
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetrazine-based inverse-electron-demand Diels–Alder reaction: a powerful tool for fabrication and functionalization of polymeric materials
Mehmet
Arslan
 *a,
Aysun
Degirmenci
b,
Rana
Sanyal
bc and
Amitav
Sanyal
*bc
*a,
Aysun
Degirmenci
b,
Rana
Sanyal
bc and
Amitav
Sanyal
*bc
aDepartment of Polymer Materials Engineering, Faculty of Engineering, Yalova University, Yalova, 77200, Türkiye. E-mail: mehmet.arslan@yalova.edu.tr
bDepartment of Chemistry, Bogazici University, Bebek, Istanbul, 34342, Türkiye. E-mail: amitav.sanyal@bogazici.edu.tr
cCenter for Targeted Therapy Technologies, Bogazici University, Istanbul, 34684, Türkiye
First published on 10th October 2024
Abstract
The advent of the click chemistry paradigm has profoundly transformed the design of functional polymeric materials. Among the various reactions in the click chemistry toolkit, the inverse electron demand Diels–Alder (IEDDA) reaction is particularly notable for its rapid kinetics, high specificity, and bioorthogonality. Of the different electron-deficient dienes, the tetrazine (Tz) building block has become the most extensively employed component in the IEDDA reaction-based synthesis and functionalization of polymeric materials. This review begins with an overview of the IEDDA reaction involving Tz, followed by a discussion of examples where the Tz moiety has been utilized as a reactive handle for polymer synthesis and functionalization. We then focus on the application of Tz-based IEDDA reactions in the development of functional hydrogels and surface modifications. The selected examples highlight the diversity and versatility of this highly efficient chemistry. This review aims not only to survey recent progress but also to demonstrate the significant potential of IEDDA chemistry in advancing the design of functional polymeric materials.
1. Introduction
In recent years, the inverse electron demand Diels–Alder (IEDDA) reaction has witnessed an increasing utilization in the synthesis and functionalization of polymeric materials.1 While electron-rich dienes and electron-poor dienophiles are employed in the Diels–Alder reaction, reactive counterparts with the opposite electronic demand are used in the IEDDA reaction. Although its usage is relatively new in polymer science, since its inception, the IEDDA reaction has been extensively used in organic synthesis, especially in heterocyclic chemistry.2,3 Until recently, normal Diels–Alder and hetero Diels–Alder reactions have been widely used in polymer science to create a wide range of compositionally and architecturally complex macromolecular structures.4–13 Compared to the normal Diels–Alder cycloaddition reactions that have been employed over the past several decades in the synthesis and functionalization of polymeric materials, the widespread use of the IEDDA reactions has been limited due to the lack of ready availability of suitable diene and dienophile partners that could be readily incorporated into polymeric materials. Unlike the electron-rich dienes such as butadiene, isoprene, and furan, among many others, the most widely used electron-deficient diene, namely, the 1,2,4,5-tetrazine (Tz) derivatives, usually required multistep synthesis. For researchers with some synthetic experience, a relatively simpler synthetic approach for obtaining various Tz derivatives was reported in due course.14 Nonetheless, the attractive attributes of the IEDDA reaction, such as its high efficiency and selectivity, have ensured continued efforts to increase its usage in polymer science. The Tz-based diene reacts with a variety of alkenes and alkynes and generally possesses higher reactivity toward the strained derivatives (Fig. 1). The reactivity of the IEDDA can be tuned by the selection of appropriately substituted dienes and dienophiles. As expected, the presence of electron-withdrawing substituents increases the reactivity of the diene, while the presence of electron-donating groups reduces the reactivity. Thus, using an appropriately substituted Tz derivative, it is possible to dial in a desirable reactivity profile, as necessitated for the intended application. While the electronic nature of the substituents affects the reactivity of the nitrogen-rich diene, the geometric strain of the dienophiles dictates their reactivity. | ||
| Fig. 1 (a) The IEDDA cycloaddition reaction, (b) commonly employed dienophiles,1 and (c) typical examples of Tz-based dienes.1 Adapted with permission from Knall et al.1 Copyright © The Royal Society of Chemistry 2013. | ||
Although the first report of the IEDDA reaction of Tz was disclosed by Carboni and Lindsey,15 its usage had been primarily limited to synthetic organic chemistry.16,17 Also, most of the early reports of the utilization of the IEDDA reactions, after the introduction of the concept of “click” chemistry by Sharpless and coworkers in 2001,18 primarily focused on its usage in areas such as bioconjugation and in vitro and in vivo applications, due to its biorthogonal nature.19,20
In this review, while we summarize the pertinent milestones in utilizing the IEDDA reaction in polymer science, the versatility this chemistry offers for creating functional polymeric materials is highlighted through various examples (Scheme 1). First, examples of the Tz-based IEDDA in synthesizing and functionalizing polymers are discussed. The section surveys polymers incorporating the Tz group as side-chain or chain end residues and highlights examples of polymers that can undergo efficient post-polymerization functionalization with Tz-containing molecules. This discussion is followed by an illustration of how the IEDDA reaction has been used to fabricate and functionalize crosslinked networks. Finally, examples of functionalization of polymer-coated surfaces using the IEDDA reactions are discussed. While the article would provide a thorough overview of this area to the beginner, it is expected that the survey of examples will promote researchers already working in the design and application of polymers in various areas of materials and biomedical sciences to consider utilizing this highly efficient chemistry.
 | ||
| Scheme 1 Schematic illustration of functionalization of polymers, synthesis, and modification of crosslinked networks, as well as functionalization of solid surfaces via IEDDA “click” reaction. | ||
2. Tetrazine-IEDDA reaction in the synthesis and functionalization of polymers
2.1. Polymers bearing Tz groups as reactive handles
Among the several methodologies of obtaining IEDDA-reactive Tz-containing polymers, a much-sought strategy relies on incorporating Tz-bearing small molecules onto existing polymer scaffolds via various post-polymerization reactions.21 While synthesis of polymers involving the formation of the Tz group in the backbone are rare, suitably modified Tz group containing monomers has been reported to obtain step-growth polymers.22 In an interesting approach, Sayed and Wiggins utilized a dipolar addition mediated step-growth polymerization to obtain poly(1,4-diphenyl-1,4-dihydro-1,2,4,5-tetrazine-3,6-diyl-2,6-pyridinediyl) from bis-nitrilimine.23 Other applications include the utilization of Tz functional ROP (ring-opening polymerization) initiators for polymerization of cyclic monomers and chain transfer agents for radical polymerization of methacrylates. Albeit the conjugated polymers with Tz groups on polymer backbones have widespread utilization in the fabrication of photovoltaic, energetic, and electrochromic materials,24–26 in limited examples, IEDDA functionalizations were reported on polymers obtained from Tz-based monomers. The following paragraphs summarize several studies that exploited the abovementioned strategies to synthesize polymers with IEDDA-clickable tetrazine groups as reactive handles.In an early example of post-polymerization modification-based incorporation of Tz unit onto a polymer scaffold, Panek and coworkers reported IEDDA reactions of resin-bound tetrazines with various electron-rich dienophiles.27 The Tz-immobilized polystyrene complexes were prepared by esterifying a hydroxyl-containing Tz analog onto acyl chloride groups of carboxylated polystyrene resin. Subsequently, elaborated azadiene groups were reacted with various electron-rich enamine, dihydrofuran, and dihydropyran dienophiles to yield resin-bound 1,2-diazines with increased regioselectivity. The obtained cycloadducts were efficiently removed from the resins by basic hydrolysis, granting the practical nature of the IEDDA reaction on solid supports.
Bioorthogonal methodologies for preparing polymeric conjugates in vivo have recently gained intense interest since this class of reactions proceeds with rapid kinetics and high chemical selectivity in biological environments without interfering with biological processes.28,29 The bioorthogonal nature of these chemical reactions minimizes the side reactions towards endogenous functional groups present in the cellular systems. Especially in the last decade, bioorthogonal reactions have played pivotal roles in preparing polymeric conjugates that serve in biomedical imaging, genetic code expansion, drug delivery, metabolic engineering, and drug target identification.30 Of these only a handful of reactions, such as the IEDDA-based transformations have been the method of choice due to their unmatchable reaction kinetics, superior orthogonality, and biocompatibility.1,20
In an impressive example, to demonstrate in vivo labeling in the tumor microenvironment, Weissleder, and coworkers have used Tz-modified and 18F-labeled dextran polymers (Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) in targeting A33 glycoproteins, which are overexpressed in colorectal cancers (Fig. 2a).31 In this study, amino-dextran polymers were partially reacted with fluorescent succinimidyl ester fluorophore, and subsequently, the remaining amine groups were modified by adding Tz-NHS. The conjugate labeling was done by reacting the Tz groups with a [18F] labeled trans-cyclooctene (TCO) derivative. The in vivo conjugation studies performed on LS174T tumor xenografts, which were modified on cell surfaces with excess anti-A33 TCO monoclonal antibodies, revealed that the dextran polymers were able to accumulate in the TCO-tagged tumors. In a later study, Meyer et al. conceptualized a TCO scavenger approach based on the utilization of Tz-modified small dextran polymers (DP-Tz) to mask the remaining TCO-moieties on the circulating monoclonal antibodies which did not interact with antigen-expressing tumor tissue (Fig. 2b).32 The pre-targeting experiments employing DP-Tz demonstrated significantly improved tumor delineation, reduced uptake in vital organs, and enhanced PET image quality.
000 g mol−1) in targeting A33 glycoproteins, which are overexpressed in colorectal cancers (Fig. 2a).31 In this study, amino-dextran polymers were partially reacted with fluorescent succinimidyl ester fluorophore, and subsequently, the remaining amine groups were modified by adding Tz-NHS. The conjugate labeling was done by reacting the Tz groups with a [18F] labeled trans-cyclooctene (TCO) derivative. The in vivo conjugation studies performed on LS174T tumor xenografts, which were modified on cell surfaces with excess anti-A33 TCO monoclonal antibodies, revealed that the dextran polymers were able to accumulate in the TCO-tagged tumors. In a later study, Meyer et al. conceptualized a TCO scavenger approach based on the utilization of Tz-modified small dextran polymers (DP-Tz) to mask the remaining TCO-moieties on the circulating monoclonal antibodies which did not interact with antigen-expressing tumor tissue (Fig. 2b).32 The pre-targeting experiments employing DP-Tz demonstrated significantly improved tumor delineation, reduced uptake in vital organs, and enhanced PET image quality.
 | ||
| Fig. 2 (a) Displaying of in vivo bioorthogonal reaction and in vivo multistep delivery of imaging agent. First, a slow-clearing targeting agent (green) is administered and is given 24 hours for localization. This is followed by injecting a low molecular weight secondary imaging agent (red), which gets localized through an IEDDA reaction with the targeting agent. Adapted with permission from Devaraj et al.31 Copyright (2012) National Academy of Sciences, and (b) illustration of the IEDDA-based small molecule radioligand labeling of TCO-functional antibodies following injection of the masking agent before the radioligand conjugation. Adapted with permission from Meyer et al.32 Copyright © 2018, American Chemical Society. | ||
In an elegant study to demonstrate the potential of bioorthogonal tetrazine ligation chemistry in preparation for smart cancer theranostics, Wang and coworkers designed a pH-responsive polymer containing tertiary amine and Tz groups at side chains.33 In the synthesis step, the Tz groups were installed onto the side chains of an atom transfer radical polymerization (ATRP)-generated copolymer (Mn = 15 kDa) via post-polymerization amidation reaction, with a yield of 98%. These polymers were shown to self-assemble into micelles under neutral conditions (pH 7.4), concurrently enclosing Tz groups and shielding their activities. In the acidic tumor microenvironment (pH 6.5), these micelles collapsed rapidly, therefore promoting the accessibility of Tz groups. A polymeric prodrug (CyPVE), a vinyl ether functional hemicyanine dye (CyPOH) with a poly(ethylene glycol) (PEG) chain was utilized to impart a bioorthogonal prodrug activation. Under acidic microenvironment conditions, Tz functional collapsed micelles were reacted with vinyl ether groups of CyPVE, leading to ligation-elimination of vinyl ether groups. The phototoxicity and NIR fluorescence properties were activated after removing the vinyl ether group from latent phenolic hemicyanine dye. In vivo, experiments on multicellular spheroids (MCSs) derived from 4T1 cells revealed that, while the fluorescence intensity of dye conjugate was only lit up around the periphery at pH 7.4, it could reach a 100 mm depth at pH 6.5 conditions. These findings were reported as a potential application of bioorthogonal Tz IEDDA chemistry to prepare smart cancer theranostics.
Fox, Jia, and coworkers employed the post-polymerization installation of tetrazine groups at chain ends of PEG to prepare hybrid multiblock copolymers based on complementary bifunctional monomers.34 The interfacial copolymerization via Tz ligation was carried out using hydrophilic bis-Tz monomers with PEG spacers and a bis-TCO monomer (Fig. 3). The results showed that the judicious selection of IEDDA-clickable monomers allows the synthesis of high molecular weight polymers at the immiscible solvent interface and enables incorporating functional peptides into the multiblock copolymer structure to tune the polymer properties. In a subsequent study, the Fox group employed bis Tz end-functional PEG polymer (Mn = 7500 g mol−1) and a bis-TCO containing monomer tethering dihydrotetrazine unit at the center for interfacial polymerization.35 After obtaining fibrous copolymers by continuous pulling, the latent Tz functionalities on polymer side chains were converted to IEDDA-clickable Tz groups by photooxidation. Several peptidic ligands and imaging agents were shown to be efficiently conjugated to the synthesized polymers via post-polymerization modification with TCO derivatives. Selectively, RGD-TCO and RGD-TCO-attached polymers displayed promoted cell adhesion and contact guidance.
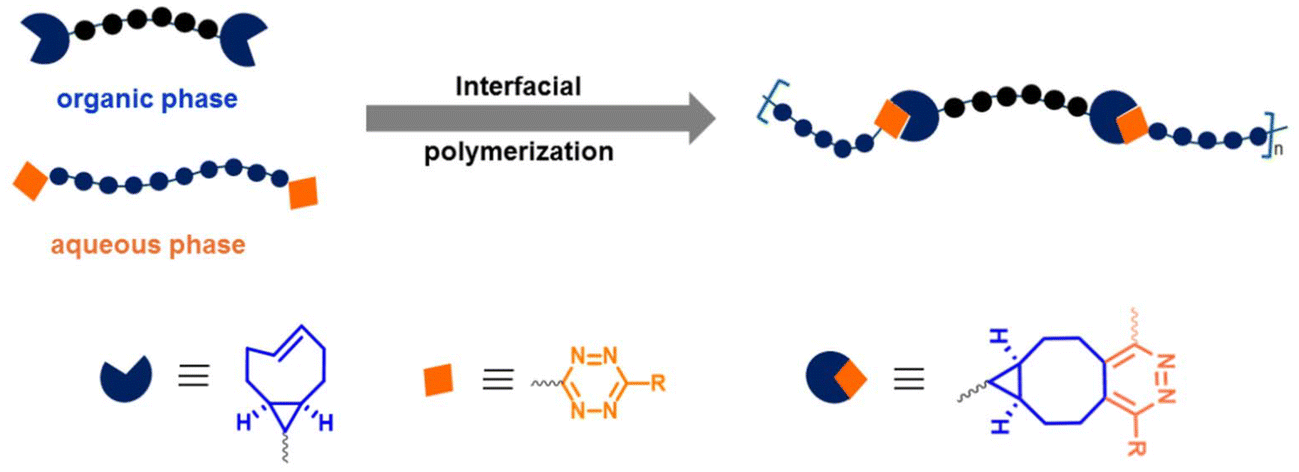 | ||
| Fig. 3 Synthesis of multiblock copolymer fibers through interfacial bioorthogonal polymerization.34 | ||
Maynard and coworkers reported the synthesis of bis-Tz end-functional poly(N-isopropropylacrylamide) (pNIPAAm) and PEG polymers that underwent Tz ligation with a TCO-functionalized lysozyme to generate protein dimers.36 Telechelic Tz functional polymer was obtained by reversible addition–fragmentation chain-transfer (RAFT) polymerization of NIPAAm monomer with a bis carboxylic acid containing chain transfer agent and subsequent post-polymerization esterification of a hydroxyl bearing Tz molecule to carboxylic end groups. In the study, the protein homodimerization of Tz telechelic polymers was compared with a bis-maleimide end-functional pNIPAAm polymer (Mn = 2000 g mol−1). Accordingly, the IEDDA-based “click” reaction demonstrated higher protein conjugation efficiency and dimer formation for both telechelic Tz functional pNIPAAm (38%) and PEG (37%) polymers in a 1 h conjugation duration, compared to thiol-maleimide Michael addition (<1%). In a screening study, O'Reilly and coworkers extensively studied several important bioconjugation reactions in the organic solvent medium for the efficient covalent conjugation of DNA to one chain end of pNIPAAm.37 Among the amide coupling, thiol–ene, and Tz ligation reactions, the IEDDA reaction between Tz and norbornene (NB) groups complementarily found on reacting molecules was found to be the most effective ligation tool to give DNA-polymer conjugates.
One of the critical drawbacks of the post-polymerization installation of Tz groups onto polymers is the requirement for a separate synthesis of reactive polymer and anchoring Tz derivative. In most cases, cumbersome reaction procedures, several side reactions, and non-orthogonal reacting functional groups can lead to tedious purification steps and overall diminished reaction yields. In an attempt to directly install the Tz functionalities onto the side chains of poly(N-isopropylacrylamide-co-acrylonitrile) (p(NIPAAm-co-AN) and p(PEGMA-co-acrylonitrile) copolymers, Kahveci and coworkers have employed an elegant one-pot two-step post-polymerization modification approach.38 In the first step, side chain nitrile groups of copolymers were cross-coupled with an aromatic nitrile-containing molecule in Lewis acid-catalyzed conditions. In the following step, NaNO2 oxidized the 1,4-dihydropyridazine groups to Tz functionalities in acidic conditions. Modification efficiency determination via1H NMR for p(NIPAAm-co-AN) and p(PEGMA-co-acrylonitrile) copolymers revealed 7.9% and 17.9% transformation yields (based on present nitrile groups on copolymers), respectively. It was also shown that Tz-modified copolymers could be efficiently functionalized with a TCO-containing small molecule in less than a minute. In another study, the same group reported the preparation of three-arm star block copolymers via Tz ligation.39 First, three-arm polylactide (PLA) polymers with Tz end groups were prepared by ring opening polymerization of D,L-lactide monomer, and subsequent Tz end group installation. Then, a mono methoxy PEG polymer with a TCO end group was reacted with a three-arm Tz functional PLA polymer under exact stoichiometric conditions. It was demonstrated that a high polymer–polymer coupling efficiency (up to 95.8%) was achieved in about an hour, leading to 3-arm star PLA-b-PEG copolymers (Mn = 23400 g mol−1). The post-polymerization introduction of Tz groups onto poly(styrene-co-acrylonitrile) polymer and subsequent TCO functionalization was also reported in this context.40
The importance of synthesizing functional polymers that allow site-specific attachment of desired molecules is prevalent in preparing polymeric conjugates for several biomedical applications. For example, in the preparation of polymer–protein conjugates, site-specific conjugation of proteins to polymers is imperative because the interaction of bioconjugates with target proteins, receptors, or cells is strongly related to the size, shape, and binding ligand density.41–43 Precise and well-defined polymeric systems are also crucial for quantitatively assessing such conjugates. In the preparation of polymer bioconjugates, end functional polymers provide both the opportunities of controlling position and quantifying attached biomolecules.44,45 Synthesis of such polymers are generally performed by choosing suitable functional initiators or chain transfer agents that allow the covalent attachment of the desired molecules.
To synthesize polymers containing the Tz groups at polymer chain ends, a preferred strategy is to use functional initiators to start the polymerization. The judicious choice of an appropriate initiator allows the installation of IEDDA-clickable Tz units at the specific end groups of polymers. This concept was initially reported by Hansell et al. to synthesize Tz-terminated PEG, poly(δ-valerolactone) (PVL) for IEDDA-based conjugation to NB-terminated polymers.46 A Tz-containing alcohol was utilized with an organo-phosphorus catalyst, which yielded a polymer with 1.7 kDa molecular weight and 1.27 dispersity, using ring opening polymerization (ROP). In a recent study, Barz and coworkers employed the functional ROP initiator strategy to prepare both Tz and TCO-functional polypeptides, polypeptoids, and polypeptide-b-polypeptoids polymers by using cyclic N-carboxyanhydride monomers (Fig. 4a).47 Using Tz or TCO-functional amine initiators, polymers with low dispersity and high-end group integrity were obtained as was accounted by several spectroscopic techniques. These functional polymers were employed in block copolymers synthesis, modification with small molecules, and preparation of polymeric micelles and organic colloids. Results showed that IEDDA-based conjugation chemistry can be very effectively and quickly implemented among the molecules bearing complementary Tz and TCO functionalities. Functional ROP initiator strategy was also used by Van den Berg et al. under continuous flow conditions to prepare Tz functional well-defined PLA polymers with low organo-catalyst loadings (0.25–1.2%) and reaction times (seconds) yielding to polymers with low dispersity (∼1.2) and monomer conversions (95–100%).48 Tz functionalized PLA polymers were efficiently coupled with a cyclooctyne end group containing PLA polymer and a NB functional small molecule.
 | ||
| Fig. 4 (a) Block copolymer synthesis and nanoparticle preparation using ROP-generated TCO- and Tz end functional polymers. Adapted with permission from Johann et al.47 Copyright © 2020, The Royal Society of Chemistry, and (b) reaction between Tz-functionalized polymeric nanoparticles and trans-cyclooctene functionalized antibodies. Adapted with permission from Kramer et al.50 Copyright © 2019 American Chemical Society. | ||
Among the several controlled radical polymerization procedures that would allow site-specific placement of Tz groups at polymer chain ends, RAFT polymerization offers a convenient method. RAFT polymerization is an excellent opportunity for synthesizing polymers in a controlled manner without needing undesired metal catalysts that might interfere with strong metal chelating Tz groups.49 In the RAFT process, introducing a Tz functional group to polymer chain ends is maintained by choosing a Tz functional group containing chain transfer agent (CTA). This concept was demonstrated by Kramer et al. for the RAFT-mediated synthesis of pentafluorophenyl methacrylate to prepare polymeric nanoparticles for bioconjugation (Fig. 4b).50 The successful polymerization with the Tz functional CTA was conducted at a relatively low temperature (40 °C) to prevent degradation of the end groups. The resulting polymer was subsequently employed as a macro-CTA for polymerizing several methacrylate monomers. The results showed that block copolymers with average molecular weights (Mn) 11–22 kDa and dispersities 1.2–1.5 could be successfully obtained under RAFT polymerization conditions with the retention of reactive Tz groups at polymer chain ends.
Functional copolymers that incorporate Tz units along their backbone have been widely explored since these polymers find applications in fabricating semi-conductive, photovoltaic, and electrochromic materials.51 Since Tz groups have highly electron-deficient aromatic structures, the Tz units are usually responsible for decreasing the HOMO level of polymer structure, thus resulting in higher open-circuit voltages.52 To incorporate Tz units on the polymer backbone, preferred approaches include the employment of dihalide functional Tz monomers in nucleophilic aromatic substitution and metal-mediated cross-coupling polymerizations. Post-polymerization modification of such Tz group containing polymers via IEDDA-based ligation allows desired functionalization. In a recent study, Adronov and coworkers employed a bisbromofuran group containing Tz compound and a fluorene diboronate-based monomer to prepare soluble poly(Tz) polymers.53 The polymerization reactions employed a palladium-catalyzed Suzuki polycondensation reaction, which yielded high molecular weight conjugated polymers containing reactive Tz units along the backbones. Quantitative functionalization of poly(Tz)s with a series of TCO derivatives and subsequent oxidation resulted in corresponding conjugated poly(pyridazine) polymers. The analytic measurements via absorption and emission studies revealed that the functionalized polymers display substantial hypsochromic shifts in absorption and increased emission intensity relative to the parent poly(Tz) polymer. In another study that employs a dichloro functional Tz monomer, Carrillo and coworkers fabricated polymer microspheres that display tailored dynamic covalent networks.54 Polymers were obtained by nucleophilic aromatic substituting chloro groups of Tz monomer with either multi-phenol or multi-thiol-containing compounds (70% to quantitative yield). Taking advantage of dynamic covalent chemistry reactions based on the nucleophilic aromatic substitution of Tz,55 the authors were able to prepare degradable and recyclable networks. IEDDA-based conjugation of Tz networks with a dicarboxylic acid functional NB derivative resulted in the tuning of polymer properties, such as water solubility. Notably, these networks were concomitantly locked to the exchange reaction after Tz ligation, thus stabilizing the polymer against depolymerization. Recently, the post-polymerization modification of the main-chain Tz group containing copolymers with aryl alkynes was reported.56
2.2 Polymer functionalization using Tz-containing functional molecules
The applications of IEDDA reaction in the fabrication and functionalization of polymeric materials principally stem from the course of the reaction that proceeds on “click” chemistry principles. On the other hand, for efficient implementation of this cycloaddition reaction on a macromolecular construct, the electronic natures of diene and dienophile functionalities present on the counterpart reacting moieties have the utmost importance in attaining high coupling efficiencies and fast reaction kinetics. In the case of IEDDA-based Tz ligation with electron-rich dienophiles, the latter reaction components are usually chosen from strained alkenes or vinylic compounds neighboring electron-donating groups. Since Tz groups have limited compatibility with nucleophiles, radical sources, redox species, and metal donors that might impede the retention of Tz structure during polymerization,57 a commonly practiced approach for IEDDA-based polymer functionalization relies on the post-polymerization attachment of Tz containing small molecules onto polymeric scaffolds that bear counterpart reacting dienophiles. Among the various IEDDA-clickable groups that can be orthogonally conjugated with Tz units, strained NB alkenes have widespread utilization in modifying polymeric materials. The suitability of NBs in thermal and UV-initiated thiol–ene radical additions,58,59 and their archetypal nature in ring-opening metathesis polymerizations60 is prominent. Due to their highly strained ring structure, NB derivatives are ideal reacting partners in the IEDDA reactions.Building on the ROP initiator strategy of placing NB groups at polymer chain ends, Dove and coworkers demonstrated post-polymerization functionalization of NB-modified polylactic acid via IEDDA “click” reaction.61 The ligation reaction of NB units with 3,6-di-2-pyridyl-1,2,4,5-Tz resulted in complete end group modification without degradation of the polymer backbone. In the same study, side chain NB functional lactide polymer poly(spiro[6-methyl-1,4-dioxane-2,5-dione-3,20-bicyclo-[2.2.1]hept[5]ene]) was also synthesized and modified similarly with Tz-based compound. In another study, Dove, O'Reilly, and coworkers adopted a similar strategy to obtain NB-functional poly(carbonate) scaffolds.62 After obtaining degradable polymers via organo-catalyzed ROP of NB-containing cyclic carbonate monomer, post-polymerization modification of pendent NB units was successfully conducted by various complementary reactive groups, including azides via a 1,3-dipolar cycloaddition, Tzs in IEDDA reaction and thiols via radical thiol–ene reaction. The orthogonal nature of these chemistries was also demonstrated by one-pot sequential modification of NB functional polymer. Synthesis and Tz ligation modification of degradable polyesters with NB functional side chains was reported within this scope.63 Other than the NB partner, Tz-based IEDDA reaction was also successfully implemented in the functionalization of allyl ether side groups containing anionic ROP copolymers.64
Due to its modular, fast, efficient, and biorthogonal nature, Tz-IEDDA chemistry is attractive for fabricating polymeric bioconjugates. In an early study reported by Bawendi and coworkers, polymeric imidazole ligands were prepared by RAFT polymerization and used to coat quantum dots (QDs) through ligand exchange.65 NB groups were subsequently installed on the surface of QDs via amidation reaction. To illustrate the application of NB-decorated QDs for live-cell imaging, they successfully targeted A431 human carcinoma cells, previously decorated with a Tz-modified epidermal growth factor. In another work by Zhang et al., side-chain NB functional polymers with desired molecular weights (7–30 kDa) with dispersities between 1.2–1.8 were synthesized by RAFT polymerization of a NB containing acrylamide monomer (Fig. 5).66 In polymerization, metal affinity chromatography deliberately employed a histidine-tagged chain transfer agent to allow straightforward purification of later-prepared polymer–antibody conjugates. The polymer bearing reactive NB groups and a histidine tag was conjugated to antibody Herceptin via amidation to prepare a polymeric bioconjugate amenable to Tz ligation. After the IEDDA-based attachment of a Tz-containing fluorescent dye molecule to the bioconjugate, the polymer–antibody scaffold provided an 83-fold increase in fluorescence intensity compared to the control antibody labeled with a standard small molecule fluorophore. In a recent study, biorthogonal Tz-NB and aminoxy-aldehyde ligation reactions were concomitantly employed in the in situ generation of synthetic dendrimers on live cells.67 The authors achieved cancer cell targeting on live tumor tissue by growing the dendritic structure on antibody Herceptin.
 | ||
| Fig. 5 Preparation of Tz-IEDDA reaction-mediated polymer-protein conjugates for fluorescent signal amplification. Adapted with permission from Zhang et al.66 Copyright © 2020, The Royal Society of Chemistry. | ||
The application of IEDDA-clickable side chain NB functional polymers in the preparation of polymeric micelles and single-chain polymer nanoparticles (SCNPs) was investigated by the O'Reilly group.68,69 In the self-assembly generated formation of micellar scaffolds, a RAFT-mediated amphiphilic diblock copolymer carrying two orthogonal “click”-compatible functionalities in the polystyrene block core and PEG-based shell was utilized. Upon the formation of micellar structures in water, functionalizations of NB groups in the core structure and terminal alkynes in the shell of aggregates have been carried out by Tz-NB chemistry and copper-catalyzed azide–alkyne reaction. It was shown that this single micellar scaffold can be used either in separate core–shell functionalizations or in a one-pot sequential manner that allows both core- and shell functionalization using two orthogonal “click” reactions. Later, the same group employed the Tz-NB ligation reaction to prepare single-chain polymer nanoparticles. Using polystyrene polymers decorated with pendent NB groups and a bifunctional Tz crosslinker, SCNPs of diameters below 10 nm were prepared. In an elegant work by Li and coworkers, IEDDA-based Tz-NB chemistry was also employed to prepare polymeric micelles from bacteria templating.70 In this study, either doxorubicin (DOX) or α-tocopheryl succinate (TOS) anticancer agents were attached to the one-chain end of a PEG polymer through redox-responsive disulfide linkers. The DOX and TOS loading levels in MDOX and MTOS micelles were approximately 18.9% and 14.9%. A NB derivative was installed through an acid-labile linker to the other end of polymers. These polymeric platforms were later IEDDA-conjugated to Escherichia coli Nissle 1917 (EcN) bacterial carrier decorated with Tz groups on the cellular surface. Upon introducing the bacterial microbot drug carrier system to the slightly acidic tumor microenvironment, drug-loaded copolymers were released from the bacterial surface and concomitantly arranged into hybrid micellar structures. It was demonstrated that after intravenous administration of bacterial carrier system into tumor-bearing mice, selective tumor accumulation and prolonged retention of synergistic anticancer drugs at target tissue were achieved.
The Tz-based IEDDA chemistry has been recently employed to end chain norbornadiene group-containing polymers, in which, in this case, the ligation chemistry allowed the unveiling of latent reactive cyclopentadiene functionalities. In a report by Hawker, De Alaniz, and coworkers, norbornadiene groups, previously installed at the chains ends of polymers during polymerization via ROP, ATRP, and RAFT processes, were employed in polymer functionalization and block copolymer synthesis through one-pot, sequential Tz-NB IEDDA and cyclopentadiene-maleimide Diels–Alder reactions (Fig. 6).71 It was shown that initial Tz cycloaddition with norbornadienes reveals a cyclopentadiene derivative selectively at polymer chain end, which can subsequently participate in Diels–Alder cycloaddition with maleimide. In a later work, the same group employed this cascade “click” modification methodology to the norbornadiene end group containing polymers in which the norbornadiene groups were installed at polymer chains via post-polymerization modification.72
 | ||
| Fig. 6 One-pot functionalization of norbornadiene end-functional polymer with maleimides. Adapted with permission from Amant et al.71 Copyright © 2019, American Chemical Society. | ||
Besides NB, one of the most potent reactive dienophiles is TCO and its derivatives for the Tz-based IEDDA reactions. The ligation reactions between TCO and Tz groups could reach the fastest known reaction kinetics (rate constants up to 106 M−1 s−1), making them exceptionally useful tools for bioconjugation studies.73,74 Important drawbacks of TCO compounds, however, are their low commercial availability and complex synthesis.75 The applications of Tz-TCO ligation chemistry in polymeric material synthesis and functionalization predominantly include preparing polymer bioconjugates suited to cell imaging, protein modification, and cell targeting.
Herth, Radchenko and coworkers reported radiolabeling of polyglutamic acid (pGlu) (Mn = 23300 g mol−1) with 177Lu and 225Ac radioligands using pGlu modified with pendant TCO groups and radiolabeled Tz chelates.76 The method yielded conjugates with excellent stability in human plasma. In another study, Zeglis and coworkers combined two bioorthogonal chemistries, strain-promoted azide–alkyne cycloaddition (SPAAC) and Tz-TCO ligation, to produce antibody-attached dendrimer scaffolds, that can be used in signal amplification in pre-targeted positron-emission tomography (PET) imaging.77 After site-specific SPAAC conjugation of peripheral TCO-functional dendrimer to huA33 antibody, in vivo tumor targeting was carried out using a murine model of colorectal carcinoma. The labeling of resulting TCO-bearing immunoconjugates by a Tz-modified radioligand demonstrated highly enhanced PET imaging efficiency compared to an analogous dendrimer lacking immunoconjugate. By Herth and coworkers, the pretargeted imaging strategy was extended to the TCO polypeptide-g-polypeptoid polymers that were utilized in nuclear imaging.78 After tumor accumulation of the ROP-generated TCO-functional ‘peptobrush’ (polyglutamic acid-g-polysarcosine) polymers, biorthogonal conjugation of a Tz modified [111In] contrast agent provided efficient tumor radiolabeling. In a follow-up study, the Herth group employed TCO functional peptobrush polymers for radiolabeling tumor tissues with [11C]-Tz markers.79 A pretargeting strategy developed for the passive delivery of side chain TCO-functional RAFT star polymers to tumor tissue was reported by Goos and coworkers.80 In the study, while star-shaped polymeric carriers loaded with Gd-based MRI components provided passive tumor targeting, a Tz functional [18F] radiolabel conjugated to the TCO groups allowed nuclear imaging.
Xu et al. have broadened the applications of Tz-TCO ligation chemistry to the site-selective protein modification established on disulfide rebridging strategy.81 A Tz-based disulfide reagent was employed for site-selective installation of the Tz group into both disulfide-containing hormone somatostatin and the antigen-binding fragment of human immunoglobulin G. A set of TCO-containing substrates such as dyes, polymers, and enzymes were conjugated to the Tz modified proteins. The fast and efficient conjugation results demonstrated that IEDDA reaction based on Tz and TCO substrates could be concurrently applied with other bioconjugation tools to prepare the bioconjugates in a site-selective manner.
Although, due to its biorthogonal nature and fast reaction kinetics, Tz-IEDDA chemistry has found extensive utilization in preparing polymer bioconjugates, the applications have also been directed to the covalent modification of industrial polymers. These polymers are commercial and produced on massive scales; thus, their covalent modifications via highly efficient “click” reactions have attracted attention to preparing polymers with unique properties.82–84 In an initial report, Bagge et al. demonstrated the modification of polybutadienes with 3,6-dichloro-1,2,4,5-Tz.85 Polymeric foams with enhanced chain rigidities were obtained by the liberation of nitrogen gas during the cycloaddition process. The incorporation of 1,4-dihydropyridazine group to the polymers also enhanced the antioxidant properties of polymers as deduced by the free radical inhibition experiments. Similarly, Li, Wu, and coworkers applied 3,6-di(2-pyridyl)-1,2,4,5-Tz to modify and introduce nitrogen-containing groups into styrene–butadiene rubber chains covalently.86 These metal chelating groups were further demonstrated to coordinate Cu2+ ions, allowing fabrication of mechanically robust and reversibly processable materials. By the same groups, the same chemistry was applied to nitrile-butadiene rubber to obtain tough, recyclable and healable polymers.87
3. Tetrazine-IEDDA mediated synthesis and functionalization of crosslinked networks
The distinguished characteristics of IEDDA reactions, such as fast reaction kinetics, high monomer conversions, and catalyst-free conditions, make them ideal reaction tools for fabricating crosslinked homogenous polymer networks.88–91 The Tz-IEDDA reaction has been utilized in fabricating hydrogels, nanogels, organogels, and metal-crosslinked polymer scaffolds for diverse applications.92 Particularly, the biorthogonal and biocompatible nature of the Tz-IEDDA reaction serves utility for fabricating hydrogels for various biomedical applications, including cell encapsulation, bioconjugation, tissue engineering, and sustained drug/protein delivery.One of the earlier examples of Tz-IEDDA reaction in hydrogel synthesis was reported by Johnson and coworkers in which A2 + B3 type cycloaddition of bisnorbornene end functional PEG (Mn = 6 kDa and 10 kDa) and a three-Tz containing crosslinker resulted in gel networks.93 The study demonstrated that the Tz-IEDDA reaction could provide an efficient and additive-free crosslinking process by complementing the reaction procedure with other “click” chemistry methodologies. Besides several hydrogelation systems that employed end group functional polymer precursors,94 Tz-IEDDA reaction has also been exemplified in smooth crosslinking hydrogelation using side chain Tz functional polymers.95 The rapid reaction kinetics of Tz-IEDDA reaction has been extended to highly efficient interfacial crosslinking at oil-water96–98 and liquid-gel interface,99,100 as well as annealing hydrogel microparticles.101–105 Although Tz-IEDDA has primarily served in preparation of cast and injectable hydrogel-based materials, this chemistry also has the edge on synthesizing patterned hydrogels,106,107 nanogels108–110 and cryogels.111
The orthogonal nature of Tz-IEDDA reaction to various other “click” chemistry-based methodologies benefits dual-click crosslinking for generating functional networks. Along with the IEDDA reaction with Tzs, NBs, and strained alkynes are reaction partners in thiol–ene and thiol–yne-based synthetic approaches. In the case of targeting mechanically robust 3D structures, which is especially desirable in biomedical applications, the dual-click crosslinking strategy might allow the generation of rigid and stiff crosslinked networks. Dove and coworkers exploited this approach to fabricate high-compressive strength hydrogels with two interpenetrating networks in the structure.112 In a one-pot tandem “click” procedure that combines all the reaction components, the initial formation of the loose network was achieved by Tz-IEDDA reaction of NB-functionalized chitosan with a linear PEG-Tz crosslinker (Fig. 7). Subsequently, the dense network was constructed by thiol–yne reaction of a 4-arm PEG-tetra alkyne with a linear PEG-dithiol. It was demonstrated that these mechanically strong and biocompatible hydrogels are ideal platforms for encapsulation of human cells. In another report that shares a similar dual crosslinking methodology, photo-initiated thiol-NB addition was subsequently followed by Tz-IEDDA reaction to fabricate dual crosslinked networks.113 Incorporation of a mono Tz end functional polymer into thiol-NB partially crosslinked networks also enhanced the network stiffening through supramolecular interactions.114
 | ||
| Fig. 7 Schematic of double network hydrogel formation via dual “click” approach. Adapted with permission from Truong et al.112 Copyright © 2015, American Chemical Society. | ||
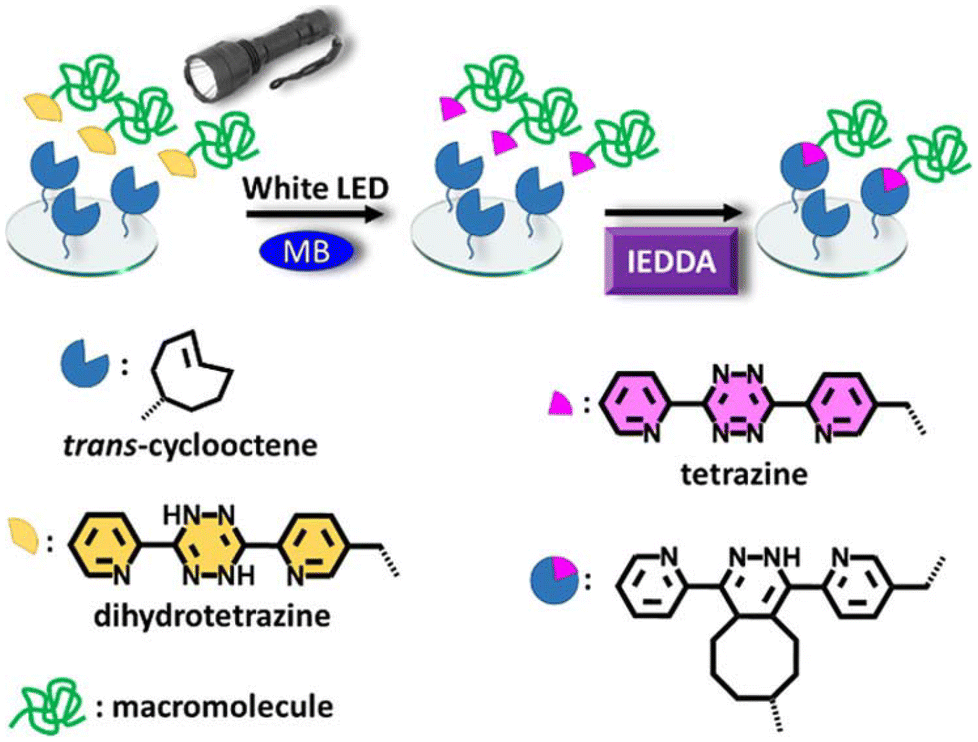
While the Tz-IEDDA reaction offers an attractive way to prepare biorthogonal hydrogels, the susceptibility of the Tz group towards different chemical environments should be considered during precursor preparation and cross-linking processes. For example, Tzs with electron-withdrawing carboxylate substitutions are not stable in an aqueous environment despite their high reactivity with dienophiles.115 Tzs might also display cross-reactivity towards free thiol groups, which are abundant in biological media.30 To address such challenges, a preferred strategy is the on-demand generation of Tz groups by oxidation of dihydrotetrazine (dHTz) precursors. Forsythe, Truong, and coworkers utilized in situ generation of Tz to obtain hydrogels using light-triggered chemistry.116 Activation of dHTz, which is more stable against reductants, in the presence of methylene blue and red-light (625 nm) furnishes hydrogel via the in situ IEDDA reaction (Fig. 8). Impressively, the gelation occurred behind a dermal tissue model with a thickness of 1 cm. The non-toxic nature allows the encapsulation of hMSC within the gel matrix. The same group also reported the fabrication of PEG-gelatin hydrogels using an enzyme-activated Tz-IEDDA reaction. The addition of horse radish peroxidase enzyme to an aqueous solution of 4-arm PEG-dHTz (Mn = 10 kDa) and gelatin-NB induced gelation. Obtained hydrogels were highly stable under physiological conditions, allowing 3D culture of human mesenchymal stem cells (hMSCs) for 32 days.117 In another study, Fox and co-workers reported an efficient method for mild oxidation of dihydrogen Tz to Tz using silicon-rhodamine dyes as cytocompatible far-red (660 nm) photocatalysts.118
 | ||
| Fig. 8 The schematic illustration of hydrogel fabrication using red-light catalyzed Tz-IEDDA reaction. Adapted with permission from Truong et al.116 Copyright © 2017, American Chemical Society. | ||
The structural tailoring of the network matrix can be conveniently carried out to induce hydrolytic stability or degradability in Tz-IEDDA hydrogels. Goepferich and coworkers prepared hydrogels that show long-term hydrolytic stability and controlled protein release over time to obtain structurally stable crosslinked networks or promote hydrolytic matrix degradation over time.119 PEG-based hydrogels prepared using 8-arm poly(ethylene glycol)s (Mn = 10, 20, or 40 kDa) of Tz and NB end functionalities showed complete degradation in vitro in a span of ∼150 to 500 days, depending on the molecular weight of PEG used. These depots allowed a long-term control release (over 250 days) for glucose oxidase protein encapsulated within hydrogels. In a subsequent study, the group placed hydrolyzable ester structures in the PEG-NB polymer precursor to increase the hydrolytic degradation of the hydrogels.120 Among the various carbonate, carbamate, and tetrahydrophthalimide hydrolyzable linkers, considerable degradation acceleration was accounted for in the case of phenyl carbonate ester inclusion. Instead of changing the nature of the linkages to the precursor polymers, it is also possible to tune the degradability of the hydrogels by varying the structure of the reactive groups. This was recently demonstrated by Lin and coworkers, who investigated hydrolytically degradable PEG-based hydrogels prepared via the IEDDA reaction using substituted Tz and NB derivatives (Fig. 9).121 For example, carbic anhydride was introduced to 5-norbornene-2-carboxylic acid functionalized multiarm poly(ethylene glycol) to achieve fast degradation. In addition to carbic anhydride, utilization of (methyl)tetrazine-modified macromers to form hydrogel provided highly tunable degradation. The work demonstrates that introducing small changes in the functional groups profoundly affects the stability of obtained materials. The authors postulated that such PEG-based degradable hydrogel systems may be helpful in tissue regeneration and delivery applications.
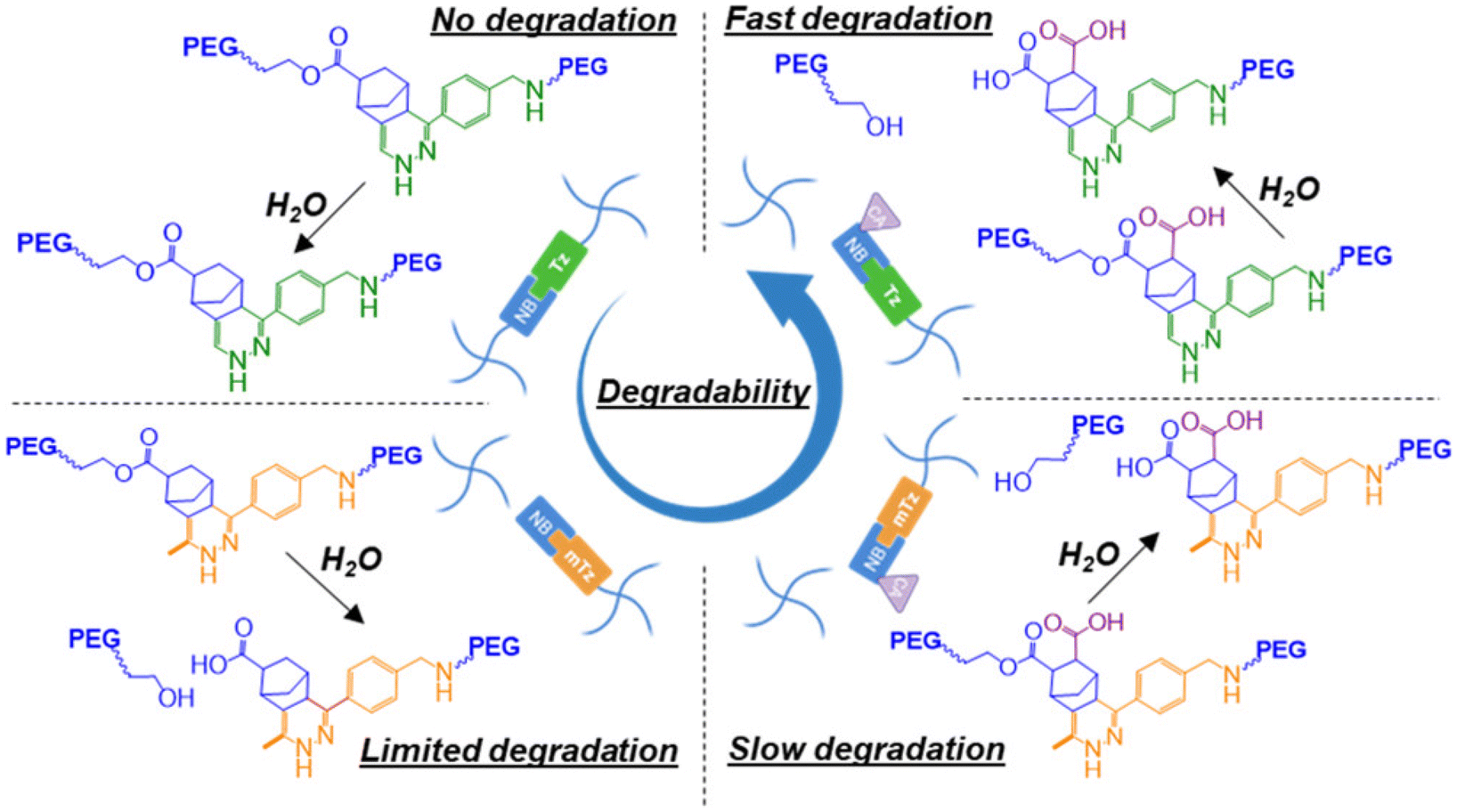 | ||
| Fig. 9 Hydrolytic degradation of PEG-based Tz-IEDDA “click” hydrogels. Adapted with permission from Dimmitt et al.121 Copyright © 2022, American Chemical Society. | ||
Apart from the installation of crosslinks through the reaction of Tz with appropriate dienophiles, the nitrogen-rich Tz building block and the resultant pyridazine moieties are known to coordinate with metal ions in which this attribute has been utilized in the fabrication of various metal-coordinated polymer networks.122 In one of their studies, Johnson and coworkers utilized the Tz-IEDDA reaction to prepare a drug-containing polymer precursor for the hydrogel, as well as harnessed the metal coordination of the Tz group to obtain supramolecular hydrogels.123 Briefly, 3,6-bis(2-pyridyl)-1,2,4,5-Tzs (bptz) groups were conjugated to the end of the PEG chains (Mn = 2000 g mol−1) with a yield of 71%, which were utilized to obtain hydrogels in the presence of Fe2+ and Ni2+ ions (Fig. 10). The bptz-terminated PEG chains could be functionalized using NB-containing functional molecules. In particular, an exo-norbornene-imide linked to doxorubicin with an ortho-nitrobenzyl alcohol unit and to tryptamine glycinamide unit through a chymotrypsin-cleavable peptide linker, to impart photo- and enzymatic-responsive release, respectively, were introduced into the gels through mono-functionalization of the precursor polymers using the IEDDA reaction. Obtained gels demonstrated photo- and enzymatically stimulated release of the conjugated drugs. Moreover, the gels were found to undergo complete degradation after one week, which is an attractive feature warranting the clearance of these materials for in vivo applications. In an alternative approach, Knall and coworkers reported the functionalization of macroporous poly(dicyclopentadiene) foams to obtain metallo-gels.124 The macroporous poly(dicyclopentadiene) monoliths were functionalized with di(pyridyl)-pyridazines using the IEDDA reaction to introduce dipyridyl(pyridazine) units, which were used to coordinate europium(III) nitrate to impart these materials with bright red long-lived emission.
 | ||
| Fig. 10 The schematic illustration of metallo-hydrogel using IEDDA reaction and metal coordination. Adapted with permission from Kawamoto et al.123 Copyright © 2015, American Chemical Society. | ||
Hydrogels are indispensable materials for fabricating 3D macroscopic tissue scaffolds for encapsulation of cells. For cell encapsulation, it is essential to maintain a suitable chemical environment to promote their proliferation and function. Thus, the chemo-selective and bioorthogonal nature of the Tz-IEDDA reaction has been extensively employed in fabricating hydrogel scaffolds that efficiently incorporate cells with high post-encapsulation viability. In a seminal contribution, Anseth and coworkers reported one of the earlier examples of Tz-IEDDA “click” crosslinked hydrogels in which simple mixing of a Tz functionalized multi-armed PEG (Mn ≈ 20 000 g mol−1, 75% Tz functionalization) and dinorbornene-conjugated degradable peptide yielded hydrogels125 (Fig. 11). Mononorbornene-functionalized peptides introduced during gelation provided hydrogels suitable for cell encapsulation and spatially-controlled protein patterning. Notably, the cytocompatible nature of the system led to high post-encapsulation viability of human mesenchymal stem cells.
 | ||
| Fig. 11 Overview of hydrogel formation by Tz-NB IEDDA reaction using multi-functional PEG-Tz and cell degradable dinorbornene crosslinker peptide. Adapted with permission from Alge et al.125 Copyright © 2013, American Chemical Society. | ||
Mooney and coworkers extensively utilized the Tz-IEDDA reaction to fabricate structurally diverse hydrogel scaffolds in cell proliferation studies. In a study, the group reported the fabrication of alginate-based hydrogels by crosslinking individually prepared Tz and NB functionalized alginate polymers.126 To promote cell adhesion, a thiol-modified cell adhesion peptide (cysteine bearing RGD) was also conjugated to the network structure via thiol–ene reaction by employing the NB groups. The alginate-based platform was reported to allow good cell adhesion and proliferation, bringing minimal inflammatory response and high structural stability. In a subsequent study, the group extended the Tz-IEDDA-based crosslinking to hydrolytically-degradable alginate hydrogels.127 A similar thiol–ene-based cell adhesion peptide conjugation to the network structure promoted high cellular growth and viability.
Tailorable functionalization of natural polymers or biopolymers with Tz groups is a convenient strategy to tune the network properties for efficient cell encapsulation and spreading. Since gel porosity and mechanical properties are essential to achieve a suitable scaffold for cell adhesion and spreading, tailorable swelling profiles, rheological, and morphological properties could be attained by changing the total polymer concentration and amount of complementary “click” functional groups. The biocompatible natures of these polymers also have a pivotal role in assisting enhanced cell viability and proliferation. Accordingly, Tz-modified hyaluronic acid,128–132 methylcellulose,133 gelatin134,135 and chondroitin sulfate136,137 have been reported in IEDDA-based hydrogel synthesis for cell culture, neural stem cells delivery, and oxygen-releasing microparticle generation.
The catalyst-free reaction conditions and bioorthogonal nature of the Tz-IEDDA reaction provide ideal cross-linking chemistry that generates sustained release depots for therapeutic proteins and drugs. The enhanced chemoselectivity of Tz-based ligation chemistry is especially advantageous in encapsulating therapeutic proteins in hydrogel scaffolds by minimizing the possible protein-matrix chemical interactions. Famili and Rajagopal exploited Tz-IEDDA reaction with NBs to fabricate hydrogels for encapsulation and release of a model protein, Fab1.138 Hydrogels were obtained using Tz-appended hyaluronic acid (200 kg mol−1) and NB-conjugated PEG (2 kg mol−1) for in situ encapsulation of Fab1 (Fig. 12). Complete release of Fab1 from the hydrogel scaffold occurred over several weeks, and the protein was released in pristine form, without any effect on antigen binding capacity. Raghavan and coworkers reported that IEDDA-generated hyaluronic acid hydrogels can induce antibacterial activity by the slow release of BSA-conjugated silver nanoparticles, which were encapsulated in situ in the gel matrix.139
 | ||
| Fig. 12 Schematic illustration of fabricating protein-releasing hydrogels using Tz-IEDDA-based crosslinking of hyaluronic acid and PEG polymers. Adapted with permission from Famili et al.138 Copyright © 2017, American Chemical Society. | ||
Injectable hydrogels are essential in designing deliverable drug formulations, local tissue engineering scaffolds, and dermal fillers on site. Due to fast reaction kinetics under mild conditions with high chemo-selectivity between the reactive partners, the Tz-IEDDA “click” reaction provides an attractive avenue for the fabrication of injectable hydrogels.140 Since the network physicochemical properties are paramount to attain the desired application features, easy installation of Tz and complementary alkene functionalities to a diverse set of polymer structures allows fine-tuning of matrix properties after gel formation.141,142 The bioorthogonal and biocompatible nature of this reaction has proven especially useful in 3D cell encapsulation studies.143,144 In injectable systems, cells can be pre-suspended in the hydrogel precursor solutions and locally administered to the target body. This approach was demonstrated in Tz-IEDDA based in situ encapsulation and delivery of stem cells for osteogenic differentiation145,146 and cartilage tissue engineering.147,148 Tz-IEDDA injectable hydrogels have been efficiently utilized for in vivo delivery of chemical or protein therapeutic agents to articular joints,149 inner ear,150 intervertebral discs,151 bone tissue152 and dorsal skin.153,154
Tz-IEDDA-based in situ gelation could provide a suitable platform for the delivery of anticancer agents to the target tumor tissue.155 In a study to prepare injectable polypeptide hydrogels for localized cisplatin release, Chen and coworkers employed NB-modified poly(L-glutamic acid) (5.4% degree of substitution) and Tz-functionalized four-arm PEG (Mn = 10000 g mol−1) (Fig. 13).156 The antitumor agent cisplatin was attached to the network structure through polymer-metal complexation with the carboxylic acid groups of poly(L-glutamic acid). A rapid hydrogel formation was attained after subcutaneous injection of the gel formulation in rats, and good stability was observed in vivo. This cisplatin-loaded delivery platform exhibited reduced systemic toxicity and improved antitumor efficacy in an MCF-7-bearing mice model.
 | ||
| Fig. 13 Schematic representation of preparation and delivery of injectable hydrogel system loaded with antitumor agent. Adapted with permission from Zhang et al.156 Copyright © 2020, Molecular Diversity Preservation International (MDPI). | ||
Stimuli-responsive hydrogels have emerged as an important subclass of hydrogels, where the triggered release of bioactive agents in the presence of internal or external stimuli provides increased control.157,158 In several studies, Tz-IEDDA reaction has been prompted for incorporation of stimuli-responsive linkages into hydrogels for controlled drug release applications. For example, Lim and coworkers reported an injectable redox-responsive hydrogel by utilizing alginate functionalized with pendant NB units and a Tz-based PEG cross-linker where the Tz group is linked through disulfide linkages (Fig. 14).159 The authors demonstrated that the encapsulated doxorubicin was efficiently (∼92%) released in the presence of glutathione (GSH) (10 mM) with minimal release (<25%) in the physiological buffer.
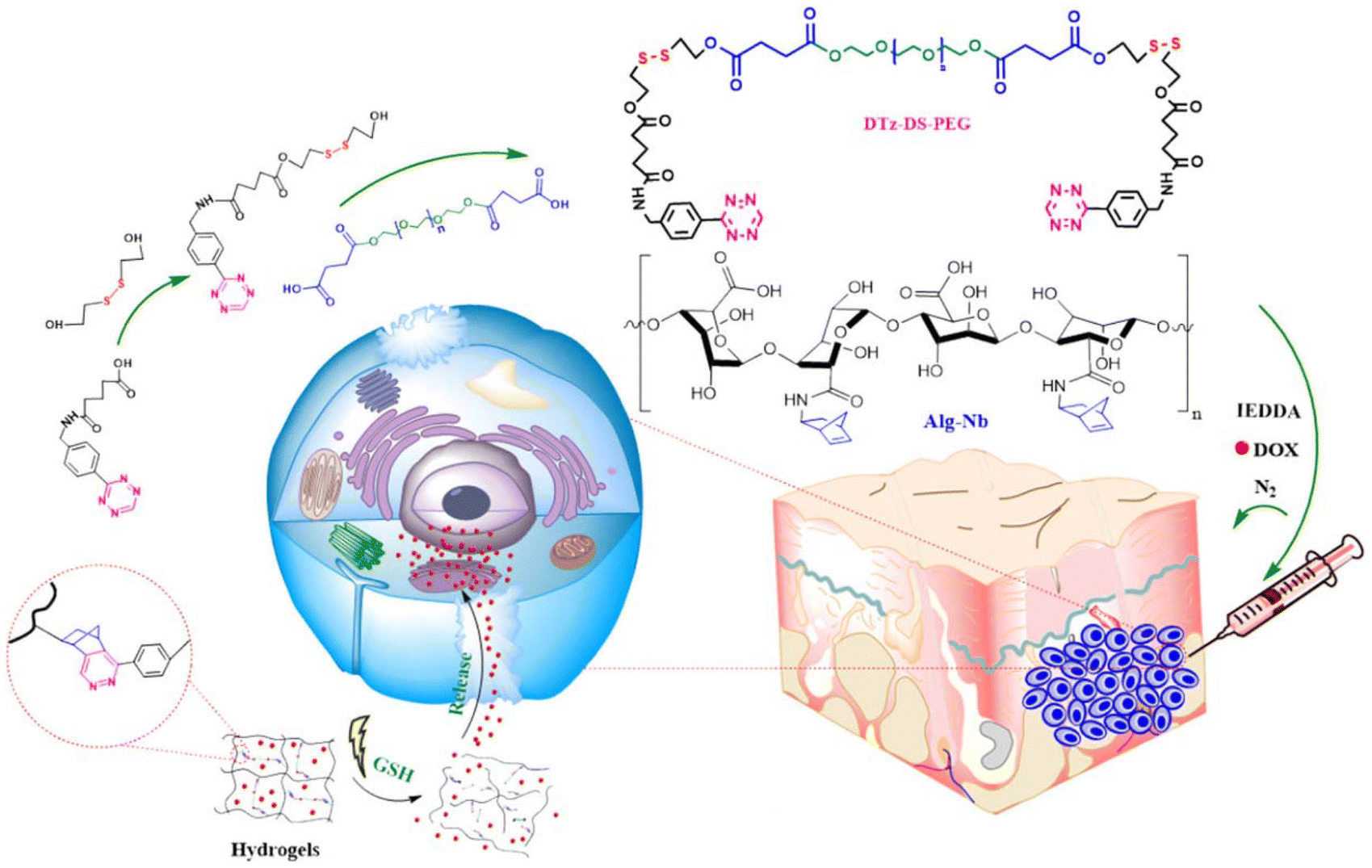 | ||
| Fig. 14 Fabrication of an alginate-based injectable and reduction-responsive hydrogels. Adapted with permission from Vu et al.159 Copyright © 2021 Elsevier. | ||
In another related work, Lim and coworkers reported Tz-IEDDA generated diselenide-containing hydrogels for stimuli-responsive drug release responsive to near-infrared (NIR) irradiation.160 Hydrogels were obtained by using NB-functionalized alginate polymer (25.53% NB functionalization) and a diselenide-containing Tz crosslinker with high conversions of 92–96% (Fig. 15). NIR-sensitive indocyanine green (ICG) dye and DOX were loaded into the hydrogel matrix during gelation. ICG generated reactive oxygen species (ROSs) upon NIR-light irradiation, which triggered hydrogel degradation through cleavage of the diselenide bonds and allowed the release of the encapsulated drug. In another work, the group reported reduction-responsive and biorthogonal carboxymethyl cellulose hydrogels obtained using a similar approach.161 In this work, DOX release from hydrogels was achieved through the cleavage of diselenide bonds in the presence of reducing agent GSH. In a subsequent study, alginate-based biocompatible hydrogels containing redox-responsive disulfide linkers for GSH-responsive DOX release were reported.162 Recently, the group extended their scope to hyaluronic acid-based Tz-IEDDA hydrogels that show multi-stimuli responsive drug release.163 It was demonstrated that diselenide crosslinked networks can undergo increased DOX release triggered by exposure to DTT, H2O2, and NIR irradiation.
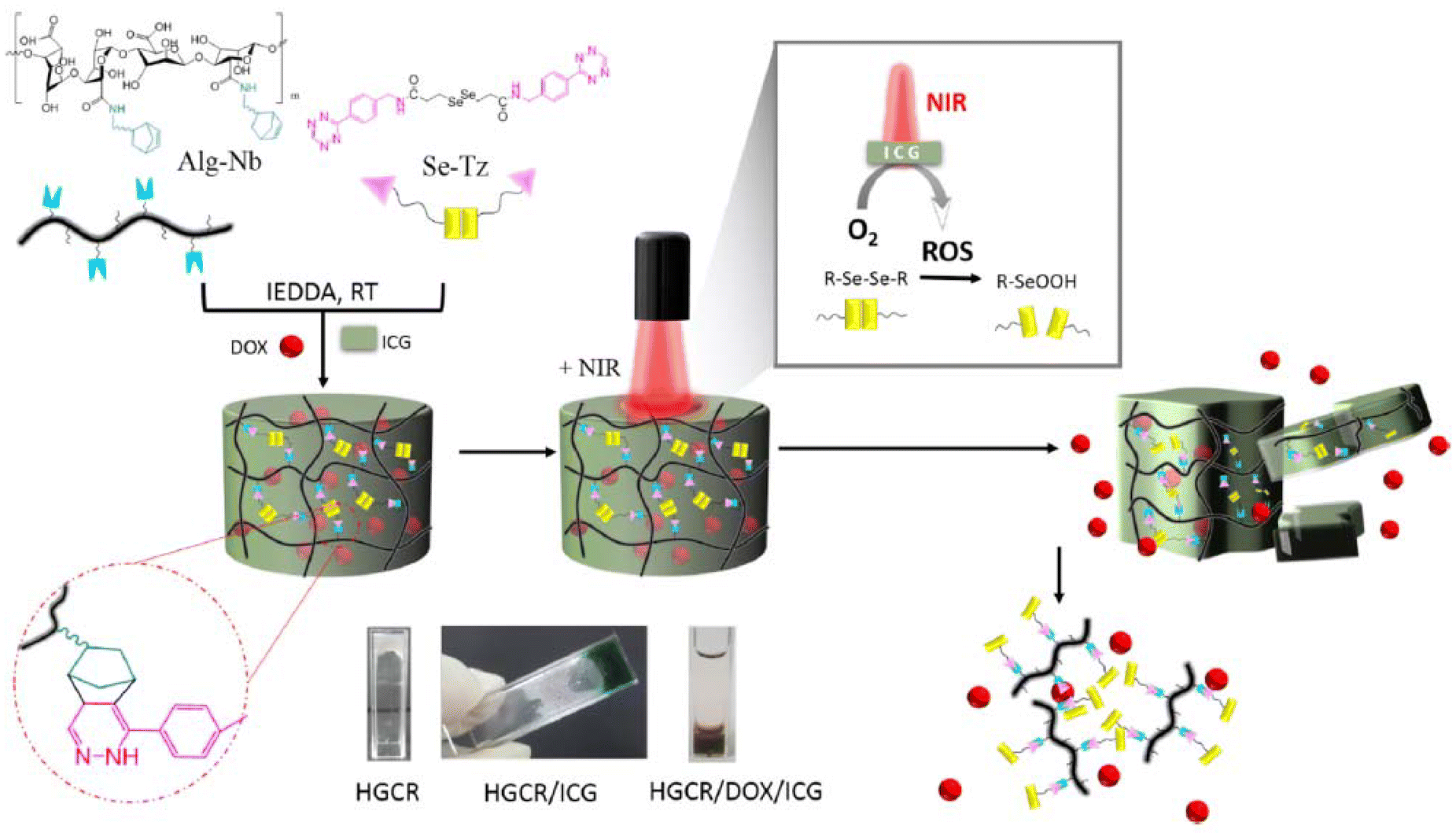 | ||
| Fig. 15 Fabrication of Tz-IEDDA generated NIR-responsive alginate hydrogels. Adapted with permission from Anugrah et al.160 Copyright © 2019, Elsevier. | ||
In addition to approaches that utilize the Tz-IEDDA reaction for chemical crosslinking of polymers, this chemistry has also been exploited in the post-polymerization functionalization of pre-fabricated hydrogels. In a report, Alge and coworkers demonstrated the preparation and functionalization of hydrogel microparticles via sequential thiol–ene addition and Tz-IEDDA “click” reactions.164 Hydrogels were synthesized by photo-polymerizing tetra-functional PEG-NB macromer/dithiothreitol mixtures emulsified in the dextran phase. Hydrogel microparticles with controlled sizes were obtained by adjusting the stoichiometry between NB and thiol groups. It was shown that the unreacted strained alkene functionalities can efficiently be conjugated with Tz-containing model proteins such as fluorescein-labeled ovalbumin, as well as bioactive proteins alkaline phosphatase (ALP) and glucose oxidase (GOx). After functionalization, both ALP and GOx maintained their bioactivity, as demonstrated by enzyme activity assays. In a later study, the group employed a similar procedure to functionalize nucleophilic/radical thiol–ene-generated peptide hydrogels via a Tz-based IEDDA reaction.165 Installation of Tz groups onto pre-fabricated hydrogels is another strategy for further functionalization of the templates by using appropriately modified bioactive molecules. This was demonstrated by Yi and coworkers to install Tz groups onto chitosan-polyacrylamide-166 and chitosan-poly(ethylene glycol)167 -based hydrogel microspheres.
4. Tetrazine-mediated polymer functionalization on solid surfaces
Reactive polymeric coatings that are applied on organic/inorganic solid platforms including metal surfaces, polymer substrates, glass slips or wafers are indispensable platforms in various biomedical applications.168 While polymeric coatings can bestow range of important properties to applied surfaces, such as anti-biofouling, wear resistance, corrosion protection and biocompatibility; deliberately installed reactive units on these coatings might endow specific covalent attachment of desired molecules.169–171 Such functional platforms have especially held promise in cell culture, aligned cell growth, peptide purification, enzyme immobilization applications, and microarray technologies.172,173In the past decade, Tz-based IEDDA reaction has found a growing interest in modifying reactive polymeric coatings that are applied on solid surfaces. In an early report, Ravoo and coworkers used a microcontact printing technique and surface-initiated ATRP polymerization to obtain patterned glass and silicon surfaces with reactive Tz groups.174 By using a Tz functional ATRP initiator, poly(methyl acrylate) brushes were efficiently grown in grafting from the approach on 10-undecyl trichlorosilane-modified surfaces. These reactive patterned surfaces were subsequently modified with both NB and cyclooctyne-containing substrates via IEDDA reaction in short reaction times and room temperature conditions. In a recent study, Kahveci and coworkers developed a grafting technique for light-induced IEDDA modification of glass surfaces (Fig. 16).175 TCO-decorated surfaces were grafted with latent Tz functional dihydrotetrazine end group containing p(NIPAAm) and PLA polymers. It was demonstrated that the conversion of dihydrotetrazine groups to IEDDA clickable Tz functionalities could be achieved by a light-promoted photooxidation process allowing on-demand modification of glass surfaces with relevant polymers or biomolecules.
 | ||
| Fig. 16 Schematic illustration of a surface grafting approach based on light-induced inverse electron demand Diels–Alder (photo-IEDDA) reaction. Adapted with permission from Ozbek et al.175 Copyright © 2021 American Chemical Society. | ||
Titanium has become the material of choice in many implant applications originating from unique advantages that this metal can offer i.e. outstanding mechanical properties, resistance to erosion, and durability. On the other hand, biocompatibility to promote cell adhesion or osseointegration on biomaterial is essential for the long-term success of titanium-based implants. To enhance the surface adhesive properties and proliferation of cells, a practiced strategy is to attach specific cell-binding moieties on the metal surface covalently. Pagel et al. employed a mussel-derived peptide containing strong metal affinity rendering DOPA (L-3,4-dihydroxylphenylalanine) groups and two cell binding motifs to increase the cell adhesion on the titanium surface.176 Two “click” chemistry-based transformations, IEDDA and copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction, were utilized to conjugate a cyclic RGD peptide and a heparin-binding peptide to the reactive platform. It was shown that the conjugation of two cell-binding moieties on the peptide allowed the improved spreading, proliferation, and viability of cells on the titanium surface, rendering well-developed actin cytoskeleton formation. In a similar approach, Czuban et al. used a DOPA-containing Tz functional dendritic polyglycerol polymer to coat titanium surfaces.177 An antimicrobial agent with a carbamate-TCO linker was then coupled onto the polymeric coating to facilitate the slow drug release. Bacterial experiments demonstrated that coated titanium surfaces can activate the release of anti-infective medication after IEDDA modification. This bioorthogonally reactive polymeric coating was conceptualized to prevent orthopedic implant-associated infections. In another work, Sanyal and coworkers demonstrated the fabrication of titanium surface-bound hydrogels amenable to functionalization with several “click” chemistry reactions (Fig. 17).178 Hydrogel layers incorporating furan-protected maleimide groups as reactive handles were prepared by UV-based free radical crosslinking approach. In the study, oxanorbornene groups were efficiently functionalized with IEDDA and radical thiol–ene reactions by covalent attachment of molecular cues with complementary Tz and thiol functionalities. It was also demonstrated that, after furan deprotection and liberation of maleimide groups, nucleophilic thiol-maleimide addition and Diels–Alder chemistries could also be applied to modify these surface-bound hydrogel platforms covalently. In another work, the same group extended the fabrication of a multi-functionalizable bioconjugation platform concept to surface-immobilized nanofibers.179 Electrospun PLA-based polymers carrying orthogonal oxanorbornene and azide groups were efficiently functionalized with Tz and cyclooctyne-bearing bioactive cues via sequential SPAAC and IEDDA bioorthogonal “click” reactions. Recently, sequential SPAAC and IEDDA functionalization strategy was used by Huskens and coworkers on gold and silicon dioxide surfaces coated with either Tz or cyclooctyne functional poly-L-lysine (PLL) polymers.180 It was shown that DNA probe molecules with complementary azide and TCO groups can be selectively conjugated to the surfaces within less than 20 min incubation period. Notably, while modified surfaces showed hybridization with entirely complementary DNA sequences, they did not respond to non-complementary DNA sequences.
 | ||
| Fig. 17 Multi-functionalization of titanium surface bound hydrogels using different “click” chemistry reactions, including the IEDDA reaction. Adapted with permission from Gevrek et al.178 Copyright © 2020, Molecular Diversity Preservation International (MDPI). | ||
Dankers and coworkers investigated the supramolecular elastomeric materials based on ureido pyrimidinone matrices as anti-fouling coating materials.181 The supramolecular polymer scaffolds facilitating Tz groups as IEDDA reactive handles were surface modified by cyclooctyne end group containing PEG polymers with different topologies. The protein adsorption experiments revealed that the efficiency of anti-fouling behavior is directly related to the PEG polymer applied, i.e., the best performance was accounted for by using a star PEG polymer (Mn = 10000 g mol−1) rendering four cyclooctyne groups, then monofunctional and bifunctional PEG-cyclooctyne derivatives (Mn = 2000 g mol−1 and 5000 g mol−1). In a follow-up study, using a TCO-functional peptide derivative, these Tz functional supramolecular substrates fabricated as cast films or electrospun fiber mats were employed in specific protein immobilization.182
5. Outlook and conclusions
The preceding sections highlight the significant potential of IEDDA cycloaddition reactions in designing and synthesizing functional polymeric materials. While IEDDA reactions follow the core principles of “click” chemistry, their biorthogonal nature extends their utility to new applications. Notably, in vivo studies have demonstrated the reaction's effectiveness in living systems. Furthermore, the high efficiency of IEDDA reactions makes them invaluable for post-polymerization modifications, particularly when rapid transformations are needed, such as the conjugation of short-lived species like radioisotopes.Current research predominantly focuses on incorporating Tz moieties through post-polymerization modifications. However, developing polymerization methods that directly yield polymers with these reactive handles would be advantageous. This approach poses challenges with vinylic monomers due to the reactivity of electron-deficient dienes with double bonds, but it may be more feasible with non-vinylic monomers, such as those used in ROP. As reviewed, the excellent selectivity and high yield of IEDDA reactions also make them suitable for fabricating network structures. Although numerous examples of such applications exist, integrating inorganic components into hybrid materials remains limited. Advancing the development of biomolecule-polymer hybrid materials could lead to innovative functional materials for biomedical applications. Nonetheless, challenges related to the multi-step synthesis of reactive counterparts persist. Continued progress in synthesizing Tz-bearing linkers and their complementary reactive partners is expected. Ultimately, the remarkable efficiency of the IEDDA reactions in addressing specific challenges in fabricating functional polymeric materials for targeted applications will continue to drive their broader adoption and utilization.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.Data availability
No primary research results have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.References
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- A. M. Prokhorov and D. N. Kozhevnikov, Chem. Heterocycl. Compd., 2012, 48, 1153–1176 CrossRef CAS.
- N. Saracoglu, Tetrahedron, 2007, 63, 4199–4236 CrossRef CAS.
- Z. Geng, J. J. Shin, Y. Xi and C. J. Hawker, J. Polym. Sci., 2021, 59, 963–1042 CrossRef CAS.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145 RSC.
- A. Sanyal, Macromol. Chem. Phys., 2010, 211, 1417–1425 CrossRef CAS.
- G. Hizal, U. Tunca and A. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4103–4120 CrossRef CAS.
- M. Cetin, C. Esen, O. Daglar, S. Luleburgaz, G. Hizal, H. Durmaz and U. Tunca, Polym. Chem., 2016, 7, 7094–7100 RSC.
- A. Dag, H. Durmaz, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2008, 47, 178–187 CrossRef.
- H. Sun, C. P. Kabb, Y. Dai, M. R. Hill, I. Ghiviriga, A. P. Bapat and B. S. Sumerlin, Nat. Chem., 2017, 9, 817–823 CrossRef CAS PubMed.
- P. Espeel and F. E. Du Prez, Macromolecules, 2015, 48, 2–14 CrossRef CAS.
- K. K. Oehlenschlaeger, N. K. Guimard, J. Brandt, J. O. Mueller, C. Y. Lin, S. Hilf, A. Lederer, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Polym. Chem., 2013, 4, 4348–4355 RSC.
- A. Bousquet, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1773–1781 CrossRef CAS.
- J. Yang, M. R. Karver, W. Li, S. Sahu and N. K. Devaraj, Angew. Chem., Int. Ed., 2012, 51, 5222–5225 CrossRef CAS PubMed.
- R. A. Carboni and R. V. Lindsey, J. Am. Chem. Soc., 1959, 81, 4342–4346 CrossRef CAS.
- D. L. Boger, Chem. Rev., 1986, 86, 781–793 CrossRef CAS.
- J. Zhang, V. Shukla and D. L. Boger, J. Org. Chem., 2019, 84, 9397–9445 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816–827 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- S. A. Busche, S. Peplau, L. Zuhse, D. Steimer, D. D. August and H. G. Börner, Polym. Chem., 2023, 14, 2888–2892 RSC.
- A. R. Sayed and J. S. Wiggins, J. Heterocycl. Chem., 2024, 61, 118–126 CrossRef CAS.
- A. R. Sayed and J. S. Wiggins, J. Appl. Polym. Sci., 2011, 120, 623–630 CrossRef CAS.
- Z. Li and J. Ding, Macromol. Chem. Phys., 2011, 212, 2260–2267 CrossRef CAS.
- L. Peng, Y. Yu, J. Lu, P. He, G. Wang, M. Huang, B. Zhao, Y. Pei and S. Tan, Dyes Pigm., 2019, 171, 107717 CrossRef CAS.
- J. Ding, Z. Li, Z. Cui, G. P. Robertson, N. Song, X. Du and L. Scoles, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3374–3386 CrossRef CAS.
- J. S. Panek and B. Zhu, Tetrahedron Lett., 1996, 37, 8151–8154 CrossRef CAS.
- S. L. Scinto, D. A. Bilodeau, R. Hincapie, W. Lee, S. S. Nguyen, M. Xu, C. W. Am Ende, M. G. Finn, K. Lang, Q. Lin, J. P. Pezacki, J. A. Prescher, M. S. Robillard and J. M. Fox, Nat. Rev. Methods Primers, 2021, 1, 30 CrossRef CAS PubMed.
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825–858 CrossRef CAS PubMed.
- R. E. Bird, S. A. Lemmel, X. Yu and Q. A. Zhou, Bioconjugate Chem., 2021, 32, 2457–2479 CrossRef CAS PubMed.
- N. K. Devaraj, G. M. Thurber, E. J. Keliher, B. Marinelli and R. Weissleder, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4762–4767 CrossRef CAS PubMed.
- J.-P. Meyer, K. M. Tully, J. Jackson, T. R. Dilling, T. Reiner and J. S. Lewis, Bioconjugate Chem., 2018, 29, 538–545 CrossRef CAS PubMed.
- Y. Dong, Y. Tu, K. Wang, C. Xu, Y. Yuan and J. Wang, Angew. Chem., Int. Ed., 2020, 59, 7168–7172 CrossRef CAS PubMed.
- S. Liu, H. Zhang, R. A. Remy, F. Deng, M. E. Mackay, J. M. Fox and X. Jia, Adv. Mater., 2015, 27, 2783–2790 CrossRef CAS PubMed.
- H. Zhang, W. S. Trout, S. Liu, G. A. Andrade, D. A. Hudson, S. L. Scinto, K. T. Dicker, Y. Li, N. Lazouski, J. Rosenthal, C. Thorpe, X. Jia and J. M. Fox, J. Am. Chem. Soc., 2016, 138, 5978–5983 CrossRef CAS PubMed.
- M. M. Lorenzo, C. G. Decker, M. U. Kahveci, S. J. Paluck and H. D. Maynard, Macromolecules, 2016, 49(1), 30–37 CrossRef CAS PubMed.
- T. R. Wilks and R. K. O'Reilly, Sci. Rep., 2016, 6, 39192 CrossRef CAS PubMed.
- S. S. Kara, M. Y. Ateş, G. Deveci, A. Cetinkaya and M. U. Kahveci, J. Polym. Sci., Part A: Polym. Chem., 2018, 57, 673–680 CrossRef.
- G. Bakirdogen, E. L. Sahkulubey Kahveci and M. U. Kahveci, Eur. Polym. J., 2020, 140, 110027 CrossRef CAS.
- E. Meyvacı and T. Öztürk, ChemistrySelect, 2022, 7, e202200668 CrossRef.
- M. Ambrosi, N. R. Cameron and B. G. Davis, Org. Biomol. Chem., 2005, 3, 1593–1608 RSC.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef PubMed.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Angew. Chem., Int. Ed., 2006, 45, 2348–2368 CrossRef CAS PubMed.
- C. Timperanza, H. Jensen, T. Bäck, S. Lindegren and E. Aneheim, Pharmaceuticals, 2023, 16, 595 CrossRef CAS PubMed.
- I. Lepori, Y. Oz, J. Lm, N. Ghosh, M. Paul, U. S. Schubert and S. Fedeli, Reactions, 2024, 5, 231–245 CrossRef CAS.
- C. F. Hansell, P. Espeel, M. M. Stamenovic, I. A. Barker, A. P. Dove, F. E. Du Prez and R. K. O'Reilly, J. Am. Chem. Soc., 2011, 133, 13828–13831 CrossRef CAS PubMed.
- K. Johann, D. Svatunek, C. Seidl, S. Rizzelli, T. A. Bauer, L. Braun, K. Koynov, H. Mikula and M. Barz, Polym. Chem., 2020, 11, 4396–4407 RSC.
- S. A. van den Berg, H. Zuilhof and T. Wennekes, Macromolecules, 2016, 49, 2054–2062 CrossRef CAS.
- O. Stetsiuk, A. Abherve and N. Avarvari, Dalton Trans., 2020, 49, 5759–5777 RSC.
- S. Kramer, D. Svatunek, I. Alberg, B. Gräfen, S. Schmitt, L. Braun, A. H. A. M. van Onzen, R. Rossin, K. Koynov, H. Mikula and R. Zentel, Biomacromolecules, 2019, 20, 3786–3797 CrossRef CAS PubMed.
- K. R. Aswani, S. Joshi, R. Ghosh and R. R. Malakalapalli, Polym. Adv. Technol., 2023, 34, 1331–1340 CrossRef.
- L. Xu, W. Tao, M. Guan, X. Yang, M. Huang, H. Chen, J. Zhang, B. Zhao and S. Tan, ACS Appl. Energy Mater., 2021, 4, 11624–11633 CrossRef CAS.
- V. Kardelis, M. M. Denk and A. Adronov, Angew. Chem., Int. Ed., 2021, 60, 2980–2986 CrossRef CAS PubMed.
- D. S. Rivero, R. E. Paiva-Feener, T. Santos, E. Martín-Encinas and R. Carrillo, Macromolecules, 2021, 54, 10428–10434 CrossRef CAS.
- T. Santos, D. S. Rivero, Y. Pérez-Pérez, E. Martín-Encinas, J. Pasán, A. H. Daranas and R. Carrillo, Angew. Chem., Int. Ed., 2021, 60, 18783–18791 CrossRef CAS PubMed.
- Z. M. Png, X. Y. D. Soo, J. X. D. Liew, M. H. Chua, S. Xiong, Q. Zhu and J. Xu, Polym. Chem., 2023, 14, 3347–3351 RSC.
- R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302–6337 CrossRef CAS PubMed.
- N. B. Cramer, S. K. Reddy, A. K. O'Brien and C. N. Bowman, Macromolecules, 2003, 36, 7964–7969 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- I. A. Barker, D. J. Hall, C. F. Hansell, F. E. Du Prez, R. K. O'Reilly and A. P. Dove, Macromol. Rapid Commun., 2011, 32, 1362–1366 CrossRef CAS PubMed.
- R. J. Williams, I. A. Barker, R. K. O'Reilly and A. P. Dove, ACS Macro Lett., 2012, 1, 1285–1290 CrossRef CAS PubMed.
- R. Baumgartner, Z. Song, Y. Zhang and J. Cheng, Polym. Chem., 2015, 6, 3586–3590 RSC.
- S. Jain, K. Neumann, Y. Zhang, J. Geng and M. Bradley, Macromolecules, 2016, 49, 5438–5443 CrossRef CAS.
- H.-S. Han, N. K. Devaraj, J. Lee, S. A. Hilderbrand, R. Weissleder and M. G. Bawendi, J. Am. Chem. Soc., 2010, 132, 7838–7839 CrossRef CAS PubMed.
- Y. Zhang, A. Gambardella, M. Üçüncü, J. Geng, J. Clavadetscher, M. Bradley and A. Lilienkampf, Chem. Commun., 2020, 56, 13856–13859 RSC.
- Y. Zhang, M. Üçüncü, A. Gambardella, A. Baibek, J. Geng, S. Zhang, J. Clavadetscher, I. Litzen, M. Bradley and A. Lilienkampf, J. Am. Chem. Soc., 2020, 142, 21615–21621 CrossRef CAS PubMed.
- C. F. Hansell and K. O'Reilly, ACS Macro Lett., 2012, 1, 896–901 CrossRef CAS PubMed.
- C. F. Hansell, A. Lu, J. P. Patterson and R. K. O'Reilly, Nanoscale, 2014, 6, 4102–4107 RSC.
- S. Xie, M. Chen, X. Song, Z. Zhang, Z. Zhang, Z. Chen and X. Li, Acta Biomater., 2018, 78, 198–210 CrossRef CAS PubMed.
- A. H. St Amant, E. H. Discekici, S. J. Bailey, M. S. Zayas, J.-A. Song, S. L. Shankel, S. N. Nguyen, M. W. Bates, A. Anastasaki, C. J. Hawker and J. R. de Alaniz, J. Am. Chem. Soc., 2019, 141, 13619–13624 CrossRef CAS PubMed.
- S. J. Bailey, E. H. Discekici, S. M. Barbon, S. N. Nguyen, C. J. Hawker and J. Read de Alaniz, Macromolecules, 2020, 53, 4917–4924 CrossRef CAS PubMed.
- R. Rossin, S. M. van den Bosch, W. ten Hoeve, M. Carvelli, R. M. Versteegen, J. Lub and M. S. Robillard, Bioconjugate Chem., 2013, 24, 1210–1217 CrossRef CAS PubMed.
- F. Thalhammer, U. Wallfahrer and J. Sauer, Tetrahedron Lett., 1990, 31, 6851–6854 CrossRef CAS.
- D. Svatunek, C. Denk, V. Rosecker, B. Sohr, C. Hametner, G. Allmaier, J. Fröhlich and H. Mikula, Monatsh. Chem., 2016, 147, 579–585 CrossRef CAS PubMed.
- V. Shalgunov, G. Engudar, L. Bohrmann, L. Wharton, K. Maskell, K. Johann, M. Barz, P. Schaffer, M. M. Herth and V. Radchenko, Nucl. Med. Biol., 2022, 104–105, 11–21 CrossRef CAS PubMed.
- B. E. Cook, R. Membreno and B. M. Zeglis, Bioconjugate Chem., 2018, 29, 2734–2740 CrossRef CAS PubMed.
- E. J. L. Stéen, J. T. Jørgensen, K. Johann, K. Nørregaard, B. Sohr, D. Svatunek, A. Birke, V. Shalgunov, P. E. Edem, R. Rossin, C. Seidl, F. Schmid, M. S. Robillard, J. L. Kristensen, H. Mikula, M. Barz, A. Kjær and M. M. Herth, ACS Nano, 2020, 14, 568–584 CrossRef PubMed.
- R. García-Vázquez, U. M. Battisti, V. Shalgunov, G. Schäfer, M. Barz and M. M. Herth, Macromol. Rapid Commun., 2021, 43, 2100655 CrossRef PubMed.
- J. A. C. M. Goos, M. Davydova, T. R. Dilling, A. Cho, M. A. Cornejo, A. Gupta, W. S. Price, S. Puttick, M. R. Whittaker, J. F. Quinn, T. P. Davis and J. S. Lewis, Nucl. Med. Biol., 2020, 84–85, 63–72 CrossRef CAS PubMed.
- L. Xu, M. Raabe, M. M. Zegota, J. C. F. Nogueira, V. Chudasama, S. L. Kuan and T. Weil, Org. Biomol. Chem., 2020, 18, 1140–1147 RSC.
- M. Arslan, G. Acik and M. A. Tasdelen, Polym. Chem., 2019, 10, 3806–3821 RSC.
- M. Arslan and M. A. Tasdelen, Polymers, 2017, 9, 499 CrossRef PubMed.
- M. Arslan and M. A. Tasdelen, Chem. Afr., 2019, 2, 195–214 CrossRef.
- R. E. Bagge, T. C. Mauldin, D. J. Boday, B. M. Kobilka and D. A. Loy, Chem. Mater., 2017, 29, 7953–7960 CrossRef CAS.
- Q. Wang, W. Wang, Q. Li and C. Wu, Ind. Eng. Chem. Res., 2021, 60, 2163–2177 CrossRef CAS.
- Q. Wang, Y. Shi, Q. Li and C. Wu, Eur. Polym. J., 2021, 150, 110415 CrossRef CAS.
- H. Zhou and J. A. Johnson, Angew. Chem., Int. Ed., 2013, 52, 2235–2238 CrossRef CAS PubMed.
- H. Zhou, J. Woo, A. M. Cok, M. Wang, B. D. Olsen and J. A. Johnson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19119–19124 CrossRef CAS PubMed.
- H. Zhou, E.-M. Schön, M. Wang, M. J. Glassman, J. Liu, M. Zhong, D. D. Díaz, B. D. Olsen and J. A. Johnson, J. Am. Chem. Soc., 2014, 136, 9464–9470 CrossRef CAS PubMed.
- A. Degirmenci, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2024, 35, 433–452 CrossRef CAS PubMed.
- S. A. Busche, M. Traxler, A. Thomas and H. G. Börner, ChemSusChem, 2024, 17, e202301045 CrossRef CAS PubMed.
- A. M. Cok, H. Zhou and J. A. Johnson, Macromol. Symp., 2013, 329, 108–112 CrossRef CAS.
- S. E. Holt, A. Rakoski, F. Jivan, L. M. Pérez and D. L. Alge, Macromol. Rapid Commun., 2020, 41, 2000287 CrossRef CAS PubMed.
- D. S. B. Anugrah, M. P. Patil, X. Li, C. M. Q. Le, K. Ramesh, G.-D. Kim, K. Hyun and K. T. Lim, eXPRESS Polym. Lett., 2020, 14, 248–260 CrossRef CAS.
- S. Liu, A. C. Moore, A. B. Zerdoum, H. Zhang, S. L. Scinto, H. Zhang, L. Gong, D. L. Burris, A. K. Rajasekaran, J. M. Fox and X. Jia, Biomaterials, 2018, 180, 24–35 CrossRef CAS PubMed.
- O. J. George, J. Song, J. M. Benson, Y. Fang, H. Zhang, D. L. Burris, J. M. Fox and X. Jia, Biomacromolecules, 2022, 23, 3017–3030 CrossRef CAS PubMed.
- R. P. Tello, S. Wang, F. Fontana, A. Correia, G. Molinaro, S. L. Cerdà, S. Hietala, J. Hirvonen, G. Barreto and H. A. Santos, Biomater. Sci., 2023, 11, 4972–4984 RSC.
- H. Zhang, K. T. Dicker, X. Xu, X. Jia and J. M. Fox, ACS Macro Lett., 2014, 3, 727–731 CrossRef CAS PubMed.
- K. T. Dicker, A. C. Moore, N. T. Garabedian, H. Zhang, S. L. Scinto, R. E. Akins, D. L. Burris, J. M. Fox and X. Jia, ACS Appl. Mater. Interfaces, 2019, 11, 16402–16411 CrossRef CAS PubMed.
- N. J. Darling, W. Xi, E. Sideris, A. R. Anderson, C. Pong, S. T. Carmichael and T. Segura, Adv. Healthcare Mater., 2020, 9, 1901391 CrossRef CAS PubMed.
- K. L. Wilson, S. C. L. Pérez, M. M. Naffaa, S. H. Kelly and T. Segura, Adv. Mater., 2022, 34, 2201921 CrossRef CAS PubMed.
- A. Isaac, F. Jivan, S. Xin, J. Hardin, X. Luan, M. Pandya, T. G. H. Diekwisch and D. L. Alge, ACS Biomater. Sci. Eng., 2019, 5, 6395–6404 CrossRef CAS PubMed.
- F. Jivan and D. L. Alge, Adv. Ther., 2020, 3, 1900148 CrossRef PubMed.
- T. J. Tigner, G. Dampf, A. Tucker, Y.-C. Huang, V. Jagrit, A. J. Clevenger, A. Mohapatra, S. A. Raghavan, J. N. Dulin and D. L. Alge, Adv. Healthcare Mater., 2024, e2303912 CrossRef PubMed.
- A. Lueckgen, D. S. Garske, A. Ellinghaus, D. J. Mooney, G. N. Duda and A. Cipitria, Acta Biomater., 2020, 115, 185–196 CrossRef CAS PubMed.
- A. L. Rutz, E. S. Gargus, K. E. Hyland, P. L. Lewis, A. Setty, W. R. Burghardt and R. N. Shah, Acta Biomater., 2019, 99, 121–132 CrossRef CAS PubMed.
- A. Oehrl, S. Schötz and R. Haag, Macromol. Rapid Commun., 2020, 41, 1900510 CrossRef CAS PubMed.
- A. Oehrl, S. Schötz and R. Haag, Colloid Polym. Sci., 2020, 298, 719–733 CrossRef CAS.
- S. Schötz, F. Reisbeck, A.-C. Schmitt, M. Dimde, E. Quaas, K. Achazi and R. Haag, Pharmaceutics, 2021, 13, 1276 CrossRef PubMed.
- S. T. Koshy, D. K. Y. Zhang, J. M. Grolman, A. G. Stafford and D. J. Mooney, Acta Biomater., 2018, 65, 36–43 CrossRef CAS PubMed.
- V. X. Truong, M. P. Ablett, S. M. Richardson, J. A. Hoyland and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 1618–1622 CrossRef CAS PubMed.
- M. R. Arkenberg, N. H. Dimmitt, H. C. Johnson, K. R. Koehler and C.-C. Lin, Adv. Biosyst., 2020, 4, 2000129 CrossRef CAS PubMed.
- S. E. Holt, J. Arroyo, E. Poux, A. Fricks, I. Agurcia, M. Heintschel, A. Rakoski and D. L. Alge, Biomacromolecules, 2021, 22, 3040–3048 CrossRef CAS PubMed.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2011, 22, 2263–2270 CrossRef CAS PubMed.
- V. X. Truong, K. M. Tsang, F. Ercole and J. S. Forsythe, Chem. Mater., 2017, 29, 3678–3685 CrossRef CAS.
- J. Carthew, J. E. Frith, J. S. Forsythe and V. X. Truong, J. Mater. Chem. B, 2018, 6, 1394–1401 RSC.
- C. Wang, H. Zhang, T. Zhang, X. Zou, H. Wang, J. E. Rosenberger, R. Vannam, W. S. Trout, J. B. Grimm, L. D. Lavis, C. Thorpe, X. Jia, Z. Li and J. M. Fox, J. Am. Chem. Soc., 2021, 143, 10793–10803 CrossRef CAS PubMed.
- C. E. Ziegler, M. Graf, M. Nagaoka, H. Lehr and A. M. Goepferich, Biomacromolecules, 2021, 22, 3223–3236 CrossRef CAS PubMed.
- C. E. Ziegler, M. Graf, M. Nagaoka and A. M. Goepferich, Macromol. Biosci., 2022, 22, 2200226 CrossRef CAS PubMed.
- N. H. Dimmitt, M. R. Arkenberg, M. M. de Lima Perini, J. Li and C.-C. Lin, ACS Biomater. Sci. Eng., 2022, 8, 4262–4273 CrossRef CAS PubMed.
- Z. Yao, Z. Wang, Y. Yu, C. Zeng and K. Cao, Polymer, 2017, 119, 98–106 CrossRef CAS.
- K. Kawamoto, S. C. Grindy, J. Liu, N. Holten-Andersen and J. A. Johnson, ACS Macro Lett., 2015, 4, 458–461 CrossRef CAS PubMed.
- A.-C. Knall, S. Kovacic, M. Hollauf, D. Reishofer, R. Saf and C. Slugovc, Chem. Commun., 2013, 49, 7325–7327 RSC.
- D. L. Alge, M. A. Azagarsamy, D. F. Donohue and K. S. Anseth, Biomacromolecules, 2013, 14, 949–953 CrossRef CAS PubMed.
- R. M. Desai, S. T. Koshy, S. A. Hilderbrand, D. J. Mooney and N. S. Joshi, Biomaterials, 2015, 50, 30–37 CrossRef CAS PubMed.
- A. Lueckgen, D. S. Garske, A. Ellinghaus, R. M. Desai, A. G. Stafford, D. J. Mooney, G. N. Duda and A. Cipitria, Biomaterials, 2018, 181, 189–198 CrossRef CAS PubMed.
- V. Delplace, P. E. B. Nickerson, A. Ortin-Martinez, A. E. G. Baker, V. A. Wallace and M. S. Shoichet, Adv. Funct. Mater., 2020, 30, 1903978 CrossRef CAS.
- K. A. Gultian, R. Gandhi, T. W. B. Kim and S. L. Vega, Macromol. Biosci., 2023, 23, 2200425 CrossRef CAS PubMed.
- J. Song, H. Gao, H. Zhang, O. J. George, A. S. Hillman, J. M. Fox and X. Jia, ACS Appl. Mater. Interfaces, 2022, 14, 51669–51682 CrossRef CAS PubMed.
- Y. Hao, J. Song, A. Ravikrishnan, K. T. Dicker, E. W. Fowler, A. B. Zerdoum, Y. Li, H. Zhang, A. K. Rajasekaran, J. M. Fox and X. Jia, ACS Appl. Mater. Interfaces, 2018, 10, 26016–26027 CrossRef CAS PubMed.
- M. Pol, H. Gao, H. Zhang, O. J. George, J. M. Fox and X. Jia, Biomaterials, 2023, 299, 122180 CrossRef CAS PubMed.
- V. Delplace, A. J. Pickering, M. H. Hettiaratchi, S. Zhao, T. Kivijärvi and M. S. Shoichet, Biomacromolecules, 2020, 21, 2421–2431 CrossRef CAS.
- N. C. Negrini, A. A. Volponi, P. T. Sharpe and A. D. Celiz, ACS Biomater. Sci. Eng., 2021, 7, 4330–4346 CrossRef PubMed.
- M. R. Arkenberg, K. Koehler and C.-C. Lin, Biomacromolecules, 2022, 23, 4141–4152 CrossRef CAS PubMed.
- E. de Sousa Araújo, T. D. Stocco, G. F. de Sousa, S. Afewerki, F. R. Marciano, M. A. F. Corat, M. M. M. de Paula, T. F. C. L. Verde, M. C. M. Silva and A. O. Lobo, Colloids Surf., B, 2021, 205, 111850 CrossRef PubMed.
- G. F. Sousa, S. Afewerki, D. Dittz, F. E. P. Santos, D. O. Gontijo, S. R. A. Scalzo, A. L. C. Santos, L. C. Guimaraes, E. M. Pereira, L. S. Barcelos, S. J. H. Do Monte, P. P. G. Guimaraes, F. R. Marciano and A. O. Lobo, J. Funct. Biomater., 2022, 13, 45 CrossRef CAS PubMed.
- A. Famili and K. Rajagopal, Mol. Pharmaceutics, 2017, 14, 1961–1968 CrossRef CAS PubMed.
- B. Zewde, O. Atoyebi, A. Gugssa, K. J. Gaskell and D. Raghavan, ACS Omega, 2021, 6, 11614–11627 CrossRef CAS PubMed.
- C. E. Ziegler, M. Graf, M. Nagaoka, J. Groner, R. Mietzner, M. Breunig and A. M. Goepferich, Mol. Pharm., 2023, 20, 2465–2476 CrossRef CAS PubMed.
- S. Li, J. Zhou, Y. Huang, J. Roy, N. Zhou, K. Yum, X. Sun and L. Tang, Acta Biomater., 2020, 110, 95–104 CrossRef CAS PubMed.
- S. Hong, J. Carlson, H. Lee and R. Weissleder, Adv. Healthcare Mater., 2016, 5, 421–426 CrossRef CAS PubMed.
- S. T. Koshy, R. M. Desai, P. Joly, J. Li, R. K. Bagrodia, S. A. Lewin, N. S. Joshi and D. J. Mooney, Adv. Healthcare Mater., 2016, 5, 541–547 CrossRef CAS PubMed.
- H. Zhan, H. de Jong and D. W. P. M. Löwik, ACS Appl. Bio Mater., 2019, 2, 2862–2871 CrossRef CAS PubMed.
- Y. Zhang, H. Chen, T. Zhang, Y. Zan, T. Ni, M. Liu and R. Pei, Biomater. Sci., 2018, 6, 2578–2581 RSC.
- S. H. Park, J. Y. Park, Y. B. Ji, H. J. Ju, B. H. Min and M. S. Kim, Acta Biomater., 2020, 117, 108–120 CrossRef CAS PubMed.
- S. H. Park, J. Y. Seo, J. Y. Park, Y. B. Ji, K. Kim, H. S. Choi, S. Choi, J. H. Kim, B. H. Min and M. S. Kim, NPG Asia Mater., 2019, 11, 1–16 CrossRef.
- Y. Zhang, M. Liu and R. Pei, Mater. Adv., 2021, 2, 4733–4742 RSC.
- J. Seo, S. H. Park, M. J. Kim, H. J. Ju, X. Y. Yin, B. H. Min and M. S. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 24984–24998 CrossRef CAS PubMed.
- H. J. Ju, M. Park, J. H. Park, G. R. Shin, H. S. Choi, M.-W. Suh and M. S. Kim, Materials, 2020, 13, 3070 CrossRef CAS PubMed.
- J. Luo, A. Darai, T. Pongkulapa, B. Conley, L. Yang, I. Han and K.-B. Lee, Bioact. Mater., 2023, 23, 551–562 CAS.
- X. Liu, Y. Zhang, Z. Hussain, P. Zheng, M. Xu, H. Zhao, Y. Liu, Y. Cao, I. Ullah, A. Osaka and R. Pei, Appl. Mater. Today, 2023, 30, 101693 CrossRef.
- J. Y. Heo, J. H. Noh, S. H. Park, Y. B. Ji, H. J. Ju, D. Y. Kim, B. Lee and M. S. Kim, Pharmaceutics, 2019, 11, 438 CrossRef CAS PubMed.
- M. Czuban, S. Srinivasan, N. A. Yee, E. Agustin, A. Koliszak, E. Miller, I. Khan, I. Quinones, H. Noory, C. Motola, R. Volkmer, M. Di Luca, A. Trampuz, M. Royzen and J. M. M. Oneto, ACS Cent. Sci., 2018, 4, 1624–1632 CrossRef CAS PubMed.
- T. T. Vu, S. Yadav, O. S. Reddy, S.-H. Jo, S.-B. Joo, B. K. Kim, E. J. Park, S.-H. Park and K. T. Lim, Pharmaceuticals, 2023, 16, 841 CrossRef CAS PubMed.
- Z. Zhang, C. He and X. Chen, Polymers, 2020, 12, 884 CrossRef CAS PubMed.
- I. Ali, A. Rizwan, T. T. Vu, S.-H. Jo, C.-W. Oh, Y. H. Kim, S.-H. Park and K. T. Lim, Int. J. Biol. Macromol., 2024, 260, 129549 CrossRef CAS PubMed.
- A. Rizwan, M. Gulfam, S.-H. Jo, J.-W. Seo, I. Ali, T. T. Vu, S.-B. Joo, S.-H. Park and K. T. Lim, Eur. Polym. J., 2023, 191, 112019 CrossRef CAS.
- T. T. Vu, M. Gulfam, S.-H. Jo, S.-H. Park and K. T. Lim, Carbohydr. Polym., 2022, 278, 118964 CrossRef CAS PubMed.
- D. S. B. Anugrah, K. Ramesh, M. Kim, K. Hyun and K. T. Lim, Carbohydr. Polym., 2019, 223, 115070 CrossRef CAS PubMed.
- I. Ali, M. Gulfam, S.-H. Jo, J.-W. Seo, A. Rizwan, S.-H. Park and K. T. Lim, Int. J. Biol. Macromol., 2022, 219, 109–120 CrossRef CAS PubMed.
- S. A. P. Siboro, D. S. B. Anugrah, K. Ramesh, S.-H. Park, H.-R. Kim and K. T. Lim, Carbohydr. Polym., 2021, 260, 117779 CrossRef CAS PubMed.
- Y.-J. Jo, M. Gulfam, S.-H. Jo, Y.-S. Gal, C.-W. Oh, S.-H. Park and K. T. Lim, Carbohydr. Polym., 2022, 286, 119303 CrossRef CAS PubMed.
- F. Jivan, R. Yegappan, H. Pearce, J. K. Carrow, M. McShane, A. K. Gaharwar and D. L. Alge, Biomacromolecules, 2016, 17, 3516–3523 CrossRef CAS PubMed.
- F. Jivan, N. Fabela, Z. Davis and D. L. Alge, J. Mater. Chem. B, 2018, 6, 4929–4936 RSC.
- S. Jung, J. H. Abel, J. L. Starger and H. Yi, Biomacromolecules, 2016, 17, 2427–2436 CrossRef CAS PubMed.
- E. Y. Liu, S. Jung, D. A. Weitz, H. Yi and C.-H. Choi, Lab Chip, 2018, 18, 323–334 RSC.
- D. Mirta, E.-T. Kang and K. G. Neoh, ACS Appl. Polym. Mater., 2021, 3, 2233–2263 CrossRef.
- T. N. Gevrek, I. Kosif and A. Sanyal, ACS Appl. Mater. Interfaces, 2017, 9, 27946–27954 CrossRef CAS PubMed.
- T. N. Gevrek, K. Yu, J. N. Kizhakkedathu and A. Sanyal, ACS Appl. Polym. Mater., 2019, 1, 1308–1316 CrossRef CAS.
- A. Degirmenci, G. Y. Bas, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2022, 33, 1672–1684 CrossRef CAS PubMed.
- J. Escorihuela, A. T. M. Marcelis and H. Zuilhof, Adv. Mater. Interfaces, 2015, 2, 1500135 CrossRef.
- A. Shahrokhtash and D. S. Sutherland, ACS Appl. Mater. Interfaces, 2024, 16, 21534–21545 CrossRef CAS PubMed.
- O. Roling, A. Mardyukov, S. Lamping, B. Vonhören, S. Rinnen, H. F. Arlinghaus, A. Studera and B. J. Ravoo, Org. Biomol. Chem., 2014, 12, 7828–7835 RSC.
- N. Ozbek, E. L. Sahkulubey Kahveci and M. U. Kahveci, ACS Appl. Polym. Mater., 2021, 3, 3721–3732 CrossRef CAS.
- M. Pagel, R. Hassert, T. John, K. Braun, M. Wießler, B. Abel and A. G. Beck-Sickinger, Angew. Chem., Int. Ed., 2016, 55, 4826–4830 CrossRef CAS PubMed.
- M. Czuban, M. W. Kulka, L. Wang, A. Koliszak, K. Achazi, C. Schlaich, I. S. Donskyi, M. Di Luca, J. M. M. Oneto, M. Royzen, R. Haag and A. Trampuz, Mater. Sci. Eng., C, 2020, 116, 111109 CrossRef CAS PubMed.
- T. N. Gevrek, A. Degirmenci, R. Sanyal and A. Sanyal, Polymers, 2020, 12, 1211 CrossRef CAS PubMed.
- O. I. Kalaoglu-Altan, R. Sanyal and A. Sanyal, ACS Omega, 2019, 4, 121–129 CrossRef CAS PubMed.
- D. D. Iorio, A. Marti, S. Koeman and J. Huskens, RSC Adv., 2019, 9, 35608–35613 RSC.
- O. J. G. M. Goor, J. E. P. Brouns and P. Y. W. Dankers, Polym. Chem., 2017, 8, 5228–5238 RSC.
- M. Putti, S. M. J. de Jong, O. M. J. A. Stassen, C. M. Sahlgren and P. Y. W. Dankers, ACS Appl. Polym. Mater., 2019, 1, 2044–2054 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |