
Solvent quality shape control in continuous flow block copolymer self-assembly†
Axel-Laurenz
Buckinx
a,
Lakshani J.
Weerarathna
 a,
Anna
Sokolova
b and
Tanja
Junkers
*a
a,
Anna
Sokolova
b and
Tanja
Junkers
*a
aSchool of Chemistry, Monash University, 19 Rainforest Walk, Building 23, Clayton, Vic 3800, Australia. E-mail: tanja.junkers@monash.edu
bAustralian Centre for Neutron Scattering, Australian Nuclear Science and Technology Organisation, Lucas Heights, NSW 2234, Australia
First published on 26th October 2024
Abstract
Effects of solvent quality on block copolymer (BCP) self-assembly in flow are investigated. Stable kinetically trapped nanoaggregates are created using a continuous flow technique with turbulent mixing under systematically changing THF/water ratios. To elucidate particles morphologies, small angle neutron scattering (SANS) is used as an online analytical method. At high organic solvent contents, elongated particles are observed while at low contents shorter particles are formed. The method can hence be used in a versatile way to control particle morphologies without changing the BCP block lengths used for self-assembly. This offers a more efficient and flexible approach in drug delivery and biomedical applications where particle morphologies directly influence the cell uptake capabilities and reduces the need to resynthesize BCPs with varying block lengths to control size and morphology.
Nanoaggregates formed via self-assembly of amphiphilic block copolymers have been extensively studied in the past decades.1–3 One of the main focusses of research towards these structures is to reliably and reproducibly control particle morphology. Classically the key to adjusting morphology is given by tuning the molecular weight of the hydrophobic block in relation to the hydrophilic block, where an increase in ratio results in changes from spherical, to rod-like shapes to vesicles, respectively.4–6 A plethora of studies have shown these effects for solvent based self-assembly, bulk self-assembly and polymerization-induced self-assembly.4,7,8 However, this approach presumes thermodynamic control over the formation of particles, which is not necessarily present. In fact, changing morphologies and particle sizes upon variation of the exact procedure of mixing block copolymers into the dispersed phase are a common observation, showing that kinetics have a profound impact on the process in many cases.4 Hence, another way of controlling particle morphologies is given by controlling the mixing of solutions precisely, removing the need to use a series of block copolymer compositions to access certain desired shapes. Amphiphilic polymers with polystyrene as the hydrophobic block have been shown to self-assemble into different morphologies via alteration of parameters such as BCP composition, solvent type and concentration of added ions.1,4 Another recent work2 has also shown that particle shape can be altered by preparing the nanoaggregates in continuous flow and adjusting the mixing shear forces by changing flowrates. Interestingly, in this flow study the two phases (dispersed water phase and organic phase containing the BCP) are mixed in a small volume on millisecond scale under high shear mixing forces. This results into kinetic trapping of thermodynamically unstable aggregates under highly reproducible conditions. Logically, upon faster flow velocities, BCPs have less time to rearrange, and smaller more spherical particles are observed as a result. When the flow velocity is decreased, particles tend to become larger and more ellipsoidal in shape as was shown in our previous work.2 However, it can be easily seen that other parameters besides flow velocity would influence the assembly process, and trapping of kinetically stable aggregates. In here we focus on these aspects of continuous flow micelle formation.
One parameter that immediately comes to mind to combine with our flow micelle formation process is solvent quality (ratio of organic solvent to water). When the quality of the solvent is good for the hydrophobic polymer block, the chains will have a higher degree of flexibility due to plasticisation. These effects were studied by Eisenberg and coworkers,9 when they altered the water content from 0 wt% to 45 wt% of micelle (poly(acrylic acid)-polystyrene) solutions after the self-assembly process. In this study it was concluded that adding water to the micelle mixture caused a transition from sphere to rods to vesicles with solvent quality as the determining factor. However, only the effects of solvent quality after initial self-assembly had been studied. Furthermore, Jones et al. have investigated the effect of solvent composition of solution self-assembly during PISA, where increasing the water: alcohol content limited the formation of higher order nanoparticles.10 More importantly, these effects were only studied for batch experiments, which generally suffer from reproducibility issues.
Flow self-assembly via turbulent mixing units is an interesting alternative for particle formation.2,11–13 The high shear forces in the mixing process in flow combined with the varying degrees of freedom of the polymer chains as a consequence of solvent quality will affect the final aggregates. This would be unachievable using the classical batch method. Further, committing to continuous flow allows to reproduce particle formations with high precision, and allows at the same time for scaling of particle formation.14 The advantages of flow mixing should drastically enhance solvent quality effects and offer more stream-lined approach to study kinetically controlled self-assembly. Also, this approach allows to keep the water content comparatively high, which is important if particles are aimed for biomedical applications.11,15 Another benefit of performing the self-assembly in flow is the ability to use online monitoring for fast data screening.
Online monitoring and continuous flow processes are ideal partners, paving the way to autonomous synthesis platforms.16 The immediate feedback of online monitoring combined with the flexibility and adaptability of flow processes allow for comprehensive optimization of reactions. Techniques such as mass spectrometry, infrared spectroscopy and 1H NMR have emerged as commonplace online monitoring techniques.16–18 However, online monitoring techniques are still scarce for nanoparticles, due to the difficulties of investigating effects on soft matter systems with commonly available analysis techniques. Parkinson et al. have used 1H NMR as an online monitoring tool in continuous flow nanoparticle synthesis via dispersion polymerization.19 In addition, Dynamic Light Scattering (DLS), in principle applicable as online monitor, bases its evaluation on the assumption of particles being perfect spheres, which renders morphology studies impossible.20 Conventional (dry-state) electron microscopy on the other hand, whilst being a powerful technique for analysis of the nano structures shows only a small area of a dried and stained sample in 2D. This inevitably alters the state of the soft matter materials especially for aggregates that are not in thermodynamic equilibrium. Moreover, the sample preparation procedures make online monitoring impossible.21 Small angle neutron scattering (SANS) and small angle X-ray scattering (SAXS) are other complementary techniques that could be used in the characterization of nanoparticles. SAXS measurements use high flux X-ray beams and allow fast detections.22 Guild et al. have used laboratory small angle X-ray scattering (SAXS) as an in situ online monitoring tool to observe changes in size and shape of nanoparticles in flow-PISA reactions.23 Yet, considering the contrast variations and particle size range that could be studied in the concentration regimes we are working on, a more suitable system to perform these investigations is Small Angle Neutron Scattering (SANS). SANS allows to study a sufficient volume (∼0.4–0.8 ml) of a sample in its hydrated state providing information about morphology and size parameters.24 In this study we present one of the first studies using SANS as on online monitoring tool for micelle self-assembly using a continuous flow system to produce the nanoaggregates. A series of BCPs with different block ratios are self-assembled in flow under systematically changing solvent qualities (see Fig. 1). In these experiments, the flow velocity was kept constant to not to change the applied shear forces excluding their potential influence on the shape of the resulting aggregate. BCPs were synthesised using RAFT (reversible addition–fragmentation chain transfer) polymerization. For such study, ideally a BCP with a hydrophobic block with a high glass transition temperature (Tg of PS is 107 °C)25 is required since upon reaching the critical water content, a kinetically frozen core will be formed. Therefore a perfect candidate for these investigations is poly(2-hydroxy ethyl acrylate)-b-poly(styrene) (PHEA-b-PS) for its known behaviour in the previous flow self-assembly studies and the high Tg of styrene.2,9,24 In order to test a variety of polymers, four macroRAFT polymers were synthesised and chain extended with styrene to obtain different block ratios. Details for the polymerisations can be found in the SI. The SEC traces of the polymers used are shown in Fig. 2. Well controlled polymers were obtained as expected: block lengths and molecular weights were determined via1H NMR since SEC is prone to erroneous results when analysing BCPs (see SI for details).
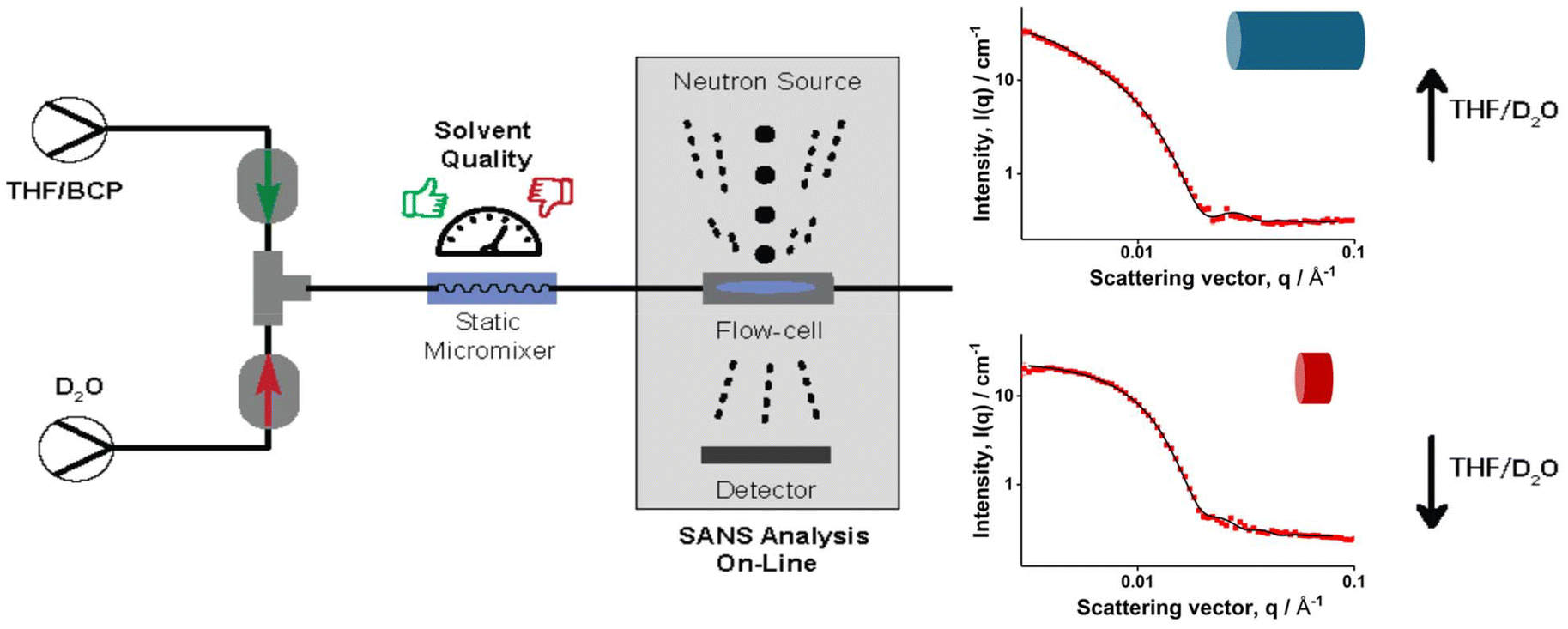 | ||
| Fig. 1 Schematic representation of online SANS measurements to study the effects of solvent quality with self-assembly in flow. Organic and aqueous phases are mixed in a static micromixer and subsequently flown into a flow cell where SANS analysis takes place. | ||
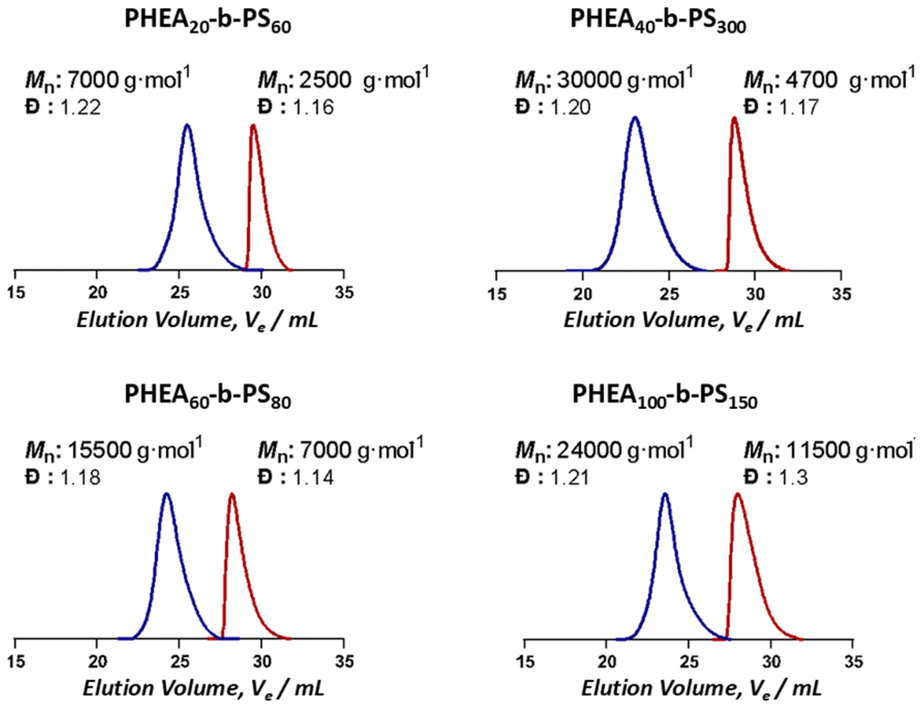 | ||
| Fig. 2 Elution volume of synthesised BCPs. Blue chromatograms represent the BCPs while red ones depict the homopolymers. Molecular weights as given were calculated theoretically from 1H NMR conversion since SEC calibration for amphiphilic BCPs is inherently difficult. | ||
With these polymers, self-assembly was performed and then studied using SANS on the Bilby instrument26,27 at the Australian Centre for Neutron Scattering (Australian Nuclear Science and Technology Organization). To facilitate this process, Bilby was setup as on online monitoring tool as depicted in Fig. 1. This allowed to measure effects of mixing immediately without requirement of offline sampling. At the same time, it also enabled studying how the particles would change over time right after the formation process was stopped. Two Azura Knauer HPLC pumps were used to feed the dispersed phase and D2O. For the polymer solution, 5 mg mL−1 of the BCP was dissolved in THF and fed into the mixer, a U-466 static mixer from Upchurch Scientific. The micromixer was connected to a flow cell containing 1 mm quartz flow cell with a spacer with a thickness of 0.95mm. The pumps were controlled via a custom software to control flowrates from outside of the sample environment. This facilitates the process of changing flowrates of the pumps without the need to approach the sample environment which in turn allows for a significant increase of data acquisition over time. In total, four THF/D2O ratios were tested for the self-assembly process, these being: 3.2/0.8, 2.5/1.5, 2/2 and 1.5/2.5. With these ratios, the solvent ratios were altered without changing the overall flow velocity, which is already known to affect the self-assembly process. It should also be noted that due to the changing of the THF content, the polymer concentration inevitably changes as well. Most importantly though, the changes upon changing BCP concentration are expected to be minimal and should not drastically affect the self-assembly process itself.4
SANS data for the self-assembled BCPs is shown in Fig. 3. The scattering data was modelled by the SasView software with the sphere, cylinder and ellipsoid models presented by Guinier and Fournet.28Table 1 gives a summary of the geometric parameters for the nanoaggregates as determined by SANS data fitting. More information on the fitting parameters used for SANS data in the determination of these final particle sizes are given in ESI.† In addition, the equations for each model that were used to fit the SANS data at different solvent ratios, pair-distance distribution functions and SLD calculations are also provided in the ESI.† Assumption of monodisperse aggregates led to reasonable fits. As seen in Fig. 3 all samples show a similar trend upon changing solvent ratio. The cartoons placed next to the scattering profiles are the nanoaggregate shapes and sizes fitted to the data.
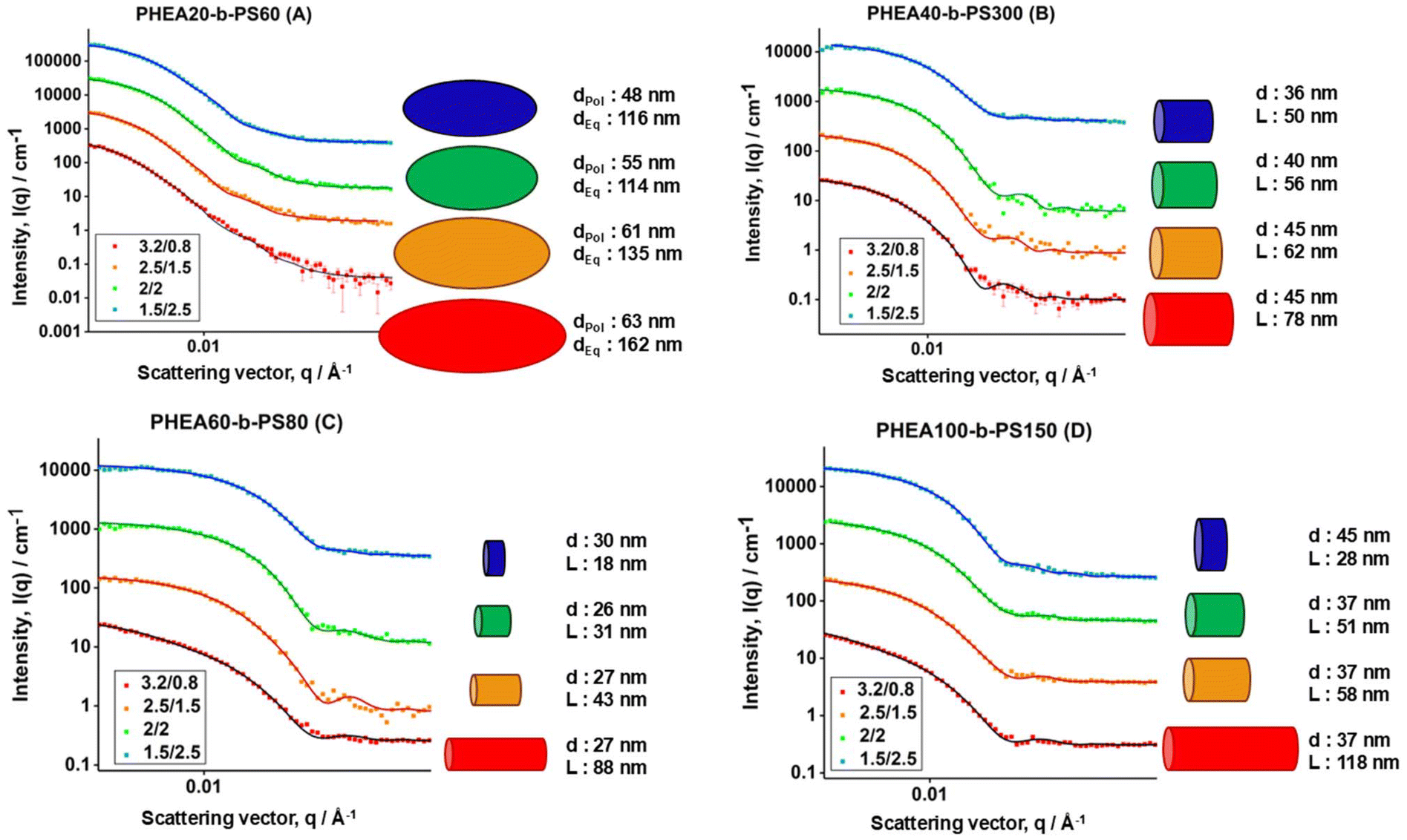 | ||
| Fig. 3 SANS data of BCP nanoaggregates made in flow using different solvent ratios at similar flow velocities with background subtracted. Ratios are depicted in THF/D2O form, blue 1.5/2.5, green 2/2, yellow 2.5/1.5 and red 3.2/0.8. Error bars are added on the plots. Plots are spaced on y axis by 1×, 10×, 100×, 1000× for visual clarity. Schematic representations of the particles with accurate relative dimensions are shown for each fit for easier interpretation of the data (dPol = polar diameter, dEq = equatorial diameter, d = diameter, L = length). A–D denotes different individual block copolymers as indicated in the figure. | ||
| Block copolymer | THF/D2O ratio | Shape | Radius (polar)/nm | Radius (equatorial)/nm |
|---|---|---|---|---|
| PHEA20-b-PS60 (A) | 3.2/0.8 | Ellipsoid | 31.7 ± 0.1 | 81.1 ± 0.5 |
| 2.5/1.5 | Ellipsoid | 30.7 ± 0.1 | 67.5 ± 0.3 | |
| 2/2 | Ellipsoid | 27.6 ± 0.1 | 56.8 ± 0.2 | |
| 1.5/2.5 | Ellipsoid | 24.1 ± 0.1 | 57.8 ± 0.2 | |
| Radius (nm) | Length (nm) | |||
| PHEA40-b-PS300 (B) | 3.2/0.8 | Cylinder | 22.3 ± 0.15 | 77.8 ± 2 |
| 2.5/1.5 | Cylinder | 22.3 ± 0.2 | 62.1 ± 1.5 | |
| 2/2 | Cylinder | 19.8 ± 0.15 | 56.1 ± 1.2 | |
| 1.5/2.5 | Cylinder | 18.2 ± 0.15 | 50.3 ± 1.1 | |
| PHEA60-b-PS80 (C) | 3.2/0.8 | Cylinder | 13.5 ± 0.1 | 87.8 ± 3 |
| 2.5/1.5 | Cylinder | 13.4 ± 0.1 | 43.3 ± 0.7 | |
| 2/2 | Cylinder | 13.1 ± 0.1 | 31.3 ± 0.6 | |
| 1.5/2.5 | Cylinder | 15.2 ± 0.1 | 18.2 ± 0.3 | |
| PHEA100-b-PS150 (D) | 3.2/0.8 | Cylinder | 18.5 ± 0.1 | 118 ± 6 |
| 2.5/1.5 | Cylinder | 18.7 ± 0.1 | 58 ± 0.8 | |
| 2/2 | Cylinder | 18.5 ± 0.1 | 51 ± 0.6 | |
| 1.5/2.5 | Cylinder | 22.5 ± 0.14 | 27.8 ± 0.3 |
At higher THF contents, particles tend to be more elongated while at lower contents the particle takes a shorter, closer to spherical shape. For polymer A (PHEA20-b-PS60), all particles keep an ellipsoidal morphology which is in agreement with studies on a similar polymer system made in this way.2 At the highest THF content, the ellipsoid has an equatorial radius of 80 nm and a polar radius of about 30 nm. The polar radius as well as equatorial radius gradually show a decrease upon a decrease of organic solvent during the self-assembly process, which is likely the consequence of the limited freedom the styrene chains have during the self-assembly process with reduced solvent quality. Polymer B (PHEA40-b-PS300) is even more interesting. Here, at high THF contents cylindrical particles are formed with a length of 78 nm and radii of 22 nm. Upon increase of the water content, the length as well as radii decrease making the cylinders shorter. At the lowest THF content, the SANS data could be fitted with a sphere too, yet the better fit was shown with a very short cylinder.
Polymers C (PHEA60-b-PS80) and D (PHEA100-b-PS150) show similar trends, where at high organic solvent content during mixing, elongated cylindrical shapes are clearly observed and on the increase of water content, the radii do not vastly change, but the length of the cylinders decreases by more than half. At the lowest THF content, they become very short cylinders, which could also be fitted as spheres in SasView, yet cylinder model showing better fit in both the instances. It is important to note that despite the drastic difference in styrene chain length all these systems are self-assembled in similar morphologies. This indicates that moving the process from batch to flow might, to a certain extent, bypass the commonly accepted rules of making different morphology polymers by extending their hydrophobic block.
Clearly, kinetic effects are dominant over thermodynamic effects in the investigated system. There, kinetic trapping of the morphologies due to the flow self-assembly using micromixer plays a major role.11,13 Considering the different THF/water solvent ratios used in self-assembly, at higher THF content environments, the solvent quality for both the hydrophobic PS and hydrophilic PHEA blocks of the polymer is higher and this results in PS chains in the core getting more stretched, swollen and flexible. When there is a higher degree of stretching in the core forming block, they can relax by changing to morphologies such as cylinders, minimizing free energy.9,29 This could be a major cause for the nanoaggregates synthesized at higher THF content to be larger in size and to be self-assembled into cylinders instead of spheres compared to those at lower THF content.30,31
When there is a higher water content in the environment (lower THF ratio), the solvent quality for core polystyrene inherently gets poorer since water is a non-solvent for PS and the plasticizing effect of chains decrease. With the slow chain mobility and lesser swollen and stretched core, the nanoaggregates get frozen in the achieved morphology, with smaller dimensions than those at higher THF content.4,29,30 Similar trends, but different morphologies have been observed by Wong et al. in the studies done with PEO44-b-PS86 BCP.30
Some thermodynamic control is, however, still observed from effects of the polystyrene chain length on nanoaggregation. Polymer C shows thinner and shorter cylinders when compared to polymers B and D due to the shorter polystyrene chains present in this system, which results in a less bulky interior in the particle. In addition, polymer A never formed cylinders but remained ellipsoidal which is happening likely due to its smaller overall size. Similar trends with changing hydrophobic length of BCPs could be identified in previous studies.32
Moreover, it is also apparent that an increase in hydrophilic chain length influences the morphology via inducing stronger interchain repulsion in the corona.4 This is confirmed via the formation of very short cylinders which are closer to spherical particles at a lower THF content in samples B, C and D compared to A. These approximately closer spherical particles are formed by B, C and D which are polymers with comparably longer PHEA chains, at THF/water content of 1.5/2.5, and it is not observed for polymer A with a smaller hydrophilic chain at the same THF/water ratio. Most likely here, at longer hydrophilic chain lengths, the polar groups on the corona are so strong, inter-coronal chain repulsions get more dominant than the stretching of core block,29 hence, the polystyrene chains lose the ability to define particle morphology. Therefore, they are trying to achieve a morphology which is closer to thermodynamically favoured spherical shape at 1.5/2.5 ratio. Considering all these effects, and the enhanced freedom of the polymer chains during the formation of particles due to the high Tg of styrene, the BCPs are kinetically trapped in these states upon leaving the static mixing unit, which results in the observed structures.9
Scanning Transmission Electron Microscopy (STEM) was performed to verify if the particles would remain stable after being dried and stained on a grid (see ESI†). STEM was chosen over conventional Transmission Electron Microscopy (TEM) to obtain higher spatial resolution at relatively less harsh imaging conditions. Moreover, with the use of low acceleration voltages compared to TEM, knock-on damage could be reduced, leading to reduced sample damage. Interestingly, particles on STEM all appear spherical and much larger (up to factor 2) than observed via SANS. While this might seem odd at first, it is likely that further bulk self-assembly occurs upon drying of the samples which brings the micelles closer to their thermodynamically favoured state. Micelles were made in the presence of a large amount of organic solvent, hence when placed on a grid to dry it is likely their polystyrene chains rearrange during the drying process forming large spherical structures. A test was done trying to fit SANS data with parameters obtained from STEM data. No match of SANS data was found, allowing for the conclusion that STEM samples preparation procedures are altering the shape of particles. There are studies in literature discussing similar scenarios due to the drying and staining procedures in imaging techniques resulting in clustering, array formation and collapsing of structures.33,34 Nonetheless, there are advanced techniques that could be utilized to reduce these events, such as freeze-fracture electron microscopy and plunge freezing in cryo-TEM.33 Further, sophisticated detector calibration-less, quantitative 4D STEM techniques can be used in imaging for more accurate results.35 Yet, in studying the real-time morphology of a nanoparticle sample in solution, still they can be tedious techniques which need more careful sample preparation. Hence, a technique such as SANS still stands out as a better candidate in real-time online monitoring of these particle synthesis processes.
Using SANS as an online detector, in principle it is possible that the data given in Fig. 3 only depicts a snapshot right after particle formation, and that a change in morphology might occur with time, relaxing the kinetically trapped state into the thermodynamically stable aggregate. Following particle evolution over time in online SANS allowed to study the stability of the nanoaggregates by acquiring data over a period of 10 min directly after the self-assembly process in stop flow experiments. Comparison of the scattering on 100sec intervals in the very beginning and the end of these 10 minutes exhibit no difference from each other, meaning the particles were relatively stable and trapped in their kinetically frozen state further validating the usefulness of the flow approach (data slices are shown in the ESI in Fig. S4†). Further investigation will be needed on the effects of post-processing (such as dialysis) and the formed nanoparticles. These effects however should remain minimal due to the high Tg of polystyrene, and in a previous study we had seen that dialysis, when carried out in flow, has only miniscule effects on the particle size.14 It can also be assumed that particles remain in their stable state for extended times, not only for the timeframe covered in here. Since flow allows for on-demand synthesis of micelles, and also allows for direct introduction of payloads, a 20 min timeframe is fully sufficient to exploit, for example, biomedical properties of the aggregates.2
In summary, the effect of the solvent ratio during the continuous flow mixing on the self-assembly of BCPs was investigated using SANS as an online detector. Altering the amount of organic solvent drastically influences on the particle morphology and size of residual nanoaggregates at a much lower final organic solvent content than used previously. Generally, at higher organic solvent ratio the particles were significantly elongated due to the increased freedom of the polymer chain during the self-assembly process, allowing for comprehensive shape control of micelles independently from their respective polymer block ratio. However, particles were shorter when water content was higher. This result offers new pathways to control particle shape outside of the currently established paradigm of purely thermodynamically motivated BCP nanoparticle design principles.
Author contributions
ALB: investigation, methodology, formal analysis, data curation, visualization, writing – original draft; LJW: writing – revision, data curation, visualization; AS: supervision, methodology, investigation, formal analysis, data curation; TJ: conceptualization, supervision, methodology, funding acquisition, writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.† Primary data can be obtained from DOI: 10.26180/27300630.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for funding from the ARC in form of a discovery grant (DP240100120). They further acknowledge funding from the Australian Science and Technology Organization (proposal 16692 and 9382).References
- L. M. Bergström, Thermodynamics of Self-Assembly, Appl. Thermodyn. Biol. Mater. Sci., 2011, 11, 289–314 Search PubMed.
- A. Buckinx, K. Verstraete, E. Baeten, R. F. Tabor, A. Sokolova, N. Zaquen and T. Junkers, Kinetic Control of Aggregation Shape in Micellar Self–Assembly, Angew. Chem., Int. Ed., 2019, 58(39), 13799–13802 CrossRef CAS PubMed.
- A. Jahn, W. N. Vreeland, M. Gaitan and L. E. Locascio, Controlled Vesicle Self-Assembly in Microfluidic Channels with Hydrodynamic Focusing, J. Am. Chem. Soc., 2004, 126(9), 2674–2675 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Self-Assembly of Block Copolymers, Chem. Soc. Rev., 2012, 41(18), 5969–5985 RSC.
- O. Terreau, L. Luo and A. Eisenberg, Effect of Poly (Acrylic Acid) Block Length Distribution on Polystyrene-b-Poly (Acrylic Acid) Aggregates in Solution. 1. Vesicles, Langmuir, 2003, 19(14), 5601–5607 CrossRef CAS.
- T. Chang, M. S. Lord, B. Bergmann, A. Macmillan and M. H. Stenzel, Size Effects of Self-Assembled Block Copolymer Spherical Micelles and Vesicles on Cellular Uptake in Human Colon Carcinoma Cells, J. Mater. Chem. B, 2014, 2(19), 2883–2891 RSC.
- F. S. Bates and G. H. Fredrickson, Block Copolymers—Designer Soft Materials, Phys. Today, 1999, 52(2), 32–38 CrossRef CAS.
- C. György and S. P. Armes, Recent Advances in Polymerization–Induced Self–Assembly (PISA) Syntheses in Non–Polar Media, Angew. Chem., Int. Ed., 2023, 62(42), e202308372 CrossRef PubMed.
- H. Shen and A. Eisenberg, Morphological Phase Diagram for a Ternary System of Block Copolymer PS310-b-PAA52/Dioxane/H2O, J. Phys. Chem. B, 1999, 103(44), 9473–9487 CrossRef CAS.
- E. R. Jones, M. Semsarilar, P. Wyman, M. Boerakker and S. P. Armes, Addition of Water to an Alcoholic RAFT PISA Formulation Leads to Faster Kinetics but Limits the Evolution of Copolymer Morphology, Polym. Chem., 2016, 7(4), 851–859 RSC.
- R. Thiermann, R. Bleul and M. Maskos, Kinetic Control of Block Copolymer Self–Assembly in a Micromixing Device–Mechanistical Insight into Vesicle Formation Process, Macromol. Chem. Phys., 2017, 218(2), 1600347 CrossRef.
- R. Karnik, F. Gu, P. Basto, C. Cannizzaro, L. Dean, W. Kyei-Manu, R. Langer and O. C. Farokhzad, Microfluidic Platform for Controlled Synthesis of Polymeric Nanoparticles, Nano Lett., 2008, 8(9), 2906–2912 CrossRef CAS PubMed.
- R. Thiermann, W. Mueller, A. Montesinos-Castellanos, D. Metzke, P. Löb, V. Hessel and M. Maskos, Size Controlled Polymersomes by Continuous Self-Assembly in Micromixers, Polymer, 2012, 53(11), 2205–2210 CrossRef CAS.
- K. Verstraete, A.-L. Buckinx, N. Zaquen and T. Junkers, Micelle Purification in Continuous Flow via Inline Dialysis, Macromolecules, 2021, 54(8), 3865–3872 CrossRef CAS.
- R. Bleul, R. Thiermann and M. Maskos, Techniques to Control Polymersome Size, Macromolecules, 2015, 48(20), 7396–7409 CrossRef CAS.
- J. J. Haven and T. Junkers, Online Monitoring of Polymerizations: Current Status, Eur. J. Org. Chem., 2017, 2017(44), 6474–6482, DOI:10.1002/ejoc.201700851.
- M. Rubens, J. Van Herck and T. Junkers, Automated Polymer Synthesis Platform for Integrated Conversion Targeting Based on Inline Benchtop NMR, ACS Macro Lett., 2019, 8(11), 1437–1441, DOI:10.1021/acsmacrolett.9b00767.
- B. Zhang, A. Mathoor and T. Junkers, High Throughput Multidimensional Kinetic Screening in Continuous Flow Reactors, Angew. Chem., Int. Ed., 2023, 62(38), e202308838 CrossRef CAS PubMed.
- S. Parkinson, S. T. Knox, R. A. Bourne and N. J. Warren, Rapid Production of Block Copolymer Nano-Objects via Continuous-Flow Ultrafast RAFT Dispersion Polymerisation, Polym. Chem., 2020, 11(20), 3465–3474 RSC.
- B. J. Ree, J. Lee, Y. Satoh, K. Kwon, T. Isono, T. Satoh and M. Ree, A Comparative Study of Dynamic Light and X-Ray Scatterings on Micelles of Topological Polymer Amphiphiles, Polymers, 2018, 10(12), 1347 CrossRef PubMed.
- L. E. Franken, E. J. Boekema and M. C. A. Stuart, Transmission Electron Microscopy as a Tool for the Characterization of Soft Materials: Application and Interpretation, Adv. Sci., 2017, 4(5), 1600476 CrossRef PubMed.
- D. Lombardo, P. Calandra and M. A. Kiselev, Structural Characterization of Biomaterials by Means of Small Angle X-Rays and Neutron Scattering (SAXS and SANS), and Light Scattering Experiments, Molecules, 2020, 25(23), 5624 CrossRef CAS PubMed.
- J. D. Guild, S. T. Knox, S. B. Burholt, E. M. Hilton, N. J. Terrill, S. L. M. Schroeder and N. J. Warren, Continuous-Flow Laboratory SAXS for In Situ Determination of the Impact of Hydrophilic Block Length on Spherical Nano-Object Formation during Polymerization-Induced Self-Assembly, Macromolecules, 2023, 56(16), 6426–6435 CrossRef CAS PubMed.
- R. Lund, L. Willner and D. Richter, Kinetics of Block Copolymer Micelles Studied by Small-Angle Scattering Methods, Control. Polym. Polym. Struct. flow microreactor Polym. micelles Kinet. Polypept. ordering, Light Emit. Nanostructures, 2013, 51–158 CAS.
- J. Rieger, The Glass Transition Temperature of Polystyrene: Results of a Round Robin Test, J. Therm. Anal. Calorim., 1996, 46(3–4), 965–972 CrossRef CAS.
- A. Sokolova, J. Christoforidis, A. Eltobaji, J. Barnes, F. Darmann, A. E. Whitten and L. de Campo, BILBY: Time-of-Flight Small Angle Scattering Instrument, Neutron News, 2016, 27(2), 9–13 CrossRef.
- A. Sokolova, A. E. Whitten, L. de Campo, J. Christoforidis, A. Eltobaji, J. Barnes, F. Darmann and A. Berry, Performance and Characteristics of the BILBY Time-of-Flight Small-Angle Neutron Scattering Instrument, J. Appl. Crystallogr., 2019, 52(1), 1–12 CrossRef CAS.
- A. Guinier, G. Fournet and K. L. Yudowitch, Small-Angle Scattering of X-Rays, 1955 Search PubMed.
- L. Zhang and A. Eisenberg, Formation of Crew–cut Aggregates of Various Morphologies from Amphiphilic Block Copolymers in Solution, Polym. Adv. Technol., 1998, 9(10–11), 677–699 CrossRef CAS.
- C. K. Wong, R. Y. Lai and M. H. Stenzel, Dynamic Metastable Polymersomes Enable Continuous Flow Manufacturing, Nat. Commun., 2023, 14(1), 6237 CrossRef CAS PubMed.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, Theory of Self-Assembly of Hydrocarbon Amphiphiles into Micelles and Bilayers, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- A. Takashima, Y. Maeda and S. Sugihara, Morphology Control via RAFT Emulsion Polymerization-Induced Self-Assembly: Systematic Investigation of Core-Forming Blocks, ACS Omega, 2022, 7(30), 26894–26904 CrossRef CAS PubMed.
- H. Friedrich, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Imaging of Self–assembled Structures: Interpretation of TEM and Cryo–TEM Images, Angew. Chem., Int. Ed., 2010, 49(43), 7850–7858 CrossRef CAS PubMed.
- Y. Zhang, T. Huang, D. M. Jorgens, A. Nickerson, L.-J. Lin, J. Pelz, J. W. Gray, C. S. López and X. Nan, Quantitating Morphological Changes in Biological Samples during Scanning Electron Microscopy Sample Preparation with Correlative Super-Resolution Microscopy, PLoS One, 2017, 12(5), e0176839 CrossRef PubMed.
- R. Skoupy and V. Krzyzanek, Calibration-Less Quantitative 4D-STEM Imaging of Amorphous Samples, Microsc. Microanal., 2021, 27(S1), 1602–1603 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, characterization of block copolymers and (S)TEM images of nanoaggregates. See DOI: https://doi.org/10.1039/d4py01174k |
| This journal is © The Royal Society of Chemistry 2024 |