
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmploying a template synthesis to access diastereopure Np(IV) and U(IV) complexes and analysis of their 5f orbitals in bonding†
Stephanie H.
Carpenter‡
 a,
María J.
Beltrán-Leiva‡
b,
Shikha
Sharma
a,
Michael L.
Tarlton
a,
James T.
Moore
a,
Andrew J.
Gaunt
a,
Enrique R.
Batista
*b,
Aaron M.
Tondreau
*a and
Ping
Yang
*b
a,
María J.
Beltrán-Leiva‡
b,
Shikha
Sharma
a,
Michael L.
Tarlton
a,
James T.
Moore
a,
Andrew J.
Gaunt
a,
Enrique R.
Batista
*b,
Aaron M.
Tondreau
*a and
Ping
Yang
*b
aChemistry Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA. E-mail: tondreau_a@lanl.gov
bTheoretical Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA. E-mail: erb@lanl.gov; pyang@lanl.gov
First published on 10th June 2024
Abstract
Development of new synthetic methods to employ with transuranium organometallic complexes will help further the field and may provide a stronger understanding of their distinct reactivity, electronic structures, and chemical bonding. Although the organoneptunium field has grown over the last decade, just a handful of complexes have been synthesized, most of which are found in the trivalent oxidation state. Few reactions with neptunium (Np) have considered the stereochemical outcome, thus we employed a diastereoselective template protocol to synthesize meso (or: C2) complexes of neptunium(IV). Following previous success with the diastereoselective synthesis of ((tBu2P)ONO)UCl2(dtbpy) (1), adaptation to NpCl4(dme)2 led to the isolation of diastereopure ((tBu2P)ONO)NpCl2(dtbpy) (2), ((tBu2P)ONO = 2,6-bis((di-tert-butylphosphino)-methanolato)pyridine). The addition of a bulky indenide ligand, derived from the in situ deprotonation of 4,7-dimethyl-1,3-bis(1-methylethyl)-1H-indene (1,3-(iPr)2-4,7-Me2-C9H3), was pursued to synthesize the organoactinide complexes ((tBu2P)ONO)U(1,3-(iPr)2-4,7-Me2-C9H3)Cl (3) and ((tBu2P)ONO)Np(1,3-(iPr)2-4,7-Me2-C9H3)Cl (4). Bonding analyses consisting of orbital localization and energy decomposition methods show resemblances in covalent character between uranium (1 and 3) and neptunium (2 and 4) systems. However, an increased orbital overlap was identified in the metal – indenide with respect to the metal–dtbpy interaction, mainly driven by 5f electrons. This was attributed to the better symmetry match between the organic ligand and metal orbitals that allowed the participation of the 5f orbitals.
Introduction
Neptunium coordination chemistry has experienced rapid growth in recent years,1–11 which has been facilitated by its lower activity, cost, and regulatory burden relative to plutonium with respect to the most common isotopes of these elements used in synthetic chemistry. However, the specialized facilities required for the radiological hazards of 237Np and its 233Pa daughter are more significant than those of uranium. Although progress is reflected in the increasing number of complexes that have been reported, the gap with respect to organoactinide chemistry is pronounced. Traversal of the actinide series is marked by a gradual energetic stabilization of the 5f shell, unlike the 6d shell which remains almost at constant energy. This, added to the more contracted nature of the 5f with respect to the 6d shell, makes the latter more prone to participate in metal–ligand interactions.12,13 However, by taking advantage of the unique shape of the f-orbitals, in combination with the diverse symmetry-adapted interactions that aromatic ligands such as arene or indene ligands can provide, a preferential interaction that exposes the 5f over the 6d shell can be favored, encouraging their participation in covalent interactions.Recent developments in organoneptunium chemistry can be traced back to well-defined neptunium starting materials for air- and moisture-free synthesis, NpCl4(thf)3, NpCl3(py)4, NpBr3(thf)4, NpI3(thf)4 and NpCl4(dme)2 (thf = tetrahydrofuran, dme = 1,2-dimethoxyethane),14–18 all of which circumvent the need for Np0 metal that was required previously.19 In addition to neptunium complexes bearing cyclopentadienyl ligands,20–22 η6-arene complexes and hydrocarbyl-coordinated Np4+ centers have also been described.23–25 These systems have set the foundations of organoneptunium chemistry and analysis of its chemical bonds. Investigating novel coordination motifs in organoactinide complexes may provide further understanding of f-orbital participation in neptunium–ligand bonding interactions. Accordingly, we expanded upon our prior efforts and introduced template techniques to neptunium chemistry.
Template synthesis protocols are reactions that use a metal center to arrange reactive fragments prior to reactive product generation. Templating is pursued when mixtures of products, or facile deleterious side reactions, are expected during a chemical reaction. Accordingly, templating has often been employed for the formation of macromolecules and molecular knots.26–29 Additional work has shown that template synthesis can aid in the activation of inert pre-ligand moieties to enable reactivity that is unobserved with traditional synthesis.30–32 Our interests lie in developing synthetic procedures for use with actinide elements, where fundamental research efforts are often limited to small scales due to low availability and radiological hazards, persistent problems when researching the chemistry of neptunium. Here, employing template protocols to increase product yields and minimize product mixtures was sought during the requisite use of small scales of neptunium.
We previously reported a template procedure that constructed diastereopure complexes of both iron and uranium.33 Coordination of 2,6-pyridine dicarboxaldehyde ((CHO)2Py) to the metal center followed by the addition of two equivalents of ditertbutyl(trimethylsilyl)phosphine (tBu2PSiMe3) generated the meso complexes. Synthesis of the free ligand without a template generated a 63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :37 rac:meso (C2:CS) ratio favoring slightly the formation of the rac isomer, which required fractional crystallization to purify the diastereoisomers following coordination to FeCl2.33 Remarkably, pre-coordination of (CHO)2Py to a metal halide using either FeCl2 or UCl4 prior to addition of tBu2PSiMe3 resulted in substantial stereospecificity (>90%) for the meso diastereoisomers of the respective products. The reaction of the uranium congener produced a pyridine-based dianionic [ONO]2− complex arising from the loss of TMS-Cl and U–O bond formation (Scheme 1, top right).33 Both the improved stereospecificity and higher yields suggested that adaptation of this method to neptunium would be successful.
:37 rac:meso (C2:CS) ratio favoring slightly the formation of the rac isomer, which required fractional crystallization to purify the diastereoisomers following coordination to FeCl2.33 Remarkably, pre-coordination of (CHO)2Py to a metal halide using either FeCl2 or UCl4 prior to addition of tBu2PSiMe3 resulted in substantial stereospecificity (>90%) for the meso diastereoisomers of the respective products. The reaction of the uranium congener produced a pyridine-based dianionic [ONO]2− complex arising from the loss of TMS-Cl and U–O bond formation (Scheme 1, top right).33 Both the improved stereospecificity and higher yields suggested that adaptation of this method to neptunium would be successful.
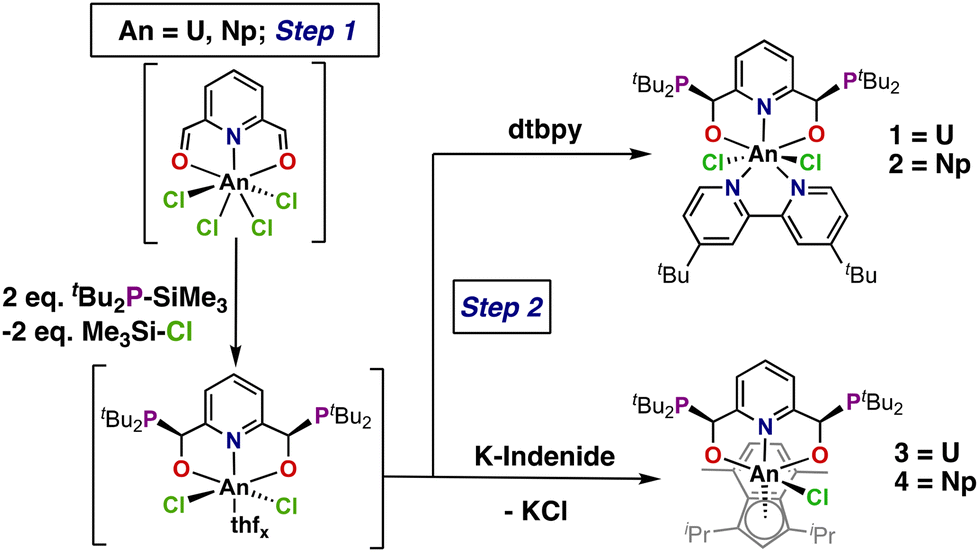 | ||
| Scheme 1 Proposed reactivity of the template synthesis from either UCl4 or (dme)2NpCl4 (dme = 1,2-dimethoxyethane) with the loss of TMS-Cl (TMS = trimethylsilyl). | ||
Similar bond dissociation energies34 for U–O (755 kJ mol−1) and Np–O (731 kJ mol−1) suggest a successful diastereoselective synthesis of a neptunium complex can be achieved via this synthetic route. The ease and high purity of this method lends itself to the generation of unique starting materials for subsequent chemistry. In this report, organometallic derivatization was pursued to establish chemistry beyond the synthesis of halide materials and to generate the respective diastereopure organoactinide complexes. An indene derivative was employed to generate the corresponding organoactinide derivatives (Scheme 1). Characterization of the resulting complexes via single-crystal X-ray diffraction (SC-XRD), UV-Vis-NIR, and NMR spectroscopies was performed alongside natural bond orbital (NBO) analysis, time-dependent density functional theory (TD-DFT) and multi-reference wavefunction calculations to explore the electronic structure and bonding of these systems. This analysis allowed for direct comparisons between homologous neptunium and uranium complexes.
Experimental results
Applying a diastereoselective synthesis to neptunium
Scale-down reactions using UCl4 were performed prior to work using NpCl4(dme)2 in order to identify optimal reaction and crystallization conditions. Coordination of (CHO)2Py to a stirring solution of NpCl4(dme)2 in THF at room temperature resulted in little color change of the pale-pink solution (Fig. S18†). Following the coordination of the dialdehyde to the metal center, tBu2PSiMe3 (2 equivalents) was added to the reaction solution, resulting in a color change to yellow (Fig. S19†). Removal of TMS-Cl was performed by holding the reaction solution under reduced pressure to a constant mass. The addition of 4,4′-ditertbutyl-2,2′-bipyridyl (dtbpy) in fluorobenzene to the tacky yellow solid resulted in the formation of a pale-peach solution (Fig. S20†). Pale-peach rectangular plates were observed after leaving the filtered reaction solution undisturbed for 10 days at −35 °C (Fig. S21†). These crystals were isolated in 46% yield and were assigned as ((tBu2P)ONO)NpCl2(dtbpy), 2.Complex 2 was successfully characterized by NMR and UV-Vis-NIR spectroscopies. The 1H NMR spectrum of 2 has a broad signal range, as expected for a formally paramagnetic 5f3 Np4+ complex, with resonances found over a 70 ppm window. The resonances attributed to the tBu2P groups were found in the 1H NMR spectrum at δ 13.05 ppm and δ −1.86 ppm as broadened doublets with a resolved coupling of ∼10 Hz. A diagnostic set of downfield resonances centered at δ 65.50 ppm and δ 54.43 ppm in 2 is reminiscent for those observed for 1 (δ 67.16 ppm and δ 24.30 ppm), which are tentatively assigned as the m-pyridine C–H and the two methine C–H groups. The 31P NMR spectrum of 2 contained a single resonance at δ 79.3 ppm, significantly shifted from the resonance of the free ligand, meso-OTMSPNPtBu (δ 50.2 ppm), and slightly shifted from the observed resonance of 1 (δ 86.0 ppm).33 The presence of a single phosphorus resonance suggests formation of a single diastereoisomer, a result consistent with the U-based analogue. The chemical shift difference between 1 and 2 likely arises from the inherent disparities of their expected magnetic moments based on electron count; U4+ is formally a 5f2 system with an inherently lower magnetic moment when compared to the 5f3 Np4+ center. Previously established Evans method protocols for transuranium complexes were followed,24,35 and the magnetic moment of 2 was found to be 2.32μB, lower than previously observed solution values for a 5f3 Np4+ complexes.24
SC-XRD experiments were performed to unambiguously assign the identity of the complex as the desired meso diastereoisomer (Fig. 1). The solid-state structure of 2 was solved in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with unit cell parameters that were significantly different from our original report of 1 (solved in the monoclinic P21/c space-group). The differences in the structures arise from a different arrangement of the solvent molecules in the unit cells. In the reported P21/c cell of 1, the asymmetric unit cell contained four discrete fluorobenzene molecules. The P cell of 2 contained three disordered fluorobenzene molecules that were modelled as partially occupied. Additional crystallization attempts produced a polymorph of 1 that was solved in P, allowing for a direct comparison to 2 (Table 1, ESI section 5†).
with unit cell parameters that were significantly different from our original report of 1 (solved in the monoclinic P21/c space-group). The differences in the structures arise from a different arrangement of the solvent molecules in the unit cells. In the reported P21/c cell of 1, the asymmetric unit cell contained four discrete fluorobenzene molecules. The P cell of 2 contained three disordered fluorobenzene molecules that were modelled as partially occupied. Additional crystallization attempts produced a polymorph of 1 that was solved in P, allowing for a direct comparison to 2 (Table 1, ESI section 5†).
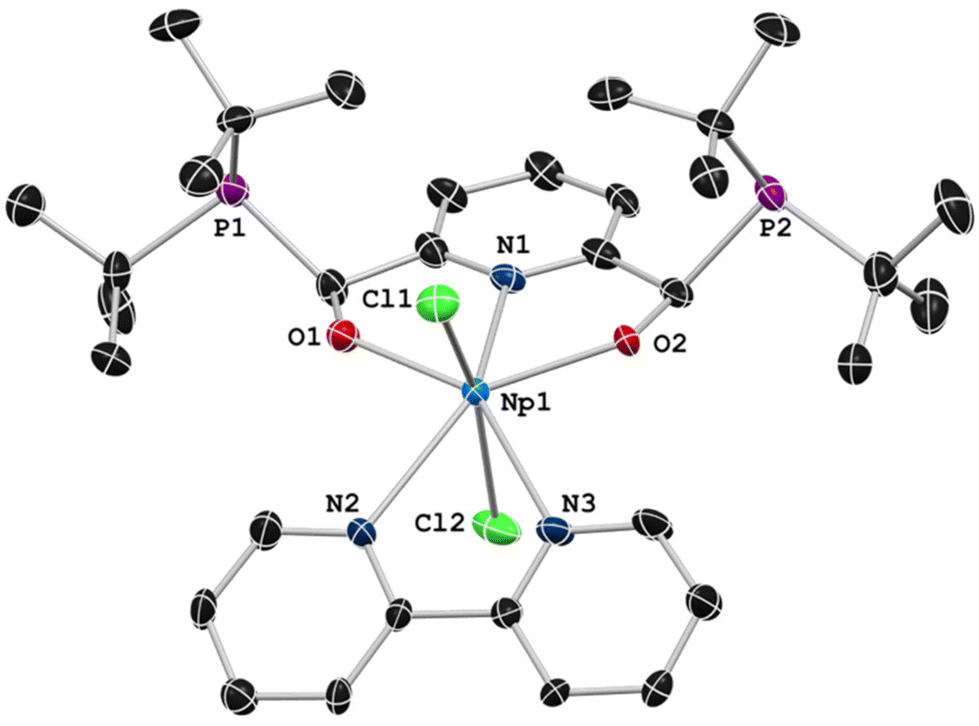 | ||
| Fig. 1 Solid-state structure of 2 presented at 50% probability ellipsoids. Hydrogen atoms, disordered fluorobenzene solvent molecules (Fig. S25†), and para-tertbutyl groups of the dtbpy ligand have been omitted for clarity. | ||
), 1 (P21/c), and 2
| Distance (Å), ∠ (°) | 1 (P21/c)a |
1 (P) |
2 (P) |
|---|---|---|---|
| a Previously reported in ref. 33. | |||
| M–O (avg) | 2.165(4) | 2.154(4) | 2.150(4) |
| M–Cl (avg) | 2.670(4) | 2.653(3) | 2.642(3) |
| M–N1pyr | 2.481(2) | 2.484(3) | 2.461(4) |
| M–Ndtbpy (avg) | 2.621(7) | 2.602(4) | 2.586(6) |
| Cl1–M–Cl2 | 163.69(7) | 163.41(4) | 162.45(4) |
| O1–M–O2 | 129.2(2) | 129.21(10) | 129.68(10) |
| N1pyr–M–O1 | 65.0(2) | 64.85(10) | 66.35(10) |
| N1pyr–M–O2 | 65.02(2) | 64.64(10) | 65.84(10) |
Little difference between the core molecule of 1 was found between the two different unit cells. The bond lengths and angles between 1 and 2 are nearly identical, though the Np–O bond distances are consistently slightly contracted than the U–O bond distances as expected (Table 1), although the values remain within 3σ of ESD values (ESD = estimated standard deviation). The M–N bond is found to be 0.02 Å shorter in the neptunium congener, also within 3σ of ESD values. Complex 2 represents a rare [ONO]2− ligand type for neptunium, with dipicolinate [ONO]2− representing the previously known complexes.36–42 Additionally, there is recently a single report of an enantioselective synthesis on neptunium bearing optically active (chiral) amidinates,43 yet complex 2, to our knowledge, describes the first diastereoselective reaction of neptunium by employing the benefits of template methods.
Beyond dtbpy: synthesis of actinide-indenyl complexes
Complexes 1 and 2 were isolated in good yields and high purity. However, the lability of the dtbpy ligand of 1 was problematic for subsequent experiments, contaminating resultant products. An alternative route to substitution was pursued to circumvent the use of dtbpy in the synthesis of these complexes. The addition of an anionic ligand derived from in situ deprotonation of 4,7-dimethyl-1,3-bis(1-methylethyl)-1H-indene (1,3-(iPr)2-4,7-Me2-C9H3) was employed following U–O bond formation and TMS-Cl release. The indenide ligand was chosen for electronic and coordination variability and reported high-degree of crystallinity,44 and was expected to provide steric protection and stability to the [(ONO)An]-core of these complexes. Furthermore, steric hindrance was expected to induce coordination to the metal trans to the tBu2P groups.The organometallic synthesis was optimized on uranium, with adjustments to the synthetic protocol of 1 to include the use of [K][(1,3-(iPr)2-4,7-Me2-C9H3)]. (CHO)2Py (1 equivalent) was initially added to UCl4 to form a tan slurry in THF. The addition of two equivalents of tBu2PSiMe3 to the reaction slurry resulted in the formation of a vibrant green solution and the loss of TMS-Cl. Removal of all volatiles under reduced pressure left a tacky, green material, which was then redissolved in THF. A THF solution of [K][(1,3-(iPr)2-4,7-Me2-C9H3)] was added dropwise to the uranium solution, resulting in a rapid color change to dark red. After work-up, crystalline material was isolated overnight from a concentrated pentane solution at −35 °C in 61% crystalline yield and assigned as the desired product (1,3-(iPr)2-4,7-Me2-C9H3)((tBu2P)ONO)UCl (3). Analysis of the 1H NMR spectrum suggested a paramagnetic complex, the spectrum comprising broadened, featureless peaks over a wide range (∼δ −64 ppm to δ 79 ppm). The resonances attributed to the tBu2P groups were found with a broad singlet at δ 32.21 ppm and a broadened doublet (J = 6 Hz) at δ −2.97 ppm, also with diagnostic downfield resonances centered at δ 78.90 ppm and δ 44.75 ppm. The 31P NMR spectrum of 3 contained a single resonance centered at δ 81.8 ppm, slightly shifted from that of complex 1 (δ 86.0 ppm).33 Magnetic studies performed via Evans method NMR gave a magnetic moment of 2.71μB, for 3 consistent with both complex 1 (2.33μB) and other previously reported U4+ complexes.33,45
Crystals of 3 grew as well-defined blocks but the crystals produced diffraction patterns suggestive of twinning. Following several attempts and careful crystal splitting, smaller single crystals produced sufficient, high-quality data with suggestions of minor twinning of the data. SC-XRD analysis of these crystals confirmed the assignment of the product as (1,3-(iPr)2-4,7-Me2-C9H3)((tBu2P)ONO)UCl (Fig. 2). Complex 3 was solved in the monoclinic P21/n space group, where the asymmetric unit cell contained a single molecule of 3, as well as pentane solvent disordered over two positions.
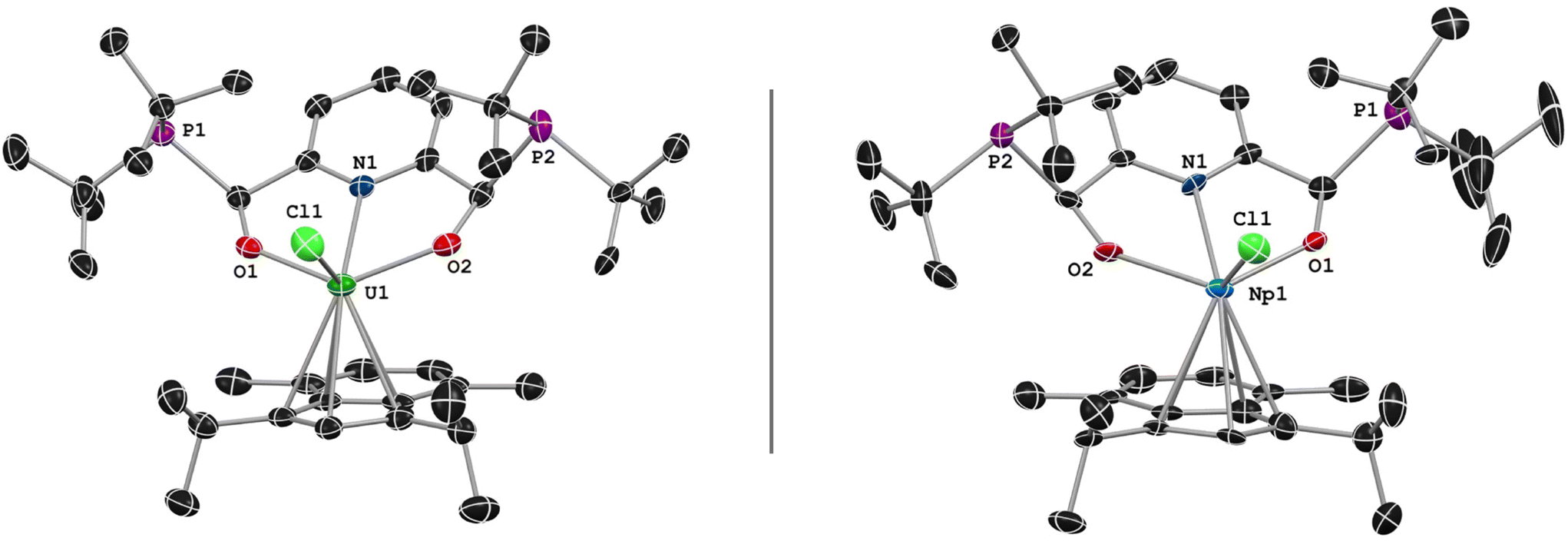 | ||
| Fig. 2 General solid-state structure of 3 (left) and 4 (right) presented at 50% probability ellipsoids. Hydrogen atoms and pentane solvent molecule have been omitted for clarity. | ||
The coordination environment around the uranium center consists of a single η5-indenide ligand, the [ONO]2− chelate, and one Cl−. As anticipated, the installation of the indenide occurred on the sterically accessible side, and the remaining Cl− is nestled between the steric bulk of the tBu2P moieties. The solid-state metrical parameters of 1 and 3 show little change to the chelate between the two metals, with the U–N and U–O bond distances found slightly contracted in 3. The O–U–O angle of the chelate is effectively unchanged, found at 129.2(2)° in 1 and 130.66(2)° in 3. The average U–centroid bond of 3 (2.477(1) Å) is consistent with, if slightly contracted, tris(indenyl)uranium chloride, U(Cl)(C9H7)3, which is reported at 2.49 Å.46 Additional metrical parameters can be found in Table 2. Compound 3 joins a small number of U-indenide complexes that have been reported47–53 in the larger established field of organouranium chemistry.
| Distance (Å), ∠ (°) | 3 | 4 |
4
Calc.a |
|---|---|---|---|
| a Structural data obtained from a geometry optimization employing scalar ZORA-B3LYP/TZP level of theory. | |||
| M–O1 (avg) | 2.130(2) | 2.126(11) | 2.146 |
| M–Cl1 | 2.603(1) | 2.574(3) | 2.585 |
| M–N1pyr | 2.465(1) | 2.449(6) | 2.479 |
| M–Centroid | 2.477(1) | 2.443(6) | 2.458 |
| Cl1–M–Cent. | 118.96(1) | 116.33(15) | 112 |
| O1–M–O2 | 130.66(2) | 130.6(4) | 128 |
| N1pyr–M–O1 | 65.63(2) | 64.8(4) | 65 |
| N1pyr–M–O2 | 65.44(2) | 66.4(4) | 65 |
Synthesis of the Np-indenide homologue
The synthesis employed for 3 was adapted to neptunium using (dme)2NpCl4. Following TMS-Cl elimination and formation of the [ONO]2− chelate, a THF solution of [K][(1,3-(iPr)2-4,7-Me2-C9H3)] was added. The color of the solution rapidly turned dark from the yellow color (Fig. S22†). Following work-up, the dark residue was taken into pentane and crystallized at −35 °C. Dark, diamond-shaped crystals formed and were assigned as (1,3-(iPr)2-4,7-Me2-C9H3)((tBu2P)ONO)NpCl, 4 (Fig. S23†). Characterization of 4 using NMR and UV-Vis-NIR spectroscopies and SC-XRD conclusively show the generation of the desired complex 4 in low yields (12% crystalline yield).The 1H NMR spectrum of 4 exhibited a wide chemical shift range (∼60 ppm) that is consistent with those of 1, 2, and 3. Resonances attributed to the tBuP moieties were found at δ 8.80 and δ 4.56 ppm as doublets (J = 7 Hz, 5 Hz, respectively), and again containing resonances downfield at δ 51.91 and δ 24.67 ppm. The 31P NMR spectrum reveals a single resonance at δ 72.71 ppm, similar to the observed 31P NMR spectrum of both 2 and 3. Evans method NMR was used to calculate the solution phase magnetic moment of 4, which was found at 1.21μB, uncharacteristically low for the expected magnetic moment for a 5f3 ion of Np4+ of 3.62μB.54 The 1H NMR spectrum of 4 gave evidence for the presence of diamagnetic indenide integrating to roughly 40% as compared to 4, likely causing the significantly low magnetic value and complicating the 1H NMR characterization of the complex. This diamagnetic material is attributed to the difficulties in small scale synthesis, sensitivity of the isolated complex (decomposition) or unreacted starting material, though the exact cause remains unknown.
SC-XRD studies of 4 were marred by significant twinning. The most promising crystal mounted during the SC-XRD experiments contained twinned data, which was modelled as a two-component, non-merohedral twin. Complex 4 was solved in the monoclinic P21/n space group, with unit cell parameters consistent with those of 2 (Fig. 2). As in 3, a disordered pentane molecule was present in the unit cell, and one of the tBu2P-units was disordered. Many of the bond distances between 3 and 4 were within 3σ of the ESD values, but in certain cases, such as the An–centroid distances, 4 displayed the contraction of the bonds expected for neighbors in the actinide series (Table 2). Despite the technical challenges in the synthesis and characterization of 4, we were able to confirm the first synthesis of an organometallic, diastereopure neptunium complex.
Electronic structure studies of uranium- and neptunium-ONO complexes
The twinning issue observed with crystals of 4 hampered precise metrical analysis between complexes 3 and 4. Accordingly, 1–4 were studied computationally, whereby both geometry optimizations and electronic structure studies were performed to assist with the comparison of the differences in geometry, electronic structure, and bonding. Detailed descriptions of the metal-to-supporting ligand bonding and the differences in f-orbital participation between uranium and neptunium was pursued. UV-Vis-NIR and computational geometry optimizations provide further insights for the comparison of the uranium and neptunium homologues.:C6D6 solutions with ratio 60:40) of 2 and 4 are relatively similar, both exhibiting several sharp, weak features from 500 nm through 1100 nm. These features are likely attributable to Laporte forbidden 5f → 5f transitions and 5f → 6d transitions for a Np4+ ion.24 The molar absorptivity of the spectral features of 2 are smaller than those observed for 4, consistent with the observed dark coloration of 4 (Fig. S14 and S17†). The spectral features observed for 3 are more pronounced and contain similar features to those of the previously reported spectrum of 1 (Fig. S15†).33 These results suggest minimal change in the electronic structure of the An4+ center upon substitution of dtbpy for 1,3-(iPr)2-4,7-Me2-C9H3.
The U4+ and Np4+ systems have formal 5f2 and 5f3 configurations, respectively and according to Hund's rule, they possess 3H and 4I ground terms. Under the intermediate coupling scheme, the ground state with spin–orbit coupling (SO–GS) is expected to be dominated by these terms, with additional contributions from some excited terms. This was supported by SO-CAS(n,7)SCF/NEVPT2 (n = 2, 3 for U4+, Np4+, respectively) calculations, where the ground-state term contributed between ∼76 and 86% to the total composition of the SO–GS (Tables S4–S7†). The substitution of dtbpy with the indenide ligand causes negligible differences in the composition of the SO–GS wavefunction in both the uranium and neptunium systems. For instance, the SO–GS of the uranium systems 1 and 3 respectively have ∼88 and 84% contribution from the 3H RS term, and ∼8 and 7% from the excited 1G. An extension of the active space was performed by including indenide orbitals in complexes 3 and 4 to evaluate correlation effects between the metal and ligand orbitals (see ESI for Computational descriptions†). Similarities were observed in terms of the SO–GS composition (Tables S6 and S7†), where excitations involving ligand orbitals only contribute about ∼1%. Comparable observations were found for the spin–orbit excited states (SO–ES) of all systems 1–4 (Tables S4–S7†).
Charge transfer bands and 5f → 6d transitions. TD-DFT calculations show that the region between 300 and 500 nm is dominated by inter-ligand, charge transfer (CT) and 5f → 6d bands in all the complexes. The most intense spectroscopic features are ascribed to LMCT and MLCT transitions, which are mixed together making it difficult to assign their categorization by energy ranges (see energy level orbital diagrams in Fig. 3). The 6d orbitals are split between ∼250–500 nm showing a significant mixing with ligand orbitals. Therefore, the 5f → 6d transitions that are observed in this range are not pure and are found overlapping with CT transitions. In the uranium systems 1 and 3, most of these excitations involve the dtbpy ligand and the [ONO]2− chelate (Tables S8 and S10†). Similar observations are found in the neptunium complexes (2 and 4), although a shift associated to the stabilization of the 5f orbitals is found, consistent with a bathochromic shift observed in some of the calculated transitions (Tables S9 and S11†).
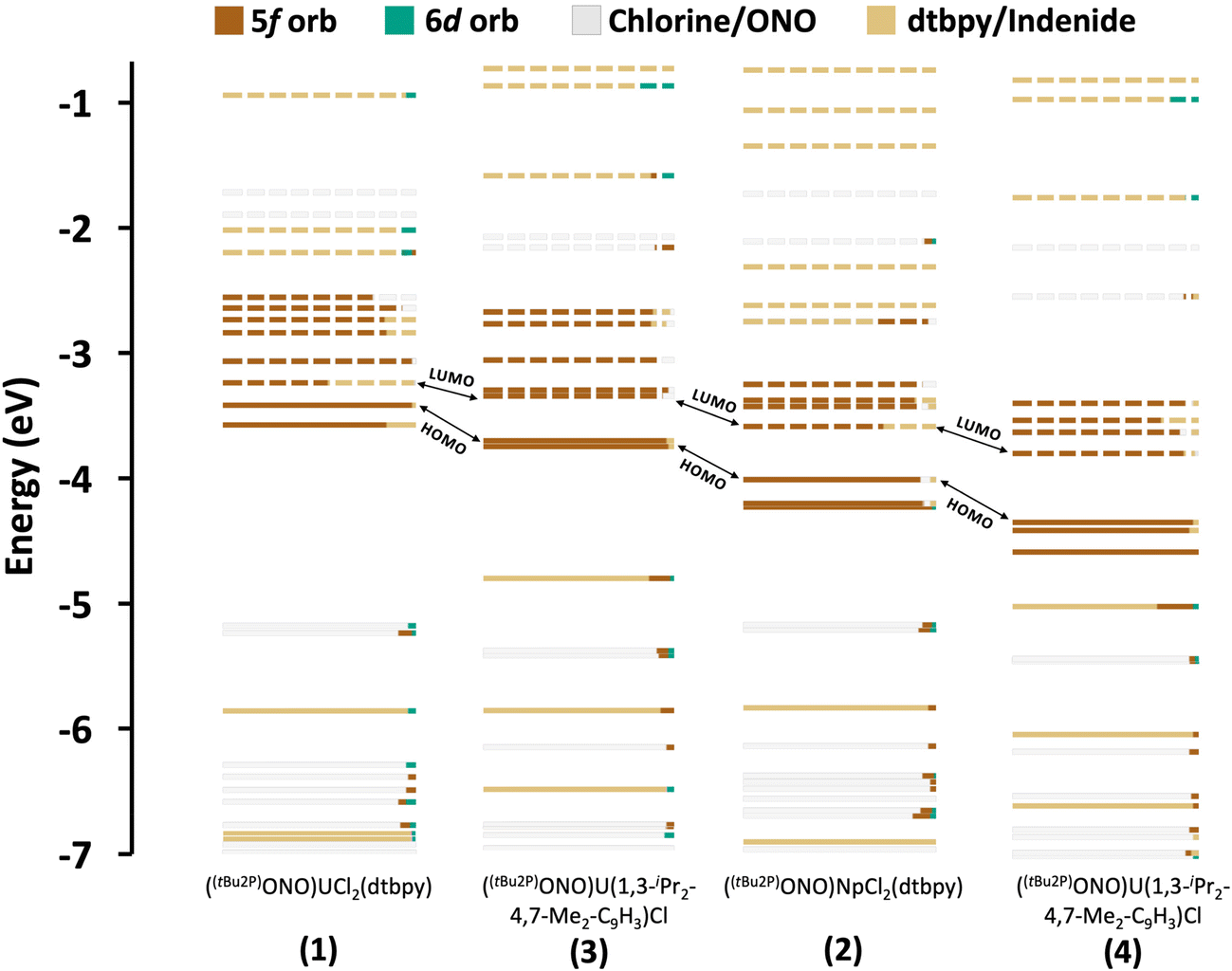 | ||
| Fig. 3 Energy level diagrams of systems ((tBu2P)ONO)UCl2(dtbpy) (1), ((tBu2P)ONO)NpCl2(dtbpy) (2), ((tBu2P)ONO)U(1,3-(iPr)2-4,7-Me2-C9H3)Cl (3) and ((tBu2P)ONO)Np(1,3-(iPr)2-4,7-Me2-C9H3)Cl (4) calculated at ZORA/B3LYP-TZP level of theory. The solid and dashed lines indicate occupied and empty orbitals, respectively. Each orbital is depicted in different colours, depending on its most predominant contributing fragment. Since the percentage of metal participation in the orbitals found in the range of −4 to −7 eV is small, the raw data with all the compositions and energies is available in the ESI.† | ||
f–f transitions. The assignment of the absorption bands was based on the analysis of low-lying excited states calculations (500 and 2000 nm range for complexes 1 and 3; 500 and 1500 nm range for complexes 2 and 4). The energy of each state along with its composition is shown in Tables S4–S7.† For the sake of clarity, each band in the experimental and theoretical spectra was labeled accordingly (Fig. 4). Generally speaking, the substitution of dtbpy by the indenide ligand does not significantly affect the positions of the absorption peaks for both the uranium and neptunium systems. Small differences were observed in terms of the splitting and intensities of some bands and attributed to the differential ligand fields.
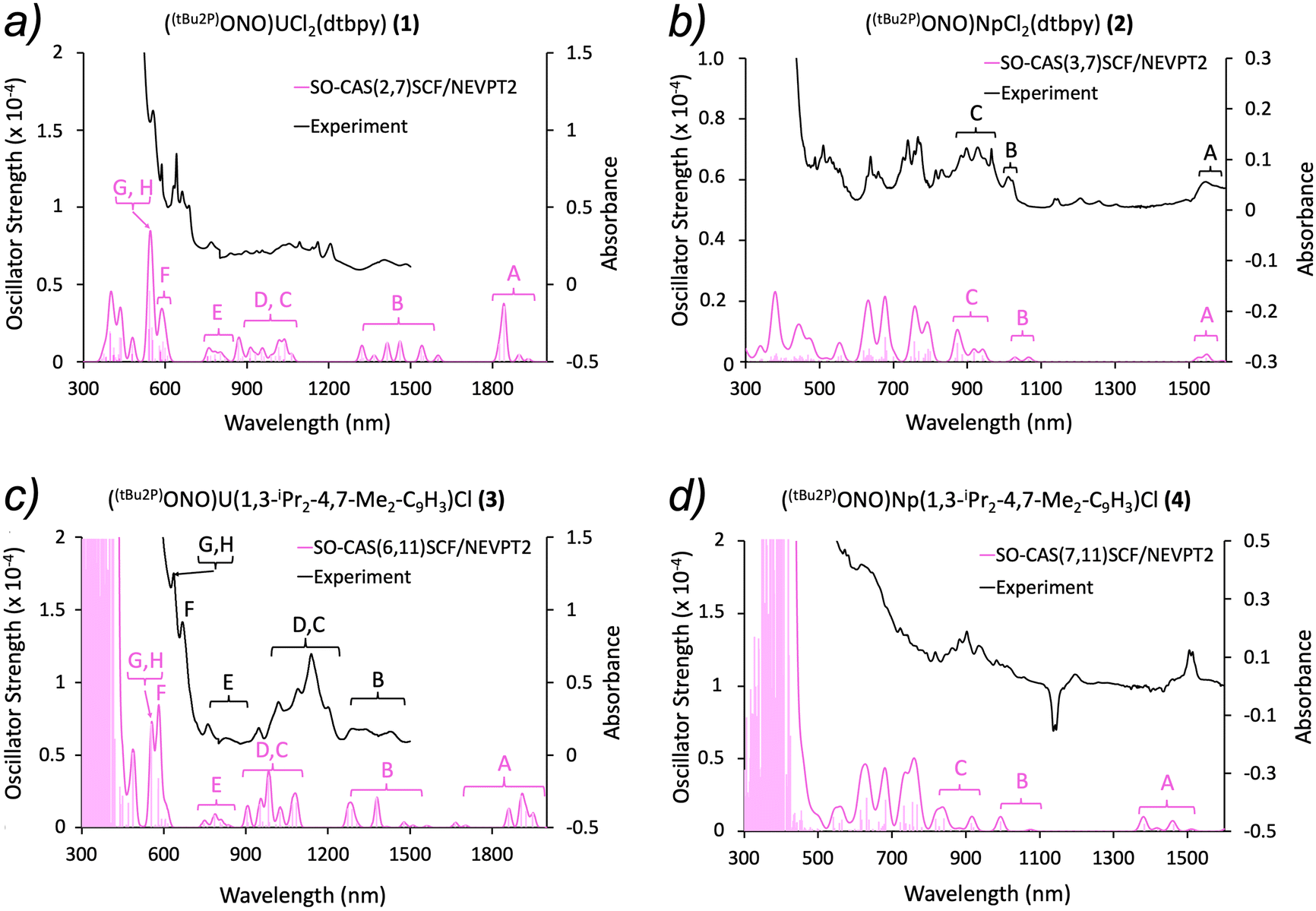 | ||
| Fig. 4 Experimental (black) and simulated (pink) UV-vis-NIR spectra for complexes 1–4. The vertical bars correspond to vertical transitions calculated at SO-CASSCF/NEVPT2 level of theory. (a) The simulated spectrum for 1 was labelled from A to H to show the different assignments of the bands. (b) The experimental and simulated spectra for 2 were labelled from A to C. The remaining states and respective energies are detailed in terms of J in Table S6.† (c) The experimental and simulated spectra for 3 were labelled from A to H to show the different assignments of the bands. (d) The simulated spectra for 4 was labelled from A to C; note the large negative and positive features at ∼1130 nm and 1510 nm are known artefacts from the instrument. The remaining states and respective energies are detailed in terms of J in Table S7.† | ||
Eight well defined absorption bands were predicted between 500 and 2000 nm for the uranium complexes (1 and 3, Fig. 4a and c). The first absorption band predicted (peak A) was not observed experimentally, although previous reports on [UCl5(THF)]− have identified a band in similar energy ranges.56,57 Due to the limited resolution in the experimental spectrum of complex 1, the band assignment was performed only for the predicted bands (Fig. 4a). Conversely, a comparison between experimental and theoretical data was possible in complex 3. Whereas the results are in good agreement with the experimental spectrum, the calculated spectrum appears blue shifted with respect to experiment (Fig. 4c and Fig. S5†). This shift is likely due to an incomplete treatment of the electronic correlation as consequence of a limited active space. Previous reports show comparable spectroscopic features with same assignments for U4+ complexes.56–58
The region between ∼680 and 1600 nm was assigned in terms of major contributions of RS terms to each state for the neptunium systems 2 and 4, as done above with the uranium complexes. Theoretical results are in good agreement with the experimental peaks, particularly in 2, where the spectrum is better resolved (Fig. 4b and d). A continuum of states was observed from ∼680 nm and up, which rendered the analysis of its composition difficult. Since all these states appear close in energy to each other, some of them overlap, causing multiple contributions to the same spectroscopic band. Additionally, finding the predominant RS term for a specific state became even more challenging since the contribution of other terms increased. Therefore, the assignment was performed only in terms of J in the range of ∼500 to 680 nm. Theoretical studies on Np4+ spectra are scarce, though the prediction and assignment of the spectra in complexes 2 and 4 are in good agreement with reported NpF4 and [NpCl6]2− systems.59,60
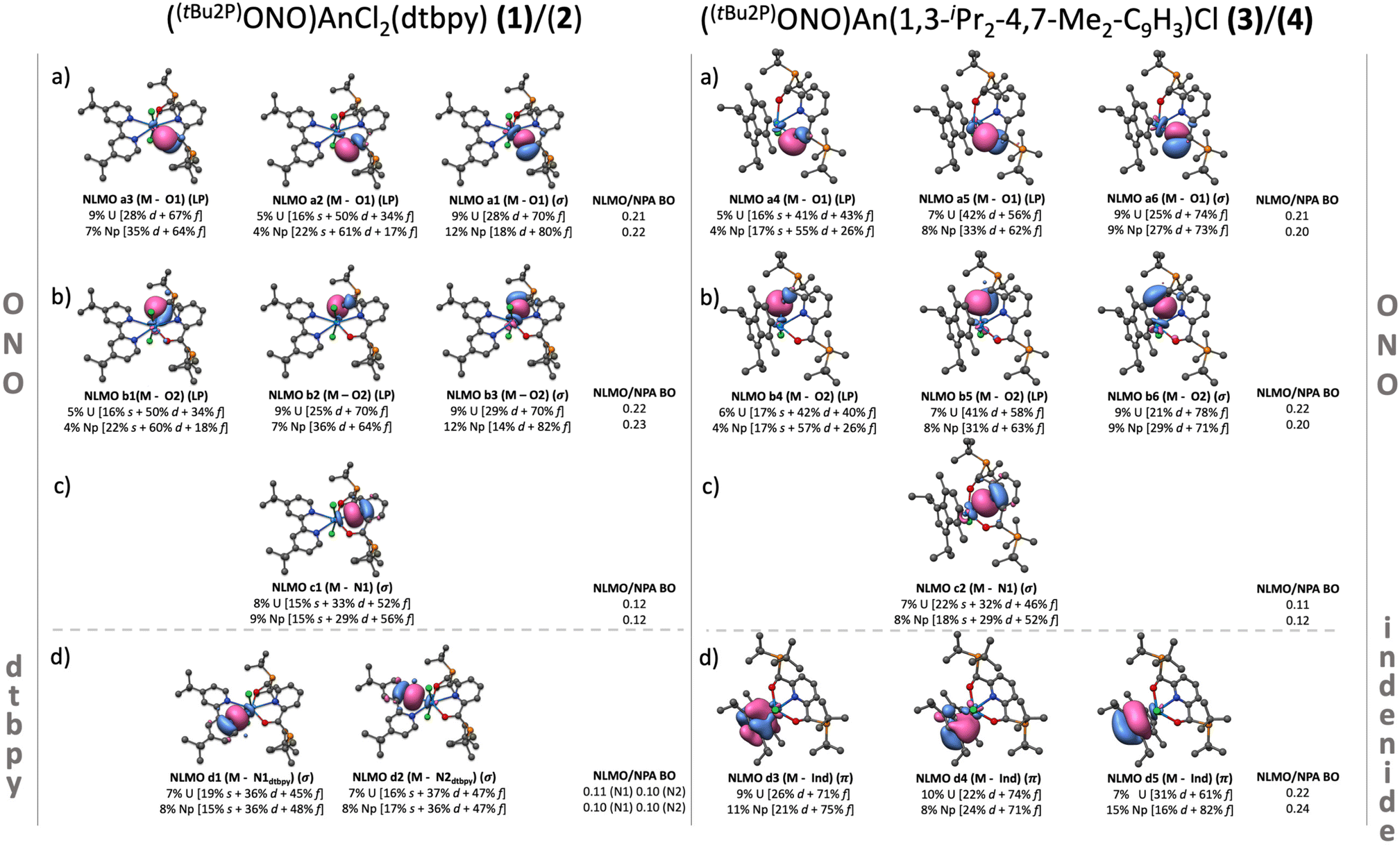 | ||
| Fig. 5 Left: NLMO composition of the most important interactions describing the An–ONO (above the gray dashed line) and An–dtbpy (below the gray dashed line) bonding in 1 and 2. The level of theory employed for these calculations was ZORA-B3LYP/TZP (isovalue = 0.03). Right: NLMO composition of the most important interactions describing the An–ONO (above the gray dashed line) and An–indenide (below the gray dashed line) bonding in 3 and 4. All NLMOs were plotted using an isovalue = 0.03. Additionally, the type of interaction σ, π, or lone-pair (LP) plus the NLMO/NPA bond orders (NLMO/NPA BO) for the M–O, M–N and M–C bonds are specified. In the case of the indenide ligand, the M–C bond-orders correspond to an average of all carbons in the 5-member ring. The plots in this figure correspond to systems 1 (left) and 3 (right). | ||
In a second step, the extended transition state (ETS) method for the energy decomposition analysis (EDA) combined with the natural orbitals for chemical valence (NOCV) theory was employed. The EDA allows to quantify contributions to the total energy in terms of electrostatic and orbital interactions along with the Pauli repulsion. Two fragments, corresponding to the metal (charge = 4+) and surrounding ligands (charge = 4−), were considered in this analysis. Thus, each component of the total interaction energy can be quantified between the defined fragments, and the orbital interactions can be further explored through the deformation energies obtained from the NOCV analysis.
Overall, the NLMO analysis describes the An–ligand interaction as predominantly ionic with a non-negligible degree of covalency (see below). It was also noticed that the An–indenide interaction displays a greater overlap compared to the An–dtbpy species. These results are consistent with conclusions coming from the EDA (Table 3). Here, it was observed that the dominance of the electrostatic component is common for all the systems, as expected considering the charged nature of the fragments employed in this calculation. When comparing uranium and neptunium analogs (1vs.2 and 3vs.4), similarities were observed in terms of the contribution of the electrostatic and orbital components. The most prominent differences emerge from changes in the ligand environment, where the highest orbital contributions to the total energy were found for the indenide systems 3 and 4 (∼6% more than in the dtbpy compounds).
| Energy component | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Electrostatic | −1911.1 (71%) | −1913.1 (71%) | −1696.2 (66%) | −1702.9 (65%) |
| Pauli | 388.2 | 376.6 | 368.8 | 365.7 |
| Steric | −1522.9 | −1536.5 | −1327.4 | −1335.2 |
| Orbital | −777.9 (29%) | −789.1 (29%) | −885.8 (34%) | −899.8 (35%) |
| Total | −2300.8 | −2325.5 | −2213.1 | −2230.9 |
Since the general picture of the bonding shows complementary conclusions between both methods, a more detailed discussion of the metal–ligand interactions is presented below. To facilitate this discussion, an analysis based on the orbital composition as well as the associated stabilization energies was performed. Because the conclusions reached from both the NOCV and the perturbative analysis analyses are similar, the results of the former are discussed while the ones coming from the perturbative analysis in NBO basis can be found in Tables S20–S23.† To simplify the comparison across the four systems (1–4), the discussion is organized with a focus on each type of metal–ligand interaction.
An–chlorine bonding. The An–Cl interaction is represented through four NLMOs, three of which display Cl → An donation. One NLMO describes a σ (An–Cl) covalent bond, while the other three correspond to electron lone-pairs (LP) (Tables S12–S15†). This σ bond is the strongest ligand–metal donation for all the systems, judging by its bond order (0.29–0.30) and orbital composition. In complexes 1 and 2, the average metal contribution to the An–Cl bond is ∼10%, primarily by 5f orbitals, while the rest comes from the chlorine atom 3p orbitals. The metal contribution to the bond stayed constant in complexes 3 and 4, however the average 5f contribution decreased ∼6% with respect to 1 and 2, which was mainly counterbalanced by a small 7s and 6d increment. These observations are consistent with the analysis of the deformation densities, where σ donations involving the Cl(3p) and An(5f/6d) orbitals were found to provide the most pronounced stabilization energies (between −48 and −52 kcal mol−1) (Fig. S7, a1 and b1†).
An–[ONO]2− bonding. In terms of individual interactions, each An–O bond is represented by three NLMOs displaying O → An donation (Fig. 5, 5a and 5b). One corresponds to a distorted σ (An–O) bond, while the other two correspond to lone pairs. Conversely, the An–N interaction was represented by one NLMO displaying σ N → An donation (NLMO 6a, Fig. 5). These σ bonds are the second strongest ligand–metal donations among all the systems. The average metal contribution to the bond is between 7–9% while the orbital contributions showed a negligible change between 1 and 3, and 2 and 4. The deformation densities show stabilization energies in the range of −49 and −28 kcal mol−1, where the most stabilizing interactions are σ in nature, similar for both 1 and 3, and 2 and 4. These results suggest that the An–[ONO]2− core is not significantly affected by the coordination of the dtbpy or indenide ligand (Fig. S7, a2–a4 and b3–b6†).
An–dtbpy bonding. Two σ (An–N) bonds (Fig. 5d, left) describe the interactions between the actinide metal and dtbpy, with 7% and 8% metal contribution to the bond for complexes 1 and 2, respectively. A bond order of 0.10 was found for both 1 and 2. Besides these interactions, corresponding to donations from the dtbpy ligand to the An center, an interaction consistent with a small degree of back-donation was found in 1 (Fig. 6). Since no significant differences in bond lengths and bond orders were found between 1 and 2, this back-bonding was attributed to orbital mixing due to a better energy match between the virtual orbitals in the dtbpy ligand and uranium f-orbitals (Fig. 6). From the NOCV analysis, σ donations from the 2p (dtbpy) orbitals were found at lower energies (∼−30 and −21 kcal mol−1), compared to those of the other ligands (Fig. S7, a5 and a6†). Additionally, the back-bonding interaction in 1 was found to provide a small stabilization energy of ∼2 kcal mol−1.
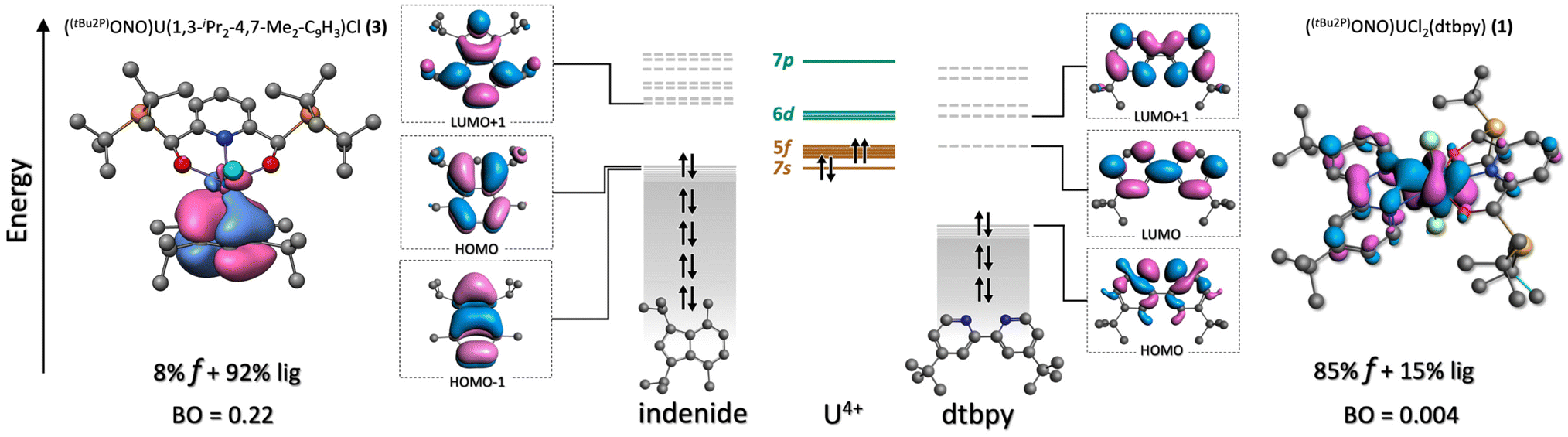 | ||
| Fig. 6 Qualitative molecular orbital diagram of 1 and 3. The brown lines/blocks indicate occupied orbitals while the green lines correspond to empty orbitals. As observed, the LUMO and LUMO+1 of the dtbpy ligand are close in energy to the 5f manifold. In consequence, an interaction displaying a small degree of back-donation (depicted in the right side) takes place. In the case of 3, the energetics is different and no back-bonding is observed. However, focusing on the HOMO of the indenide ligand, it is noteworthy how the 5-member ring orbital symmetry has a better match with the 5f orbitals than that of the 6-member ring. This interaction forms the dative interaction depicted in the left side. Isovalue = 0.03. | ||
An–indenide bonding. In complexes 3 and 4, the metal is located perpendicular to the plane of the 5-member ring in the indenide ligand. The metal preference towards this ring over the 6-member ring is attributed to the better symmetry match between its π orbitals and the 5f orbitals of the metal. The metal–ring interaction is via three NLMOs displaying donation towards the metal center. These correspond to π-type bonds with significant metal and 5f contributions (Fig. 5d right, Fig. 6). The uranium complex 3 exhibits an average metal contribution of ∼9%, out of which ∼68% corresponds to 5f orbitals. In the neptunium analog, 4, a small 2% increase in the metal contribution was observed. Interestingly, this increase was accompanied by an increment in the 5f instead of 6d contribution, the more common occurrence in actinide–ligand bonding. The increased 5f contribution, along with the calculated bond orders, indicates a differential and more covalent Np–indenide interaction. In terms of the deformation densities obtained from the NOCV analysis, the stabilization energies were found between −57 and −46 kcal mol−1, a similar energy range where the An–Cl interactions were also identified (Fig. S7, b1–b3†), but stronger than the An-dtbpy motif.
In summary, the bonding analysis suggests that the An–ligand interactions are mostly ionic with a minor component of covalency that is consistent with the conclusions obtained from previous reports on other An(IV) compounds.65–67 The dtbpy ligand shows a better energy match with respect to the metal orbitals. However, based on orbital composition and bond orders, the metal–dtbpy interactions also display the weakest overlap, consistent with the experimentally observed lability of this ligand. Conversely, the indenide derivative exhibits a greater orbital overlap, which overall suggests stronger interactions. Expanding the discussion to other An–L interactions, it is noteworthy that both An–indenide and An–Cl bonds show similar magnitudes of metal contribution, an interesting result considering that organic ligands are historically considered more covalent than halides when bound to metals. Though, a slight but important difference was observed for these two interactions in terms of the 5f involvement being higher in the An–indenide bond with respect to the An–Cl bond. This was compensated by an increase in 7s and 6d contribution in the latter. The differences associated with coordination of the indenide highlights the role of organic ligands in enhancing the 5f participation in chemical bonding, which is already established with cyclooctatetraene (COT) or cyclopentadienyl ligands.68,69 This feature emerges from the diversity of symmetry–permitted combinations between these orbitals and the π system of the ligand; by including organic π-based orbitals of appropriate symmetries, the 5f orbitals can better participate in bonding interactions.
Finally, a synergistic combination of wavefunction and DFT approximations has enabled an accurate analysis of the electronic structure, spectroscopy, and bonding of the complexes 1–4. This combination of methods has proven successful when applied to the f-block, yet a careful analysis and interpretation of the results is required. In this case, the different approaches led to complementary conclusions shedding light on the similarities between uranium and neptunium complexes, and differences in f-orbital participation arising from the coordination of dtbpy or indenide ligands.
Conclusions
This report shows the benefits of pursuing template methodology for preferentially forming diastereopure products with uranium and neptunium, increasing the versatility of this method for the small-scale synthesis necessary for neptunium chemistry. The template method employed here is selective for the meso diastereoisomer. This is a stark improvement over the rac:meso mixture of diastereoisomers that form in a 63:37 ratio when the free-ligand synthesis is performed without a metal template. The meso-[ONO]2− supported products stabilize the metal center and allow for further chemistry. The use of 1,3-(iPr)2-4,7-Me2-C9H3 provided homologous organometallic uranium and neptunium complexes for comparison. Not only is this the first diastereoselective synthesis for neptunium, but also the first indenide-coordinated neptunium structure reported, contributing to the small but growing number of established organoneptunium complexes.
Detailed computational exploration of complexes 1–4 provides a complete picture of the electronic structure, spectroscopy, and bonding of these compounds. The geometry optimization process shed light on the importance of the steric effects, concluding that the An–[ONO]2− contraction observed upon coordination of the indenide ligand was a consequence of lower steric hindrance, allowing for tighter metal–ligand coordination. Spectroscopic analysis performed through the CASSCF/NEVPT2 methodology was able to reproduce the UV-Vis-NIR spectra of the systems, where similarities in terms of position of the bands were detected after replacing the dtbpy by the indenide ligand. Differential splitting and intensities were identified in some bands, which was attributed to the intrinsic field produced by these ligands.
Finally, the NLMO analysis revealed similarities in composition and bond orders between the An–[ONO]2− and An–dtbpy interactions for both uranium and neptunium complexes. The most pronounced differences resulted from the substitution of the dtbpy by the indenide ligand, where an increased metal and 5f orbital contribution associated to the An–indenide interaction was found. In general terms, the bonding analysis suggests that the dtbpy ligand has a better energy match with respect to the metal orbitals, while the indenide ligand possesses a better orbital overlap. This is consistent with experimental observations describing the dtbpy ligand as extremely labile. The ETS-NOCV and second order perturbation theory analysis in NBO basis are consistent with the conclusions discussed above, suggesting a differential coordination for the indenide ligand. Additionally, this work highlights the importance of organic ligands in exposing the 5f orbital participation in An–L bonding.
Rational experimental design targeting homologous actinide complexes and implementation of multidisciplinary investigations resulted in the study of the electronic structure and bonding of diastereopure organoactinide complexes. The template protocol employed warrants further investigation in terms of generating new organometallic complexes of uranium and neptunium, as the specificity for meso-complexes is rather rare, and the singular steric profile may provide unique reactivity beyond the regiospecificity observed here. Further comparison of novel homologous complexes of uranium and neptunium based on complexes arising from the use of template protocols may provide further insight into differences in the electronic structure and bonding of these metals.
Author contributions
S.H.C and M.J.B.-L. contributed equally to this work, respectively performing the technical synthetic and computational tasks. A.M.T. conceived the synthetic chemistry and supervised the synthetic effort. E.R.B. and P.Y. supervised the computational effort. A.J.G. assisted with the execution of neptunium experimental work, providing guidance to S.H.C. and A.M.T. throughout. S.S. provided the necessary organic ligands for the organometallic chemistry. M.L.T. performed supplementary uranium chemistry, while J.T.M. assisted with data analysis and resolving twinned SC-XRD data. All authors contributed to manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was conceived and executed at Los Alamos National Laboratory. Los Alamos National Laboratory is operated by Triad National Security, LLC, for the National Nuclear Security Administration of the U.S. Department of Energy (contract no. 89233218CNA000001). A.M.T., A.J.G., P.Y., E.R.B., S.H.C., M.L.T., and M.J.B-L. acknowledge the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Heavy Element Chemistry program (2020LANLE372, DE-AC52-06NA25396). S.S. was supported by an LDRD Early Career Research project awarded to A.M.T (20190570ECR). We are grateful for postdoctoral support provided in part by the Glenn T. Seaborg Institute for S.H.C. and M.J.B.-L.References
- J. E. Niklas, K. S. Otte, C. M. Studvick, S. R. Chowdhury, B. Vlaisavljevich, J. Bacsa, F. Kleemiss, I. A. Popov and H. S. La Pierre, A tetrahedral neptunium(V) complex, Nat. Chem., 2024 DOI:10.1038/s41557-024-01529-6 . Online ahead of print.
- C. A. P. Goodwin, R. W. Adams, A. J. Gaunt, S. K. Hanson, M. T. Janicke, N. Kaltsoyannis, S. T. Liddle, I. May, J. L. Miller, B. L. Scott, J. A. Seed and G. F. S. Whitehead, N-Heterocyclic Carbene to Actinide d-Based π-bonding Correlates with Observed Metal-Carbene Bond Length Shortening Versus Lanthanide Congeners, J. Am. Chem. Soc., 2024, 146, 10367–10380 CrossRef CAS PubMed.
- B. N. Long, J. M. Sperling, C. J. Windorff, Z. K. Huffman and T. E. Albrecht-Schoenzart, Expanding Transuranium Organoactinide Chemistry: Synthesis and Characterization of (Cp′3M)2(μ-4,4′-bpy) (M = Ce, Np, Pu), Inorg. Chem., 2023, 62, 6368–6374 CrossRef CAS PubMed.
- M. S. Dutkiewicz, C. A. P. Goodwin, M. Perfetti, A. J. Gaunt, J.-C. Griveau, E. Colineau, A. Kovács, A. J. Wooles, R. Caciuffo, O. Walter and S. T. Liddle, A terminal neptunium(V)–mono(oxo) complex, Nat. Chem., 2022, 14, 342–349 CrossRef CAS PubMed.
- M. Černá, J. A. Seed, S. G. Fernandez, M. T. Janicke, B. L. Scott, G. F. S. Whitehead, A. J. Gaunt and C. A. P. Goodwin, Isostructural r-hydrocarbyl phospholide complexes of uranium, neptunium, and plutonium, Chem. Commun., 2022, 58, 13278–13281 RSC.
- C. A. P. Goodwin, A. J. Wooles, J. Murillo, E. Lu, J. T. Boronski, B. L. Scott, A. J. Gaunt and S. T. Liddle, Carbene Complexes of Neptunium, J. Am. Chem. Soc., 2022, 144, 9764–9774 CrossRef CAS PubMed.
- J. Su, T. Cheisson, A. McSkimming, C. A. P. Goodwin, I. M. DiMucci, T. Albrecht-Schönzart, B. L. Scott, E. R. Batista, A. J. Gaunt, S. A. Kozimor, P. Yang and E. J. Schelter, Complexation and redox chemistry of neptunium, plutonium and americium with a hydroxylaminato ligand, Chem. Sci., 2021, 12, 13343–13359 RSC.
- S.-X. Hu, E. Lu and S. Liddle, Prediction of High Bond-Order Metal-Metal Multiple-Bonds in Heterobimetallic 3d−4f/5f Complexes [TM-M{N(o-[NCH2P(CH3)2]C6H4)3}] (TM = Cr, Mn, Fe; M = U, Np, Pu, and Nd), Dalton Trans., 2019, 48, 12867–12879 RSC.
- J. T. Brewster II, D. N. Mangel, A. J. Gaunt, D. P. Saunders, H. Zafar, V. M. Lynch, M. A. Boreen, M. E. Garner, C. A. P. Goodwin, N. S. Settineri, J. Arnold and J. L. Sessler, In-Plane Thorium(IV), Uranium(IV), and Neptunium(IV) Expanded Porphyrin Complexes, J. Am. Chem. Soc., 2019, 141, 17867–17874 CrossRef PubMed.
- M. S. Dutkiewicz, J. H. Farnaby, C. Apostolidis, E. Colineau, O. Walter, N. Magani, M. G. Gardiner, J. B. Love, N. Kaltsoyannis, R. Caciuffo and P. L. Arnold, Organometallic neptunium(III) complexes, Nat. Chem., 2016, 8, 797–802 CrossRef CAS PubMed.
- J. L. Brown, E. R. Batista, J. M. Boncella, A. J. Gaunt, S. D. Reilly, B. L. Scott and N. C. Tomson, A linear trans-bis(imido)neptunium(V) actinyl analog: NpV (NDipp)2(tBu2bipy)2Cl (Dipp=2, 6-iPr2C6H3), J. Am. Chem. Soc., 2015, 137, 9583–9586 CrossRef CAS PubMed.
- M. L. Neidig, D. L. Clark and R. L. Martin, Covalency in f-element complexes, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS.
- N. Kaltsoyannis, Does Covalency Increase or Decrease across the Actinide Series? Implications for Minor Actinide Partitioning, Inorg. Chem., 2013, 52, 3407–3413 CrossRef CAS PubMed.
- P. L. Arnold, M. S. Dutkiewicz and O. Walter, Organometallic Neptunium Chemistry, Chem. Rev., 2017, 117, 11460–11475 CrossRef CAS PubMed.
- S. D. Reilly, J. L. Brown, B. L. Scott and A. J. Gaunt, Synthesis and characterization of NpCl4(DME)2 and PuCl4(DME)2 neutral transuranic An(IV) starting materials, Dalton Trans., 2014, 43, 1498–1501 RSC.
- M. A. Whitefoot, D. Perales, M. Zeller and S. C. Bart, Synthesis of Non-Aqueous Neptunium(III) Halide Solvates from NpO2, Chem. – Eur. J., 2021, 27(72), 18054–18057 CrossRef CAS PubMed.
- C. A. P. Goodwin, M. T. Janicke, B. L. Scott and A. J. Gaunt, Carbene Complexes of Neptunium, J. Am. Chem. Soc., 2021, 143, 20680–20696 CrossRef CAS PubMed.
- S. A. Pattenaude, N. H. Anderson, S. C. Bart, A. J. Gaunt and B. L. Scott, Non-aqueous neptunium and plutonium redox behaviour in THF – access to a rare Np(III) synthetic precursor, Chem. Commun., 2018, 54, 6113–6116 RSC.
- L. R. Avens, S. G. Bott, D. L. Clark, A. P. Sattelberger, J. G. Watkin and B. D. Zwick, A Convenient Entry into Trivalent Actinide Chemistry: Synthesis and Characterization of AnI3(THF)4 and An[N(SiMe3)2]3 (An = U, Np, Pu), Inorg. Chem., 1994, 33, 2248–2256 CrossRef CAS.
- M. S. Dutkiewicz, C. Apostolidis, O. Walter and P. L. Arnold, Reduction chemistry of neptunium cyclopentadienide complexes: from structure to understanding, Chem. Sci., 2017, 8, 2553–2561 RSC.
- J. Su, C. J. Windorff, E. R. Batista, W. J. Evans, A. J. Gaunt, M. T. Janicke, S. A. Kozimor, B. L. Scott, D. H. Woen and P. Yang, Identification of the Formal +2 Oxidation State of Neptunium: Synthesis and Structural Characterization of {NpII[C5H3(SiMe3)2]3}1−, J. Am. Chem. Soc., 2018, 140, 7425–7428 CrossRef CAS PubMed.
- F. Baumgaertner, E. O. Fischer, B. Kanellakopulos and P. Laubereau, Tetrakis(cyclopentadienyl)neptunium(IV), Angew. Chem., Int. Ed. Engl., 1968, 7, 634 CrossRef CAS.
- A. J. Myers, M. L. Tarlton, S. P. Kelley, W. W. Lukens and J. R. Walensky, Synthesis and Utility of Neptunium(III) Hydrocarbyl Complex, Angew. Chem., Int. Ed., 2019, 58, 14891–14895 CrossRef CAS PubMed.
- S. L. Staun, L. M. Stevens, D. E. Smiles, C. A. P. Goodwin, B. S. Billow, B. L. Scott, G. Wu, A. M. Tondreau, A. J. Gaunt and T. W. Hayton, Expanding the Nonaqueous Chemistry of Neptunium: Synthesis and Structural Characterization of [Np(NR2)3Cl], [Np(NR2)3Cl]−, and [Np{N(R)(SiMe2CH2)}2(NR2)]− (R = SiMe3), Inorg. Chem., 2021, 60, 2740–2748 CrossRef CAS PubMed.
- J. Murillo, C. A. P. Goodwin, L. Stevens, S. Fortier, A. Gaunt and B. L. Scott, Synthesis and comparison of iso-structural f-block metal complexes (Ce, U, Np, Pu) featuring η6-arene interactions, Chem. Sci., 2023, 14, 7438–7446 RSC.
- T. J. McMurry, K. N. Raymond and P. H. Smith, Molecular Recognition and Metal Ion Template Synthesis, Science, 1989, 244, 938–943 CrossRef CAS PubMed.
- A. F. Williams, Oligopolydentate ligands for helices and metallacycles, Pure Appl. Chem., 1996, 68, 1285–1289 CrossRef CAS.
- T. J. Hubin and D. H. Busch, Template routes to interlocked molecular structures and orderly molecular entanglements, Coord. Chem. Rev., 2000, 200–202, 5–52 CrossRef CAS.
- P. A. Vigato, S. Tamburini and L. Bertolo, The development of compartmental macrocyclic Schiff bases and related polyamine derivatives, Coord. Chem. Rev., 2007, 251, 1311–1492 CrossRef CAS.
- M. A. Esteruelas, A. M. Lopez, L. Mendez, M. Olivan and E. Oñate, Preparation, Structure, and Ethylene Polymerization Behavior of Bis(imino)pyridyl Chromium(III) Complexes, Organometallics, 2003, 22, 395–406 CrossRef CAS.
- A. M. Archer, M. W. Bouwkamp, M.-P. Cortez, E. Lobkovsky and P. J. Chirik, Arene Coordination in Bis(imino)pyridine Iron Complexes: Identification of Catalyst Deactivation Pathways in Iron-Catalyzed Hydrogenation and Hydrosilation, Organometallics, 2006, 25, 4269–4278 CrossRef CAS.
- A. Mikhailine, A. J. Lough and R. H. Morris, Efficient asymmetric transfer hydrogenation of ketones catalyzed by an iron complex containing a P-N-N-P tetradentate ligand formed by template synthesis, J. Am. Chem. Soc., 2009, 131, 1394–1395 CrossRef CAS PubMed.
- S. H. Carpenter, B. S. Billow and A. M. Tondreau, Diastereoselective Template Synthesis on Iron and Uranium, Organometallics, 2021, 40, 2389–2393 CrossRef CAS.
- Y.-R. Luo, Calculated diatomic bond strengths serve as an approximation, CRC Press, Boca Raton, Florida, 2007 Search PubMed.
- C. A. P. Goodwin, J. Su, T. E. Albrecht-Schmitt, A. V. Blake, E. R. Batista, S. R. Daly, S. Dehnen, W. J. Evans, A. J. Gaunt, S. A. Kozimor, N. Lichtenberger, B. L. Scott and P. Yang, [Am(C5Me4H)3]: An Organometallic Americium Complex, Angew. Chem., Int. Ed., 2019, 58, 11659–11699 Search PubMed.
- G. Tian, L. Rao and S. J. Teat, Thermodynamics, Optical Properties, and Coordination Modes of Np(V) with Dipicolinic Acid, Inorg. Chem., 2009, 48, 10158–10164 CrossRef CAS PubMed.
- A. B. Yusov, M. S. Grigor'ev, A. M. Fedoseev, P. Moisy, V. P. Shilov and A. V. Gogolev, Behavior of Np(V), Pu(V), and Am(V) in solutions of pyridine-2,6-dicarboxylic (dipicolinic) acid and synthesis of crystalline compounds, Radiochemistry, 2015, 57, 6–19 CrossRef CAS.
- C. Xu, G. Tian, S. J. Teat, G. Liu and L. Rao, Thermodynamic and Structural Trends in Hexavalent Actinyl Cations: Complexation of Dipicolinic Acid with NpO22+ and PuO22+ in Comparison with UO22+, Chem. – Eur. J., 2013, 19, 16690–16698 CrossRef CAS PubMed.
- N. A. Budantseva, G. B. Andreev, A. M. Fedoseev, M. Y. Antipin and J.-C. Krupa, Interaction of neptunium(V) with picolinic, nicotinic, and isonicotinic acids, Radiochim. Acta, 2006, 94, 69–74 CrossRef CAS.
- G. B. Andreev, M. Y. Antipin, A. M. Fedoseev and N. A. Budantseva, Synthesis and crystal structure of bis(carbamide)chromatoneptunyl NpO2CrO4 · 2[OC(NH2)2], Crystallogr. Rep., 2001, 46, 383–384 CrossRef.
- G. B. Andreev, V. N. Khrustalev, M. Y. Antipin, A. M. Fedoseev, N. A. Budantseva, Z.-K. Krupa and C. Madic, Synthesis andcrystal structure of neptunyl pyridine-2,6-dicarboxylate pentahydrate (NpO2)2[2,6-(OOC)2C5H3N]·5H2O, Russ. J. Coord. Chem., 2000, 26, 825–827 CAS.
- A. B. Yusov, V. I. Mishkevich, A. M. Fedoseev and M. S. Grigor'ev, Complexation of An(VI) (An = U, Np, Pu, Am) with 2,6-pyridinedicarboxylic acid in aqueous solutions. Synthesis and structures of new crystalline compounds of U(VI), Np(VI), and Pu(VI), Radiochemistry, 2013, 55, 269–278 CrossRef CAS.
- S. Fichter, S. Kaufmann, P. Kaden, T. S. Brunner, T. Stumpf, P. W. Roesky and J. März, nantiomerically Pure Tetravalent Neptunium Amidinates: Synthesis and Characterization, Chem. – Eur. J., 2020, 26, 8867–8870 CrossRef CAS PubMed.
- G. P. McGovern, F. Hung-Low, J. W. Tye and C. A. Bradley, Synthesis and Reactivity Studies of Benzo-Substituted Bis (indenyl) Iron and Zirconium Complexes: The Difference a Methyl Group Can Make, Organometallics, 2012, 31, 3865–3879 CrossRef CAS.
- S. Fortier, B. C. Melot, G. Wu and T. W. Hayton, Homoleptic uranium(IV) alkyl complexes: synthesis and characterization, J. Am. Chem. Soc., 2009, 131, 15512–15521 CrossRef CAS PubMed.
- J. H. Burns and P. G. Laubereau, Crystal structure of triindenyluranium chloride, Inorg. Chem., 1971, 10, 2789 CrossRef CAS.
- J. Rebizant, M. R. Spirlet, S. Bettonville and J. Goffart, Structure of bis [(1, 2, 3, 3a, 7a-η)-indenyl] bis (tetrahydroborato) uranium(IV), Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, C45, 1509 CrossRef CAS.
- R. J. Kahan, J. H. Farnaby, N. Tsoureas, F. G. N. Cloke, P. B. Hitchcock, M. P. Coles, S. M. Roe and C. Wilson, Sterically encumbered mixed sandwich compounds of uranium(III): Synthesis and reactivity with small molecules, J. Organomet. Chem., 2018, 857, 110–122 CrossRef CAS.
- M. R. Spirlet, J. Rebizant, S. Bettonville and J. Goffart, Structure of chlorotris (1, 2, 4, 5, 6, 7-hexamethylindenyl) uranium(IV), Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, C48, 1221 CrossRef CAS.
- M.-R. Spirlet, J. Rebizant, S. Betonville and J. Goffart, The crystal structure of tris (indenyl) uranium (β, β, β-trifluoroethanolate), J. Organomet. Chem., 1993, 460, 177 CrossRef CAS.
- J. Rebizant, M. R. Spirlet and J. Goffart, Structure of tris (1–3-η-indenyl) iodouranium, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, C41, 334 CrossRef CAS.
- J. Meunier-Piret, J. P. Declercq, G. Germain and M. Van Meerssche, Structure du complexe triindenyl-uranium, Bull. Soc. Chim. Belg., 1980, 89, 121–124 CrossRef CAS.
- J. Rebizant, M. R. Spirlet and J. Goffart, Structure of indenyluranium trichloride-bis(tetrahydrofuran), [U(C9H7)]Cl3⋅2C4H8O, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, C39, 1041 CrossRef CAS.
- T. F. Wall, S. Jan, M. Autillo, K. L. Nash, L. Guerin, C. Le Naour, P. Moisy and C. Berthon, Paramagnetism of Aqueous Actinide Cations. Part I: Perchloric Acid Media, Inorg. Chem., 2014, 53, 2450–2459 CrossRef CAS PubMed.
- P. S. Bagus, E. S. Ilton, R. L. Martin, H. J. A. Jensen and S. Knecht, Spin–orbit coupling in actinide cations, Chem. Phys. Lett., 2012, 546, 58–62 CrossRef CAS.
- E. Hashem, A. N. Swinburne, C. Schulzke, R. C. Evans, J. A. Platts, A. Kerridge, L. S. Natrajan and R. J. Baker, Emission spectroscopy of uranium(IV) compounds: a combined synthetic, spectroscopic and computational study, RSC Adv., 2013, 3, 4350–4361 RSC.
- M. Godsall and N. F. Chilton, Investigation of the Electronic Structure and Optical Spectra of Uranium(IV), (V), and (VI) Complexes Using Multiconfigurational Methods, J. Phys. Chem. A, 2022, 126, 6059–6066 CrossRef CAS PubMed.
- A. T. Chemey, C. Celis-Barros, K. Huang, J. M. Sperling, C. J. Windorff, R. E. Baumbach, D. E. Graf, D. Paez-Hernandez, M. Ruf, D. E. Hobart and T. E. Albrecht-Schmitt, Electronic, Magnetic, and Theoretical Characterization of (NH4)4UF8, a Simple Molecular Uranium(IV) Fluoride, Inorg. Chem., 2019, 58, 637–647 CrossRef CAS PubMed.
- W. T. Carnall, G. K. Liu, C. W. Williams and M. F. Reid, Analysis of the crystal–field spectra of the actinide tetrafluorides. I. UF4, NpF4, and PuF4, J. Chem. Phys., 1991, 95, 7194 CrossRef CAS.
- J. Jung, M. Atanasov and F. Neese, Ab initio ligand-field theory analysis and covalency trends in actinide and lanthanide free ions and octahedral complexes, Inorg. Chem., 2017, 56, 8802–8816 CrossRef CAS PubMed.
- T. S. Grimes, C. R. Heathman, S. Jansone-Popova, A. S. Ivanov, S. Roy, V. S. Bryantsev and P. R. Zalupski, Influence of a Heterocyclic Nitrogen-Donor Group on the Coordination of Trivalent Actinides and Lanthanides by Aminopolycarboxylate Complexants, Inorg. Chem., 2018, 57, 1373–1385 CrossRef CAS PubMed.
- C. Celis-Barros, T. Albrecht-Schonzart and C. J. Windorff, Computational Investigation of the Bonding in [(η5−Cp′)3(η1−Cp′)M]1− (M = Pu, U, Ce), Organometallics, 2021, 40, 1577–1587 CrossRef CAS.
- D.-C. Sergentu, F. Gendron, E. D. Walter, S. Park, C. Capan, R. G. Surbella, C. Z. Soderquist, G. B. Hall, S. I. Sinkov, J. Autschbach and H. Cho, Equatorial Electronic Structure in the Uranyl Ion: Cs2UO2Cl4 and Cs2UO2Br4, Inorg. Chem., 2022, 61, 3821–3831 CrossRef CAS PubMed.
- Y. Liu, C.-Z. Wang, Q.-Y. Wu, J.-H. Lan, Z.-F. Chai, W.-S. Wu and W.-Q. Shi, Theoretical Probing of Size-Selective Crown Ether Macrocycle Ligands for Transplutonium Element Separation, Inorg. Chem., 2022, 61, 4404–4413 CrossRef CAS PubMed.
- M. J. Tassel and N. Kaltsoyannis, Covalency in AnCp4 (An = Th–Cm): a comparison of molecular orbital, natural population and atoms-in-molecules analyses, Dalton Trans., 2010, 39, 6719–6725 RSC.
- J. Su, E. R. Batista, K. S. Boland, S. E. Bone, J. A. Bradley, S. K. Cary, D. L. Clark, S. D. Conradson, A. S. Ditter, N. Kaltsoyannis, J. M. Keith, A. Kerridge, S. A. Kozimor, M. W. Löble, R. L. Martin, S. G. Minasian, V. Mocko, H. S. La Pierre, G. T. Seidler, D. K. Shuh, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, Energy-Degeneracy-Driven Covalency in Actinide Bonding, J. Am. Chem. Soc., 2018, 140, 17977–17984 CrossRef CAS PubMed.
- T. Cantat, C. R. Graves, K. C. Jantunen, C. J. Burns, B. L. Scott, E. J. Schelter, D. E. Morris, P. J. Hay and J. L. Kiplinger, Evidence for the Involvement of 5f Orbitals in the Bonding and Reactivity of Organometallic Actinide Compounds: Thorium(IV) and Uranium(IV) Bis(hydrazonato) Complexes, J. Am. Chem. Soc., 2008, 130, 17537–17551 CrossRef CAS PubMed.
- A. Kerridge, f-Orbital covalency in the actinocenes (An = Th–Cm): multiconfigurational studies and topological analysis, RSC Adv., 2014, 4, 12078–12086 RSC.
- O. Walter, Actinide Organometallic Complexes with π-Ligands, Chem. – Eur. J., 2019, 25, 2927–2934 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, pictures, spectra, crystallographic tables, CCDC deposition numbers, and theoretical calculations are provided. CCDC 2297381–2297385. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi00810c |
| ‡ S.H.C and M.J.B.-L. contributed equally to this work, respectively performing the synthetic and computational tasks. |
| This journal is © the Partner Organisations 2024 |