
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntroducing dibenzocyclooctatetraene into actinide chemistry: isolation of rare trivalent uranium sandwich complexes†
Ernesto
Castellanos
 ,
Wei
Su
and
Selvan
Demir
*
,
Wei
Su
and
Selvan
Demir
*
Department of Chemistry, Michigan State University, 578 South Shaw Lane, East Lansing, Michigan 48824, USA. E-mail: sdemir@chemistry.msu.edu
First published on 15th October 2024
Abstract
Peerless actinide complexes containing π-conjugated dibenzocyclooctatetraene (dbCOT) dianions were isolated. These three uranium molecules have the tripositive oxidation state of the metal ion in common and vary in ligand arrangement where the difference originates from the presence of coordinating solvent molecules and encapsulating chelating agents, respectively. The first compound [K(DME)2][U(dbCOT)2] (where DME = dimethoxyethane), 1, features a potassium ion interacting with one of the dbCOT ligands and was obtained from a salt metathesis reaction employing UI3(dioxane)1.5 and K2dbCOT. The second compound [K(crypt-222)][U(dbCOT)2], 2, arose from treating 1 with 2.2.2-cryptand, yielding an outer-sphere potassium ion. The third compound [K(crypt-222)][U(dbCOT)2(THF)], 3, was generated through the exposure of 1 to THF solvent and 2.2.2-cryptand, resulting in the coordination of THF to uranium(III) along with η8-ligation of each dbCOT ligand while placing the potassium ion outer sphere, encased in the chelating agent. Notably, the compounds present the largest uranocene derivatives, considering the presence of fused aromatic rings to the central COT framework, which bear a total of 36 π-electrons. The three uranium molecules were thoroughly studied via single-crystal X-ray diffraction and UV-Vis-NIR, IR, and NMR spectroscopy. DFT computations on 1–3 shed light on their divergent electronic structure.
Congratulations on the 10th anniversary of Inorganic Chemistry Frontiers!No other journal, covering all areas of inorganic chemistry, has gained so much momentum during the last decade and received such high acclaim from the community. Marking an outstanding success story, ICF has risen to a top, high-impact inorganic chemistry journal, with an impressive, global visibility, attracting also early career researchers to their reader- and authorship. The breadth of themed collections is particularly striking. I enjoy being a reader, reviewer, and author for this outstanding journal. I am also excited about my new role as an Early Career Advisory Board Member of ICF and look forward to offering guidance and support. My two publications on rare earth chemistry were featured on outside front covers, and one was chosen by readers as one of the three best ICF covers of 2023. I think that artistic graphics greatly enrich science and captivate even the non-specialists. This current publication has a special meaning to me, as it marks the first uranium paper from my group, and we are particularly delighted to have it published in ICF. |

Introduction
The discovery of the landmark uranocene, U(COT)2,1,2 (where COT = cyclooctatetraene), molecule ushered in a new class of organometallic sandwich complexes that invoked covalent bonding interactions between metal-based 5f-orbitals and ligand-centred π-orbitals.3–6 The captivating electronic structure and metal–ligand covalency found in such uranium complexes have inspired scientists to study uranocene derivatives with functionalized COT ligands, ranging from alkyl,6–8 aryl,9 to silyl10–13 groups. This has vastly enriched actinide organometallic chemistry and provided invaluable insight into electronic structure descriptions, where a thorough understanding of actinide complexes is seminal to advancing nuclear waste remediation technologies and actinide separation methods.14,15Further functionalization of the central COT ring can be envisioned by fusing annulated carbocyclic rings to the central COT framework. While theoretically possible, such systems are scarce and hitherto known with only one carbocylic ring fused to the COT moiety.16–18 Among those are four- and five-membered alkyl ring-fused uranium(IV)–COT complexes, U(C8H6(CH2)2)216 and U(C8H6(CH2)3)2,17 respectively, and the first uranocene complex containing fused aromatic rings, U(C8H6(C4H4))2.18 The cyclobuto- and cyclopento-COT ligands adopt staggered geometries with respect to the central COT rings to generate C2v-symmetric complexes featuring trans-oriented annulated rings. In contrast, the benzo-fused COT complex, U(C8H6(C4H4))2,16 is approximately eclipsed with benzo moieties nearly superimposable. Thus, an eclipsed or staggered outcome may be directed through COT use that contains or lacks fused aromatic rings.
Notably, homoleptic uranocene complexes with a UIII ion are rare and the only known examples contain methyl-,8 1,4-bis(trimethylsilyl)-,13 and 1,4-bis(tert-butyldimethylsilyl)11-substituted COT ligands. The scarcity is attributed to the highly reductive and reactive metal center,19,20 rendering synthesis difficult. Notably, trivalent uranocene molecules with any fused rings on the central COT are unknown, which may be ascribed to both the lack of readily available π-conjugated cyclooctatetraene ligands and the synthetic challenge of handling highly reactive organouranium complexes.
The dibenzocyclooctatetraene (dbCOT) dianion, dbCOT2−, is planar and contains 18 π-electrons rendering it aromatic according to Hückel's rule.21 Its coordination chemistry is heavily underexplored and unknown with d-block metals beyond group III and actinides. In 2022, we isolated the first rare earth (RE) dbCOT complexes which were sterically congested heteroleptic RE metallocenes. Each metallocene was composed of two tetramethylcyclopentadienyl and one η2-dbCOT ligand, yielding anionic metallocenes charge-balanced by an outer sphere [K(crypt-222)]+ cation (where crypt-222 = 2.2.2-cryptand).22 Thereafter, we successfully employed dbCOT in the synthesis of homoleptic RE sandwich complexes, initially with YIII,23 followed by ErIII, affording the first erbium-based single-molecule magnet comprising a dbCOT ligand.24 RE compounds comprising anionic and cationic dbCOT metallocenes are also accessible.25
Taking into account the recent developments in RE chemistry, the dianionic dbCOT ligand serves as an excellent candidate for the generation of the first actinide complexes containing dbCOT in any oxidation state. The complexation of redox-active actinide ions with the expanded π-surface of the dbCOT ligands holds great potential towards applications in switchable,26 and conductive materials.27 Herein, we report the synthesis and characterization of three uranium dbCOT sandwich complexes, [K(DME)2][U(dbCOT)2] (where DME = dimethoxyethane), 1, [K(crypt-222)][U(dbCOT)2], 2, and [K(crypt-222)][U(dbCOT)2(THF)] (where THF = tetrahydrofuran), 3. Notably, each uranocene complex exhibits similar spectroscopic properties despite pronounced differences in molecular geometries. This suggests that the electronic structure is governed by the predominantly ionic interaction between the COT ligands and UIII centre. However, computational characterisation uncovered differences in the f-orbital contribution to the frontier molecular orbitals.
Experimental methods
General information
Caution! 238U is radioactive and an α emitter with a half-life of approximately 4.5 × 109 years. The handling of uranium-containing samples should only be performed in laboratories appropriately equipped to safely handle radioactive materials.All manipulations were performed in either an argon- or nitrogen-filled MBraun glovebox with an atmosphere of <0.1 ppm O2 and <0.1 ppm H2O. House nitrogen was purified using an MBraun HP-500-MO-OX gas purifier. Tetrahydrofuran (THF) was refluxed over potassium for several days and was subsequently dried further over the Na/K alloy. Dimethoxyethane (DME) and toluene were dried over potassium. nHexane and npentane were dried over calcium hydride. All solvents were distilled under N2 from their drying agents, and water/oxygen absence was confirmed via benzophenone solution as an indicator within a glovebox prior to use. Deuterated solvents were purchased from Cambridge Isotope Laboratories, dried over Na/K alloy for several days, and filtered prior to use. Ul3(dioxane)1.5,28 UI3(THF)4,28 dibenzocyclooctatetraene (dbCOT),29 potassium dibenzocyclooctatetraenide (K2dbCOT),24 and potassium graphite (KC8),30 were prepared according to literature procedures. 2.2.2-Cryptand (crypt-222) was crystallised from a hot nhexane solution prior to use. 1,4-Dioxane and 2-methyltetrahydrofuran (2-MeTHF) were purchased from Sigma-Aldrich, stirred over Na/K alloy for several days, and filtered before use. Elemental analysis was performed at Michigan State University, using a PerkinElmer 2400 Series II CHNS/O analyser.
Synthesis of [K(DME)2][U(dbCOT)2], 1
In a 20 mL vial equipped with a stir bar, UI3(dioxane)1.5 (95.8 mg, 0.128 mmol, 1 equiv.) was suspended in a 4 mL DME solution. To this stirring dark purple suspension, K2dbCOT (69.0 mg, 0.244 mmol, 1.9 equiv.) was added portionwise as a solid over 1 min, and the colour rapidly changed to dark brown, followed by the precipitation of colourless insoluble solids, presumably potassium iodide. The reaction was allowed to stir for 4 h at room temperature, after which colourless insoluble solids were removed via filtration over Celite, and the resulting clear, dark brown solution was evaporated under reduced pressure yielding a tacky dark brown residue. The product was triturated in nhexane and yielded a fine, dark brown powder after drying. The resulting dark brown powder was extracted in THF, filtered over Celite, and dried under dynamic vacuum to give a dark brown solid (85.7 mg). Brown, needle-like crystals, suitable for single-crystal X-ray diffraction analysis, were obtained from a concentrated DME solution after layering with nhexane and stored at −35 °C for 24 h. The crystals were separated from the mother liquor and dried under dynamic vacuum before crushing to a fine powder, washed with npentane, and further dried under reduced pressure for the elemental and spectroscopic analysis of 1. Crystalline yield: 85%, 80.2 mg, 0.103 mmol. 1H NMR (500 MHz, THF-d8, 25 °C) δ: 7.14 (br s, 8 H, COT(C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) (CH)2C)2), 4.53 (br s, 8 H, COT(CH(C)2CH)2), 3.49 (s, 4 H, H3CO(C2)2OCH3), 3.31 (s, 6 H, 3CO(CH2)2OC3), −35.99 (br s, 8 H (COT–)). 13C NMR (126 MHz, THF-d8, 25 °C) δ: 165.24 (
(CH)2C)2), 4.53 (br s, 8 H, COT(CH(C)2CH)2), 3.49 (s, 4 H, H3CO(C2)2OCH3), 3.31 (s, 6 H, 3CO(CH2)2OC3), −35.99 (br s, 8 H (COT–)). 13C NMR (126 MHz, THF-d8, 25 °C) δ: 165.24 (![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) 8H4), 126.46 (8H4), 117.32 (6H4) 73.02 (H3CO(H2)2OCH3), 62.99 (6H4), 59.12 (H3O(CH2)2OH3). IR spectrum (FTIR, cm−1): 3217w, 3065w, 3029w, 2998w, 2978w, 2893w, 2822w, 1743w, 1611w, 1522w, 1470w, 1446w, 1428w, 1383s, 1281w, 1263w, 1243w, 1215w, 1192w, 1143m, 1126m, 1086s, 1031w, 1015m, 936w, 852m, 768s, 758s, 709m, 656w. Anal. calcd for C36H34O2KU: C, 55.74; H, 4.42; N, 0.00. Found: C, 56.17; H 4.33; N 0.23.
8H4), 126.46 (8H4), 117.32 (6H4) 73.02 (H3CO(H2)2OCH3), 62.99 (6H4), 59.12 (H3O(CH2)2OH3). IR spectrum (FTIR, cm−1): 3217w, 3065w, 3029w, 2998w, 2978w, 2893w, 2822w, 1743w, 1611w, 1522w, 1470w, 1446w, 1428w, 1383s, 1281w, 1263w, 1243w, 1215w, 1192w, 1143m, 1126m, 1086s, 1031w, 1015m, 936w, 852m, 768s, 758s, 709m, 656w. Anal. calcd for C36H34O2KU: C, 55.74; H, 4.42; N, 0.00. Found: C, 56.17; H 4.33; N 0.23.
Synthesis of [K(DME)2][U(dbCOT)2], 1 from UI3(THF)4
Following the analogous synthetic procedure for 1 described above, brown needle-like crystals were grown from a concentrated DME solution layered with nhexane after storing at −35 °C for 24 h in 56% crystalline yield (45.8 mg, 0.059 mmol). Masses used: UI3(THF)4 (100.5 mg, 0.111 mmol, 1 equiv.) and K2dbCOT (59.6 mg, 0.211 mmol, 1.9 equiv.).Synthesis of [K(crypt-222)][U(dbCOT)2], 2
In a 7 mL vial equipped with a stir bar, [K(DME)][U(dbCOT)2] (58.5 mg, 0.075 mmol, 1 equiv.) was suspended in a 1 mL DME solution at 60 °C. To this dark brown solution, 2.2.2-cryptand (29.8 mg, 0.079 mmol, 1.05 equiv.) was added directly as a solid, after which the formation of dark insoluble solids, presumably ‘[K(crypt-222)][U(dbCOT)2]’, was observed. The reaction was allowed to stir for 10 min at 60 °C, and the dark brown solution was carefully removed from the insoluble solids and filtered over Celite. The resulting clear, dark brown solution was subsequently stored at −35 °C. The remaining insoluble solids were extracted in 10 mL of hot DME (60 °C), filtered over Celite, and stored at −35 °C. After 16 h at −35 °C, the solutions were carefully layered with nhexane, and brown, block-shaped crystals, suitable for single crystal X-ray diffraction analysis, were obtained from the layered solutions after storing at −35 °C for 2 d. The crystals were separated from the mother liquor and dried under dynamic vacuum before crushing to a fine powder, washing with DME, and drying further under reduced pressure for the elemental and spectroscopic analysis of 2. Crystalline yield: 79%, 63.2 mg, 0.060 mmol. 1H NMR (500 MHz, THF-d8, 25 °C) δ: 7.03 (br s, 8 H, COT(C(CH)2C)2), 4.73 (s, 12 H, OC2CH2N), 4.69 (s, 12 H, O(C2)2O), 4.56 (br s, 8 H, COT(CH(C)2CH)2), 3.72 (s, 12 H, OCH2C2N), −35.61 (br s, 8 H (COT–)). 13C NMR (126 MHz, THF-d8, 25 °C) δ: 163.78 (8H4), 129.54 (8H4), 115.89 (6H4) 72.68 (O(H2)2O), 70.01 (OH2CH2N), 61.14 (6H4), 56.36 (OCH2H2N). IR spectrum (FTIR, cm−1): 3024w, 2945w, 2873m, 2807m, 1476m, 1442m, 1386s, 1352s, 1292m, 1260m, 1238w, 1213w, 1171w, 1145w, 1128m, 1100s, 1074s, 1057m, 1016m, 948m, 930m, 829w, 818w, 770s, 705m, 685w, 656w. Anal. calcd for C50H60N2O6KU: C, 56.54; H, 5.69; N, 2.64. Found: C, 56.12; H, 5.90; N, 2.61.
Synthesis of [K(crypt-222)][U(dbCOT)2(THF)], 3
In a 7 mL vial equipped with a stir bar, [K(DME)][U(dbCOT)2] (126.1 mg, 0.168 mmol, 1 equiv.) was suspended in a 2 mL THF solution. To this dark brown solution, 2.2.2-cryptand (64.5 mg, 0.171 mmol, 1.05 equiv.) was added directly as a solid, gradually yielding a clear, dark brown solution. The reaction was diluted with 1 mL THF to allow for adequate stirring and was subsequently allowed to stir for 10 min at room temperature, after which, the solution was filtered over Celite and concentrated to approximately 1.5 mL. Dark brown, needle-like crystals, suitable for single crystal X-ray diffraction analysis, were obtained after storing at −35 °C for 24 h. Crystalline yield: 76%, 140.2 mg, 0.124 mmol. 1H NMR (500 MHz, THF-d8, 25 °C) δ: 7.02 (br s, 8 H, COT(C(CH)2C)2), 4.70 (s, 12 H, OC2CH2N), 4.67 (s, 12 H, O(C2)2O), 4.56 (br s, 8 H, COT(CH(C)2CH)2), 3.70 (s, 12 H, OCH2C2N), 3.62 (br, t, 4 H, THF–2COC2), 1.78 (br, t, 4 H, THF–H2C(C2)2CH2), −35.60 (br s, 8 H (COT–)). 13C NMR (126 MHz, THF-d8, 25 °C) δ: 129.68 (8H4), 115.86 (6H4) 72.73 (O(H2)2O), 70.06 (OH2CH2N), 68.39 (THF–H2OH2), 61.11 (6H4), 56.39 (OCH2H2N), 28.58 (THF–H2C(H2)2CH2). IR spectrum (FTIR, cm−1): 3024w, 2946w 2874m, 2807m, 1476w, 1459w, 1442w, 1387s, 1353m, 1294m, 1261m, 1238w, 1171w, 1146w, 1129m, 1102s, 1076s, 1058m, 1017m, 949m, 930m, 830s, 819w, 772s, 706m, 686w, 659w. Anal. calcd for C50H60N2O6KU: C, 56.54; H, 5.69; N, 2.64. Found: C, 55.97; H, 6.15; N, 2.66.
Single-crystal X-ray diffraction
Brown crystals of 1 (needle-like), 2 (block-shaped), and 3 (needle-like) with dimensions of 0.110 × 0.080 × 0.060 mm3, 0.156 × 0.060 × 0.055 mm3, and 0.250 × 0.080 × 0.080 mm3, respectively, were mounted on a nylon loop using Paratone oil. Data for 1–3 were recorded on an XtaLAB Synergy, Dualflex, HyPix diffractometer equipped with an Oxford Cryosystems low-temperature device, operating at T = 100.00(10), 100.01(10), and 99.98(10) K, for 1–3, respectively. Data were measured using ω scans with Cu Kα radiation (microfocus sealed X-ray tube, 50 kV, 1 mA). The total number of runs and images were based on the strategy calculation using the CrysAlisPro program (Rigaku, V1.171.41.90a, 2020), which was used to retrieve and refine the cell parameters, as well as for data reduction. A numerical absorption correction based on Gaussian integration over a multifaceted crystal model empirical absorption correction using spherical harmonics was implemented in the SCALE3 ABSPACK scaling algorithm. The structures were solved in the P21/n, P21/n, and P21/c space groups for 1–3, respectively, by intrinsic phasing using the ShelXL structure solution program.31 The structure was refined by least squares using version 2018/2 of XL31 incorporated in Olex2.32 All nonhydrogen atoms were refined anisotropically. Hydrogen atom positions were calculated geometrically and refined using the riding model.NMR spectroscopy
NMR spectra were recorded between −40 °C and 50 °C on a Bruker Avance NEO 500 spectrometer and calibrated to the residual solvent signals (THF-d8: δH = 3.58 ppm, δC = 67.57 ppm). Signal multiplicities are abbreviated as follows: s (singlet), t (triplet), and br (broad). Air-sensitive NMR samples were prepared in a nitrogen-filled glovebox using J. Young NMR tubes outfitted with an inner Teflon liner for double containment.UV-Vis-NIR spectroscopy
The UV-Vis spectra were recorded with an Agilent Cary 60 spectrophotometer at ambient temperature from 230 to 800 nm. NIR data were collected on a PerkinElmer Lambda 1050 spectrometer at ambient temperature from 1000 to 1600 nm. Samples were prepared in a nitrogen-filled glove box and filtered into 1 cm quartz cuvettes outfitted with Teflon screw caps. The spectra were baseline corrected from a sample of dry 2-MeTHF.Infrared spectroscopy
The IR spectra were recorded with an Agilent Cary 630 FTIR spectrometer on crushed crystalline solids under a nitrogen atmosphere.Computational methods
All calculations were performed using the ORCA 5.0.4 program suite.33,34 Owing to the presence of a capping [K(DME)2]+ motif in 1, the complete contact ion pair, [K(DME)2][U(dbCOT)2], was used for all calculations of 1, whereas the charge-separated complexes, [K(crypt-222)][U(dbCOT)2], 2, and [K(crypt-222)][U(dbCOT)2(THF)], 3, were calculated considering only the anionic uranium metallocene moieties, [U(dbCOT)2]− and [U(dbCOT)2(THF)]−, respectively. All calculations were performed with the hybrid TPPSh functional (10% HF exchange) and the D3 dispersion correction using the BJ damping scheme (“d3bj” keyword).35,36 A finer integration grid (“defgrid3” keyword) was also employed. The trust-region augmented Hessian procedure was turned off and the calculations were accelerated using the “notrah” and “rijcosx” keywords, respectively. Due to the presence of open-shell nature of 1–3, the “veryslowconv” keyword was employed to mitigate poor convergence behaviour. Auxiliary basis sets were generated using the “autoaux” keyword.37The initial geometry optimization was carried out on the respective crystallographic coordinates considering a quartet electronic ground state. For 1–3, the def2-SVP basis set38 was used to describe K, O, C, and H atoms, where applicable. To reduce computational costs, the small-core 60-electron effective core potential (ECP60MDF)39 and the inherent basis set39 were used to describe U atoms. The exact Hessian values were calculated in the first optimization step to improve convergence behaviour. To obtain a more accurate electronic structure description of 1–3, the optimized geometry was subjected to a single point calculation considering scalar relativistic effects via the Douglas–Kroll method, where the all-electron SARC-DKH-TZVP basis set40 was used for U atoms and density-fitting was accomplished via the “dkh” keyword. The DKH-def2-SVP basis set38 was used for K, O, C, and H atoms, where applicable, and the frozen core approximation was turned off utilizing the “nofrozencore” keyword. All subsequent energy calculations were executed at this level of theory. Natural Bond Orbital (NBO) calculations were conducted using the NBO 6 program.41 Molecular orbitals were generated using the Orca_plot module as cube files and the graphical representation of orbital surfaces were produced using the VMD software package.42
Results and discussion
Synthesis and structural characterisation
Isolation of the π-expanded uranocene derivative, [K(DME)2][U(dbCOT)2], 1, proceeded from the stoichiometric reaction of UI3(dioxane)1.5 and K2dbCOT in DME for 4 hours (Fig. 1A). Brown, needle-like crystals of 1 suitable for single-crystal X-ray diffraction analysis were grown in 24 h from a concentrated DME solution layered with nhexane at −35 °C in 85% yield. The homoleptic uranocene complex, 1, crystallizes in the P21/n space group and features a trivalent uranium ion asymmetrically ligated by two dbCOT2− ligands (Fig. 1B and S1†). The absence of alkyl or silyl substituents from the central COT rings severely limits the solubility of 1 in ethereal solvents, adding to the synthetic rigor required to isolate and purify organouranium complexes. The asymmetric coordination mode of the (dbCOT)2− ligands is attributed to the presence of a contact ion pair, where a capping potassium ion exhibits a canted interaction with one of the central COT rings of a dbCOT2− fragment in 1. Here, the potassium ion is coordinated with two DME molecules, generating a distorted square pyramidal geometry with the COT ring centroid at the apex. The K–CCOT distances range between 3.056(4) and 3.419(4) Å and are similar to the those reported for K(diglyme)[U(COTMe)2] (where diglyme = 1-methoxy-2-(2-methoxyethoxy)ethane, and COTMe = C8H7Me) and [K(DME)2][U(COTTBDMS2)2] (where COTTBDMS2 = C8H6(tBuMe2Si)2-1,4), of 3.13(2)–3.34(2) Å and 3.042(6)–3.360(5), respectively.11,19 The elongated K–CCOT distances with respect to the free dbCOT dianion, [K(DME)]2[dbCOT] (2.934(2)–3.042(2) Å), may arise from a pronounced increase in electronic repulsion owing to the presence of a charge-dense UIII ion. In addition, the electron-rich dbCOT2− ligand bridges two Lewis acidic ions in an inverse sandwich motif, smearing the electron density between U and K ions. Within the [K(DME)2]+ motif of 1, one of the K–ODME distances is elongated (2.808(5) Å) by comparison to the other K–O interactions (2.675(4)–2.705(6) Å), enabling the desolvation of one DME molecule upon exposure to dynamic vacuum.11 The anionic uranocene moiety, [U(dbCOT)2]−, in 1 is oriented in a nearly perfect eclipsed geometry with a deviation of only 2.2° from the idealized eclipsed conformation, leading to a pseudo D2h symmetry within the [U(dbCOT)2]− anion, Fig. 2. This remarkable orientation remains elusive in RE metal complexes bearing dbCOT dianions, suggesting that the ∼3.9 Å separation between C6H4 rings of opposing dbCOT2− ligands in 1 may be outside the range of electrostatic repulsion or may experience weak π–π stacking interactions. In fact, the Cnt–U–Cnt angle is 177°, the nearest to perfect linearity observed for any trivalent uranocene complex. The U–Cnt distances of 2.055 Å and 2.004 Å differ only slightly, where the longer U–Cnt distance belongs to the sandwiched dbCOT ligand between U and K ions. Here, the coordination of two Lewis acidic ions to a single (dbCOT)2− ligand likely minimizes the electron density accumulated at the central COT ring, resulting in a weaker coordination to the trivalent U centre. Furthermore, the planarity of the bridging (dbCOT)2− ligand sandwiched between U and K ions is in stark contrast to the concave (inwardly bent) geometry of the opposite (dbCOT)2− ligand (Fig. 2D). This deviation from planarity in the coordinating dbCOT ligand has been observed when complexed to highly Lewis RE metals and arises from a combination of crystal packing effects and weak electrostatic attraction between the vacant metal orbitals and electron-rich ligand π-orbitals.23 Notably, the U–CCOT distances in 1 (2.698(4)–2.856(4) Å) are consistent with the presence of a UIII ion, albeit are elongated with respect to known trivalent uranocene complexes, as tetravalent uranocene derivatives exhibit shortened U–CCOT contacts owing to the contracted ionic size of UIV compared to UIII.8,13,43 The exceedingly large U–C contacts arise between the shared C atoms of the central COT rings and the annulated benzo motifs, which are on average longer in the bridging dbCOT2− ligand by 0.07 Å.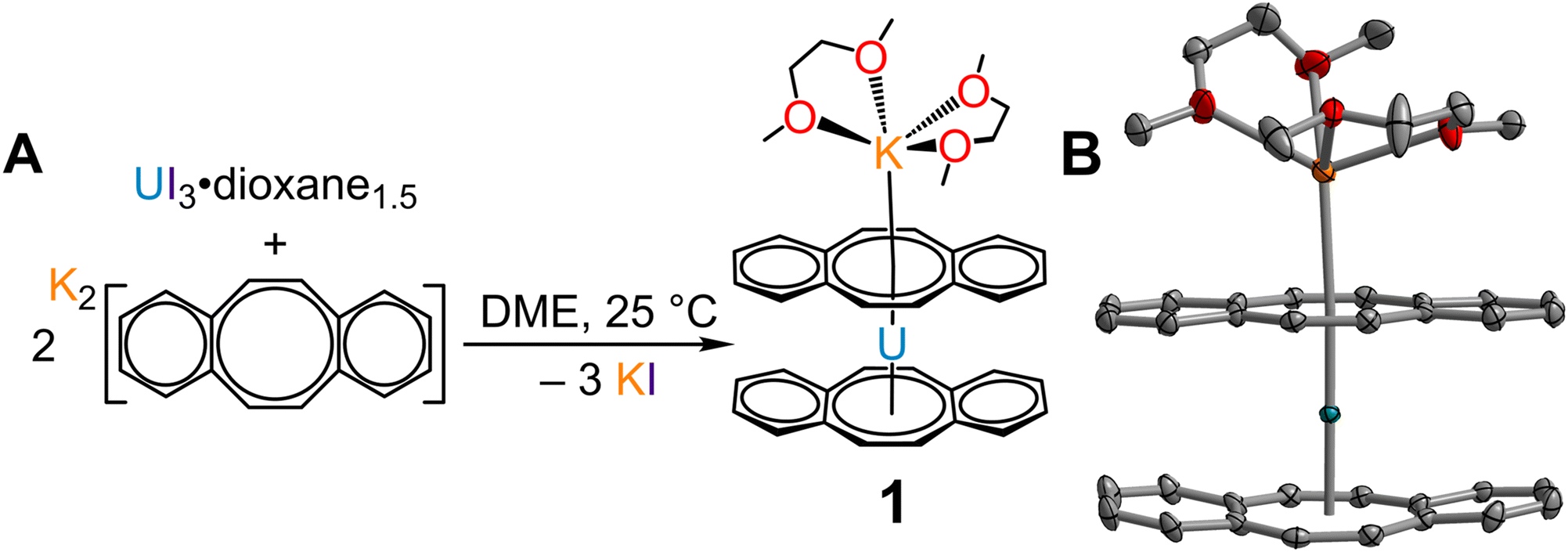 | ||
| Fig. 1 (A) Synthetic route to [K(DME)2][U(dbCOT)2], 1. (B) Structure of 1, with thermal ellipsoids drawn at the 50% probability level. Teal, orange, red, and grey ellipsoids represent U, K, O, and C atoms, respectively. H atoms are omitted for clarity. Select interatomic distances (Å) and angles (°) for 1: U–CCOT = 2.698(4)–2.856(4); U–Cnt = 2.004, 2.055, Cnt–U–Cnt: 177.1. We define that the circles in each dbCOT drawing represent in total 18 π-electrons. | ||
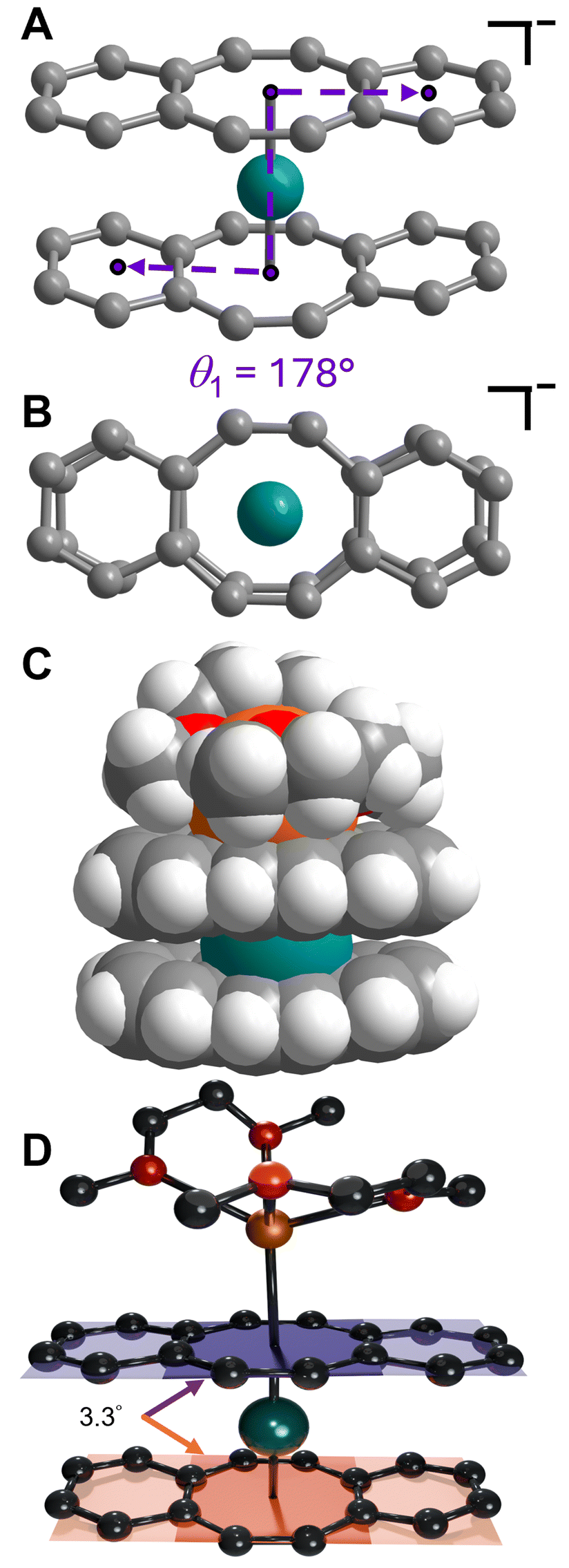 | ||
| Fig. 2 (A) Structure of the [U(dbCOT)2]− anion in a crystal of [K(DME)2][U(dbCOT)2], 1, with corresponding atom labels. The dihedral angles (Θ1 = 178° and Θ2 = 2°) were calculated using the benzo– and COT ring centroids. (B) Arial perspective of 1. Teal and grey spheres represent U and C atoms, respectively. H atoms and the coordinating [K(DME)2]+ countercation are omitted for clarity. (C) Space filling model of 1. Teal, orange, red, grey, and white-grey spheres represent U, K, O, C, and H atoms, respectively. (D) Structure of 1 with planes bisecting the central COT rings of each dbCOT2− ligand, where the angle between planes is 3.3°. Teal, orange, red, and black spheres represent U, K, O, and C atoms, respectively. H atoms are omitted for clarity. | ||
Excitingly, 1 can be transformed into a charge-separated ion pair upon exposure to 2.2.2-cryptand in hot DME (60 °C) and crystallised from a concentrated DME solution layered with nhexane at −35 °C after 2 days (Fig. 3A). Single-crystal X-ray diffraction analysis confirmed the solid-state structure of [K(crypt-222)][U(dbCOT)2], 2, which differs dramatically in the geometry of the primary coordination sphere (Fig. 3 and S2–S6†). In fact, the removal of the capping potassium ion yields a pseudo D2d symmetric [U(dbCOT)2]− fragment, where both the central CCOT atoms and annulated benzo rings are markedly staggered (107°) (Fig. 3B). Surprisingly, the U–CCOT distances decrease slightly (2.690(3)–2.803(3) Å), although comparable to 1, despite the significant decrease in Cnt–U–Cnt angle (167°) and pronounced twisting within the dbCOT2− ligands (Fig. S4†). The large change in bite angle strongly contrasts to that monitored for the analogous RE metal complexes bearing encapsulated potassium ions, [K(crypt-222)][RE(dbCOT)2],23,24 which exhibit perfectly linear Cnt–RE–Cnt angles (180°). However, unprecedented bending of the Cnt–U–Cnt angle in a UIV COT metallocene was first reported for the super bulky U (COTBIG)2 complex (where COTBIG = C8H6(SiPh3)2-1,4), which manifested in an angle of 168.7°.
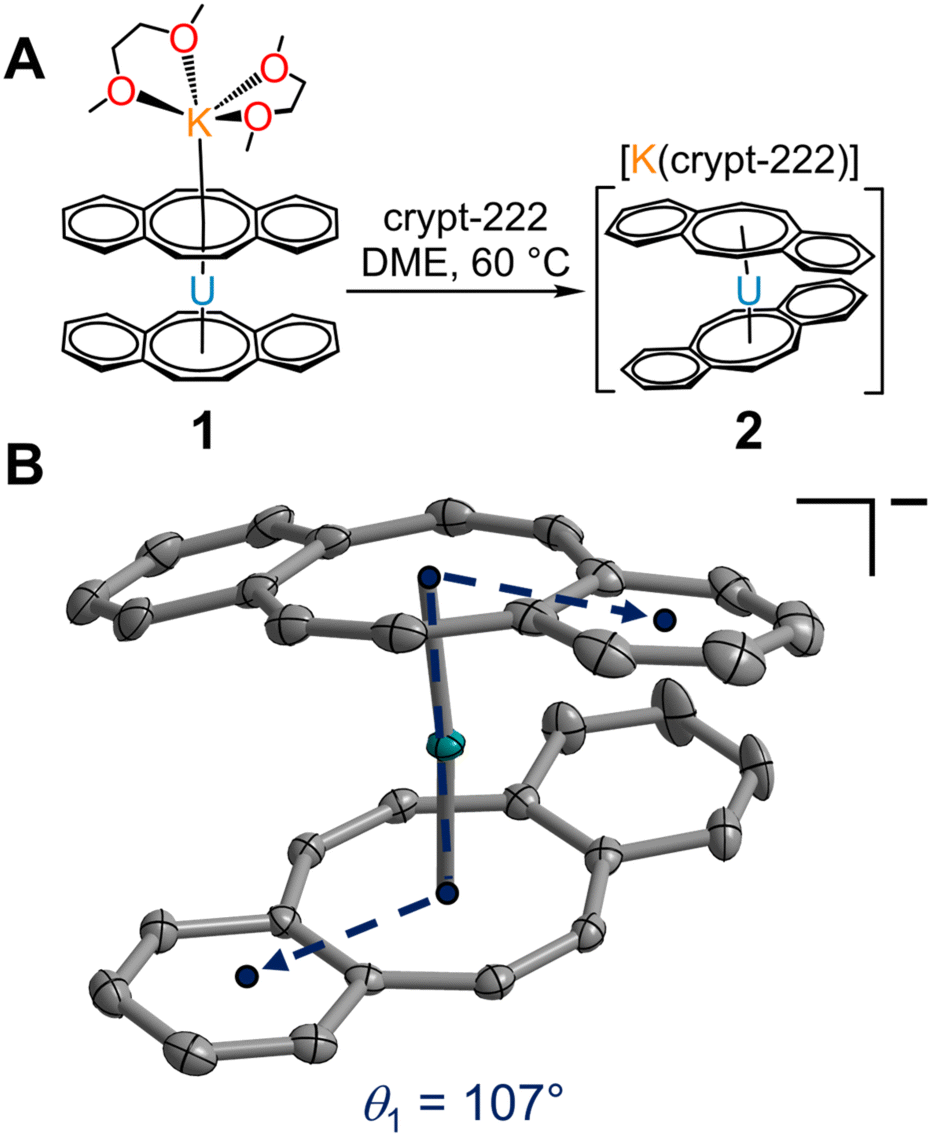 | ||
| Fig. 3 (A) Synthetic route to [K(crypt-222)][U(dbCOT)2], 2. (B) Structure of the [U(dbCOT)2]− anion in a crystal of [K(crypt-222)][U(dbCOT)2], 2, with thermal ellipsoids drawn at the 50% probability level. Teal and grey ellipsoids represent U and C atoms, respectively. H atoms and the [K(crypt-222)]+ countercation are omitted for clarity. Select interatomic distances (Å) and angles (°) for 2: U–CCOT = 2.690(3)–2.803(3); U–Cnt = 2.000, 2.003; Cnt–U–Cnt: 167.2. We define that the circles in each dbCOT drawing represent in total 18 π-electrons. | ||
To probe the accessibility of a more sterically encumbered UIII centre, the reaction of 1 with 2.2.2-cryptand was carried out in THF, yielding the THF-solvated uranium complex, [K(crypt-222)][U(dbCOT)2(THF)], 3 (Fig. 4 and S7–S9†). Brown, needle-like crystals, suitable for single-crystal X-ray diffraction analysis were obtained from a concentrated THF solution at −35 °C after 1 day, which confirmed the presence of the sterically congested trivalent uranocene derivative. The 9-coordinate UIII ion exhibits a U–OTHF distance of 2.618(2) Å, exceeding that of organouranium complexes featuring neutral O-donor ligands.44 Liberation of 1 from the coordinating K ion may invoke the necessary structural flexibility to accommodate coordination of THF. The elongated U–O distance in 3 is attributed to the presence of a bulky bis-COT framework, which accommodates the coordinating THF through a pronounced rearrangement of dbCOT2− ligands within 3 (Fig. 4C). In fact, THF coordination engenders an evident increase of the U–Cnt distances to 2.106 and 2.128 Å, corresponding to an increase of 0.102 and 0.073 Å (1) and 0.106 and 0.125 Å (2). Expectedly, the U–CCOT distances are also elongated, exceeding 2.9 Å (2.692(3)–2.939(3) Å). In contrast to 1, both dbCOT2− ligands in 2 and 3 deviate from planarity and exhibit small Cnt–U–Cnt angles. The Cnt–U–Cnt angle of 150.4° in 3 is the smallest bite angle monitored for any homoleptic uranocene derivative, while 2 retains the smallest angle among homoleptic uranocene complexes bearing no equatorial ligands.
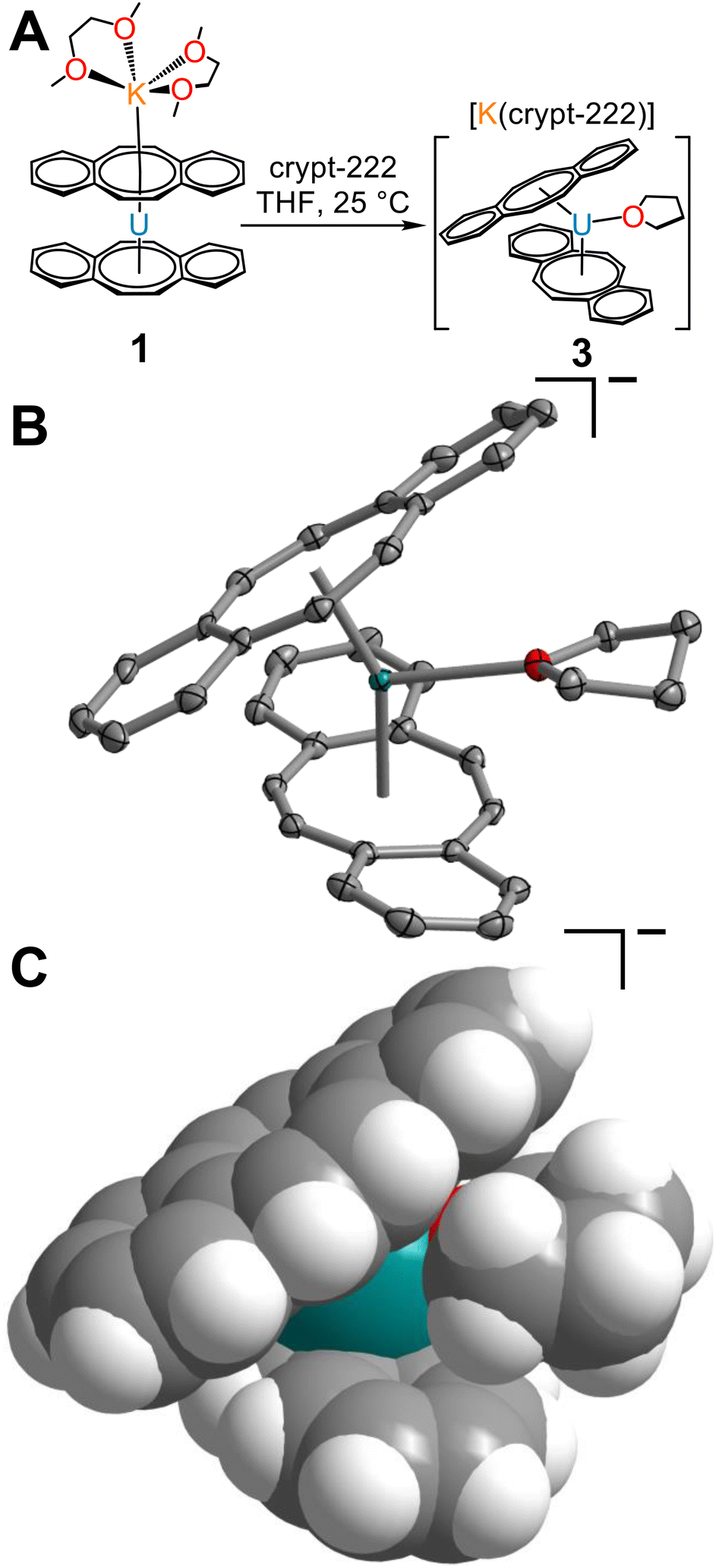 | ||
| Fig. 4 (A) Synthetic route to [K(crypt-222)][U(dbCOT)2(THF)], 3. (B) Structure of the [U(dbCOT)2(THF)]− anion in a crystal of [K(crypt-222)][U(dbCOT)2(THF)], 3, with thermal ellipsoids drawn at the 50% probability level. Teal, red, and grey ellipsoids represent U, O, and C atoms, respectively. H atoms and the [K(crypt-222)]+ countercation are omitted for clarity. (C) Space filling model of the [U(dbCOT)2(THF)]− anion in a crystal of [K(crypt-222)][U(dbCOT)2(THF)], 3. Teal, red, grey, and white-grey spheres represent U, O, C, and H atoms, respectively. The [K(crypt-222)]+ countercation is omitted for clarity. Select interatomic distances (Å) and angles (°) for 3: U–CCOT = 2.692(3)–2.939(3); U–Cnt = 2.106, 2.128; Cnt–U–Cnt: 150.4. We define that the circles in each dbCOT drawing represent in total 18 π-electrons. | ||
The stoichiometric reaction of UI3(THF)4 and K2dbCOT was additionally pursued to evaluate the accessibility of a new sterically congested THF-solvated complex, [K(DME)2][U(dbCOT)2(THF)]. However, despite the presence of coordinating THF in the trivalent uranium source, only the unsolvated complex, 1, was crystallized in 59% yield. This suggests that the capping [K(DME)2]+ motif prevents the necessary structural reorganization of the [U(dbCOT)2]− geometry to enable THF coordination, whereas the generation of a charge-separated [K(crypt-222)]+ countercation allows sufficient flexibility in the uranocene coordination sphere. Intriguingly, 2 and 3 constitute the first examples of charge-separated potassium salts of reduced uranocene derivatives. Only the lithium salt, [Li(DME)3][U(COT′′)2]13 (where COT′′ = C8H6(SiMe3)2-1,4), has been isolated as charge-separated ion pairs, suggesting that the ability to generate a given structural motif could only be accomplished by selecting the appropriate alkali metal reductant. However, as portrayed herein, this tunability is also feasible with potassium using the encapsulating chelate 2.2.2-cryptand.
Spectroscopic characterisation
The solution state structures of 1–3 were probed via 1- and 2D NMR spectroscopy, as well as UV-Vis-NIR spectroscopy. The 1H NMR spectrum of 1 in THF-d8 showcases three well-integrating broad singlets ranging between 7.14 and −35.99 ppm, consistent with the presence of a paramagnetic uranium(III) ion (Fig. S10–S17†). The room temperature 1H resonance monitored at −35.99 ppm is most significantly broadened, with a half width at half maximum (HWHM) of 115.9 Hz, indicating greater proximity of the 1H nuclei to the UIII ion relative to the other protons within the [U(dbCOT)2]− scaffold. Thus, this resonance can be readily ascribed to the protons of the central COT rings, whereas the broad singlets between 4 and 7 ppm originate from the ancillary benzo moieties of the dbCOT ligands. Specifically, the protons of the α- and β-carbons to the central COT ring should experience different paramagnetic contributions, which is supported by the difference in the observed linewidth of 37.5 and 22.6 Hz, respectively. Consequently, the narrower resonance at 4.53 ppm can be attributed to the peripheral β-protons and the shift at 7.14 ppm is subsequently ascribed to the protons at the α-carbon position directly bound to the COT ring. Assignment of the 1H resonances, coupled with the 2D 1H–13C spectra greatly facilitates the assignment of the 13C NMR spectrum, except for the COT ring carbons. The rapid relaxation induced by the U(III) centre, as well as the direct one-bond connectivity coupled with the reduced abundance and lower sensitivity of the 13C nucleus hampered unambiguous determination of the 13C resonances.45 However, the relative intensities, linewidths, and 2D data provide meaningful insight, where the resonance at 165.24 ppm (HWHM = 122.9 Hz) is tentatively assigned to the protonated C atoms of the COT ring, and the narrow resonance at 129.54 ppm (HWHM = 62.4 Hz) stems from the ipso C atoms of the dbCOT framework.Similarly, the 1H NMR spectra of 2 and 3 in THF-d8 exhibit equally paramagnetically shifted 1H resonances between 7.03 and −35.61 ppm, and 7.02 and −35.60 ppm, respectively. Despite the descent in molecular symmetry traversing from 1 to 3, the number of 1H or 13C resonances remains unchanged, suggesting a dynamic solution state structure without any barrier to rotation at room temperature. The NMR spectra of 1 also confirm the presence of only one coordinating DME molecule, which agrees well with the elemental analysis values obtained, suggesting that one DME molecule is labile and removed upon prolonged exposure to reduced pressure.11 Importantly, the NMR spectra of 1 and 2 (Fig. S18–S25†) show no indication of the coordination of THF, given the presence of narrow THF linewidths. By contrast, 3 clearly exhibits well-defined resonances for the coordinated THF at 3.62 and 1.78 ppm (Fig. S26–S33†).
Excitingly, the distribution of spin density, originating from a paramagnetic metal such as the 5f3 U(III) ion, onto the ligand framework can be readily evaluated through paramagnetic NMR (pNMR) experiments.45 In pNMR, the observed chemical shift, δobs, is comprised of diamagnetic and hyperfine contributions, δHF, where the hyperfine shift is further decomposed into sum components of the Fermi contact (through-bond) shift and pseudocontact (through-space) shift. The diamagnetic component can be readily approximated from Curie plots (by fitting δ (ppm) versus T−1) in the absence of an isostructural, diamagnetic complex.46 Since δobs is sensitive to the local electron density of a given nucleus, the hyperfine contribution can be utilized to evaluate the spin density distribution of a given paramagnetic complex. Thus, to corroborate the proton assignments of 1–3, and gain insight into the spin delocalisation within the [U(dbCOT)2]− manifold, variable-temperature 1H NMR data were collected between 50 °C and −40 °C in THF–d8 (Fig. 5 and S34–S39†). Notably, the resonances caused by the C8H4 ring of 1–3, are linearly dependent on temperature, exhibiting Curie–Weiss behaviour within the entire experimental temperature range, typical of uranocene complexes.18,19,47 The proximity of these protons to the UIII ion enables a spin delocalisation from the 5f3 manifold, leading to a sizable change in the chemical shift across the 90 °C temperature range of 12.15, 11.87, and 11.75 ppm for 1–3, respectively. By contrast, the α- and β-protons of the [8]-annulene shift marginally with temperature, albeit still exhibiting a strong temperature dependence, with an average range of 0.55 and 1.55 ppm, respectively. Although the peripheral α-protons are positioned closer to the U centre relative to the neighbouring β-protons, the α-protons exhibit a weaker Fermi contact shift contribution consistent with that monitored for dibenzouranocene (where dibenzouranocene = U(C8H6(C4H4))2).18 This weaker hyperfine contribution to the ancillary proton chemical shift supports the abovementioned proton assignment, such that the COT-ring protons remain closest to the U centre and feature a stronger paramagnetic shift. Expectedly, the observed 1H resonances of the C8H4 rings are predominantly affected by the hyperfine shift owing to the relatively short U–H distances. By contrast, the protons of the benzo moieties exhibit a significantly greater U–H distance and demonstrate markedly different temperature dependence. In sum, this suggests that the ancillary protons may be more influenced by the pseudocontact (dipolar) contribution.45 This is evident in the case of 1, which shows an estimated diamagnetic contribution of −1.90 ppm for the C8H4 protons, yet the observed chemical shift at 293.15 K is −35.99, yielding a paramagnetic contribution of −34.09 ppm. The linear fit expressions (Fig. 5) reveal a more distinct hyperfine shift relative to the diamagnetic contribution for 1–3. Specifically, the hyperfine shift accounts for 95%, 94%, and 93% of the observed chemical shifts of the central COT rings in 1, 2, and 3, respectively. Note: the role of the pseudocontact and Fermi contact cannot be parsed from these approximations alone. Interestingly, the low temperature 1H data of 2 and 3 between −40 and −20 °C show subtle deviations from a linear temperature dependence, which may be an effect of an emerging energy barrier to spin rotation owing to the small Cnt–U–Cnt angles monitored in the solid state. Rotational barriers in uranocenes have been reported, albeit for the bulky tetratertbutyluranocene complex, U(C8H6((CCH3)2)-1,4)2, and at temperatures below −60 °C.47
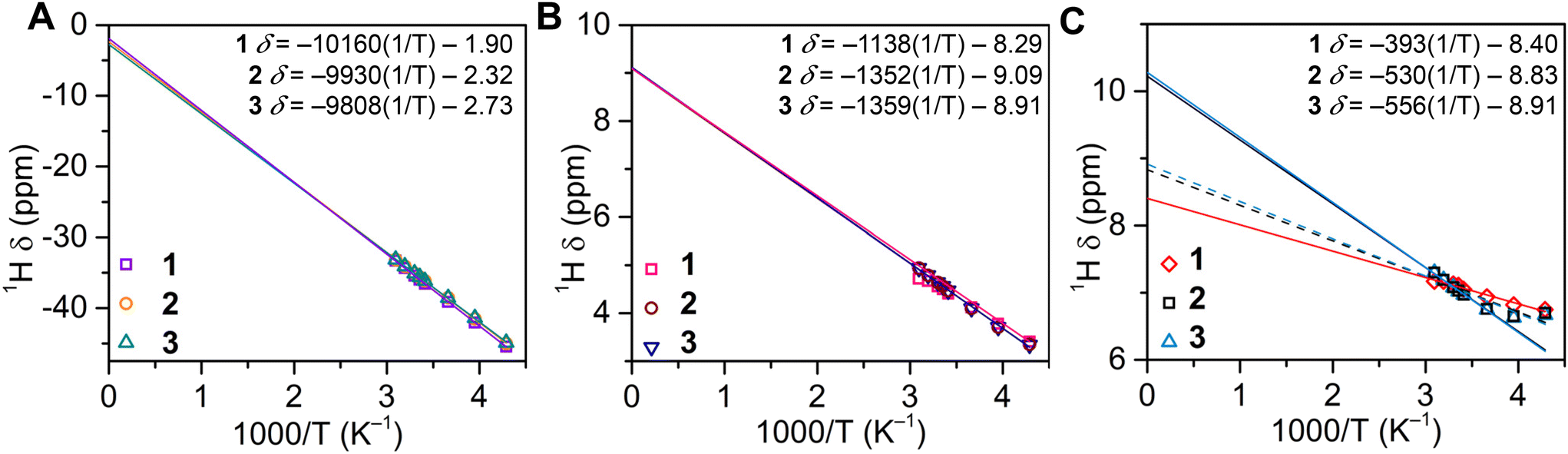 | ||
| Fig. 5 (A) Temperature dependence of the 1H chemical shifts for the central COT ring protons in [K(DME)2][U(dbCOT)2], 1 (purple squares), [K(crypt-222)][U(dbCOT)2], 2 (orange circles), and [K(crypt-222)][U(dbCOT)2(THF)], 3 (teal triangles). Linear fits to experimental data between 50 °C (323.15 K) and −40 °C (233.15 K) for 1–3 are represented as purple, orange, and teal lines, respectively. (B) Temperature dependence of the 1H chemical shifts for the β-protons on the annulated benzo-rings in [K(DME)2][U(dbCOT)2], 1 (pink squares), [K(crypt-222)][U(dbCOT)2], 2 (maroon circles), and [K(crypt-222)][U(dbCOT)2(THF)], 3 (dark blue triangles). Linear fits to experimental data between 50 °C (323.15 K) and −40 °C (233.15 K) for 1–3 are showcased as pink, maroon, and dark blue lines, respectively. (C) Temperature dependence of the 1H chemical shifts for the α-protons on the annulated benzo-rings in [K(DME)2][U(dbCOT)2], 1 (red diamonds), [K(crypt-222)][U(dbCOT)2], 2 (black squares), and [K(crypt-222)][U(dbCOT)2(THF)], and 3 (light blue triangles). Linear fits to experimental data between 50 °C (323.15 K) and −40 °C (233.15 K) for 1 and between 50 °C (323.15 K) and 0 °C (273.15 K) for 2 and 3 are represented as red, black, and light blue solid lines, respectively. The dashed black and blue lines constitute linear fits to the experimental data between 50 °C (323.15 K) and −40 °C (233.15 K) for 2 and 3, respectively. A linear fit of the data affords an estimate of the diamagnetic contribution to δobs at x = 0 K. For 2 and 3, the diamagnetic contribution was calculated employing the fit between 50 °C and 0 °C. | ||
The electronic absorption spectra of 1–3 were monitored between 230 and 1600 nm (4.35 × 104 and 0.625 × 104 cm−1) and are qualitatively similar and are dominated by ligand-based π–π* transitions below 400 nm (>25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1). Notably, the absent transitions in the visible region reinforce the trivalent character of 1–3.19 In fact, the optical spectra of 1–3 are markedly distinct from that of U(COT)2 (Fig. 6A). Moreover, 2-MeTHF solutions of 1–3 undergo rapid changes in the respective absorption spectra, more so for 1 and 3 (Fig. S41, S42, S47 and S48†). This may be tentatively attributed to the destabilization or displacement of coordinating solvent molecules (DME, 1; THF, 3), which is supported by the relatively unchanged UV-Vis spectra of 2 over the course of six scans (Fig. S44 and S45†), which bears no coordinating solvent molecules in the inner or outer coordination spheres. At lower energies, the observation of weak and broad f–f transitions in the NIR regime supports the identity of the UIII oxidation state48–52 (Fig. 6B and S50–S53†). While the NIR regime of 1–3 remains qualitatively similar, 3 features less intense transitions, possibly owing to the presence of a coordinating THF molecule, giving rise to a potential perturbation of the 5f-manifold. The solid-state structures of 1–3 were also examined via IR spectroscopy. Notably, the measured vibrational absorption spectra are dominated by C–C and C–H stretching and bending modes in the fingerprint region (Fig. S54–S56†). While the IR spectra of 2 and 3 are nearly identical, the vibrational spectrum of 1 contrasts strongly between 1400 and 2000 cm−1, which can be ascribed to the coordination of [K(DME)2]+ with the central COT ring in 1.
000 cm−1). Notably, the absent transitions in the visible region reinforce the trivalent character of 1–3.19 In fact, the optical spectra of 1–3 are markedly distinct from that of U(COT)2 (Fig. 6A). Moreover, 2-MeTHF solutions of 1–3 undergo rapid changes in the respective absorption spectra, more so for 1 and 3 (Fig. S41, S42, S47 and S48†). This may be tentatively attributed to the destabilization or displacement of coordinating solvent molecules (DME, 1; THF, 3), which is supported by the relatively unchanged UV-Vis spectra of 2 over the course of six scans (Fig. S44 and S45†), which bears no coordinating solvent molecules in the inner or outer coordination spheres. At lower energies, the observation of weak and broad f–f transitions in the NIR regime supports the identity of the UIII oxidation state48–52 (Fig. 6B and S50–S53†). While the NIR regime of 1–3 remains qualitatively similar, 3 features less intense transitions, possibly owing to the presence of a coordinating THF molecule, giving rise to a potential perturbation of the 5f-manifold. The solid-state structures of 1–3 were also examined via IR spectroscopy. Notably, the measured vibrational absorption spectra are dominated by C–C and C–H stretching and bending modes in the fingerprint region (Fig. S54–S56†). While the IR spectra of 2 and 3 are nearly identical, the vibrational spectrum of 1 contrasts strongly between 1400 and 2000 cm−1, which can be ascribed to the coordination of [K(DME)2]+ with the central COT ring in 1.
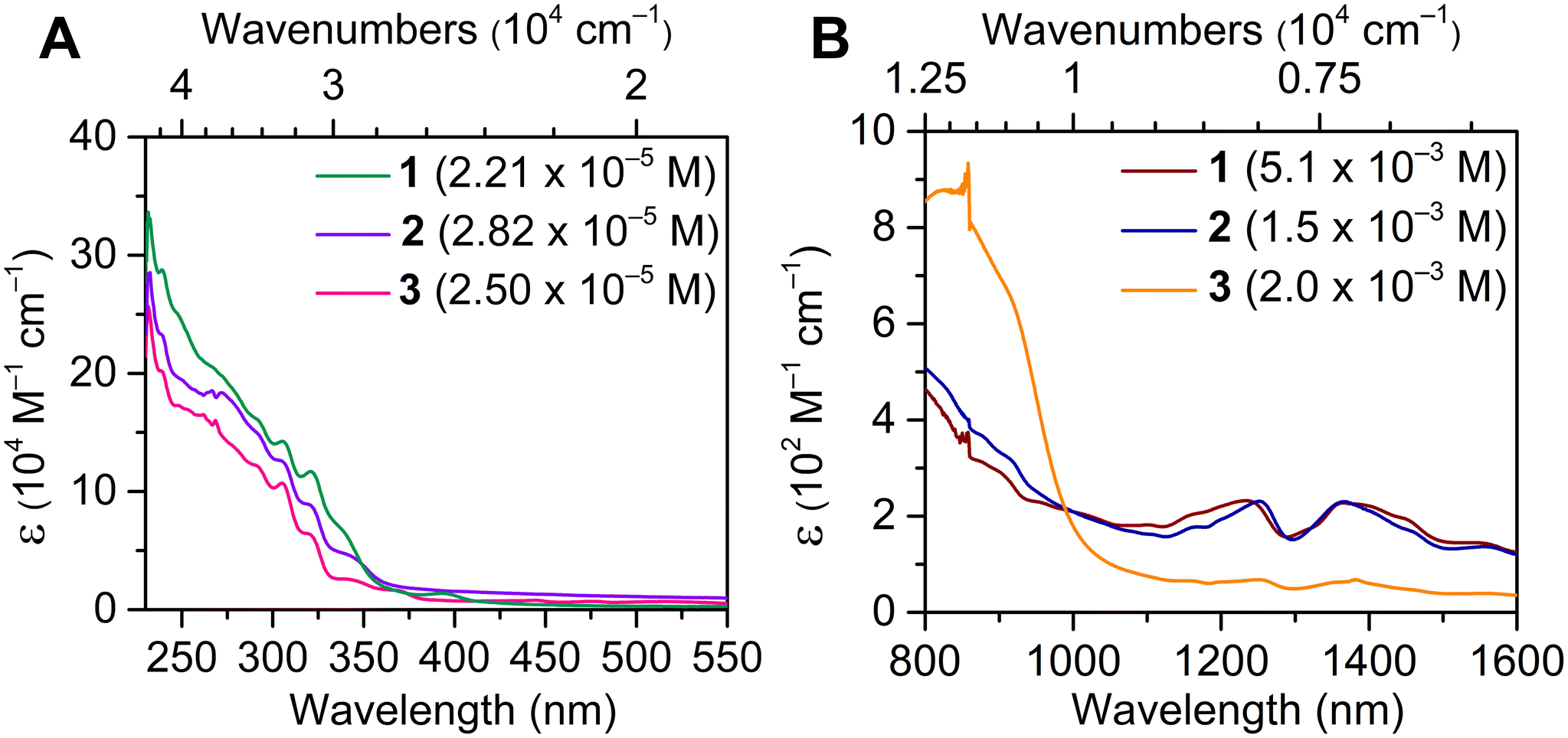 | ||
| Fig. 6 (A) UV-Vis spectra of [K(DME)2][U(dbCOT)2], 1 (green line), [K(crypt-222)][U(dbCOT)2], 2 (purple line), and [K(crypt-222)][U(dbCOT)2(THF)], 3 (pink line), in 2-MeTHF solution. (B) NIR spectra of [K(DME)2][U(dbCOT)2], 1 (maroon line), [K(crypt-222)][U(dbCOT)2], 2 (blue line), and [K(crypt-222)][U(dbCOT)2(THF)], 3 (orange line). | ||
Computational characterisation
To gain further insight into the electronic structures of 1–3, density functional theory (DFT) calculations were undertaken on the optimised crystal coordinates considering quartet ground state configurations (see the Experimental methods for details). Examination of the relative orbital energies uncovered that among 1–3, 1 possesses greatly stabilized frontier orbitals, which may be attributed to the coordinating potassium ion, as the generation of an inverse sandwich motif may lead to better stabilization of the electron-rich dbCOT2− ligand. For 2, two singly occupied molecular orbitals (SOMOs) are nearly degenerate, of which one experiences a destabilization upon the coordination of THF in 3 (Fig. 7). An NBO analysis of 1–3 revealed a 5fIII electronic ground state for each complex, with almost exclusive 5f-character for the alpha-singly-occupied molecular orbitals (αSOMOs) (Fig. 7, S57, and Table S9†). Here, the contribution of the U-character is more distinct in 1 and 2, however, 1–3 still show more pronounced 5f-orbital character within the frontier molecular orbitals (MOs) in comparison to their rare earth [RE(dbCOT)2]− counterparts, which contribute negligible metal–orbital character (Tables S6–S8†).23 Specifically, in 1 and 2, vacant metal orbitals constitute 15% of the highest occupied molecular orbital, where the uranium contribution in 3 is 13%. Similarly, for 3, merely 11% of the lowest unoccupied molecular orbitals is of U character, yet 19% U–character is calculated for 1 and 2 (Tables S6–S8†). Early 5f ions such as U can exhibit greater metal–ligand covalency than the later 5f ions and all 4f ions, owing to the more diffuse 5f- and 6d-valence orbitals. A definitive assessment of the extent of 5f-orbital participation in bonding may be possible through comprehensive spectroscopic methods such as EPR,53 NMR,54,55 or X-ray absorption spectroscopy in the future. Importantly, the qualitative bonding picture revealed herein confirms the predominant ionic bonding interaction, which is supplemented with nonnegligible 5f contributions. A second order perturbation theory analysis of 1–3 identified weak, repulsive interactions on the order of 4 kcal mol−1 between dbCOT ligands as one reason for the long U–CCOT distances. For comparison, the interaction between accepter vacant U orbitals and the donor OTHF lone pair is 48 kcal mol−1. The O–C bonding orbitals within the THF molecule and unoccupied metal orbitals also result in an additional donor–accepter interaction of 17 kcal mol−1, however, no molecular orbital comprising significant U–O character (>5%) was realized computationally. Collectively, the comparable involvement of 5f-orbitals in the frontier MOs of 1 and 2 alludes to the potential accessibility of a reduced, low-valent and highly reactive UII species upon chemical reduction.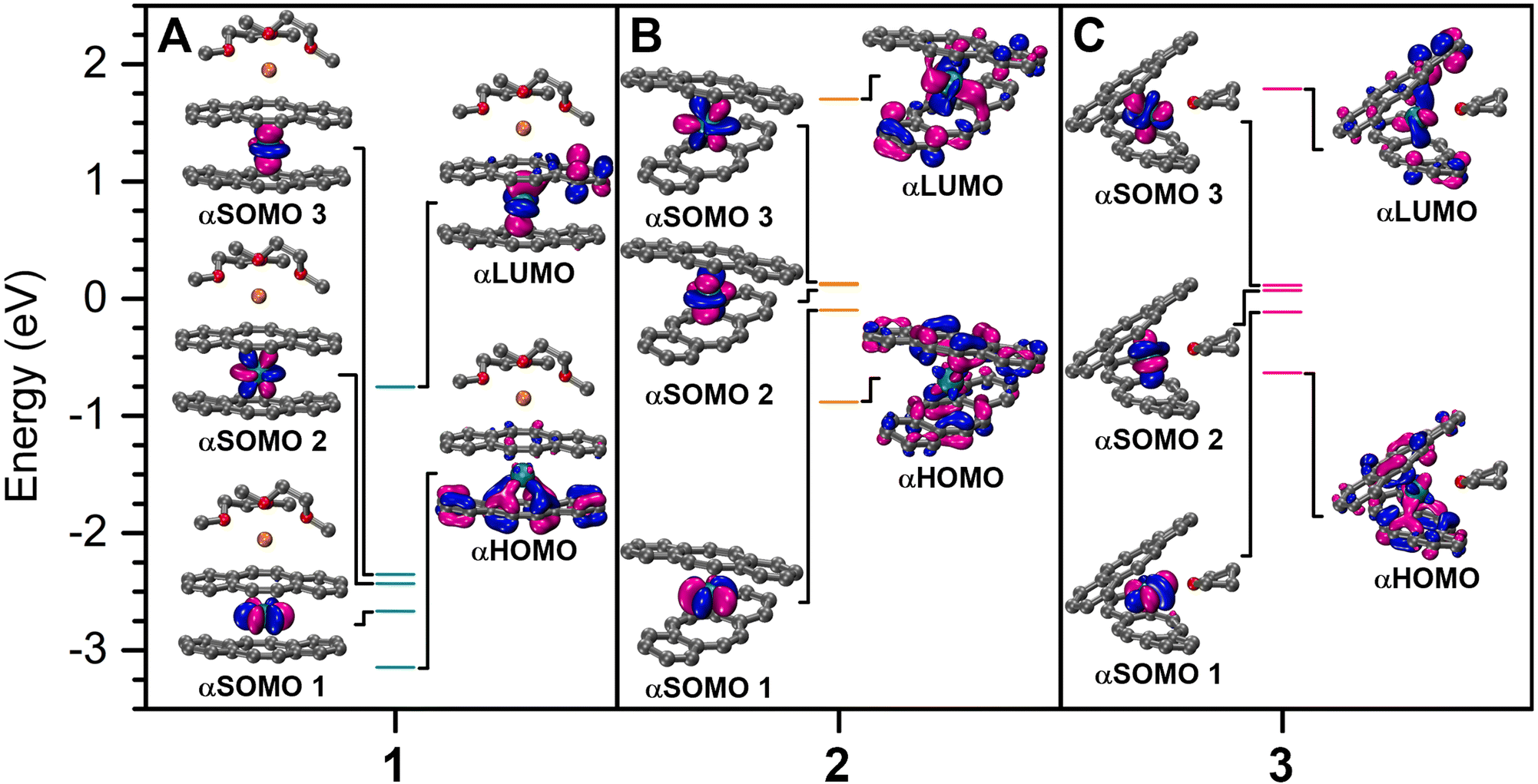 | ||
| Fig. 7 DFT-calculated frontier α-molecular orbitals of the optimized structures of (A) [K(DME)2][U(dbCOT)2], 1, as well as the [U(dbCOT)2]− and [U(dbCOT)2(THF)]− anions in (B) [K(crypt-222)][U(dbCOT)2], 2, and (C) [K(crypt-222)][U(dbCOT)2(THF)], 3. Teal, orange, red, and grey spheres represent U, K, O, and C atoms, respectively. H atoms and the [K(crypt-222)]+ countercation (in 2 and 3) have been omitted for clarity. Energy levels are shown to scale. | ||
Conclusions
A series of new trivalent uranocene derivatives, [K(DME)2][U(dbCOT)2], 1, [K(crypt-222)][U(dbCOT)2], 2, and [K(crypt-222)][U(dbCOT)2(THF)], 3, has been isolated and unambiguously confirmed. The expanded π-surface of the dbCOT ligands gives rise to electron-rich uranocene complexes each bearing 36 π-electrons, and simultaneously constitute the largest examples of uranocene derivatives. The annulated π-surfaces may enable non-covalent π–π interactions, useful for the development of supramolecular assemblies containing redox-active U ions. Upon chelation of the capping K+ ion, 1 undergoes significant structural reorganization, yielding the first examples of charge-separated UIII bis-COT potassium salt complexes as 2 and 3. Simultaneously, the solid-state structure of 3 revealed the smallest Cnt–U–Cnt angle across all crystallographically known uranocene type frameworks. The geometric differences manifest into subtle electronic structure differences, as evidenced through spectroscopic and computational methods. Specifically, variable-temperature 1H NMR experiments allude to the onset of a rotational barrier in 2 and 3, as well as pronounced paramagnetic contributions to the observed chemical shifts for 1–3. Excitingly, 1 and 2 hold promise as candidates for the future isolation of rare, low-valent UII species upon a one-electron chemical reduction, which would mark the first example of a divalent uranium complex in a COT-based scaffold.Author contributions
E. C. prepared and characterised all compounds. W. S. provided experimental assistance. S. D. assisted with data analysis, formulated, and directed the research, and wrote the manuscript with input from all authors.Data availability
CCDC 2371693–2371695 contains the supplementary crystallographic data for this paper. The other characterisation data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded in part through the National Science Foundation (NSF) Faculty Early Career Development Program (CAREER) CHE-2339595. S. D. is grateful to the Department of Chemistry at Michigan State University (MSU) for generous start-up funds. The authors are grateful to Dr Daniel Holmes (MSU) for assistance with NMR data collection and analysis and to the group of Prof. James K. McCusker (MSU) for providing access to the UV-Vis-NIR spectrometer. This work was supported in part through computational resources and services provided by the Institute for Cyber-Enabled Research, as well as the Max T. Rodgers NMR Facility at MSU. We thank Francis Delano IV and Saroshan Deshapriya for assistance with elemental analysis. Funding for the single-crystal X-ray diffractometer was provided through the MRI program of the National Science Foundation under Grant No. CHE-1919565.References
- A. Streitwieser, U. Muller-Westerhoff, G. Sonnichsen, F. Mares, D. G. Morrell, K. O. Hodgson and C. A. Harmon, Preparation and properties of uranocene, di-.pi.-cyclooctatetraeneuranium(IV), J. Am. Chem. Soc., 1973, 95, 8644–8649 CrossRef CAS.
- A. Streitwieser and U. Mueller-Westerhoff, Bis(cyclooctatetraenyl)uranium (uranocene). A new class of sandwich complexes that utilize atomic f orbitals, J. Am. Chem. Soc., 1968, 90, 7364–7364 CrossRef CAS.
- A. Zalkin and K. N. Raymond, Structure of di-.pi.-cyclooctatetraeneuranium (uranocene), J. Am. Chem. Soc., 1969, 91, 5667–5668 CrossRef CAS.
- A. Avdeef, K. N. Raymond, K. O. Hodgson and A. Zalkin, Two isostructural actinide .pi. complexes. Crystal and molecular structure of bis(cyclooctatetraenyl)uranium(IV), U(C8H8)2, and bis(cyclooctatetraenyl)thorium(IV), Th(C8H8)2, Inorg. Chem., 1972, 11, 1083–1088 CrossRef CAS.
- N. Edelstein, G. N. Lamar, F. Mares and A. Streitwieser, Proton magnetic resonance shifts in (bis(cyclooctatetraenyl)uranium(IV), Chem. Phys. Lett., 1971, 8, 399–402 CrossRef CAS.
- A. Streitwieser, D. Dempf, G. N. La Mar, D. G. Karraker and N. Edelstein, Bis(1,3,5,7-tetramethylcyclooctatetraene)uranium(IV) and bis(1,3,5,7-tetramethylcyclooctatetraene)-neptunium(IV). Proton magnetic resonance spectrum and the question of f-orbital covalency, J. Am. Chem. Soc., 1971, 93, 7343–7344 CrossRef CAS.
- K. O. Hodgson and K. N. Raymond, Rotomeric configurations of a methyl-substituted cyclooctatetraene dianion complex of uranium(IV). Crystal and molecular structure of bis(1,3,5,7-tetramethylcyclooctatetraenyl)uranium(IV), U(C8H4(CH3)4)2, Inorg. Chem., 1973, 12, 458–466 CrossRef CAS.
- T. R. Boussie, D. C. Eisenberg, J. Rigsbee, A. Streitwieser and A. Zalkin, Structures of organo-f-element compounds differing in the oxidation state of the central metal: crystal structures of bis([8]annulene) complexes of cerium(IV), ytterbium(III), and uranium(III), Organometallics, 1991, 10, 1922–1928 CrossRef CAS.
- L. K. Templeton, D. H. Templeton and R. Walker, Molecular structure and disorder in crystals of octaphenyluranocene, U[C8H4(C6H5)4]2, Inorg. Chem., 1976, 15, 3000–3003 CrossRef CAS.
- V. Lorenz, B. M. Schmiege, C. G. Hrib, J. W. Ziller, A. Edelmann, S. Blaurock, W. J. Evans and F. T. Edelmann, Unprecedented Bending and Rearrangement of f-Element Sandwich Complexes Induced by Superbulky Cyclooctatetraenide Ligands, J. Am. Chem. Soc., 2011, 133, 1257–1259 CrossRef CAS PubMed.
- J. S. Parry, F. G. N. Cloke, S. J. Coles and M. B. Hursthouse, Synthesis and Characterization of the First Sandwich Complex of Trivalent Thorium: A Structural Comparison with the Uranium Analogue, J. Am. Chem. Soc., 1999, 121, 6867–6871 CrossRef CAS.
- J. Rausch, C. Apostolidis, O. Walter, V. Lorenz, C. G. Hrib, L. Hilfert, M. Kühling, S. Busse and F. T. Edelmann, One ligand fits all: lanthanide and actinide sandwich complexes comprising the 1,4-bis(trimethylsilyl)cyclooctatetraenyl (=COT′′) ligand, New J. Chem., 2015, 39, 7656–7666 RSC.
- J. J. Le Roy, S. I. Gorelsky, I. Korobkov and M. Murugesu, Slow Magnetic Relaxation in Uranium(III) and Neodymium(III) Cyclooctatetraenyl Complexes, Organometallics, 2015, 34, 1415–1418 CrossRef CAS.
- N. Kaltsoyannis, Does Covalency Increase or Decrease across the Actinide Series? Implications for Minor Actinide Partitioning, Inorg. Chem., 2013, 52, 3407–3413 CrossRef CAS.
- P. J. Panak and A. Geist, Complexation and Extraction of Trivalent Actinides and Lanthanides by Triazinylpyridine N-Donor Ligands, Chem. Rev., 2013, 113, 1199–1236 CrossRef CAS PubMed.
- A. Zalkin, D. H. Templeton, S. R. Berryhill and W. D. Luke, Crystal and molecular structure of bis[.pi.-(cyclobutenocyclooctatetraene)]uranium(IV), U[C8H6(CH2)2]2, Inorg. Chem., 1979, 18, 2287–2289 CrossRef CAS.
- A. Zalkin, D. H. Templeton, W. D. Luke and A. Streitwieser, Synthesis and structure of dicyclopentenouranocene, U[C8H6(CH2)3]2, Organometallics, 1982, 1, 618–622 CrossRef CAS.
- A. Zalkin, D. H. Templeton, R. Kluttz and A. Streitwieser, Disordered structure of dibenzouranocene, [U(C12H10)2], Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 327–329 CrossRef.
- F. Billiau, G. Folcher, H. Marquet-Ellis, P. Rigny and E. Saito, Reduction of bis(cyclooctatetraene)uranium, J. Am. Chem. Soc., 1981, 103, 5603–5604 CrossRef CAS.
- J. N. Cross, E. M. Villa, V. R. Darling, M. J. Polinski, J. Lin, X. Tan, N. Kikugawa, M. Shatruk, R. Baumbach and T. E. Albrecht-Schmitt, Straightforward Reductive Routes to Air-Stable Uranium(III) and Neptunium(III) Materials, Inorg. Chem., 2014, 53, 7455–7466 CrossRef CAS PubMed.
- M. Solà, Aromaticity rules, Nat. Chem., 2022, 14, 585–590 CrossRef PubMed.
- E. R. Pugliese, F. Benner, E. Castellanos, F. Delano and S. Demir, Heteroleptic Rare-Earth Tris(metallocenes) Containing a Dibenzocyclooctatetraene Dianion, Inorg. Chem., 2022, 61, 2444–2454 CrossRef CAS PubMed.
- E. Castellanos and S. Demir, Linear, Electron-Rich, Homoleptic Rare Earth Metallocene and Its Redox Activity, Inorg. Chem., 2023, 62, 2095–2104 CrossRef CAS PubMed.
- E. Castellanos, F. Benner and S. Demir, Linear, Electron-Rich Erbium Single-Molecule Magnet with Dibenzocyclooctatetraene Ligands, Inorg. Chem., 2024, 63, 9888–9898 CrossRef CAS PubMed.
- Y. Zhu, J. Mahoney, A. J. Babson, Z. Zhou, Z. Wei, M. Gakiya-Teruya, J. McNeely, A. Yu. Rogachev, M. Shatruk and M. A. Petrukhina, Homoleptic Rare-Earth-Metal Sandwiches with Dibenzo[a,e]cyclooctatetraene Dianions, Inorg. Chem., 2024, 63, 9579–9587 CrossRef CAS PubMed.
- M. Tasić, J. Ivković, G. Carlström, M. Melcher, P. Bollella, J. Bendix, L. Gorton, P. Persson, J. Uhlig and D. Strand, Electro-mechanically switchable hydrocarbons based on [8]annulenes, Nat. Commun., 2022, 13, 860 CrossRef PubMed.
- L. Barluzzi, S. P. Ogilvie, A. B. Dalton, P. Kaden, R. Gericke, A. Mansikkamäki, S. R. Giblin and R. A. Layfield, Triply Bonded Pancake π-Dimers Stabilized by Tetravalent Actinides, J. Am. Chem. Soc., 2024, 146, 4234–4241 CrossRef CAS PubMed.
- M. J. Monreal, R. K. Thomson, T. Cantat, N. E. Travia, B. L. Scott and J. L. Kiplinger, UI4(1,4-dioxane)2, [UCl4(1,4-dioxane)]2, and UI3(1,4-dioxane)1.5: Stable and Versatile Starting Materials for Low- and High-Valent Uranium Chemistry, Organometallics, 2011, 30, 2031–2038 CrossRef CAS.
- G. Franck, M. Brill and G. Helmchen, Dibenzo[a,e]cyclooctene: Multi-gram Synthesis of a Bidentate Ligand, Org. Synth., 2012, 89, 55–65 CrossRef CAS.
- D. E. Bergbreiter and J. M. Killough, Reactions of potassium-graphite, J. Am. Chem. Soc., 1978, 100, 2126–2134 CrossRef CAS.
- G. M. Sheldrick, SHELXT – Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- F. Neese, Software update: The ORCA program system—Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104–154104 CrossRef PubMed.
- G. L. Stoychev, A. A. Auer and F. Neese, Automatic Generation of Auxiliary Basis Sets, J. Chem. Theory Comput., 2017, 13, 554–562 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3297 RSC.
- M. Dolg and X. Cao, Accurate Relativistic Small-Core Pseudopotentials for Actinides. Energy Adjustment for Uranium and First Applications to Uranium Hydride, J. Phys. Chem. A, 2009, 113, 12573–12581 CrossRef CAS PubMed.
- D. A. Pantazis and F. Neese, All-Electron Scalar Relativistic Basis Sets for the Actinides, J. Chem. Theory Comput., 2011, 7, 677–684 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, NBO 6.0: Natural bond orbital analysis program, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- N. Tsoureas, A. Mansikkamäki and R. A. Layfield, Synthesis, bonding properties and ether activation reactivity of cyclobutadienyl-ligated hybrid uranocenes, Chem. Sci., 2021, 12, 2948–2954 RSC.
- M. Hiller, M. Maier, H. Wadepohl and M. Enders, Paramagnetic NMR Analysis of Substituted Biscyclooctatetraene Lanthanide Complexes, Organometallics, 2016, 35, 1916–1922 CrossRef CAS.
- J. Novotný, D. Přichystal, M. Sojka, S. Komorovsky, M. Nečas and R. Marek, Hyperfine Effects in Ligand NMR: Paramagnetic Ru(III) Complexes with 3-Substituted Pyridines, Inorg. Chem., 2018, 57, 641–652 CrossRef PubMed.
- W. D. Luke and A. Streitwieser, Barriers to ring rotation in 1,1′,4,4′-tetra-tert-butyluranocene and 1,1′,3,3′-tetra-tert-butylferrocene, J. Am. Chem. Soc., 1981, 103, 3241–3243 CrossRef CAS.
- D. Cohen and W. T. Carnall, Absorption Spectra or Uranium(III) and Uranium(IV) in DClO4 Solution, J. Phys. Chem., 1960, 64, 1933–1936 CrossRef CAS.
- B. S. Billow, B. N. Livesay, C. C. Mokhtarzadeh, J. McCracken, M. P. Shores, J. M. Boncella and A. L. Odom, Synthesis and Characterization of a Neutral U(II) Arene Sandwich Complex, J. Am. Chem. Soc., 2018, 140, 17369–17373 CrossRef CAS.
- D. Perales, S. A. Ford, S. R. Salpage, T. S. Collins, M. Zeller, K. Hanson and S. C. Bart, Conversion of Trivalent Uranium Anilido to Tetravalent Uranium Imido Species via Oxidative Deprotonation, Inorg. Chem., 2020, 59, 11910–11914 CrossRef CAS.
- C. J. Tatebe, Z. Tong, J. J. Kiernicki, E. J. Coughlin, M. Zeller and S. C. Bart, Activation of Triphenylphosphine Oxide Mediated by Trivalent Organouranium Species, Organometallics, 2018, 37, 934–940 CrossRef CAS.
- B. Vlaisavljevich, P. L. Diaconescu, W. L. Lukens, L. Gagliardi and C. C. Cummins, Investigations of the Electronic Structure of Arene-Bridged Diuranium Complexes, Organometallics, 2013, 32, 1341–1352 CrossRef CAS.
- A. Formanuik, A.-M. Ariciu, F. Ortu, R. Beekmeyer, A. Kerridge, F. Tuna, E. J. L. McInnes and D. P. Mills, Actinide covalency measured by pulsed electron paramagnetic resonance spectroscopy, Nat. Chem., 2017, 9, 578–583 CrossRef CAS PubMed.
- G. T. Kent, X. Yu, G. Wu, J. Autschbach and T. W. Hayton, Synthesis and electronic structure analysis of the actinide allenylidenes, [{(NR2)3 }An(CCCPh2)]− (An = U, Th; R = SiMe3), Chem. Sci., 2021, 12, 14383–14388 RSC.
- O. Ordoñez, X. Yu, G. Wu, J. Autschbach and T. W. Hayton, Quantifying Actinide–Carbon Bond Covalency in a Uranyl–Aryl Complex Utilizing Solution 13C NMR Spectroscopy, Inorg. Chem., 2024, 63, 9427–9433 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Other characterisation data. CCDC 2371693–2371695. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi02102a |
| This journal is © the Partner Organisations 2025 |