
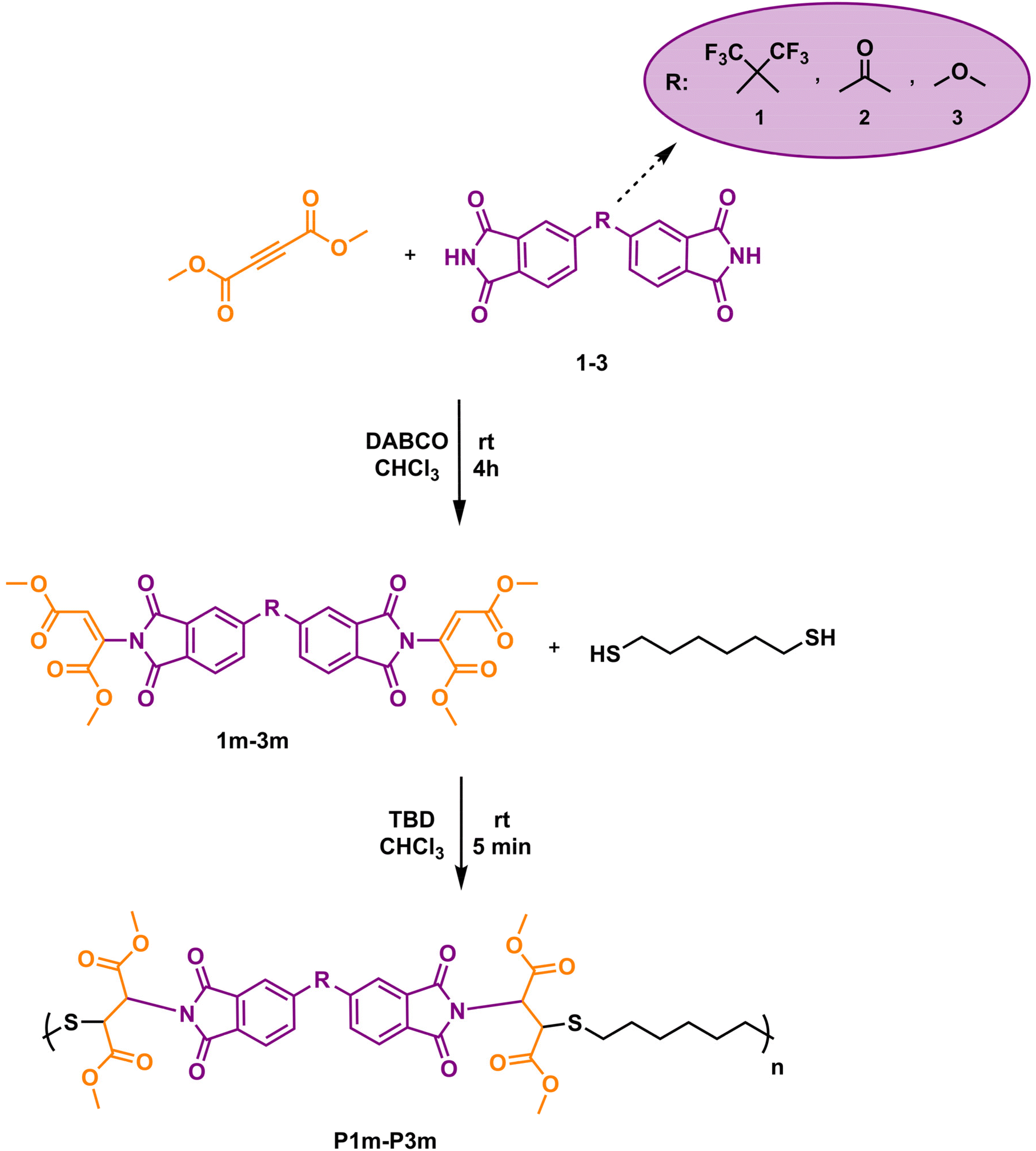
Imide-yne click polymerization: a new and versatile tool for the toolbox of X-yne click polymerization†
Oguzhan
Aslanturk
 a,
Gokhan
Sagdic
a,
Emrah
Cakmakci
b,
Hakan
Durmaz
*a and
Ufuk Saim
Gunay
*a
a,
Gokhan
Sagdic
a,
Emrah
Cakmakci
b,
Hakan
Durmaz
*a and
Ufuk Saim
Gunay
*a
aDepartment of Chemistry, Istanbul Technical University, 34469 Istanbul, Türkiye. E-mail: durmazh@itu.edu.tr; gunayuf@itu.edu.tr
bDepartment of Chemistry, Marmara University, 34722 Istanbul, Türkiye
First published on 13th September 2024
Abstract
The Michael reaction, a cornerstone in organic chemistry, continues to revolutionize the field with its unparalleled versatility in forming carbon–carbon, carbon–oxygen, carbon–nitrogen, and carbon–sulfur bonds, paving the way for groundbreaking advancements in complex molecule and macromolecule construction. In this study, imide-yne reaction was employed at the macromolecular level for the first time to prepare linear poly(imide ester)s. A wide range of bisimides and dipropiolates were reacted through imide-yne click polymerization in the presence of 1,4-diazabicyclo[2.2.2] octane (DABCO) at room temperature. The polymerizations proceed in an anti-Markovnikov fashion, yielding the E-isomer as the major product. Polymers were obtained in high yields and their molecular weights were found to be in the range of 5.64–12.67 kDa. The remaining unreacted double bonds in the linear polymers were found to undergo further functionalization with thiols using a strong organocatalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), which was also supported by a model study. Post-polymerization modification study prompted us to prepare imide-yne monomers that can react with dithiols to synthesize poly(imide thioether)s through nucleophilic thiol–ene click reaction using TBD as the catalyst. The obtained polymers displayed a wide range of glass transition temperatures and thermal stability. Thus, it can be said that the proposed method enables the synthesis of new polyimide-based structures with tailorable thermal properties. It is believed that the proposed strategy will make a significant contribution to expanding the versatility of active alkyne chemistry at the macromolecular level.
Introduction
A class of organic molecules known as imides exhibit remarkable properties that make them valuable in various industrial, commercial, and scientific research applications. The imide functional group is highly resistant to thermal degradation, even at very high temperatures, due to strong resonance stabilization. Polyimides are used to produce high-performance materials with many exceptional features, including high solvent resistance, high mechanical and electrical properties, and thermooxidative and thermal stability.1–6 The majority of poly(imide)s are insoluble and have rigid, planar aromatic, and heteroaromatic structures. Polyimides are typically synthesized by reacting aromatic diamines with dianhydrides to produce poly(amic acid), which is subsequently dehydrated either by high temperatures (160–350 °C)7–11 or through the chemical imidization method (rt–160 °C), which requires an excess quantity of the dehydrating reagent (Ac2O/pyridine).12–14 Alternatively, polyimides can be synthesized directly by reacting dianhydrides with diisocyanates through a process that releases carbon dioxide.15 Some studies utilized the use of diester-diacid derivatives of tetracarboxylic dianhydrides, which are hydrolytically more stable and possess a higher degree of solubility than the parent dianhydrides.16,17 Nucleophilic substitution was also employed between dinitro-substituted bisimide monomers and bisphenolates to obtain polyetherimides.18–20 The Diels–Alder reaction was also studied as an alternative route.21–25Michael addition reactions are well-known reactions and have found tremendous applications in both synthetic organic chemistry and polymer science.26–30 This reaction is highly compatible with several nucleophiles, reaction media, and conditions. Moreover, the reaction mostly proceeds at ambient temperature and affords the final products with high yields and/or efficiencies, emphasizing operation simplicity and energy-saving. All these features put Michael reactions in a special place in “click” chemistry, a term coined by Sharpless and coworkers.31 In Michael reactions, amine, thiol, and oxygen-based nucleophiles are mostly studied compounds to react with electron-deficient double and triple bonds.32 Among them, the amine-based Michael reaction, namely the aza-Michael reaction, has some privileges such as the reaction taking place mostly without the use of any additives or catalysts.27,33–35 Similarly, the addition of imide to a reactive double or a triple bond is a typical aza-Michael addition reaction and has been employed successfully in organic chemistry to construct various nitrogen-based heterocyclic compounds.36–39 Unlike amines, a catalyst is required to activate imide for the Michael addition reaction. Although the participation of imide in a Michael addition reaction was first examined in the late 1940s,36 this chemistry has rarely been tested at the macromolecular level. Park et al., prepared poly(imide-aramid-sulfone)s by reacting divinyl sulfone and bis(4-(vinylsulfonyl)phenyl)terephthalamide with pyromellitic diimide in the presence of tetrabutylammonium hydroxide; moderate molecular weight polymers in good yields were obtained in these polymerizations.40
Although it has not been studied as much as reactive double bonds, the chemistry of activated alkynes in Michael addition reactions has been rejuvenated in the last two decades and has attracted much attention from every discipline of chemistry since the reactive triple bond has been shown to undergo rapid and efficient reactions with the nucleophiles. This chemistry, also known as nucleophile-yne or X-yne, has become particularly important in polymer and material science as it enables ultra-fast polymer synthesis, functionalization, gelation, network preparation, etc.32,41,42 In this regard, thiol–yne,43–50 amino-yne,33,51–57 hydroxyl-yne,30,58–61 phospho-yne,62 and finally imine-yne63 reactions have been proven to be robust and versatile strategies for the preparation of the above-mentioned polymer-based structures. As emphasized above, imides can also be added to the activated alkynes via aza-Michael reactions and this chemistry has been used several times in organic studies.64–68 For instance, in one of the early studies, the Trost group performed an α-addition to the alkynoates using phthalimide and sulfonamide in the presence of phosphine catalysts. The reactions proceeded at high temperatures and the target products were obtained in good to high yields.64 Later, Mola et al., reported the preparations of lactams, imides, and nucleosides via nucleophilic addition to activated triple bonds and 1,4-diazabicyclo[2.2.2]octane (DABCO) and 4-(dimethylamino)-pyridine (DMAP) were found to be the best catalysts for the synthesis of imides.66 Shahraki et al. reported the N-vinylation of heterocyclic compounds, including phthalimide, using ethyl and methyl esters of acetylenedicarboxylic acid using an equimolar amount of pyridine over 24 hours.67 Petit et al., used tosylacetylene as a protecting group for various N–H group-bearing compounds, including imides, in the presence of a base or a catalyst, and observed an excellent stereochemistry control depending on the base or catalyst.68 Despite all these nice organic examples, the imide-Michael reaction involving activated alkynes has not been extensively implemented as an alternative polymer synthesis method. Herein we offer a new and versatile method to add to the toolkit of X-yne click polymerization reactions and expand the library of activated alkyne-based polymer synthesis. In this current study, we demonstrate for the first time that imide-yne click polymerization could be a new synthetic method and energy-efficient approach for the preparation of a series of poly(imide ester)s using various bisimides and dipropiolates as monomers at room temperature, and in the presence of DABCO as a catalyst (Scheme 1).
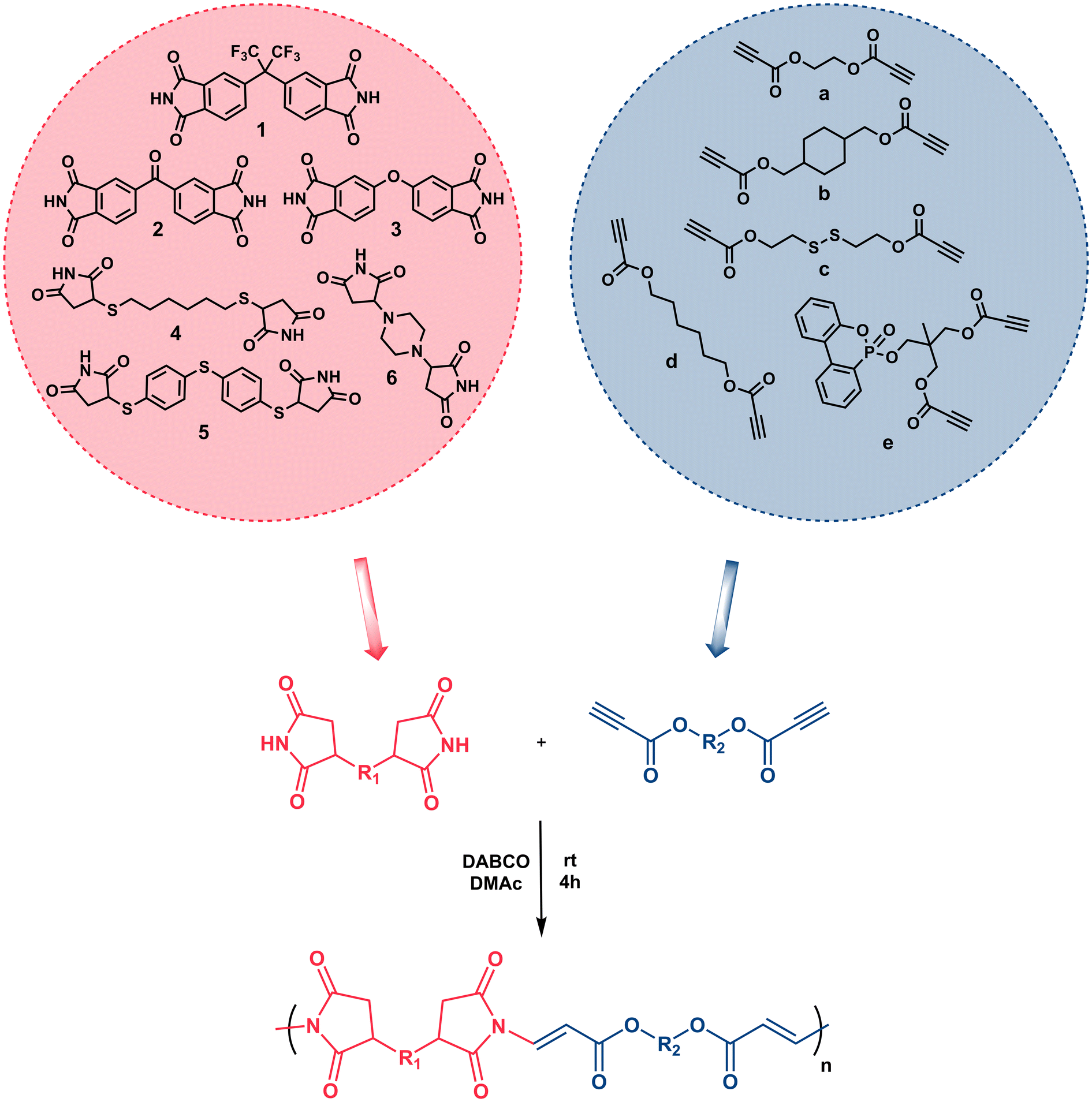 | ||
| Scheme 1 Depiction of imide-yne click polymerization. | ||
Experimental part
Materials
Propiolic acid (95%), ethylene glycol (99.8%), 1,4-cyclohexanedimethanol (mixture of cis and trans, 99%), bis(2-hydroxyethyl)disulfide (technical grade), 1,6-hexanediol (99%), p-toluenesulfonic acid monohydrate (p-TsOH, 98%), 1,1,1-tris(hydroxymethyl)ethane (98%), carbon tetrachloride (CCl4, 99%), sodium sulfate (Na2SO4), 4,4′-(hexafluoroisopropylidene) diphthalic anhydride (6FDA, 99%), benzophenone-3,3′,4,4′-tetracarboxylic dianhydride (BTDA, 98%), 4,4′-oxydiphthalic anhydride (ODPA, 97%), urea (98%), 1,6-hexanedithiol (HDT, 97%), 4,4′-thiobisbenzenethiol (98%), piperazine (99%), maleimide (99%), methyl propiolate (99%), dimethyl acetylenedicarboxylate (DMAD, 99%), cis-1,2,3,6-tetrahydrophthalimide (96%), 1-propanethiol (99%), triethylamine (TEA, 99.5%), 1,4-diazabicyclo[2.2.2] octane (DABCO, 99%), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 98%), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 98%), 4-(dimethylamino)pyridine (DMAP, 99%), benzene (99%), chloroform (CHCl3, 99.8%, amylene stabilized), dichloromethane (DCM, 99%), ethanol (99%), tetrahydrofuran (THF, 99%) were all purchased from Aldrich and used as received. Dimethyl sulfoxide (DMSO, 99.9%, Aldrich), N,N-dimethylacetamide (DMAc, 99.8%, Aldrich), N,N-dimethylformamide (DMF, 99.8%, Aldrich), N-methyl-2-pyrrolidone (NMP, 99.5%, Aldrich) were anhydrous and high-performance liquid chromatography (HPLC) quality and used without further purification. Methanol and hexane were of reagent grade and used as received. 9,10-Dihydro-9-oxa-10-phosphaphenanthrene-10-oxide (DOPO) was purchased from TCI.Instrumentation
1H NMR (500 MHz), 13C NMR (125 MHz), 19F NMR (470 MHz), and 31P NMR (202 MHz) spectra were recorded using an Agilent VNMRS 500 instrument in CDCl3 and d6-DMSO. Gel permeation chromatography (GPC) measurements were carried out with an Agilent instrument (model 1100) with a pump, refractive index and UV detectors, and four Waters Styragel columns (HR 5E, HR 4E, HR 3, HR 2) (4.6 mm internal diameter, 300 mm length, packed with 5 μm particles). The effective molecular weight ranges of the columns were 2000–4000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 50–100000, 500–30000, and 500–20000 g mol−1, respectively. THF was used as an eluent at a flow rate of 0.3 mL min−1 at 30 °C, and 2,6-di-tert-butyl-4-methylphenol (BHT) was used as an internal standard. The weight-average molecular weights (Mw) and dispersities (Đ) of the polymers were calculated based on linear polystyrene (PS) standards (Polymer Laboratories). Thermogravimetric analyses (TGA) of the synthesized polymers were performed by using a PerkinElmer thermogravimetric analyzer (Pyris 1 TGA model). Differential scanning calorimetry (DSC) measurements were performed under a nitrogen atmosphere on the TA Q1000 DSC apparatus. Synthesized polymers were heated to 250 °C with a heating rate of 10 °C min−1. After holding for 2 min at this temperature, samples were cooled to 25 °C with a cooling rate of 10 °C min−1, followed by keeping at this temperature for 2 min. Finally, they were reheated to 250 °C with a heating rate of 10 °C min−1. Data from the second heating cycle were reported. Mass spectra were obtained using a Thermo Fisher Scientific LC-HRMS spectrometer. Fourier transform infrared (FT-IR) spectra were recorded on a Cary 630 FT-IR instrument (Agilent Technologies) in the 4000–400 cm−1 range.
000, 50–100000, 500–30000, and 500–20000 g mol−1, respectively. THF was used as an eluent at a flow rate of 0.3 mL min−1 at 30 °C, and 2,6-di-tert-butyl-4-methylphenol (BHT) was used as an internal standard. The weight-average molecular weights (Mw) and dispersities (Đ) of the polymers were calculated based on linear polystyrene (PS) standards (Polymer Laboratories). Thermogravimetric analyses (TGA) of the synthesized polymers were performed by using a PerkinElmer thermogravimetric analyzer (Pyris 1 TGA model). Differential scanning calorimetry (DSC) measurements were performed under a nitrogen atmosphere on the TA Q1000 DSC apparatus. Synthesized polymers were heated to 250 °C with a heating rate of 10 °C min−1. After holding for 2 min at this temperature, samples were cooled to 25 °C with a cooling rate of 10 °C min−1, followed by keeping at this temperature for 2 min. Finally, they were reheated to 250 °C with a heating rate of 10 °C min−1. Data from the second heating cycle were reported. Mass spectra were obtained using a Thermo Fisher Scientific LC-HRMS spectrometer. Fourier transform infrared (FT-IR) spectra were recorded on a Cary 630 FT-IR instrument (Agilent Technologies) in the 4000–400 cm−1 range.
Synthetic procedures for monomers
To a 50 mL round bottom flask equipped with a magnetic stir bar was added maleimide (1.5 g, 15.45 mmol) and dissolved in 30 mL of DCM. Upon dissolution of maleimide, HDT (945 μL, 6.18 mmol) and TEA (215 μL, 1.545 mmol) were added to the flask, respectively, and the mixture was allowed to stir at room temperature for 1 hour. As the reaction progressed, the reaction medium turned into a suspension containing a solid precipitate. After the specified time, the solids were filtered with a sintered glass filter and washed with DCM. The resultant product was dried in a vacuum oven at 40 °C overnight, yielding a white solid product (yield: 1.80 g, 85%). 1H NMR (d6-DMSO, δ): 11.30 (s, 2H, NH), 3.87 (t, 2H, SCH), 3.12 (m, 2H, C
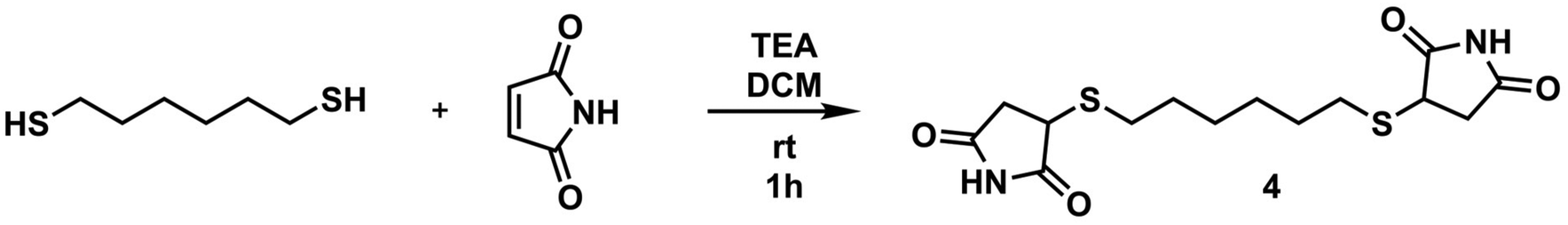
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OCH2), 2.71–2.66 (m, 4H, SCH2), 2.44–2.40 (m, 2H, COCH2), 1.54 (br, 4H, SCH2CH2), 1.34 (br, 4H, SCH2CH2CH2). 13C NMR (d6-DMSO, δ): 178.66, 177.08, 40.93, 37.70, 30.79, 28.86, 28.12. ESI-MS, m/z C14H20N2O4S2 calculated: 344.086; found: 345.093 [M + H]+.
OCH2), 2.71–2.66 (m, 4H, SCH2), 2.44–2.40 (m, 2H, COCH2), 1.54 (br, 4H, SCH2CH2), 1.34 (br, 4H, SCH2CH2CH2). 13C NMR (d6-DMSO, δ): 178.66, 177.08, 40.93, 37.70, 30.79, 28.86, 28.12. ESI-MS, m/z C14H20N2O4S2 calculated: 344.086; found: 345.093 [M + H]+.
To a 250 mL round bottom flask equipped with a magnetic stir bar was added 1,1,1-tris(hydroxymethyl)ethane (6.67 g, 55.5 mmol) and partially dissolved in 60 mL of DCM. Then, TEA (19.34 mL, 138.75 mmol) and DOPO (6.00 g, 27.76 mmol) were added to the flask, respectively. Upon complete dissolution of DOPO, the mixture was cooled to 0 °C in an ice bath. After that, CCl4 (5.35 mL, 55.5 mmol) was added to the reaction medium dropwise, then the flask was removed from the ice bath and allowed to stir overnight at room temperature. After the specified time, solid fragments were filtered and the mixture was diluted with 40 mL of DCM and extracted with 100 mL of distilled water three times. The organic layers were dried over Na2SO4, filtered, and evaporated to dryness to give DOPO-Diol as a viscous brown liquid (yield: 5.85 g, 63%). 1H NMR (CDCl3, δ) 7.96–7.26 (m, 8H, Ar–H), 4.43 (t, 1H, P
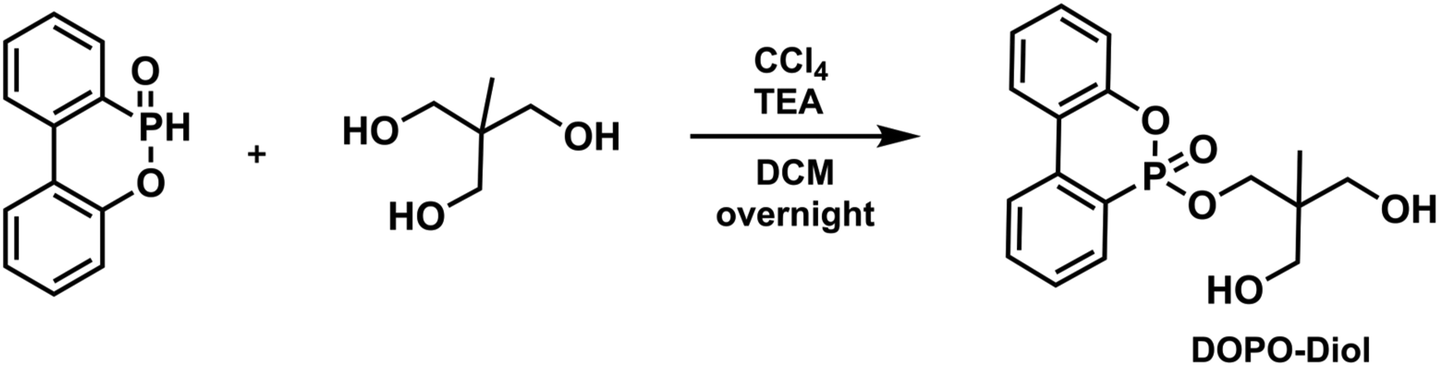 OOCH2), 4.08 (t, 1H, POOCH2), 3.62–3.57 (m, 4H, CCH2OH), 3.27 (br, 2H, –OH), 0.72 (s, 3H, CCH3). 13C NMR (CDCl3, δ) 149.62, 137.07, 133.84, 130.66, 130.12, 128.47, 128.34 125.29, 124.97, 124.15, 122.48, 122.23, 120.78, 120.18, 68.63, 67.56, 67.17, 41.35, 15.65. 31P NMR (CDCl3, δ) 13.27, 11.66. ESI-MS, m/z C17H19O5P calculated: 334.097; found: 335.104 [M + H]+.
OOCH2), 4.08 (t, 1H, POOCH2), 3.62–3.57 (m, 4H, CCH2OH), 3.27 (br, 2H, –OH), 0.72 (s, 3H, CCH3). 13C NMR (CDCl3, δ) 149.62, 137.07, 133.84, 130.66, 130.12, 128.47, 128.34 125.29, 124.97, 124.15, 122.48, 122.23, 120.78, 120.18, 68.63, 67.56, 67.17, 41.35, 15.65. 31P NMR (CDCl3, δ) 13.27, 11.66. ESI-MS, m/z C17H19O5P calculated: 334.097; found: 335.104 [M + H]+.
To a 100 mL round bottom flask equipped with a magnetic stir bar were added DOPO-Diol (3.50 g, 10.47 mmol) and 30 mL of benzene. Propiolic acid (2.6 mL, 41.88 mmol) and p-TsOH (0.2 g, 1.05 mmol) were then added to the flask, respectively, and the mixture was allowed to stir under a reflux system equipped with a Dean–Stark apparatus at 105 °C overnight. After the specified time, the reaction mixture was concentrated under reduced pressure, and the crude product was dissolved in 100 mL of DCM and extracted with 100 mL of distilled water three times. The organic layers were collected and dried over Na2SO4, filtered, and evaporated to dryness to give compound e (DOPO-Dipropiolate) as a viscous brown liquid (yield: 4.3 g, 93%). 1H NMR (CDCl3, δ) 7.95–7.27 (m, 8H, Ar–H), 4.10 (s, 2H, P
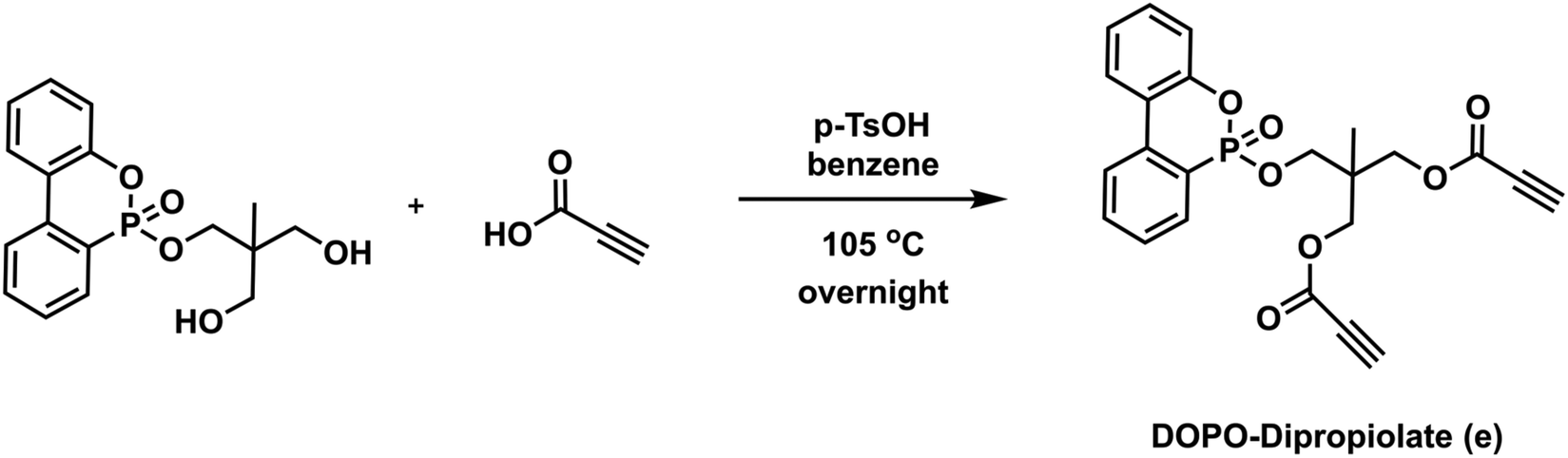 OOCH2), 3.94 (m, 4H, COOCH2), 2.92 (s, 2H, C
OOCH2), 3.94 (m, 4H, COOCH2), 2.92 (s, 2H, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 0.93 (s, 3H, CCH3). 13C NMR (CDCl3, δ): 152.07, 149.69, 137.13, 133.80, 130.72, 130.25, 128.47, 124.97, 124.15, 122.50, 122.24, 120.79, 120.12, 77.02, 75.59, 74.08, 66.92, 66.54, 38.98, 16.34. 31P NMR (CDCl3, δ) 10.64, 5.84. ESI-MS, m/z C23H19O7P calculated: 438.087; found: 439.093 [M + H]+.
CH), 0.93 (s, 3H, CCH3). 13C NMR (CDCl3, δ): 152.07, 149.69, 137.13, 133.80, 130.72, 130.25, 128.47, 124.97, 124.15, 122.50, 122.24, 120.79, 120.12, 77.02, 75.59, 74.08, 66.92, 66.54, 38.98, 16.34. 31P NMR (CDCl3, δ) 10.64, 5.84. ESI-MS, m/z C23H19O7P calculated: 438.087; found: 439.093 [M + H]+.
General procedure for imide-yne click polymerization
To a 10 mL round bottom flask equipped with a magnetic stir bar was added dipropiolate monomer (0.3 mmol, 1 equiv.) and dissolved in DMAc (1.2 mL). Bisimide monomer (0.3 mmol, 1 equiv.) was then added and upon complete dissolution, DABCO (0.03 mmol, 0.1 equiv.) was finally added to the flask. After the addition of DABCO, a color change was observed. The reaction was allowed to stir at room temperature for 4 hours. After the specified time, the solution was precipitated into acidified methanol and filtered. The dissolution-precipitation procedure (CHCl3-acidified methanol) was repeated two times. The resultant polymers were dried in a vacuum oven at 40 °C for 24 hours. The linear poly(imide ester)s were named based on the monomer codes. The synthetic procedures and characterization details of obtained linear poly(imide ester)s can be found in the ESI.†Post-polymerization modification of P1a with 1-propanethiol
To a 10 mL round bottom flask equipped with a magnetic stir bar was added P1a (182.5 mg, 0.3 mmol) and dissolved in 0.6 mL of DMAc. Then, 1-propanethiol (81.6 μL, 0.9 mmol) and TBD (10.4 mg, 0.075 mmol) were added to the reaction medium, respectively. Upon the addition of TBD, the pale yellow solution turned to dark brown instantly. The flask was allowed to stir at room temperature for 15 minutes. The solution was diluted with 1 mL of CHCl3 and precipitated into acidified methanol and filtered. The dissolution-precipitation procedure (CHCl3-acidified methanol) was repeated two times. The resultant polymer was obtained as a pale-yellow powder and dried in a vacuum oven at 40 °C for 24 hours (yield: 223.5 mg, 89%). 1H NMR (d6-DMSO, δ): 8.04–7.70 (m, 6H, Ar–H), 5.56 (t, 2H, CONCH), 4.18 (br, 4H, COOCH2), 3.34 (t, 4H, SCH2), 3.13–2.57 (m, 4H, CONCHCH2), 1.52 (br, 4H, SCH2CH2), 0.85 (t, 6H, SCH2CH2CH3). 13C NMR (d6-DMSO, δ): 169.47, 166.19, 137.90, 136.59, 132.81, 132.42, 124.68, 124.06, 64.96, 62.78, 50.88, 49.04, 37.79, 33.58, 22.59, 13.44.
Synthesis of imide-yne monomer 1m
To a 10 mL round bottom flask equipped with a magnetic stir bar was added compound 1 (774 mg, 1.75 mmol) and suspended in 7 mL of CHCl3. DMAD (648.7 μL, 5.25 mmol) and DABCO (49 mg, 0.44 mmol) were added to the reaction medium, respectively, and the mixture was allowed to stir at room temperature for 4 hours. After the specified time, the solution was diluted with 30 mL of DCM and extracted with 30 mL of acidified water three times. The organic layers were collected and dried over anhydrous Na2SO4, filtered and concentrated in vacuo. After that, the solution was precipitated into hexane and filtered. The dissolution-precipitation (CHCl3–hexane) procedure was repeated two times. The resultant product (1m) was dried in a vacuum oven at 40 °C overnight and obtained as an off-white solid (yield: 1.1 g, 86%). 1H NMR (CDCl3, δ): 8.01–7.77 (m, 6H, Ar–H), 7.22–6.72 (s, 2H, NCCH), 3.91–3.76 (m, 12H, OCH3). 13C NMR (CDCl3, δ): 164.31, 163.04, 162.01, 139.22, 136.33, 132.93, 131.44, 129.19, 125.41, 124.44, 119.60, 65.20, 53.60, 52.57. 19F NMR (CDCl3, δ): −63.19. ESI-MS, m/z C31H20F6N2O12 calculated: 726.092; found: 744.125 [M + NH4]+.
Imide-yne monomers 2m and 3m were prepared using the same procedure described for 1m, and their experimental procedures and characterization data can be found in the ESI.†
General procedure for nucleophilic thiol–ene polymerization of imide-yne monomers
To a 10 mL round bottom flask equipped with a magnetic stir bar was added imide-yne monomer (1 mmol, 1 equiv.) and dissolved in CHCl3 (1 mL). Upon complete dissolution of imide-yne monomer, HDT (1 mmol, 1 equiv.) and TBD (0.25 mmol, 0.25 equiv.) were added to the flask in that order. After the addition of TBD, a color change and an increase in viscosity was observed. The reaction was allowed to stir at room temperature for 5 minutes. After the specified time, the reaction mixture was diluted with 1 mL of CHCl3 and precipitated into acidified methanol and filtered. The dissolution-precipitation procedure (CHCl3-acidified methanol) was repeated two times. The resultant polymers were dried in a vacuum oven at 40 °C for 24 hours. The synthetic procedures and characterization details of the polymers obtained linear poly(imide thioether)s can be found in the ESI.†Results and discussion
The structures of monomers used in this study are depicted in Scheme 1. Here, bisimide monomers 1–3 were prepared according to published procedures.69,70 Due to the poor solubility of monomers 2 and 3, we also designed new bisimide monomers 4–6, which were synthesized via thiol- and amine-maleimide click reactions using commercially available dithiols, piperazine, and maleimide. The Michael addition reactions simply proceeded at room temperature in the presence of TEA as a catalyst. All imide compounds were obtained as white solids in good to high yields and fully characterized by FT-IR, NMR and mass spectrometry. The experimental and spectroscopic details regarding these compounds can be found in the ESI.† Dipropiolate structures were synthesized by conventional esterification reactions using Dean–Stark apparatus at high temperatures. While compounds a–d were prepared in single-step reactions using commercially available diols and excess of propiolic acid, compound e was prepared in two steps; first, DOPO-Diol was synthesized via Atherton-Todd reaction followed by an esterification reaction with excess propiolic acid. The experimental and spectroscopic details regarding dipropiolate structures are also provided in the ESI.†Next, the optimum conditions for the proposed imide-yne polymerization were examined. For this purpose, bisimide 1, chosen for its high solubility, and dipropiolate a, chosen for its simplicity and easy monitoring by 1H NMR, were used as model monomers, and DABCO was used as the catalyst. Also, monomer concentrations were set to 0.25 M, and the mole ratios of bisimide:dipropiolate:DABCO was set to be 1:1:0.1. The results for the obtained polymer, namely P1a, are located in Table 1. Initially, we kinetically screened the effect of solvent on polymerization. Since monomer 1 and the other imide monomers have apparent solubility issues, we could have tested only NMP, DMSO, and DMAc together with DMF as solvents (entries 1–4, Table 1). As seen in this table, all solvents afforded polymer within 1 h, and a slight increase in molecular weights was observed over time. Among them DMF (entry 1, Table 1) and DMAc (entry 4, Table 1) gave little higher molecular weights; the former produced Mw = 8.3 kDa in 1 h, increased to 10.2 in 4 h and 10.8 in 24 h, and the latter gave Mw = 8.04 kDa in 1 h, increased to 11.8 in 4 h and 12.6 in 24 h. The other two solvents (i.e., NMP and DMSO) produced little lower molecular weights compared to these solvents (entries 2 and 3, Table 1). According to the results obtained from the solvent trials, we decided to use DMAc as the polymerization solvent, and since no distinct change was observed in the molecular weights after 4 h, the polymerization duration was fixed as 4 h for other experiments. Temperature effect on polymerization was next examined; however, increasing the reaction temperature to 40 °C and 80 °C did not improve molecular weights (entries 5 and 6, Table 1). As expected, lowering monomer concentrations to 0.1 M yielded a low molecular weight polymer (entry 7, Table 1); however, increasing monomer concentrations to 0.5 M gave an insoluble polymer (entry 8, Table 1). The same insoluble polymer was also observed when the catalyst concentration was increased to 0.25 equivalent (per monomer) while keeping the monomer concentrations at 0.25 M (entry 9, Table 1). It is worth mentioning here that either increasing the amount of dipropiolate or DABCO leads to a black precipitate appearing in a gel-like form, which might be attributed to a competitive enyne reaction that can take place in the presence of dipropiolate and DABCO.74 It should also be noted here that organocatalysts other than DABCO, such as TEA, TBD, DBU, and DMAP, were also tested in the optimum conditions but none of them yielded polymer. As such, we did not change the catalyst (i.e., DABCO) or its equivalent (0.1 equivalent per monomer) or increase the monomer concentrations, and kept the aforementioned optimum conditions for the rest of the studies.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | M w (kDa)b/Đb | |||||
| 1 h | 2 h | 4 h | 6 h | 8 h | 24 h | ||
| a Reaction conditions: 0.3 mmol of bisimide (1), 0.3 mmol of dipropiolate (a) and 0.03 mmol of DABCO in 1.2 mL of solvent at room temperature. b Determined by GPC calibrated based on linear PS standards in THF. c Reaction was carried out at 40 °C. d Reaction was carried out at 80 °C. e Reaction was carried out using monomer concentrations of 0.1 M. f Reaction was carried out using monomer concentrations of 0.5 M and upon the addition of DABCO, an insoluble polymer was obtained. g 0.25 equivalent of DABCO was utilized and an insoluble polymer was obtained. | |||||||
| 1 | DMF | 8.3/2.47 | 9.6/2.36 | 10.2/2.37 | 10.5/2.39 | 10.6/2.40 | 10.8/2.00 |
| 2 | NMP | 6.92/2.25 | 9.02/2.33 | 9.66/2.44 | 10.3/2.43 | 10.1/2.39 | 10.53/1.85 |
| 3 | DMSO | 3.6/1.71 | 4.16/1.81 | 6.4/2.16 | 7.94/2.40 | 8.96/2.55 | 9.17/2.44 |
| 4 | DMAc | 8.04/2.28 | 10.04/2.52 | 11.8/2.48 | 12.3/2.59 | 12.4/2.77 | 12.6/2.13 |
| 5c | DMAc | 10.57/2.41 | |||||
| 6d | DMAc | 9.19/2.06 | |||||
| 7e | DMAc | 7.54/1.69 | |||||
| 8f | DMAc | Insoluble | |||||
| 9g | DMAc | Insoluble | |||||
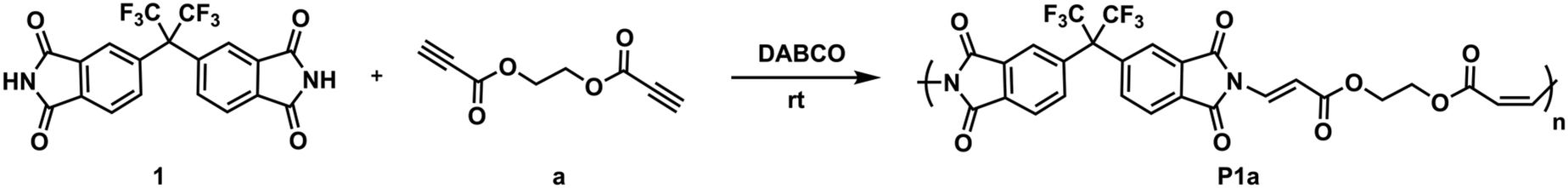
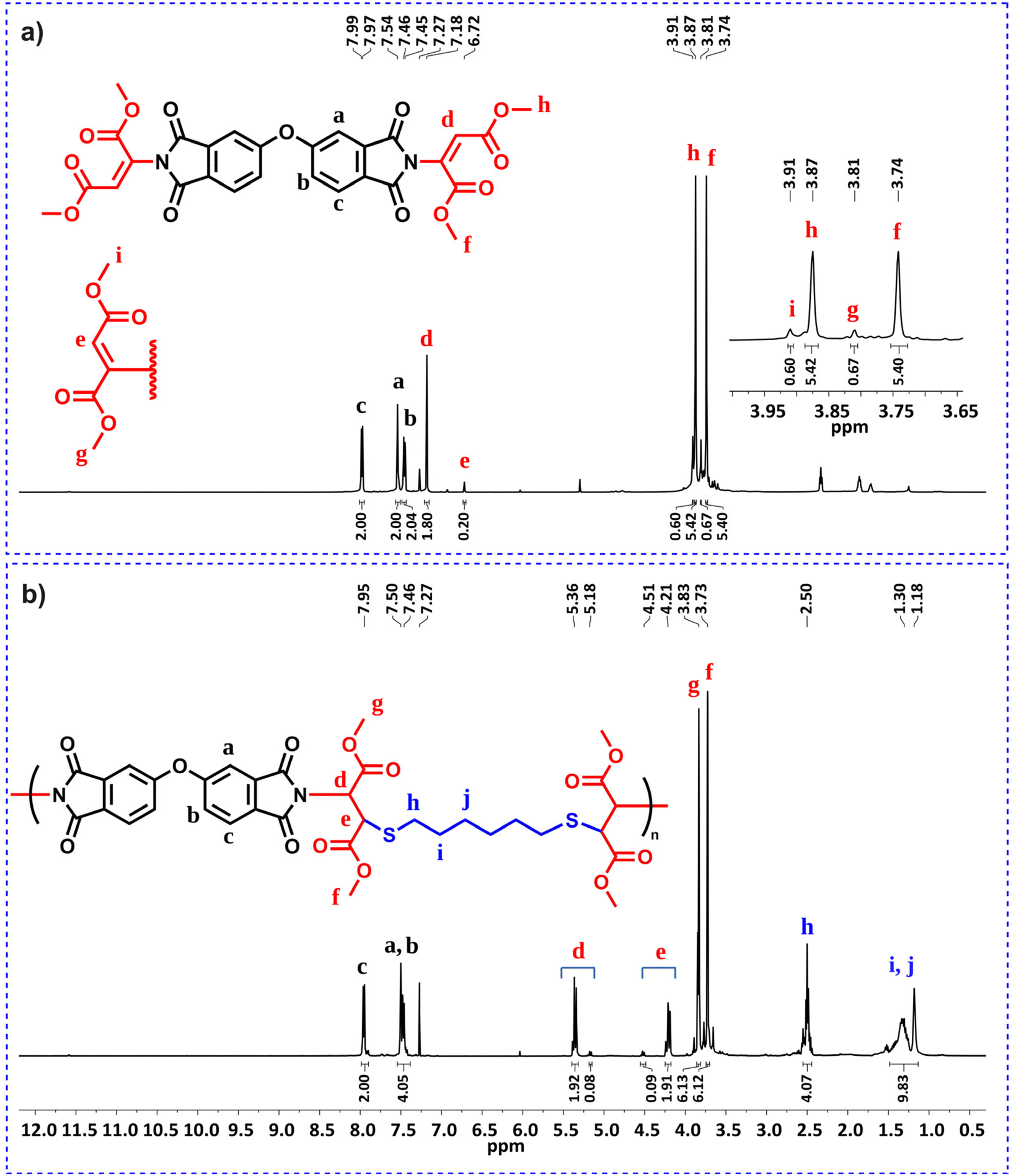
Before performing other reactions to create a polymer library, the obtained polymer, P1a, was analyzed by NMR (Fig. 1). From the 1H NMR spectra, it can be seen that the alkyne proton of propiolate unit at δ 2.96 ppm disappeared and the integral ratio of the N–H proton of the imide structure reduced to a large extent. Importantly, the characteristic N-vinyl proton signals after the imide-yne reaction were detected as doublets and found to resonate at around δ 8.5–5.5 ppm with some of them overlapping with the aromatic proton signals. Here, NCHCH for the E- and the Z-isomers were found to resonate at δ 6.75–6.72 and 5.70–5.53 ppm, respectively. Aromatic protons were also detected between δ 8.16–7.59 ppm. When the integral ratios of the vinyl protons were compared, the ratio for the E-isomer was found to be 91% for P1a. The structure of P1a was further validated by 13C NMR analysis (Fig. S55†). As seen in this figure, propiolate carbons resonated at around δ 75 ppm completely disappeared and new vinylic carbons stemming from imide-yne addition were detected at δ 160.71 and 100.38 ppm. NMR results show that the expected product (i.e., P1a) was successfully prepared and the imide-yne polymerization reaction proceeded smoothly.
 | ||
| Fig. 1 Overlaid 1H NMR spectra of bisimide 1 (top) in d6-DMSO (500 MHz), dipropiolate a (middle) in CDCl3 (500 MHz), and P1a (bottom) in d6-DMSO (500 MHz). | ||
To diversify the poly(imide ester)s, different bisimide structures along with different dipropiolates given in Scheme 1 were reacted under the optimum conditions, and the obtained results are collected in Table 2. Unfortunately, bisimides 2 and 3 were found to have limited solubility in DMAc, so they were not utilized directly in imide-yne click polymerizations; however, these two compounds were used in the preparation of imide-yne monomers (see below). Bisimide 1 was combined with all dipropiolates (entries 1–5, Table 2), and for dipropiolate b, the reaction time was reduced to 1 h due to solubility problems after that time (entry 2, Table 2). Low to moderate molecular weight polymers ranging from Mw = 6.87–12.67 kDa were obtained from these polymerizations in good to high yields. Also, bisimides 4 and 5 that were synthesized by thiol-maleimide reactions were found to yield lower molecular weight polymers (entries 6 and 7, Table 2) and were less reactive towards imide-yne click reaction which could be due to the limited resonance stabilization of the imide anion. Moreover, bisimide 6 synthesized by amino-maleimide reaction yielded THF insoluble polymer so GPC data is not available for this polymer (entry 8, Table 2).
| Entry | Bismide | Dipro-piolate | Polymer |
M
wb (kDa) |
Đ |
T
gc (°C) |
Isolated yieldd (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 0.3 mmol of bisimide, 0.3 mmol of dipropiolate and 0.03 mmol of DABCO in 1.2 mL of DMAc at room temperature for 4 h. b Determined by GPC calibrated based on linear PS standards in THF. c Determined by DSC from the second heating cycle. d Gravimetric yield obtained after first precipitation. e Polymerization was carried out for 1 h. f The resulting polymer was observed to be insoluble in THF and therefore GPC data could not be obtained. | |||||||
| 1 | 1 | a | P1a | 12.43 | 2.48 | 199 | 90 |
| 2e | 1 | b | P1b | 12.67 | 1.96 | 193 | 82 |
| 3 | 1 | c | P1c | 7.82 | 1.95 | 121 | 72 |
| 4 | 1 | d | P1d | 12.65 | 1.68 | 147 | 84 |
| 5 | 1 | e | P1e | 6.87 | 1.88 | 162 | 80 |
| 6 | 4 | a | P4a | 6.55 | 2.96 | 61 | 62 |
| 7 | 5 | a | P5a | 5.64 | 2.83 | 130 | 70 |
| 8f | 6 | a | P6a | — | — | 205 | 94 |
The poly(imide ester)s were also characterized via FT-IR spectroscopy (Fig. S60, S67, S74, S81, S89, S95, S101, and S106†). The FT-IR spectra of the poly(imide ester)s displayed the absence of the vibration bands for propiolate triple bonds (–CC–) at around 2100 cm−1 and the presence of the characteristic imide carbonyl stretching vibration bands at around 1775 cm−1 and 1710 cm−1. Besides, a new band appeared at around 1640 cm−1 which was attributed to the newly formed double bonds (–N–CHCH–CO).
The Tg values of the synthesized poly(imide ester)s are collected in Table 2. It can be seen that the polyimides synthesized via imide-yne click polymerization displayed a wide range of Tg values ranging from 61 °C to 205 °C. This shows that the Tg values; i.e., the processability and flexibility of the resulting polyimides can be tuned, highlighting the modularity of the proposed method. Among the polyimides based on 1, P1a and P1b displayed the highest Tg values. P1a and P1b contain short chains between their propiolate end groups, respectively. When these short chains were replaced with a relatively long flexible hexanediol the Tg value decreased (P1d). When the propiolate was changed to a more flexible disulfide bond-containing one (P1c), the molecular weight was found to be lower and the Tg value decreased significantly. P1e which has rigid DOPO units, displayed a lower Tg value than expected which could be ascribed to its lower molecular weight compared to P1a.
P1a, P4a, and P5a polymers are produced from the reaction of the same dipropiolate but using different bisimides. When the Tg values of the homologous P1a, P4a, and P5a are compared, the Tg values were found to decrease in the following order: P1a > P5a > P4a. Bisimides 4 and 5 have flexible thioether linkages in their structures and therefore they displayed significantly lower Tg values than P1a which is composed of a rigid aromatic bisimide (1). Finally, the highest Tg value was obtained for the polymer P6a which has a rigid piperazine core. The nitrogen groups in these piperazine units create additional dipolar interactions which increase the intramolecular interactions and thus limit the degree of rotation and lead to reduced free volumes.
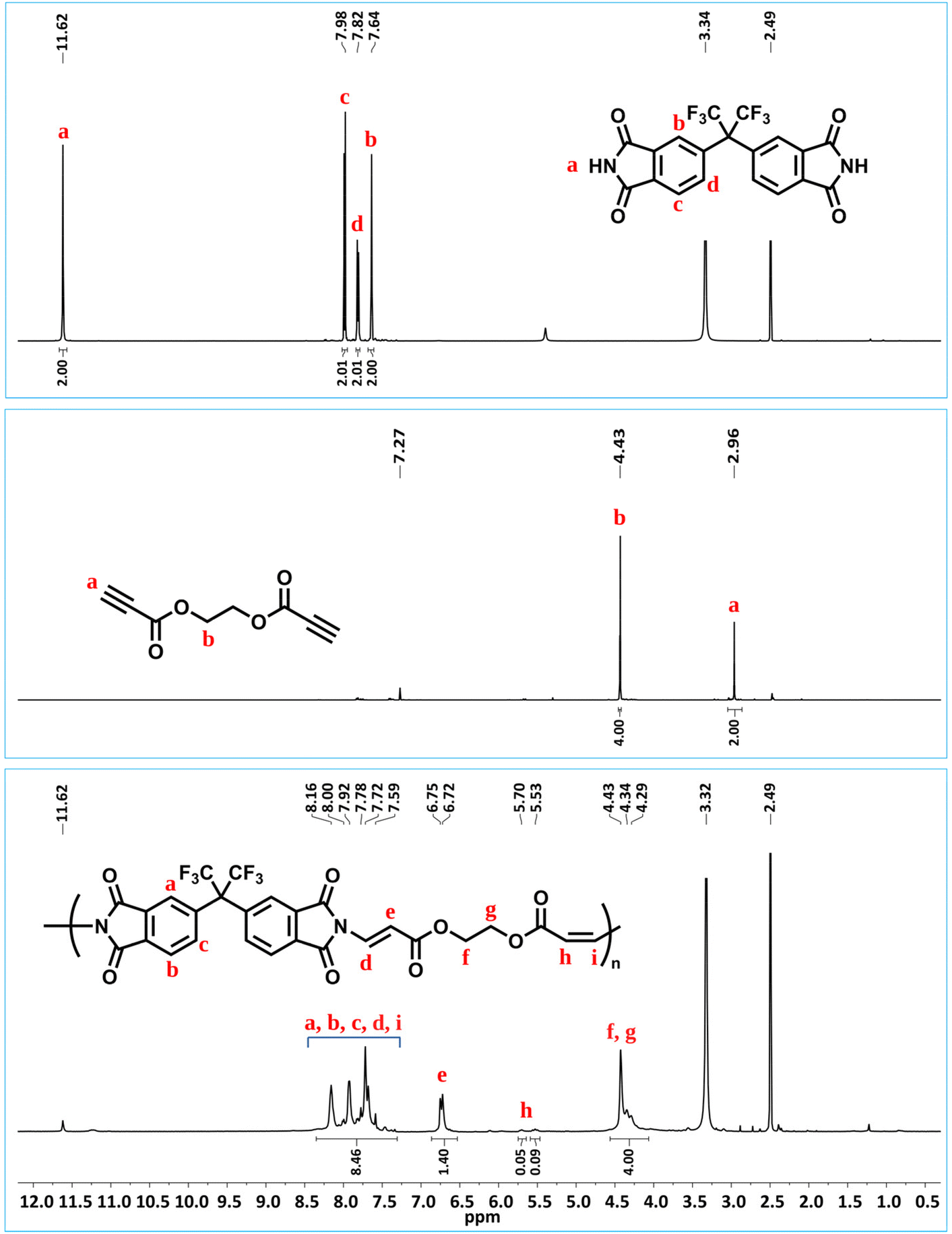
Since reactive double bonds are still present in the resulting imide-yne polymers, we hypothesized that these bonds can be further functionalized by thiol–ene click chemistry. To test this foresight, we designed a model study as depicted in Scheme 2a. In this experiment, cis-1,2,3,6-tetrahydrophthalimide was reacted with methyl propiolate in the presence of DABCO, resulting in N-vinylated product, which was characterized by 1H NMR. Fig. S47† indicates the full disappearance of alkyne signal δ 2.98 ppm and the appearance of vinyl signals between δ 7.68–5.65 ppm, full agreement with the integral ratios confirms the expected product. This product was then reacted with 1-propanethiol in CHCl3 at room temperature. It is known that nucleophilic thiol–ene reactions require a suitable catalyst to drive the reaction. It has been shown previously by our group47–49 and others50,75,76 that TBD is a robust organocatalyst due to its stronger basicity and nucleophilicity and can easily provoke a thiol compound to a reactive double bond. Here, we implemented this catalyst in the second step for further functionalization, and the reaction proceeded for 5 min at room temperature. After purification, the modified compound was characterized by NMR. Full disappearance of vinyl protons (Fig. S50†) and carbons (Fig. S51†) and the appearance of acetal signal (NCHS) at δ 5.48 ppm indicate that nucleophilic thiol–ene click reaction occurred quantitatively.
 | ||
| Scheme 2 Model imide-yne reaction and the following nucleophilic thiol–ene click reaction (a) and post-polymerization modification of P1a with 1-propanethiol (b). | ||
The results obtained from model experiments indicate that the main chain reactive double bonds of an imide-yne polymer could be readily functionalized in the same way described in the model reaction. To examine this, a post-polymerization modification reaction was employed on P1a and the reactive double bonds on the main chain were further reacted with 1.5 equiv. of 1-propanethiol (per alkene unit) in the presence of 0.25 equiv. of TBD for 15 min (Scheme 2b). According to the 1H NMR spectrum of modified P1a (Fig. 2), the addition was found to be quantitative. N-Vinyl proton peaks were diminished and a new peak emerged at δ 5.56 ppm corresponding to the acetal proton. Also, the expected signals arising from 1-propanethiol were detected at δ 3.30 (SCH2), partially overlapping with the H2O peak at 3.33 ppm as well as at δ 1.52 (SCH2CH2) and 0.85 (SCH2CH2CH3) ppm. Finally, aromatic protons appeared as singlets at δ 8.04, 7.85 & 7.70 ppm. 13C NMR spectrum also displayed the full disappearance of main chain double bond carbons and the formation of corresponding acetal carbon at around δ 50 ppm after the reaction (Fig. S125†). Collectively, the NMR results indicate a successful post-modification reaction. It should also be noted that modified P1a exhibited a lower Tg value, decreasing from 199 °C to 118 °C (see Fig. S126 and S128†). This decrease can be attributed to the increased flexibility and free volume after modification.
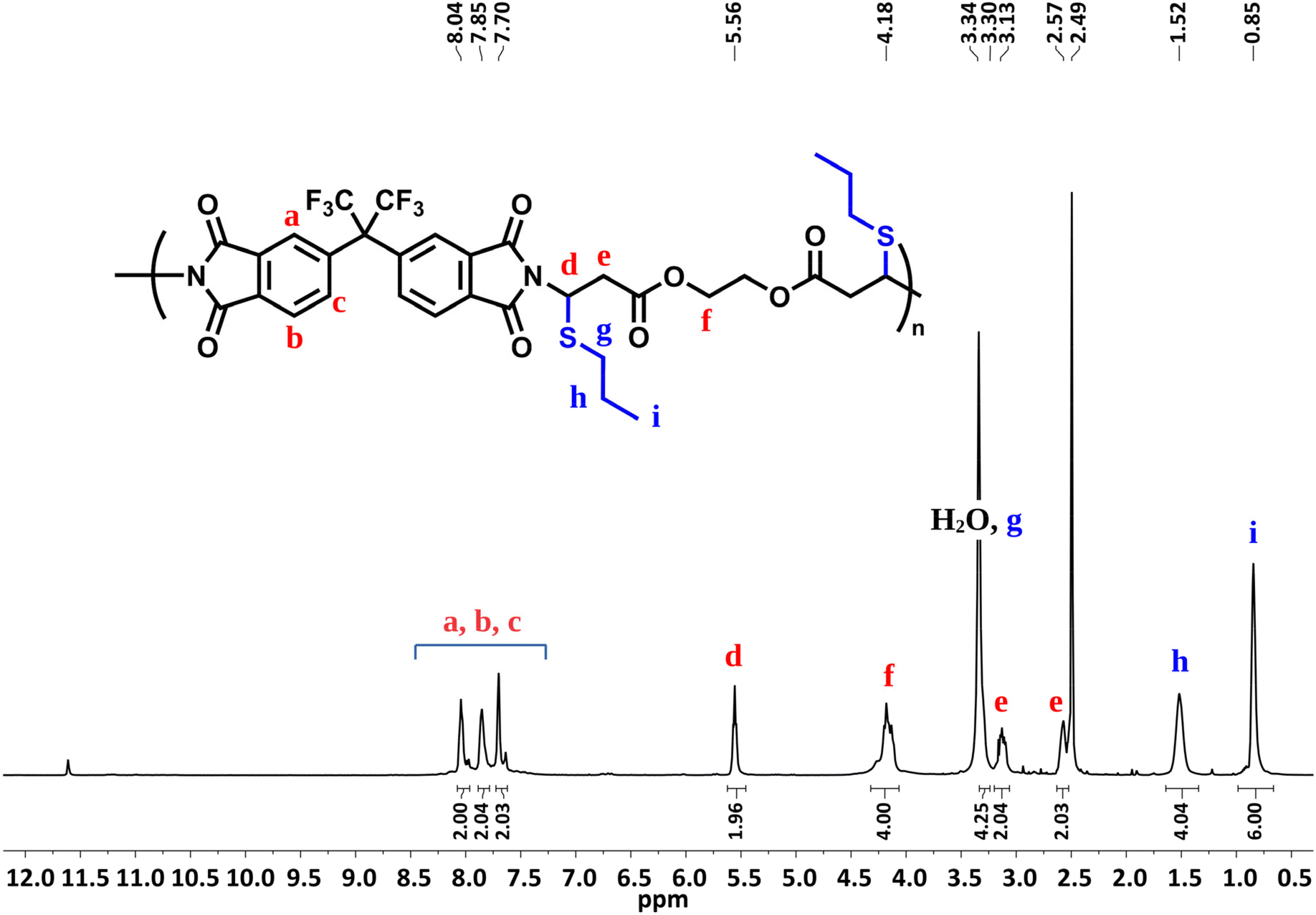 | ||
| Fig. 2 1H NMR spectrum of modifiedP1a in d6-DMSO (500 MHz). | ||
It is worth mentioning here that we found that we could only add the imide groups to the propiolate's carbon once, using DABCO as the catalyst. We also attempted a second imide addition but failed; however, we were able to achieve a second addition using thiols in the presence of TBD. Following this observation, we decided that the selective mono addition of imides to the triple bonds could be utilized for the preparation of imide-yne monomers and these monomers can undergo further thiol-Michael addition reactions to produce poly(imide thioether)s. In our previous publications, we demonstrated that when activated alkynes are reacted with dithiol compounds, the outcomes of the reactions are typical polythioethers that proceed via double thiol-Michael addition.47–49 We also indicated in these studies that dialkyl acetylenedicarboxylates are more reactive and offer better results than dipropiolates in terms of molecular weight and yield, as the alkyne unit is connected to two carbonyl groups, making the alkyne more electron-deficient and therefore more reactive towards thiols. As such, in this study, when preparing imide-yne monomers, we purposely reacted the imide compounds with DMAD. Scheme 3 depicts the synthesis of imide-yne monomers and the following poly(imide thioether)s. In the first step, bisimide compounds 1–3 were reacted with excess DMAD in CHCl3 at room temperature using DABCO as the catalyst. All three monomers were isolated by precipitation in hexane. It should be noted here that bisimide monomers 2 and 3 are not highly soluble in organic solvents and therefore are not used in imide-yne polymerizations as mentioned previously. The same solubility issue is also valid in the preparation of monomers using CHCl3 as the reaction solvent. Moreover, when compounds 2 and 3 were mixed with CHCl3, a suspension was formed, and we observed that this situation did not change when DMAD and DABCO were added to the reaction medium. However, a clear solution started to form when stirring was continued, indicating the expected monomer formation.
 | ||
| Scheme 3 Depiction of the reactions between bisimides 1–3 and DMAD using DABCO as the catalyst. The scheme also illustrates the polymerization of imide-yne monomers 1m–3m with HDT in the presence of TBD via nucleophilic thiol–ene polymerization. | ||
The obtained monomers could be easily characterized by NMR, Fig. 3a shows the 1H NMR spectrum of representative monomer 3m. As seen in this figure, both E- (δ 7.18 ppm) and Z-isomer (δ 6.72 ppm) products are formed simultaneously, with the E-isomer in excess (∼90%). Next, nucleophilic thiol–ene step-growth polymerization was performed using 3m and HDT. The polymerization proceeded for 5 min at room temperature in CHCl3 and low molecular weight polymer (Mw = 7.44 kDa) was obtained in high yield, almost the same results were found for the other two monomers (Table 3). Notably, increasing polymerization duration did not improve molecular weight or yield (data not shown). Fig. 3b displays the corresponding 1H NMR spectrum of polymer (P3m). As can be seen in this figure, vinyl signals completely disappeared and new peaks regarding the polymeric structure could be detected. Here, NCH protons appear at δ 5.36 ppm, SCH protons at δ 4.21 ppm, and SCH2 protons at δ 2.50 ppm, respectively, confirming poly(imide thioether) formation.
 | ||
| Fig. 3 Overlaid 1H NMR spectra of 3m (a) and P3m (b) in CDCl3 (500 MHz). | ||
| Entry | Polymer |
M
wb (kDa) |
Đ |
T
gc (°C) |
Isolated yieldd (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 1 mmol of imide-yne monomer, 1 mmol of HDT, and 0.25 mmol of TBD in 1 mL of CHCl3 at room temperature for 5 minutes. b Determined by GPC calibrated based on linear PS standards in THF. c Determined by DSC from the second heating cycle. d Gravimetric yield obtained after first precipitation. | |||||
| 1 | P1m | 8.24 | 1.80 | 97 | 81 |
| 2 | P2m | 8.73 | 1.81 | 98 | 88 |
| 3 | P3m | 7.44 | 1.93 | 91 | 85 |
The Tg values of the synthesized poly(imide thioether)s were found to be similar (Table 3) and no significant change was observed regarding their structures. As noted above for polymers P1c, P4a, and P5a which contain sulfur linkages, the poly(imide thioether)s exhibited lower Tg values compared to poly(imide ester)s.
Traditional polyimides are thermally stable and display exceptional thermal properties. The TGA thermograms of all polyimides synthesized herein are given in the ESI† and the results are collected in Table 4. All poly(imide ester)s based on bisimide 1, displayed similar TGA spectra. The 10% weight loss temperatures (T10) were above 310 °C for all samples (P1a, P1b, P1c, P1d, and P1e). The presence of the disulfide linkages led to a remarkable decrease in T10 temperatures (P1c). This early decomposition in P1c is also reflected in the Tmax temperatures. While the Tmax values were over 400 °C in P1a, P1b, P1d, and P1e, in P1c, the Tmax value was shifted to lower temperatures and besides, instead of one Tmax temperature, two maximum weight loss temperatures were observed for P1c. Nevertheless, the early decomposition positively affected the char yield of P1c, and a high char yield of 15.8% was obtained at 750 °C under a nitrogen atmosphere. As expected, the highest char yield (21.4%) was obtained for the P-containing poly(imide ester), i.e., P1e, among the poly(imide ester)s synthesized by using the bismide 1. P4a, which contains thioether bonds-containing bisimide, displayed lower T10 and Tmax temperatures but a significant amount of char similar to P1c compared to other poly(imide ester)s based on bisimide 1. Again, in this case, the presence of thiol groups led to early degradation but in turn, produced a remarkable char yield. P5a displayed a higher char yield compared to P4a due to the presence of the aromatic groups in its structure. P6a displayed the lowest T10 temperature among the studied poly(imide ester)s which could be ascribed to the presence of unreacted monomers and solvent. Nevertheless, P6a produced a high amount of char yield due to the additional thermal stability brought by piperazine rings.
| Entry |
T
10a (°C) |
T
maxb (°C) |
Charc (%) |
|---|---|---|---|
| a T 10 is the 10% weight loss temperature. b T max values were obtained from the derivative weight loss curves. c Char value at 750 °C under a nitrogen atmosphere. | |||
| P1a | 399 | 457 | 15.2 |
| P1b | 392 | 442 | 10.5 |
| P1c | 311 | 324/387 | 15.8 |
| P1d | 367 | 434 | 14.8 |
| P1e | 391 | 426 | 21.4 |
| P4a | 314 | 361 | 17.6 |
| P5a | 258 | 384 | 25.4 |
| P6a | 163 | 273/423 | 24.0 |
| P1m | 195 | 353/405 | 7.4 |
| P2m | 295 | 346/408 | 11.5 |
| P3m | 304 | 344/407 | 8.8 |
Moreover, poly(imide thioether)s displayed relatively lower T10 and Tmax temperatures due to the flexible and thermally weak thioether linkages and displayed two-step degradation profiles. The first maximum weight loss temperature was attributed to the decomposition of the thiol linkages while the second maximum weight loss temperature was ascribed to the degradation of the imide groups. The Tmax temperatures of the synthesized poly(imide thioether)s were found to be similar regardless of their structures but P2m produced relatively higher char yield compared to P1m and P3m, which could be ascribed to the presence of thermally stable benzophenone units in P2m.
Conclusions
In summary, the imide-yne reaction was demonstrated to be a new and robust tool for activated alkyne-based click polymerization methods and proved to expand the toolkit of X-yne click polymerization. Using this strategy, a wide range of poly(imide ester)s were synthesized by reacting a variety of bisimide structures with various dipropiolates under benign conditions using a mild catalyst DABCO. Polymerizations provided low to moderate molecular weight polymers in high yields. Post-polymerization modification was shown to be possible, and for this purpose, the main chain double bonds of a representative imide-yne polymer (P1a) were modified using 1-propanethiol by switching the catalyst to a stronger base, TBD, and quantitative yield was obtained within 15 minutes of reaction time. The selective mono-addition nature of imides to activated alkynes further enabled us to develop imide-yne monomers and their nucleophilic thiol–ene step-growth polymerizations were carried out with HDT in the presence of TBD within 5 minutes. According to DSC and TGA analysis results, it was revealed that the obtained polymers have tunable Tg values and thermal stability. Collectively, in this study, a facile time- and energy-saving strategy for polyimide synthesis was demonstrated. We believe the imide-yne polymerization routes introduced herein will be greeted with interest by the polymer community and will be utilized for the synthesis of novel polymers for a wide range of applications in the future.Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research Fund of the Istanbul Technical University (Project Number: TGA-2024-45713).References
- D. Wilson, H. D. Stenzenberger and P. M. Hergenrother, Polyimides, Blackie & Son, Glasgow and London, 1990 Search PubMed.
- C. E. Sroog, Prog. Polym. Sci., 1991, 16, 561–694 CrossRef CAS.
- M. K. Ghosh and K. L. Mittal, Polyimides: Fundamentals and Applications, Marcel Dekker, Inc., New York, 1996 Search PubMed.
- E. Cakmakci, A. Güngör and A. C. Gören, J. Fluor. Chem., 2016, 186, 66–78 CrossRef CAS.
- H. Birtane, A. Beyler-Cigil, E. Cakmakci and M. V. Kahraman, Polym.-Plast. Technol. Mater., 2017, 56, 443–452 CrossRef CAS.
- A. Beyler-Cigil, E. Cakmakci and M. V. Kahraman, Polym. Compos., 2016, 37, 2285–2292 CrossRef CAS.
- M. I. Bessonov and V. A. Zubkov, in Polyamic Acids and Polyimides: Synthesis, Transformations, and Structure, CRC Press, Inc., Boca Raton, 1993, p. 2 Search PubMed.
- K. L. Mittal, Polyimides and Other High-Temperature Polymers: Synthesis, Characterization and Applications, BRILL, Lei-den, 2009, vol. 5, p. 25 Search PubMed.
- H. F. Mark, Encyclopedia of Polymer Science and Technology, Concise, John Wiley & Sons, 2013, p. 914 Search PubMed.
- X. Li, X. Wang, Q. Wang and Q. Wei, J. Eng. Fibers Fabr., 2014, 9, 33–91438 Search PubMed.
- C. P. Wong, Polymers for Electronic & Photonic Application, Academic Press, Inc., Boston, 1992, p. 252 Search PubMed.
- M. H. Kailani and C. S. P. Sung, Macromolecules, 1998, 31, 5771–5778 CrossRef CAS.
- H. Yu, J. Jung, J. Choi and C. Chung, J. Polym. Sci., Part A: Polym. Chem., 2015, 54, 1593–1602 CrossRef.
- M. A. B. Meador, E. J. Malow, R. Silva, S. Wright, D. Quade, S. L. Vivod, H. Guo, J. Guo and M. Cakmak, ACS Appl. Mater. Interfaces, 2012, 4, 536–544 CrossRef CAS.
- R. A. Meyers, J. Polym. Sci., Part A-1: Polym. Chem., 1969, 7, 2757–2762 CrossRef.
- H. R. Lubowitz, Polym. Prepr., 1971, 12, 329 CAS.
- T. M. Moy, C. D. DePorter and J. E. McGrath, Polymer, 1993, 34, 819–824 CrossRef CAS.
- T. Takekoshi, J. G. Wirth, D. R. Heath, J. E. Kodhznowski, J. S. Manello and M. J. Webber, J. Polym. Sci., Polym. Chem. Ed., 1980, 18, 3069–3080 CrossRef CAS.
- T. Takekoshi, in Polyimides, ed. D. Wilson, H. D. Stenzenberger and P. M. Hergenrother, Blackie, New York, 1990, p. 38 Search PubMed.
- T. Takekoshi, in Polyimides, ed. M. K. Ghosh and K. L. Mittal, Marcel Dekker, 1996, p. 7 Search PubMed.
- L. S. Tan and F. E. Arnold, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 1819–1834 CrossRef CAS.
- R. W. Lenz, Organic Chemistry of Synthetic High Polymers, Interscience, New York, 1967 Search PubMed.
- H. Laita, S. Boufi and A. Gandini, Eur. Polym. J., 1997, 33, 1203–1211 CrossRef CAS.
- D. G. Harwthorne, J. H. Hodgkin, M. B. Jackson and T. C. Morton, High Perform. Polym., 1994, 6, 249–256 CrossRef.
- C. Glousse and A. Gandini, Polym. Int., 1999, 48, 723–731 CrossRef.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- A. Genest, D. Portinha, E. Fleury and F. Ganachaud, Prog. Polym. Sci., 2017, 72, 61–110 CrossRef CAS.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- K. Ratzenböck, S. M. Fischer and C. Slugovc, Monatsh. Chem., 2023, 154, 443–458 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- J. C. Worch, C. J. Stubbs, M. J. Price and A. P. Dove, Chem. Rev., 2021, 121, 6744–6776 CrossRef CAS PubMed.
- J. Zhang, Z. Zhang, J. Wang, Q. Zang, J. Z. Sun and B. Z. Tang, Polym. Chem., 2021, 12, 2978–2986 RSC.
- X. Chen, R. Hu, C. Qi, X. Fu, J. Wang, B. He, D. Huang, A. Qin and B. Z. Tang, Macromolecules, 2019, 52, 4526–4533 CrossRef CAS.
- B. He, S. Zhen, Y. Wu, R. Hu, Z. Zhao, A. Qin and B. Z. Tang, Polym. Chem., 2016, 7, 7375–7382 RSC.
- O. A. Moe and D. T. Warner, J. Am. Chem. Soc., 1949, 71, 1251–1253 CrossRef CAS.
- G. H. Imanzadeh, M. M. Tavana, M. R. Zamanloo and Y. Mansoori, Chin. J. Chem., 2009, 27, 389–396 CrossRef CAS.
- G. Imanzadeh, F. Ahmadi, M. Zamanloo and Y. Mansoori, Molecules, 2010, 15, 7353–7362 CrossRef CAS PubMed.
- S. Ma, L. Wu, M. Liu, Y. Huang and Y. Wang, Tetrahedron, 2013, 69, 2613–2618 CrossRef CAS.
- H. S. Park and M. S. Gong, Polymer, 2012, 36, 427–433 CAS.
- X. Fu, A. Qin and B. Z. Tang, Aggregate, 2023, 4, e350 CrossRef CAS.
- B. He, J. Huang, X. Liu, J. Zhang, J. W. Y. Lam and B. Z. Tang, Prog. Polym. Sci., 2022, 126, 101503 CrossRef CAS.
- B. Yao, J. Sun, A. Qin and B. Z. Tang, Chin. Sci. Bull., 2013, 58, 2711–2718 CrossRef CAS.
- O. Daglar, S. Luleburgaz, E. Baysak, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Eur. Polym. J., 2020, 137, 109926 CrossRef CAS.
- J. Du, D. Huang, H. Li, A. Qin, B. Z. Tang and Y. Li, Macromolecules, 2020, 53, 4932–4941 CrossRef CAS.
- J. C. Worch, A. C. Weems, J. Yu, M. C. Arno, T. R. Wilks, R. T. R. Huckstepp, R. K. O'Reilly, M. L. Becker and A. P. Dove, Nat. Commun., 2020, 11, 3250 CrossRef CAS.
- O. Daglar, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2019, 52, 3558–3572 CrossRef CAS.
- O. Daglar, E. Cakmakci, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2020, 53, 2965–2975 CrossRef CAS.
- U. S. Gunay, U. Tunca and H. Durmaz, J. Macromol. Sci., Part A: Pure Appl. Chem., 2024, 61, 143–154 CrossRef CAS.
- H. He, B. Liu, M. Wang, R. Vachet and S. Thayumanavan, Polym. Chem., 2018, 10, 187–193 RSC.
- B. He, H. Su, T. Bai, Y. Wu, S. Li, M. Gao, R. Hu, Z. Zhao, A. Qin, J. Ling and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 5437–5443 CrossRef CAS.
- R. Jiang, M. Liu, H. Huang, L. Mao, Q. Huang, Y. Wen, Q. Cao, J. Tian, X. Zhang and Y. Wei, Dyes Pigm., 2018, 151, 123–129 CrossRef CAS.
- L. Yu, Z. Zhang, Y. Z. You and C. Y. Hong, Eur. Polym. J., 2018, 103, 80–87 CrossRef CAS.
- B. Alkan, O. Daglar, B. A. Temel, H. Durmaz and G. Temel, Polym. Chem., 2022, 13, 2442–2449 RSC.
- E. Akar, S. Luleburgaz, U. S. Gunay, V. Kumbaraci, U. Tunca and H. Durmaz, Eur. Polym. J., 2023, 199, 112470 CrossRef CAS.
- B. Celik, D. Kandemir, S. Luleburgaz, E. Cakmakci, U. S. Gunay, V. Kumbaraci and H. Durmaz, ACS Sustainable Chem. Eng., 2023, 11, 831–841 CrossRef CAS.
- O. Yildirimkaraman, S. Özenler, U. S. Gunay, H. Durmaz and U. H. Yıldız, Langmuir, 2021, 37, 10902–10913 CrossRef CAS.
- H. Si, K. Wang, B. Song, A. Qin and B. Z. Tang, Polym. Chem., 2020, 11, 2568–2575 RSC.
- Q. M. Jiang, H. Y. Zhang, Q. L. Jiang, S. Y. Zhang, S. Y. Guan, W. Y. Huang, X. Q. Xue, H. J. Yang, L. Jiang, B. B. Jiang and S. Komarneni, Polym. Chem., 2023, 14, 1232–1240 RSC.
- L. D. Thai, J. A. Kammerer, P. Théato, H. Mutlu and C. Barner-Kowollik, ACS Macro Lett., 2024, 13, 681–687 CrossRef.
- Q. Jiang, S. Zhang, Z. Liu, S. Guan, W. Huang, H. Yang, L. Jiang, X. Xue, S. Komarneni and B. Jiang, ACS Appl. Polym. Mater., 2024, 6, 4127–4137 CrossRef CAS.
- G. Sagdic, O. Daglar, E. Cakmakci, V. Findik, S. S. Erdem, U. Tunca, U. S. Gunay and H. Durmaz, Macromolecules, 2023, 56, 7006–7022 CrossRef CAS.
- J. Chen, X. Xu, Q. Xia, J. Wang, A. Qin and B. Z. Tang, Macromolecules, 2024, 57, 1970–1978 CrossRef CAS.
- B. M. Trost and G. R. Dake, J. Am. Chem. Soc., 1997, 119, 7595–7596 CrossRef CAS.
- A. Ilin, A. F. Gubaev, D. R. Islamov, K. R. Islamov and V. I. Galkin, Chem. Heterocycl. Compd., 2021, 57, 175–180 CrossRef CAS.
- L. Mola, J. Font, L. Bosch, J. Caner, A. M. Costa, G. Etxebarría-Jardí, O. Pineda, D. Vicente and J. Vilarrasa, J. Org. Chem., 2013, 78, 5832–5842 CrossRef CAS.
- A. Shahraki and A. Hassanabadi, J. Chem. Res., 2014, 38, 586–588 CrossRef CAS.
- E. Petit, L. Bosch, J. Font, L. Mola, A. M. Costa and J. Vilarrasa, J. Org. Chem., 2014, 79, 8826–8834 CrossRef CAS.
- G. C. Crockett, B. J. Swanson, D. R. Anderson and T. H. Koch, Synth. Commun., 1981, 11, 447–454 CrossRef CAS.
- C. B. Yoon and H. K. Shim, J. Mater. Chem., 1999, 9, 2339–2344 RSC.
- C. A. Bell, J. Yu, I. A. Barker, V. X. Truong, Z. Cao, A. V. Dobrinyin, M. L. Becker and A. P. Dove, Angew. Chem., Int. Ed., 2016, 55, 13076–13080 CrossRef CAS.
- X. Wang, R. Hu, Z. Zhao, A. Qin and B. Z. Tang, Sci. China: Chem., 2016, 59(12), 1554–1560 CrossRef CAS.
- I. E. Kakas, G. Sagdic, V. Kumbaraci, H. Durmaz and U. S. Gunay, J. Macromol. Sci., Part A: Pure Appl.Chem., 2024, 61, 630–639 CrossRef CAS.
- T. Han, S. Chen, X. Wang, X. Fu, H. Wen, Z. Wang, D. Wang, A. Qin, J. Yang and B. Z. Tang, Adv. Sci., 2022, 9, 2105395 CrossRef CAS.
- E. Fritz-Langhals, Org. Process Res. Dev., 2022, 26, 3015–3023 CrossRef CAS.
- J. G. Kim, G. S. Lee and A. Lee, J. Polym. Sci., 2024, 62, 42–91 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00918e |
| This journal is © The Royal Society of Chemistry 2024 |