
Diversified synthesis of poly(sulfonyl isourea)s by multicomponent polymerizations of isocyanides, sulfonyl azides, and alcohols/phenols†
Dongyang
Fan‡
a,
Fenggang
Chen‡
a,
Dong
Wang
 a,
Ting
Han
*a and
Ben Zhong
Tang
*b
a,
Ting
Han
*a and
Ben Zhong
Tang
*b
aCenter for AIE Research, Guangdong Provincial Key Laboratory of New Energy Materials Service Safety, College of Materials Science and Engineering, Shenzhen University, Shenzhen, Guangdong 518060, China. E-mail: hanting@szu.edu.cn
bSchool of Science and Engineering, Shenzhen Institute of Aggregate Science and Technology, The Chinese University of Hong Kong, Shenzhen (CUHK-Shenzhen), Guangdong 518172, China. E-mail: tangbenz@cuhk.edu.cn
First published on 4th November 2024
Abstract
Multicomponent polymerization (MCP) has become a powerful tool for the preparation of polymers with complex or well-defined structures. The development of MCPs that can take full advantage of the great diversity of monomer combinations is attractive but still remains challenging. Herein, we report a facile MCP method using isocyanides, sulfonyl azides, and alcohol/phenols as monomers, in which the monomer combination can be readily tuned to achieve the diversity-oriented synthesis of poly(sulfonyl isourea)s in excellent atom economy. By systematically optimizing the polymerization conditions and changing the functionalities and combinations of monomers, a series of heteroatom-rich polymers with sulfonyl isourea units and high molecular weights (Mw values of up to 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500) were successfully prepared. All the obtained polymers exhibit good thermal stability, and the variation in polymer backbone structures led to an obvious difference in their morphological stability. This diversity-oriented MCP strategy may provide inspiration and new possibilities for the development of heterochain polymers with advanced functionalities.
500) were successfully prepared. All the obtained polymers exhibit good thermal stability, and the variation in polymer backbone structures led to an obvious difference in their morphological stability. This diversity-oriented MCP strategy may provide inspiration and new possibilities for the development of heterochain polymers with advanced functionalities.
Introduction
The development of new polymerization tools for the preparation of functional polymeric materials with novel structures and excellent properties is an important research topic in the field of polymer science. Most polymerizations are developed from small molecular organic reactions. Among various strategies, multicomponent reactions (MCRs) have attracted much attention in organic chemistry due to their high atom economy, simple operation, good functional group tolerance, and high efficiency.1–5 Taking advantage of MCRs, polymer chemists have devoted great efforts to introducing them into the field of polymer synthesis. The derived multicomponent polymerization (MCP) strategies provide new possibilities for the efficient preparation of structurally complex and well-defined polymers from simple precursors.6–12 In comparison with traditional homopolymerizations and two-component polymerizations, one remarkable and unique advantage of MCPs is the excellent product diversity. By tuning the combinations of monofunctional and bifunctional monomers, a vast variety of polymer structures can be facilely generated by one MCP strategy.11,13–16 However, the main focus of reported MCPs is still on the development of an MCP with a single type of monomer combination. There have been few studies that fully exploit the applicability of different monomer combinations to facilitate diversity-oriented polymer synthesis.As a representative group of MCPs, MCPs based on triple-bond-containing monomers can yield polymers with conjugated linkers such as double bonds, triple bonds and aromatic heterocycles or special structural units.17–25 The unique structures can impart attractive thermal, mechanical, optical, electrical, magnetic and biological properties to the polymers.26–29 By combining the advantages of the classical click chemistry of copper-catalyzed alkyne–azide cycloadditions and MCP strategies, MCPs derived from the MCRs of alkynes and sulfonyl azides have become a powerful toolbox for the synthesis of heterochain or heterocyclic polymers.25,30–35 For example, a series of MCPs of alkynes, sulfonyl azides, and nucleophilic reagents have been developed for the preparation of diverse polyamidines and polyimidates.31,32 As depicted in Scheme 1A, MCPs based on alkynes and sulfonyl azides mainly work through the formation of ketenimine as the key intermediate to give heteroatom-rich target products. With amines as nucleophilic reagents, polyamidines can be readily generated.32 When alcohols were employed as nucleophiles, polyimidates were produced with high efficiency.31
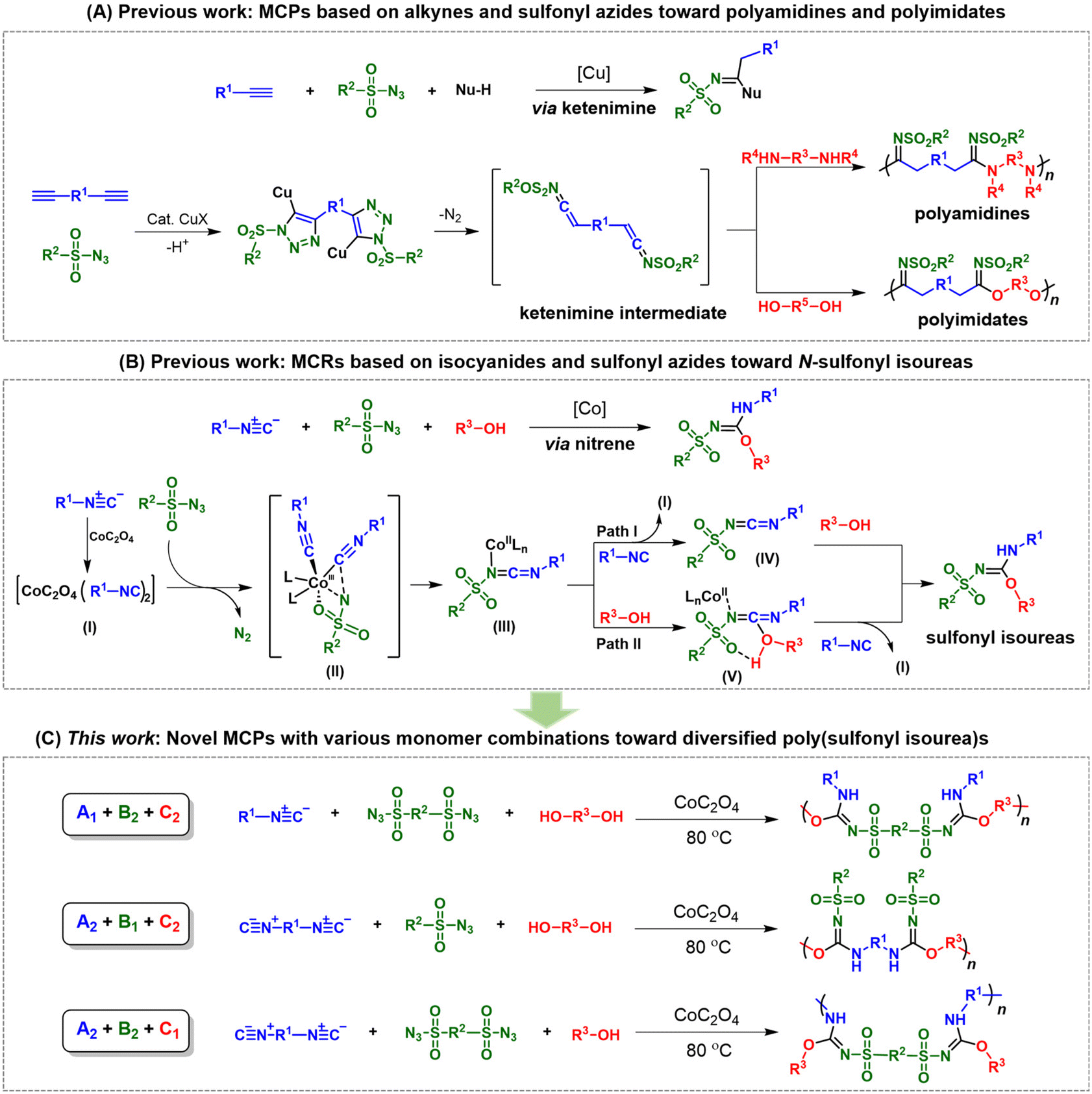 | ||
| Scheme 1 (A) Multicomponent polymerizations (MCPs) based on alkynes and sulfonyl azides; (B) multicomponent reactions (MCRs) based on isocyanides and sulfonyl azides toward N-sulfonyl isoureas; (C) MCPs reported in this work with various monomer combinations. | ||
Apart from acetylenic monomers, isocyanides, another class of triple-bond-containing monomers, have also been introduced to MCPs as highly reactive monomers.22,24,36 They are capable of reacting with almost any type of reagent such as electrophiles, nucleophiles and even free radicals to construct heterocyclic and spirocyclic compounds with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N unit in polymer backbones. The high reactivity of isocyanide substrates greatly reduces the harsh requirements for synthetic conditions, which makes them widely used in the preparation of heteroatom-rich compounds and pharmaceutical molecules. Classical isocyanide-based MCPs mainly include Passerini three-component polymerizations and Ugi multicomponent polymerizations.13,22,37–40 In recent years, some fascinating MCPs with isocyanide and sulphur or selenium as monomers have also been developed to synthesize multifunctional heteroatom-rich polymers in a facile, economical and highly efficient manner.41–46 However, despite the attractive merits of click-type MCPs and isocyanide-based MCPs, MCPs with both isocyanides and sulfonyl azides as monomers have rarely been reported.
N unit in polymer backbones. The high reactivity of isocyanide substrates greatly reduces the harsh requirements for synthetic conditions, which makes them widely used in the preparation of heteroatom-rich compounds and pharmaceutical molecules. Classical isocyanide-based MCPs mainly include Passerini three-component polymerizations and Ugi multicomponent polymerizations.13,22,37–40 In recent years, some fascinating MCPs with isocyanide and sulphur or selenium as monomers have also been developed to synthesize multifunctional heteroatom-rich polymers in a facile, economical and highly efficient manner.41–46 However, despite the attractive merits of click-type MCPs and isocyanide-based MCPs, MCPs with both isocyanides and sulfonyl azides as monomers have rarely been reported.
Recently, Ji et al. reported a cobalt-catalyzed MCR of isocyanides, sulfonyl azides, and alcohols for the preparation of N-sulfonyl isoureas (Scheme 1B).47 This MCR has the advantages of high efficiency, wide substrate scope and ease of operation. Unlike the MCRs of alkynes and sulfonyl azides, and nucleophilic reagents, the reaction mechanism of this isocyanide-based MCR involves nitrene as the key intermediate. First, the cobalt complex activates two isocyanide molecules to form intermediate I. Then, the sulfonyl azide substrate releases one molecule of N2 and coordinates with intermediate I via transition state II to afford the reactive intermediate (III, nitrene) with the NCN structure. There exist two paths in the following procedure. In path I, intermediate III reacts with isocyanide to give intermediate IV, which is then attacked by the alcohol to give the target molecule. In path II, intermediate III coordinates with alcohol to form intermediate V, which then undergoes exchange with isocyanide to form the final product. The mechanism of this MCR is relatively lucid and the structure of the product is well-defined. The produced sulfonyl isoureas exhibit the structural features of both amidines and imidates. The introduction of such structures into polymer backbones may impart the corresponding polymers with the properties and functionalities of both polyamidines and polyimidates.
Inspired by this MCR, herein we developed a novel MCP strategy with isocyanides, sulfonyl azides and alcohols/phenols for the preparation of poly(sulfonyl isourea)s (Scheme 1C). To take full advantage of the facile monomer tunability of this MCP, we thoroughly explored the possibility of this MCP for diversity-oriented polymer synthesis. The combination of isocyanide, sulfonyl azide, and alcohol/phenol monomers with different functionalities resulted in the realization of three routes of MCPs (A1 + B2 + C2, A2 + B1 + C2, and A2 + B2 + C1), which greatly improved the diversity of polymerization products. The MCPs displayed relatively good tolerance to different functional groups and all polymerizations can be carried out under mild conditions. The obtained heteroatom-rich poly(sulfonylisourea)s exhibited molecular weight (Mw) values of up to 120500 g mol−1, with isolated yields ranging from 25% to 87%. All of the resulting polymers displayed good solubility and thermal stability.
Results and discussion
Polymerization
To advance MCPs for the efficient synthesis of poly(sulfonyl isourea)s, a series of isocyanide, sulfonyl azide, and alcohol/phenol monomers were systematically designed and synthesized based on the literature.14,48,49 The readily available compounds 1-((isocyanomethyl)sulfonyl)-4-methylbenzene (1a), 4,4′-oxydibenzenesulfonyl azide (2a), and hexane-1,6-diol (3a) were selected as model monomers to optimize the polymerization conditions in the combination “A1 + B2 + C2”.Initially, polymerization was conducted in air using DCE at 80 °C with 10 mol% CoC2O4 and a monomer concentration of [2a] = 0.2 M for 24 hours. The monomer feed ratio of 1a, 2a, and 3a was 2:1:1. Under such polymerization conditions, the product with an Mw of 8700 g mol−1 was obtained in only 21% yield. To enhance both yield and molecular weight, various solvents were tested (Table 1, entries 1–5). The results with THF were similar to those with DCE, while using a small amount of polar toluene, the molecular weight was increased, but the yield was reduced to 12%. Polymerization in DMF led to undesired products, while MeCN, a solvent with higher polarity, yielded slightly better results than DCE and a significantly improved molecular weight. Therefore, MeCN was selected as the optimal solvent. Further optimization focused on catalyst loading (Table 1, entries 5–8). The yield and molecular weight increased continuously as the amount of CoC2O4 was increased to 30 mol% and remained constant when the loading was further increased to 40 mol%. Additionally, no enhancement in yield or molecular weight was observed when the monomer concentration was increased (Table 1, entries 9 and 10). Considering both activity and cost, 30 mol% CoC2O4 with a 0.2 M monomer concentration was chosen for further optimization.
a
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | CoC2O4 (mol%) | [2a] (M) | [1a]:[2a]:[3a] |
Yield (%) |
M
nb (g mol−1) |
M
wb (g mol−1) |
Đ |
| a The polymerizations were carried out at 80 °C for 24 h under air. b Estimated by gel permeation chromatography (GPC) with DMF containing 0.05 M LiBr as the mobile phase on the basis of a linear polystyrene calibration. Dispersity (Đ) = Mw/Mn. c Product with undesired and unidentified structures. | ||||||||
| 1 | DCE | 10 | 0.2 | 2:1:1 |
21 | 6800 | 8700 | 1.3 |
| 2 | THF | 10 | 0.2 | 2:1:1 |
20 | 7700 | 8900 | 1.2 |
| 3 | Toluene | 10 | 0.2 | 2:1:1 |
12 | 10300 |
13300 |
1.3 |
| 4 | DMF | 10 | 0.2 | 2:1:1 |
UDc | — | — | — |
| 5 | MeCN | 10 | 0.2 | 2:1:1 |
24 | 9800 | 12500 |
1.3 |
| 6 | MeCN | 20 | 0.2 | 2:1:1 |
48 | 10200 |
13100 |
1.3 |
| 7 | MeCN | 30 | 0.2 | 2:1:1 |
58 | 10600 |
14000 |
1.3 |
| 8 | MeCN | 40 | 0.2 | 2:1:1 |
57 | 10700 |
14000 |
1.3 |
| 9 | MeCN | 30 | 0.3 | 2:1:1 |
45 | 10400 |
13400 |
1.3 |
| 10 | MeCN | 30 | 0.4 | 2:1:1 |
48 | 10600 |
13200 |
1.3 |
| 11 | MeCN | 30 | 0.2 | 3:1:1 |
68 | 13300 |
17200 |
1.3 |
| 12 | MeCN | 30 | 0.2 | 4:1:1 |
75 | 11800 |
15300 |
1.3 |
| 13 | MeCN | 30 | 0.2 | 5:1:1 |
55 | 12400 |
15600 |
1.3 |
Excess isocyanide may enhance polymerization based on the mechanism of small molecules.47,50,51 As depicted in Scheme 1B, reactive intermediate I was formed by combining CoC2O4 with two isocyanide monomers, though this process was slow and challenging. Thus, a surplus of isocyanide might be necessary to produce sufficient intermediate I for stoichiometric balance with the sulfonyl azide monomer. Results in entries 11–13 of Table 1 showed that increasing the ratio of 1a improved the polymerization efficiency. When the monomer ratio of [1a]:[2a]:[3a] was 4:1:1, the yield increased to 75% with an Mw of 15300 g mol−1. Further increasing the ratio led to a noticeable decrease in yield. Consequently, the optimal monomer ratio for the polymerization reaction was established as 4:1:1. In summary, the optimal conditions for this MCP are conducting the polymerizations in MeCN at 80 °C for 24 h in air in the presence of 30 mol% CoC2O4 with a monomer ratio of 1a]:[2a]:[3a] = 0.8:0.2:0.2.
Based on the optimized conditions, we explored the monomer scope. Various disulfonyl azides, isocyanides, and alcohols/phenols were used in the polymerizations (Table S1, entries 1–4†). Polymerization did not occur with monomer 1b, probably due to activation of the C–H bonds adjacent to the isocyanide group under the polymerization conditions, which led to a side reaction rather than polymerization. Monomer 1c, which has steric hindrance adjacent to the isocyanide group, resulted in polymer P1c2a3a with a molecular weight (Mw) of 19900 g mol−1 and a yield of 44%. When the aliphatic monomer 1d containing a morpholine structure was employed, polymer P1d2a3a was obtained with a yield of 62% and an Mw of 15500 g mol−1. However, both polymers exhibited lower purity. We then examined the scope of sulfonyl azide monomers. Monomer 2b yielded a gelatinous product (Table S1, entry 5†). Even under adjusted polymerization conditions, such as shorter reaction time, the target polymer remained insoluble, indicating the high reactivity of monomer 2b. The use of monomer 2c, which contains flexible alkoxy chains, produced unidentified byproducts (Table S1, entry 6†). Finally, we examined the reactivity of the third component. In addition to the aliphatic diol 3a, the reactivity of diphenol was also explored using monomer 3b (Table S1, entry 7†). The polymerization reaction could proceed smoothly to give P1a2a3b.
Structural characterization
To gain insight into the structures of the synthesized polymers, model compound model-1 was prepared according to Scheme S1,† in accordance with the reported literature.47 The structure of model-1 was fully characterized using high-resolution mass spectrometry (HRMS), NMR techniques (Fig. S1 and S2†), and FT-IR spectroscopy (Fig. 1D).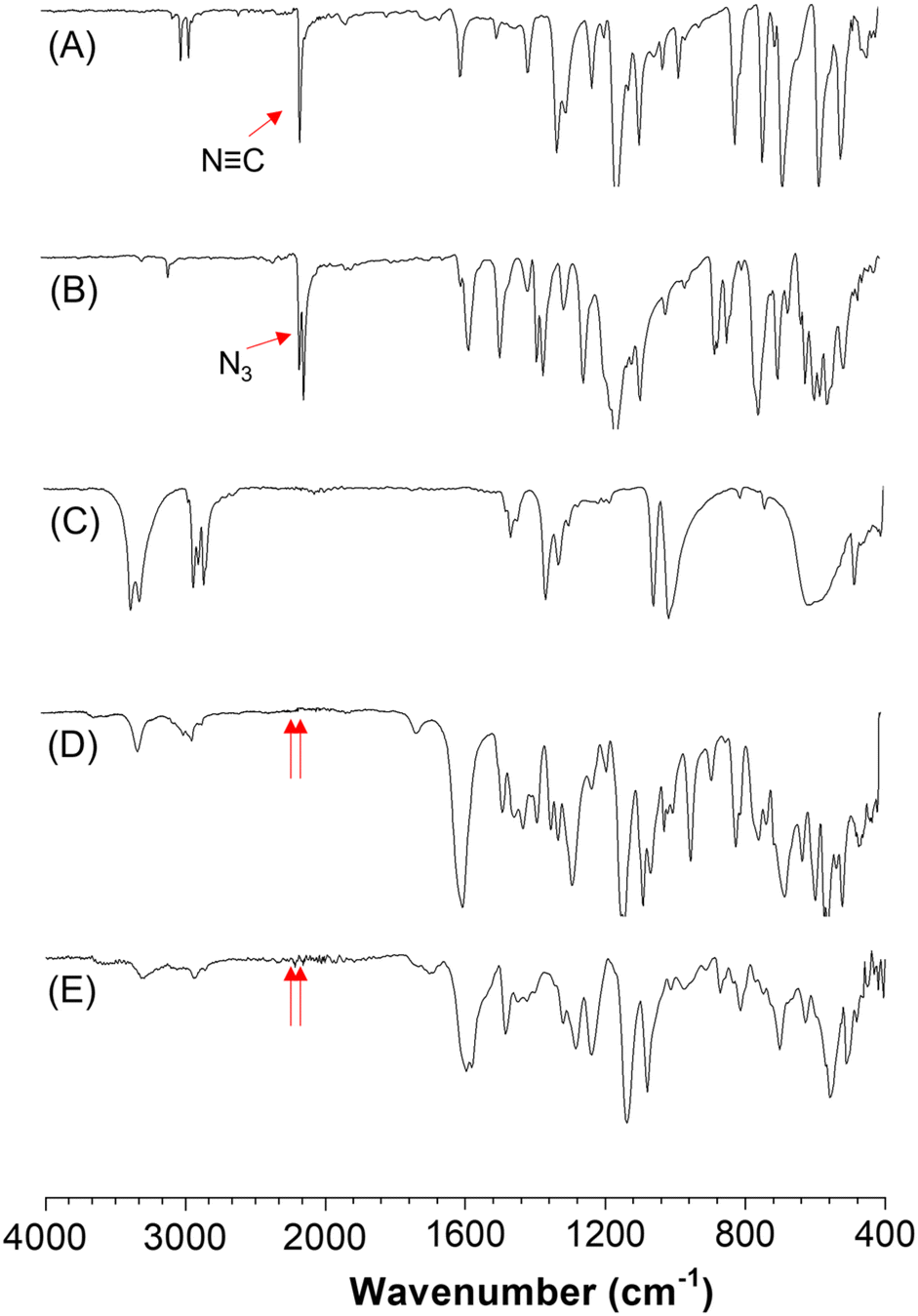 | ||
| Fig. 1 IR spectra of (A) 1a, (B) 2a, (C) 3a, (D) model-1, and (E) P1a2a3a. | ||
The characterization results of P1a2a3a were compared with those of model-1 and the corresponding monomers, as illustrated in Fig. 1. The characteristic peaks associated with the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C and N3 stretching vibrations at 2152 cm−1 and 2125 cm−1 for monomers 1a and 2a, respectively, nearly disappeared in both model-1 and P1a2a3a. This observation indicated the consumption of isocyanide and azide by MCP.
C and N3 stretching vibrations at 2152 cm−1 and 2125 cm−1 for monomers 1a and 2a, respectively, nearly disappeared in both model-1 and P1a2a3a. This observation indicated the consumption of isocyanide and azide by MCP.
1H NMR analysis was performed to gain more detailed information about the polymer structure. As illustrated in Fig. 2A–E, the resonance peaks corresponding to the methyl and methylene groups of 1a at positions “a” and “b” were observed in similar regions of the 1H NMR spectra for both model-1 and P1a2a3a. The OH proton (position “c”) of monomer 1c that resonated at δ 1.70 ppm disappeared, while the resonance peak of the proton at position “d” was shifted to low-field positions e and e′ after the reaction. New peaks at positions “f” and “f′” in the products were attributed to the NH protons of the sulfonyl isourea units. These observations confirm that polymerization successfully occurred and that new sulfonyl isourea structures were formed via the MCP processes.
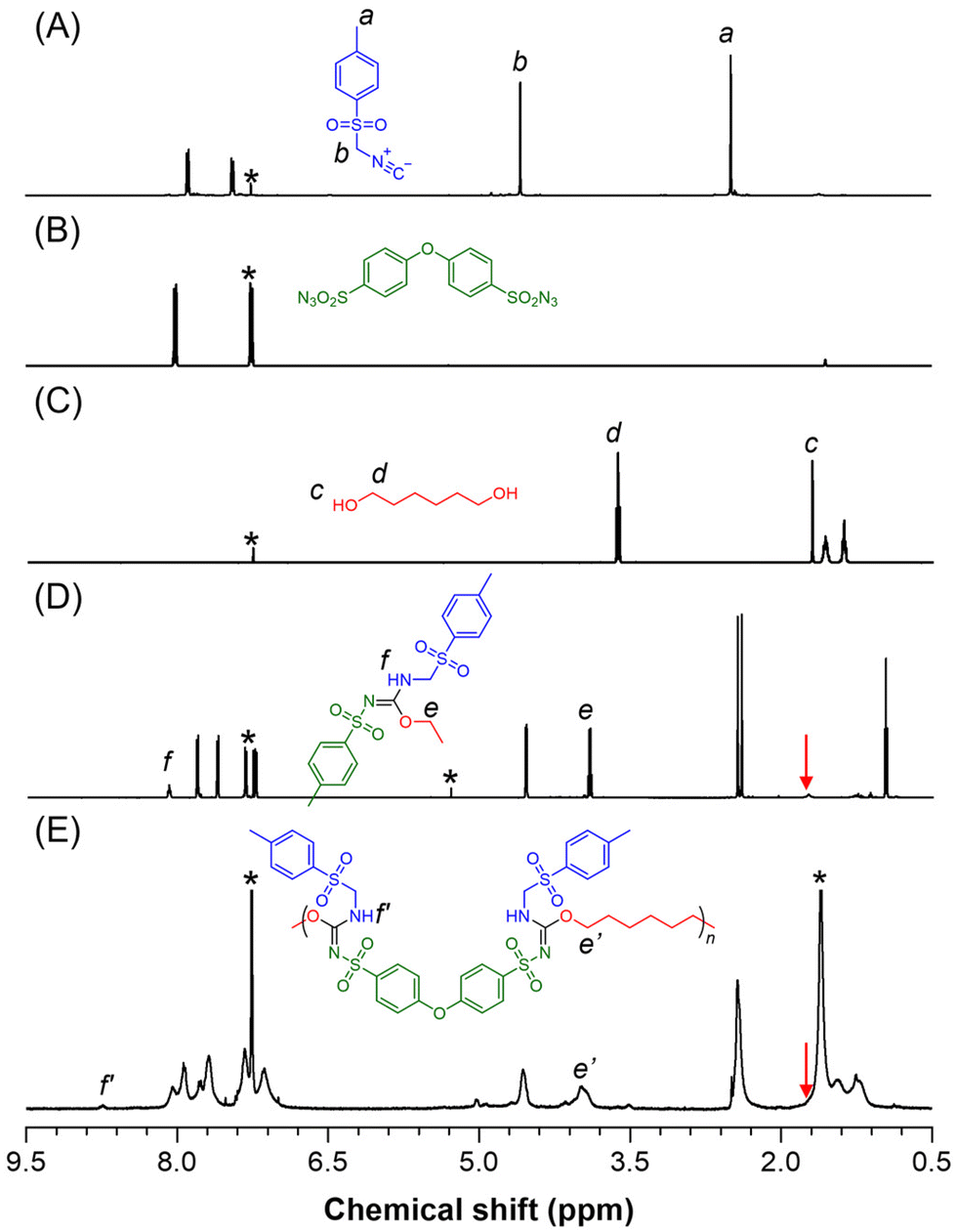 | ||
| Fig. 2 1H NMR spectra of (A) 1a, (B) 2a, (C) 3a, (D) model-1, and (E) P1a2a3a in CDCl3. The solvent peaks are marked with asterisks. | ||
The polymer structure was further validated through 13C NMR analysis (Fig. 3A–E). The carbon associated with the isocyanide group (“a”) in monomer 1a disappeared after the reaction, while the methylene carbon (“b”) was detected at similar positions (“c” and “c′”) in both model-1 and P1a2a3a. The signal at position “d” in 2a was retained in P1a2a3a at position “e”. The resonance peak at position “f” in 3a shifted to lower fields, appearing at positions “g” and “g′” in model-1 and P1a2a3a, respectively. Carbon signals corresponding to the newly formed imine groups (“h” and “h′”) were detected in both model-1 and P1a2a3a, indicating the formation of sulfonyl isourea groups. Similar features were observed in the NMR results of other polymers (Fig. S3–S5†), confirming that the MCP processes successfully produced the target polymers.
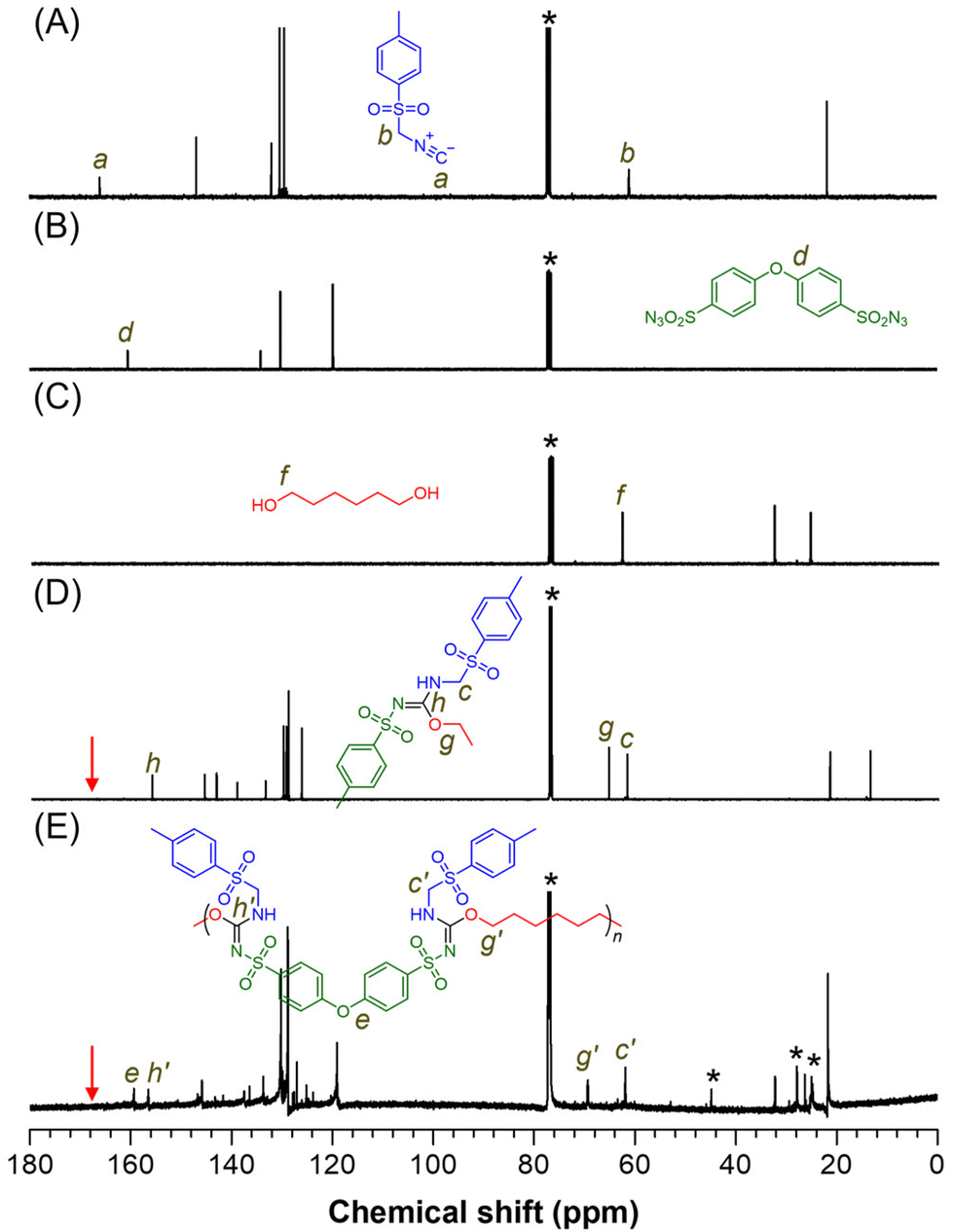 | ||
| Fig. 3 13C NMR spectra of (A) 1a, (B) 2a, (C) 3a, (D) model-1, and (E) P1a2a3a in CDCl3. The solvent peaks are marked with asterisks. | ||
MCPs of other monomer combinations
Although the applicability of monomers for route A1 + B2 + C2 was not fully satisfactory, the results still demonstrated the potential for multi-route polymerization. Consequently, we next investigated MCPs using the monomer combinations of A2 + B1 + C2 (Table 2).a
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Polymer | Solvent | [1] (M) | [1]:[2]:[3] |
Yield (%) |
M
nb (g mol−1) |
M
wb (g mol−1) |
Đ |
| a The polymerizations were carried out using CoC2O4 (30 mol%) at 80 °C for 24 h under air. b Estimated by gel permeation chromatography (GPC) with DMF containing 0.05 M LiBr as the mobile phase on the basis of a linear polystyrene calibration. Dispersity (Đ) = Mw/Mn. c Product with undesired and unidentified structures. d Product with the coexistence of the desired structure and impure structure. | ||||||||
| 1 | P1e2d3a | DCE | 0.2 | 1:2:1 |
20 | 12100 |
15800 |
1.3 |
| 2 | P1e2d3a | DCE | 0.2 | 1:3:1 |
25 | 20500 |
22600 |
1.1 |
| 3 | P1e2d3a | DCE | 0.2 | 1:4:1 |
23 | 19900 |
22500 |
1.1 |
| 4 | P1e2d3a | DMF | 0.2 | 1:3:1 |
UDc | — | — | — |
| 5 | P1e2d3a | MeCN | 0.2 | 1:3:1 |
26 | 19500 |
21100 |
1.1 |
| 6 | P1f2d3a | DCE | 0.2 | 1:3:1 |
60d | 19200 |
23000 |
1.2 |
| 7 | P1f2d3c | DCE | 0.2 | 1:3:1 |
61d | 19900 |
24700 |
1.2 |
Initially, polymerization was performed using DCE as the solvent under the optimized conditions previously described. The first attempt of MCP yielded a product with a molecular weight (Mw) of 15800 g mol−1 but only achieved a 20% yield (Table 2, entry 1). Due to this low efficiency, we subsequently explored variations in monomer ratios and solvents. Adjusting the monomer ratio to 1:3:1 resulted in a slight increase in yield, while using DCE as the solvent slightly improved the molecular weight (Table 2, entries 2–5). The low yield may be attributed to the significant steric hindrance of monomer 1e, which likely impeded the efficient formation of diimide intermediates with sulfonyl azide monomers. Consequently, we employed the less hindered diisocyanide monomer 1f, which led to a notable increase in yield to approximately 60% (Table 2, entry 6). Additionally, when ethylene glycol monomer 3c was polymerized with 1f and 2d, both yield and molecular weight improved slightly (Table 2, entry 7). However, small amounts of impurities were detected in the structures of the resulting products. Detailed characterization data for this route are available in the ESI (Fig. S6–S10†).
Based on the reaction mechanism, the polymerization of difunctionalized isocyanide, difunctionalized sulfonyl azide, and alcohol (A2 + B2 + C1) initially forms polymer main chains, which then react with the third component to generate branched chains. This process differs significantly from the previous two routes. Initial polymerization attempts under the optimal conditions of the first route yielded only 20% (Table 3, entry 1), with the formation of a large amount of orange insolubles. These insolubles, probably resulting from the reaction between isocyanide and sulfonyl azide, exhibited poor solubility in MeCN, which negatively impacted subsequent polymerizations. Switching solvents to DCE and DMF improved the solubility and subsequently the polymerization yields. In DCE, the yield of P1f2a3d increased to 76% with a molecular weight (Mw) of 120500 g mol−1, making DCE the optimal solvent (Table 3, entries 2 and 3). The yield of P1e2a3d reached 87% with a Mw of 78300 g mol−1 (Table 3, entry 4). To explore the potential for fluorescence applications, we conducted MCPs with a TPE-containing phenolic monomer 3e (Table 3, entries 5 and 6). Despite achieving a bright yellowish-green fluorescence, the polymer structure was incorrect. Even upon changing the solvent and other conditions, we failed to achieve the target product. The differences in electronegativity and chemical properties between phenols and alcohols may have contributed to the polymerization failure. Detailed characterization data for this route are available in the ESI (Fig. S10–S14†).
a
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Polymer | Solvent | [1] (M) | Amount of 3 | Yield (%) |
M
nb (g mol−1) |
M
wb (g mol−1) |
Đ |
| a The polymerizations were carried out using CoC2O4 (30 mol%) at 80 °C for 24 h under an air atmosphere and a monomer molar ratio of [1] = [2] = 0.2 M. b Estimated by gel permeation chromatography (GPC) with DMF containing 0.05 M LiBr as the mobile phase on the basis of a linear polystyrene calibration. Dispersity (Đ) = Mw/Mn. c Product with undesired and unidentified structures. | ||||||||
| 1 | P1f2a3d | MeCN | 0.2 | 0.5 mL | 20 | 13800 |
20700 |
1.5 |
| 2 | P1f2a3d | DCE | 0.2 | 0.5 mL | 76 | 36300 |
120500 |
3.3 |
| 3 | P1f2a3d | DMF | 0.2 | 0.5 mL | 73 | 32100 |
60600 |
1.9 |
| 4 | P1e2a3d | DCE | 0.2 | 0.5 mL | 87 | 44000 |
78300 |
1.8 |
| 5 | P1e2a3e | DCE | 0.2 | 0.5 M | UDc | — | — | — |
| 6 | P1f2a3e | DCE | 0.2 | 0.5 M | Gel | — | — | — |
Solubility and thermal properties
The prepared heteroatom-rich poly(sulfonyl isourea)s in this work all exhibit good solubility and can be dissolved in commonly used organic solvents such as DCM, CHCl3, THF, DMSO, and DMF.The thermal properties of representative polymers were evaluated for each MCP route. Fig. 4A presents the specific structures and molecular weights of the polymers. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed to assess thermal and morphological stability. As shown in Fig. 4B, the thermal degradation temperature (Td) at 5% weight loss of these polymers reached 220–278 °C. Among the polymers, P1a2a3a with long alkyl chains in the main chains and sulfonamide side chains showed the lowest Td values. In contrast, P1e2d3a, which also contained alkyl chains in the main chains but had sulfonimide side chains, demonstrated excellent thermal stability with a Td value of 274 °C. P1e2a3d and P1f2a3d with more rigid main chains and higher molecular weights showed superior thermal stability. It is worth noting that polymer structures greatly affect char residue values. In general, polymers with rigid structures and high molecular weights tend to exhibit higher char residue values. For example, P1a2a3a and P1e2d3a, which have flexible alkyl chains and low molecular weights, exhibited low char residue values of approximately 9% and 15% at 800 °C, respectively. In contrast, P1e2a3d and P1f2a3d with aromatic conjugated structures and high molecular weights exhibited a higher char residue value of around 27%. As shown in Fig. 4C, the glass transition temperature (Tg) of these polymers ranged from 86 °C to 164 °C. P1a2a3a and P1e2d3a had the lowest Tg values, attributed to the presence of long alkyl chains in their backbones, while P1e2a3d and P1f2a3d exhibited higher Tg values. This suggests that polymers with more rigid main chains have higher Tg values, and vice versa.
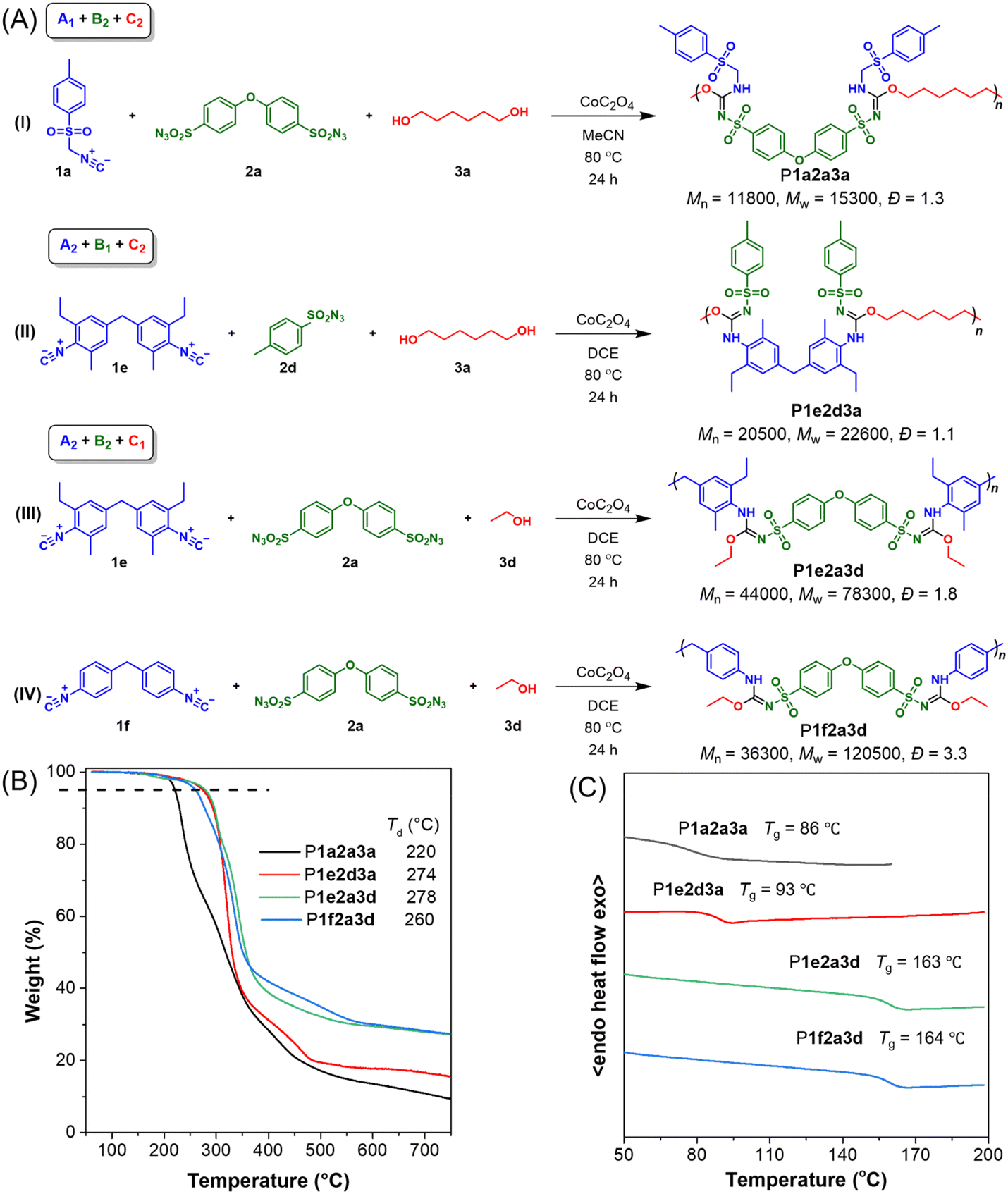 | ||
| Fig. 4 (A) The specific structures and molecular weights of P1a2a3a, P1e2d3a, P1f2a3d and P1e2a3d. (B) TGA thermograms of polymers recorded under nitrogen at a heating rate of 10 °C min−1. (C) DSC thermograms of polymers recorded under nitrogen during the second heating cycle at a heating rate of 10 °C min−1. | ||
Conclusions
In summary, we have successfully developed a new MCP strategy of isocyanides, sulfonyl azides, and alcohols/phenols for the preparation of heteroatom-rich poly(sulfonyl isourea)s. Based on the facile preparation of the difunctionalized monomers, possible monomeric combinations are fully achieved to realize the diversity of polymer structures of MCPs. A series of poly(sulfonyl isourea)s with different main chains and branched chains were conveniently prepared under mild reaction conditions. The obtained polymers showed high Mw values of up to 120500 g mol−1 with high yields of up to 87%. These polymers have good solubility, and thermal and morphological stability. It is foreseeable that the present MCP strategy could provide new inspiration and possibilities for the facile and efficient preparation of heteroatom-rich multifunctional polymers and may facilitate the development of diversity-oriented polymerization strategies.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding from the National Natural Science Foundation of China (22271197), the Guangdong Basic and Applied Basic Research Foundation (2023A1515011578), the Shenzhen Science and Technology Program (RCYX20221008092924059 and JCYJ20220531102601003), the Open Fund of Guangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates (2023B1212060003), Shenzhen Key Laboratory of Functional Aggregate Materials (ZDSYS20211021111400001), and the 2035 Research Excellence Program of Shenzhen University (2023C001). The authors also acknowledge the Instrumental Analysis Center of Shenzhen University.References
- A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef.
- I. Ugi, A. Dömling and W. Hörl, Endeavour, 1994, 18, 115–122 CrossRef CAS.
- E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS PubMed.
- T. Zarganes-Tzitzikas, A. L. Chandgude and A. Dömling, Chem. Rec., 2015, 15, 981–996 CrossRef CAS PubMed.
- J. Zhu, Eur. J. Org. Chem., 2003, 1133–1144 CrossRef CAS.
- C. V. Robotham, C. Baker, B. Cuevas, K. Abboud and D. L. Wright, Mol. Divers., 2003, 6, 237–244 CrossRef CAS PubMed.
- O. Kreye, T. Tóth and M. A. R. Meier, J. Am. Chem. Soc., 2011, 133, 1790–1792 CrossRef CAS PubMed.
- O. Kreye, C. Trefzger, A. Sehlinger and M. A. R. Meier, Macromol. Chem. Phys., 2014, 215, 2207–2220 CrossRef CAS.
- O. Kreye, O. Türünç, A. Sehlinger, J. Rackwitz and M. A. R. Meier, Chem. – Eur. J., 2012, 18, 5767–5776 CrossRef CAS PubMed.
- A. Sehlinger, K. Ochsenreither, N. Bartnick and M. A. R. Meier, Eur. Polym. J., 2015, 65, 313–324 CrossRef CAS.
- P. Stiernet and A. Debuigne, Prog. Polym. Sci., 2022, 128, 101528 CrossRef CAS.
- Z. Zhang, Z.-B. Tan, C.-Y. Hong, D.-C. Wu and Y.-Z. You, Polym. Chem., 2016, 7, 1468–1474 RSC.
- N. Gangloff, D. Nahm, L. Döring, D. Kuckling and R. Luxenhofer, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1680–1686 CrossRef CAS.
- A. Sehlinger, P.-K. Dannecker, O. Kreye and M. A. R. Meier, Macromolecules, 2014, 47, 2774–2783 CrossRef CAS.
- X. Wang, T. Han, J. Gong, P. Alam, H. Zhang, J. W. Y. Lam and B. Z. Tang, Macromolecules, 2022, 55, 4389–4401 CrossRef CAS.
- H. Xue, Y. Zhao, H. Wu, Z. Wang, B. Yang, Y. Wei, Z. Wang and L. Tao, J. Am. Chem. Soc., 2016, 138, 8690–8693 CrossRef CAS PubMed.
- J. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799–5867 CrossRef CAS PubMed.
- E. S. Alsolami, H. S. Alorfi, K. A. Alamry and M. A. Hussein, RSC Adv., 2024, 14, 1757–1781 RSC.
- X. Fu, A. Qin and B. Z. Tang, Aggregate, 2023, 4, e350 CrossRef CAS.
- R. Hu, W. Li and B. Z. Tang, Macromol. Chem. Phys., 2015, 217, 213–224 CrossRef.
- D. Huang, Y. Liu, A. Qin and B. Z. Tang, Polym. Chem., 2018, 9, 2853–2867 RSC.
- M. Li, X. Fu, J. Wang, A. Qin and B. Z. Tang, Macromol. Chem. Phys., 2022, 224, 2200352 CrossRef.
- X. Liu, T. Han, J. W. Y. Lam and B. Z. Tang, Macromol. Rapid Commun., 2020, 42, 2000386 CrossRef PubMed.
- Y. Liu, A. Qin and B. Z. Tang, Prog. Polym. Sci., 2018, 78, 92–138 CrossRef CAS.
- M. A. R. Meier, R. Hu and B. Z. Tang, Macromol. Rapid Commun., 2021, 42, 2100104 CrossRef CAS PubMed.
- T. Han, D. Yan, Q. Wu, N. Song, H. Zhang and D. Wang, Chin. J. Chem., 2021, 39, 677–689 CrossRef CAS.
- L. Liu, M. Zhu, J. Feng, H. Peng, Y. Shi, J. Gao, L. C. Tang and P. Song, Aggregate, 2024, 5, e494 CrossRef CAS.
- J. Yan, W. Yu, Z. Wang, L. Wu, Y. Wang and T. Xu, Aggregate, 2024, 5, e527 CrossRef CAS.
- Y. Zhang, Z. Li, Z. Xu, M. Xiao, Y. Yuan, X. Jia, R. Shi, L. Zhang and P. Wan, Aggregate, 2024, e566, DOI:10.1002/agt2.566 , early view.
- A. K. Agrahari, P. Bose, M. K. Jaiswal, S. Rajkhowa, A. S. Singh, S. Hotha, N. Mishra and V. K. Tiwari, Chem. Rev., 2021, 121, 7638–7956 CrossRef CAS PubMed.
- H. Kim and T.-L. Choi, ACS Macro Lett., 2014, 3, 791–794 CrossRef CAS PubMed.
- I.-H. Lee, H. Kim and T.-L. Choi, J. Am. Chem. Soc., 2013, 135, 3760–3763 CrossRef CAS PubMed.
- M. Li, X. Duan, Y. Jiang, X. Sun, X. Xu, Y. Zheng, W. Song and N. Zheng, Macromolecules, 2022, 55, 7240–7248 CrossRef CAS.
- P. Saini, N. Sonika, G. Singh, G. Kaur, J. Singh and H. Singh, Mol. Catal., 2021, 504, 111432 CrossRef CAS.
- N. V. Sokolova and V. G. Nenajdenko, RSC Adv., 2013, 3, 16212–16242 RSC.
- Z. Cai, Y. Ren, X. Li, J. Shi, B. Tong and Y. Dong, Acc. Chem. Res., 2020, 53, 2879–2891 CrossRef CAS PubMed.
- S. Kumar, A. Arora, S. Kumar, R. Kumar, J. Maity and B. K. Singh, Eur. Polym. J., 2023, 190, 11204 CrossRef.
- B. T. Tuten, L. De Keer, S. Wiedbrauk, P. H. M. Van Steenberge, D. R. D'Hooge and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2019, 58, 5672–5676 CrossRef CAS PubMed.
- X. Wang, C. Liu, Z. Xing, H. Suo, R. Qu, Q. Li and Y. Qin, Macromolecules, 2022, 55, 8857–8865 CrossRef CAS.
- J. Xie, N. Niu, X. Fu, X. Su, D. Wang, A. Qin, T. Han and B. Z. Tang, Chem. Sci., 2023, 14, 903–915 RSC.
- T. Tian, R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 6156–6163 CrossRef CAS PubMed.
- B. T. Tuten, F. R. Bloesser, D. L. Marshall, L. Michalek, C. W. Schmitt, S. J. Blanksby and C. Barner-Kowollik, ACS Macro Lett., 2018, 7, 898–903 CrossRef CAS PubMed.
- X. Wu, J. He, R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2021, 143, 15723–15731 CrossRef CAS PubMed.
- X. Wu, H. Lin, F. Dai, R. Hu and B. Z. Tang, CCS Chem., 2020, 2, 191–202 CrossRef CAS.
- T.-J. Yue, W.-M. Ren and X.-B. Lu, Chem. Rev., 2023, 123, 14038–14083 CrossRef CAS PubMed.
- J. Zhang, Q. Zang, F. Yang, H. Zhang, J. Z. Sun and B. Z. Tang, J. Am. Chem. Soc., 2021, 143, 3944–3950 CrossRef CAS PubMed.
- T. Jiang, Z.-Y. Gu, L. Yin, S.-Y. Wang and S.-J. Ji, J. Org. Chem., 2017, 82, 7913–7919 CrossRef CAS PubMed.
- W. Fu, L. Kong, J. Shi, B. Tong, Z. Cai, J. Zhi and Y. Dong, Macromolecules, 2019, 52, 729–737 CrossRef CAS.
- A. Qin, Y. Liu and B. Z. Tang, Macromol. Chem. Phys., 2015, 216, 818–828 CrossRef CAS.
- T. Han, Z. Zhao, H. Deng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Polym. Chem., 2017, 8, 1393–1403 RSC.
- T. Han, Z. Zhao, J. W. Y. Lam and B. Z. Tang, Polym. Chem., 2018, 9, 885–893 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00953c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |