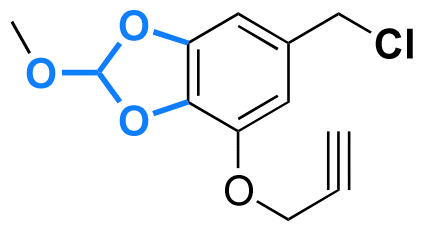
|
161 | |
| Docetaxel (DTX) |
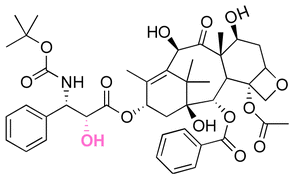
|
Hydroxyl | Thioether ester |
|
165 | |
| Curcumin |
|
|
Hydroxyl | Acetal |
|
166 |
| Dasatinib |
|
Hydroxyl | 5-Membered ring orthoester |
|
167 | |
| Cholesterol |
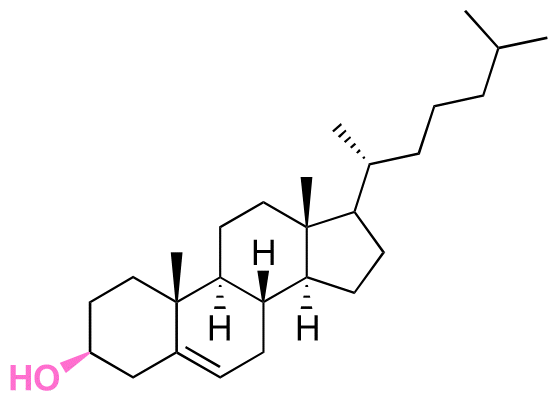
|
Hydroxyl | 5-Membered ring orthoester |
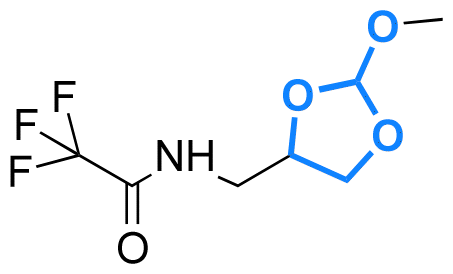
|
160 | |
| (3-Hydroxypropyl)-cholesteryl carbamate |
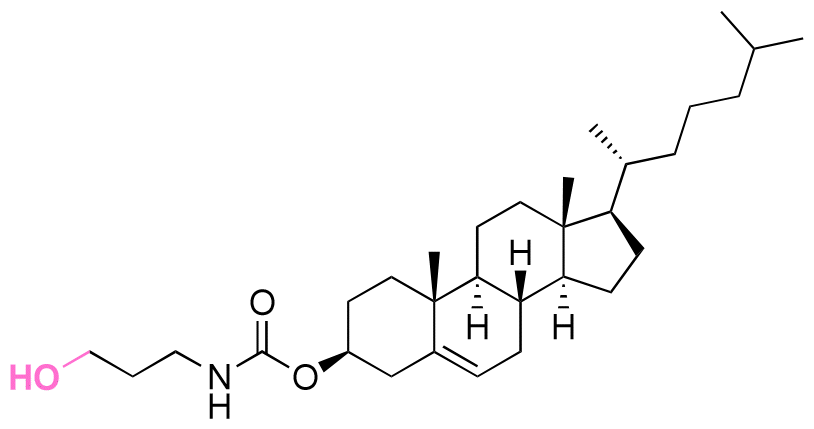
|
Hydroxyl | 6-Membered ring orthoester |
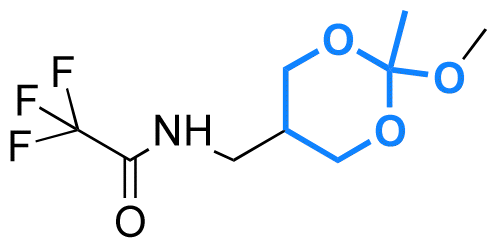
|
160 | |
| 8-Hydroxyquinoline (8HQ) |
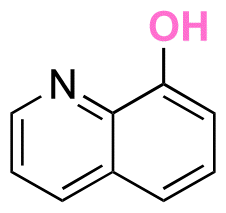
|
Hydroxyl | Thioether ester |
|
42 and 168–173 | |
| 1-Pyrenemethanol |
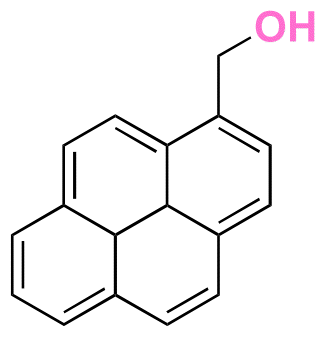
|
Hydroxyl | Thioether ester |
|
147 | |
| Camptothecin |
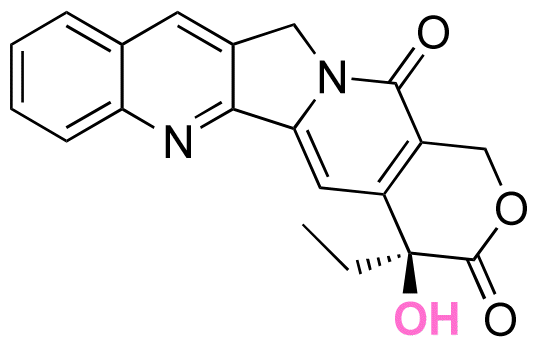
|
Hydroxyl | Thioether ester |
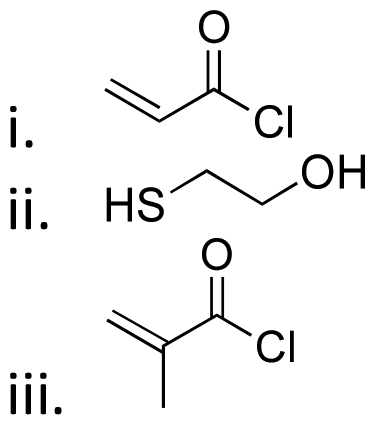
|
16 and 163 | |
| Cyrene |
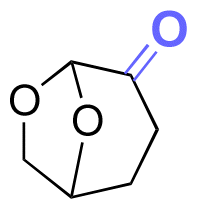
|
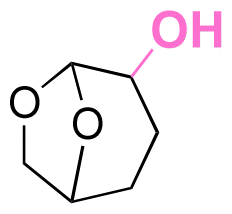
|
Hydroxyl | Thioether ester |
|
174 |
| Tetraglycol |
|
Hydroxyl | Thioether ester |
|
174 | |
| tert-Butanol |
|
Hydroxyl | Thioether ester |
|
174 |
Precursors for acetal or cyclic acetal linkages were synthesized by converting the aldehyde group of 6-bromohexanal to an enol ether by a Witting reaction using 1-methoxyethyl-1-triphenylphosphonium salt or methoxymethyl-triphenyl phosphonium as well as by reacting 4-hydroxy-2,6-dimethoxybenz aldehyde with 1,8-dibromooctane.155 Then, the bromide group of functionalized 6-bromohexanol and 1,8 dibromooctane were reacted with potassium phthalimide to provide I-F21, IV-F21, or VII-F21.155 The enol group of I-F21 or IV-F21 was subsequently reacted with a primary alcohol (5-fluoro-2′-deoxyuridine) or a secondary alcohol (estradiol derivative) to form acetal linkages in compounds II and V. The syn-1,2 diol of 5-fluorouridine was converted to a cyclic acetal by reaction with the aldehyde group of VII-F21 to form VIII-F21. The phthalimide groups of II-F21, V-F21, or VII-F21 were reacted with hydrazine to obtain amine groups, which were further reacted with the activated carbonate of poly(ethylene glycol) to provide III-F21, VI-F21, or VIII-F21 (Fig. 21A–C). 5-Fluoro-2′-deoxyuridine was released from III-F21 more significantly than estradiol was released from VI-F21 after immersing the polymers at pH 5.0 for 20 min, due to the lower inductive effects of the acetal linkages in VI-F21. Indeed, the slower release rate of 5-fluorouridine from VIII-F21 compared with the release rate of 5-fluoro-2′-deoxyuridine from III at pH 5.0 was attributed to torsional strain associated with the 5-membered ring of cyclic acetal in VIII-F21. After immersion at pH 7.4 for 1 h, less than 10% 5-fluoro-2′-deoxyuridine, estradiol, and 5-fluorouridine were released from III-F21, VI-F21, and VIII-F21, respectively.
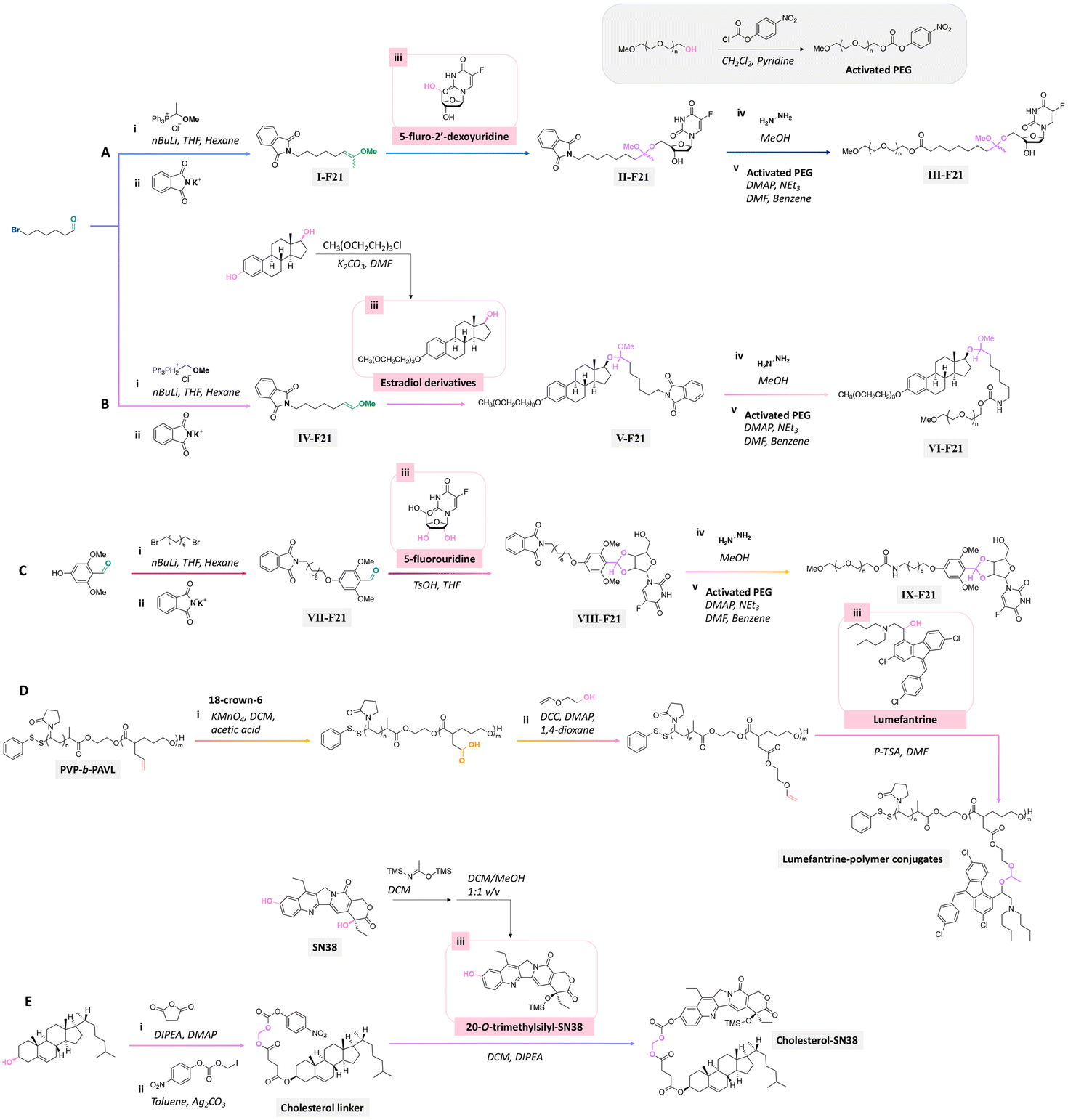 | ||
| Fig. 21 Scheme of the preparation of precursors using (A and B) 6-bromohexanal, (C) 4-hydroxy-2,6-dimethoxybenzaldehyde,155 (D) poly(N-vinylpyrrolidone)-block-poly(α-allylvalerolactone) (PVP-b-PAVL),159 and (E) cholesterol157,158 as reagents for further conjugation with either the primary alcohol group of (A) 5-fluoro-2′-deoxyuridine, secondary alcohol group of (B) estradiol derivative or (D) lumefantrine, or syn-1,2 diol group of (C) 5-fluorouridine, or tertiary alcohol group of (E) 7-ethyl-10-hydroxycamptothecin (SN38) through the formation of acetal or cyclic acetal bonds. | ||
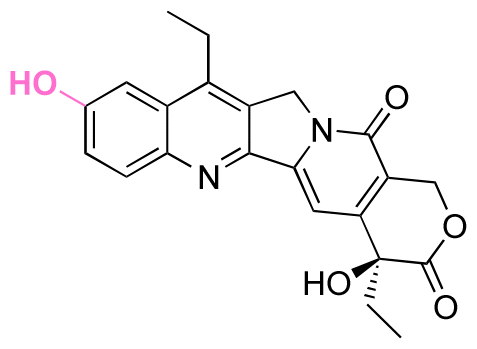
Lumefantrine, an antimalarial agent against intraerythrocytic P. falciparum parasites, was conjugated with poly(N-vinylpyrrolidone)-block-poly(α-allylvalerolactone) (PVP-b-PAVL) via acetal linkages (Fig. 21D).159 First, the allyl groups in PVP-b-PVAL were oxidized to carboxylic acid groups in a biphasic medium composed of DCM and acetic acid with KMnO4 in the presence of 18-crown-6. The carboxylic groups were further reacted with ethylene glycol vinyl ether by a Steglich esterification to obtain functionalized PVP-b-PAVL. Then, the hydroxyl group of lumefantrine was reacted with the vinyl ether group of functionalized PVP-b-PAVL to form acetal linkages. After incubating the polymer for 72 h at 37 °C, the amount of released lumefantrine from the polymer conjugates at pH 5.0 was 4 times larger than at pH 7.4. Also, the conjugation of cholesterol to 7-ethyl-10-hydroxycamptothecin (SN38, an antitumor agent) via acetal linkages was achieved by the reaction of a cholesterol linker containing acetal linkages with a phenol group of the SN-38 derivative (Fig. 21E).157,158 The cholesterol-SN38 prodrug was then loaded in to the cores of hydrophilic oxaliplatin nanoparticles. The growth of colorectal tumor in a mouse model was reduced by 6.0 and 4.9 times compared with the experimental results with free drugs.
The orthoester bond, another acid-labile bond, is widely used as a linkage for conjugating active agents containing hydroxyl groups to substrates, because they can be easily prepared by a nucleophilic substitution or Pinner reaction (see Fig. 22A).160
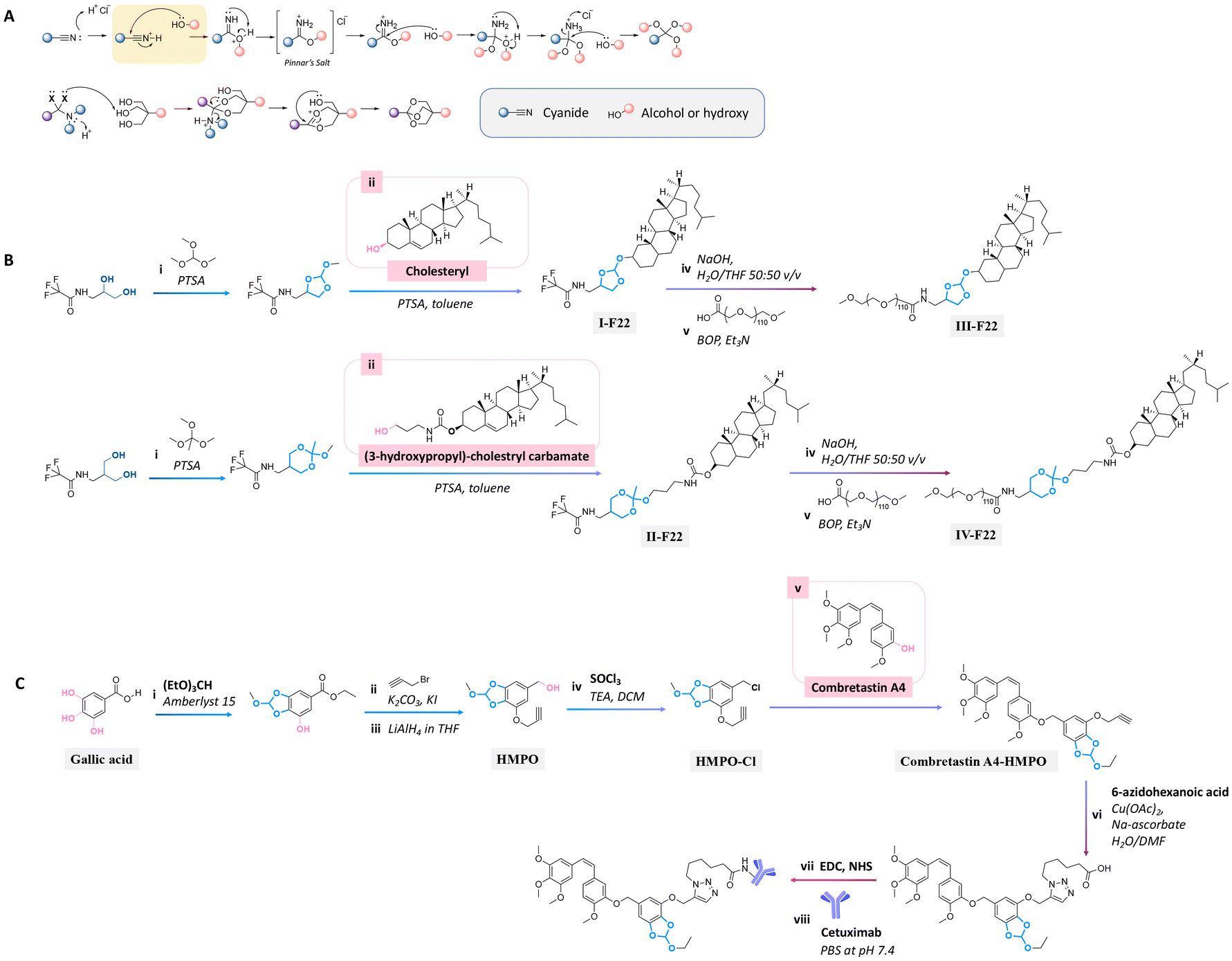 | ||
| Fig. 22 Mechanism of orthoester formation via a nucleophilic substitution or Pinner reaction (A) and schematic of the preparation of precursors containing orthoester bonds for conjugating with the secondary alcohol group of (B) cholesteryl or primary alcohol group of (3-hydroxypropyl)-cholestryl carbamate160 and (C) tertiary alcohol group of combretastatin A4,161 followed by reacting with (B) polyoxyethylene stearyl ether (monomer unit ∼ 100, PEG100) or (C) Cetuximab. | ||
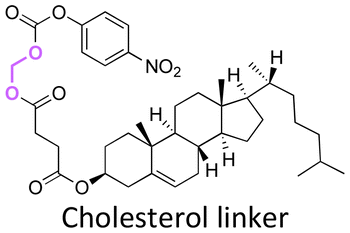
Cholesterol in lipid formulation induces an increase in transfection efficiency, leading to the effective delivery of prodrugs to tumors via endocytosis.158,175 Cholesterol conjugation to a polyoxyethylenestearyl ether (degree of polymerization ∼ 100, PEG100) via five-/six-membered orthoester linkages could be performed either by the reaction of N-protected 1-amino-propanediol with trimethyl orthoformate or N-protected serinol with trimethyl orthoacetate.160 These precursors were then reacted with the hydroxyl groups of cholesterol or (3-hydroxypropyl)-cholestryl carbamate to provide compounds I-F22 and II-F22 (Fig. 22B). The trifluoroacetate groups of I-F22 and II-F22 (Fig. 22B) were deprotected by sodium hydroxide in THF, and the products were subsequently reacted with the carboxylic group of α-carboethoxy-ω-methoxy-PEG in the presence of benzotriazole-1-yloxytris(dimethylamino) phosphonium hexa fluorophosphate (BOP) as a coupling agent to yield III-F22 and IV-F22. The hydrolysis rate at pH 4 of the six-membered ring orthoester in IV-F22 was 10 times faster than the hydrolysis rate of the five-membered ring orthoester in III-F22. The faster hydrolysis rate of IV-F22 was attributed to the ideal chair conformation of the six-membered ring, inducing a higher basicity of the exocyclic oxygen atoms.
Gallic acid, a natural tannin component containing three hydroxyl groups, was conjugated with an anticancer agent through orthoester linkages.161 Two hydroxy groups of gallic acid were reacted with triethyl orthoformate by an esterification reaction to form a five-membered ring orthoester. The free hydroxy group and ether group of gallic acid were reacted with propargyl bromide and LiAlH4 to obtain a 5-(hydroxymethyl) pyrogallol orthoester derivative (HMPO). Thionyl chloride was reacted with HMPO to obtain HMPO-Cl, which was then reacted with the hydroxyl group of combretastatin A4, an anticancer agent, to produce a combretastatin A4-HMPO prodrug (Fig. 22C). The prodrug was then conjugated with Cetuximab, a monoclonal antibody specific for the epidermal growth factor receptor, in the presence of N-hydroxysulfosuccinimide and 1-ethyl-3-(3-dimethyl aminopropyl)carbodiimide as coupling agents to form a prodrug conjugated with an antibody. The release of combretastatin from the antibody-prodrug decreased the cell viability of A431 and A549 cancer cells more than compared with the use of Cetuximab alone.
Thioether ester bonds are typically incorporated in materials for inducing a pH-responsive property. The mechanism of formation of thioether ester from active agents containing hydroxy groups is provided in Fig. 23A. Camptothecin, derivatives of 20-O-trimethysilliyl-SN38, paclitaxel, and docetaxel were conjugated on materials via thioether ester bonds by esterification followed by a Michael-addition reaction or esterification reaction with dithiodipropionic acid or thiodipropionic acid (Fig. 23B).162–165 Interestingly, the burst release of these anticancer drugs was not observed. After incubating the polymer conjugates for 7 days, the amounts of released drugs from the conjugates were 50% and 20% at pH 5.5 and 7.4.
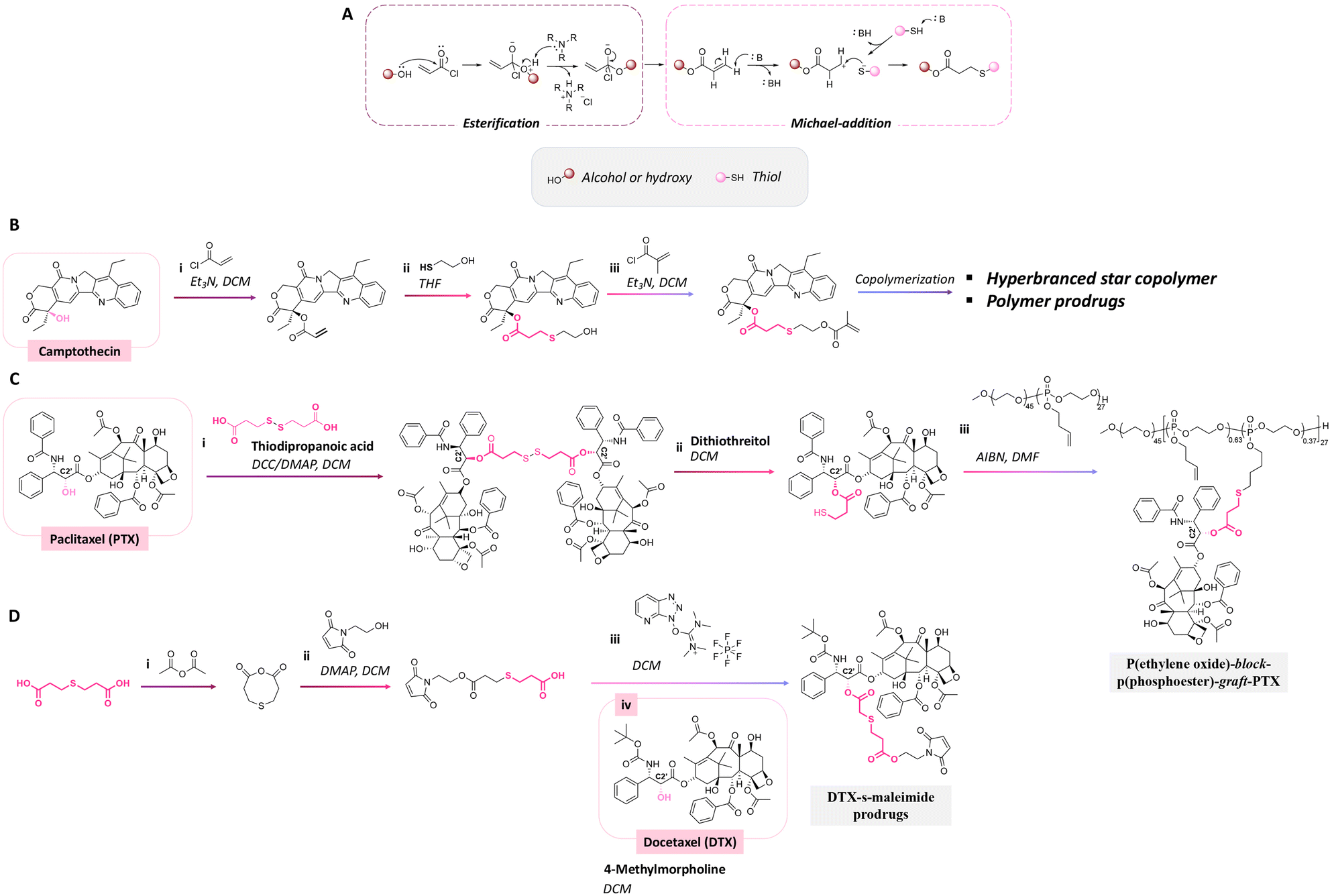 | ||
| Fig. 23 Mechanism of thioether ester bond formation via esterification and Michael-addition reaction (A). Scheme of the conjugation of (B) camptothecin162,163 and (C) paclitaxel164 with a hyperbranched star copolymer or poly(ethylene oxide)-block-poly(phosphoester) via a thioether ester bond and (D) the synthesis of a docetaxel prodrug containing thioether ester bonds.165 | ||
While thioether ester bonds are typically introduced in the side chains of the polymers, they can be also present in the main chain.176 Their cleavage by hydrolysis leads then to the release of active substances used as monomer building blocks, along with the degradation of the polymer.
For long-term application, a slow release of payloads is required. On the contrary, a fast release is needed for situations requiring the immediate presence of a high concentration of payloads, such as in severe corrosion. Thus, corrosion inhibitors were conjugated to polymers with linkages that can be cleaved upon changes of the local pH value induced by corrosion, to yield a sustained release of inhibitors.8,177 Derivatives of 8-hydroxyquinoline (8HQ), which have applications as drugs, antibacterial agents, and corrosion inhibitors, were processed as nanoparticles or polymer coatings. Recently, the rate and selective release of 8HQ from 8HQ-polymer conjugates via thioether ester bonds were investigated.42,168–173 For instance, 8HQ was esterified with acryloyl chloride or methacryloyl chloride, followed by the addition of mercaptans through a Michael-addition reaction to obtain molecules with thioether ester linkages. In some experiments, the sulfur of the thioether group of 8HQ derivatives was oxidized to obtain sulfoxide groups (Fig. 24A).42 The steric hindrance effects on the thioether ester bond induced a significant decrease in the release rate of 8HQ, whereas the release rate of 8HQ was accelerated by the oxidation of sulfur atom on the thioether ester linkages.
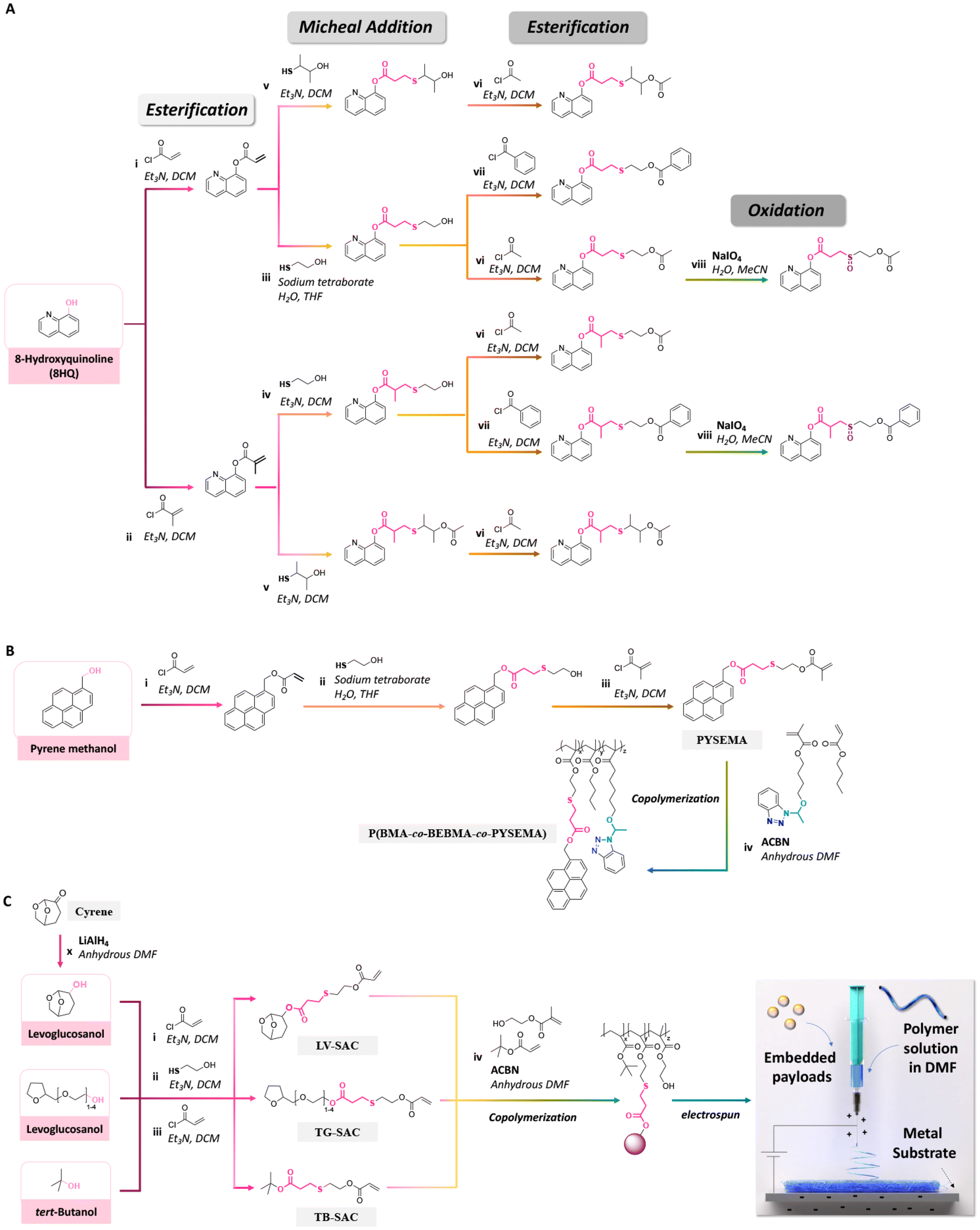 | ||
| Fig. 24 Schematic description of the synthesis of (A) 8HQ derivatives containing different thioether ester bonds;42 (B) pyrene-sulfide-ethyl methacrylate and its copolymerization with n-butyl methacrylate and 4-(1-(benzotriazole-N-yl)ethoxy)butyl methacrylate;147 and (C) polymerizable levoglucosanol, tetraglycol and tert-butanol, their copolymerization with tert-butylacrylate and 2-(hydroxyethyl)methacrylate, and their electrospinning.174 | ||
An effective and cost-efficient strategy to detect corrosion on metals is provided by coating metals with sensing molecules yielding optical changes upon corrosion of the metal. Pyrene methanol, a hydrophobic fluorescent dye, was conjugated with a methacrylate monomer via a thioether ester bond to obtain pyrene-sulfide-ethyl methacrylate (PYSEMA). The latter was copolymerized with n-butyl methacrylate (BMA) and 4-(1-(benzotriazole-N-yl)ethoxy)butyl methacrylate to afford P(BMA-co-BEBMA-co-PYSEMA) (Fig. 24B).147 P(BMA-co-BEBMA-co-PYSEMA) was then processed as a polymer coating for investigating the release of pyrene methanol. Confocal laser scanning microscopy was carried out to measure the fluorescence intensity and track the localization of pyrene methanol before and after immersion of the polymer coating in acidic solution. The changes in fluorescence intensity and appearance of fluorescence in the background were attributed to the release and diffusion of pyrene methanol from the coatings by hydrolysis of the thioether ester bonds.
Recently, polymerizable hydrotropes (levoglucosanol as a cyrene derivative and tetraglycol) and kosmotrope (tert-butanol) containing thioether ester linkages were synthesized via esterification and Michael-addition reactions. The functional monomers were then copolymerized with tert-butylacrylate or 2-(hydroxyethyl)methacrylate (Fig. 24C).174 As demonstrated by 1H NMR spectroscopy, steric hindrance in the conjugated hydrotropes and kosmotropes in the copolymers significantly decreased the acid hydrolysis rate of the thioether ester linkages. Then, the copolymers were electrospun to produce nanofibres for embedding ibuprofen, levofloxacin, rhodamine B, or FITC. Upon cleavage of the thioether ester linkages, the hydrotropes and kosmotropes were released from the nanofibres, hence leading to a sustained and large amount of release of the embedded payloads due to the influence of the local concentration of the released hydrotropes and kosmotrope, which acted as solubilizers, near hydrolyzed fibres during the release process.
3.4 Active agents containing carboxylic groups
Benzoic has also been used as a corrosion inhibitor in emulsions and paints for steel.178 Recently, benzoic acid was conjugated to polymers for investigating the anticorrosion performance of coatings on steel substrates.179 Polymerizable benzoic acid was synthesized by a Markovnikov addition reaction between the carboxylic group of benzoic acid and alkene group of 2-(vinyloxy)ethyl methacrylate to obtain 2-(1-(benzoyloxy)-ethyoxyl)ethyl methacrylate (BEEMA).179 BEEMA was further copolymerized with MMA and 2-((4-((((2-(1H-indol-3-yl)ethyl)carbamoyl)oxy)-methyl)phenyl)disulfanyl) ethylmeth acrylate (IDEMA) to obtain P(MMA-co-BEEMA-co-IDEMA) (Fig. 25). The released rate of benzoic acid from the copolymers in acidic conditions was ∼2 times faster than in neutral conditions. The copolymers were then processed as coatings on steel substrates, leading to a decrease in the corrosion rate of steel by 800 and 19 times in comparison with uncoated steel and steel coated with PMMA.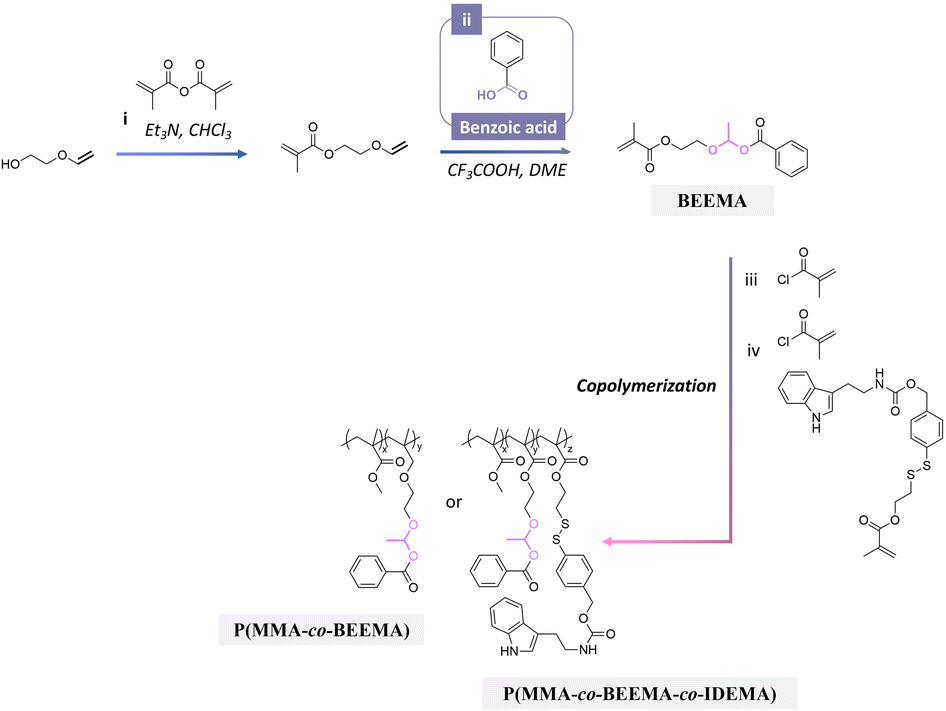 | ||
| Fig. 25 Scheme of the synthesis of 2-(1-(benzoyloxy)-ethyoxyl)ethyl methacrylate (BEEMA) and its copolymerization with either methyl methacrylate or 2-((4-((((2-(1H-indol-3-yl)ethyl)carbamoyl)oxy)-methyl)phenyl)disulfanyl)ethylmethacrylate (IDEMA) and methyl methacrylate.179 | ||
Cisplatin-demethylcantharidin, an anticancer agent capable of inhibiting serine/threonine protein phosphastase 2A in cancer cells, was prepared by the oxidation reaction of cisplatin, followed by a conjugation between the oxidized cisplatin and demethylcantharidin.180 The two carboxylic groups of cisplatin-demethylcantharidin were reacted with the amine groups of a diamine monomer containing othoester linkages by a condensation reaction to produce the polyprodrug cisplatin-demethylcantharidin.181 The carboxylic acid end group of the polyprodrug was terminated with the active ester group of poly(ethylene glycol) monomethyl ether in the presence of EDC and NHS as coupling agents to obtain an amphipathic polyprodrug. More cisplatin was released from the micelle polyprodrug at pH 6.8 than at pH 7.4, attributed to the faster hydrolysis rate of the othoester linkage.
4. Conclusions
The uncontrolled leaching of encapsulated drugs, corrosion inhibitors, antibacterial agents, or dyes from materials reduces their efficiency and can cause negative impacts on healthy cells or the environment. Therefore, the precise delivery of payloads in specific environments and at targeted sites is of paramount importance for biomedical and anticorrosion applications. Indeed, incorporating active agents in the chemical structure of materials through labile linkages is a suitable strategy for controlling the amount and rate of released active agents.Key factors for designing labile linkages are their hydrolytic stability, hydrolysis selectivity of the conjugated linkages, and functional groups of the organic active agents to be released. Generally, acetal/ketal, imine, hydrazine, and orthoester linkages are readily hydrolyzed in very mild acidic and basic conditions, whereas thioether esters are quite stable. The structural fine tuning of pH-sensitive linkers allows access to large variations of the hydrolysis rate and hydrolysis selectivity under neural, acidic, and basic conditions. Longer half-lives of acetal linkages are induced by a higher stability of carboxonium ions. The presence of electron-withdrawing groups on acetal or ketal linkages induces a decrease in the hydrolysis rate, whereas this could be increased with electron-donating groups on the linkages.
Imines with aryl groups exhibited higher stability towards hydrolysis than alkyl imines groups due to electron delocalization with π-bond conjugations between aromatic rings and the imine bonds. The hydrolytic rate of hydrazones is typically slower in acid conditions than for their imine analogues, which is attributed to electron delocalization between the N–N atoms in the hydrazone structures. The hydrolytic rate of hydrazone can be decreased using hydrazones with electron-withdrawing groups. Steric hindrance significantly reduces the hydrolytic rate of thioether ester bonds, while the hydrolysis rate can be dramatically increased by oxidation of the sulfur atom. The presence of steric hindrance and stronger van de Waals repulsion induces a higher selectivity of the hydrolysis rate of thioether ester bonds.
The incorporation of labile linkages in the main chain of polymers allows the fabrication of polyprodrugs or more generally polypro-active substances, which can be released in large quantities and lead to the degradation of the polymer backbone.12 The presence of functional groups in active substances is the most crucial point to take into account for effectively and simply designing polymer prodrugs with pH-labile linkages. Active agents bearing aldehyde, ketal, amine, or hydrazine groups can be conjugated with substrates through imine, hydrazone, or hemiaminal ether linkages, while active agents containing hydroxyl or carboxylic groups can be functionalized to yield compounds with acetal, orthoester, or thiopropionate linkages. Finally, the concept of structurally fine-tuning pH-cleavable linkages is not limited to the modification of drugs but has been extended to fragrances, solubilizers, phytosanitary products, self-healing agents, and corrosion inhibitors.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. N. K. and D. C. designed and conceptualized the review. N. K. wrote the original draft. D. C. supervised the project and reviewed and edited the manuscript.Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. C. acknowledges funding support from the Thailand Science Research and Innovation agency.References
- Q. Leng, Z. Imtiyaz, M. C. Woodle and A. J. Mixson, Pharmaceutics, 2023, 15, 1482 CrossRef CAS PubMed
.
- S. Binauld and M. H. Stenzel, Chem. Commun., 2013, 49, 2082–2102 RSC
- J. Liu, Y. Huang, A. Kumar, A. Tan, S. Jin, A. Mozhi and X.-J. Liang, Biotechnol. Adv., 2014, 32, 693–710 CrossRef CAS PubMed
- X. Lv, J. Zhang, D. Yang, J. Shao, W. Wang, Q. Zhang and X. Dong, J. Mater. Chem. B, 2020, 8, 10700–10711 RSC
- Z. Han, M. Yuan, L. Liu, K. Zhang, B. Zhao, B. He, Y. Liang and F. Li, Nanoscale Horiz., 2023, 8, 422–440 RSC
- H. Ding, P. Tan, S. Fu, X. Tian, H. Zhang, X. Ma, Z. Gu and K. Luo, J. Controlled Release, 2022, 348, 206–238 CrossRef CAS PubMed
- D. Borisova, D. Akçakayıran, M. Schenderlein, H. Möhwald and D. G. Shchukin, Adv. Funct. Mater., 2013, 23, 3799–3812 CrossRef CAS
- F. Seidi and D. Crespy, Chem. Commun., 2020, 56, 11931–11940 RSC
- R. H. Staff, M. Gallei, K. Landfester and D. Crespy, Macromolecules, 2014, 47, 4876–4883 CrossRef CAS
- S. Behzadi, M. Gallei, J. Elbert, M. Appold, G. Glasser, K. Landfester and D. Crespy, Polym. Chem., 2016, 7, 3434–3443 RSC
- K. Bhise, S. Sau, H. Alsaab, S. K. Kashaw, R. K. Tekade and A. K. Iyer, Ther. Delivery, 2017, 8, 1003–1018 CrossRef CAS PubMed
- F. Seidi, Y. Zhong, H. Xiao, Y. Jin and D. Crespy, Chem. Soc. Rev., 2022, 51, 6652–6703 RSC
- F. Seidi, R. Jenjob and D. Crespy, Chem. Rev., 2018, 118, 3965–4036 CrossRef CAS PubMed
- G. K. Such, Y. Yan, A. P. R. Johnston, S. T. Gunawan and F. Caruso, Adv. Mater., 2015, 27, 2278–2297 CrossRef CAS PubMed
- L. A. Wallace, L. Gwynne and T. Jenkins, Ther. Delivery, 2019, 10, 719–735 CrossRef CAS PubMed
- A. Jobdeedamrong, M. Theerasilp, N. Wongsuwan, N. Nasongkla and D. Crespy, Chem. Commun., 2020, 56, 12725–12728 RSC
- P. Phoungtawee, W. Jaroonwatana and D. Crespy, ACS Appl. Polym. Mater., 2023, 5, 2374–2381 CrossRef CAS
- S. J. Sonawane, R. S. Kalhapure and T. Govender, Eur. J. Pharm. Sci., 2017, 99, 45–65 CrossRef CAS PubMed
- Z. Su, D. Xiao, F. Xie, L. Liu, Y. Wang, S. Fan, X. Zhou and S. Li, Acta Pharm. Sin. B, 2021, 11, 3889–3907 CrossRef CAS PubMed
- J. Zhang, F. Hu, O. Aras, Y. Chai and F. An, ChemMedChem, 2024, 19, e202300720 CrossRef CAS PubMed
- Y. Xue, H. Bai, B. Peng, B. Fang, J. Baell, L. Li, W. Huang and N. H. Voelcker, Chem. Soc. Rev., 2021, 50, 4872–4931 RSC
- S. Shahi, H. Roghani-Mamaqani, S. Talebi and H. Mardani, Coord. Chem. Rev., 2022, 455, 214368 CrossRef CAS
- N. Deirram, C. Zhang, S. S. Kermaniyan, A. P. R. Johnston and G. K. Such, Macromol. Rapid Commun., 2019, 40, 1800917 CrossRef PubMed
- S. A. Jacques, G. Leriche, M. Mosser, M. Nothisen, C. D. Muller, J.-S. Remy and A. Wagner, Org. Biomol. Chem., 2016, 14, 4794–4803 RSC
- D. P. N. Satchell and R. S. Satchell, Chem. Soc. Rev., 1990, 19, 55–81 RSC
- E. H. Cordes and H. G. Bull, Chem. Rev., 1974, 74, 581–603 CrossRef CAS
- Substituent Effects on the pH Sensitivity of Acetals and Ketals and Their Correlation with Encapsulation Stability in Polymeric Nanogels | Journal of the American Chemical Society, https://pubs.acs.org/doi/full/10.1021/jacs.6b11181, (accessed May 21, 2024).
- R. W. Layer, Chem. Rev., 1963, 63, 489–510 CrossRef CAS
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1963, 85, 2843–2848 CrossRef CAS
- A. Xavier and N. Srividhya, IOSR-JAC, 2014, 7, 06–15 CrossRef
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825–858 CrossRef CAS PubMed
- P. Chytil, E. Koziolová, T. Etrych and K. Ulbrich, Macromol. Biosci., 2018, 18, 1700209 CrossRef PubMed
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed
- X. Zhang, Y. Huang, M. Ghazwani, P. Zhang, J. Li, S. H. Thorne and S. Li, ACS Macro Lett., 2015, 4, 620–623 CrossRef CAS PubMed
- H. Chen, H. Zhang, C. M. McCallum, F. C. Szoka and X. Guo, J. Med. Chem., 2007, 50, 4269–4278 CrossRef CAS PubMed
- P. Deslongchamps, R. Chênevert, R. J. Taillefer, C. Moreau and J. K. Saunders, Can. J. Chem., 1975, 53, 1601–1615 CrossRef CAS
- J. Cheng, R. Ji, S.-J. Gao, F.-S. Du and Z.-C. Li, Biomacromolecules, 2012, 13, 173–179 CrossRef CAS PubMed
- H. Löw, E. Mena-Osteritz and M. von Delius, Chem. Sci., 2018, 9, 4785–4793 RSC
- A. E. Rydholm, K. S. Anseth and C. N. Bowman, Acta Biomater., 2007, 3, 449–455 CrossRef CAS PubMed
- R. G. Schoenmakers, P. van de Wetering, D. L. Elbert and J. A. Hubbell, J. Controlled Release, 2004, 95, 291–300 CrossRef CAS PubMed
- B. J. Crielaard, C. J. F. Rijcken, L. Quan, S. van der Wal, I. Altintas, M. van der Pot, J. A. W. Kruijtzer, R. M. J. Liskamp, R. M. Schiffelers, C. F. van Nostrum, W. E. Hennink, D. Wang, T. Lammers and G. Storm, Angew. Chem., Int. Ed., 2012, 51, 7254–7258 CrossRef CAS PubMed
- N. Kongkatigumjorn, P. Srikamut, F. Seidi, S. Bureekaew and D. Crespy, Chem. Mater., 2022, 34, 2842–2852 CrossRef CAS
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC
- M. Ciaccia and S. D. Stefano, Org. Biomol. Chem., 2014, 13, 646–654 RSC
- S. Gambaro, C. Talotta, P. Della Sala, A. Soriente, M. De Rosa, C. Gaeta and P. Neri, J. Am. Chem. Soc., 2020, 142, 14914–14923 CrossRef CAS PubMed
- Z. Wang, D. Pei, Y. Zhang, C. Wang and J. Sun, Molecules, 2012, 17, 5151–5163 CrossRef CAS PubMed
- J. M. Sayer, M. Peskin and W. P. Jencks, Imine-Forming Elimination Reactions. I. General Base Acid Catalysis and Influence of the Nitrogen Substituent on Rates and Equilibria for Carbinolamine Dehydration, https://pubs.acs.org/doi/epdf/10.1021/ja00794a600, (accessed October 6, 2023).
- S. Han, J. Lee, E. Jung, S. Park, A. Sagawa, Y. Shibasaki, D. Lee and B.-S. Kim, ACS Appl. Bio Mater., 2021, 4, 2465–2474 CrossRef CAS PubMed
- H. Kamogawa, H. Mukai, Y. Nakajima and M. Nanasawa, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 3121–3129 CrossRef CAS
- H. Morinaga, H. Morikawa, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4529–4536 CrossRef CAS
- B. Levrand, Y. Ruff, J.-M. Lehn and A. Herrmann, Chem. Commun., 2006, 2965–2967 RSC
- M. Stroescu, A. Stoica-Guzun, G. Isopencu, S. I. Jinga, O. Parvulescu, T. Dobre and M. Vasilescu, Food Hydrocolloids, 2015, 48, 62–71 CrossRef CAS
- G. Godin, B. Levrand, A. Trachsel, J.-M. Lehn and A. Herrmann, Chem. Commun., 2010, 46, 3125–3127 RSC
- T. Kuhnt, A. Herrmann, D. Benczédi, C. Weder and E. J. Foster, RSC Adv., 2014, 4, 50882–50890 RSC
- D. Berthier, A. Trachsel, C. Fehr, L. Ouali and A. Herrmann, Helv. Chim. Acta, 2005, 88, 3089–3108 CrossRef CAS
- Y. Wang, H. Morinaga, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1881–1886 CrossRef CAS
- Y. Wang, H. Morinaga, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 596–602 CrossRef CAS
- H. Q. N. Gunaratne, P. Nockemann and K. R. Seddon, Chem. Commun., 2015, 51, 4455–4457 RSC
- H. Morinaga, H. Morikawa and T. Endo, Polym. Bull., 2018, 75, 197–207 CrossRef CAS
- T. Tree-udom, S. P. Wanichwecharungruang, J. Seemork and S. Arayachukeat, Carbohydr. Polym., 2011, 86, 1602–1609 CrossRef CAS
- G. Nicastro, L. M. Black, P. Ravarino, S. d'Agostino, D. Faccio, C. Tomasini and D. Giuri, Int. J. Mol. Sci., 2022, 23, 3105 CrossRef CAS PubMed
- J. Wahbeh and S. Milkowski, SLAS Technol., 2019, 24, 161–168 CrossRef CAS PubMed
- Q. Song, X. Zhou, C. Xue, Z.-A. Zhang, J. Feng, M. Ji, L. Wang, X. Liu and J. Han, Colloids Surf., A, 2022, 655, 130243 CrossRef CAS
- S. Wang, D. Gurav, O. P. Oommen and O. P. Varghese, Chem. – Eur. J., 2015, 21, 5980–5985 CrossRef CAS PubMed
- Z. Zhang, C. He and X. Chen, Mater. Chem. Front., 2018, 2, 1765–1778 RSC
-
G. T. Hermanson, in Bioconjugate Techniques, ed. G. T. Hermanson, Academic Press, Boston, 3rd edn., 2013, pp. 229–258 Search PubMed
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed
- D. C. Kennedy, C. S. McKay, M. C. B. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester and J. P. Pezacki, J. Am. Chem. Soc., 2011, 133, 17993–18001 CrossRef CAS PubMed
- C. X. Thang, H. Vu-Quang, V.-D. Doan and V. C. Nguyen, Colloid Polym. Sci., 2021, 299, 675–683 CrossRef
- S. B. Santhosh, D. Dutta, L. K. Nath, M. J. Nanjan and M. J. N. Chandrasekar, J. Drug Delivery Sci. Technol., 2020, 59, 101856 CrossRef CAS
- M. Ashjari, M. Kazemi, M. N. Abi, M. Mohammadi and S. Rafiezadeh, J. Drug Delivery Sci. Technol., 2020, 59, 101914 CrossRef CAS
- P. Liu, Q. Wu, Y. Li, P. Li, J. Yuan, X. Meng and Y. Xiao, Colloids Surf., B, 2019, 181, 1012–1018 CrossRef CAS PubMed
- N. Rahoui, B. Jiang, N. Taloub, M. Hegazy and Y. D. Huang, J. Biomater. Sci., Polym. Ed., 2018, 29, 1482–1497 CrossRef CAS PubMed
- F. Wang, L. Li, W. Sun, L. Li, Y. Liu, Y. Huang and Z. Zhou, J. Drug Targeting, 2018, 26, 231–241 CrossRef CAS PubMed
- T. Etrych, T. Mrkvan, P. Chytil, Č. Koňák, B. Říhová and K. Ulbrich, J. Appl. Polym. Sci., 2008, 109, 3050–3061 CrossRef CAS
- Y. Zhu, X. Yao, X. Chen and L. Chen, J. Appl. Polym. Sci., 2015, 132, 42778 CrossRef
- S. V. Lale, A. Kumar, F. Naz, A. C. Bharti and V. Koul, Polym. Chem., 2015, 6, 2115–2132 RSC
- Z. Zhou, L. Li, Y. Yang, X. Xu and Y. Huang, Biomaterials, 2014, 35, 6622–6635 CrossRef CAS PubMed
- T. Jiang, Y.-M. Li, Y. Lv, Y.-J. Cheng, F. He and R.-X. Zhuo, Colloids Surf., B, 2013, 111, 542–548 CrossRef CAS PubMed
- M. Xu, J. Qian, A. Suo, H. Wang, X. Yong, X. Liu and R. Liu, Carbohydr. Polym., 2013, 98, 181–188 CrossRef CAS PubMed
- X. Chen, S. S. Parelkar, E. Henchey, S. Schneider and T. Emrick, Bioconjugate Chem., 2012, 23, 1753–1763 CrossRef CAS PubMed
- W. Xiong, W. Wang, Y. Wang, Y. Zhao, H. Chen, H. Xu and X. Yang, Colloids Surf., B, 2011, 84, 447–453 CrossRef CAS PubMed
- K. Ulbrich, T. Etrych, P. Chytil, M. Jelínková and B. Ríhová, J. Drug Targeting, 2004, 12, 477–489 CrossRef CAS PubMed
- T. Etrych, J. Strohalm, L. Kovář, M. Kabešová, B. Říhová and K. Ulbrich, J. Controlled Release, 2009, 140, 18–26 CrossRef CAS PubMed
- T. Etrych, P. Chytil, M. Jelínková, B. Říhová and K. Ulbrich, Macromol. Biosci., 2002, 2, 43–52 CrossRef CAS
- R. Tan, D. Tian, J. Liu, C. Wang and Y. Wan, Int. J. Nanomed., 2021, 16, 4527–4544 CrossRef PubMed
- H. Zhang, M. Pei and P. Liu, J. Ind. Eng. Chem., 2019, 80, 739–748 CrossRef CAS
- G. Hou, J. Qian, W. Xu, T. Sun, Y. Wang, J. Wang, L. Ji and A. Suo, Carbohydr. Polym., 2019, 212, 334–344 CrossRef CAS PubMed
- S. E. Park, K. Shamloo, T. A. Kristedja, S. Darwish, M. Bisoffi, K. Parang and R. K. Tiwari, Int. J. Mol. Sci., 2019, 20, 3291 CrossRef CAS PubMed
- A. Suo, J. Qian, M. Xu, W. Xu, Y. Zhang and Y. Yao, Mater. Sci. Eng., C, 2017, 76, 659–672 CrossRef CAS PubMed
- O. Lidický, O. Janoušková, J. Strohalm, M. Alam, P. Klener and T. Etrych, Molecules, 2015, 20, 19849–19864 CrossRef PubMed
- M. Huan, B. Zhang, Z. Teng, H. Cui, J. Wang, X. Liu, H. Xia, S. Zhou and Q. Mei, PLoS One, 2012, 7, e44116 CrossRef CAS PubMed
- T. Etrych, L. Daumová, E. Pokorná, D. Tušková, O. Lidický, V. Kolářová, J. Pankrác, L. Šefc, P. Chytil and P. Klener, J. Controlled Release, 2018, 289, 44–55 CrossRef CAS PubMed
- H. Guan, M. J. McGuire, S. Li and K. C. Brown, Bioconjugate Chem., 2008, 19, 1813–1821 CrossRef CAS PubMed
- L. Deng, H. Dong, A. Dong and J. Zhang, Eur. J. Pharm. Biopharm., 2015, 97, 107–117 CrossRef CAS PubMed
- V. Rao, N. Shivshankar, R. Mane, A. Kishore, J. Das Sarma and R. Shunmugam, Biomacromolecules, 2012, 13, 221–230 CrossRef PubMed
- C. Lu, M. M. Q. Xing and W. Zhong, Nanomedicine, 2011, 7, 80–87 CrossRef CAS PubMed
- T. Etrych, T. Mrkvan, B. Říhová and K. Ulbrich, J. Controlled Release, 2007, 122, 31–38 CrossRef CAS PubMed
- A. Lau, G. Bérubé, C. H. J. Ford and M. Gallant, Bioorg. Med. Chem., 1995, 3, 1305–1312 CrossRef CAS PubMed
- X.-B. Xiong and A. Lavasanifar, ACS Nano, 2011, 5, 5202–5213 CrossRef CAS PubMed
- T. Yin, Y. Wang, X. Chu, Y. Fu, L. Wang, J. Zhou, X. Tang, J. Liu and M. Huo, ACS Appl. Mater. Interfaces, 2018, 10, 35693–35704 CrossRef CAS PubMed
- C. Fu, H. Li, N. Li, X. Miao, M. Xie, W. Du and L.-M. Zhang, Carbohydr. Polym., 2015, 128, 163–170 CrossRef CAS PubMed
- M. Xu, J. Qian, X. Liu, T. Liu and H. Wang, Mater. Sci. Eng., C, 2015, 50, 341–347 CrossRef CAS PubMed
- G. Yilmaz, E. Guler, C. Geyik, B. Demir, M. Ozkan, D. O. Demirkol, S. Ozcelik, S. Timur and C. R. Becer, Mol. Syst. Des. Eng., 2018, 3, 150–158 RSC
- A. Sangtani, E. Petryayeva, M. Wu, K. Susumu, E. Oh, A. L. Huston, G. Lasarte-Aragones, I. L. Medintz, W. R. Algar and J. B. Delehanty, Bioconjugate Chem., 2018, 29, 136–148 CrossRef CAS PubMed
- G. Yilmaz, B. Demir, S. Timur and C. R. Becer, Biomacromolecules, 2016, 17, 2901–2911 CrossRef CAS PubMed
- D. Willner, P. A. Trail, S. J. Hofstead, H. D. King, S. J. Lasch, G. R. Braslawsky, R. S. Greenfield, T. Kaneko and R. A. Firestone, Bioconjugate Chem., 1993, 4, 521–527 CrossRef CAS PubMed
- X. Li, M. Takashima, E. Yuba, A. Harada and K. Kono, Biomaterials, 2014, 35, 6576–6584 CrossRef CAS PubMed
- D.-W. Dong, B. Xiang, W. Gao, Z.-Z. Yang, J.-Q. Li and X.-R. Qi, Biomaterials, 2013, 34, 4849–4859 CrossRef CAS PubMed
- D.-W. Dong, S.-W. Tong and X.-R. Qi, J. Biomed. Mater. Res., Part A, 2013, 101, 1336–1344 CrossRef PubMed
- J. Prakash, E. de Jong, E. Post, A. S. H. Gouw, L. Beljaars and K. Poelstra, J. Controlled Release, 2010, 145, 91–101 CrossRef CAS PubMed
- L. Zhu, L. Zhao, X. Qu and Z. Yang, Langmuir, 2012, 28, 11988–11996 CrossRef CAS PubMed
- X.-M. Liu, L.-D. Quan, J. Tian, Y. Alnouti, K. Fu, G. M. Thiele and D. Wang, Pharm. Res., 2008, 25, 2910–2919 CrossRef CAS PubMed
- N. Chauhan, P. Gupta, L. Arora, D. Pal and Y. Singh, Mater. Sci. Eng., C, 2021, 130, 112463 CrossRef CAS PubMed
- M. Li, S. Jiang, A. Haller, S. Wirsching, M. Fichter, J. Simon, M. Wagner, V. Mailänder, S. Gehring, D. Crespy and K. Landfester, Nanoscale Horiz., 2021, 6, 791–800 RSC
- T. Etrych, M. Šírová, L. Starovoytova, B. Říhová and K. Ulbrich, Mol. Pharmaceutics, 2010, 7, 1015–1026 CrossRef CAS PubMed
- G. Mo, X. Hu, S. Liu, J. Yue, R. Wang, Y. Huang and X. Jing, Eur. J. Pharm. Sci., 2012, 46, 329–335 CrossRef CAS PubMed
- H. Nakamura, E. Koziolová, T. Etrych, P. Chytil, J. Fang, K. Ulbrich and H. Maeda, Eur. J. Pharm. Biopharm., 2015, 90, 90–96 CrossRef CAS PubMed
- H. Lai, X. Ding, J. Ye, J. Deng and S. Cui, Colloids Surf., B, 2021, 198, 111455 CrossRef CAS PubMed
- O. Sedláček, M. Hrubý, M. Studenovský, D. Větvička, J. Svoboda, D. Kaňková, J. Kovář and K. Ulbrich, Bioorg. Med. Chem., 2012, 20, 4056–4063 CrossRef PubMed
- S. Han, J. Lee, E. Jung, S. Park, A. Sagawa, Y. Shibasaki, D. Lee and B.-S. Kim, ACS Appl. Bio Mater., 2021, 4, 2465–2474 CrossRef CAS PubMed
- K. Boonpavanitchakul, L. K. Bast, N. Bruns and R. Magaraphan, Bioconjugate Chem., 2020, 31, 2312–2324 CrossRef CAS PubMed
- Y. Liu, K. Liu, M. Zhao, S. Wang, Z. Zhou, Y. Shen and L. Jiang, React. Funct. Polym., 2018, 132, 138–144 CrossRef CAS
- M. Ye, Y. Zhao, Y. Wang, N. Yodsanit, R. Xie and S. Gong, Adv. Funct. Mater., 2020, 30, 2002655 CrossRef CAS
- W. Huang, Y. Wang, S. Zhang, L. Huang, D. Hua and X. Zhu, Macromolecules, 2013, 46, 814–818 CrossRef CAS
- V. Tchakalova, E. Lutz, S. Lamboley, E. Moulin, D. Benczédi, N. Giuseppone and A. Herrmann, Chem. – Eur. J., 2021, 27, 13468–13476 CrossRef CAS PubMed
- E. T. Kool, D.-H. Park and P. Crisalli, J. Am. Chem. Soc., 2013, 135, 17663–17666 CrossRef CAS PubMed
- S. E. López and J. Salazar, J. Fluorine Chem., 2013, 156, 73–100 CrossRef
-
W. P. Jencks, in Progress in Physical Organic Chemistry, John Wiley & Sons, Ltd, 1964, pp. 63–128 Search PubMed
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1962, 84, 826–831 CrossRef CAS
- P. Crisalli and E. T. Kool, Org. Lett., 2013, 15, 1646–1649 CrossRef CAS PubMed
- D. Larsen, A. M. Kietrys, S. A. Clark, H. S. Park, A. Ekebergh and E. T. Kool, Chem. Sci., 2018, 9, 5252–5259 RSC
- D. Larsen, M. Pittelkow, S. Karmakar and E. T. Kool, Org. Lett., 2015, 17, 274–277 CrossRef CAS PubMed
- X. Liu, M. Zhu and J. Xie, Toxicol. Mech. Methods, 2010, 20, 36–44 CrossRef CAS PubMed
- S. A. Marchitti, R. A. Deitrich and V. Vasiliou, Pharmacol. Rev., 2007, 59, 125–150 CrossRef CAS PubMed
- C. B. M. Tulen, S. J. Snow, P. A. Leermakers, U. P. Kodavanti, F. J. van Schooten, A. Opperhuizen and A. H. V. Remels, Toxicology, 2022, 469, 153129 CrossRef CAS PubMed
- L. H. Yuen, N. S. Saxena, H. S. Park, K. Weinberg and E. T. Kool, ACS Chem. Biol., 2016, 11, 2312–2319 CrossRef CAS PubMed
- M. Y. Wang, Y. Qu, D. R. Hu, L. J. Chen, K. Shi, Y. P. Jia, Y. Y. Yi, Q. Wei, T. Niu and Z. Y. Qian, Int. J. Pharm., 2019, 557, 74–85 CrossRef CAS PubMed
- A. Libánská, T. Špringer, L. Peštová, K. Kotalík, R. Konefał, A. Šimonová, T. Křížek, J. Homola, E. Randárová and T. Etrych, Commun. Chem., 2023, 6, 1–12 CrossRef PubMed
- A. Libánská, E. Randárová, S. Skoroplyas, M. Bartoš, J. Luňáčková, F. Lager, G. Renault, D. Scherman and T. Etrych, J. Controlled Release, 2023, 353, 30–41 CrossRef PubMed
- E. T. Kool, P. Crisalli and K. M. Chan, Org. Lett., 2014, 16, 1454–1457 CrossRef CAS PubMed
- F. Seidi, A. Couffon, M. Prawatborisut and D. Crespy, Macromol. Chem. Phys., 2019, 220, 1900236 CrossRef
- N. Dararatana, F. Seidi, J. Hamel and D. Crespy, Polym. Chem., 2020, 11, 1752–1762 RSC
- J. Fickert, C. Wohnhaas, A. Turshatov, K. Landfester and D. Crespy, Macromolecules, 2013, 46, 573–579 CrossRef CAS
- F. Seidi, V. Druet, N. Huynh, T. Phakkeeree and D. Crespy, Chem. Commun., 2018, 54, 13730–13733 RSC
- M. Chen, S. Xie, J. Wei, X. Song, Z. Ding and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 36814–36823 CrossRef CAS PubMed
- N. Dararatana, F. Seidi and D. Crespy, Polymer, 2020, 194, 122346 CrossRef CAS
- A. K. Galande, R. Weissleder and C.-H. Tung, Bioconjugate Chem., 2006, 17, 255–257 CrossRef CAS PubMed
- S. Li, D. Cohen-Karni, E. Kallick, H. Edington and S. Averick, Polymer, 2016, 99, 59–62 CrossRef CAS
- K. Zuwala, A. A. A. Smith, M. Tolstrup and A. N. Zelikin, Chem. Sci., 2016, 7, 2353–2358 RSC
- I. Ali, M. N. Lone and H. Y. Aboul-Enein, MedChemComm, 2017, 8, 1742–1773 RSC
- P. Phoungtawee, F. Seidi, A. Treetong, C. Warin, A. Klamchuen and D. Crespy, ACS Macro Lett., 2021, 10, 365–369 CrossRef CAS PubMed
- R. Hennig, A. Veser, S. Kirchhof and A. Goepferich, Mol. Pharmaceutics, 2015, 12, 3292–3302 CrossRef CAS PubMed
- M. Shamis and D. Shabat, Chem. – Eur. J., 2007, 13, 4523–4528 CrossRef CAS PubMed
- E. R. Gillies, A. P. Goodwin and J. M. J. Fréchet, Bioconjugate Chem., 2004, 15, 1254–1263 CrossRef CAS PubMed
- E. R. Gillies and J. M. J. Fréchet, Chem. Commun., 2003, 1640–1641 RSC
- X. Jiang, M. Lee, J. Xia, T. Luo, J. Liu, M. Rodriguez and W. Lin, ACS Nano, 2022, 16, 21417–21430 CrossRef CAS PubMed
- X. Jiang, W. Han, J. Liu, J. Mao, M. J. Lee, M. Rodriguez, Y. Li, T. Luo, Z. Xu, K. Yang, M. Bissonnette, R. R. Weichselbaum and W. Lin, Adv. Sci., 2022, 9, e2201614 CrossRef PubMed
- L. Fortuin, M. Leshabane, R. Pfukwa, D. Coertzen, L.-M. Birkholtz and B. Klumperman, ACS Biomater. Sci. Eng., 2020, 6, 6217–6227 CrossRef CAS PubMed
- C. Masson, M. Garinot, N. Mignet, B. Wetzer, P. Mailhe, D. Scherman and M. Bessodes, J. Controlled Release, 2004, 99, 423–434 CrossRef CAS PubMed
- F. Migliorini, E. Cini, E. Dreassi, F. Finetti, G. Ievoli, G. Macrì, E. Petricci, E. Rango, L. Trabalzini and M. Taddei, Chem. Commun., 2022, 58, 10532–10535 RSC
- L. Qiu, J.-W. Li, C.-Y. Hong and C.-Y. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 40887–40897 CrossRef CAS PubMed
- L. Qiu, Q. Liu, C.-Y. Hong and C.-Y. Pan, J. Mater. Chem. B, 2016, 4, 141–151 RSC
- J. Zou, F. Zhang, S. Zhang, S. F. Pollack, M. Elsabahy, J. Fan and K. L. Wooley, Adv. Healthcare Mater., 2014, 3, 441–448 CrossRef CAS PubMed
- W. Wei, C. Luo, J. Yang, B. Sun, D. Zhao, Y. Liu, Y. Wang, W. Yang, Q. Kan, J. Sun and Z. He, J. Controlled Release, 2018, 285, 187–199 CrossRef CAS PubMed
- M. Li, M. Gao, Y. Fu, C. Chen, X. Meng, A. Fan, D. Kong, Z. Wang and Y. Zhao, Colloids Surf., B, 2016, 140, 11–18 CrossRef CAS PubMed
- J. Gao, Z. Qiao, S. Liu, J. Xu, S. Wang, X. Yang, X. Wang and R. Tang, Eur. J. Pharm. Biopharm., 2021, 163, 188–197 CrossRef CAS PubMed
- N. Dararatana, F. Seidi and D. Crespy, ACS Appl. Mater. Interfaces, 2018, 10, 20876–20883 CrossRef CAS PubMed
- N. Dararatana, F. Seidi and D. Crespy, Polym. Chem., 2020, 11, 4723–4728 RSC
- K. Auepattana-Aumrung and D. Crespy, Chem. Eng. J., 2023, 452, 139055 CrossRef CAS
- N. Kongkatigumjorn and D. Crespy, Macromolecules, 2024, 57, 3502–3514 CrossRef CAS
- X. Li, K. Auepattana-Aumrung, H.-J. Butt, D. Crespy and R. Berger, J. Chem. Phys., 2023, 158, 144901 CrossRef CAS PubMed
- C. Tirayaphanitchkul, K. Auepattana-Aumrung, D. Crespy and M. Ogawa, ACS Appl. Polym. Mater., 2024, 6, 2129–2138 CrossRef CAS
- N. Kongkatigumjorn and D. Crespy, Macromolecules, 2024, 57, 3502–3514 CrossRef CAS
- D. Pozzi, C. Marchini, F. Cardarelli, H. Amenitsch, C. Garulli, A. Bifone and G. Caracciolo, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 2335–2343 CrossRef CAS PubMed
- B. Klahan, F. Seidi and D. Crespy, Macromol. Chem. Phys., 2018, 219, 1800392 CrossRef
- T. Yimyai, D. Crespy and M. Rohwerder, Adv. Mater., 2023, 35, 2300101 CrossRef CAS PubMed
- S. Bilgiç, Mater. Chem. Phys., 2002, 76, 52–58 CrossRef
- P. Srikamut, T. Phakkeeree, F. Seidi, S. Iamsaard and D. Crespy, ACS Appl. Polym. Mater., 2021, 3, 5425–5433 CrossRef CAS
- Y. Cong, H. Xiao, H. Xiong, Z. Wang, J. Ding, C. Li, X. Chen, X.-J. Liang, D. Zhou and Y. Huang, Adv. Mater., 2018, 30, 1706220 CrossRef PubMed
- L. Yang, G. Yan, S. Wang, J. Xu, Q. Fang, Y. Xue, L. Yang, X. Xu and R. Tang, Acta Biomater., 2021, 129, 209–219 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |