
Dinitrogen activation by a titanium hydride complex supported by 2-butene ligand†
Xianghui
Shi
 a,
Yongliang
Zhang
a,
Mingdong
Zhong
a,
Rui
Feng
a,
Yuanjin
Chen
a,
Lei
Yu
a,
Yue
Wu
a,
Junnian
Wei
*a and
Zhenfeng
Xi
ab
a,
Yongliang
Zhang
a,
Mingdong
Zhong
a,
Rui
Feng
a,
Yuanjin
Chen
a,
Lei
Yu
a,
Yue
Wu
a,
Junnian
Wei
*a and
Zhenfeng
Xi
ab
aBeijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China. E-mail: jnwei@pku.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Shanghai, 200032, China
First published on 9th November 2023
Abstract
The activation of dinitrogen by a titanium hydride complex with two bridging hydrides located between metal Ti and Li was investigated. Exposing cyclic bis-alkylidene titanium complex 1 to H2 in THF solution yielded titanium hydride 2, {[(2-butene)(C5H5)Ti](μ-H)2Li(THF)3}. 2 aggregated into dimer 3 in hexane, accompanied by the release of LiH. Treatment of 2 with N2 (1 atm) in THF resulted in ionic end-on bridged dinitrogen dititanium complex 4, {[((2-butene)(C5H5)Ti)2(μ2-η1,η1-N2)][Li(THF)4]}, without using external reducing agents. Furthermore, 4 could be oxidized to yield the binuclear complex 5, {[(2-butene)(C5H5)Ti]2(μ2-η1,η1-N2)}. The solid-state structures of 4 and 5, as revealed by X-ray crystallographic studies, exhibit relatively short N–N bond distances of 1.198(2) Å and 1.179(2) Å, respectively. These results align with the observed N–N Raman stretching frequencies at 1699 cm−1 and 1704 cm−1. The N2 moiety bridging two titanium atoms could be converted into ammonia and hydrazine upon treatment with a proton source and reductant.
Introduction
N2 gas is the predominant and most cost-effective nitrogen source on the Earth. However, its chemical inertness, attributed to its non-polarity, large HOMO–LUMO gap, high bond dissociation energy (942 kJ mol−1), and negative electron affinity, hinders its direct utilization. The activation and transformation of N2 have been long-standing challenges, drawing considerable attention over the past few decades.1 Among various strategies for dinitrogen fixation, metal hydride complexes have already shown great potential. Hydridic hydrogen (H−) can serve as an electron and/or hydrogen donor, facilitating the dinitrogen activation and reduction in heterogeneous, biological, and homogeneous reaction fields via more accessible pathways.2 For example, the main group metal hydrides (LiH, CaH2 and BaH2) have been reported to effectively convert N2 gas into ammonia (NH3)3 and diverse N-containing organic compounds.4For transition metal hydrides, they inextricably served as a platform for dinitrogen activation since the discovery of the dinitrogen-containing cobalt complex [(Ph3P)3Co(N2)H], which reacted with H2 to obtain the cobalt hydride complex [(Ph3P)3CoH3] reversibly.5 To date, many molecular transition metal hydride complexes, especially titanium hydrides, have been found to activate the N2 molecule to varying extents.2b Mach and co-workers reported the synthesis of titanocene monohydride species [(C5HMe4)2TiH], which reduced N2 to produce a dinuclear end-on dinitrogen complex.6 By modifying the substituents on the cyclopentadienyl (Cp) ligand, the corresponding side-on coordination mode [(η5-C5H2-1,2,4-Me3)2Ti]2(μ2-η2,η2-N2) could be prepared by H2 reductive elimination from the in situ generated titanocene hydride derivative.7 Recently, Hou and co-workers disclosed many elegant titanium polyhydride complexes, capable of cleaving the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds and facilitating dinitrogen derivatization.8 Despite the significant interest and progress in this field, the study of titanium hydride activation of N2 is still in its early stages.
N bonds and facilitating dinitrogen derivatization.8 Despite the significant interest and progress in this field, the study of titanium hydride activation of N2 is still in its early stages.
For over 25 years, our research group has been dedicated to the study of bimetallic reagents, with a particular emphasis on 1,4-dilithio-1,3-butadiene species.9 As an extension of our work, we continue to expand this field to prepare the corresponding titanium hydrides and explore their reactivities with N2 gas. In this work, by hydrogenating titanacyclopentatriene complex 1,10 stemming from the 1,4-dilithio-1,3-butadiene precursor, we obtained Ti-hydride complex 2, {[(2-butene)(C5H5)Ti](μ-H)2Li(THF)3}. The subsequent reaction of 2 with dinitrogen led to the formation of Ti-N2 complexes 4 and 5, {[((2-butene)(C5H5)Ti)2(μ2-η1,η1-N2)][Li(THF)4]} and {[(2-butene)(C5H5)Ti]2(μ2-η1,η1-N2)}, respectively. It is noteworthy that 4 has a [TiN2Ti] unit with the odd-electron reduction, which is rare in the literature.11 Furthermore, NH3 and N2H4 generation from the titanium dinitrogen complex with the addition of acids and KC8 was also discussed.
Results and discussion
Synthesis and characterization
Exposing our previously reported cyclic bis-alkylidene titanium complex 1 to H2 in THF solution resulted in the formation of 2. When 2 was crystallized from Et2O, dark-blue crystals were obtained with 68% yield (Scheme 1). The 1H NMR spectrum of 2 shows a well-resolved singlet at 1.57 ppm in d6-benzene, which integrates as 2 H per 2-butene or Cp ligand, assigned to the titanium-hydride ligand. To further confirm the attribution of the bridged Ti–H signal, we carried out the HSQC 2D spectroscopy test. Additionally, the corresponding deuterated analogue 2-D was synthesized and exhibited a resonance at 1.35 ppm in the 2H NMR spectra. The hydride chemical shift in 2 is slightly more upfield than those observed in the trinuclear titanium heptahydride complex {[(C5Me4SiMe3)Ti]3(μ3-H)(μ2-H)6} (δ 2.66 ppm)8a and mononuclear dihydride [1,3-(Me3)2C5H3TiH2] (δ 2.62 ppm).12 Under an inert atmosphere, 2 remains stable for two days in its solid state at 25 °C. But it is unstable in solution, as observed by generation of unknown by-products in the 1H NMR spectra.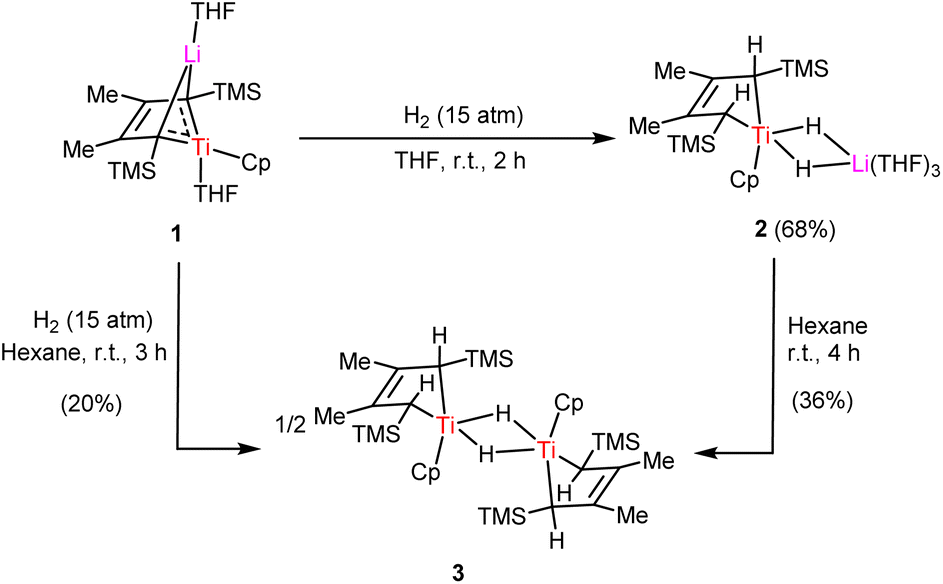 | ||
| Scheme 1 Syntheses of 2 and 3. | ||
The solid-state structure of 2 was determined by X-ray crystallographic analysis. As shown in Fig. 1, the Ti center is coordinated with the Cp, the dianionic 2-butene ligand and two bridging hydride units. The geometry of {Ti(μ-H)2Li(THF)3} is similar to those reported in {[(C5Me5)2Ti(μ-H)2]2Mg}13 and [(C5Me5)2Ti(μ-H)2Li(TMEDA)],14 where the hydride ligands bridged between the Ti and other metal atom. Ti–H bond distances of 1.65(4) Å and 1.70(2) Å in 2 are found to be comparable to that in the titanium hydride complex {[(PNP)Ti]2(μ2-NH)(μ2-N)H} (PNP = N(C6H3-2-PiPr2-4-CH3)2, 1.68(5) Å).8c However, they are notably shorter than the distance of 1.84(2) Å in the cationic titanium hydride [(C5Me5)(tBu3PN)TiH(THF)][B(C6F5)4].15 The C2–C3 bond distance of 1.408(3) Å is shorter than C1–C2 and C3–C4 bond distances (1.438(3) Å and 1.439(3) Å) in 2, suggestive of double-bond character.
 | ||
| Fig. 1 ORTEP drawing of 2 with thermal ellipsoids drawn at the 35% probability level. Except for the bridging hydrides, all the hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Ti1–H1 1.70(2), Ti1–H2 1.65(4), Ti1–Cp(cent) 2.051, C1–C2 1.438(3), C2–C3 1.408(3), C3–C4 1.439(3), H1–Ti1–H2 73(1). (Cent: centre of the Cp ring.) | ||
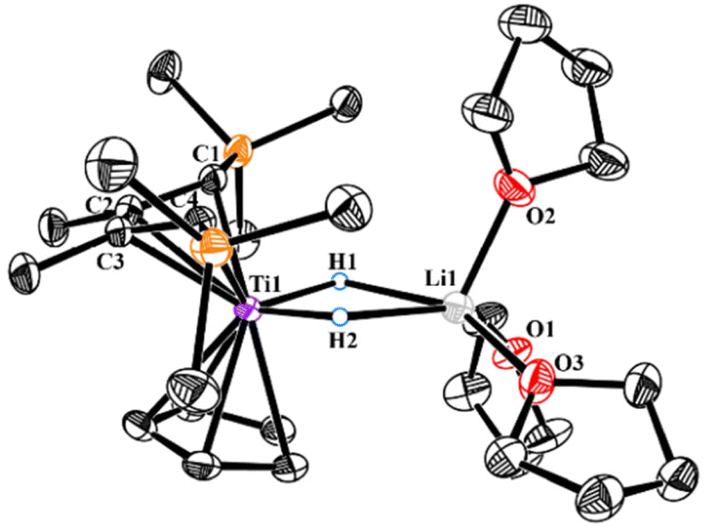
Stirring the hexane solution of 2 led to a gradual colour change over 4 h from dark-blue to brown. After removing all volatiles and re-dissolving the residual solids in Et2O, purple 3 could be crystallized in 36% isolated yield. It is worth noting that the alternative ways to prepare 3 were involved in hydrogenation of 1 in hexane with low yield, or in aromatic solvents to get the mixture of 2 and 3. In the 1H NMR spectrum, newly formed hydride signals of Ti–H and methine C–H were displayed at 2.11 ppm and −5.18 ppm, respectively. X-ray analysis of 3 revealed a dimeric centro-symmetrical molecule (Fig. 2) The planar, rhombic [Ti(μ-H)2Ti] core consists of two 3-center-2e {Ti–H–Ti} bonds with Ti–H distances of 1.88(2) Å and 1.87 (2) Å, falling into the range (1.71–2.08 Å) of those previously reported in titanium bridging hydrides.16 Attempts to generate the corresponding dinitrogen complex by reacting 3 with N2 in THF were unsuccessful. Only unidentified products were detected from interval NMR experiments.
 | ||
| Fig. 2 ORTEP drawing of 3 with thermal ellipsoids drawn at the 35% probability level. Except for the bridging hydrides, all the hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Ti1–H1 1.88(2), Ti1–H1i 1.87(2), Ti1–Cp(cent) 2.044, H1–Ti1–H1i 65.3(8). (Cent: centre of the Cp ring.) | ||
Treatment of 2 with an atmosphere of N2 in THF for 2 days afforded dinitrogen 4. A moderate yield of 45% was achieved, hindered by the decomposition of 2 in THF. The 1H NMR spectrum of 4 in d6-benzene displayed a series of broad signals, typical of the paramagnetic complex with a solution magnetic moment of 2.03μB (measured by Evans’ method in d6-benzene) at 25 °C. Additionally, when crystals of 4 were redissolved in toluene at 77 K, an EPR resonance showed at g = 2.006. In the 15N NMR spectrum of 15N-4, a broad signal was presented at δ 394.31 ppm (using NH3 as a standard reference). The N–N stretching frequencies of 4 and 15N-4 were observed at 1699 cm−1 and 1642 cm−1 respectively, in agreement with the marked mass difference between 14N2 and 15N2 (Scheme 2).
 | ||
| Scheme 2 Synthesis of 4. | ||
The crystal structure of 4 contains two crystallographically independent but chemically identical units. The bridging end-on N2 ligand is ligated to two Ti centers, and the THF-coordinated lithium cation (Li+) serves as a counterion (Fig. 3). To the best of our knowledge, just one well-defined titanium dinitrogen complex with a separate alkali–metal cationic fragment has been reported in the complex {[(O3C)Ti]2(μ-N2)K3(THF)3}[K(THF)6] ([O3C] = [(3,5-tBu2-2-O-C6H2)3C]4−).17 It should be noted that 4 contains an odd-electron reduction of the [TiN2Ti] unit. A parallel instance has only been documented by the Ohki group, who reported a trace titanium dinitrogen complex {[(C5Me5)3Mo3S4Ti]2(μ-N2)[K3(THF)5]}.11 The N–N bond distances are 1.198(2) Å and 1.200(2) Å for each independent molecule in 4, comparable to those seen in similar titanium derivatives {[Cp2Ti(PMe3)]2(μ-N2)} (1.191 (8) Å)18 and {[(TrenTMS)Ti]2(μ-N2)} (TrenTMS = N(CH2CH2NSiMe3)3), 1.121 (6) Å.19 However, they are shorter than those in {[(Me2NC(NiPr)2)2Ti]2(μ-N2)} (1.28(1) Å),20 {[((Nacnac)CpTi)2(μ-N2)]} (Nacnac = [HC(C(Me)N(C6H3)(iPr)2)2], 1.273(6) Å)21 and {(C5Me5)Ti[N(iPr)C(Me)N(iPr)]2(μ-N2)} (1.270(2) Å).22 The bond distance of Ti1–N1 (1.903 (2) Å) is longer than that of {[(TrenTMS)Ti]2(μ-N2)} (2.022 (3) Å), but shorter than that of {[(TrenTMS)Ti]2(μ-η1,η1,η2,η2-N2K2)} (1.812 (2) Å) (TrenTMS = N(CH2CH2NSiMe3)3), inferring some Ti-imido character.19 In view of the corresponding d(NN) values of free N2 and N2H2 at 1.098(1) Å and 1.45 Å, along with structural and Raman spectroscopic data of 4, it has been suggested that the dinitrogen bond was moderately activated. Moreover, the Ti–Cp(centroid) and Ti–C(Cp ring) distances in 4 are slightly longer than the corresponding bond distances in titanium hydride complexes 2 and 3.
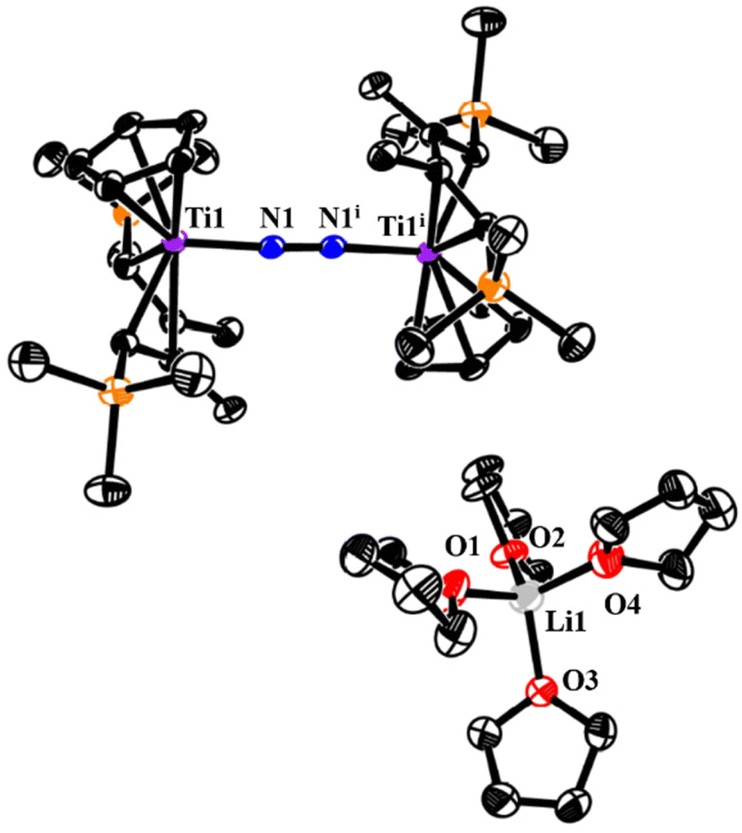 | ||
| Fig. 3 ORTEP drawing of 4 with thermal ellipsoids drawn at the 35% probability level. All the hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: N1–N1i 1.198(2), Ti1–N1 1.903(2), Ti1–Cent1 2.098, Ti1–N1–N1i 176.3(2). (Cent: centre of the Cp ring.) | ||
The oxidation of 4 in the presence of [(Cp2Fe)(BArF4)] led to the formation and isolation of a new purple species 5 in 95% yield Scheme 31H and 13C NMR characterization of 5 revealed that it was a diamagnetic complex with resonances assignable to intact 2-butene and Cp ligands. The 15N NMR spectrum of 15N-5 depicts one apparent singlet at δ 656.61 ppm. Raman spectra of 5 and 15N-5 demonstrate strong vibration peaks (1704 cm−1 for 5; 1647 cm−1 for 15N-5), assigned to the bridging N2 ligands. Notably, in the absence of air and moisture, the dinitrogen complex 5 was quite stable in the solid state or d6-benzene solution for up to two weeks without decomposition at room temperature.
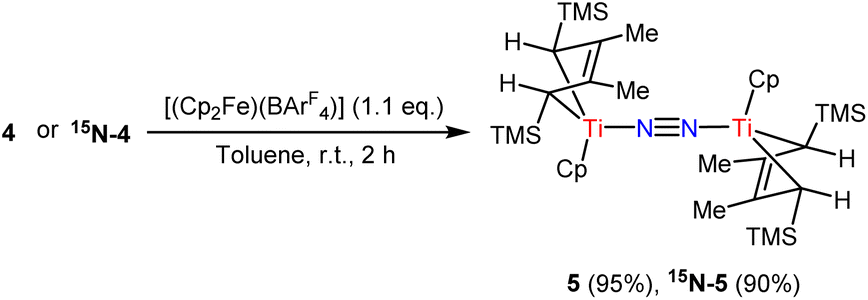 | ||
| Scheme 3 Synthesis of 5. | ||
Single crystals of 5 suitable for X-ray analysis were grown from slow volatilization of a concentrated hexane/Et2O solution at room temperature (Fig. 4). Crystallographic studies confirm it a centrosymmetric dinuclear complex, in which the middle of the N–N bond acts as the inversion center. The Ti–N–N–Ti skeleton is nearly linear with the Ti1–N1–N1i bond angle of 175.9(1)°. The N–N bond distance of 1.179(2) Å is consistent with a weakly activated N2 ligand and compares well with other reported titanocene dinitrogen complexes such as {[Cp′2Ti]2(μ-N2)} (Cp′ = (C5Me4H), 1.170(4) Å;6 (C5Me5), 1.160(14) Å;23 [C5H3-1,3-(SiMe3)2], 1.164(5) Å (ref. 24)), {[(C5H5)2Ti(p-CH3C6H4)]2(μ-N2)} (1.162(12) Å)25 and [{(C5Me5)Ti(η6-C5H4CR2)}2(μ-N2)], (R = p-CH3C6H4, 1.160(3) Å; CR2 = adamantylidene, 1.160(5) Å).26 The relatively low activation degree of the N2 moiety in 5 was mainly attributed to the donor/acceptor properties of Cp and 2-butene co-ligands as well as the oxidation state of Ti centers.26b
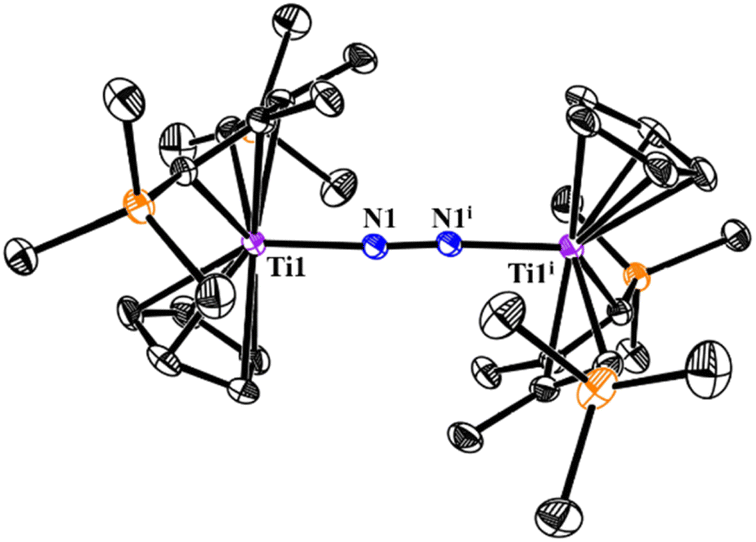 | ||
| Fig. 4 ORTEP drawing of 5 with thermal ellipsoids drawn at the 35% probability level. All the hydrogen atoms are omitted for clarity. Selected interatomic distances [Å] and angles [°]: N1–N1i 1.179(2), Ti1–N1 1.945(1), Ti1–Cent1 2.050, Ti1–N1–N1i 175.9(1). (Cent: centre of the Cp ring.) | ||
The reaction of 4 with commonly used electrophilic reagents including CO2, methyl triflate, pinacolborane, and phenylsilane resulted in either no reaction or the formation of 5 along with other unknown products. From cyclic voltammetry measurements of 4, the reversible peak at −2.923 V indicates a chemically accessible one-electron reduction of 4. However, after many trials, treating 4 with less or equivalent amount of different reducing agents in various solvents only gave intractable products.
Next, we investigated the reactivity of 4 and 5 in the presence of reductants and weak proton sources under a N2 atmosphere (Scheme 4). The data are summarized in Table S1.† Utilizing KC8 (72 equv.) as the reductant and [(Ph2NH2)(OTf)] (78 equv.) as the proton source resulted in a mixture of ammonia and hydrazine (92% and 7% based on 5 respectively, in Table S1,† entry 14). No significant enhancement of the yields was observed even with larger amounts of reductants and proton sources. This result suggests that 5 is ineffective as a catalyst toward N2 reduction, although a stoichiometric transformation of the bridged-dinitrogen ligand is achieved. 4 can also be converted into ammonia and hydrazine under similar conditions (see Table S1† for more details). Inspired by the above results, we replaced proton sources with several electrophilic reagents, in hopes of getting nitrogenous organic compounds. Unfortunately, no clear target products were detected by the GC-MS method.
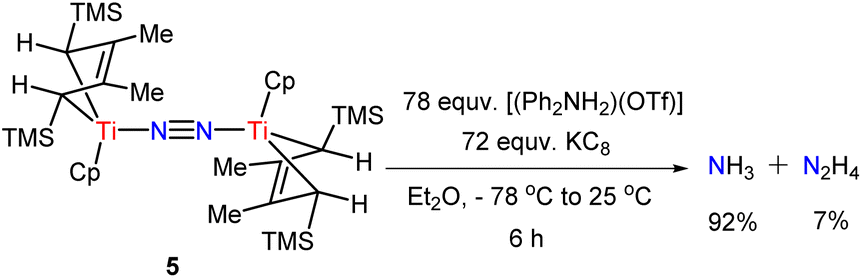 | ||
| Scheme 4 Protonation to produce a stoichiometric amount of N2H4 and NH3 with a proton source and reductant. | ||
Electronic structure
To gain further insight into the electronic structure of 4, DFT calculations have been performed. The computations were performed using ORCA 5.0.3.27,28 Geometric structures were optimized employing the r2SCAN-3c level of theory.29–31 Doublet state calculation was carried out for 4. The optimized structure showed good agreement with the crystal structure. Wave function analysis was performed by the Multiwfn software.32The Singly Occupied Molecular Orbital (SOMO) of 4 is depicted in Fig. 5. Utilizing the Fuzzy atom partitioning method for spin population analysis, we observed that the unpaired electron is predominantly delocalized over the [Ti–N2–Ti] core. Specifically, two titanium atoms account for 68%, while the dinitrogen unit contributes 22%. For other related molecular orbitals of 4, see the ESI.† Fuzzy Bond Order (FBO) analysis is a reputable bond order index developed by Mayer and Salvador, which agrees well with classical chemical concepts and shows minimal basis sensitivity.33 For 4, the FBO of Ti–N bonds is 1.58, while that of the N–N bond has a bond order of 1.96. These results affirm the partial multiple-bond character of Ti–N and a moderate degree of dinitrogen reduction.
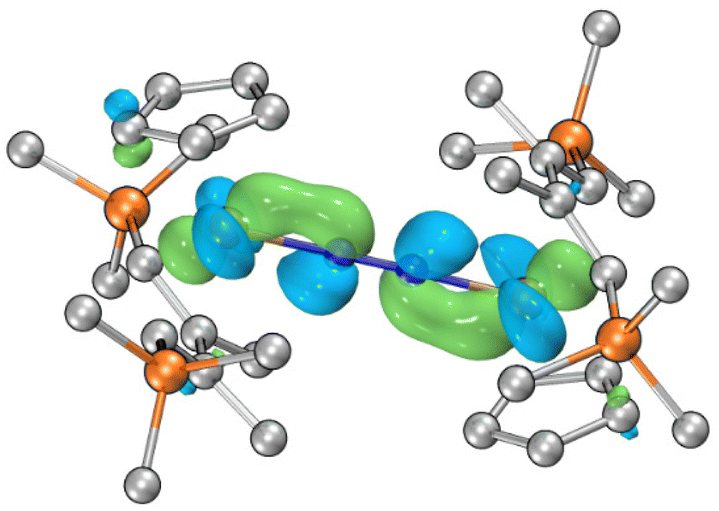 | ||
| Fig. 5 SOMO of 4. | ||
Conclusions
In summary, we report the synthesis of a well-defined titanium hydride 2 supported by both 2-butene and Cp ligands, resulting from hydrogenolysis of the corresponding precursor. 2 can react with N2 gas to yield anionic end-on dinitrogen-bridged titanium complex 4. 4 readily undergoes a one-electron oxidation to give neutral titanium dinitrogen complex 5. The resulting products 4 and 5 achieve moderate reduction of dinitrogen as evidenced by N–N bond distances and Raman vibrational frequencies of the N–N stretching band. In addition, we made preliminary efforts to investigate the reactivity of the N2 unit in 4 and 5. In the presence of an excess of protons and KC8, the bridged dinitrogen ligand was able to afford stoichiometric quantities of ammonia and hydrazine under mild conditions.Experimental
General remarks
All reactions were carried out under an atmosphere of dinitrogen or argon by using standard Schlenk techniques or glovebox techniques. Toluene, hexane, and Et2O were purified using an Mbraun SPS-800 Solvent Purification System and dried over fresh Na chips and molecular sieves in a glovebox. THF was distilled from Na/benzophenone ketyl and dried over fresh Na chips and molecular sieves in the glovebox. Cyclic bis-alkylidene titanium complex 1 was synthesized according to the published procedures.10 Other commercially available reagents were used without purification.Samples for NMR spectroscopic measurements were prepared in a glovebox by the use of J. Young valve NMR tubes. 1H, 13C, and 15N NMR spectra were recorded on a Bruker ARX400 spectrometer or a Bruker AVENCE 600 MHz spectrometer at room temperature unless otherwise noted. All chemical shifts were reported in units of ppm with references to the residual protons of the deuterated solvents for proton chemical shifts and the 13C of deuterated solvents for carbon chemical shifts. 15N chemical shifts were reported in ppm relative to liquid NH3 at 0 ppm. UV/Vis absorption spectra were obtained on an Agilent Cary 60 UV-Vis spectrophotometer. Magnetic moments were measured by the method originally described by Evans and experimental solutions containing a known amount of a TMSOTMS standard.34 Elemental analyses were conducted on a Vario EL elemental analyzer at the Analytical Center of Peking University. Raman Spectra were recorded on a Thermofisher DXRxi Micro Raman imaging spectrometer. X-ray data collections were performed at 180 K on a Rigaku diffractometer, using monochromated Mo Kα radiation (λ = 0.71073 Å). The structures were solved by using the SHELXTL program35 in Olex2.36 Refinement was performed on F2 anisotropically for all the non-hydrogen atoms by the full-matrix least-squares method with the XL refinement package.37 The positions of the H atoms were placed in calculated positions using an appropriate riding model.
Cyclic voltammetry experiment of 4
The oxidation–reduction properties of 4 were studied using a cyclic voltammetry experiment in THF with [(Bu4N)(PF6)] as the supporting electrolyte. All potentials are reported vs. FeCp2. 4 had one reversible reduction peak at E1/2 = −2.923 V and one oxidation peak at −1.709 V (Fig. S22:†E1/2(Fc+/Fc) = 0.14 V) at the beginning of the experimental test. However, attempts to isolate the reduction product of complex 4 were hampered due to the poor stability.General procedure for reduction of N2 into NH3 and N2H4 by 5
Under an atmosphere of dinitrogen, 5 (0.01 mmol), reductant KC8 and proton sources ([(Ph2NH2)(OTf)], [H(Et2O)2(BArF4)] or [(HPCy3)(BF4)]) were mixed in a 50 mL Schlenk flask at −78 °C, and then pre-cooled Et2O (5 mL, −78 °C) was added. The mixture was stirred −78 °C for 1 h and warmed slowly to room temperature. After the solution was stirred for 1 h at room temperature, and HCl (100 eq., 1 M in 1,4-dioxane) was added. The resulting solvent was removed under reduced pressure and the residue was extracted with H2O (10.0 mL). The amounts of NH3 and N2H4 were precisely determined by the indophenol method38 and the p-(dimethylamino)benzaldehyde method,39 respectively.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CMe) ppm. UV-vis (THF): 480, 600 nm. Anal. calcd for C29H57O3Si2Li1Ti1: C, 61.68; H, 10.17. Found: C, 61.34; H, 10.03. The synthesis of 2-D was analogous to that of complex 2 except that the deuteride gas (D2) was used (1 atm D2, 6 h). 2H NMR (77 MHz, C6H6): δ −1.20 (br, TiCD), 1.35 (s, TiD) ppm.
CMe) ppm. Anal. calcd for C34H64Si4Ti2: C, 59.97; H, 9.47. Found: C, 59.73; H, 9.22.
CMe) ppm. UV-vis (THF): 480, 600 nm. Anal. calcd for C29H57O3Si2Li1Ti1: C, 61.68; H, 10.17. Found: C, 61.34; H, 10.03. The synthesis of 2-D was analogous to that of complex 2 except that the deuteride gas (D2) was used (1 atm D2, 6 h). 2H NMR (77 MHz, C6H6): δ −1.20 (br, TiCD), 1.35 (s, TiD) ppm.
CMe) ppm. Anal. calcd for C34H64Si4Ti2: C, 59.97; H, 9.47. Found: C, 59.73; H, 9.22.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to 15 atm. The mixture was left stirring at room temperature for 48 h, then quickly moved into the glovebox. After evaporation of all the volatiles in vacuo, the residue was washed twice with hexane, and then extracted with Et2O. The solvent was removed completely under reduced pressure to give 4 (145 mg, 0.14 mmol, 29% yield). 1H NMR (400 MHz, C6D6): δ −0.06 (br, 18H, Si(CH3)3), 1.40 (m, 16H, THF), 2.19 (br, 6H, CH3), 3.57 (m, 16H, THF), 6.59 (br, 5H, Cp) ppm. 13C NMR (101 MHz, C6D6): 0.41 (s, Si(CH3)3), 4.42 (s, CH3), 26.33 (s, THF), 68.82 (s, THF), 102.93 (s, Cp), 129.90 (s, MeCCMe) ppm. (Due to the paramagnetism of 4, not all signals were obtained.) Raman (νN–N): 1699 cm−1. μeff (Evans’ method, 400 MHz, 17% v/v HMDSO in C6D6): 2.03μB. UV-vis (THF): 460 nm. Anal. calcd for C50H94O4N2Si4Li1Ti2: C, 59.92; H, 9.45; N, 2.79. Found: C, 59.29; H, 9.17; N, 2.67.
CMe) ppm. Raman (νN–N): 1704 cm−1. UV-vis (THF): 495, 540 nm. Anal. calcd for C34H62N2Si4Ti2: C, 57.76; H, 8.84; N, 3.96. Found: C, 57.27; H, 8.69; N, 3.72.
:1) to 15 atm. The mixture was left stirring at room temperature for 48 h, then quickly moved into the glovebox. After evaporation of all the volatiles in vacuo, the residue was washed twice with hexane, and then extracted with Et2O. The solvent was removed completely under reduced pressure to give 4 (145 mg, 0.14 mmol, 29% yield). 1H NMR (400 MHz, C6D6): δ −0.06 (br, 18H, Si(CH3)3), 1.40 (m, 16H, THF), 2.19 (br, 6H, CH3), 3.57 (m, 16H, THF), 6.59 (br, 5H, Cp) ppm. 13C NMR (101 MHz, C6D6): 0.41 (s, Si(CH3)3), 4.42 (s, CH3), 26.33 (s, THF), 68.82 (s, THF), 102.93 (s, Cp), 129.90 (s, MeCCMe) ppm. (Due to the paramagnetism of 4, not all signals were obtained.) Raman (νN–N): 1699 cm−1. μeff (Evans’ method, 400 MHz, 17% v/v HMDSO in C6D6): 2.03μB. UV-vis (THF): 460 nm. Anal. calcd for C50H94O4N2Si4Li1Ti2: C, 59.92; H, 9.45; N, 2.79. Found: C, 59.29; H, 9.17; N, 2.67.
CMe) ppm. Raman (νN–N): 1704 cm−1. UV-vis (THF): 495, 540 nm. Anal. calcd for C34H62N2Si4Ti2: C, 57.76; H, 8.84; N, 3.96. Found: C, 57.27; H, 8.69; N, 3.72.
Author contributions
X. S., Y. Z., M. Z., L. Y., and Y. W. carried out the syntheses and characterization of 2–5. R. F. and Y. C. helped the analyses of crystallographic data. J. W. performed DFT calculations and analysed the data. Z. X. and J. W. conceived and conceptualized this project. X. S., J. W., and Z. X. prepared and revised the manuscript. All authors participated in discussions of the research and the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21988101 and 22201013) and the Beijing Natural Science Foundation (No. 2222008). We thank Dr Qiong Yuan and Mr Peng-Xiang Fu for EPR analysis. We thank the Analytical Instrumentation Center at Peking University for the NMR measurements and the High-performance Computing Platform of Peking University for the DFT calculations.References
- (a) P. L. Holland, Nitrogen Fixation, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2003, p. 569 Search PubMed; (b) Transition Metal-Dinitrogen Complexes: Preparation and Reactivity, ed. Y. Nishibayashi, Wiley-VCH, Weinheim, 2019 Search PubMed; (c) Z.-J. Lv, J. Wei, W.-X. Zhang, P. Chen, D. Deng, Z.-J. Shi and Z. Xi, Direct Transformation of Dinitrogen: Synthesis of N-containing Organic Compounds via N–C Bond Formation, Natl. Sci. Rev., 2020, 7, 1564 CrossRef CAS PubMed.
- (a) J. Ballmann, R. F. Munha and M. D. Fryzuk, The Hydride Route to the Preparation of Dinitrogen Complexes, Chem. Commun., 2010, 46, 1013 RSC; (b) T. Shima and Z. Hou, Dinitrogen Fixation by Transition Metal Hydride Complexes, in Nitrogen Fixation, ed. Y. Nishibayashi, Springer, 2017, p. 23 Search PubMed; (c) Q. Wang, Y. Guan, J. Guo and P. Chen, Hydrides Mediate Nitrogen Fixation, Cell Rep. Phys. Sci., 2022, 3, 100779 CrossRef CAS.
- (a) P. Wang, F. Chang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Breaking Scaling Relations to Achieve Low-Temperature Ammonia Synthesis Through LiH-Mediated Nitrogen Transfer and Hydrogenation, Nat. Chem., 2017, 9, 64 CrossRef CAS PubMed; (b) W. Gao, P. Wang, J. Guo, F. Chang, T. He, Q. Wang, G. Wu and P. Chen, Barium Hydride-Mediated Nitrogen Transfer and Hydrogenation for Ammonia Synthesis: A Case Study of Cobalt, ACS Catal., 2017, 7, 3654 CrossRef CAS; (c) F. Chang, Y. Guan, X. Chang, J. Guo, P. Wang, W. Gao, G. Wu, J. Zheng, X. Li and P. Chen, Alkali and Alkaline Earth Hydrides-Driven N2 Activation and Transformation over Mn Nitride Catalyst, J. Am. Chem. Soc., 2018, 140, 14799 CrossRef CAS PubMed.
- (a) X. Shi, Q. Wang, C. Qin, L.-J. Wu, Y. Chen, G.-X. Wang, Y. Cai, W. Gao, T. He, J. Wei, J. Guo, P. Chen and Z. Xi, Synthesis of Pyrimidines from Dinitrogen and Carbon, Natl. Sci. Rev., 2022, 9, nwac168 CrossRef CAS PubMed; (b) L.-J. Wu, Q. Wang, J. Guo, J. Wei, P. Chen and Z. Xi, From Dinitrogen to N-Containing Organic Compounds: Using Li2CN2 as a Synthon, Angew. Chem., Int. Ed., 2023, 62, e202219298 CrossRef CAS PubMed.
- (a) A. Yamamoto, S. Kitazume, L. S. Pu and S. Ikeda, Study of the Fixation of Nitrogen. Isolation of Tris(triphenylphosphine)cobalt Complex Co-ordinated with Molecular Nitrogen, Chem. Commun., 1967, 79 RSC; (b) A. Yamamoto, L. S. Pu, S. Kitazume and S. Ikeda, Reverisble Combination of Mmolecular Nitrogen with a Cobalt Complex. Exchange Reactions of Nitrogen-Tris(triphenylphosphine)cobalt with Hydrogen, Ethylene, and Ammonia, J. Am. Chem. Soc., 1967, 89, 3071 CrossRef CAS; (c) A. Sacco and M. Rossi, Hydride and Nitrogen Complexes of Cobalt, Chem. Commun., 1967, 316 RSC.
- J. M. de Wolf, R. Blaauw, A. Meetsma, J. H. Teuben, R. Gyepes, V. Varga, K. Mach, N. Veldman and A. L. Spek, Bis(tetramethylcyclopentadienyl)titanium Chemistry. Molecular Structures of [(C5HMe4)(μ-η1: η5-C5Me4)Ti]2 and [(C5HMe4)2Ti]2N2, Organometallics, 1996, 15, 4977 CrossRef CAS.
- T. E. Hanna, W. H. Bernskoetter, M. W. Bouwkamp, E. Lobkovsky and P. J. Chirik, Bis(cyclopentadienyl) Titanium Dinitrogen Chemistry: Synthesis and Characterization of a Side-on Bound Haptomer, Organometallics, 2007, 26, 2431 CrossRef CAS.
- (a) T. Shima, S. Hu, G. Luo, X. Kang, Y. Luo and Z. Hou, Dinitrogen Cleavage and Hydrogenation by a Trinuclear Titanium Polyhydride Complex, Science, 2013, 340, 1549 CrossRef CAS PubMed; (b) T. Shima, G. Luo, S. Hu, Y. Luo and Z. Hou, Experimental and Computational Studies of Dinitrogen Activation and Hydrogenation at a Tetranuclear Titanium Imide/Hydride Framework, J. Am. Chem. Soc., 2019, 141, 2713 CrossRef CAS PubMed; (c) B. Wang, G. Luo, M. Nishiura, S. Hu, T. Shima, Y. Luo and Z. Hou, Dinitrogen Activation by Dihydrogen and a PNP-Ligated Titanium Complex, J. Am. Chem. Soc., 2017, 139, 1818 CrossRef CAS PubMed; (d) Z. Mo, T. Shima and Z. Hou, Synthesis and Diverse Transformations of a Dinitrogen Dititanium Hydride Complex Bearing Rigid Acridane-Based PNP-Pincer Ligands, Angew. Chem., Int. Ed., 2020, 59, 8635 CrossRef CAS PubMed; (e) Y.-M. So, C. T. To, M. M. Guru, T. Shima and Z. Hou, Hydrodeoxygenative Coupling and Transformation of Aldehydes at a N2-Derived Tetranuclear Titanium Imide/Hydride Framework, J. Am. Chem. Soc., 2023, 145, 16906 CrossRef CAS PubMed.
- (a) Z. Xi, 1,4-Dilithio-1,3-Dienes: Reaction and Synthetic Applications, Acc. Chem. Res., 2010, 43, 1342 CrossRef CAS PubMed; (b) W.-X. Zhang and Z. Xi, Organometallic Intermediate-Based Organic Synthesis: Organo-Di-lithio Reagents and Beyond, Org. Chem. Front., 2014, 1, 1132 RSC; (c) J. Wei, W.-X. Zhang and Z. Xi, The Aromatic Dianion Metalloles, Chem. Sci., 2018, 9, 560 RSC; (d) Z. Xi, Discovery and Development of Organo-di-lithio Reagents: Serendipity & Persistence, Sci. Sin.: Chim., 2020, 50, 1398 Search PubMed.
- Y. Zhang, B. Wu, M. Zhong, W.-X. Zhang and Z. Xi, Cyclic Bis-alkylidene Complexes of Titanium and Zirconium: Synthesis, Characterization, and Reaction, Chem. – Eur. J., 2020, 26, 16472 CrossRef CAS PubMed.
- Y. Ohki, K. Uchida, M. Tada, R. E. Cramer, T. Ogura and T. Ohta, N2 Activation on a Molybdenum-Titanium-Sulfur Cluster, Nat. Commun., 2018, 9, 3200 CrossRef PubMed.
- M. D. Walter, C. D. Sofield and R. A. Andersen, Preparation and Reactions of Base-Free Bis(1,3-di-tert-butylcyclopentadienyl)titanium, Cp′2Ti, and Related Compounds, Organometallics, 2008, 27, 2959 CrossRef CAS.
- R. Gyepes, K. Mach, I. Cisarova, J. Loub, J. Hiller and P. Sindelar, Easy Formation of Titanocene Hydride-Magnesium Complexes in the (C5H5−nMen)2TiCl2 (n = 3–5)-Dibutylmagnesium Systems, J. Organomet. Chem., 1995, 497, 33 CrossRef CAS.
- W. W. Lukens, P. T. Matsunaga and R. A. Andersen, Synthesis and Structure of Cp*2TiH, Cp*2TiH2Li(tmed), and [Cp*2TiOLi(THF)]2, Organometallics, 1998, 17, 5240 CrossRef CAS.
- K. Ma, W. E. Piers, Y. Gao and M. Parvez, Isolation and Characterization of a Monomeric Cationic Titanium Hydride, J. Am. Chem. Soc., 2004, 126, 5668 CrossRef CAS PubMed.
- T. Matsuo and H. Kawaguchi, Triple-Hydrogen-Bridged Dititanium(III) and Dizirconium(IV) Aryloxide Complexes, Organometallics, 2003, 22, 5379 CrossRef CAS.
- Y. Nakanishi, Y. Ishida and H. Kawaguchi, Nitrogen-Carbon Bond Formation by Reactions of a Titanium-Potassium Dinitrogen Complex with Carbon Dioxide, tert-Butyl Isocyanate, and Phenylallene, Angew. Chem., Int. Ed., 2017, 56, 9193 CrossRef CAS PubMed.
- D. H. Berry, L. J. Procopio and P. J. Carroll, Molecular Structure of {Cp2Ti(PMe3)}2(μ-N2), a Titanocene Dinitrogen Complex, Organometallics, 1988, 7, 570 CrossRef CAS.
- L. R. Doyle, A. J. Wooles, L. C. Jenkins, F. Tuna, E. J. L. McInnes and S. T. Liddle, Catalytic Dinitrogen Reduction to Ammonia at a Triamidoamine-Titanium Complex, Angew. Chem., Int. Ed., 2018, 57, 6314 CrossRef CAS PubMed.
- S. M. Mullins, A. P. Duncan, R. G. Bergman and J. Arnold, Reactivity of a Titanium Dinitrogen Complex Supported by Guanidinate Ligands: Investigation of Solution Behavior and a Novel Rearrangement of Guanidinate Ligands, Inorg. Chem., 2001, 40, 6952 CrossRef CAS PubMed.
- G. Bai, P. Wei and D. W. Stephan, Reductions of β-Diketiminato-Titanium(III) Complexes, Organometallics, 2006, 25, 2649 CrossRef CAS.
- P. P. Fontaine, B. L. Yonke, P. Y. Zavalij and L. R. Sita, Dinitrogen Complexation and Extent of NN Activation within the Group 6 “End-On-Bridged” Dinuclear Complexes, {(η5-C5Me5)M[N(i-Pr)C(Me)N(i-Pr)]}2(μ-η1: η1-N2) (M = Mo and W), J. Am. Chem. Soc., 2010, 132, 12273 CrossRef CAS PubMed.
- R. D. Sanner, D. M. Duggan, T. C. Mckenzie, R. E. Marsh and J. E. Bercaw, Structure and Magnetism of .mu.-Dinitrogen-Bis(bis(pentamethylcyclopentadienyl)titanium(II)), {(.eta.5-C5(CH3)5)2Ti}2N2, J. Am. Chem. Soc., 1976, 98, 8358 CrossRef CAS.
- T. E. Hanna, I. Keresztes, E. Lobkovsky, W. H. Bernskoetter and P. J. Chirik, Synthesis of a Base-Free Titanium Imido and a Transient Alkylidene from a Titanocene Dinitrogen Complex. Studies on Ti=NR Hydrogenation, Nitrene Group Transfer, and Comparison of 1,2-Addition Rates, Organometallics, 2004, 23, 3448 CrossRef CAS.
- J. D. Zeinstra, J. H. Teuben and F. Jellinek, Structure of μ-Dinitrogen Bis[p-Tolyldicyclopentadienyl-Titanium(III)], [(C5H5)2Ti(p-CH3C6H4)]2N2, J. Organomet. Chem., 1979, 170, 39 CrossRef CAS.
- (a) A. Scherer, K. Kollak, A. Lützen, M. Friedemann, D. Haase, W. Saak and R. Beckhaus, Low-Valent Titanium-Pentafulvene Complexes-Formation of Dinuclear Titanium-Nitrogen Complexes, Eur. J. Inorg. Chem., 2005, 2005, 1003 CrossRef; (b) F. Studt, N. Lehnert, B. E. Wiesler, A. Scherer, R. Beckhaus and F. Tuczek, Spectroscopic Comparison of Dinuclear Ti+ and Ti2+ μ-η1: η1 Dinitrogen Complexes with Cp*/Pentafulvene and Amine/Amide Ligation: Moderate versus Strong Activation of N2, Eur. J. Inorg. Chem., 2006, 2006, 291 CrossRef.
- F. Neese, The ORCA Program System, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CAS.
- F. Neese, Software Update: The ORCA Program System, Version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew and J. Sun, Accurate and Numerically Efficient r2SCAN Meta-Generalized Gradient Approximation, J. Phys. Chem. Lett., 2020, 11, 8208 CrossRef CAS PubMed.
- E. Caldeweyher, C. Bannwarth and S. Grimme, Extension of the D3 Dispersion Coefficient Mode, J. Chem. Phys., 2017, 147, 034112 CrossRef PubMed.
- S.-H. Chong and S. Ham, Aqueous Interaction Site Integral-equation Theory that Exactly Takes into Account Intramolecular Correlations, J. Chem. Phys., 2012, 137, 154101 CrossRef PubMed.
- T. Lu and F. Chen, Multiwfn: A Multifunctional Wavefunction Analyzer, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- I. Mayer and P. Salvador, Overlap Populations, Bond Orders and Valences for ‘Fuzzy’ Atoms, Chem. Phys. Lett., 2004, 383, 368 CrossRef CAS.
- (a) D. F. Evans, The Determination of the Paramagnetic Susceptibility of Substances in Solution by Nuclear Magnetic Resonance, J. Chem. Soc., 1959, 2003 RSC; (b) S. K. Sur, Measurement of Magnetic Susceptibility and Magnetic Moment of Paramagnetic Molecules in Solution by High-Field Fourier Transform NMR Spectroscopy, J. Magn. Reson., 1989, 82, 169 CAS.
- G. M. Sheldrik, A Short History of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: A Complete Structure Solution, Refinement and Analysis Pprogram, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrik, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- M. W. Weatherburn, Phenol-Hypochlorite Reaction for Determination of Ammonia, Anal. Chem., 1967, 39, 971 CrossRef CAS.
- G. W. Watt and J. D. Chrisp, Spectrophotometric Method for Determination of Hydrazine, Anal. Chem., 1952, 24, 2006 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectroscopic, and crystallographic details. CCDC 2284338–2284341. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi01911j |
| This journal is © the Partner Organisations 2024 |