
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControl over membrane fluidity and biophysical properties of synthetic terpolymer stabilized complex coacervates†
Sebastian
Novosedlik
,
Alexander B.
Cook
 ,
Tim J. F. M.
Voermans
,
Henk M.
Janssen
and
Jan C. M.
van Hest
*
,
Tim J. F. M.
Voermans
,
Henk M.
Janssen
and
Jan C. M.
van Hest
*
Bio-Organic Chemistry, Department of Chemical Engineering and Chemistry, Institute for Complex Molecular Systems (ICMS), Eindhoven University of Technology, 5600 MB Eindhoven, the Netherlands. E-mail: j.c.m.v.hest@tue.nl
First published on 31st October 2024
Abstract
Cell membranes are vital barriers that regulate the composition of the intracellular environment and facilitate communication processes essential for cellular function and survival. In comparison to lipid membranes, artificial polymeric membranes generally offer enhanced stability due to their higher molecular weight and greater variability in the nature of the macromolecular building blocks, which provides access to a broad chemistry toolbox to regulate important features such as fluidity and permeability. We recently developed an artificial cell platform based on a complex coacervate, in which a terpolymer, composed of a hydrophilic poly(ethylene glycol) segment, a hydrophobic poly(caprolactone-g-trimethylene carbonate) domain and a polyglutamate anchor (PEG-PCLgTMC-PGA) was used for stabilization. These membranized structures showed excellent permeability, due to the high fluidity of the membrane. However, the polymer membrane proved to be unselective with regard to the molecular weight of guest molecules that were exchanged with the environment. To advance this platform, a series of terpolymers with distinctive features were synthesized to further refine their regulatory features of the polymer membrane. Through investigation of structural terpolymer variants, including those in which the hydrophobic domain was based on PCLgTMC, poly(D,L-lactic acid) or polystyrene, their influence on membrane permeability, fluidity, and sequestration of hydrophobic molecules, such as cholesterol, was determined. With this extended range of membrane-forming building blocks, this coacervate platform is equipped with tailored permeability through interactions with the coacervate lumen and facilitates sequestration of hydrophobic molecules into the membrane and controlled fluidity.
1. Introduction
Cell membranes serve as crucial barriers, controlling the composition of the intracellular environment essential for function and survival, while facilitating communication through signalling, adhesion, and transport processes.1,2 Artificial membranes based on the self-assembly of amphiphilic block copolymers can be utilised as an alternative, exhibiting superior strength and stability relative to lipid membranes, all while retaining biologically relevant features like the incorporation of proteins.3,4 Furthermore, polymeric building blocks offer a wide variety of chemical modifications, making them increasingly favourable for biomedical and engineering applications,5,6 allowing precise control over thickness, fluidity, permeability and responsiveness.7A well-established polymer-membrane platform are the polymersomes, polymer vesicles composed of a bilayer membrane, which show in their composition much similarity to liposomes. They have been successfully engineered with regulated permeability, for example via the incorporation of membrane proteins.8 Semi-permeable polymersomes have been loaded with a range of (bio)catalysts which allowed their use as nanoreactors.9Via their introduction into living cells and even organisms, they adopted the role as artificial organelles. Furthermore, also multicompartment polymersomes have been developed which emulate the build-up of eukaryotic cells.10 Control over membrane permeability was essential for their functioning. Polymersomes however, from an artificial cell perspective, are less effective in mimicking the crowded cytosol as they typically comprise a diluted aqueous solution as their lumen.
Coacervates, arising from liquid–liquid phase separation of oppositely charged polyelectrolytes, provide an intriguing platform to study the influences of a crowded, cell-like environment.11–16 A number of papers have been published in which for example lipid-based membranes were introduced to regulate the exchange of molecules with the environment.17–20 Our group has developed a method to stabilize the dynamic coacervate–water interface by encapsulating coacervate microdroplets within a polymeric membrane. This approach utilizes a tailored triblock terpolymer (TP) based on a hydrophilic poly(ethylene glycol) segment, a hydrophobic poly(caprolactone-trimethylene carbonate) domain and a polyglutamate anchor (PEG–PCLgTMC–PGA) which interacts with the net positively charged coacervates.21 These assemblies are unique in the sense that the coacervate droplets and their surrounding continuous phase are water-based, giving rise to a very low interfacial tension, typically 100–1000 fold smaller than for regular water/oil interphases which is the common way for the preparation of closely packed polymeric membranes. This leads to boundary domains for stabilized coacervates that are highly dynamic.22,23
While studies on the membrane properties of terpolymer stabilized coacervates are limited, preliminary findings reported a monolayer membrane with a thickness of approximately 19 nm. It was demonstrated that our terpolymer-stabilized coacervate system allowed the partitioning of both small molecules and macromolecules such as proteins and dextran, mainly dependent on charge and other physicochemical properties, such as hydrophobicity or hydrogen bonding, but much less on size. The high permeability of the membrane was attributed to the highly disordered nature of the PCLgTMC hydrophobic block having a glass transition temperature below 0 °C.24
Tailoring permeability and fluidity as well as interactions with biomolecules are key features for an artificial cell platform and can be designed by changes in the composition of the membrane. Membrane permeability and fluidity are closely related, in which increased fluidity typically correlates with increased permeability. Membrane fluidity predominantly depends on the molecular weight, hydrophobicity, flexibility, or branching of the polymer constituting the membrane.25,26 The balance between hydrophobic and hydrophilic segments of polymers profoundly influences membrane fluidity; higher proportions of hydrophobic segments tend to yield more rigid membranes due to stronger hydrophobic interactions, whereas a more hydrophilic character enhances fluidity by promoting hydration and reducing intermolecular interactions. Likewise, polymers possessing more flexible backbones or side chains generally enhance membrane fluidity relative to their rigid counterparts.27–32
Another way to modulate membrane properties is by the addition of pores or membrane-regulating molecules. For example, cholesterol is a crucial component of biological membranes in eukaryotic cells and plays several key roles in modulating their structure and function. Cholesterol helps maintain the fluidity and stability of lipid membranes. In fluid membranes, cholesterol fills the gaps between phospholipid molecules, reducing their mobility and preventing excessive movement. Conversely, in rigid membranes, cholesterol disrupts the tight packing of lipids, increasing fluidity. This ability to modulate membrane fluidity is vital for various cellular processes, including cell signalling, membrane trafficking, and cell–cell interactions.1,33,34
In this contribution, to advance the scope of terpolymer-stabilised coacervates, we investigated how the fluidity and permeability can be affected by changes in the terpolymer composition. For this reason, we synthesized a diverse library of terpolymers featuring varied hydrophobic backbones and side chains. Terpolymers based on PCLgTMC were tailored with different small organic molecules as side chains to assess the impact of their hydrophobic characteristics on terpolymer membrane properties. For the same reason, we synthesized two terpolymers with backbones derived from poly(D,L-lactide) (PDLLA) and poly(styrene) (PS). The propensity of coacervate stabilization with the newly synthesized terpolymers was verified through brightfield microscopy. Permeability was assessed by monitoring the permeation of fluorescently labelled dextran of varying sizes via confocal microscopy. Fluorescence recovery after photobleaching (FRAP) was used to determine lateral diffusion coefficients of the various terpolymer membranes. Finally, we characterized the ability of the terpolymer membranes to sequester hydrophobic molecules and elucidated whether cholesterol could regulate the fluidity of the terpolymer membrane, just as in eukaryotic cells. This study aims to understand the interfacial stabilisation of coacervates with a terpolymer and unravels how membrane fluidity and permeability are influenced by hydrophobic interactions.
2. Results and discussion
2.1. Polymer synthesis and coacervate stabilisation
Membranised coacervates serve as compelling platforms for investigating membrane properties within the context of crowded cellular lumens. Recently, our group introduced terpolymer-stabilized coacervates, exhibiting exceptional permeability owing to their high membrane fluidity. However, the lack of molecular selectivity in the uptake process necessitated modifications to the terpolymers to finely control both fluidity and permeability.Here, we hypothesized that augmenting hydrophobicity would enhance attractive forces between the polymer constituents, thereby augmenting packing density and reducing both permeability and fluidity (Fig. 1). To test this hypothesis, we synthesized a library of terpolymers with varied hydrophobic characteristics (polymer structures and characterisation data summarised in Table 1). Terpolymers based on PCLgTMC were systematically tailored with diverse small organic molecules as side chains (designated 1–5) to elucidate the impact of their hydrophobic profiles on membrane properties. Additionally, we synthesized two terpolymers with different hydrophobic and thermal characteristics, having backbones derived from poly(D,L-lactide) (PDLLA, 6) and polystyrene (PS, 7).
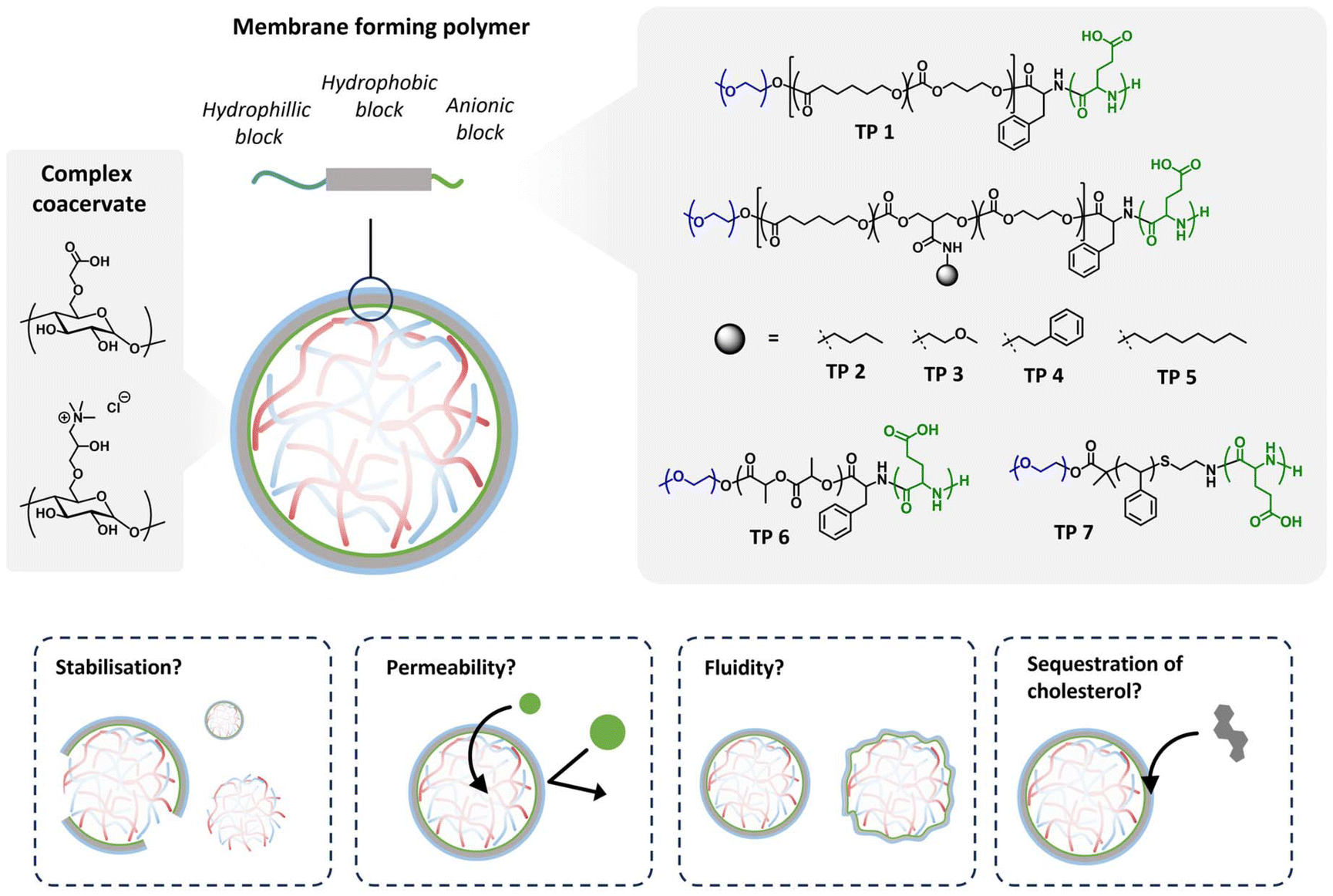 | ||
| Fig. 1 Schematic overview of the current study, where the effect of terpolymer hydrophobic block structure (TP 1–7, with either PCL–TMC, PDLLA, or PS based polymer backbones) is investigated for its impact on membrane physical properties and cargo uptake, when used as a self-assembled stabilising layer around charged amylose-based complex coacervate droplets. | ||
| Terpolymer | Compositiona | M n (kDa) | Đ | T g (°C) | T m (°C) |
|---|---|---|---|---|---|
| a Number of repeating units and number average molecular weight were determined with 1H NMR spectroscopy. b Molecular weight dispersities (Đ) were measured by size-exclusion chromatography (SEC) using PS standards in THF. c Thermal properties were measured by differential scanning calorimetry. | |||||
| 1 | PEG44(PCL57gTMC48)PGA7 | 14.42 | 1.13 | −50.55 | 25.92 |
| 2 | PEG44(PCL55gTMC37gTMCbutyl12)PGA9 | 15.97 | 1.36 | −42.35 | — |
| 3 | PEG44(PCL54gTMC37gTMCmethoxypropyl12)PGA10 | 16.19 | 1.21 | −43.14 | — |
| 4 | PEG44(PCL61gTMC45gTMCphenyl13)PGA7 | 18.01 | 1.24 | −37.69 | — |
| 5 | PEG44(PCL55gTMC37gTMCoctyl12)PGA8 | 16.49 | 1.43 | −45.86 | — |
| 6 | PEG44PDLLA95PGA7 | 16.87 | 1.08 | 29.40 | — |
| 7 | PEG44PS150PGA12 | 11.61 | 1.07 | 59.09 | — |
The synthesis of the poly(ethylene glycol)-b-poly(ε-caprolactone-g-trimethylene carbonate)-b-poly(L-glutamate) (PEG-PCLgTMC-PGA) terpolymer TP 1 was carried out as described in our previous publications (Scheme S1†).21 The differently functionalised PCLgTMC terpolymers TP 2–5 were synthesized via a post-polymerization modification strategy adapted from literature (Scheme S2†).35 First, the cationic ring opening polymerisation of caprolactone and trimethylene carbonate was performed according to the protocol for the original terpolymer 1, with the addition of the copolymerization of trimethylene carbonate pentafluorophenyl ester (PFP–TMC), yielding the copolymer poly(ethylene glycol)-b-poly(ε-caprolactone-g-trimethylene carbonate-g-pentafluorophenyl 5-methyl-2-oxo-1,3-dioxane-5-carboxylate) PEG–PCLgTMCgTMCPFP. For all terpolymers we used 2 kg mol−1 PEG as initiator and aimed for approximately 100 repeating units in the hydrophobic block. After co-polymerization, the different functionalities were introduced by reacting the pentafluorophenyl ester incorporated in the backbone of the polymer with the respective primary amine. Quantitative conversion occurred within one hour as determined by the disappearance of the fluorine peaks of the pentafluoro phenol in the 19F-NMR spectrum. Subsequently, the terpolymer synthesis was completed with the amine chain end modification viaL-phenylalanine (Phe–OH), followed by the ring opening polymerisation of (4S)-2,5-dioxo-4-oxazolidinepropanoic acid phenylmethyl ester (BLG–NCA) and the respective deprotection step, according to the original procedure. The resulting terpolymers 2–5 had the expected degree of polymerization and displayed monomodal molecular weight distributions with dispersities (Đ) between 1.21–1.43.
Poly(ethylene glycol)-b-poly(D,L-lactide) (PEG–PDLLA) copolymer was synthesized according to literature by a quasi-anionic ring-opening polymerization (ROP) with the organic base 1,8-diazabicycloundec-7-ene (DBU) as a catalyst (Scheme S3†).36,37 The subsequent steps encompassing the amine chain end modification with Phe–OH, a subsequent ring opening polymerisation of BLG–NCA and the respective deprotection step were performed according to the same protocol as described above. The composition of the final product was determined from the NMR spectra with the resulting terpolymer 6 having a Đ of 1.06, as determined by size exclusion chromatography (SEC).
The synthesis of the polystyrene terpolymer followed an adjusted protocol. First, bromide-terminated poly(ethylene glycol)-b-poly(styrene) (PEG–PS) was prepared according to literature via a bulk atom transfer radical polymerization (ATRP) catalysed with CuBr/PMDETA and anisole as a co-solvent (Scheme S4†).38 Using an SN2 reaction with 2-(Boc-amino)ethanethiol and a subsequent deprotection with TFA in DCM yielded the amine terminated PEG–PS. The glutamic acid anchor was introduced as described for the other polymers via the ring opening polymerisation of NCA–BLG, and subsequent hydrogenation on Pd/C yielded terpolymer 7.
Next, the thermal properties of terpolymers 1–7 were characterized by differential scanning calorimetry (DSC). Only terpolymer 1 showed a melting endotherm, with a melting temperature (Tm) of 26 °C. In contrast, no Tm was observed for the functional PCLgTMC terpolymers 2–5. Probably the bulkier functionalised TMC units disrupted the CL enriched block to a greater extent preventing the CL sections from crystallizing. All hydrophobic blocks of the PCLgTMC gradient polymer displayed a Tg as expected. The Tg was barely influenced by the structural variations and was in the range of −37 to −50 °C for terpolymers 2–5. The amorphous terpolymers based on PDLLA 6 and PS 7 had a Tg of 29 °C and 59 °C, respectively.
Coacervates were prepared with the different terpolymers according to the same procedure as established by our group21 and their propensity for stabilization was determined by bright-field microscopy. For terpolymers 1–6 discrete populations of coacervates with minimal amount of aggregation were observed (Fig. 2). Only terpolymer 7 failed to stabilize coacervates and formed irregular aggregates. It is remarkable that all terpolymer variants except for terpolymer 7 successfully stabilized coacervates despite notable differences in polymer composition. Terpolymer 7, characterized by a glass transition temperature well above room temperature (59 °C), exhibited solid-like behaviour characterised by a low fluidity at room temperature. Consequently, the fluidic attributes of the terpolymer were deemed essential for membrane formation around coacervates, with polymer composition variations being widely tolerated.
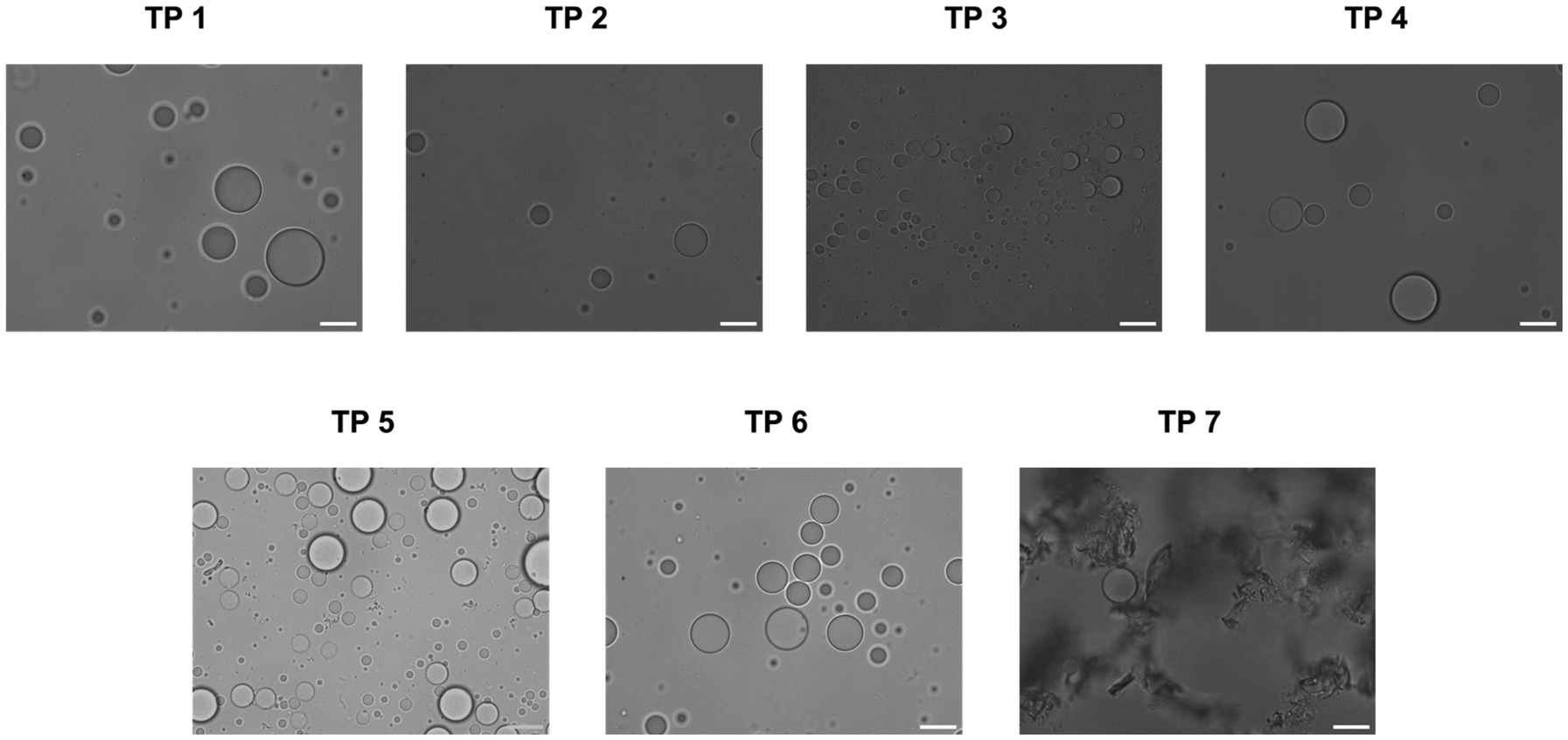 | ||
| Fig. 2 Brightfield micrographs of coacervates stabilised with terpolymers 1–7. Scale bar, 20 μm. | ||
2.2. Permeability
First, we assessed how structural variations in the terpolymers influenced the molecular weight cut-off of the semipermeable terpolymer membrane. We studied the permeability towards a range of differently sized dextran. Stabilized coacervates were first formed, followed by the addition of the fluorescein isothiocyanate-labelled dextran (4, 70, 500 kDa). The degree of permeability was then determined by confocal microscopy by quantifying the encapsulation efficiency of the fluorescent dextran. Remarkably, membrane permeability remained consistent across all terpolymer variations, as illustrated in Fig. 3a–c. This was also the case for coacervates containing no terpolymer membrane (no TP) demonstrating that the terpolymer membrane had mostly a stabilising effect but did not influence the permeability significantly. Sequestration of the small 4 kDa dextran was observed to be quantitative in all instances. Conversely, for the medium-sized 70 kDa dextran, median encapsulation efficiencies ranged between 51% to 60%, while for the larger 500 kDa dextran, median encapsulation efficiencies ranged between 19% to 32%. Although we noted a size-dependent effect on the sequestration of macromolecules into coacervates, this phenomenon was consistent across all polymer variations.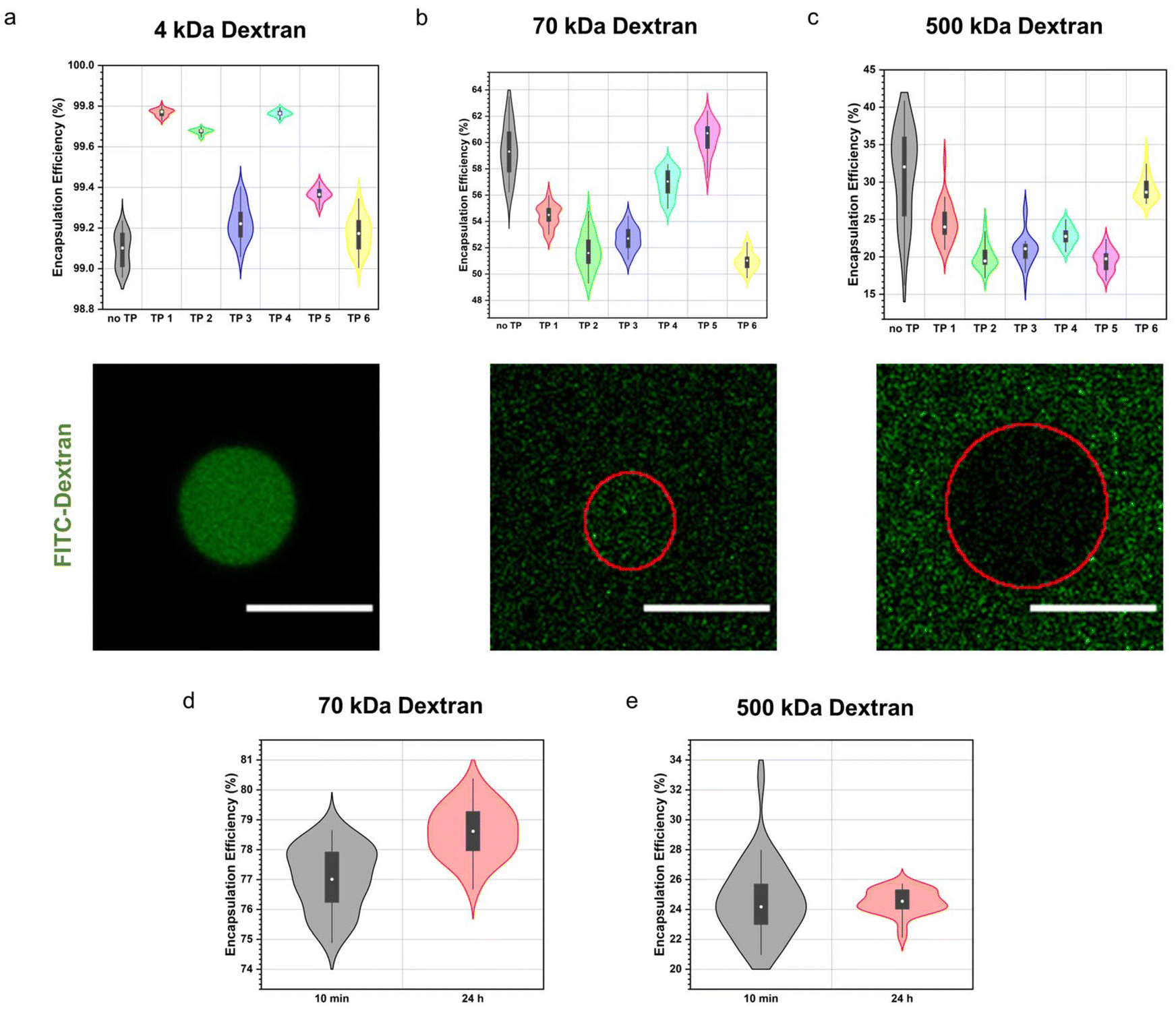 | ||
| Fig. 3 Terpolymer stabilized coacervates’ permeability towards a range of differently sized dextran. Encapsulation efficiency (top) and confocal micrograph (bottom) of terpolymer stabilized coacervates containing (a) 4 kDa dextran, (b) 70 kDa dextran and (c) 500 kDa dextran. Scale bar, 20 μm. Encapsulation efficiency of coacervates stabilized by terpolymer 1 over time for (d) 70 kDa dextran and (e) 500 kDa dextran. For all experiments n = 30 coacervates (for no TP samples n ≥ 15) were measured across multiple imaging positions in the same sample. For all violin plots the black box represents the interquartile range (IQR), black line represents 1.5× IQR and white circle represents median. | ||
Furthermore, we monitored the uptake of macromolecules over a 24 hour period. Intriguingly, we observed no significant variance in the encapsulation efficiency between the 10 minute and 24 hour time points, indicative of rapid uptake kinetics characteristic for coacervates (Fig. 3d and e).
Permeability assays utilizing different molecular weight dextran revealed consistent membrane permeability across all terpolymer variations, albeit with variations in encapsulation efficiencies correlated with dextran molecular weight. The observed size-based variations in sequestration are likely due to differences in favourable interactions with the coacervate core attributable to differences in conformation or persistence length among dextran molecules, rather than being influenced by the terpolymer composition.
2.3. Fluidity
The fluidity of the terpolymer membrane was measured using fluorescence recovery after photobleaching (FRAP) on a confocal microscope (Fig. 4). For this measurement, coacervates stabilized with the different terpolymers 1–6 were prepared. Each assembly contained 15 wt% of the fluorescently labelled PEG–PCLgTMC copolymer which was used to monitor the diffusion in the terpolymer membrane. Each population was embedded in a 1 wt% agarose gel to prevent the droplets from moving which would negatively influence the measurements. For each condition three terpolymer stabilized coacervates were probed. A small region of the membrane was bleached, and the fluorescence recovery was monitored over time. FRAP curves were fitted to a single component exponential decay and diffusion coefficients were calculated according to published methods.39,40 For all cases we observed a full fluorescence recovery within five minutes indicating that the polymer membranes are fluid. The lowest membrane fluidity was observed for the unmodified terpolymer 1 with an apparent diffusion coefficient Dapp = 0.0253 ± 0.0036 μm2 s−1. A gradual increase for the PCLgTMC terpolymer variations 2–5 in diffusivity was observed for increasing side group size with terpolymer 5 being the most fluidic with a Dapp = 0.0500 ± 0.0038 μm2 s−1. As hypothesized, we observed a higher mean Dapp for the less hydrophobic, and higher Tg, PDLLA terpolymer 6 compared to terpolymer 1 (Fig. 4d).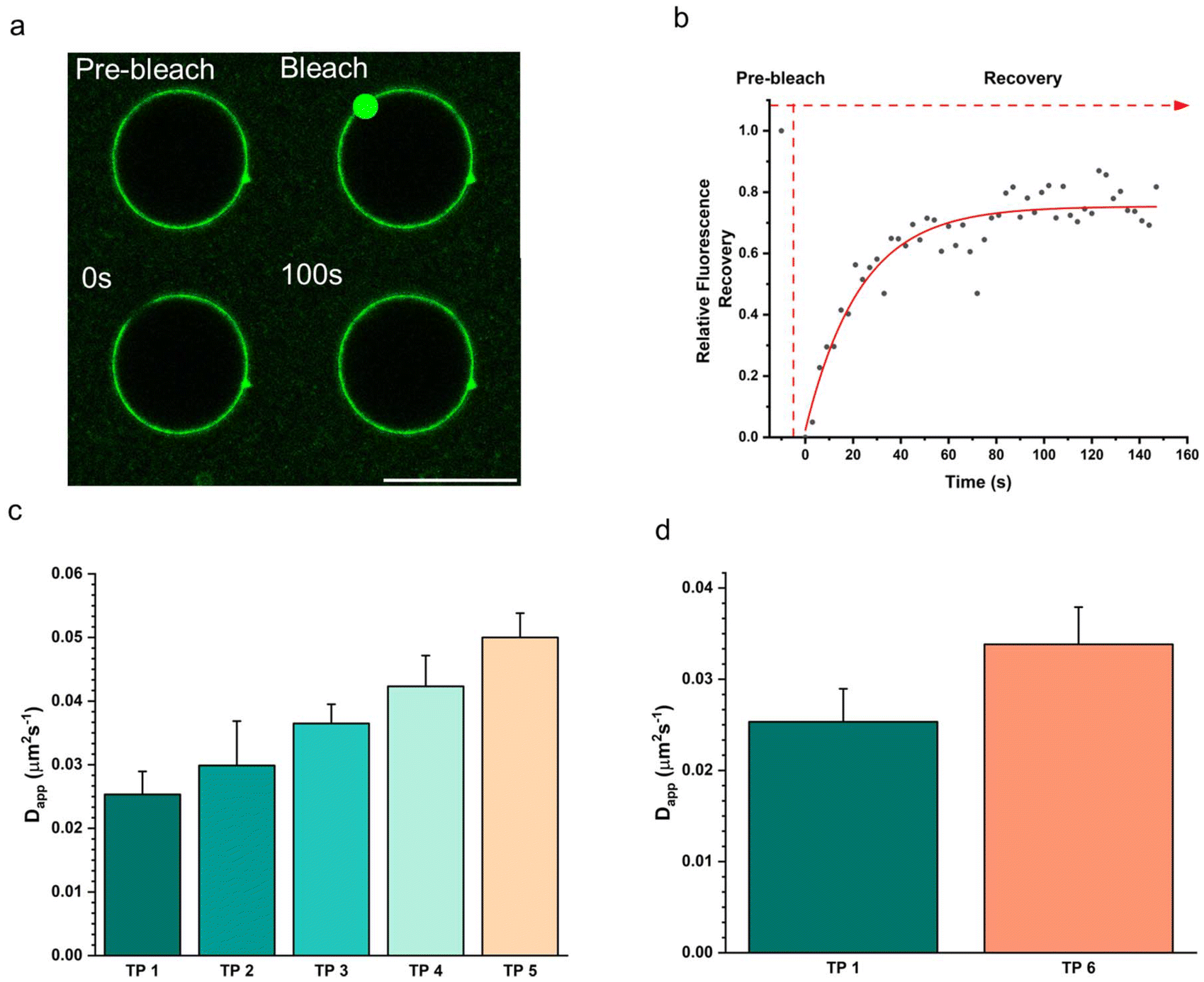 | ||
| Fig. 4 Fluorescence recovery after photobleaching (FRAP) analysis of terpolymer membrane for terpolymer 1–6. (a) FRAP measurement showing laser-mediated bleaching of a circular spot on the membrane and subsequent fluorescence recovery of the bleached spot. Scale bar, 20 μm. (b) Exemplary FRAP recovery curve for TP 1 with black dots representing experimental data and red curve exponential fitting. (c) Apparent diffusion coefficient for PCLgTMC based terpolymers 1–5. (d) Apparent diffusion coefficient for PCLgTMC and PDLLA based terpolymer 1 and 6. | ||
Interestingly, contrary to expectations, increasing hydrophobicity did not consistently correlate with reduced fluidity. While the PDLLA terpolymer displayed increased fluidity compared to the PCLgTMC terpolymer, terpolymers 1–5 exhibited a distinct trend. Specifically, the most hydrophobic terpolymer 5, featuring an octyl side chain, demonstrated the highest fluidity, suggesting that the hydrophobic effect is overruled by the increased side group chain size, which disrupted membrane packing, leading to heightened fluidity.
2.4. Integration of cholesterol in terpolymer membranes
As discussed in the introduction cholesterol is a crucial element of the eukaryotic cell membrane, regulating its fluidity. In order to assess the biomimetic properties of our platform in this respect we investigated how cholesterol interacted with the terpolymer membrane. First, we confirmed if cholesterol had an affinity for the terpolymer membrane. Therefore, we used the fluorescently labelled TopFluor® Cholesterol which enabled fluorescent detection via confocal microscopy. We prepared coacervates and added TopFluor® Cholesterol to the suspension and imaged them with confocal microscopy. Immediately after addition the coacervates appeared homogenously covered with cholesterol (Fig. 5a). Barely any cholesterol was detected in the supernatant, demonstrating a strong affinity between cholesterol and the terpolymer. This behaviour was observed for all of the terpolymer variants. Contrary, coacervates without a membrane had no affinity for cholesterol. Similar behaviour was observed for the small hydrophobic molecule Nile red and larger hydrophobic macromolecules like the fluorescently labelled AF488–PEG–PCLgTMC copolymer without the PGA anchor (Fig. 5b and c). This suggested that hydrophobic interactions are the driving force for the sequestration of hydrophobic molecules into the terpolymer membrane.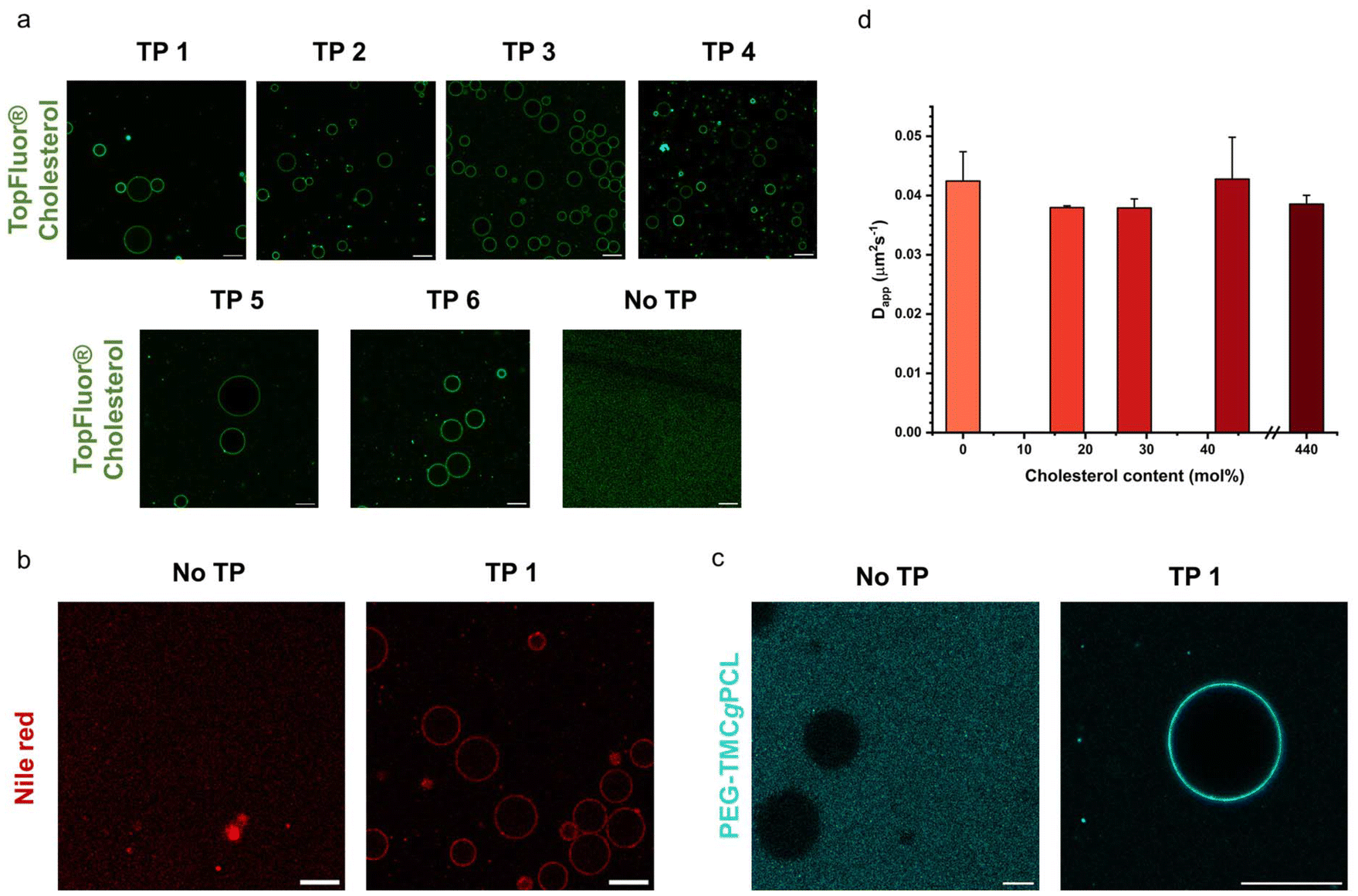 | ||
| Fig. 5 Interaction of terpolymer membrane with hydrophobic molecules. (a) Confocal micrographs of non-stabilised (no TP) and terpolymer stabilised coacervates (TP 1–6) containing TopFluor® Cholesterol. (b) Confocal micrographs of non-stabilised (no TP) and terpolymer stabilised coacervates (TP 1) containing Nile red. (c) Confocal micrographs of non-stabilised (no TP) and terpolymer stabilised coacervates (TP 1) containing AF488–PEG–PCLgTMC. (d) Apparent diffusion coefficient for terpolymer TP 4 in membrane stabilized coacervates containing different amounts of cholesterol (0–440 mol% in relation to terpolymer concentration). Scale bar, 20 μm. | ||
After having confirmed the incorporation of cholesterol into the terpolymer membrane we investigated the influence of cholesterol on the fluidity of the membrane composed of terpolymer 4 by using FRAP. Several batches of coacervates were prepared with terpolymer 4 containing 17, 29, 44 and 440 mol% cholesterol in relation to terpolymer. The FRAP experiments were performed as described above by tracking the fluorescently labelled PEG–PCLgTMC copolymer. In all cases no major difference in mean membrane diffusivity for the different amounts of cholesterol was observed (Fig. 5d). The mean apparent diffusivity remained consistently around 0.04 μm2 s−1.
Overall, we confirmed the ability of terpolymer membranes to sequester hydrophobic molecules. Investigation of a wide range of cholesterol concentrations revealed that cholesterol had minimal impact on terpolymer membrane fluidity. Unlike lipids, which exhibit “soft” properties within bilayers due to their low molecular weight and high fluidity, amphiphilic block copolymers manifest as bulky and coiled macromolecules, yielding more complex and disordered structures that are less influenced by cholesterol.
3. Conclusions
Recently, our group introduced terpolymer-stabilized coacervate artificial cells, exhibiting high stability and semi permeability to certain cargo internalisation. To investigate potential improved molecular selectivity during the uptake process, in this article we synthesised a range of modified terpolymers to finely control both fluidity and permeability. A library of terpolymers with varied hydrophobic characteristics was synthesised to evaluate the effect of enhanced attractive forces between the polymer constituents. A series of terpolymers based on PCLgTMC were systematically tailored with modified side chains (TP 1–5) as well as two terpolymers were synthesized with different hydrophobic backbones derived from poly(D,L-lactide) (TP6) and polystyrene (TP7). The terpolymer (and associated changes in packing density) impact on coacervate droplet stabilisation was assessed through brightfield microscopy, with all polymers apart from the polystyrene based TP7, being able to successfully stabilise droplets. Permeability of membranised complex coacervates was assessed through uptake of fluorescently labelled 4, 70, and 500 kDa dextran with confocal microscopy. Permeability assays revealed consistent membrane permeability across different terpolymer compositions, with some variations in encapsulation efficiencies correlated with dextran molecular weight. Terpolymer membrane fluidity was assessed with FRAP studies, and showed that the less hydrophobic PDLLA terpolymer TP 6, had a higher mean Dapp compared to TP1, and terpolymers TP2–5 exhibited a trend of increasing fluidity with increasing side group size due to disrupted membrane packing. Finally, cholesterol membrane partitioning studies showed that terpolymer membranes can successfully sequester cholesterol, but that the synthetic polymer membranes are more complex and disordered structures than lipid membranes, therefore the fluidity is less influenced by cholesterol.Our findings align with existing literature, emphasizing the high mobility and flexibility of the terpolymer stabilized coacervate membrane, which do not impede protocell permeability. We successfully manipulated membrane fluidity; bulkier groups disrupted stacking, leading to higher fluidities, while more hydrophobic backbones lead to lower fluidities. However, to significantly decrease membrane fluidity and restrict permeability, additional measures such as crosslinking or the formation of supportive structures like a cortical cytoskeleton would be interesting routes for future research.
4. Materials and methods
4.1. Materials
Monomethoxy poly(ethylene glycol) 2 kDa and azido–PEG (3 kDa) were purchased from Rapp Polymere, trimethylene carbonate was purchased from TCI Europe. Bis(pentafluorophenyl)carbonate was purchased from Apollo Scientific Limited. For the preparation of modified amylose derivatives: amylose (12–16 kDa) was supplied by Carbosynth and 3-chloro-2-hydroxypropyltrimethyl ammonium chloride (65 wt% in water) was supplied by TCI Europe. DBCO-AF488 was purchased from Lumiprobe. ε-Caprolactone (CL), n-butylamine, n-octylamine, 3-methoxypropylamine, phenethylamine, methane sulfonic acid (MSA), D,L-lactide, 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU), poly(ethylene glycol)methyl ether 2-bromoisobutyrate (mPEG45-Br), styrene, CuBr, N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA), N-carboxyanhydride γ-benzyl L-glutamate, 2-(Boc-amino)ethanethiol, triethyl amine (TEA) and Nile red were purchased from Merck. All other chemicals and solvents were purchased from Merck if not otherwise stated. Pentafluorophenyl 5-methyl-2-oxo-1,3-dioxane-5-carboxylate (TMC–PFP) was synthesised according to previously reported procedures.41 Charged amyloses, quaternised amino amylose (Q-Amylose) and carboxy methyl amylose (CM-Amylose), were synthesised according to previously reported procedures.21 Labeled dextran FITC-Dex-4 kDa, FITC-Dex-70 kDa, and FITC-Dex-500 kDa were purchased from Merck. Labeled cholesterol (TopFluor® Cholesterol) was purchased from Merck.4.2. Characterisation
1H nuclear magnetic resonance (NMR) spectra were conducted on a Bruker Avance 400 MHz spectrometer in either deuterated chloroform (CDCl3) or in case of the amyloses deuterated water (D2O). Size exclusion chromatography (SEC) was recorded by using a Shimadzu Prominence-I SEC system. The system was configured with a pLgel-mixed D column and a Shimadzu RID-20A differential refractive index detector. The used eluent was tetrahydrofuran (THF) with a flow rate of 1 mL min−1, and polystyrene calibration standards were used. Brightfield microscopy was conducted with a Leica TCS SP8, and the images analysed with Fiji (ImageJ). Coacervates were further imaged with a Leica TCS SP8 (63× water immersion objective) confocal laser scanning microscope equipped with laser lines of 405 nm, 488 nm, 552 nm, 638 nm, the pinhole was set to 1 Airy Unit. 18-well μ-slide (Ibidi) were used to image coacervate suspensions. For imaging of the fluorescein derived fluorophores, a laser set at 488 nm and emission of 510–580 nm was used. Images were analyzed using Fiji (ImageJ). Differential scanning calorimetry (DSC) data were recorded on a DSC Q2000, TA instruments, with an indium standard calibration. The thoroughly dried polymer samples, all of 2–3 mg were weighed directly into zero hermetic aluminum pans and sealed. Samples were initially heated to 150 °C, subsequently two cooling/heating cycles between −90 °C to 150 °C with a rate of 10 °C per min were performed.4.3. Synthesis of PEG–(PCL–TMC)–PGA terpolymer 1
The terpolymer was synthesized in accordance with previously published procedures.424.4. Step 1 – Preparation of poly(ethylene glycol)–poly(caprolactone-gradient-trimethylene carbonate) (PEG–PCLgTMC)
The organocatalyzed ring-opening polymerization of ε-caprolactone and trimethylene carbonate was performed, aiming for a composition of PEG44-PCL50-g-TMC50. Monomethoxy–PEG–OH macroinitiator (2 kDa, 0.5 mmol) was weighed into an oven-dried round bottom flask and dried at 140 °C under vacuum. After cooling down distilled ε-caprolactone (ε-CL, 25 mmol) and recrystallized trimethylene carbonate (TMC, 25 mmol) were added under argon and dissolved in dry dichloromethane (DCM, 12 mL). The reaction was initiated by the addition of methanesulfonic acid (0.25 mmol ≈ 125 μL). The reaction mixture was stirred at 25 °C in a water bath and reaction progress was regularly checked. After completion (4–6 h) the reaction was quenched with N,N-diisopropylethylamine (DIPEA, 1.5 mL) and the polymer was precipitated into ice cold methanol and lyophilized. This yielded 6.7 g of a waxy solid (67% yield). The composition of the resulting copolymer was confirmed by 1H NMR, comparing the protons of PEG (3.65–3.7 ppm), terminal methyl unit (singlet at 3.40 ppm) to PCL CH2 (multiplet at 2.40–2.25 ppm) and PTMC CH2 (multiplet at 2.2–1.8 ppm). GPC analysis (using a PL gel 5 μm mixed D column, with THF and PS standards) yielded a polydispersity of 1.2.4.5. Step 2 – Chain-end modification with Boc-L-phenylalanine and deprotection
PEG–PCLgTMC (49 μmol) was dissolved in 5 mL acetonitrile. Then 4-dimethylaminopyridine (DMAP, 25 μmol) and Boc-L-phenylalanine (0.25 mmol) were added and the solution was cooled to 0 °C. After that, N,N′-dicyclohexylcarbodiimide (DCC, 1.2 mmol) dissolved in acetonitrile (ACN, 1 mL) was added dropwise to the mixture. The reaction was stirred for 24 h at RT. After reaction completion the flask was placed in the freezer for 1 h and filtered through a plug of Celite. The filtrate was concentrated and precipitated in cold Et2O (50 mL) to obtain the pure product.The resulting copolymer was then dissolved in DCM (5 mL), to which trifluoro acetic acid (TFA, 5 mL) was added (on ice). The mixture was allowed to warm to RT and stirred for 2 h. After 2 h the solvent was evaporated and the copolymer was washed with NaHCO3, 1 M NaCl and brine. Then it was dried over MgSO4, filtered, concentrated and finally precipitated from ice cold Et2O.
1H NMR showed that the signal arising from the terminal TMC group had disappeared, due to addition of phenylalanine at the terminus, and aromatic protons were visible at around 7.2–7.3 ppm. GPC analysis before and after deprotection yielded polydispersities of 1.1, indicating that TFA treatment did not facilitate copolymer hydrolysis.
4.6. Step 3 – Polymerization and deprotection of N-carboxyanhydride γ-benzyl L-glutamate (NCA–BLG)
Phe-terminated copolymer (57 μmol) from step 2 was weighed into a Schlenk flask and dissolved in dry DMF (3 mL). Then NCA–BLG (5.7 mmol) was added under Ar and the reaction was left under a constant flow of N2 for 24 h. The product was precipitated into cold methanol and analysed by 1H NMR to confirm the overall composition and, in particular, the presence of benzylic and aromatic protons at 5.0–5.2 and 7.1–7.4 ppm, respectively.Polymer (200 mg) was dissolved in THF (16 mL) and EtOH (2.5 mL) was added. The solution was degassed by bubbling N2 through the solution for 20 min. Pd/C (10 mg) was added, the flask was filled with H2 and the solution was left stirring overnight. After completion the solution was filtered over Celite. The filtrate was concentrated, precipitated into ice cold methanol and lyophilized from dioxane. A colorless waxy solid was obtained. 1H NMR was used to confirm successful deprotection of the PBLG units (Fig. S1†).
4.7. Synthesis of functionalized PEG–(PCL–TMC–TMCf)–PGA terpolymer 2–5
The next steps for the synthesis of PEG–(PCL–TMC–TMCPFP) were performed as described above for the PEG–(PCLgTMC)–PGA terpolymer, but with the addition of TMC–PFP. The organocatalyzed ring-opening polymerization of ε-caprolactone and trimethylene carbonate was performed, aiming for a composition of PEG44-(PCL50TMC40TMCPFP10). Monomethoxy–PEG–OH macroinitiator (2 kDa, 0.5 mmol) was weighed into an oven-dried round bottom flask and dried at 140 °C under vacuum. After cooling down, distilled ε-caprolactone (ε-CL, 25 mmol), recrystallized trimethylene carbonate (TMC, 20 mmol), and vacuum dried pentafluorophenyl 5-methyl-2-oxo-1,3-dioxane-5-carboxylate (TMC–PFP, 5 mmol) were added under argon and dissolved in dry DCM (12 mL). The reaction was initiated by the addition of methane sulfonic acid (0.25 mmol ≈ 125 μL). The reaction mixture was stirred at 25 °C in a water bath and reaction progress was regularly checked by 1H NMR. After completion (4–6 h) the reaction was quenched with DIPEA (1.5 mL) and the polymer was precipitated into ice cold methanol and lyophilized. The composition of the resulting copolymer was confirmed by 1H NMR spectroscopy, comparing the protons of PEG (3.65–3.70 ppm, 176 H), terminal methyl unit (singlet at 3.40 ppm, 3 H) to PCL CH2 (multiplet at 2.40–2.25 ppm, 2 H), PTMC CH2 (multiplet at 2.2–1.8 ppm, 2 H) and CH2 PTMC–PFP (multiplet at 4.32–4.58 ppm, 4 H) (Fig. S2†).Amidation reactions of the PEG–(PCL–TMC–TMCPFP) copolymer followed the same general procedure, a typical example is shown here. PEG–(PCL–TMC–TMCPFP) (26 μmol) was dissolved in dry THF (3 mL) and placed in an ice bath. Then a mixture of the respective amine (920 μmol, 3 eq. per TMC–PFP unit) and the base triethylamine (1.84 mmol, 2 eq. per amine) in THF (0.5 mL) was added dropwise. The reaction progress was monitored by 19F NMR following the disappearance of the fluorine signals. After reaction completion (2 h) the polymer was precipitated in diethyl ether and freeze dried from dioxane. NMR spectra of purified polymers are shown in Fig. S3–S6.†
4.8. Synthesis of PEG–PDLLA–PGA terpolymer 6
The polymer PEG–PDLLA was synthesized in accordance with previously published procedures utilizing a PEG macroinitiator and DBU as a catalyst.36 Monomethoxy–PEG–OH macroinitiator (2 kDa, 0.5 mmol) and D,L-lactide (50 mmol) were weighed into an oven-dried round bottom flask and dried by azeotropic distillation with dry toluene (3 × 50 mL). The sample was put under argon and dissolved in dry DCM (50 mL). The polymerization was initiated by the addition of 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU, 0.5 mmol) and was stirred at room temperature until the starting material was fully consumed. After completion (2 h) the resulting polymer was precipitated into ice cold diethyl ether and lyophilized to obtain the product as white solid (88% yield). The composition of the resulting copolymer was confirmed by 1H NMR spectroscopy, comparing the protons of PEG (3.65–3.7 ppm), terminal methyl unit (singlet at 3.37 ppm) to PDLLA CH (multiplet at 5.09–5.27 ppm) (Fig. S7†). The next steps for the final PGA block were performed as described above for the PEG–(PCLgTMC)–PGA terpolymer. The final composition of the terpolymer 6 was: PEG44PDLLA95PGA7 (Fig. S8†).4.9. Synthesis of PEG–PS–PGA terpolymer 7
The PEG–PS block copolymer was synthesized in accordance with previously published procedures utilizing ATRP.38 Briefly, poly(ethylene glycol)methyl ether 2-bromoisobutyrate mPEG45–Br (0.05 mmol), styrene (25 mmol) and CuBr (0.05 mmol) were added into a 5 mL round bottom flask and were degassed with nitrogen for 30 min. Subsequently, N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 0.05 mmol) dissolved in toluene (0.5 mL) was added to the flask and the mixture was degassed for another 30 min with nitrogen. The reaction was stirred in an oil bath at 90 °C. After the desired monomer conversion was reached, the solution was cooled down, diluted with THF (50 mL) and passed through a neutral alumina column twice to remove the copper catalyst. Finally, the filtrate was concentrated and precipitated into cold methanol (50 mL) twice to obtain the product as white solid (84% yield) (Fig. S9†). To obtain an amine end functional PEG–PS, bromine-terminated PEG–PS (7 μmol), 2-(Boc-amino)ethanethiol (14 μmol), triethyl amine (18 μmol) and dry DMF (2.00 mL) were all added in a sealed vial and the reaction was left stirring overnight. Then the mixture was precipitated in cold MeOH. Subsequently, the polymer was dissolved in DCM (2 mL) and TFA (2 mL) was added. After 2 hours the DCM was evaporated and residues of TFA were removed by co-evaporation with toluene (3 × 5 mL). The polymerization and deprotection of N-carboxyanhydride γ-benzyl L-glutamate were performed as described above for the PEG–PCLgTMC–PGA terpolymer. The final composition of the terpolymer 7 was: PEG44PS150PGA12 (Fig. S10†).4.10. AF488–PEG–TMC–PCL 8
Fluorescently labelled polymer AF488–PEG–TMC–PCL was prepared in the same way as PEG–PCLgTMC but with azido–PEG (3 kDa) as initiator. The copolymer azido–PEG68–(PCL50-g-PTMC50) (50 mg, 3.3 μmol) was dissolved in dry dimethylformamide (DMF). DBCO-AF488 (0.33 μmol) was added and left reacting over night at 30 °C. The product was precipitated in cold Et2O, redissolved in chloroform and washed with water (5 times) to remove any unreacted dye. Successful labelling was confirmed by CLSM.4.11. Coacervate formation
Q-Am and CM-Am were dissolved separately in PBS at a concentration of 1 mg mL−1. Consecutively, Q-Am (66 μL, 1 mg mL−1, DS 1.0) and CM-Am (33 μL, 1 mg mL−1, DS = 0.5) were mixed. After shaking for 10 min at 1500 rpm 3.3 μL terpolymer (50 mg mL−1 in PEG350) was added and the sample was shaken for another 5–10 s.4.12. Permeability studies
Coacervates were prepared as described under “Coacervate formation”. The coacervate suspension (30 μL) was transferred to the imaging slide (IBIDI, 18 well glass bottom), diluted with deionized H2O (69 μL) and FITC-dextran was added (1 μL, 1 mg mL−1). The sample was imaged with CLSM and micrographs were analyzed using Fiji (ImageJ).4.13. FRAP studies
FRAP measurements were performed on a Leica TCS SP8 using the FRAP wizard in the LAS X software. Images were taken with an HC PL APO CS2 63× water immersion objective with a numerical aperture (NA) of 1.20. Images were acquired with a resolution of 512 × 512 pixels and a scanning speed of 600 Hz. Artificial cells between 20 and 30 μm were selected. 10 images were acquired prior to the bleaching. Subsequently, the ROI (d = 3.5 μm located at the top of the cell) was bleached for 10 iterations for a total of 7.8 s with 100% laser power. The recovery was monitored with a 3 s interval until full recovery was reached. The intensities of the bleached ROI, reference area, part of the membrane that was not bleached, and background were extracted from the images with FIJI.
Data was analyzed as follows with Origin following a literature procedure.39 Fluorescence intensity at time t, I(t) was background subtracted and corrected for unintentional photobleaching, which was calculated as
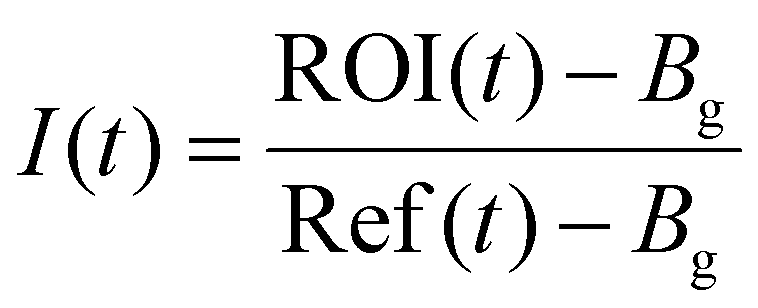
| I(t)normalized = B – Ae−τt |
Finally recovery half-time (τ1/2) and apparent diffusion coefficient (Dapp) were calculated according to following equations for a circular bleaching spot where ω is approximated as radius of the bleaching spot:
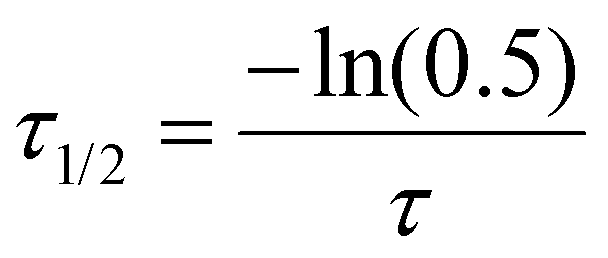
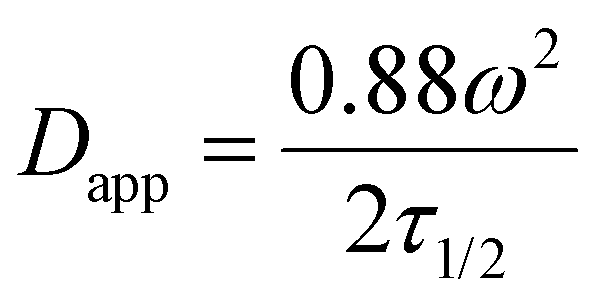
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Dutch Ministry of Education, Culture, and Science (Gravitation program IPM 024.005.020 and Spinoza premium SPI 71-259) and the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreements no. 101022398 and no. 859416.References
- I. Levental and E. Lyman, Nat. Rev. Mol. Cell Biol., 2023, 24, 107–122 CrossRef PubMed.
- T. Harayama and H. Riezman, Nat. Rev. Mol. Cell Biol., 2018, 19, 281–296 CrossRef PubMed.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef PubMed.
- A. B. Cook and S. Perrier, Adv. Funct. Mater., 2020, 30, 1901001 CrossRef.
- A. B. Cook and T. D. Clemons, Adv. NanoBiomed Res., 2022, 2, 2100087 CrossRef.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- H. Che and J. C. M. van Hest, ChemNanoMat, 2019, 5, 1092–1109 CrossRef.
- R. A. J. F. Oerlemans, J. Shao, M. H. M. E. van Stevendaal, H. Wu, T. Patiño Padial, L. K. E. A. Abdelmohsen and J. C. M. van Hest, Biomacromolecules, 2023, 24, 4148–4155 CrossRef.
- A. F. Mason, N. A. Yewdall, P. L. W. Welzen, J. Shao, M. Van Stevendaal, J. C. M. V. Hest, D. S. Williams and L. K. E. A. Abdelmohsen, ACS Cent. Sci., 2019, 5, 1360–1365 CrossRef PubMed.
- C. Xu, N. Martin, M. Li and S. Mann, Nature, 2022, 609, 1029–1037 CrossRef.
- J. Li, X. Liu, L. K. E. A. Abdelmohsen, D. S. Williams and X. Huang, Small, 2019, 15, 1902893 CrossRef PubMed.
- N. Gao, C. Xu, Z. Yin, M. Li and S. Mann, J. Am. Chem. Soc., 2022, 144, 3855–3862 CrossRef.
- M. Abbas, J. O. Law, S. N. Grellscheid, W. T. S. Huck and E. Spruijt, Adv. Mater., 2022, 34, 2202913 CrossRef PubMed.
- A. B. Cook, S. Novosedlik and J. C. M. van Hest, Acc. Mater. Res., 2023, 4, 287–298 CrossRef.
- A. B. Cook, B. D. Gonzalez and J. C. M. van Hest, Biomacromolecules, 2024, 25, 425–435 CrossRef.
- Y. Zhang, Y. Chen, X. Yang, X. He, M. Li, S. Liu, K. Wang, J. Liu and S. Mann, J. Am. Chem. Soc., 2021, 143, 2866–2874 CrossRef PubMed.
- N.-N. Deng and W. T. S. Huck, Angew. Chem., Int. Ed., 2017, 56, 9736–9740 CrossRef PubMed.
- T.-Y. Dora Tang, C. Rohaida Che Hak, A. J. Thompson, M. K. Kuimova, D. S. Williams, A. W. Perriman and S. Mann, Nat. Chem., 2014, 6, 527 CrossRef CAS.
- M. B. Lande, J. M. Donovan and M. L. Zeidel, J. Gen. Physiol., 1995, 106, 67–84 CrossRef CAS PubMed.
- A. F. Mason, B. C. Buddingh, D. S. Williams and J. C. M. Van Hest, J. Am. Chem. Soc., 2017, 139, 17309–17312 CrossRef CAS PubMed.
- S. Ali and V. M. Prabhu, Macromolecules, 2019, 52, 7495–7502 CrossRef CAS PubMed.
- N. Gao and S. Mann, Acc. Chem. Res., 2023, 56, 297–307 CrossRef CAS PubMed.
- N. A. Yewdall, B. C. Buddingh, W. J. Altenburg, S. B. P. E. Timmermans, D. F. M. Vervoort, L. K. E. A. Abdelmohsen, A. F. Mason and J. C. M. van Hest, ChemBioChem, 2019, 20, 2643–2652 CrossRef CAS PubMed.
- Y. Luo, A. B. Cook, L. K. E. A. Abdelmohsen and J. C. M. van Hest, Annu. Rev. Mater. Res., 2024, 54, 75–96 CrossRef CAS.
- J.-F. Le Meins, O. Sandre and S. Lecommandoux, Eur. Phys. J. E, 2011, 34, 14 CrossRef.
- J. C.-M. Lee, M. Santore, F. S. Bates and D. E. Discher, Macromolecules, 2002, 35, 323–326 CrossRef CAS.
- D. E. Discher, P. Janmey and Y. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS PubMed.
- A. Belluati, V. Mikhalevich, S. Yorulmaz Avsar, D. Daubian, I. Craciun, M. Chami, W. P. Meier and C. G. Palivan, Biomacromolecules, 2020, 21, 701–715 CrossRef CAS PubMed.
- F. Itel, M. Chami, A. Najer, S. Lörcher, D. Wu, I. A. Dinu and W. Meier, Macromolecules, 2014, 47, 7588–7596 CrossRef CAS.
- D. A. Los and N. Murata, Biochim. Biophys. Acta, Biomembr., 2004, 1666, 142–157 CrossRef CAS.
- K. J. Seu, L. R. Cambrea, R. M. Everly and J. S. Hovis, Biophys. J., 2006, 91, 3727–3735 CrossRef CAS PubMed.
- D. Chapman, Q. Rev. Biophys., 1975, 8, 185–235 CrossRef CAS.
- H. Borochov and M. Shinitzky, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 4526–4530 CrossRef.
- D. P. Sanders, K. Fukushima, D. J. Coady, A. Nelson, M. Fujiwara, M. Yasumoto and J. L. Hedrick, J. Am. Chem. Soc., 2010, 132, 14724–14726 CrossRef PubMed.
- L. K. E. A. Abdelmohsen, D. S. Williams, J. Pille, S. G. Ozel, R. S. M. Rikken, D. A. Wilson and J. C. M. van Hest, J. Am. Chem. Soc., 2016, 138, 9353–9356 CrossRef PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef.
- H. Che, L. N. J. de Windt, J. Zhu, I. A. B. Pijpers, A. F. Mason, L. K. E. A. Abdelmohsen and J. C. M. van Hest, Chem. Commun., 2020, 56, 2127–2130 RSC.
- D. Axelrod, D. E. Koppel, J. Schlessinger, E. Elson and W. W. Webb, Biophys. J., 1976, 16, 1055–1069 CrossRef PubMed.
- R. B. Lira, J. Steinkühler, R. L. Knorr, R. Dimova and K. A. Riske, Sci. Rep., 2016, 6, 1–12 CrossRef.
- A. C. Engler, J. M. W. Chan, D. J. Coady, J. M. O'Brien, H. Sardon, A. Nelson, D. P. Sanders, Y. Y. Yang and J. L. Hedrick, Macromolecules, 2013, 46, 1283–1290 CrossRef.
- A. F. Mason, B. C. Buddingh, D. S. Williams and J. C. M. Van Hest, J. Am. Chem. Soc., 2017, 139, 17309–17312 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00978a |
| This journal is © The Royal Society of Chemistry 2024 |