
Divergent methods for polyester and polycarbonate depolymerization with a cobalt catalyst†
Kai D.
Knight
 a and
Megan E.
Fieser
*ab
a and
Megan E.
Fieser
*ab
aDepartment of Chemistry, University of Southern California, Los Angeles, CA 90089, USA. E-mail: fieser@usc.edu
bWrigley Institute for Environment and Sustainability, University of Southern California, Los Angeles, CA 90089, USA
First published on 27th November 2023
Abstract
A pyridine diimine cobalt catalyst series is used for the cyclodepolymerization (CDP) of several different polyesters and polycarbonates. In the presence of isopropanol, a wide range of polymers can undergo solvolytic depolymerization. CDP of poly(propylene carbonate) revealed an unzip back-biting or random scission mechanism, depending on what conditions were used. The first example of a method capable of solvolysis and CDP on a polymer mixture in the same pot is identified.
Introduction
Polyesters and polycarbonates are important materials for a wide range of applications, such as CDs, water bottles, and packaging.1 Additionally, emerging methods to make these materials with a range of physical properties has established a growing movement to use these polymers as degradable, recyclable or compostable replacements for non-degradable polymers made on alarming scales.2 As technology improves to increase industrial use of these materials, it is critical to establish options for their end of life.Many polyesters and polycarbonates can be made from bio-feedstocks.3 Polyesters are often considered degradable or compostable. However, these strategies often need industrial composting, which requires an additional collection stream.3,4 While these composting methods allow for polymer degradation to CO2 to occur, the value of the chemical is not retained quickly, as a chemical that can be readily converted back to polymer is not produced. This requires vast biofeedstock sources to be grown annually.5,6 Alternatively, even if considered biodegradable, polycarbonates are often resistant to degradation, highlighting the need for different strategies to manage their end of life.7,8
These materials can also be chemically recycled, adding another strategy to recover valuable chemicals that can be used to remake the polymer directly.3,9–11 For polyesters and polycarbonates, there are four primary methods for chemical recycling: hydrogenation, transfer hydrogenation, solvolysis and cyclodepolymerization (Fig. 1 and 2).3,9,12 Each of these methods have their advantages and challenges, and some methods are better for specific polymers.
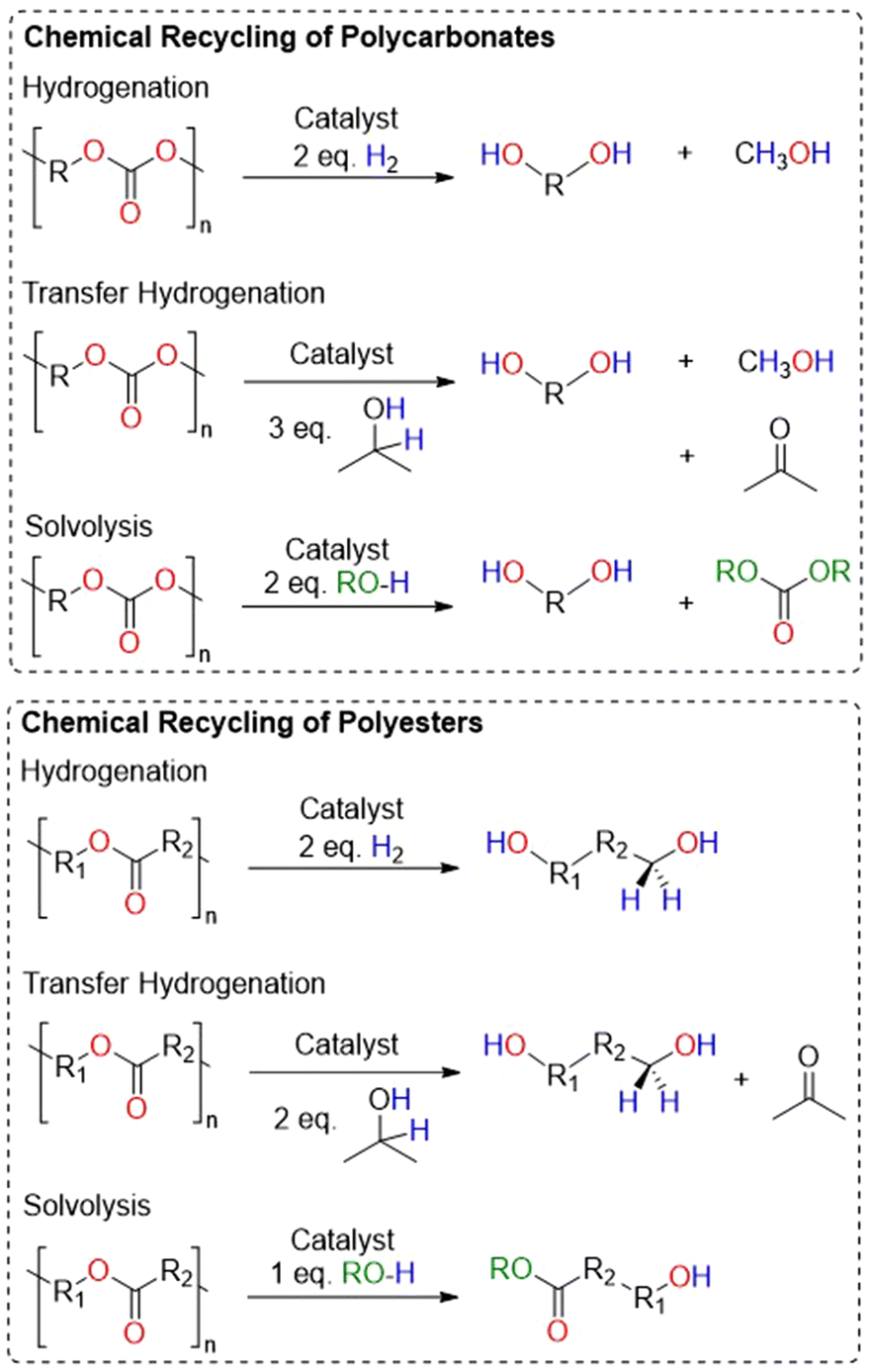 | ||
| Fig. 1 Representative methods for depolymerization of polycarbonates and polyesters. | ||
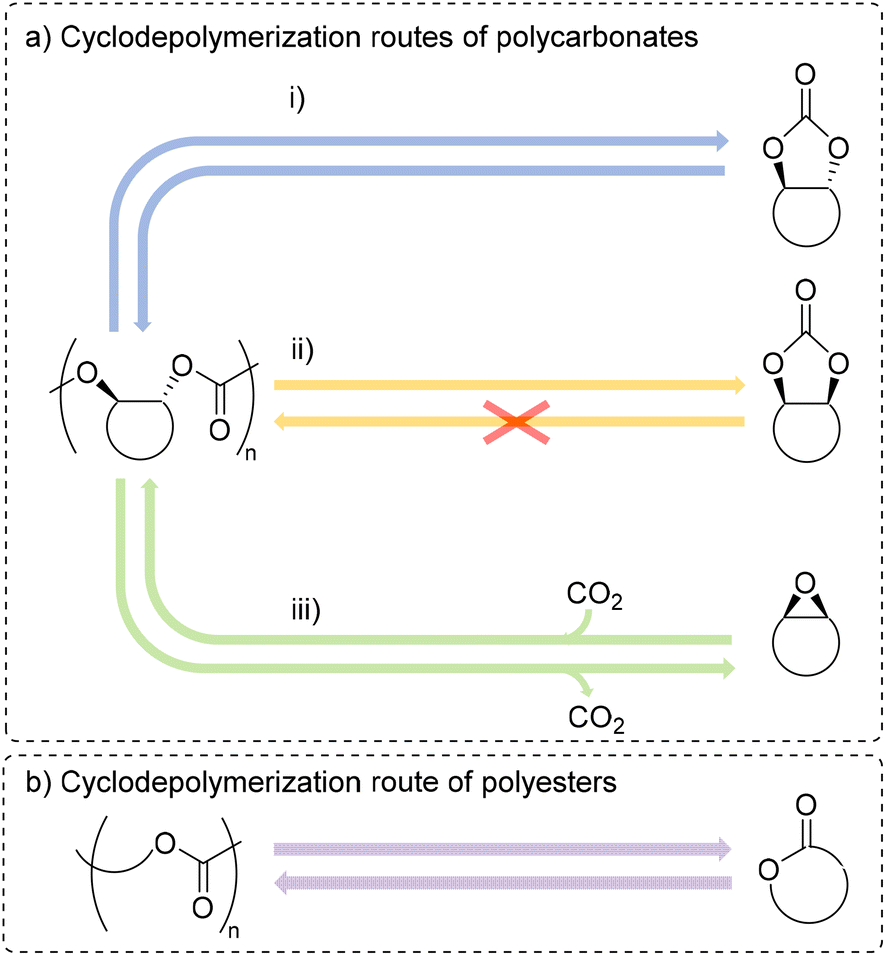 | ||
| Fig. 2 Possible cyclodepolymerization pathways for polycarbonates (a) to trans-carbonate (i), cis-carbonate (ii), and epoxide and carbon dioxide (iii) and for polyesters (b). | ||
Hydrogenation often breaks polyesters and polycarbonates down into diols, which are useful for numerous applications, including synthesizing new polymers.12–14 However, high pressures and temperatures are often used, posing safety and energy challenges. A safer alternative, albeit less atom economical, is transfer hydrogenation, which uses a hydrogen source like isopropanol to generate an equivalent of H2 and acetone as a by-product. Though apart from numerous examples of the transfer hydrogenation of lignin, few reports exist documenting the transfer hydrogenation of polymers.15–17
Solvolysis remains one of the most studied methods for the depolymerization of step-growth polyesters, particularly for poly(ethylene terephthalate) (PET).11,18 The use of water, methanol or ethylene glycol can lead to linear molecules that can be used again for polymerization.19,20 Hydrolysis is relevant to many polyesters and polycarbonates, however these materials are often insoluble in water and need harsh conditions to reach efficient depolymerization.21 Methanolysis and glycolysis may be more useful depolymerization methods, as is the case with PET, in which the product may regenerate new PET.22–24 Finally, the cyclodepolymerization (CDP) of polyesters and polycarbonates back to their cyclic precursors is an emerging area of research, as this method often applies to chain growth polymers, which are receiving much interest as replacements for non-degradable polymers. In many cases, the ceiling temperature of the polymer makes a big difference on how cyclodepolymerization can occur.25–35 Additionally, high dilution of the reaction often allows for higher conversion to cyclic monomer, albeit with the crucial flaw of requiring large amounts of solvent.36,37 Recently, reports of manipulating ceiling temperature combined with reactive distillation methods have prevented the need for high dilution.38–40 Namely, Byers and co-workers reported exciting catalysts for the CDP of many polyesters and polycarbonates at high yields using a reactive distillation method.40
CDP of polycarbonates can take two forms: either formation of cyclic carbonates or complete depolymerization to epoxides and CO2 (Fig. 2a).9 There are few examples of CDP of commercial polycarbonates and polyesters to their respective cyclic monomers.41–44 In this case, CDP of the polycarbonate often leads to a cis-cyclic carbonate, a product which lacks examples of being polymerized. Many of these examples have shown the ability to perform CDP on specially designed polymers, which have been modified or synthesized with the intention that these polymers also cyclodepolymerize to polymerizable monomers.26,28,29,31,33,34,38,45–49 Similarly, scant examples of complete CDP to epoxides and CO2 exist in the literature.50–53 To date, there is still not a clear understanding for what catalysts lead to selectivity of either form of CDP, although computational studies have helped rationalize a catalyst's ability to lower the barriers for a specific method.49 CDP of polyesters has one main pathway to generate cyclic esters (Fig. 2b). Examples of CDP for common polyesters, such as poly(ε-caprolactone) (PCL), poly(δ-valerolactone) (PVL), polydioxanone (PDO), poly(caprolactam) (nylon-6), and poly(lactic acid) (PLA) are known with metal catalysts.35,40,44,45,54
Herein, we demonstrate the use of a pyridine diimine cobalt catalyst that can catalyse the cyclodepolymerization of several different polyesters and polycarbonates. In the presence of isopropanol, these catalytic methods also facilitate solvolytic depolymerization of seven different polyesters and polycarbonates, which is attributed to the base used in the reaction. To our knowledge, this is the first example of a single catalyst system demonstrating these two depolymerization strategies. We demonstrate proof of concept for achieving both methods in the same pot, which addresses considerations of mixed polymer waste.
Results and discussion
Cobalt complexes with pyridine diimine (PDI) ligands were first targeted for the depolymerization of carbonyl-containing polymers, as these complexes are easy to make and similar complexes with chiral ligand analogues have been used for stereoselective transfer hydrogenation of small molecules.55,56 Additionally, PDI cobalt complexes have been synthesized and modified regularly for application in electrocatalysis, providing a toolbox of electronic and steric modifications for these molecules.57,58 Initially, catalysts with simple substituents were targeted, with subtle modifications in steric crowding to probe the sensitivity of these catalysts for the depolymerization of polymers (Fig. 3). The PDI ligands were varied to contain mesityl (L-MES), di-isopropyl phenyl (L-DIPP), and di-ethyl phenyl (L-DEP) substituents. The catalysts MES and DIPP were synthesized according to literature procedure while DEP was synthesized by and methods and was fully characterized (see ESI†).59,60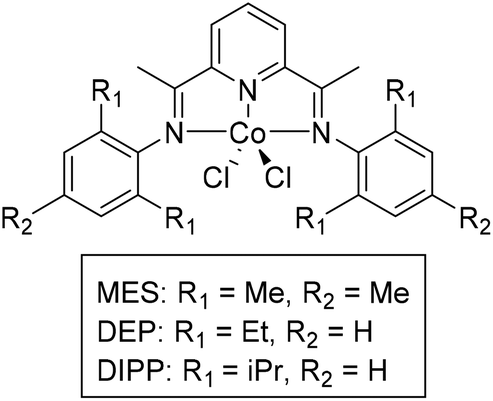 | ||
| Fig. 3 Catalysts used in depolymerization of PPC. | ||
Using transfer hydrogenation conditions of poly(propylene carbonate) (PPC), inspired by the recent work from Werner and coworkers, these cobalt complexes did not show any evidence for the formation of the expected transfer hydrogenation products: propane diol and methanol.15 Instead, exclusive formation of propylene carbonate (PC) was observed for all three catalysts, with reactions yielding PC, cyclodepolymerized from PPC, within 20 h at 140 °C (Table 1, entries 1–3).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Time (h) | KOtBu (mol%) | Solvent(s) used | Solvent volume (mL) | % Yieldb,c |
| a Conditions: 5 mol% catalyst with respect to the repeat unit molar mass of the polymer, 2 mmol PPC, run at 140 °C and 350 rpm in Parr reactor. Reactions with KOtBu were performed in an air-free, N2 environment. All other entries were performed exposed to air. b Determined by comparing 1H NMR spectroscopy using mesitylene as internal standard, taken in CDCl3. c Parentheses next to yields indicate reactions performed in duplicate; standard deviation of error in parentheses. d NMR calculation used in place of internal standard yield calculation due to neat method's challenges in collecting full product. | ||||||
| 1 | MES | 20 | 5 | iPrOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF :THF |
8:2 |
66 |
| 2 | DIPP | 20 | 5 | iPrOH:THF |
8:2 |
66 |
| 3 | DEP | 20 | 5 | iPrOH:THF |
8:2 |
64 |
| 4 | MES | 20 | 5 | THF | 6 | >99(0) |
| 5 | DIPP | 20 | 5 | THF | 6 | 78(19) |
| 6 | DEP | 20 | 5 | THF | 6 | 97(3) |
| 7 | MES | 20 | 0 | THF | 6 | 81(3) |
| 8 | DIPP | 20 | 0 | THF | 6 | 55(45) |
| 9 | DEP | 20 | 0 | THF | 6 | 99(2) |
| 10 | DEP | 10 | 5 | THF | 6 | 83(3) |
| 11 | DEP | 10 | 0 | THF | 6 | 69(4) |
| 12 | DEP | 10 | 10 | THF | 6 | 89(3) |
| 13 | DEP | 6 | 5 | THF | 6 | 87 |
| 14 | DEP | 6 | 10 | THF | 6 | 99 |
| 15 | DEP | 10 | 0 | 2-Methyl THF | 6 | 15 |
| 16 | DEP | 10 | 0 | PhCl | 6 | 25 |
| 17 | DEP | 10 | 0 | Toluene | 6 | 12 |
| 18 | DEP | 10 | 0 | THF:PC |
3:3 |
86(2) |
| 19 | DEP | 10 | 0 | PC | 6 | 93(1) |
| 20d | DEP | 10 | 0 | None | 0 | >99(0) |
| 21 | DEP | 6 | 0 | THF | 6 | 52(9) |
| 22d | DEP | 6 | 0 | None | 0 | 89(1) |
| 23 | None | 10 | 5 | THF | 6 | 41 |
Cyclodepolymerization of poly(propylene carbonate)
CDP of PPC has not been extensively studied; current reports consist of various catalysts’ ability to perform CDP on PPC to form PC.40–43 A tris(pentafluorophenyl) borane catalyst was shown to perform selective CDP with poly(cyclohexene carbonate) and PPC using CH2Cl2 and 5 mol% catalyst (with respect to the repat unit molar mass of the polymer) at 130 °C.41 The kinetics of CDP of PPC were explored with a chromium salen complex and an ammonium azide co-catalyst, which identified the importance of a nucleophilic or basic anion to initiate CDP.42 Finally, ZnEt2 has commonly been used as a CDP catalyst for many designer polymers, and it can also depolymerize PPC (see ESI†).40,61 Unfortunately, the CDP of PPC in literature is difficult to compare across works, as conditions involving catalyst, solvent, reaction times, etc. vary substantially.Notably, the PPC reaches a temperature threshold of 170 °C, at which point, the polymer is known to degrade/depolymerize into PC.61,62 For this polymer, this temperature allows for the conversion of the kinetic polymer product (PPC) to the thermodynamic cyclic carbonate (PC).7,8,42
Once CDP selectivity was identified for these cobalt catalysts, efforts to optimize the reaction conditions for this method over transfer hydrogenation was prioritized. A tetrahydrofuran (THF)/isopropanol (iPrOH) mixture with potassium tert-butoxide (KOtBu) was used to replicate Werner's transfer hydrogenation conditions.15 THF was used to solubilize the polymer, isopropanol as the hydrogen source, and KOtBu as a strong base.15 These conditions may not be necessary to produce PC. Without iPrOH, MES and DEP catalysts reached quantitative conversion within 20 hours at 140 °C, with DIPP displaying high but inconsistent yields of PC (Table 1, entries 4–6). These results indicate that the hydrogen source is unnecessary. CDP reactions conducted without this base yielded very similar results, if not slightly lower conversions, to those with base, save for that of DIPP, indicating base was not significantly impacting the rate of CDP (Table 1, entries 7–9).
When comparing the ligand substituents, the catalyst with mesityl (MES) and diisopropyl phenyl (DIPP) substituents showed slower CDP of PPC, while the catalyst with ethyl substituents (DEP) proved fastest and the most reproducible amongst the three ligands. DIPP is hypothesized to have too much steric crowding to allow for facile backbiting on the polymer. It is unclear why MES is slower than DEP, however solubility could be a challenge, as these catalysts are only mildly soluble in THF at room temperature. Given DEP's speed over DIPP and MES in the optimized conditions without iPrOH or base, this catalyst was used for further optimization. With DEP (Fig. 3), reactions shortened to 10 hours in THF showed a distinct difference when KOtBu was used (achieving 83% yield) versus when KOtBu was not used (achieving just 69% yield) (Table 1, entries 10 and 11). These results indicate that presence of base does increase the rate of CDP when considering reactions that have not reached high conversions. It is anticipated that the initiation of the reaction occurs through the deprotonation of the polymer end group, which is likely an alcohol, or through nucleophilic attack of a carbonyl on the polymer chain (Fig. 4a). An OtBu alkoxide would likely be faster than a chloride for both proposed routes, supporting the higher yield of PC with KOtBu present.
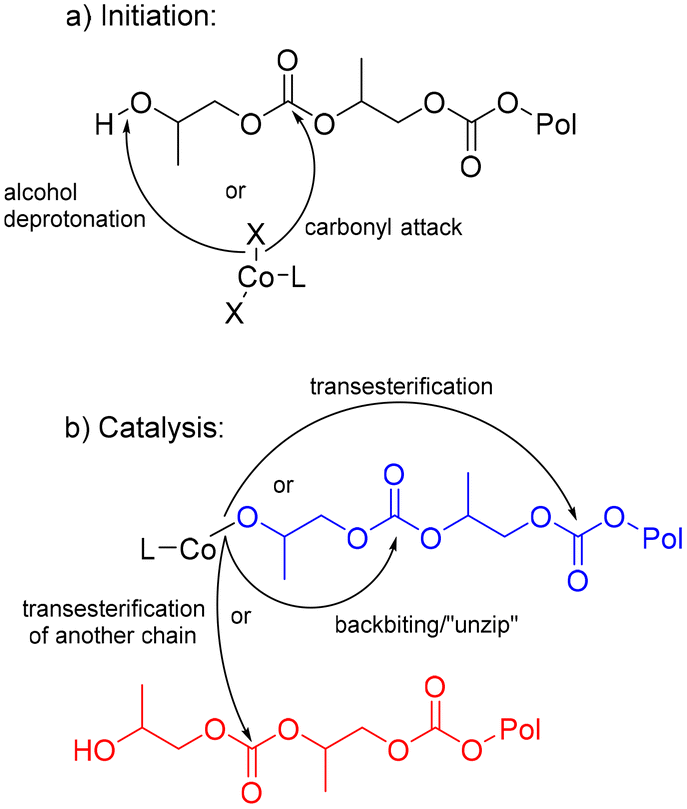 | ||
| Fig. 4 Proposed mechanisms for initiation (a), catalysis and subsequent side reactions (b) of CDP of PPC. | ||
One hypothesis is that the base could exchange the Cl ligands with the –OtBu ligands, which might enhance nucleophilic attack of carbonyls in the polymer and increase the initiation rate of depolymerization. This reaction has been conducted on similar Fe complexes with PDI ligands.63 Notably, reactions with KOtBu show a rapid color change from yellow brown to purple when solvent is added to the reaction mixture. This color change is not observed without the presence of KOtBu. In this case, two equivalents of KOtBu would be needed to exchange with all the chlorides on DEP. This indeed shows slightly faster conversion than DEP with one equivalent of KOtBu (Table 1, entry 12). Additionally, stirring DEP with one or two equivalents of KOtBu prior to the addition of PPC lead to even higher conversions, indicating the likelihood that exchanging the Cl anions for OtBu anions increases the reactivity of the DEP catalyst (Table 1, entries 13 and 14). Unfortunately, the low solubility of DEP in most solvents at room temperature prevents the ability to characterize this exchange either by NMR spectroscopy or isolation in bulk. Future directions aim to adjust the ligands for increased solubility to confirm these hypotheses.
Once the metal catalyst has the polymer chain bound, the expected mechanism of CDP with metal-based catalysts is back-biting from alkoxide or carbonate end groups bound to the metal ion. These end groups could perform consistent back-biting reactions to directly form PC, depolymerizing the polymer through a controlled “un-zip” pathway (Fig. 4b). Additionally, the end groups could perform transesterification reactions further down the polymer chain to form cyclic oligomers or on another polymer chain which changes the polymer molar mass (Fig. 4b). Indeed, these proposed mechanisms are well-documented across several PPC depolymerization studies.9,40,42,64 From these suspected routes, it was important to understand if reaction conditions could impact rate of CDP and control for the desired “un-zip” pathway.
Since KOtBu is moisture-sensitive, reactivity of the catalyst without base was investigated, with the reactions pursued in the presence of air. DEP is air stable and can be left over a month without showing decomposition. Using the standard reaction conditions taken from Table 1, entry 11, batch reactions were performed to follow the progress of the reaction (Fig. 5). Yields were characterized by 1H NMR spectroscopy. The remaining PPC was also characterized by size-exclusion chromatography (SEC). Within the first 4 hours, only slow CDP is observed, while the rate rapidly increases, demonstrating a more linear trend, over the next 8 hours to reach a total conversion of 94% after a total of 12 hours. The slow initial rate of CDP suggests an induction period, which could be due to the low basicity of the chloride anions to deprotonate chain ends of the PPC. Once all catalysts are bound to polymer chains, this could explain the increase in rate. Before all polymer chains are deprotonated, chain transfer could occur and slow productive CDP. Following the reaction by SEC showed a consistent decrease in the dispersity of the remaining polymer chains. A duplicate reaction series shows the same general trend (see ESI†). Random nucleophilic attack on the polymer would likely result in broad and/or multimodal molar mass distributions, as there would be no preference for reacting with any particular part of the polymer chain. Monomodal molar mass distributions, along with the decrease in dispersity, suggest that these side reactions are not occurring, indicating selective “un-zip” backbiting reaction for CDP of PPC with PDICoCl2 catalysts.
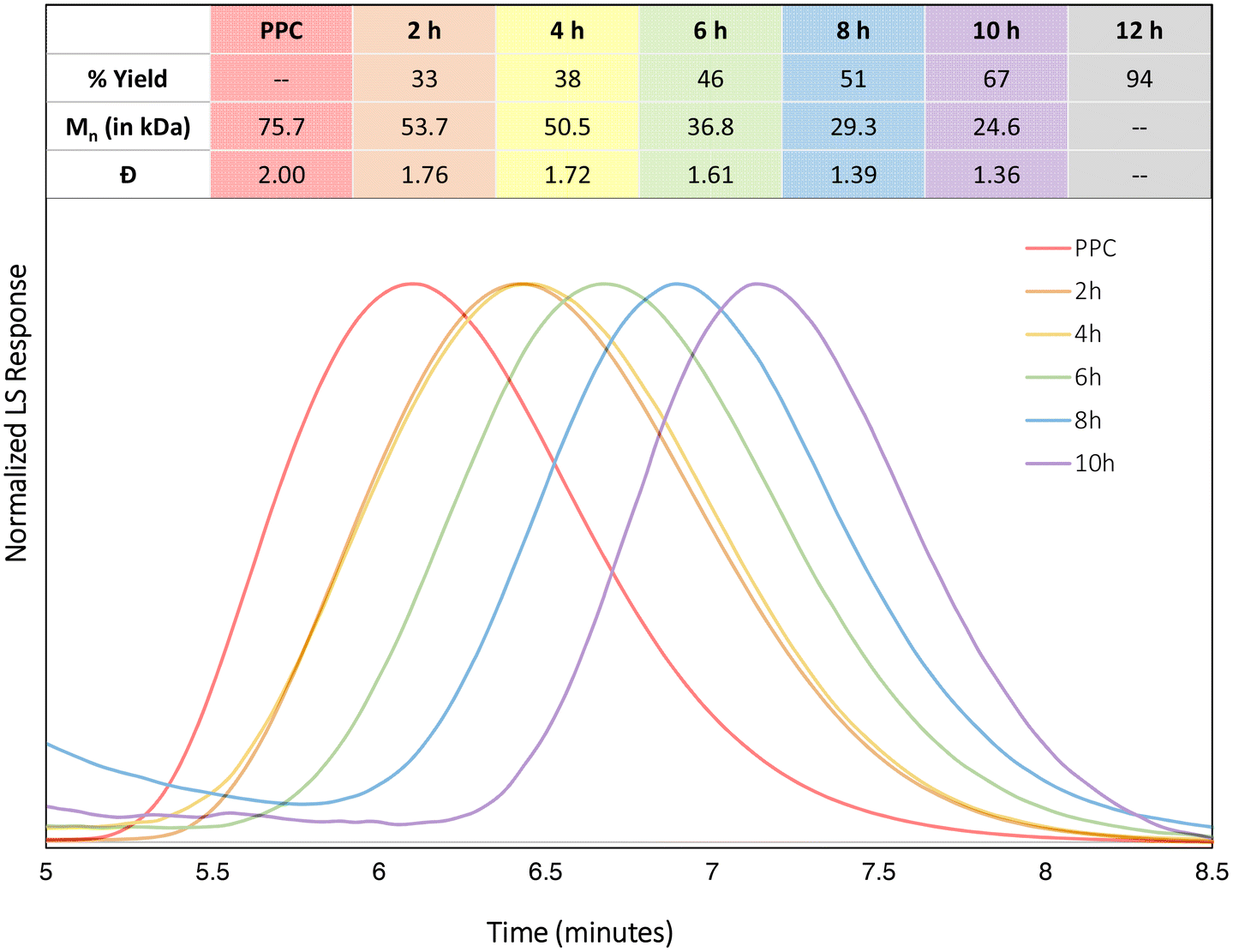 | ||
| Fig. 5 SEC traces for CDP of PPC with DEP in THF at 140 °C for different time points, using light scattering (LS). Note, the red trace labelled PPC is the SEC trace of the PPC starting polymer. | ||
It was anticipated that the presence of base could lower the induction period, which would lead to productive CDP faster than without base. Other air-stable bases in addition to KOtBu were tested (Table 2). Two of the bases, NaOH and NaHCO3, presented diminished yields in comparison to analogous conditions without base. NEt3 showed a similar yield to that without base. Two bases (KOH and NaOtBu) resulted in comparable yields to that with KOtBu, without the need for inert conditions. These results indicate that a stronger base or easier ability to exchange the chlorides on DEP may be important to improve the reaction rate. Running time points with NaOtBu, analogous to those done without base in Fig. 5, showed inconsistent yields. Notably, reactions that did not reach full conversion with NaOtBu displayed a higher dispersity than the original PPC starting polymer (ESI Table S4†). Returning to KOtBu time points showed a more consistent increase in conversion over time. However, with this base, dispersity was variable, suggesting random chain scission. Interestingly, this similar pattern of variable dispersity with CDP is seen with use of ZnEt2 from Byers et al.40 These results suggest that the base could be performing side reactions with or without the use of the catalyst, leading to random scission and less controlled CDP. Control reactions with KOtBu alone did show mild conversions of PPC to PC, indicating they can facilitate CDP with and without the catalyst (Table 1, entry 23). These conditions contrast those seen with the unzipping mechanism without base. Though the chloride from DEP alone proved better at unzipping, this may be in part due to halides being weaker nucleophiles than alkoxides, making them more likely to attack end groups because of their proximity.
|
|
|||
|---|---|---|---|
| Entry | Base | Time (h) | % Yieldb,c |
| a Conditions: 5 mol% DEP and 5 mol% base with respect to the repeat unit molar mass of polymer, 2 mmol PPC, 6 mL THF, 350 rpm, run at 140 °C in Parr reactor. Reactions performed under air-free N2 atmosphere if KOtBu used, otherwise performed in air. b Determined by 1H NMR spectroscopy using mesitylene as internal standard, taken in CDCl3. c Reactions performed in duplicate; standard deviation of error represented in parentheses. | |||
| 1 | KOH | 10 | 79(8) |
| 2 | NaOH | 10 | 58 |
| 3 | NaHCO3 | 10 | 24 |
| 4 | NEt3 | 10 | 61 |
| 5 | NaOtBu | 10 | 86(1) |
| 6 | KOtBu | 10 | 83(3) |
| 7 | NaOtBu | 2 | 40(14) |
| 8 | NaOtBu | 4 | 58(15) |
| 9 | NaOtBu | 6 | 50(12) |
| 10 | NaOtBu | 8 | 44(12) |
| 11 | NaOtBu | 12 | 65(16) |
| 12 | KOtBu | 2 | 74(0) |
| 13 | KOtBu | 4 | 85(3) |
| 14 | KOtBu | 6 | 91(2) |
| 15 | KOtBu | 8 | 92(2) |
| 16 | KOtBu | 12 | 92(2) |
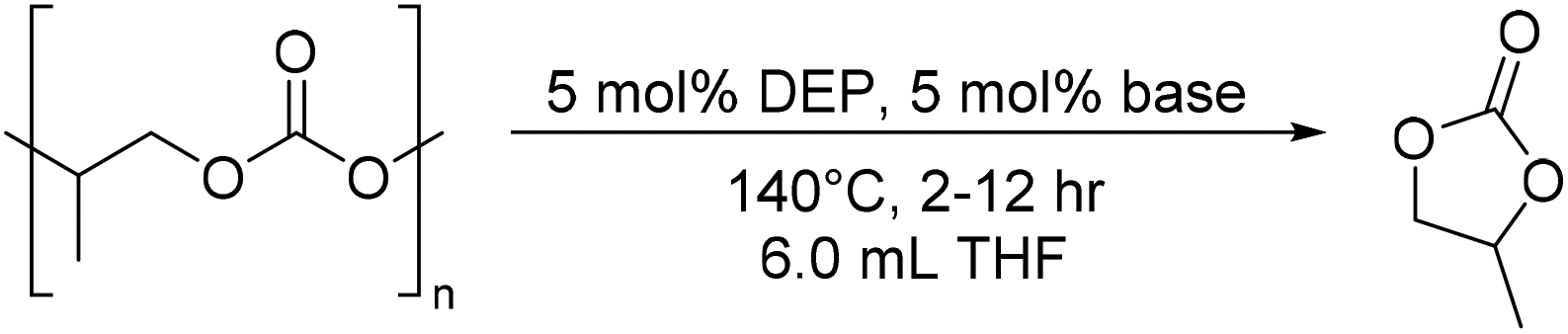
While THF was originally used, analogous to prior transfer hydrogenation reactions, the solvent scope was further tested to promote the fastest catalysis. Reactions performed in chlorobenzene, 2-methyl THF, and toluene did not facilitate rapid CDP, showing diminished yields to those conducted in THF (Table 1, entries 15–17). The use of PC or a PC/THF mixture both led to higher yields than those with just THF. This could be due to the higher boiling point of PC, which maintains solubility of the polymer and catalyst at elevated temperatures (Table 1, entries 18 and 19). These results suggested that the reaction could be done with polymer in the melt without added solvent, where the generated PC becomes the solvent over the course of the reaction. Gratifyingly, full conversion of PPC to PC was achieved within 10 hours when the reaction was conducted on PPC in the melt without solvent addition, in which no remaining PPC is observed by NMR spectroscopy (Table 1, entry 20). An 89% yield is achieved in just 6 hours with these neat conditions, compared to 52% yield with THF (Table 1, entries 21 and 22). This is the first example for CDP of PPC in the melt without a solvent. To ensure that presence of PC in the reaction does not lead to polymerizations, control reactions were conducted with DEP or KOtBu with PC as monomer and THF as solvent for 10 h at 140 °C, with no PPC formation observed (see ESI†). While reactions done with PPC in the melt could lead to greener methods for CDP, quantification of the products was more difficult if the reaction did not reach full conversion, therefore further studies were still conducted in the presence of solvent.
To identify how reaction concentration impacts CDP of PPC with DEP, the reaction concentrations were varied in two ways. In one method, the PPC and catalyst quantities were kept constant, while the THF volume was varied (Fig. 6a, blue). While this method showed no observable pressure differences on the Parr reactors as the volume of THF increased (maintaining a steady 6.0 bar), it was important to rule out any pressure contributions to the data. Therefore, the second method kept the THF volume constant, while the PPC and catalyst quantities were varied (Fig. 6a, orange). Both methods showed the same overall outcome, suggesting that pressure is not impacting the results. Four different concentrations were measured, with the most concentrated and second most dilute conditions showing the highest yields for CDP. The higher concentration reactions could show higher yields due to the catalyst initiating CDP faster, while also increasing the rate of the metal ion coming in proximity to the next carbonyl on the polymer chain. This condition also most closely matches conditions in the melt, which has shown the highest yield. The metal center could be performing chain transfer reactions between polymer chains. Dilution could prevent this side reaction. However, over-dilution could slow the metal from initiating CDP.
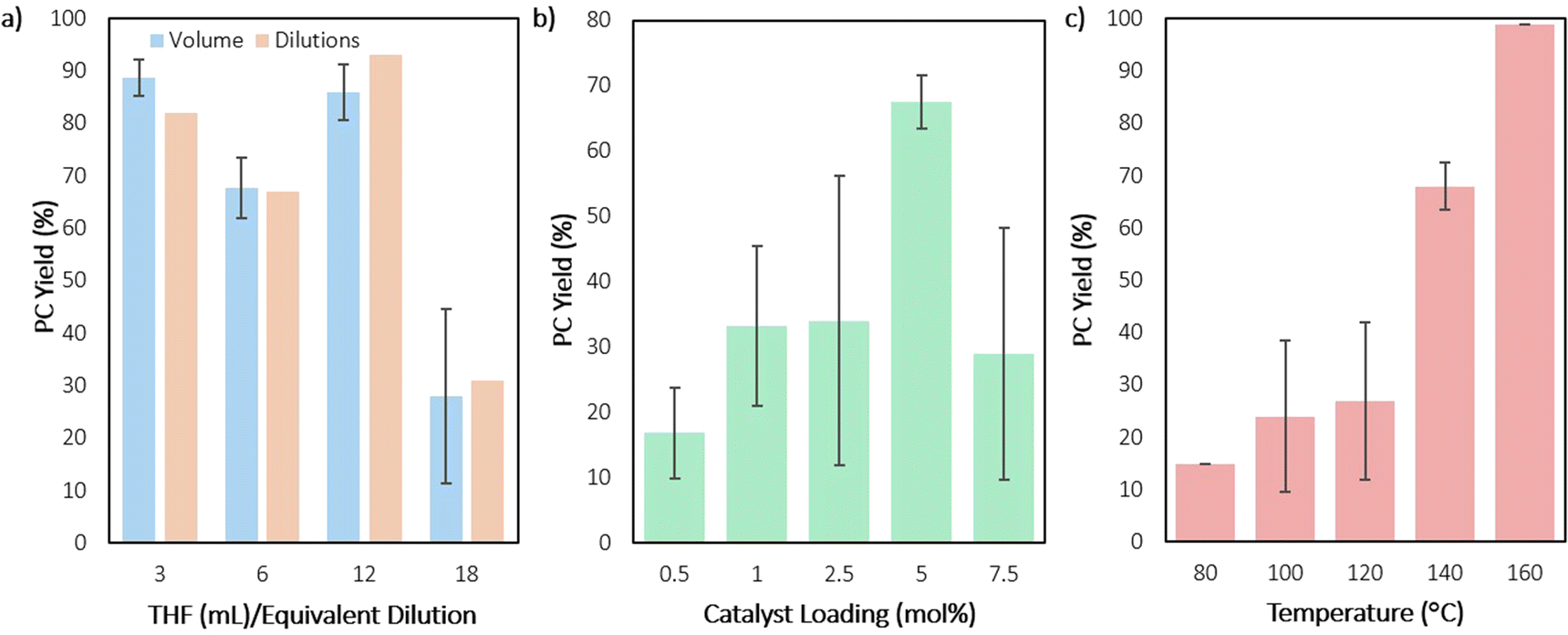 | ||
| Fig. 6 Graph of PC yield as a function of solvent dilution by varying THF volume (blue) or amount of catalyst and PPC relative to their dilution in THF while maintaining an overall volume of 6 mL THF (orange) (a), catalyst (DEP) loading (b), and temperature (c). Each experiment operated under conditions of 1–6 mmol PPC, 5 mol% DEP with respect to the repeat unit molar mass of the polymer, 0–18 mL THF, and 80–160 °C, run for 10 hours. | ||
Next, varying the catalyst loading, while maintaining all other conditions, showed an expected increase in yield as the catalyst loading is increased from 0.5 to 5 mol% (Fig. 6b). However, when catalyst loading is increased to 7.5 mol%, a drop in yield is observed. It is unclear what causes this drop in yield, though, the added concentration of metal bound to polymer chain could lead to increased presence of chain transfer or the presence of transesterification of other polymer chains, leading to lowered back-biting (Fig. 7). With smaller amounts of catalyst in fewer quantities than the polymers’ available end groups, the cobalt-alkoxide (or carbonate) may either backbite for productive PC formation or deprotonate for chain transfer, reducing efficiency of the reaction (Fig. 7a). Yet when catalyst outweighs the number of available polymer end groups, transmetalation may compete with CDP as a reaction (Fig. 7b). Based on the molar mass (Mn) of the PPC starting material, there is an approximate 10:1 ratio between DEP and polymer end groups. This would indicate that these conditions lie in the Fig. 7b region or it could suggest there is just a point at which too much DEP inhibits productive CDP. Control reactions with catalyst concentrations that are low (2.5 mol%) and high (7.5 mol%) were conducted for 2 and 6 hours, in which the remaining polymer was characterized by SEC (see ESI†). In both cases, dispersity still remained low at 2 hours, while 6-hour reactions showed an increase in dispersity. These results support the hypothesis that catalyst loading needs to match the end group concentration for the CDP to remain controlled.
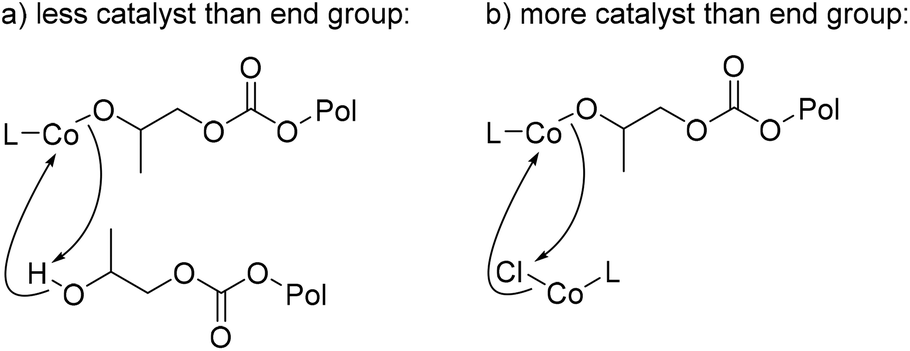 | ||
| Fig. 7 Proposed side reactions to understand catalyst loading effects when comparing lowered catalyst loading (a) to increased catalyst loading (b). | ||
The ideal temperature range for the reaction was then probed with DEP, as increased temperatures have the potential to increase CDP (but may also provoke side reactions), by nearing PPC's degradation temperature of 170 °C.61 Fortunately, high selectivity and higher conversions were maintained with increased temperature, as the reaction reached near full conversion in 10 hours at 140 °C and quantitative conversion at 160 °C (Fig. 6c). This indicated that use of DEP fully depolymerized PPC within the range of 140–160 °C, but reaction optimization conditions were maintained at 140 °C to better distinguish which conditions were more efficient. Compared to the first reported degradation of PPC to PC by Kuran et al., use of DEP requires less solvent and catalyst while producing higher yields in a shorter amount of time.43 When compared with more recent studies, like that of Kerton et al. or Darensbourg et al., the use of DEP under these conditions offers somewhat of a trade-off in certain areas.41,42 While use of DEP requires a metal catalyst, slightly higher temperatures, and longer reaction times, the use of any solvent, much less those detrimental to the environment, can be forgone.
Cyclodepolymerization of other polymers
Once CDP of PPC was better understood, extension of studies to other polycarbonates and polyesters were warranted (Fig. 8). No observable CDP was identified for nylon 6 (7a), and small amounts of CDP was observed for PCL (2a) and poly(trimethylene carbonate) (PTMC) (5a) with the DEP and THF, alone. Surprisingly, CDP of PVL was identified with DEP to form the δ-valerolactone (VL) monomer (3a). Under the standard reaction conditions provided from Table 1, PVL depolymerized with a 41% yield of VL, compared to the analogous reaction in THF alone, which yielded 7% VL. When run for 20 hours, the reaction yielded 73% product (11% by THF-only control), and when run at 180 °C for 20 h, the reaction yielded 84% product (10% by THF-only control). Prior studies have attempted depolymerization and/or thermal degradation of PVL at 250 °C, taking advantage of its relatively low ceiling temperature of 298 °C.35,45 Additionally, Byers and coworkers identify their ZnCl2/PEG600 catalyst system to achieve 94% conversion under reactive distillation at 160 °C after 16 hours.40 Using DEP, we can identify high conversion without the need for reactive distillation. With reactive distillation, analogous to reactions done by Byers and coworkers, CDP of PVL at 180 °C with PEG600 for 10 hours achieved only 85% conversion.40 The results suggest the reactive distillation of PVL performs similarly, if not somewhat worse, than mere solvent and DEP conditions. Similarly, poly(dioxanone) (PDO) displayed quantitative conversion under 180 °C after 20 hours (4a). Finally, PLA was converted to lactide (6a) in a moderate yield of 42%. While this is not optimized, and does not compete with the Sn(II)/alcohol catalyst system reported by Williams and coworkers, this represents one of only a few examples of CDP of PLA.36,39,40,44 Most notably, all polymers which were able to undergo any amount of CDP fared worse under the same conditions with KOtBu added (see ESI†). These results indicate that the DEP catalyst is more important than the presence of KOtBu for CDP.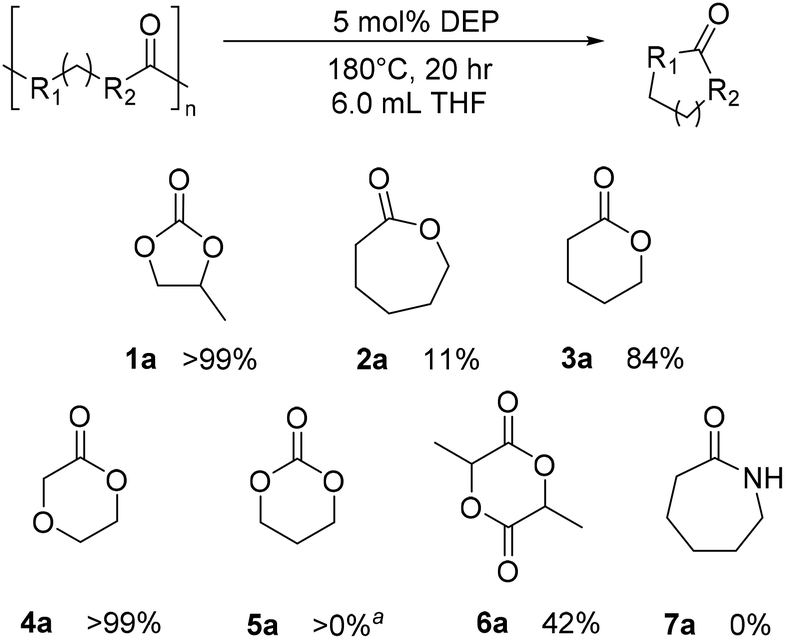 | ||
| Fig. 8 Substrate scope of CDP with DEP. Catalyst loadings made with respect to the repeat unit molar mass of polymer. aPresence of cyclic monomer is observed in small quantities in the NMR, but not enough to determine an accurate yield. | ||
Solvolysis
While DEP proved to be active for CDP of several selected polymers (under the conditions tried), it was still unclear whether transfer hydrogenation (the initial goal) was possible with this catalyst. Using the same conditions from Werner used in early PPC depolymerization trials, other polymers mentioned previously, such as PLA, PTMC, PDO, PECL, and nylon 6, in addition to poly(bisphenol A carbonate) (PBPAC) and PET, were tested.15 PET and PBPAC were not studied for CDP, as they are made through step-growth polymerization and are not known to have an accessible cyclic monomer. Almost all these plastics showed full depolymerization within 20 hours, though via a solvolysis mechanism rather than transfer hydrogenation. The polymers and their depolymerized substrates are shown in Fig. 9. Through fairly mild conditions, DEP with KOtBu achieved complete solvolysis of a wide range of polymers, demonstrating its versatility. Control reactions with KOtBu without the catalyst showed similar conversions in most cases, suggesting the base to be the primary catalyst for this solvolysis method. In the case of PBPAC, the presence of catalyst shows greatly improved solvolysis. Additionally, reactions with PTMC with just KOtBu does not perform the solvolysis identified with the DEP. Instead, it produced a still undetermined product, with no evidence of the solvolysis product. Nonetheless, this simple base has not been previously used for solvolysis of polyesters or polycarbonates. However, the versatility for high conversions of solvolysis of many polymers under these reported conditions warrant additional studies to determine the full capability of this simple base. While these reaction conditions are not optimized, these results indicate this catalytic system is active for solvolysis of numerous polymers. Since solvolysis is not commonly done with isopropanol, additional studies will be needed to compare to other methods that prioritize hydrolysis, methanolysis, and glycolysis.11,18,22–24,65–71 This shows diversity of the catalyst, as solvolysis is primarily studied for PET and PBPAC.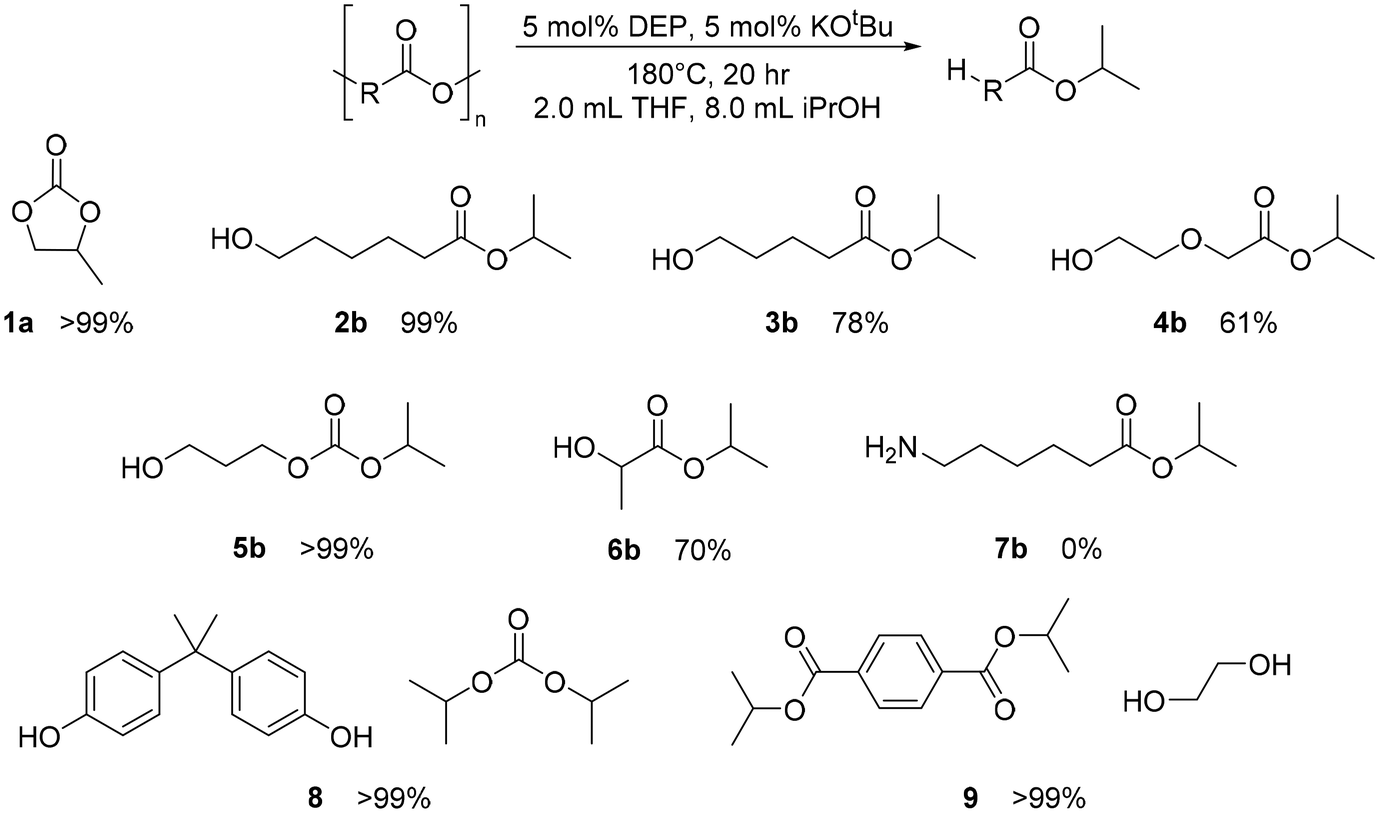 | ||
| Fig. 9 Substrate scope of solvolysis with DEP and KOtBu. Catalyst and base loadings made with respect to the repeat unit molar mass of polymer. | ||
Since these two pathways seemed compatible with each other, we questioned whether they could be used in parallel in the same reaction mixture. PPC and PBPAC were selected to mix, as they are both polycarbonates, PPC shows exclusive CDP under all conditions studied, and PBPAC does not have an accessible cyclic monomer. Notably, in a mixed pot of PBPAC and PPC in a 1:1 molar ratio with respect to the repeat unit of the polymer, under conditions of DEP at 180 °C with KOtBu and isopropanol and THF as solvents, CDP was maintained with PPC while solvolysis prevailed with PBPAC to bisphenol A (BPA) and diisopropyl carbonate (DIPC) (Fig. 10), with both pathways reaching full conversion. Similarly, under conditions of just DEP with THF as solvent at 140 °C, PBPAC remained intact while PPC depolymerized to PC at 91% yield, which bodes well for isolating depolymerization products in mixed recycling streams. On the other hand, PVL under solvolysis conditions yield solvolysis products, indicating multiple depolymerization paths for one polymer with the same catalyst under different conditions.
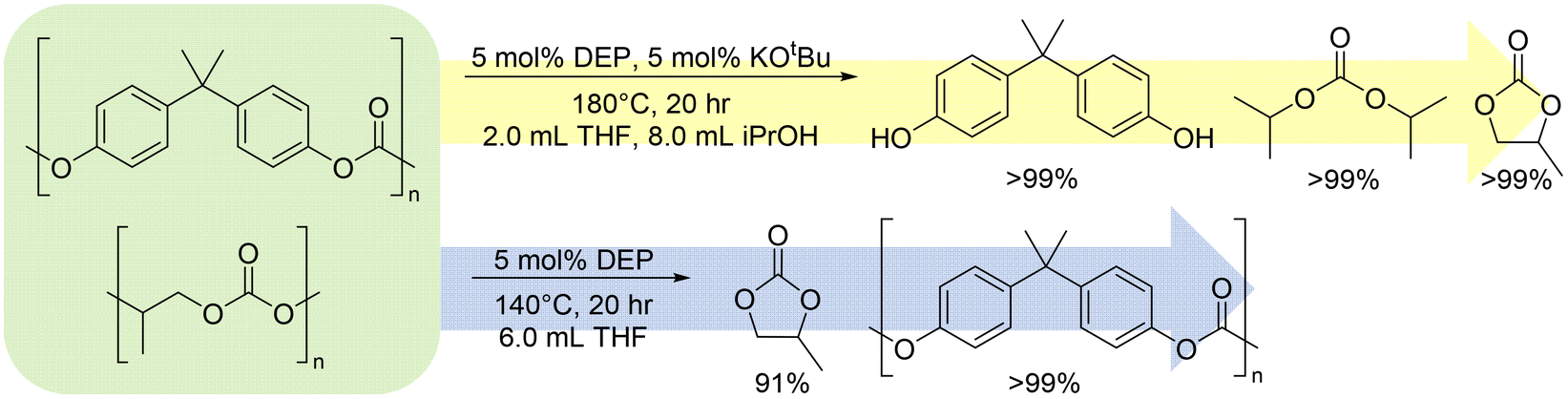 | ||
| Fig. 10 One-pot reactions of PBPAC and PPC in varying conditions leading to solvolysis of PBPAC to BPA and DIPC with CDP of PPC to PC (yellow) and CDP of PPC to PC leaving PBPAC intact (blue). PBPAC and PPC used in a 1:1 ratio, 2 mmol each, with catalyst/base loadings made with respect to repeat unit molar mass of PPC. | ||
Conclusions
A series of air-stable pyridine(diimine) cobalt dichloride catalysts were identified to be active for the selective cyclodepolymerization (CDP) of poly(propylene carbonate) (PPC) to form propylene carbonate (PC) as the exclusive product. Reaction conditions were optimized to reach high yields of PC when conducting the CDP reaction with neat polymer in the melt. Optimizations for the catalyst alone identified a controlled “un-zip” pathway to form PC, without the presence of undesirable side reactions. Alternatively, presence of a base encouraged random CDP of the polymer chain, with increases in dispersity of the polymer over the course of the reaction. Catalyst loading was found to be important, as too high or too low led to proposed inhibitive side reactions of chain transfer or transmetalation.Finally, extensions to other carbonyl-containing polymers identified these catalysts as highly active for the selective CDP of poly(δ-valerolactone) (PVL) and polydioxanone (PDO) with DEP alone. Activities of CDP for poly(trimethylene carbonate) (PTMC), poly(lactic acid) (PLA) and poly(ε-caprolactone) (PCL) were observed in smaller amounts, however optimization of the conditions or the catalyst may be needed to achieve high conversion. In the presence of KOtBu and iPrOH, DEP could promote solvolytic depolymerization of several commodity polymers. Control reactions identify the KOtBu shows comparable activity on its own, suggesting it is the active catalyst in this reaction.
The results of these studies were used in a proof of concept mixed-recycling stream of PPC and poly(bisphenol A carbonate) (PBPAC), in which solvolysis conditions lead to the full conversion of PPC CDP and PBPAC solvolysis. CDP conditions without base or iPrOH led to selective CDP of PPC to 91% conversion, with the PBPAC being left undisturbed. These results identify promising routes for tunable deconstruction of these polymers. Future work to identify optimal changes to the metal and ligands to further improve CDP of carbonyl-containing polymers are currently underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this publication comes from Cottrell Scholar Award #CS-CSA-2023-114 sponsored by Research Corporation for Science Advancement and the Wrigley Faculty Innovator Award. Instrumentation in the USC Chemistry Instrument Facility was acquired with support from the USC Research and Innovation Instrumentation Award Program. Additionally, funds were provided by the National Science Foundation (Award: CHE-2018740) to acquire the X-ray diffractometer used to determine crystal structures, the National Science Foundation (DBI-0821671, CHE-0840366) and National Institute of Health (S10 RR25432) supported the acquisition of the NMR spectrometers used in our work. Thank you to Dr Mikiyas Assefa for his assistance in crystal structure analysis.References
-
D. G. LeGrand and J. T. Bendler, Handbook of Polycarbonate Science and Technology, CRC Press, Boca Raton, FL, 1999 Search PubMed
.
- C. M. Kozak, K. Ambrose and T. S. Anderson, Copolymerization of Carbon Dioxide and Epoxides by Metal Coordination Complexes, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS
- J.-G. Rosenboom, R. Langer and G. Traverso, Bioplastics for a Circular Economy, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed
- BCC Research Report Code, https://www.bccresearch.com/market-research/plastics/global-markets-and-technologies-for-bioplastics.html, (accessed July 2023).
- R. Mülhaupt, Green Polymer Chemistry and Bio-Based Plastics: Dreams and Reality, Macromol. Chem. Phys., 2013, 214, 159–174 CrossRef
- J. Brizga, K. Hubacek and K. Feng, The Unintended Side Effects of Bioplastics: Carbon, Land, and Water Footprints, One Earth, 2020, 3, 45–53 CrossRef
- J. Geschwind and H. Frey, Poly(1,2-Glycerol Carbonate): A Fundamental Polymer Structure Synthesized from CO2 and Glycidyl Ethers, Macromolecules, 2013, 46(9), 3280–3287 CrossRef CAS
- G. W. Coates and Y. D. Y. L. Getzler, Chemical Recycling to Monomer for an Ideal, Circular Polymer Economy, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS
- H. Zhang and M. W. Grinstaff, Synthesis of Atactic and Isotactic Poly(1,2-Glycerol Carbonate)s: Degradable Polymers for Biomedical and Pharmaceutical Applications, J. Am. Chem. Soc., 2013, 135(18), 6806–6809 CrossRef CAS PubMed
- McKinsey & Company, https://www.mckinsey.com/industries/chemicals/our-insights/no-time-to-waste-what-plastics-recycling-could-offer, (accessed July 2023).
- C. Pudack, M. Stepanski and P. Fässler, PET Recycling – Contributions of Crystallization to Sustainability, Chem. Ing. Tech., 2020, 92, 452–458 CrossRef CAS
- C. Wang and O. El-Sepelgy, Reductive Depolymerization of Plastics Catalyzed with Transition Metal Complexes, Curr. Opin. Green Sustainable Chem., 2021, 32, 100547 CrossRef CAS
- A. Kumar, N. von Wolff, M. Rauch, Y.-Q. Zou, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, Hydrogenative Depolymerization of Nylons, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef CAS PubMed
- E. M. Krall, T. W. Klein, R. J. Andersen, A. J. Nett, R. W. Glasgow, D. S. Reader, B. C. Dauphinais, S. P. Mc Ilrath, A. A. Fischer, M. J. Carney, D. J. Hudson and N. J. Robertson, Controlled Hydrogenative Depolymerization of Polyesters and Polycarbonates Catalyzed by Ruthenium(II) PNN Pincer Complexes, Chem. Commun., 2014, 50, 4884–4887 RSC
- X. Liu, J. G. de Vries and T. Werner, Transfer Hydrogenation of Cyclic Carbonates and Polycarbonate to Methanol and Diols by Iron Pincer Catalysts, Green Chem., 2019, 21, 5248–5255 RSC
- R. A. Farrar-Tobar, B. Wozniak, A. Savini, S. Hinze, S. Tin and J. G. de Vries, Base-free Iron Catalyzed Transfer Hydrogenation of Esters Using EtOH as Hydrogen Source, Angew. Chem., Int. Ed., 2019, 58, 1129–1133 CrossRef CAS PubMed
- Y.-B. Huang, T. Yang, Y.-J. Luo, A.-F. Liu, Y.-H. Zhou, H. Pan and F. Wang, Simple and Efficient Conversion of Cellulose to γ-Valerolactone through an Integrated Alcoholysis/Transfer Hydrogenation System Using Ru and Aluminium Sulfate Catalysts, Catal. Sci. Technol., 2018, 8, 6252–6262 RSC
- J. Demarteau, I. Olazabal, C. Jehanno and H. Sardon, Aminolytic Upcycling of Poly(Ethylene Terephthalate) Wastes Using a Thermally-Stable Organocatalyst, Polym. Chem., 2020, 11, 4875–4882 RSC
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de Los Rios and M. E. Fieser, Catalytic Methods for Chemical Recycling or Upcycling of Commercial Polymers, Mater. Horiz., 2021, 8, 1084–1129 RSC
- H. Chen, K. Wan, Y. Zhang and Y. Wang, Waste to Wealth: Chemical Recycling and Chemical Upcycling of Waste Plastics for a Great Future, ChemSusChem, 2021, 14, 4123–4136 CrossRef CAS PubMed
- G. Grause, K. Sugawara, T. Mizoguchi and T. Yoshioka, High-Value Products from the Catalytic Hydrolysis of Polycarbonate Waste, Polym. Degrad. Stab., 2009, 94, 1119–1124 CrossRef CAS
- S. Baliga and W. T. Wong, Depolymerization of Poly(Ethylene Terephthalate) Recycled from Post-Consumer Soft-Drink Bottles, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2071–2082 CrossRef CAS
- S. R. Shukla and K. S. Kulkarni, Depolymerization of Poly(Ethylene Terephthalate) Waste, J. Appl. Polym. Sci., 2002, 85, 1765–1770 CrossRef CAS
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Chemical Recycling of Post-Consumer PET Wastes by Glycolysis in the Presence of Metal Salts, Polym. Degrad. Stab., 2010, 95, 1022–1028 CrossRef
- M. Hong and E. Y.-X. Chen, Completely Recyclable Biopolymers with Linear and Cyclic Topologies via Ring-Opening Polymerization of γ-Butyrolactone, Nat. Chem., 2018, 8, 42–49 CrossRef PubMed
- Y. Liu, H. Zhou, J.-Z. Guo, W.-M. Ren and X.-B. Lu, Completely Recyclable Monomers to Polycarbonate: Approach to Sustainable Polymers, Angew. Chem., Int. Ed., 2017, 56, 4862–4866 CrossRef CAS PubMed
- C. Li, L. Wang, Q. Yan, F. Liu, Y. Shen and Z. Li, Rapid and Controlled Polymerization of Bio-Sourced Δ-Caprolactone toward Fully Recyclable Polyesters and Thermoplastic Elastomers, Angew. Chem., Int. Ed., 2022, 61, e202201407 CrossRef CAS PubMed
- J. P. MacDonald and M. P. Shaver, An Aromatic/Aliphatic Polyester Prepared via Ring-Opening Polymerisation and Its Remarkably Selective and Cyclable Depolymerisation to Monomer, Polym. Chem., 2016, 7, 553–559 RSC
- P. Olsén, J. Undin, K. Odelius, H. Keul and A.-C. Albertsson, Switching from Controlled Ring-Opening Polymerization (CROP) to Controlled Ring-Closing Depolymerization (CRCDP) by Adjusting the Reaction Parameters That Determine the Ceiling Temperature, Biomacromolecules, 2016, 17, 3995–4002 CrossRef PubMed
- H. Nishida, Y. Andou, K. Watanabe, Y. Arazoe, S. Ide and Y. Shirai, Poly(Tetramethyl Glycolide) from Renewable Carbon, a Racemization-Free and Controlled Depolymerizable Polyester, Macromolecules, 2011, 44, 12–13 CrossRef CAS
- G. W. Fahnhorst and T. R. Hoye, Carbomethoxylated Polyvalerolactone from Malic Acid: Synthesis and Divergent Chemical Recycling, ACS Macro Lett., 2018, 7, 143–147 CrossRef CAS PubMed
- L. Cederholm, P. Olsén, M. Hakkarainen and K. Odelius, Turning Natural δ-Lactones to Thermodynamically Stable Polymers with Triggered Recyclability, Polym. Chem., 2020, 11, 4883–4894 RSC
- C. Shi, Z.-C. Li, L. Caporaso, L. Cavallo, L. Falivene and E. Y.-X. Chen, Hybrid Monomer Design for Unifying Conflicting Polymerizability, Recyclability, and Performance Properties, Chem, 2021, 7, 670–685 CAS
- D. J. Saxon, E. A. Gormong, V. M. Shah and T. M. Reineke, Rapid Synthesis of Chemically Recyclable Polycarbonates from Renewable Feedstocks, ACS Macro Lett., 2021, 10, 98–103 CrossRef CAS PubMed
- X.-L. Li, R. W. Clarke, H.-Y. An, R. R. Gowda, J.-Y. Jiang, T.-Q. Xu and E. Y.-X. Chen, Dual Recycling of Depolymerization Catalyst and Biodegradable Polyester That Markedly Outperforms Polyolefins, Angew. Chem., Int. Ed., 2023, 62, e202303791 CrossRef CAS PubMed
- L. Cederholm, J. Wohlert, P. Olsén, M. Hakkarainen and K. Odelius, “Like Recycles Like”: Selective Ring-Closing Depolymerization of Poly(L-Lactic Acid) to L-Lactide, Angew. Chem., Int. Ed., 2022, 61, e202204531 CrossRef CAS PubMed
- L. Cederholm, P. Olsén, M. Hakkarainen and K. Odelius, Chemical Recycling to Monomer: Thermodynamic and Kinetic Control of
the Ring-Closing Depolymerization of Aliphatic Polyesters and Polycarbonates, Polym. Chem., 2023, 14, 3270–3276 RSC
- B. A. Abel, R. L. Snyder and G. W. Coates, Chemically Recyclable Thermoplastics from Reversible-Deactivation Polymmerization of Cyclic Acetals, Science, 2021, 373, 783–789 CrossRef CAS PubMed
- C. Alberti and S. Enthaler, Depolymerization of End-of-life Poly(Lactide) to Lactide via Zinc-catalysis, ChemistrySelect, 2020, 5, 14759–14763 CrossRef CAS
- C. F. Gallin, W.-W. Lee and J. A. Byers, A Simple, Selective, and General Catalyst for Ring Closing Depolymerization of Polyesters and Polycarbonates for Chemical Recycling, Angew. Chem., Int. Ed., 2023, 62, e202303762 CrossRef CAS PubMed
- K. A. Andrea, M. D. Wheeler and F. M. Kerton, Borane Catalyzed Polymerization and Depolymerization Reactions Controlled by Lewis Acidic Strength, Chem. Commun., 2021, 57, 7320–7322 RSC
- D. J. Darensbourg and S.-H. Wei, Depolymerization of Polycarbonates Derived from Carbon Dioxide and Epoxides to Provide Cyclic Carbonates, Macromolecules, 2012, 45, 5916–5922 CrossRef CAS
- W. Kuran and P. Górecki, Degradation and Depolymerization of Poly(Propylene Carbonate) by Diethylzinc, Makromol. Chem., 1983, 184(5), 907–912 CrossRef CAS
- T. M. McGuire, A. Buchard and C. Williams, Chemical Recycling of Commercial Poly(L-Lactic Acid) to L-Lactide Using a High-Performance Sn(II)/Alcohol Catalyst System, J. Am. Chem. Soc., 2023, 145(36), 19840–19848 CrossRef CAS PubMed
- X.-L. Li, R. W. Clarke, J.-Y. Jiang, J.-Y. T.-Q. Xu and E. Y.-X. Chen, A Circular Polyester Platform Based on Simple Gem-Disubstituted Valerolactones, Nat. Chem., 2023, 15, 278–285 CrossRef CAS PubMed
- D. K. Schneiderman, M. E. Vanderlaan, A. M. Mannion, T. R. Panthani, D. C. Batiste, J. Z. Wang, F. S. Bates, C. W. Macosko and M. A. Hillmeyer, Chemically Recyclable Biobased Polyurethanes, ACS Macro Lett., 2016, 5, 515–518 CrossRef CAS PubMed
- J.-B. Zhu, E. M. Watson, J. Tang and E. Y.-X. Chen, A Synthetic Polymer System with Repeatable Chemical Recyclability, Science, 2018, 360, 398–403 CrossRef CAS PubMed
- R. M. Cywar, J.-B. Zhu and E. Y.-X. Chen, Selective or Living Organopolymerization of a Six-Five Bicyclic Lactone to Produce Fully Recyclable Polyesters, Polym. Chem., 2019, 10, 3097–3106 RSC
- F. N. Singer, A. C. Deacy, T. M. McGuire, C. K. Williams and A. Buchard, Chemical Recycling of Poly(Cyclohexene Carbonate) Using a Di-MgII Catalyst, Angew. Chem., Int. Ed., 2022, 61, e202201785 CrossRef CAS PubMed
- M. Scharfenberg, J. Hilf and H. Frey, Funcional Polycarbonates from Carbon Dioxide and Tailored Epoxide Monomers: Degradable Materials and Their Application Potential, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef
- D. J. Darensbourg, S.-H. Wei, A. D. Yeung and W. C. Ellis, Depolymerization of Poly(Indene Carbonate), Macromolecules, 2013, 46, 3228–3233 CrossRef CAS
- C. Li, R. J. Sablong, R. A. T. M. van Benthem and C. E. Koning, Unique Base-Initiated Depolymerization of Limonene-Derived Polycarbonates, ACS Macro Lett., 2017, 6, 684–688 CrossRef CAS PubMed
- Y. Yu, L.-M. Fang, Y. Liu and X.-B. Lu, Chemical Synthesis of CO2-Based Polymers with Enhanced Thermal Stability and Unexpected Recyclability from Biosourced Monomers, ACS Catal., 2021, 11, 8349–8357 CrossRef CAS
- L. Wursthorn, K. Beckett, J. O. Rothbaum, R. M. Cywar, C. Lincoln, Y. Kratish and T. J. Marks, Selective Lanthanide-organic Catalyzed Depolymerization of Nylon-6 to E-caprolactam, Angew. Chem., Int. Ed., 2023, 62(4) DOI:10.1002/anie.202212543
- G. Zhang, B. L. Scott and S. K. Hanson, Mild and Homogeneous Cobalt-Catalyzed Hydrogenation of C=C, C=O, and C=N Bonds, Angew. Chem., Int. Ed., 2012, 51, 12102–12106 CrossRef CAS PubMed
- P. J. Chirik, Iron- and Cobalt-Catalyzed Alkene Hydrogenation:
Catalysis with Both Redox-Active and Strong Field Ligands, Acc. Chem. Res., 2015, 48, 1687–1695 CrossRef CAS PubMed
- D. Dolui, S. Ghorai and A. Dutta, Tuning the Reactivity of Cobalt-Based H2 Production Electrocatalysts via the Incorporation of the Peripheral Basic Functionalities, Coord. Chem. Rev., 2020, 416, 213335 CrossRef CAS
- N. D. McMillion, A. W. Wilson, M. K. Goetz, M.-C. Chang, C.-C. Lin, W.-J. Feng, C. C. McCrory and J. S. Anderson, Imidazole for Pyridine Substitution Leads to Enhanced Activity under Milder Conditions in Cobalt Water Oxidation Electrocatalysis, Inorg. Chem., 2019, 58, 1391–1397 CrossRef CAS PubMed
- G. J. Britovsek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Strömberg, A. J. P. White and D. J. Williams, Iron and Cobalt Ethylene Polymerization Catalysts Bearing 2,6-Bis(Imino)Pyridyl Ligands: Synthesis, Structures, and Polymerization Studies, J. Am. Chem. Soc., 1999, 121, 8728–8740 CrossRef CAS
- H. Liu, X. Jia, F. Wang, Q. Dai, B. Wang, J. Bi, C. Zhang, L. Zhao, C. Bai, Y. Hu and X. Zhang, Synthesis of Bis(N-Arylcarboximidoylchloride)Pyridine Cobalt(II) Complexes and Their Catalytic Behavior for 1,3-Butadiene Polymerization, Dalton Trans., 2013, 42, 13723 RSC
- J.-L. Hong, X.-H. Zhang, R.-J. Wei, Q. Wang, Z.-Q. Fan and G.-R. Qi, Inhibitory Effect of Hydrogen Bonding on Thermal Decomposition of the Nanocrystalline Cellulose/Poly(Propylene Carbonate) Nanocomposite, J. Appl. Polym. Sci., 2014, 131(3), 39847 CrossRef
- G. Luinstra, Poly(Propylene Carbonate), Old Copolymers of Propylene Oxide and Carbon Dioxide with New Interests: Catalysis and Material Properties, Polym. Rev., 2008, 48(1), 192–219 CrossRef CAS
- P. O. Peterson, S. M. Rummelt, B. M. Wile, S. C. E. Stieber, H. Zhong and P. J. Chirik, Direct Observation of Transmetalation from a Neutral Boronate Ester to a Pyridine(Diimine) Iron Alkoxide, Organometallics, 2020, 39, 201–205 CrossRef CAS PubMed
-
A. Denk and B. Rieger, Biobased Synthesis and Biodegradability of CO2-Based Polycarbonates, Advances in Polymer Science, Springer Berlin Heidelberg, Berlin, Heidelberg, 2022, DOI:10.1007/12_2022_116
- Y. Liu and X.-B. Lu, Chemical Recycling to Monomers: Industrial Bisphenol-A-Polycarbonates to Novel Aliphatic Polycarbonate Materials, J. Polym. Sci., 2022, 60(24), 3256–3268 CrossRef CAS
- R. Petrus, D. Bykowski and P. Sobota, Solvothermal Alcoholysis Routes for Recycling Polylactide Waste as Lactic Acid Esters, ACS Catal., 2016, 6(8), 5222–5235 CrossRef CAS
- V. Aryan, D. Maga, P. Majgaonkar and R. Hanich, Valorisation of Polylactic Acid (PLA) Waste: A Comparative Life Cycle Assessment of Various Solvent-Based Chemical Recycling Technologies, Resour, Conserv. Recycl., 2021, 172(105670), 105670 CrossRef CAS
- B. C. Almeida, P. Figueiredo and A. T. P. Carvalho, Polycaprolactone Enzymatic Hydrolysis: A Mechanistic Study, ACS Omega, 2019, 4(4), 6769–6774 CrossRef CAS
- E. Cheung, C. Alberti, S. Bycinskij and S. Enthaler, Zinc-catalyzed Chemical Recycling of Poly(E-caprolactone) Applying Transesterification Reactions, ChemistrySelect, 2021, 6(31), 8063–8067 CrossRef CAS
- J. G. Kim, Chemical Recycling of Poly(Bisphenol A Carbonate), Polym. Chem., 2020, 11(30), 4830–4849 RSC
- R. Singh, S. Shahi and Geetanjali, Chemical Degradation of Poly(Bisphenol A Carbonate) Waste Materials: A Review, ChemistrySelect, 2018, 3(42), 11957–11962 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: X-Ray, NMR, FT-IR, and SEC data. CCDC 2292414. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi01833d |
| This journal is © the Partner Organisations 2024 |