
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of molar mass of poly(2-oxazoline) based glycopolymers on lectin binding†
Caitlin L. A.
Nutting
,
James
Lefley
,
Zivani
Varanaraja
,
Gokhan
Yilmaz
and
C. Remzi
Becer
 *
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: remzi.becer@warwick.ac.uk
First published on 25th November 2024
Abstract
Glycopolymers are a versatile polymer type employed in many applications, especially the biomedical field, due to their ability to exploit multivalent lectin–carbohydrate interactions. Understanding how to improve and manipulate the interactions between glycopolymers and lectins is crucial for their success within the pharmaceutical industry. Herein, we synthesised block copolymers via cationic ring opening polymerisation of 2-ethyl-2-oxazoline and 2-(3-butenyl-2-oxazoline) with varying quantities of 2-ethyl-2-oxazoline. These polymers were further functionalised with pendent glucose moieties to produce glycopolymers. All polymers and glycopolymers were analysed using a variety of techniques including NMR, GPC, FT-IR and MALDI-ToF MS. Their binding capabilities were evaluated by surface plasmon resonance, utilising human lectins: DC-SIGN, MBL and Langerin, to investigate how the molar mass influences lectin binding.
Introduction
Over the last decade, glycopolymers have gained significant attention due to their ability to mimic naturally occurring carbohydrate containing macromolecules which are able to bind to lectins.1,2 Lectins are crucial proteins within the body, mediating attachment and binding of carbohydrate containing molecules, allowing for their pivotal role in executing recognition events.3,4 This is essential for biological processes including modulating the inflammatory response,5 pathogen elimination,6 and regulation of glycoproteins.7The ability of glycopolymers to bind to lectins enables them to prevent bacterial infiltration,8 adhesion,9 and biofilm formation.10,11 Consequently, various biomedical applications have been developed including drug delivery systems,12 gene delivery,13,14 disease inhibition,15 and biosensors.16–19 However, development has often been hindered by their limited specificity towards targeted lectin sites, toxicity and bioavailability.1,10 The increased attention towards lectin binding glycopolymers, with their advantageous multivalency characteristics, has led to more research and in turn a greater understanding of specific site binding via the control of chemical characteristics, topology and morphology.20–25 These features can be modified and adapted to a specific site by manipulating the length of the glycopolymer,15 backbone rigidity and flexibility,26,27 molecular weight,28 and polymeric chain architecture.29,30
Poly (2-oxazoline)s (POx) are an upcoming class of biomaterials that are being recognised as promising alternatives to poly(ethylene glycol) (PEG) because of their tuneability, biocompatibility and reduced cytotoxicity.31,32 POx first gained attention in the biomedical field when Zalipsky et al.33 highlighted the impressive pharmacokinetics of poly(2-ethyl-2-oxazoline) (PEtOx). POx is synthesised via cationic ring opening polymerisation (CROP), a living polymerisation technique. POx exhibits well-defined polymers with narrow dispersion and controlled architecture34 that are highly tuneable35 and have high end-group fidelity.36 Additionally, POx have shown to be more tuneable with a greater array of monomers,37 enhanced ‘stealth’ like properties,31 and good solubility in various common solvents.38 This addresses concerns associated with PEG and PEGylated drugs.39 Presently, PEtOx based drugs and drug delivery systems are being investigated with one going through a clinical trial for Parkinson's disease.40
Molar mass is a tuneable feature of glycopolymers. The glycopolymer length and make up can lead to interesting morphological and chemical features. These have been previously investigated specifically in self-assembly and how glycopolymers can self-assemble into micelles,41 polymersomes,42 and other diverse structures. Molar mass has previously been evaluated by increasing both the polymer chain length and pendent carbohydrate moieties,28,43,44 or by how the polymer chain length influences cellular uptake.45 It has been found that there is a maximum point for increasing the glycopolymer multivalency, likely due to either all available binding sites being occupied, or coiling of the polymer backbone.46 However, it has never been investigated and distinguished whether the binding is enhanced or diminished by the glycopolymer chain length, molar mass or increasing amount of carbohydrate groups.
Herein, we evaluate how increasing the molar mass of glycopolymers, whilst keeping the carbohydrate content constant, affects the lectin binding capabilities. The glycopolymers (GP1–GP5), composed of a fixed block length of 2-(3-butenyl-2-oxazoline) (ButeneOx), and varying block length of EtOx were synthesised via CROP. Thiol–ene click chemistry was utilised for the post-polymerisation modification of PButeneOx to yield glycopolymers GP1–GP5, (Scheme 1). Thus, we were able to synthesise a small set of glycopolymers with varying molar mass from 7100 to 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 Da. From this, the binding capabilities were evaluated via surface plasmon resonance (SPR) with the biologically relevant human lectins mannose binding lectin (MBL), dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) and Langerin.
700 Da. From this, the binding capabilities were evaluated via surface plasmon resonance (SPR) with the biologically relevant human lectins mannose binding lectin (MBL), dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) and Langerin.
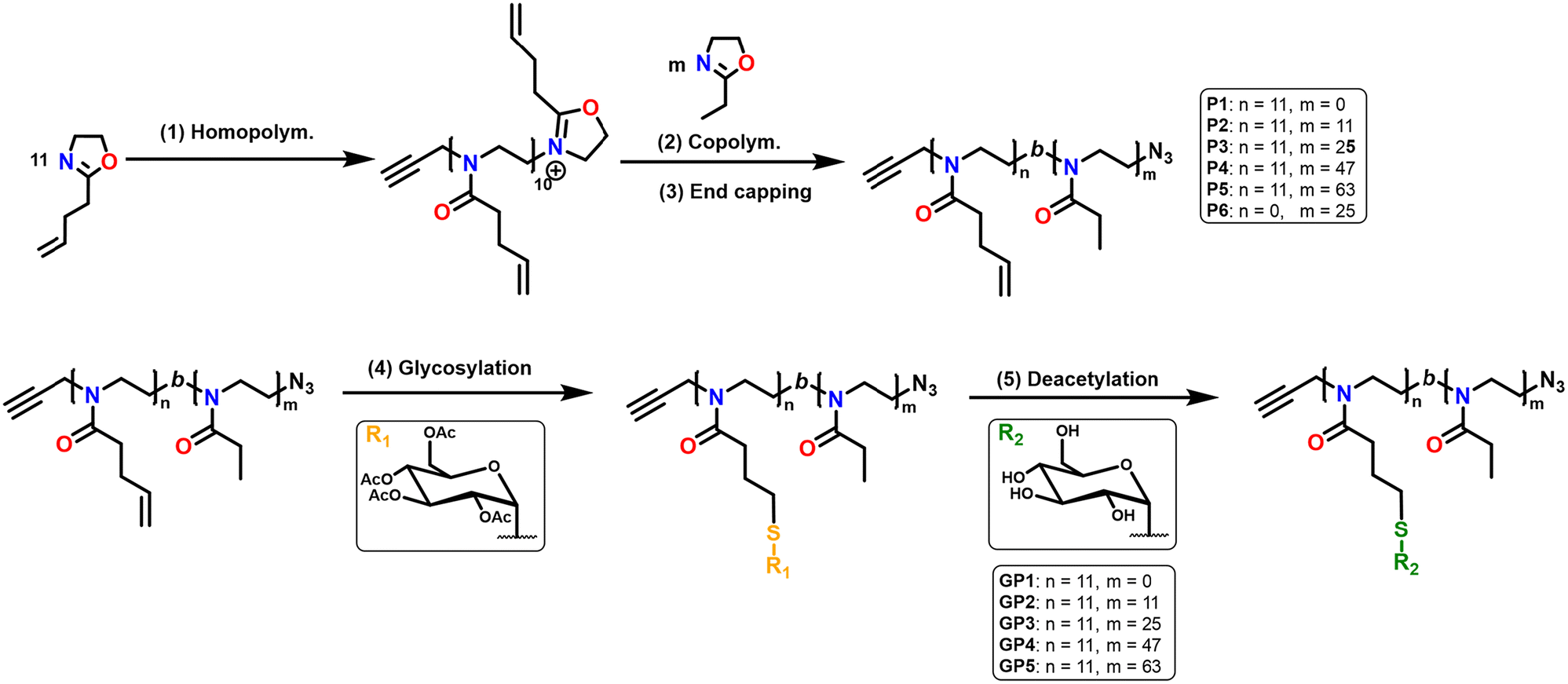 | ||
| Scheme 1 Overall scheme of the synthesis of the thio-glucose-functionalised PEtOx-b-PButeneOx glycopolymers forming GP1–GP5. Step 1. Cationic ring opening polymerisation of ButeneOx, which is initiated by propargyl p-toluenesulphonate (1 equiv.) in acetonitrile (4 M, 110 °C) and reacted for 22 minutes. Step 2. The addition of the second EtOx block in varying amounts which was then subsequently left to polymerise for the appropriate amount of time at 110 °C to obtain P1–P6. Step 3. The copolymers are subsequently terminated with sodium azide (NaN3) (3 equiv.). After being left to stir for 24 hours, the resulting polymers are precipitated into diethyl ether and dried under vacuum. Step 4. Radical mediated thiol–ene click reaction using β-thioglucose tetraacetate (1.2 equiv. per double bond) and dimethyl 2,2′-azobis(2-methylpropionate) (V601) (0.25 equiv.) in tetrahydrofuran (THF) (7 μM, 70 °C) obtaining the acetylated analogues of GP1–GP5. Step 5. Deprotection of the thio-glucose tetraacetate using sodium methoxide (NaOMe)(22 equiv.) in methanol (MeOH) (0.016 M) to yield the final glycopolymers GP1–GP5. | ||
Experimental
Materials
4-Pentenoic acid (Merck; 98%), N-hydroxysuccinamide (NHS) (Aldrich, 98%), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDAC) (ABCR; 98%), 2-chloroethylamine (Alfa Aesar; 98%), triethylamine (TEA) (Merck; 99%), potassium hydroxide (KOH) (Aldrich; ≥85%), sodium azide (NaN3) (Aldrich; ≥99.5%), β-thioglucose tetracetate (Fisher Scientific; 97%), sodium methoxide solution (25% w/t in methanol) (Aldrich, 95%), dimethyl 2,2′-azobis(2-methylpropionate) (V601) (Fujifilm; 97%), anhydrous acetonitrile (Extra Dry) (Fisher Scientific; ≥99%), dichloromethane (DCM) (Aldrich, ≥99%), tetrahydrofuran (THF) (Aldrich, ≥99%), dimethylformamide (DMF) (Aldrich, ≥99%), methanol (MeOH) (Aldrich, ≥99%), were all purchased and were used as received. Both 2-ethyl-2-oxazoline (EtOx) (Fisher Scientific; 99%) and propargyl p-toluenesulphonate (propargyl tosylate) (Fisher Scientific; 98%) were distilled over calcium hydride (CaH2) (Aldrich; for synthesis) and then stored under nitrogen. All moisture sensitive reactions were carried out under nitrogen. All lectins were purchased from Biotechne – R&D Systems.Analytical techniques
Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III AV 400 MHz at 278 K. Analysis was carried out in CDCl3 or D2O and the residual peak from CHCl3 and H2O were used as the reference chemical shift. The data analysis was performed using TopSpin 4.1.1 Software.
Gel permeation chromatography (GPC) measurements were performed using THF (2% TEA and 0.1% BHT) as the eluent. The Agilent Technologies 1260 Infinity instrument was equipped with a refractive index (RI) and 308 nm UV detector, a PLgel 5 μM guard column, and a PLgel 5 μM mixed D column (300 × 7.5 mm). Samples were run at 1 mL min−1 at 40 °C. Poly(methyl methacrylate) (PMMA) standards (Agilent PMMA calibration kits M-M-10 and M-L-10) were used for the calibration. All polymer samples were made up to 1 mg mL−1 using THF and then before injection (100 μL), the samples were filtered through a PTFE membrane with a 0.2 μL pore size. Experimental molar mass (Mn), weight-average molar mass (Mw), and dispersity (Đ) values of synthesised polymers were determined by conventional calibration using Agilent GPC/SEC software. GPC measurements in dimethylformamide (DMF) were carried out on an Agilent 1260 Infinity II instrument with two Polargel M columns and a Polargel M guard column. The mobile phase was DMF stabilised with 0.1% w/v lithium bromide (LiBr) with a flow rate of 1 mL min−1 at a 50 °C and equipped with the following detectors: differential refractive index (DRI), viscometer (VS), light scattering (LS), and variable wavelength detector (MWD). Agilent poly(methyl methacrylate) Easivials were used to create a third order calibration curve. The calibrants cover a range between 535 and 538500 g mol−1. All glycopolymer samples were made up to 1 mg mL−1 using DMF and were then passed through 0.2 μm nylon filters prior to GPC measurements. All samples were then analysed using Agilent GPC/SEC software.
:2:5 and then spotted onto the target (0.5 μL). Spectra were recorded in reflective mode, and the mass spectrometer was calibrated with a PMMA standard up to 3 kDa.
:1 mixture of 0.1 M N-hydroxy succinimide and 0.05 M N-ethyl-N′(dimethylamino propyl) carbodiimide over the chip for 5 min at 25 °C at a flow rate of 10 μL min−1 after the system equilibration with HEPES filtered buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 5 mM CaCl2). Subsequently, channels 1 (blank), 2, 3, and 4 were blocked by flowing a solution of ethanol amine (1 M pH 8.5) for 10 min at 10 μL min−1 to block the remaining reactive groups on the channels. Sample solutions were prepared at varying concentrations (20.0–1.25 μM) in the same HEPES buffer to calculate the binding kinetics. Sensorgrams for each glycopolymer concentration were recorded with a 350 seconds injection of polymer solution (on period), followed by 200 seconds of buffer alone (off period). Regeneration of the sensor chip surfaces was performed using 10 mM HEPES pH 7.4, 150 mM NaCl, 10 mM EDTA, and 0.01% Tween20 surfactant solution. A blank analyte was also used to correct for unspecific binding events. Kinetic data was evaluated using a single set of sites (1:1 Langmuir binding model) in the BIA evaluation 3.1 software.
Dynamic light scattering (DLS) measurements were carried out on an Anton Paar Litesizer instrument using a disposable cuvette. Samples were measured at 25 °C at a backscattering measuring angle of 175°. Each sample was measured in triplicate with 30 runs per measurement and 5 min equilibration time between each measurement. PEtOx refractive index used was 1.52.
Foruier transoform infrared spectroscopy (FT-IR)
FT-IR was carried out on Agilent Cary 630 with ATR sampling module running at 65 scans per sample with a speed of 0.5 cm s−1.Step 2 Product A was combined with 2-chloroethylamine hydrochloride (0.33 mol, 1 equiv.) and dissolved in DCM (150 mL). Triethylamine (TEA) (2.5 equiv.) was added dropwise to the reacting mixture which was stirred at 0 °C for 24 h. The solution was washed with water (6 × 100 mL) and then dried over MgSO4. The DCM was removed via rotary evaporation to yield product B (yellow liquid, 38 g, Fig. S2†). Step 3 Potassium hydroxide (KOH) (0.23 mol, 1 eqiuv.) was dissolved in methanol (MeOH) (135 mL) and added dropwise to product B and was then stirred at 50 °C for 24 h. The product was then filtered and MeOH was removed via rotary evaporation. The liquid was distilled twice. The first distillation was carried out under vacuum and the distillate was observed at 30 °C. This was then subsequently stirred with calcium hydride (CaH2) for 24 h and distilled a second time under vacuum and the distillate was observed at 30 °C. The system was then sparged under nitrogen to yield 2-(3-butenyl-2-oxazoline) (clear liquid, 12 g, Fig. S3†).
| Code | PButenOx (DP) | PEtOx (DP) | M n (theo) (Da) | M n (GPC) (Da) | Đ |
|---|---|---|---|---|---|
| a GPC analysis of polymers was performed using eluent: THF +2% TEA +0.1% BHT. b GPC SEC analysis of glycopolymers were performed using eluent: DMF +0.1% LiBr. | |||||
| P1 | 11 | 0 | 1500 | 1800a | 1.42 |
| P2 | 11 | 9 | 2300 | 2500a | 1.32 |
| P3 | 11 | 25 | 3900 | 3900a | 1.35 |
| P4 | 11 | 45 | 5900 | 5900a | 1.68 |
| P5 | 11 | 63 | 7700 | 7800a | 1.50 |
| P6 | 0 | 30 | 3000 | 2800a | 1.16 |
| GP1 | 11 | 0 | 4000 | 7100b | 1.29 |
| GP2 | 11 | 9 | 4700 | 8700b | 1.21 |
| GP3 | 11 | 25 | 6300 | 9500b | 1.24 |
| GP4 | 11 | 45 | 8300 | 13000b | 1.27 |
| GP5 | 11 | 63 | 10100 |
14700b | 1.17 |
Results and discussion
Synthesis and analysis of block copolymerisation via CROP
Non-functionalised block copolymers P1–P6 were synthesised via CROP using a one-pot, sequential addition method utilising propargyl tosylate as an initiator and sodium azide as the terminating agent. Optimal conditions for the copolymerisation between PButeneOx and PEtOx were implemented from a previous study carried out by Schlaad et al.,47 with the homopolymerisation conditions of PButeneOx confirmed via a kinetic study (Fig. S4†). The first block was kept the same throughout the series via a stock solution of ButeneOx, keeping the [M]/[I] ratio = 11 (Fig. S5†). The conversion of the first block was confirmed via1H NMR and GPC. Subsequently, the second block was then added in varying [M]/[I] ratios, ranging from 9 to 63. The purified polymers were then analysed by GPC and 1H NMR (Table 1, Fig. 1(A)–2, Fig. S6–S10 and S11–18†). MALDI-ToF was used to demonstrate the end group fidelity of P1 (Fig. S11†). All polymers achieved ≥99% monomer conversion apart from P6 which achieved 97%. This was confirmed by the lack of monomer present in the final 1H NMR (Fig. 2), usually observed in the region of ∼4.2 ppm and ∼3.7 ppm. Polymers P1–P5 showed a slight difference between Mn, theo (calculated from conversion) and Mn, GPC (Table 2). Polymers P1–P5Mn, theo (calculated from conversion) and Mn, GPC matched up well. High dispersity was shown in the larger blocks P4 and P5, this is likely attributed to early termination after the addition of the second block. This monomer combination has been selected due their water solubility and ability to undergo post-polymerisation functionalisation, making it a good starting point for future biomedical applications. Block copolymer structures were selected in order to keep the carbohydrate content constant, and a stock solution used to keep the first block consistent throughout the class. This was important so that we could accurately confirm that it was the molar mass influence that was affecting the binding affinity and not the increasing sugar content. Azide–alkyne end-group functionality was used for the ability to further functionalise if desired keeping the study relevant within the current literature.4,13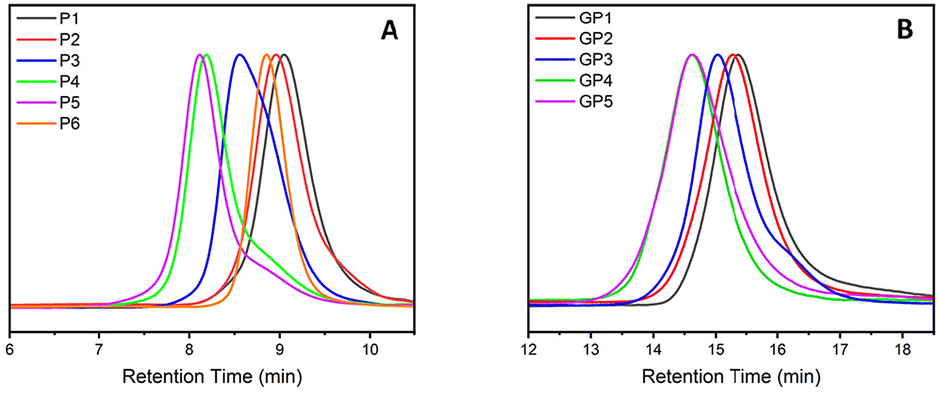 | ||
| Fig. 1 (A) GPC traces of PButeneOx with varying amounts of PEtOx: P1–P5. Measurements performed using THF (+2% TEA and +0.1% BHT) as the eluent. PMMA standards were used for calibration. (B) GPC traces of PButeneOX with varying amounts of PEtOx: GP1–GP5. Measurements performed using DMF (+0.1% LiBr) as the eluent. PMMA standards were used for calibration. | ||
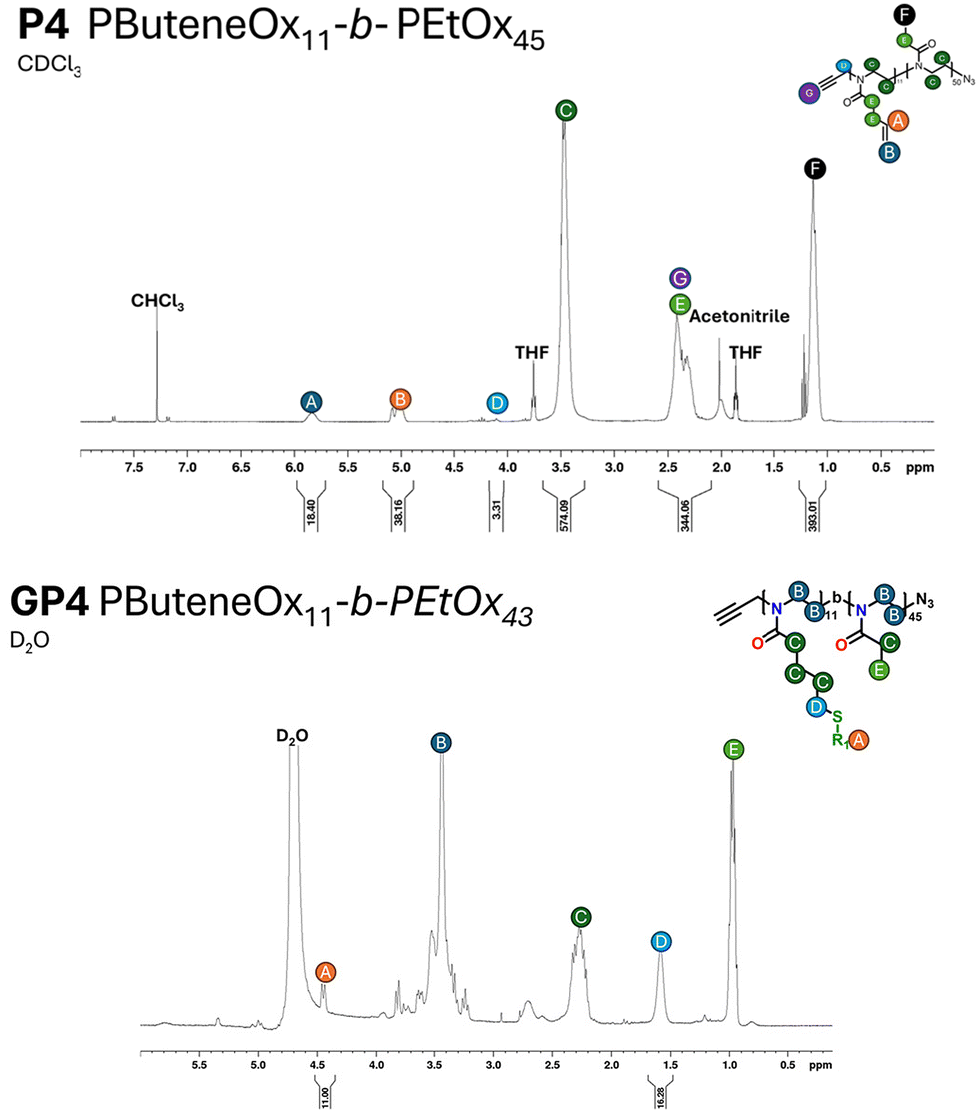 | ||
| Fig. 2 A representative example of the 1H NMR of the polymer and glycopolymer of P4 and GP4. All measurements were performed on an AV400 MHz NMR. | ||
:1 binding model
| Lectin | Code | k a (M−1 s−1) | k d (s−1) | K d (M) | R max (RU) |
|---|---|---|---|---|---|
| DC-SIGN | GP1 | 387 | 6.51 × 10−4 | 1.59 × 10−6 | 3000 |
| DC-SIGN | GP2 | 198 | 3.39 × 10−4 | 1.72 × 10−6 | 725 |
| DC-SIGN | GP3 | 274 | 2.71 × 10−4 | 9.88 × 10−7 | 750 |
| DC-SIGN | GP4 | 834 | 8.08 × 10−4 | 9.69 × 10−7 | 170 |
| DC-SIGN | GP5 | 261 | 6.57 × 10−4 | 2.52 × 10−6 | 75 |
| Langerin | GP1 | 14.2 | 3.49 × 10−5 | 2.46 × 10−6 | 430 |
| Langerin | GP2 | 1220 | 5.80 × 10−4 | 4.75 × 10−7 | 110 |
| Langerin | GP3 | 836 | 2.80 × 10−4 | 3.43 × 10−7 | 75 |
| Langerin | GP4 | 1030 | 2.82 × 10−4 | 2.73 × 10−7 | 45 |
| Langerin | GP5 | 2470 | 9.23 × 10−4 | 3.73 × 10−7 | 45 |
| MBL | GP1 | 217 | 1.42 × 10−4 | 6.54 × 10−7 | 6500 |
| MBL | GP2 | 84.3 | 15.3 × 10−4 | 18.2 × 10−6 | 205 |
| MBL | GP3 | 172 | 13.8 × 10−4 | 8.03 × 10−6 | 140 |
| MBL | GP4 | 470 | 19.7 × 10−4 | 4.20 × 10−6 | 140 |
| MBL | GP5 | 307 | 16.1 × 10−4 | 5.26 × 10−6 | 100 |
Synthesis and analysis of glycopolymers GP1–GP5
Glycosylation of all the polymers was achieved via the post-polymerisation of the pendent double bonds. These were functionalised via a ‘thiol click’ reaction using β-thioglucose tetracetate. The reaction was set up according to the following conditions: ene/thiol/V601 = 1.0/1.2/0.25. Completion of the glycosylation was monitored by 1H NMR (Fig. 2) via the disappearance of the allylic peak observed at 5.7 ppm. Subsequent addition of V601 and β-thioglucose tetraacetate was added if the allylic peak was still observed after 24 hours. After the completion of all ‘thiol–ene click’ reactions, the pendant sugars were deacetylated with NaOMe and the crude product dialysed against water to remove any remaining impurities. The dialysis cut off was 500–1000 Da and as a result likely removed some of the smaller chains, resulting in a narrower dispersity (Table 1 and Fig. 1).The final glycopolymers were analysed via GPC (Fig. 1(B)), Fourier transform infrared spectroscopy (FT-IR) (Fig. S29–S36†) and 1H NMR (Fig. 2 and Fig. S19–S22†) were used to confirm complete deacetylation. The deacetylation was confirmed by the disappearance of the acetyl peaks at ∼2.0 ppm. This was also confirmed by FT-IR, the FT-IR was taken before and after deacetylation. After deacetylation the acetyl peaks (1600 cm−1) disappeared and the broad OH peaks grew (3000 cm−1). The glycopolymers GP1–GP5 showed a difference between Mn, theo (calculated from conversion) and Mn, GPC. This result is likely due to the difference between the hydrodynamic volume of the standards used to calibrate the GPC, and the glycopolymer.
Lectin binding analysis via SPR
SPR studies were conducted by immobilising lectins on a chip surface (CM5) and then flowing GP1–GP5 over the substrate with a HBS buffer solution containing calcium (pH = 7.4). The binding assays were recorded in a concentration range of 20 μM–1.75 μM. The lectins used, MBL, DC-SIGN and Langerin, were all selected for their affinity towards glycosyl residues and their roles within the human innate immune response.48It was expected that across the series, the binding affinity towards lectins would increase as molar mass increased due to the mass effect.49,50 A general trend in affinity throughout the series was observed. Kinetic evaluation of SPR results suggests that overall, there is an increasing affinity with higher molar mass. After GP4, it seemed to reach a plateau, whereby after GP4 the binding no longer increases rather it stays the same or decreases which agrees with the literature, whereby they increased the molar mass by increasing the sugar content.46 It appears likely that these findings are caused by the increase in non-glycosylated polymer domains across the series, preventing the carbohydrates from reaching the lectin binding sites by coil formation. GP1 demonstrated a large Rmax value, likely due to the high density of carbohydrate in comparison to the other glycopolymers. It was observed that in all lectins that the Rmax value decreased as the molar mass increased.
MBL is a type of lectin within human serum, which has a range of roles within the immune system. MBL is key to the innate immune response that can bind to pathogens and other invading pathogens to activate an antibody response required for the removal of infection.51 Glycopolymers GP1–GP5 showed binding to MBL evidenced by the ka values (Table 2) obtained from SPR. GP1 has a Kd value of 6.54 × 10−7 M for MBL. This demonstrates that GP1 has the strongest binding within the class, further supported by its small kd value of 1.42 × 10−4 s−1. This low dissociation rate constant indicated that strong interactions with the glucose units on GP1 are persistent or that rebinding of released glucose units occurs more quickly than the dissociation of the complex during the buffer wash period. In comparison to the rest of the class, the Rmax of GP1 was ∼10 times greater whilst its affinity to the lectin (reflected by the kd values) was ∼100 times greater than GP2 and ∼10 times greater than the rest of the class. Overall, the kinetic values support that there is a maximum affinity in this series of polymers, as evidenced by the similar Kd values in GP3–GP5.
DC-SIGN is a C-type (calcium-dependent) transmembrane protein found predominantly in dendric cells. DC-SIGN is a cell adhesion and pathogen recognition receptor, which has high affinity for mannose containing glycoproteins. Predominantly, DC-SIGN acts as an adhesion molecule but can also initiate the innate immune response.52 All glycopolymers (GP1–GP5) demonstrated binding to DC-SIGN, as reflected by the ka values and the SPR curves (Table 2 and Fig. 3). GP4 was found to have the lowest Kd value of 9.69 × 10−7 M, indicating that it binds the most effectively within the class to DC-SIGN, because of its slow disassociation. It should be noted that GP1 has the highest Rmax value within the class, and this also follows the previous trend observed of the glycopolymers binding to MBL. GP5 demonstrated the weakest binding within the class. As discussed above, this is to be expected as polymer chains likely coil which restricts access to the pendant sugar moieties. Notably, all polymers, except for higher molar mass ones, displayed a saturated binding profile during the association phase, indicating that some lectin binding sites remained unoccupied on the chip. This could be attributed to the limited accessibility and flexibility of the carbohydrates on the polymer chain due to their bulky nature in solution.
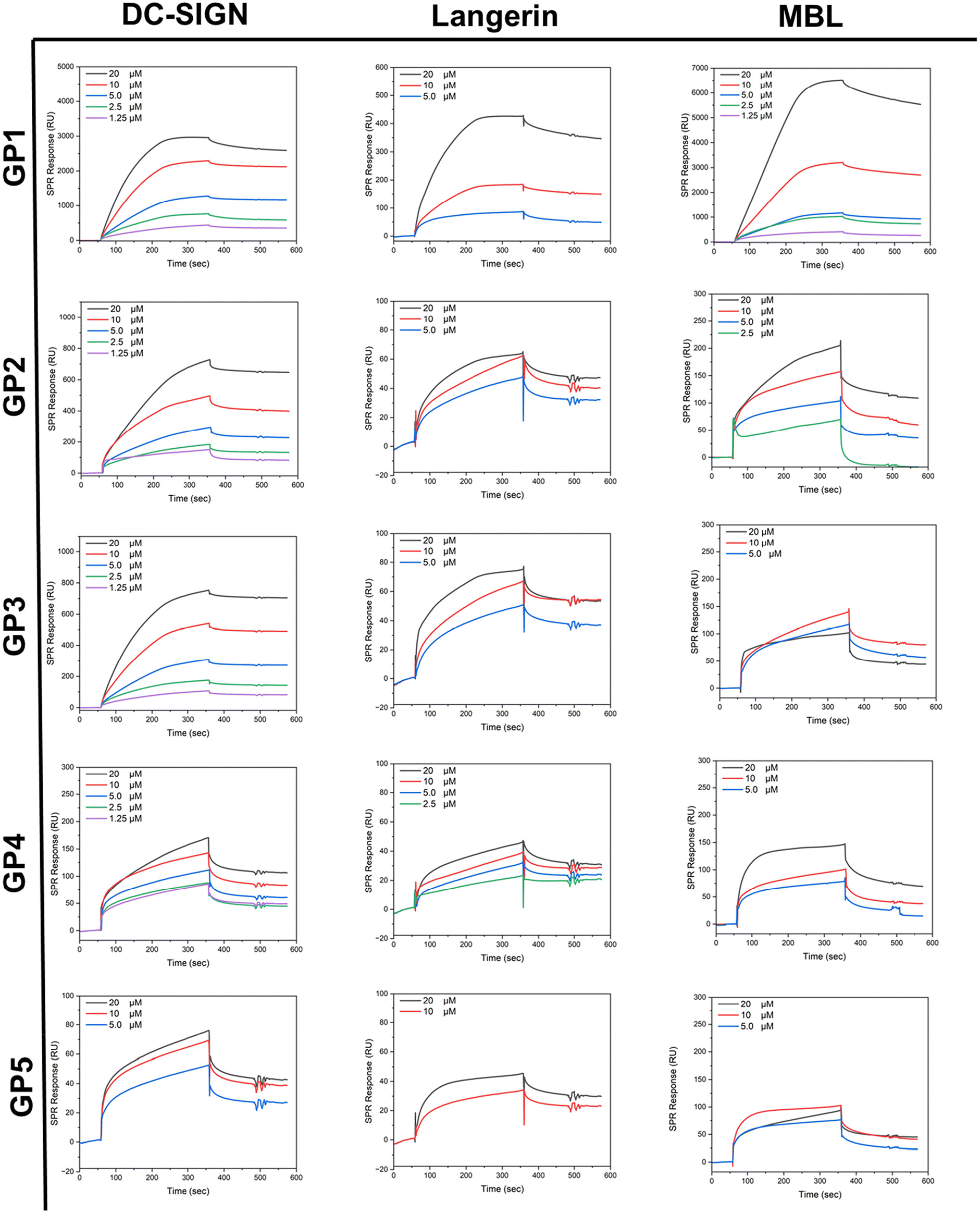 | ||
| Fig. 3 The SPR sensorgrams of glycopolymers GP1–GP5 demonstrating the binding curves associated with them. All SPR measurements were carried out in a HBS buffer against immobilised lectins: DC-SIGN, Langerin and MBL. | ||
Langerin is a transmembrane protein which binds specifically towards mannose, fucose and N-acetylglucosamine. Langerin is expressed in the Langerhans cells (LC) which are a subset of DC cells. Langerin, like other C-type lectins, are pattern recognition receptors which recognise carbohydrates expressed by pathogens.53 All glycopolymers (GP1–GP5) demonstrated a reasonable binding to Langerin. This is confirmed by the ka values (Table 2). GP4 was found to have the greatest binding reflected by the lowest Kd value of 2.73 × 10−7 M. Again, GP1 had the highest Rmax value within the class. This result follows the trend observed in the last two analysed lectins. In conclusion, as the molar mass increases the affinity increases up to a certain point. This point is the critical point at which the polymers with higher molar mass at constant glucose content likely block their own availability, reducing their binding capability.
Further investigations into GP1 and its high Rmax values were conducted via DLS in both water and buffer and TEM (Fig. 4). It should be noted that Rmax has no direct influence on the binding efficacy of the polymer. However, it can be linked to the mass of the polymer.54 This determined that GP1 self-assembled into elongated micelle morphology (Fig. 4A). This feature was not observed in the other glycopolymers. The reason for the self-assembly could be attributable to hydrophobic nature of the butyl side chain. In theory this is sufficiently hydrophobic to act as a driving force for the self-assembly of GP1.
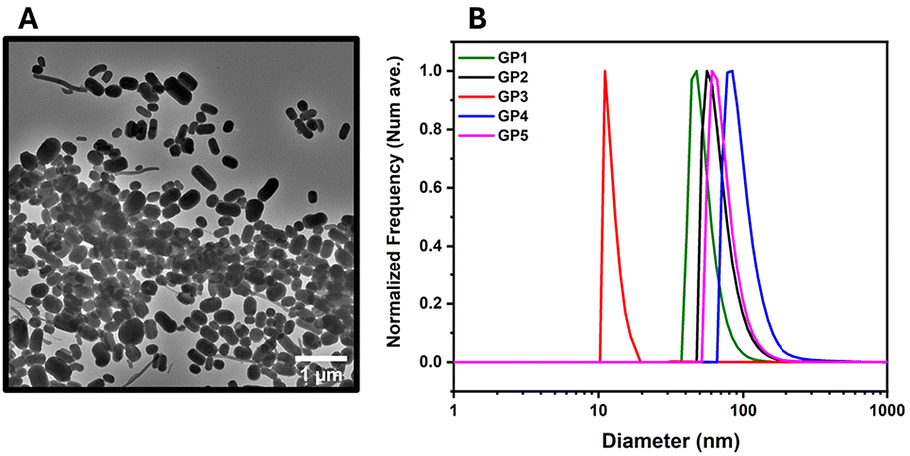 | ||
| Fig. 4 The DLS and TEM of glycopolymers GP1–GP5. (A) TEM image of GP1. (B) The DLS traces of the glycopolymers GP1–GP5 in an HBS buffer. | ||
Conclusions
In conclusion, we have demonstrated how the molar mass of glycopolymers influences their lectin binding. Initially, it was hypothesised that the polymers within the class would demonstrate increasing affinity as the molar mass increased even with the constant carbohydrate content. It was concluded that with increasing molar mass, the binding affinity did also increase up to a maximum point. With the maximum point possibly owing to the steric hindrance caused reduced availability of glucose units on the polymers. While all polymer parameters play a role in forming high affinity interactions, polymer length and epitope density are critical properties for developing effective glycopolymers against C-type human lectins.Overall, a set of poly(2-oxazolines) copolymers was synthesised and subsequently glycosylated via thiol–ene chemistry. The resulting glycopolymers were successfully investigated by SPR and their affinities to three biologically relevant lectins was obtained. Thus, these glycopolymers not only investigate the impact of molar mass on their multivalent binding properties but also deepen our fundamental understanding of carbohydrate–lectin interactions in biological systems.
Author contributions
C. R. B conceived the project. C. R. B and C. N. designed the experiments. C. N. conducted all experimental work. G. Y. helped perform SPR. J. L. did the TEM and DLS. Both J. L. and Z. V. supervised and discussed any problems associated with the project. All authors analysed the experimental data, edited, and commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the Polymer RTP for analysis of polymers and EUTOPIA for providing funding for CN.References
- M. H. Stenzel, Macromolecules, 2022, 55, 4867–4890 CrossRef CAS.
- T. Zhao, R. Terracciano, J. Becker, A. Monaco, G. Yilmaz and C. R. Becer, Biomacromolecules, 2022, 23, 543–575 CrossRef CAS PubMed.
- S. S. Pinho, I. Alves, J. Gaifem and G. A. Rabinovich, Cell. Mol. Immunol., 2023, 20, 1101–1113 CrossRef CAS.
- J. Becker, R. Terracciano, G. Yilmaz, R. Napier and C. R. Becer, Biomacromolecules, 2023, 24, 1924–1933 CrossRef CAS PubMed.
- T. E. Pauline, M. Rudd, P. Cresswell, I. A. Wilson and R. A. Dwek, Science, 2001, 291, 2370–2376 CrossRef PubMed.
- Y. van Kooyk and G. A. Rabinovich, Nat. Immunol., 2008, 9, 593–601 CrossRef CAS PubMed.
- B. Lepenies and R. Lang, Front. Immunol., 2019, 10, 2379–2382 CrossRef CAS PubMed.
- L. Zheng, Y. Luo, K. Chen, Z. Zhang and G. Chen, Biomacromolecules, 2020, 21, 5233–5240 CrossRef CAS PubMed.
- X. Yan, A. Sivignon, N. Yamakawa, A. Crepet, C. Travelet, R. Borsali, T. Dumych, Z. Li, R. Bilyy, D. Deniaud, E. Fleury, N. Barnich, A. Darfeuille-Michaud, S. G. Gouin, J. Bouckaert and J. Bernard, Biomacromolecules, 2015, 16, 1827–1836 CrossRef CAS PubMed.
- U. I. M. Gerling-Driessen, M. Hoffmann, S. Schmidt, N. L. Snyder and L. Hartmann, Chem. Soc. Rev., 2023, 52, 2617–2642 RSC.
- M. Korpidou, J. Becker, S. Tarvirdipour, I. A. Dinu, C. R. Becer and C. G. Palivan, Biomacromolecules, 2024, 25, 4492–4509 CrossRef CAS.
- F. Demir Duman, A. Monaco, R. Foulkes, C. R. Becer and R. S. Forgan, ACS Appl. Nano Mater., 2022, 5, 13862–13873 CrossRef CAS PubMed.
- G. Hayes, B. Dias-Barbieri, G. Yilmaz, R. J. Shattock and C. R. Becer, Biomacromolecules, 2023, 24, 5142–5151 CrossRef CAS PubMed.
- A. K. Blakney, R. Liu, G. Yilmaz, Y. Abdouni, P. F. McKay, C. R. Bouton, R. J. Shattock and C. R. Becer, Polym. Chem., 2020, 11, 3768–3774 RSC.
- M. Nagao, Y. Fujiwara, T. Matsubara, Y. Hoshino, T. Sato and Y. Miura, Biomacromolecules, 2017, 18, 4385–4392 CrossRef CAS PubMed.
- Y. Terada, A. Obara, H. Takamatsu, W. V. Espulgar, M. Saito and E. Tamiya, ACS Appl. Bio Mater., 2021, 4, 7913–7920 CrossRef CAS PubMed.
- Y. Miura, Y. Hoshino and H. Seto, Chem. Rev., 2016, 116, 1673–1692 CrossRef CAS PubMed.
- Y. Oz, Y. Abdouni, G. Yilmaz, C. R. Becer and A. Sanyal, Polym. Chem., 2019, 10, 3351–3361 RSC.
- G. Yilmaz, E. Guler, C. Geyik, B. Demir, M. Ozkan, D. Odaci Demirkol, S. Ozcelik, S. Timur and C. R. Becer, Mol. Syst. Des. Eng., 2018, 3, 150–158 RSC.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- J. Chen, R. Terracciano, J. Becker, G. Yilmaz and C. R. Becer, Eur. Polym. J., 2024, 211, 113006 CrossRef CAS.
- M. Hartweg, Y. Jiang, G. Yilmaz, C. M. Jarvis, H. V. T. Nguyen, G. A. Primo, A. Monaco, V. P. Beyer, K. K. Chen, S. Mohapatra, S. Axelrod, R. Gómez-Bombarelli, L. L. Kiessling, C. R. Becer and J. A. Johnson, JACS Au, 2021, 1, 1621–1630 CrossRef CAS PubMed.
- A. Monaco, V. P. Beyer, R. Napier and C. R. Becer, Biomacromolecules, 2020, 21, 3736–3744 CrossRef CAS PubMed.
- Y. Abdouni, G. Yilmaz, A. Monaco, R. Aksakal and C. R. Becer, Biomacromolecules, 2020, 21, 3756–3764 CrossRef CAS PubMed.
- F. Shamout, A. Monaco, G. Yilmaz, C. R. Becer and L. Hartmann, Macromol. Rapid Commun., 2020, 41, 1900459 CrossRef CAS PubMed.
- Y. Wang, Y. Kotsuchibashi, Y. Liu and R. Narain, ACS Appl. Mater. Interfaces, 2015, 7, 1652–1661 CrossRef CAS PubMed.
- G. Yilmaz, V. Uzunova, M. Hartweg, V. Beyer, R. Napier and C. R. Becer, Polym. Chem., 2018, 9, 611–618 RSC.
- Y. Chen, M. S. Lord, A. Piloni and M. H. Stenzel, Macromolecules, 2015, 48, 346–357 CrossRef CAS.
- F. Shamout, A. Monaco, G. Yilmaz, C. R. Becer and L. Hartmann, Macromol. Rapid Commun., 2020, 41, e1900459 CrossRef PubMed.
- G. Yilmaz, V. Uzunova, R. Napier and C. R. Becer, Biomacromolecules, 2018, 19, 3040–3047 CrossRef CAS.
- M. Bauer, C. Lautenschlaeger, K. Kempe, L. Tauhardt, U. S. Schubert and D. Fischer, Macromol. Biosci., 2012, 12, 986–998 CrossRef CAS.
- T. Lorson, M. M. Lubtow, E. Wegener, M. S. Haider, S. Borova, D. Nahm, R. Jordan, M. Sokolski-Papkov, A. V. Kabanov and R. Luxenhofer, Biomaterials, 2018, 178, 204–280 CrossRef CAS PubMed.
- S. Zalipsky, C. B. Hansen, J. M. Oaks and T. M. Allen, J. Pharm. Sci., 1996, 85, 133–137 CrossRef CAS PubMed.
- R. Hoogenboom and H. Schlaad, Polymers, 2011, 3, 467–488 CrossRef CAS.
- L. Yang, F. Wang, P. Ren, T. Zhang and Q. Zhang, Macromol. Res., 2023, 31, 413–426 CrossRef CAS.
- C. J. Waschinski and J. C. Tiller, Biomacromolecules, 2005, 6, 235–243 CrossRef CAS PubMed.
- B. Verbraeken, B. D. Monnery, K. Lava and R. Hoogenboom, Eur. Polym. J., 2017, 88, 451–469 CrossRef CAS.
- T. X. Viegas, M. D. Bentley, J. M. Harris, Z. Fang, K. Yoon, B. Dizman, R. Weimer, A. Mero, G. Pasut and F. M. Veronese, Bioconjugate Chem., 2011, 22, 976–986 CrossRef CAS PubMed.
- R. Hoogenboom, Eur. Polym. J., 2022, 179, 111521–111531 CrossRef CAS.
- R. W. Moreadith, T. X. Viegas, M. D. Bentley, J. M. Harris, Z. Fang, K. Yoon, B. Dizman, R. Weimer, B. P. Rae, X. Li, C. Rader, D. Standaert and W. Olanow, Eur. Polym. J., 2017, 88, 524–552 CrossRef CAS.
- M. Lu, Y. Y. Khine, F. Chen, C. Cao, C. J. Garvey, H. Lu and M. H. Stenzel, Biomacromolecules, 2019, 20, 273–284 CrossRef CAS PubMed.
- J. Leong, J. Y. Teo, V. K. Aakalu, Y. Y. Yang and H. Kong, Adv. Healthc. Mater., 2018, 7, 1701276–1701303 CrossRef PubMed.
- K. H. M. Motomu Kanai and L. L. Kiessling, J. Am. Chem. Soc., 1997, 119, 9931–9932 CrossRef.
- S. J. Richards and M. I. Gibson, JACS Au, 2021, 1, 2089–2099 CrossRef CAS PubMed.
- M. Ahmed and R. Narain, Biomaterials, 2012, 33, 3990–4001 CrossRef CAS PubMed.
- K. Lin and A. M. Kasko, Biomacromolecules, 2013, 14, 350–357 CrossRef CAS PubMed.
- A. V. Anja Gress and H. Schlaad, Macromolecules, 2007, 40, 7928–7933 CrossRef.
- C. D. Raposo, A. B. Canelas and M. T. Barros, Biomolecules, 2021, 11, 188–215 CrossRef CAS PubMed.
- S. G. Patching, Biochim. Biophys. Acta, 2014, 1838, 43–55 CrossRef CAS PubMed.
- R. P. Sparks, J. L. Jenkins and R. Fratti, Methods Mol. Biol., 2019, 1860, 199–210 CrossRef CAS PubMed.
- M. W. Turner, Immunobiology, 1998, 199, 327–339 CrossRef CAS PubMed.
- Y. van Kooyk and T. B. Geijtenbeek, Nat. Rev. Immunol., 2003, 3, 697–709 CrossRef CAS PubMed.
- M. van der Vlist and T. B. Geijtenbeek, Immunol. Cell Biol., 2010, 88, 410–415 CrossRef CAS PubMed.
- N. J. de Mol and M. J. E. Fischer, Surface Plasmon Resonance: A General Introduction, Springer, New York, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01135j |
| This journal is © The Royal Society of Chemistry 2024 |