
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licencemeso-Aryl substituents modify the electrochemical profile and palladium(II) coordination of redox-active tripyrrin ligands†
Iva
Habenšus
and
Elisa
Tomat
 *
*
Department of Chemistry and Biochemistry, The University of Arizona, 1306 E. University Blvd., Tucson, AZ 85721-0041, USA. E-mail: tomat@arizona.edu
First published on 12th February 2024
Abstract
The tripyrrin coordination motif, namely a conjugated tripyrrolic fragment of the porphyrin scaffold, is found in numerous metal complexes of oligopyrrolic macrocycles. Because of their typically limited stability, linear tripyrrins are underutilized in coordination chemistry; however, hexaalkyl-tripyrrindiones featuring the pyrrolinone termini characteristic of biopyrrin pigments have recently emerged as versatile redox-active ligands. Herein, we report the synthesis of the first example of meso-di(pentafluorophenyl) tripyrrin-1,14-dione through the demethylation of a stable 1,14-dimethoxytripyrrin precursor. The two tripyrrin ligands coordinate palladium(II) in square planar geometries in completely distinct ways: the dimethoxytripyrrin forms a cyclopalladate following C(sp3)–H bond activation at one of the methoxy groups, whereas the tripyrrindione binds as a trianionic ligand engaging an adventitious aqua ligand in hydrogen-bonding interactions. When compared to the hexaalkyl substituents of previous tripyrrindiones, the meso-aryl groups significantly alter the electrochemical profile of the Pd(II) tripyrrindione complex, shifting anodically by ∼500 mV the one-electron processes to attain the other redox states of the ligand. The formation of a ligand-based radical on the Pd(II)-bound meso-aryl tripyrrindione is confirmed by spectroelectrochemical and electron paramagnetic resonance (EPR) methods.
Introduction
Linear oligopyrroles have been known for several decades as naturally occurring and synthetic pigments.1 Among the tripyrrolic compounds, tripyrranes (in which the pyrrole rings are bridged by methylene groups) are important precursors for the synthesis of porphyrinoid macrocycles,2,3 including texaphyrin-based drug candidates.4,5 In contrast, conjugated tripyrrins (Fig. 1), with methine bridges between the pyrrole rings, are rather difficult intermediates prone to degradation in the presence of nucleophiles and/or bases.6 Nevertheless, the tripyrrin motif is present in numerous expanded porphyrins7 and was recently incorporated in the design of new fluorophores8,9 and double-helical assemblies.10,11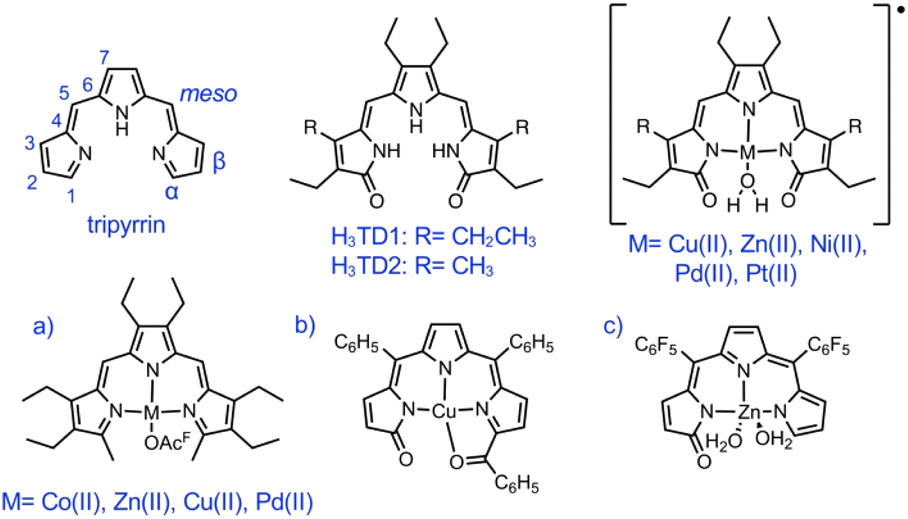 | ||
| Fig. 1 The tripyrrin and tripyrrin-1,14-dione scaffolds and examples of metal complexes featuring 1,14-dimethyltripyrrin (a) and tripyrrin-1-one (b, c) ligands. | ||
Despite the array of pyrrolic nitrogen donors for tridentate binding of metal ions, linear tripyrrins are underutilized in coordination chemistry when compared to tetrapyrrolic and dipyrrolic systems.12 The limited stability of early tripyrrin compounds featuring methine bridges and all-alkyl substitution of the pyrrole rings likely hindered the broad use of these ligands; however, a number of complexes were reported over the years (Fig. 1a),13 including several examples of coordination polymers.14 Interestingly, tripyrrin complexes featuring 5,10-diaryl (aka meso-diaryl) substitution (Fig. 1b) were isolated serendipitously following the oxidative degradation of N-confused porphyrins.15 In fact the ligand in these complexes is tripyrrin-1-one, consistent with the reported tendency of 5,10-diaryl tripyrranes and tripyrrins to oxidize at one of the terminal α position.16,17 A meso-pentafluorophenyl tripyrrin-1-one was later used for the synthesis of a fluorescent zinc(II) complex (Fig. 1c) and for the detection of zinc by fluorescence microscopy in cultured cells.18 The gentle oxidation and subsequent metal insertion of meso-phenyl tripyrrane with chloranil and Ni(OAc)2, respectively, led to a bis(5,10-diphenyl-tripyrrinato) nickel(II) complex of octahedral geometry; however, the tripyrrin intermediate could not be isolated and its oxidative degradation to a black oligomeric material was reported.19
In recent years, tripyrrin-1,14-dione was identified as a robust redox-active platform for metal coordination.20 This biopyrrin motif shares the scaffold of heme metabolites that were initially isolated as urinary pigments.21,22 The β-alkyl substituted tripyrrindione ligands (i.e., H3TD1 and H3TD2, Fig. 1) coordinate multiple divalent transition metals as stable dianionic radicals (e.g., [Pd(TD1˙)(H2O)], Fig. 1).23–27 Easily accessible one-electron processes in these complexes reversibly alter the redox state of the tripyrrindione ligand. In the case of the zinc(II) complex, the tripyrrindione ligand produced a rare example of fluorescence emission from a radical chromophore.25 Multi-center interactions between delocalized spins lead to dimers of tripyrrindione radicals in the solid state and at low temperature in solution.26,28 Although most reported tripyrrindione complexes present an aqua ligand as a monodentate donor within the primary coordination sphere, examples featuring isocyanates26 and primary amines27 underscore the ability to modify the properties of this versatile ligand.
Synthetic methods to prepare the naturally occurring biopyrrins have only become available very recently,29 and all tripyrrindione complexes reported to date have featured ethyl or methyl β-substituents on the pyrrole rings (Fig. 1).26,30 Even with this simplified structure, these syntheses required the multi-step preparation of 3,4-dialkyl pyrrole and did not allow for facile, additional functionalization of the ligand scaffold. To further extend the scope of the tripyrrindione ligand system, herein we report the synthesis and palladium(II) coordination of the first 5,10-diaryl-tripyrrin-1,14-dione. In addition, we include comparisons to a new 1,14-dimethoxy-tripyrrin precursor and its palladium complex, which are instructive in the broader context of delineating the chemistry of tripyrrin ligands.
Results and discussion
Synthesis and characterization of ligands
The previously reported tripyrrindione ligands (i.e., H3TD1 and H3TD2, Fig. 1) were synthesized through condensation of 3,4-diethyl-2,5-diformyl-1H-pyrrole and a 3-pyrrolin-2-one precursor (2 equiv.) under basic conditions.26,30 An analogous condensation strategy failed to produce the desired meso-aryl tripyrrindione when starting from aroyl diketones or benzylic diols as precursors for the central pyrrole.The oxidation of 5,10-bis(pentafluorophenyl)-tripyrrane was then pursued using common oxidants such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), hydrogen peroxide, and peroxycarboxylic acids (e.g., mCPBA). We also tested chloroauric acid, which has been used in the oxidation of meso-pentafluorophenyltetrapyrrane to yield three bilindione isomers.31 These oxidative transformations generally produced tripyrrin-1-one (as previously reported in the case of DDQ)16 and/or a complex mixture of oligomers. Photooxidation of the tripyrrin-1-one, analogous to the synthesis of dipyrrin-1,9-dione from dipyrrin-1-one,32,33 also failed to produce the desired product with two terminal pyrrolin-2-one moieties. Recent reports on the use of 1,14-dibromo-5,10-diaryltripyrrins as substrates for nucleophilic substitutions8,10 prompted us to pursue a synthetic route through this key precursor.
The meso-pentafluorophenyl-tripyrrane was synthesized from pyrrole and pentafluorobenzaldehyde under acidic conditions34,35 and then subjected to bromination with N-bromosuccinimide (NBS) at −78 °C and subsequent oxidation with DDQ to afford 5,10-bis(pentafluorophenyl)-α,α′-dibromotripyrrin (HTBC6F5, Scheme 1) as previously reported.8 Next, nucleophilic substitution with sodium methoxide (3.0 equiv.) afforded 5,10-bis(pentafluorophenyl)-α,α′-dimethoxy-tripyrrin (HTMC6F5, Scheme 1). As previously reported for meso-pentafluorophenyl porphyrins36 and dipyrrins,37 tripyrrin HTMC6F5 is susceptible to reactivity with nucleophiles: an excess of sodium methoxide results in additional methoxy substitution at the para position of the pentafluorophenyl ring through nucleophilic aromatic substitution.
 | ||
| Scheme 1 Synthesis of 5,10-di(pentafluorophenyl)-tripyrrin-1,14-dione (H3TDC6F5). | ||
The new dimethoxytripyrrin ligand HTMC6F5 was purified by chromatography and then precipitated from CH2Cl2/hexane as a red solid. Finally, demethylation of HTMC6F5 with aluminum trichloride in dry CH2Cl2 gave the desired 5,10-bis(pentafluorophenyl)-tripyrrin-1,14-dione (H3TDC6F5), which was purified by chromatography and isolated as an orange solid. Given the availability of tripyrrane precursors with a variety of substituents, this synthetic procedure is expected to allow the future preparation of several other tripyrrindione ligands.
The 1H NMR spectrum of HTMC6F5 in CDCl3 at room temperature shows five distinct signals (Fig. S1†): three resonances for the pyrrolic β-protons, a broad singlet at 12.89 ppm due to the NH proton, and a sharp tall singlet at 4.17 ppm due to the methoxy protons. The successful conversion to H3TDC6F5 was evident in the 1H NMR spectrum of the product (Fig. S2†): the methoxy resonances of the starting material disappeared and the tripyrrin NH signal shifted upfield, overlapping with the other NH protons and integrating to three. In the 13C NMR spectra, the methoxy carbon resonance for the dimethoxytripyrrin is at 57.2 ppm whereas the carbonyl carbon resonance for the tripyrrindione is at 174.8 ppm (Fig. S1 and S2†).
The small number of resonances for H3TDC6F5 indicates a symmetrical conformation of the ligand in solution but does not specify the configuration of the double (Z vs. E) and single bonds (syn vs. anti) between adjacent pyrrole rings. NOESY NMR data, however, showed through-space effects only between the β-CH protons of the terminal pyrrolic units and no interactions between the NH and β-CH protons (Fig. S3†), therefore indicating a ZZ, syn, syn configuration (as drawn in Scheme 1). In addition, the sharp resonance at 10.8 ppm is similar to those observed for β-alkyl-substituted tripyrrindiones30 and dipyrrinones38 forming hydrogen-bonded dimers in chloroform solutions. This NH signal is broadened into the baseline when collected in an acetone-d6 solution (Fig. S4†), consistent with formation of a monomer in a hydrogen-bonding, polar solvent and fast exchange with residual water as we previously observed in the case of H3TD1 in methanol.39 Notably, the pyrrolic NH remains strongly deshielded and clearly visible for HTMC6F5 in acetone (at ∼13 ppm, Fig. S4†) owing to the intramolecular hydrogen-bonding interactions with the iminic nitrogen atoms.
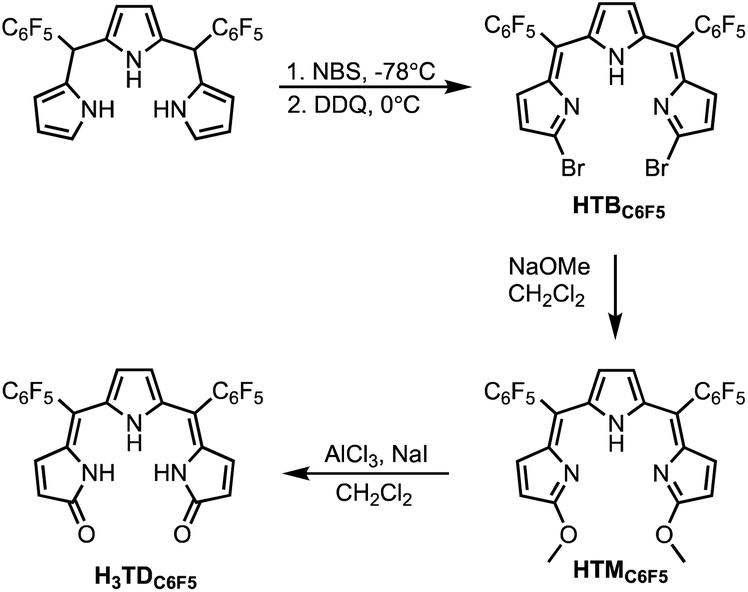
Single crystals suitable for X-ray diffraction analysis were obtained by slow diffusion of pentane into solutions of HTMC6F5 or H3TDC6F5 in CH2Cl2. The crystal structure of the dimethoxytripyrrin (Fig. 2) reveals a nearly planar tripyrrolic scaffold: one of the terminal pyrrole units is only 7.5° out of plane with respect to the other two.
 | ||
| Fig. 2 Top and side views of the crystal structure of HTMC6F5. Carbon-bound hydrogen atoms in calculated positions are omitted for clarity. The pyrrolic nitrogen-bound hydrogen atom was located on the Fourier map during refinement. Non-hydrogen atoms are displayed as thermal ellipsoids set at 50% probability level (CCDC 2300776†). | ||
Indeed, the bulky pentafluorophenyl substituents also led to an essentially planar tripyrrin unit in the case of α,α′-dianilinotripyrrins.40 Lacking the NH hydrogen-bonding donors of the aniline derivatives,10,40 however, HTMC6F5 does not adopt a double helical arrangement. Instead, the HTMC6F5 ligands pack in a herringbone pattern with pairs of ligands oriented in antiparallel fashion.
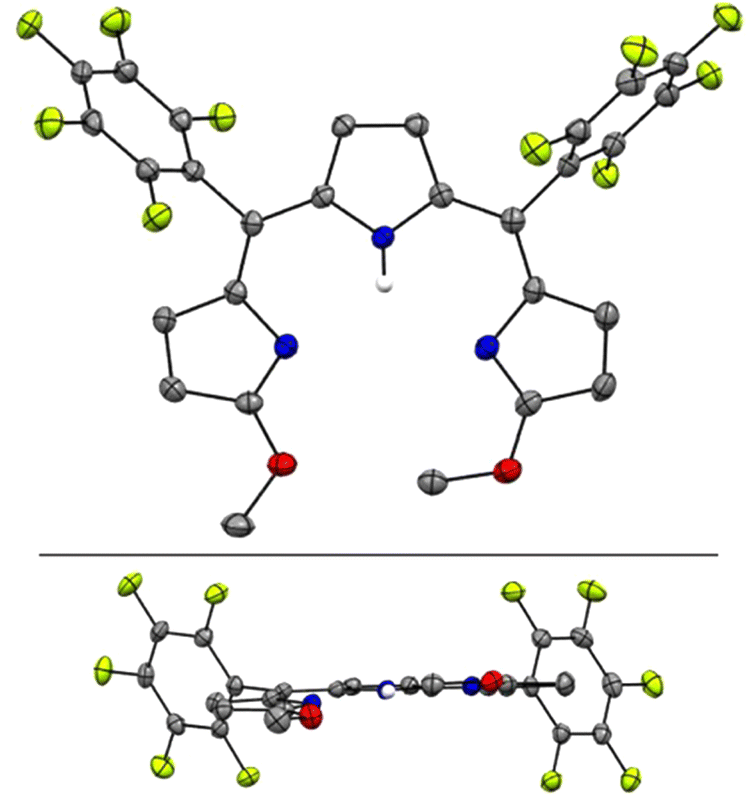
The crystal structure of H3TDC6F5 confirmed the ZZ, syn, syn conformation indicated by the NMR data (Fig. 3a) and revealed a helical dimer featuring two hydrogen-bonding interactions between the lactam units on both ends of the tripyrroles (Fig. 3b and c). This type of intermolecular hydrogen bonding was reported for hexaethyl tripyrrindione H3TD1, in which the twisting of the terminal pyrrolinones relative to the plane of the middle pyrrolic unit is about 20° on each side.30 For H3TDC6F5, the twisting is asymmetrical, with angles of ∼32° and 12° at the aryl-substituted bridges. Both M and P helices, each featuring four hydrogen-bonding interactions (Fig. 3b), are observed in the crystal structure.
 | ||
| Fig. 3 Crystal structure of 5,10-bis(pentafluorophenyl)-tripyrrin-1,14-dione H3TDC6F5, which crystallizes as a helical dimer. (a) Top view, (b) side view of the dimer showing four hydrogen bonds as dashed lines, and (c) side view of the dimers showing the relative orientation of the two tripyrrolic scaffolds. Carbon-bound hydrogens in calculated positions and aryl substituents are omitted for clarity in the dimer views. Pyrrolic nitrogen-bound hydrogen atoms were located on the Fourier map during refinement. Non-hydrogen atoms are displayed as thermal ellipsoids set at 50% probability level (CCDC 2300775†). | ||
The electrochemical profiles of HTMC6F5 and H3TDC6F5 were investigated by cyclic voltammetry in CH2Cl2 (Fig. S5†). The voltammogram of HTMC6F5 displays one quasi-reversible oxidation event at 0.634 V. Conversely, similar to the hexaethyl tripyrrindione ligand H3TD1,23 H3TDC6F5 has a rather complicated redox profile featuring at least three irreversible oxidations above −0.2 V, which in turn elicit two irreversible reduction events past −1.0 V.
Synthesis and characterization of palladium(II) complexes
The formation of square planar palladium complexes is well documented for tripyrrins6,41 and tripyrrindiones,23,24 therefore we sought to investigate the Pd(II) coordination chemistry of both HTMC6F5 and H3TDC6F5. The reactions were conducted in the presence of palladium(II) acetate at room temperature (Scheme 2) and monitored by optical absorption spectroscopy (Fig. 4). | ||
| Scheme 2 Syntheses of square planar Pd(II) complexes of tripyrrin HTMC6F5 and tripyrrindione H3TDC6F5. | ||
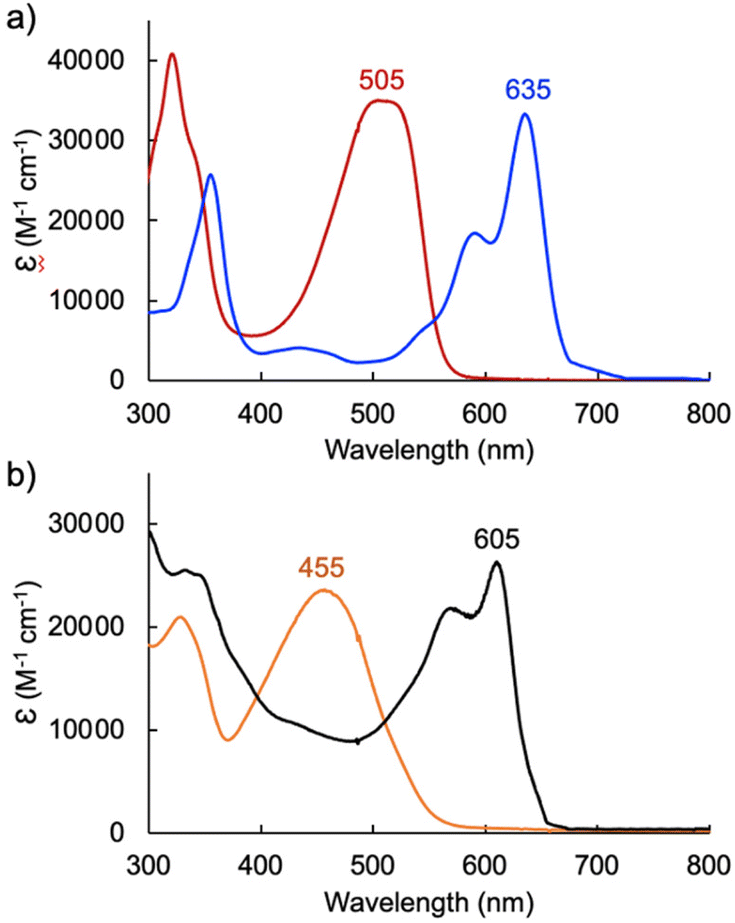 | ||
| Fig. 4 UV-visible absorption spectra of (a) tripyrrin HTMC6F5 (red) and its Pd(II) complex [Pd(TMC6F5#)] (blue) in CH2Cl2 and (b) tripyrrindione H3TDC6F5 (orange) and complex [TBA][Pd(TDC6F5)(H2O)] (black) in CH2Cl2. | ||
In the case of dimethoxytripyrrin HTMC6F5, metal coordination led to the appearance of a new peak at 635 nm, causing a color change of the reaction mixture from red to blue. Following purification by column chromatography, initial 1H NMR data indicated the formation of a complex with an asymmetric tripyrrolic ligand. A total of five peaks were observed in the aromatic region, and a singlet integrating to three protons at 4.13 ppm indicated only one methoxy group in the product (Fig. S6†). The structure was ultimately determined through X-ray diffraction analysis (vide infra) and showed a square planar cyclometallated Pd(II) complex, resulting from C(sp3)–H bond activation at one of the methoxy groups. The peak at 6.57 ppm corresponds to the Pd-bound CH2 protons as determined by 1H–13C HSQC NMR methods (Fig. S5†).
C(sp2)–Pd bonds are well-established within the coordination chemistry of N-confused porphyrins,42 carbaporphyrins,43 and several expanded porphyrins.44,45 In contrast, C(sp3)–Pd bonds, which are relevant to Pd-based organometallic catalysis,46 are not common for oligopyrrolic ligands. Relevant but distinct examples with polydentate nitrogen ligands include the intramolecular C(sp3)–H bond activation observed in Pd(II) complexes of 6-alkyl-2,2′-bipyridines47 and t-butyl-substituted 1,3-bis(aryliminio)-isoindolines.48 Neutral complex [Pd(TMC6F5#)] (Scheme 2) therefore represents a unique example of tripyrrin coordinating as a tetradentate dianionic ligand (i.e., (TM#C6F5)2−) through three pyrrolic N donors and one C(sp3) donor.
Initial coordination attempts on H3TDC6F5 using Pd(OAc)2 in THF under ambient conditions proved unsuccessful. Within an hour, the solution color changed from orange to green and a metallic film was observed, indicating slow reduction of the metal cation concurrent with oxidative degradation of the ligand. This observation is in contrast with our previous findings on hexaalkyl tripyrrindiones, which undergo one-electron oxidation in air upon coordinating Pd(II) and other divalent metals as dianionic radicals (e.g., [Pd(TD1˙)(H2O)]).23–27 To facilitate the isolation of a Pd(II) complex of the meso-aryl tripyrrindione in its most reduced state (i.e., TDC6F53−), we added tetra-n-butylammonium bromide (i.e., [(C4H9)4N]Br) to the reaction mixture so as to provide a monocationic counterion soluble in organic solvents. In these conditions, the reaction proceeded steadily, as determined by a color change from orange to blue and consumption of H3TDC6F5 in the absorption profile (Fig. 4b). The spectrum of the complex displays a main absorption band at 605 nm, and no bands were observed at longer wavelengths. The lack of weak intraligand π–π charge transfer bands, which characterize radical tripyrrindione complexes,23–27 was the first indication of a trianionic binding mode for the isolated complex. 1H NMR data confirmed the isolation of a diamagnetic species (Fig. S8†): three resonances between 5.7 and 6.5 ppm correspond to the β-CH protons, four peaks in the aliphatic regions account for the tetrabutylammonium ion, and the 2H peak at 11.16 ppm was tentatively assigned to a coordinated aqua ligand as found in other tripyrrindione complexes.
The crystal structure of [Pd(TMC6F5#)] (Fig. 5) shows a square planar cyclometallated Pd(II) complex in which a carbon atom from one of the methoxy groups is bound to the Pd(II) center following C(sp3)–H activation. The Pd1–C16 bond distance is 2.035 Å, similar to that to the C(sp3) donor in the 1,3-bis(aryliminio)isoindoline complexes (2.056 and 2.039 Å).49 Consistent with a strong trans influence of the C16 donor, the Pd1–N2 distance to the central pyrrole, which is typically the shorter of the three,6,23 is slightly elongated to 2.067 Å. In contrast, the Pd1–N3 distance is shorter (1.934 Å) because of the formation of the five-membered metallacycle. [Pd(TMC6F5#)] packs in a herringbone pattern similar to that of the free ligand and no π stacking is observed.
 | ||
| Fig. 5 Crystal structure of [Pd(TMC6F5#)]. Carbon-bound hydrogens in calculated positions are omitted for clarity. Non-hydrogen atoms are displayed as thermal ellipsoids set at 50% probability level (CCDC 2300774†). | ||
The crystal structure of [TBA][Pd(TDC6F5)(H2O)] reveals a square planar Pd(II)-bound tripyrrindione associated with a tetrabutylammonium ion (i.e., TBA) confirming the anionic nature of the complex (Fig. 6). The Pd(II) center also coordinates an aqua ligand, which is engaged in hydrogen-bonding interactions to the carbonyl groups as observed in other tripyrrindione23,24,26 and dipyrrindione complexes.50,51 The metal center lies in the plane of the three nitrogen donors while the aqua ligand is 10.3° out of plane. The observed C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond lengths (1.269 and 1.249 Å) are comparable to those found in the anionic complex [Pd(TD1)(H2O)]− (1.260 and 1.259 Å) and longer than those in the neutral radical [Pd(TD1˙)(H2O)] (1.240 and 1.238 Å), indicating a similar correspondence between the structural metrics and the ligand redox state.23 Indeed, the donor–acceptor distances for the hydrogen bonds in [Pd(TDC6F5)(H2O)]− (2.534 and 2.528 Å) are also in agreement with those found in [Pd(TD1)(H2O)]− (2.521 and 2.525 Å), indicative of stronger hydrogen bonds compared to those in neutral and cationic Pd(II) tripyrrindione complexes, which feature longer donor–acceptor distances. No stacking of the tripyrrindione units is observed: instead, the anionic complexes and the tetrabutylammonium cations alternate in a sheet-like packing.
O bond lengths (1.269 and 1.249 Å) are comparable to those found in the anionic complex [Pd(TD1)(H2O)]− (1.260 and 1.259 Å) and longer than those in the neutral radical [Pd(TD1˙)(H2O)] (1.240 and 1.238 Å), indicating a similar correspondence between the structural metrics and the ligand redox state.23 Indeed, the donor–acceptor distances for the hydrogen bonds in [Pd(TDC6F5)(H2O)]− (2.534 and 2.528 Å) are also in agreement with those found in [Pd(TD1)(H2O)]− (2.521 and 2.525 Å), indicative of stronger hydrogen bonds compared to those in neutral and cationic Pd(II) tripyrrindione complexes, which feature longer donor–acceptor distances. No stacking of the tripyrrindione units is observed: instead, the anionic complexes and the tetrabutylammonium cations alternate in a sheet-like packing.
 | ||
| Fig. 6 Crystal structure of [N(C4H9)4][Pd(TDC6F5)(H2O)]. In the top panel, the cation is omitted for clarity. In the bottom panel, the cation is shown whereas the aryl groups are omitted. Carbon-bound hydrogens in calculated positions are omitted for clarity. Non-hydrogen atoms are displayed as thermal ellipsoids set at 50% probability level (CCDC 2300777†). | ||
The electrochemical profile of [Pd(TMC6F5#)] (Fig. 7a, blue trace) presents a wide open-circuit potential window between two one-electron events: a quasi-reversible oxidation at 0.50 V and a quasi-reversible reduction at −1.26 V. There are no reports of the electrochemical profile of cyclometallated Pd(II) tripyrrins; however, the electrochemical profile resembles that of symmetrical tripyrrin complexes,41 with the first one-electron events assigned as ligand-based and occurring at similar potentials (oxidations occur between 0.36 and 0.45 V, and reductions between −1.01 and −1.37 V).
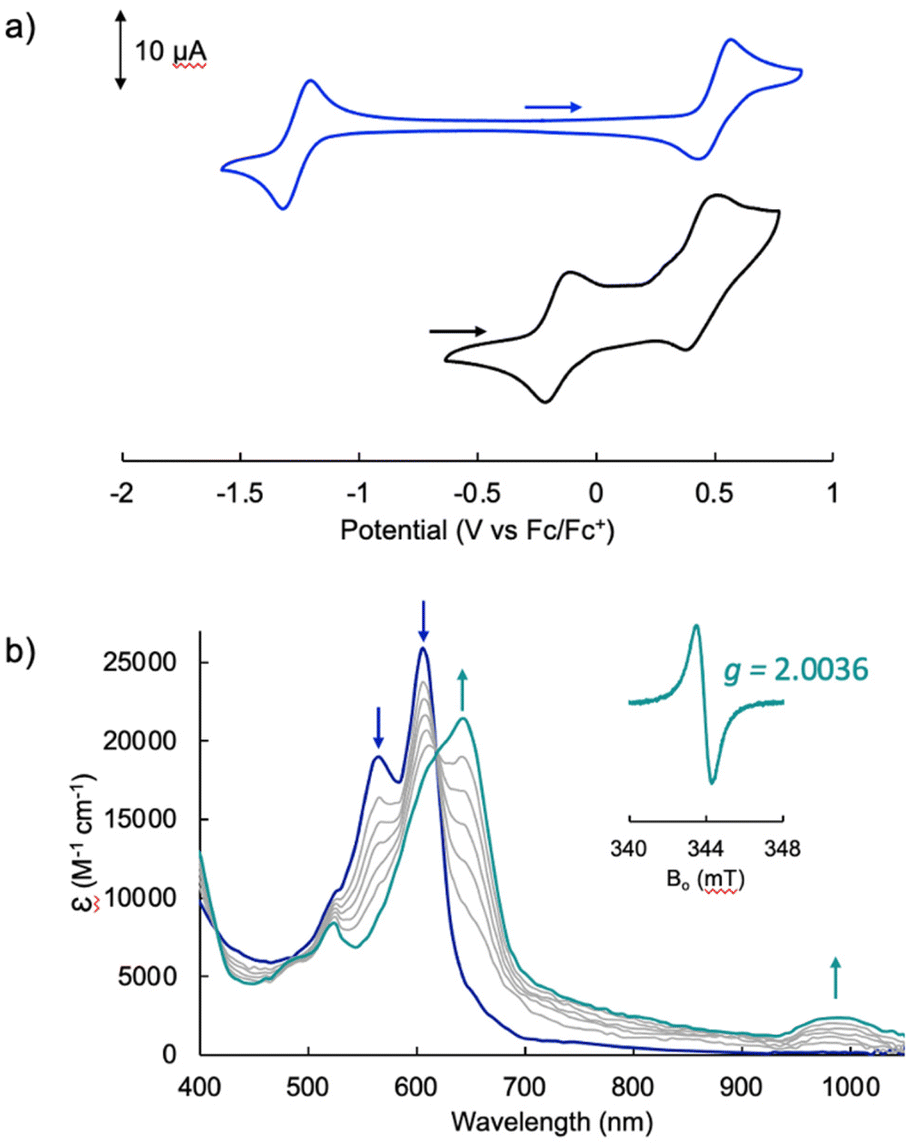 | ||
Fig. 7 Electrochemical analysis of the Pd(II) tripyrrin complexes. (a) Cyclic voltammograms of [Pd(TMC6F5#)] (blue trace) and [TBA][Pd(TDC6F5)(H2O)] (black trace) at a glassy carbon electrode in CH2Cl2 with 0.1 M [(n-Bu4)(PF6)] as a supporting electrolyte. Data collected at a 100 mV s−1 scan rate using a Ag/AgNO3 reference electrode and a Pt wire auxiliary electrode. (b) Spectral changes observed upon oxidation of [Pd(TDC6F5)(H2O)]− (CH2Cl2, 0.1 M (n-Bu4)(PF6)) by controlled potential electrolysis (at 0.2 V vs. Fc+/Fc) to give  . Inset: EPR spectrum at room temperature of . Inset: EPR spectrum at room temperature of  ; experimental conditions: mw frequency, 9.643 GHz; mw power, 3 mW; field modulation amplitude, 0.05 mT. ; experimental conditions: mw frequency, 9.643 GHz; mw power, 3 mW; field modulation amplitude, 0.05 mT. | ||
The voltammogram of [TBA][Pd(TDC6F5)(H2O)] (Fig. 7a, black trace) presents one quasi-reversible oxidation event at −0.165 V and a second quasi-reversible oxidation at 0.439 V. A small shoulder is observed around 0.33 V and is attributed to partial decomposition upon the first oxidation event. When compared to the ligand-based redox processes of the hexaethyl tripyrrindione complex [Pd(TD1˙)(H2O)], which is reduced at −0.672 V to form [Pd(TD1)(H2O)]− and oxidized at −0.052 V to [Pd(TD1)(H2O)]+, it is clear that the meso-pentafluorophenyl substitution causes a significant anodic shift (by ∼500 mV) of the redox profile of the complex. The two one-electron events are assigned as oxidations of the anionic complex [Pd(TDC6F5)(H2O)]− to give the radical  and then the cation [Pd(TDC6F5)(H2O)]+.
and then the cation [Pd(TDC6F5)(H2O)]+.
We further investigated the first oxidation event of [Pd(TDC6F5)(H2O)]− by controlled-potential electrolysis and monitored the progress via spectroelectrochemical methods (Fig. 7b). When the cell potential was held at 0.2 V (vs. Fc/Fc+), a decrease in the main absorption bands of [Pd(TDC6F5)(H2O)]− (565 and 605 nm) was accompanied by the appearance of a new main absorption band at 633 nm and a long-wavelength band at 990 nm, which is characteristic of metal-bound tripyrrindione radicals.
The formation of a tripyrrindione radical was confirmed through the chemical oxidation of [TBA][Pd(TDC6F5)(H2O)] with silver(I) tetrafluoroborate in acetone. The EPR spectrum of the oxidation product at room temperature (Fig. 7b, inset) resembles that of previously reported tripyrrindione radicals, with a single isotropic line at g = 2.0036 and a width of 0.84 mT. Whereas [Pd(TD1˙)(H2O)] forms EPR-silent π-dimers at lower temperatures through multicentered interaction between the ligand-based spins,28 no loss of EPR signal was observed for 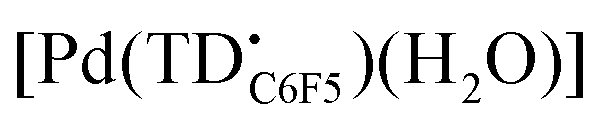 at 77 K (Fig. S9†). Differences in the spin distribution on the ligand scaffold as well as the effect of the bulky aryl substituent are likely decreasing the ability of the complexes to engage in pancake bonding. Although
at 77 K (Fig. S9†). Differences in the spin distribution on the ligand scaffold as well as the effect of the bulky aryl substituent are likely decreasing the ability of the complexes to engage in pancake bonding. Although 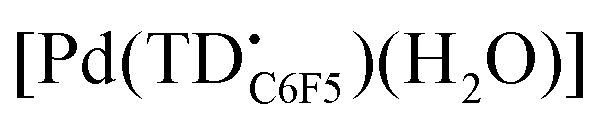 is fairly stable in solution at room temperature (Fig. S10†), when attempting to isolate or crystallize this complex we observed a tendency to demetalate and/or oligomerize at high concentrations leading to an intractable solid. Further investigations on the effects of meso-aryl substituents on the stability of tripyrrindione radical complexes are underway.
is fairly stable in solution at room temperature (Fig. S10†), when attempting to isolate or crystallize this complex we observed a tendency to demetalate and/or oligomerize at high concentrations leading to an intractable solid. Further investigations on the effects of meso-aryl substituents on the stability of tripyrrindione radical complexes are underway.
Experimental
Materials and methods
The synthesis of meso-di(pentafluorophenyl)tripyrrane35 and HTBC6F5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 52 were performed according to reported procedures. Dichloromethane (CH2Cl2), pentane and THF were dried by passage through a solvent purifier. Dry solvents were confirmed to contain <0.1 ppm H2O using a Mettler Toledo C10S Coulometric Karl Fisher Titrator. All other commercial reagents were used without further purification. Aluminum-backed silica gel plates (thickness: 200 μm) were used to monitor reactions by thin layer chromatography (TLC). Chromatographic purifications on silica gel were conducted on a Biotage Isolera One Flash instrument. 1H, 13C and 19F NMR spectra were recorded on a Bruker NEO-500 instrument at the NMR Facility of the University of Arizona (UArizona) Department of Chemistry and Biochemistry. UV-visible absorption spectra were obtained at ambient temperature using an Agilent 8453 spectrophotometer or an Agilent Cary 60 spectrophotometer. High-resolution mass spectrometry data were acquired at the UArizona Mass Spectrometry Core Facility. Elemental analyses were performed by Numega Resonance Labs, San Diego, CA. The continuous-wave (CW) EPR experiments were carried out at the UArizona Department of Chemistry and Biochemistry EPR facility on an X-band Elexsys E500 spectrometer (Bruker Biospin) equipped with a rectangular TE102 resonator.
52 were performed according to reported procedures. Dichloromethane (CH2Cl2), pentane and THF were dried by passage through a solvent purifier. Dry solvents were confirmed to contain <0.1 ppm H2O using a Mettler Toledo C10S Coulometric Karl Fisher Titrator. All other commercial reagents were used without further purification. Aluminum-backed silica gel plates (thickness: 200 μm) were used to monitor reactions by thin layer chromatography (TLC). Chromatographic purifications on silica gel were conducted on a Biotage Isolera One Flash instrument. 1H, 13C and 19F NMR spectra were recorded on a Bruker NEO-500 instrument at the NMR Facility of the University of Arizona (UArizona) Department of Chemistry and Biochemistry. UV-visible absorption spectra were obtained at ambient temperature using an Agilent 8453 spectrophotometer or an Agilent Cary 60 spectrophotometer. High-resolution mass spectrometry data were acquired at the UArizona Mass Spectrometry Core Facility. Elemental analyses were performed by Numega Resonance Labs, San Diego, CA. The continuous-wave (CW) EPR experiments were carried out at the UArizona Department of Chemistry and Biochemistry EPR facility on an X-band Elexsys E500 spectrometer (Bruker Biospin) equipped with a rectangular TE102 resonator.
Synthetic procedures
000), 505 (35000). HRMS-ESI+: [M + H]+ calcd for [C28H13F10N3O2], 614.09209; found, 614.09207.
400), 328 (20900), 455 (23500). HRMS-ESI+: [M + H]+ calcd for [C26H9F10N3O2], 586.06078; found, 586.06086.
000), 433 (4700), 590 (18600), 635 (33300). HRMS-ESI+: [M + H]+ calcd for [C28H11F10N3O2Pd], 717.98099; found, 717.98022. Anal. Calcd for [C28H11F10N3O2Pd]: C, 46.9; H, 1.5; N, 5.9%; found: C, 47.2; H, 2.1; N, 5.8%.
100), 565 (21600), 605 (25900). HRMS-ESI−: [M]− calcd for [C26H8F10N3O3Pd], 705.94463; found, 705.94427. Anal. Calcd for [C42H44F10N4O4Pd]·CH2Cl2·1.5(H2O): C, 48.7; H, 4.7; N, 5.3%; found: C, 48.8; H, 5.1; N, 5.2%.
Electrochemical measurements
Cyclic voltammograms were performed on a Gamry Reference 600 potentiostat utilizing a single-compartment cell with three electrodes: a glassy carbon working electrode, a platinum wire auxiliary electrode, and a Ag/AgCl reference electrode. Measurements were performed at ambient temperature under an inert argon atmosphere in CH2Cl2 containing 0.1 M [(n-Bu4N)(PF6)] (triply recrystallized) as a supporting electrolyte. Sample concentrations were 1–2 mM, and all electrochemical data were internally referenced to the ferrocene/ferrocenium couple (set at 0.00 V).Chemical oxidation of [Pd(TDC6F5)(H2O)]− and EPR analysis
Due to the limited stability of upon solvent removal, this species was formed in situ and analyzed as such. A sample of [Pd(TDC6F5)(H2O)]− (1 mM in acetone) was oxidized with AgBF4 (1.1 equiv.). The sample was filtered through a Celite plug to remove elemental silver. An aliquot (0.5 mL) was taken and diluted by half with toluene for analysis both at room temperature and 77 K.
upon solvent removal, this species was formed in situ and analyzed as such. A sample of [Pd(TDC6F5)(H2O)]− (1 mM in acetone) was oxidized with AgBF4 (1.1 equiv.). The sample was filtered through a Celite plug to remove elemental silver. An aliquot (0.5 mL) was taken and diluted by half with toluene for analysis both at room temperature and 77 K.
Conclusions
We report the synthesis and characterization of the first examples of two new classes of tripyrrolic ligands featuring meso-pentafluorophenyl substituents, namely 1,14-dimethoxytripyrrin HTMC6F5 and tripyrrin-1,14-dione H3TDC6F5. The synthetic route from a tripyrrane precursor is expected to allow for a variety of meso-aryl groups. Additionally, the reactivity at the para positions of the pentafluorophenyl substituents36,37 will allow for post-synthetic functionalization of HTMC6F5 and H3TDC6F5 and their further utilization as building blocks.Both of the prepared tripyrroles coordinate Pd(II) at ambient temperature to form square planar complexes. HTMC6F5 undergoes C(sp3)–H bond activation in the presence of palladium(II) acetate leading to the formation of the cyclometallated complex [Pd(TMC6F5#)], in which the ligand binds in a dianionic tetradentate fashion including a Pd-methylene bond. In contrast, tripyrrindione H3TDC6F5 binds Pd(II) as a trianionic ligand and the resulting complex [TBA][Pd(TDC6F5)(H2O)] features an aqua ligand and a tetrabutylammonium counterion.
The meso-aryl substituents changed substantially the electrochemical profile of the tripyrrindione ligand: whereas hexaalkyl tripyrrindiones coordinate divalent cations as dianionic radicals (i.e., TD1˙2− or TD2˙2−) under aerobic conditions, this meso-pentafluorophenyl analog stabilizes complexes in the most reduced state of the ligand (i.e., TDC6F53−). Spectroelectrochemical and EPR spectroscopic experiments confirmed that the radical state of the novel meso-pentafluorophenyl tripyrrindione complex remains accessible upon one-electron oxidation at −0.165 V (vs. Fc/Fc+). Notably, the shifted redox profile of the new meso-aryl tripyrrindione could accommodate coordination of metal cations of higher oxidation states, which have not been previously explored for this class of pyrrolic ligands.
Modifications of the aryl substituents and associated electronic effects are expected to modulate the coordination chemistry of these ligands as well as the electronic structure53 and reactivity of the complexes, including C–H bond activation chemistry and one-electron redox processes. The optical and/or electrochemical properties of these tripyrrinato complexes could prove advantageous in applications in photocatalysis54 and energy conversion and storage,55 including in redox flow batteries requiring wide open-circuit potential windows as observed here for [Pd(TMC6F5#)]. Furthermore, inspired by the well-established supramolecular chemistry of meso-aryl porphyrins,56,57meso-diaryl tripyrrin and tripyrrindione complexes could serve as new redox-responsive building blocks for supramolecular assemblies, with additional tunability provided by the monodentate ligand at the fourth coordination site.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Vlad Kumirov, Dr William Montfort, and Dr Andrei Astashkin for assistance with the acquisition and analysis of NMR, X-ray diffractometry, and EPR data, respectively. This work was supported by the National Science Foundation (award CHE-2203361 to E.T.). The Bruker NEO-500 spectrometer and the Bruker D8 Venture diffractometer in the UArizona Dept. of Chemistry and Biochemistry NMR Facility and X-ray Diffraction Facility, respectively, were purchased thanks to support from the National Science Foundation (MRI awards CHE-1920234 and CHE-2117516).References
- H. Falk, The Chemistry of Linear Oligopyrroles and Bile Pigments, Springer-Verlag, Vienna, 1989 Search PubMed.
- J. L. Sessler and D. Seidel, Synthetic Expanded Porphyrin Chemistry, Angew. Chem., Int. Ed., 2003, 42, 5134–5175 CrossRef CAS PubMed.
- T. Tanaka and A. Osuka, Chemistry of meso-Aryl-Substituted Expanded Porphyrins: Aromaticity and Molecular Twist, Chem. Rev., 2017, 117, 2584–2640 CrossRef CAS PubMed.
- J. F. Arambula and J. L. Sessler, Porphyrinoid Drug Conjugates, Chem, 2020, 6, 1634–1651 CAS.
- G.-Q. Jin, C. V. Chau, J. F. Arambula, S. Gao, J. L. Sessler and J.-L. Zhang, Lanthanide porphyrinoids as molecular theranostics, Chem. Soc. Rev., 2022, 51, 6177–6209 RSC.
- M. Bröring and C. D. Brandt, Tripyrrin—the missing link in the series of oligopyrrolic ligands, Chem. Commun., 2001, 499–500 RSC.
- J. L. Sessler and S. J. Weghorn, Expanded, Contracted, and Isomeric Porphyrins, Pergamon Press, New York, 1997 Search PubMed.
- J.-F. Wang, Y. Yao, Y. Ning, Y.-S. Meng, C.-L. Hou, J. Zhang and J.-L. Zhang, The design of rigid cyclic tripyrrins: the importance of intermolecular interactions on aggregation and luminescence, Org. Chem. Front., 2018, 5, 1877–1885 RSC.
- Y. Yao, C.-L. Hou, Z.-S. Yang, G. Ran, L. Kang, C. Li, W. Zhang, J. Zhang and J.-L. Zhang, Unusual near infrared (NIR) fluorescent palladium(II) macrocyclic complexes containing M–C bonds with bioimaging capability, Chem. Sci., 2019, 10, 10170–10178 RSC.
- M. Umetani, T. Tanaka and A. Osuka, Conjugated double helices via self-dimerization of α,α′-dianilinotripyrrins, Chem. Sci., 2018, 9, 6853–6859 RSC.
- K. Ueta, M. Umetani, A. Osuka, G. D. Pantoş and T. Tanaka, Single- and double-helices of α,α′-dibenzylaminotripyrrin: solution and solid state studies, Chem. Commun., 2021, 57, 2617–2620 RSC.
- E. Tomat, Coordination Chemistry of Linear Tripyrroles: Promises and Perils, Comments Inorg. Chem., 2016, 36, 327–342 CrossRef CAS.
- M. Bröring, in Handbook of Porphyrin Science, World Scientific Publishing Company, 2010, vol. 8, pp. 343–501 Search PubMed.
- M. Bröring, S. Prikhodovski, C. D. Brandt and E. Cónsul Tejero, Pillars, Layers, Pores and Networks from Nickeltripyrrins: A Porphyrin Fragment as a Versatile Building Block for the Construction of Supramolecular Assemblies, Chem. – Eur. J., 2007, 13, 396–406 CrossRef PubMed.
- H. Furuta, H. Maeda and A. Osuka, Regioselective Oxidative Liberation of Aryl-Substituted Tripyrrinone Metal Complexes from N-Confused Porphyrin, Org. Lett., 2002, 4, 181–184 CrossRef CAS PubMed.
- J.-Y. Shin, S. S. Hepperle, B. O. Patrick and D. Dolphin, Oxidized forms of a tripyrrane: α-tripyrrinone, β-tripyrrinone and a C2 symmetric hexapyrrole, Chem. Commun., 2009, 2323 RSC.
- M. Gałeęzowski, J. Jaźwiński, J. P. Lewtak and D. T. Gryko, Rational Synthesis of Tripyrranes, J. Org. Chem., 2009, 74, 5610–5613 CrossRef PubMed.
- Y. Ding, Y. Xie, X. Li, J. P. Hill, W. Zhang and W. Zhu, Selective and sensitive “turn-on” fluorescent Zn2+ sensors based on di- and tripyrrins with readily modulated emission wavelengths, Chem. Commun., 2011, 47, 5431–5433 RSC.
- P. P. Liu, Y. Q. Feng, C. Z. Gu, C. J. Li, S. X. Meng and B. Zhang, A Meso-Substituted Tripyrrin Metal Complex as the Candidate for Fluorescent Material, Adv. Mater. Res., 2012, 557–559, 665–668 CAS.
- E. Tomat and C. J. Curtis, Biopyrrin Pigments: From Heme Metabolites to Redox-Active Ligands and Luminescent Radicals, Acc. Chem. Res., 2021, 54, 4584–4594 CrossRef CAS PubMed.
- J. Beruter, J.-P. Colombo and U. P. Schlunegger, Isolation and Identification of the Urinary Pigment Uroerythrin, Eur. J. Biochem., 1975, 56, 239–244 CrossRef CAS PubMed.
- T. Yamaguchi, I. Shioji, A. Sugimoto, Y. Komoda and H. Nakajima, Chemical Structure of a New Family of Bile Pigments from Human Urine, J. Biochem., 1994, 116, 298–303 CrossRef CAS PubMed.
- R. Gautam, J. J. Loughrey, A. V. Astashkin, J. Shearer and E. Tomat, Tripyrrindione as a Redox–Active Ligand: Palladium(II) Coordination in Three Redox States, Angew. Chem., Int. Ed., 2015, 54, 14894–14897 CrossRef CAS PubMed.
- S. Bahnmüller, J. Plotzitzka, D. Baabe, B. Cordes, D. Menzel, K. Schartz, P. Schweyen, R. Wicht and M. Bröring, Hexaethyltripyrrindione (H3Et6tpd): A Non–Innocent Ligand Forming Stable Radical Complexes with Divalent Transition–Metal Ions, Eur. J. Inorg. Chem., 2016, 8 Search PubMed.
- R. Gautam, S. J. Petritis, A. V. Astashkin and E. Tomat, Paramagnetism and Fluorescence of Zinc(II) Tripyrrindione: A Luminescent Radical Based on a Redox-Active Biopyrrin, Inorg. Chem., 2018, 57, 15240–15246 CrossRef CAS PubMed.
- E. Tomat, C. J. Curtis, A. V. Astashkin, J. Conradie and A. Ghosh, Multicenter interactions and ligand field effects in platinum(II) tripyrrindione radicals, Dalton Trans., 2023, 52, 6559–6568 RSC.
- I. Habenšus, A. Ghavam, C. J. Curtis, A. V. Astashkin and E. Tomat, Primary amines as ligands and linkers in complexes of tripyrrindione radicals, J. Porphyrins Phthalocyanines, 2023, 27, 1448–1456 CrossRef.
- R. Gautam, A. V. Astashkin, T. M. Chang, J. Shearer and E. Tomat, Interactions of Metal-Based and Ligand-Based Electronic Spins in Neutral Tripyrrindione π Dimers, Inorg. Chem., 2017, 56, 6755–6762 CrossRef CAS PubMed.
- T. Mujawar, P. Sevelda, D. Madea, P. Klán and J. Švenda, A Platform for the Synthesis of Oxidation Products of Bilirubin, J. Am. Chem. Soc., 2024, 146, 1603–1611 CrossRef CAS PubMed.
- S. D. Roth, T. Shkindel and D. A. Lightner, Intermolecularly hydrogen-bonded dimeric helices: tripyrrindiones, Tetrahedron, 2007, 63, 11030–11039 CrossRef CAS PubMed.
- J.-F. Wang, F. Ma, H.-L. Sun, J. Zhang and J.-L. Zhang, Three bilindione isomers: synthesis, characterization and reactivity of biliverdin analogs, J. Biol. Inorg. Chem., 2017, 22, 727–737 CrossRef CAS PubMed.
- E. Tomat, Propentdyopents: Brief history of a family of dipyrrolic pigments, J. Porphyrins Phthalocyanines, 2019, 23, 1265–1272 CrossRef CAS.
- R. Bonnett, S. Ioannou and F. J. Swanson, Propentdyopents and related compounds. Part 4. Propentdyopent–alkanol adducts by the photo-oxygenation of pyrromethenones, J. Chem. Soc., Perkin Trans. 1, 1989, 711–714 RSC.
- C. Brückner, E. D. Sternberg, R. W. Boyle and D. Dolphin, 5,10-Diphenyltripyrrane, a useful building block for the synthesis of meso-phenyl substituted expanded macrocycles, Chem. Commun., 1997, 1689–1890 RSC.
- J.-W. Ka and C.-H. Lee, Optimizing the synthesis of 5,10-disubstituted tripyrromethanes, Tetrahedron Lett., 2000, 41, 4609–4613 CrossRef CAS.
- J. I. T. Costa, A. C. Tomé, M. G. P. M. S. Neves and J. A. S. Cavaleiro, 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin: a versatile platform to novel porphyrinic materials, J. Porphyrins Phthalocyanines, 2011, 15, 1116–1133 CrossRef CAS.
- H. R. A. Golf, H.-U. Reissig and A. Wiehe, Nucleophilic Substitution on (Pentafluorophenyl)dipyrromethane: A New Route to Building Blocks for Functionalized BODIPYs and Tetrapyrroles, Org. Lett., 2015, 17, 982–985 CrossRef CAS PubMed.
- M. T. Huggins and D. A. Lightner, Hydrogen-Bonded Dimers in Dipyrrinones and Acyldipyrrinones, Monatsh. Chem., 2001, 132, 203–221 CrossRef CAS.
- A. Swain, B. Cho, R. Gautam, C. J. Curtis, E. Tomat and V. Huxter, Ultrafast Dynamics of Tripyrrindiones in Solution Mediated by Hydrogen-Bonding Interactions, J. Phys. Chem. B, 2019, 123, 5524–5535 CrossRef CAS PubMed.
- A. Nishiyama, K. Ueta, M. Umetani, Y. Akamatsu and T. Tanaka, Substituent Effects at the 5,10-Positions of Dianilinotripyrrins on Their Dimerization Themodynamics, Chem. – Asian J., 2022, 17, e202200562 CrossRef CAS PubMed.
- J. Bley-Escrich, S. Prikhodovski, C. D. Brandt, M. Bröring and J.-P. Gisselbrecht, Electrochemical investigations of tripyrrin complexes, J. Porphyrins Phthalocyanines, 2003, 07, 220–226 CrossRef CAS.
- M. Toganoh and H. Furuta, Creation from Confusion and Fusion in the Porphyrin World—The Last Three Decades of N-Confused Porphyrinoid Chemistry, Chem. Rev., 2022, 122, 8313–8437 CrossRef CAS PubMed.
- T. D. Lash, Carbaporphyrinoid Systems, Chem. Rev., 2017, 117, 2313–2446 CrossRef CAS PubMed.
- S. Mori, S. Shimizu, R. Taniguchi and A. Osuka, Group 10 Metal Complexes of meso-Aryl-Substituted [26]Hexaphyrins with a Metal−Carbon Bond, Inorg. Chem., 2005, 44, 4127–4129 CrossRef CAS PubMed.
- T. Yoneda, Y. M. Sung, J. M. Lim, D. Kim and A. Osuka, Pd II Complexes of [44]- and [46]Decaphyrins: The Largest Hückel Aromatic and Antiaromatic, and Möbius Aromatic Macrocycles, Angew. Chem., Int. Ed., 2014, 53, 13169–13173 CrossRef CAS PubMed.
- G. C. Dickmu and I. P. Smoliakova, Cyclopalladated complexes containing an (sp3)C–Pd bond, Coord. Chem. Rev., 2020, 409, 213203 CrossRef CAS.
- A. Zucca, M. A. Cinellu, M. V. Pinna, S. Stoccoro, G. Minghetti, M. Manassero and M. Sansoni, Cyclopalladation of 6-Substituted-2,2′-bipyridines. Metalation of Unactivated Methyl Groups vs Aromatic C−H Activation, Organometallics, 2000, 19, 4295–4304 CrossRef CAS.
- M. Bröring and C. Kleeberg, Facile intramolecular C(sp3)–H bond activation with PdII, Chem. Commun., 2008, 2777 RSC.
- C. Kleeberg and M. Bröring, Palladium complexes of unsymmetrical 1,3-bis(aryliminio)isoindoline (BAI) ligands: A novel class of complexes exhibiting unusual structural features, Polyhedron, 2010, 29, 507–513 CrossRef CAS.
- C. J. Curtis and E. Tomat, Heteroleptic palladium(II) complexes of dipyrrin-1,9-dione supported by intramolecular hydrogen bonding, J. Porphyrins Phthalocyanines, 2020, 24, 112–120 CrossRef CAS.
- C. J. Curtis, A. V. Astashkin, J. Conradie, A. Ghosh and E. Tomat, Ligand-Centered Triplet Diradical Supported by a Binuclear Palladium(II) Dipyrrindione, Inorg. Chem., 2021, 60, 12457–12466 CrossRef CAS PubMed.
- C.-L. Hou, Y. Yao, D. Wang, J. Zhang and J.-L. Zhang, Orthogonally arranged tripyrrin–BODIPY conjugates with an “edge to plane” mode, Org. Chem. Front., 2019, 6, 2266–2274 RSC.
- A. Kumar, B. Thompson, R. Gautam, E. Tomat and V. Huxter, Temperature-Dependent Spin-Driven Dimerization Determines the Ultrafast Dynamics of a Copper(II)-Bound Tripyrrindione Radical, J. Phys. Chem. Lett., 2023, 11268–11273 CrossRef CAS PubMed.
- R. Costa e Silva, L. Oliveira da Silva, A. de Andrade Bartolomeu, T. J. Brocksom and K. T. de Oliveira, Recent applications of porphyrins as photocatalysts in organic synthesis: batch and continuous flow approaches, Beilstein J. Org. Chem., 2020, 16, 917–955 CrossRef PubMed.
- J. M. Park, J. H. Lee and W.-D. Jang, Applications of porphyrins in emerging energy conversion technologies, Coord. Chem. Rev., 2020, 407, 213157 CrossRef CAS.
- C. M. Drain, A. Varotto and I. Radivojevic, Self-Organized Porphyrinic Materials, Chem. Rev., 2009, 109, 1630–1658 CrossRef CAS PubMed.
- S. Durot, J. Taesch and V. Heitz, Multiporphyrinic Cages: Architectures and Functions, Chem. Rev., 2014, 114, 8542–8578 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectroscopic and electrochemical characterization data, details of X-ray diffraction data collection and analysis. CCDC 2300774–2300777. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi02597g |
| This journal is © the Partner Organisations 2024 |