
Ultrathin core–shell–satellite structured Au@PtPd@Pt nanowires for superior electrocatalytic hydrogen evolution†
Caikang
Wang‡
ac,
Xian
Jiang‡
 *a,
Qicheng
Liu
d,
Jiaqian
Ding
d,
Juan
Zhou
*c,
Yawen
Tang
d,
Gengtao
Fu
d and
Jong-Min
Lee
*b
*a,
Qicheng
Liu
d,
Jiaqian
Ding
d,
Juan
Zhou
*c,
Yawen
Tang
d,
Gengtao
Fu
d and
Jong-Min
Lee
*b
aSchool of New Energy, Nanjing University of Science and Technology, Jiangyin, 214443, China. E-mail: xianjiang@njust.edu.cn
bSchool of Chemical and Biomedical Engineering, Nanyang Technology University, Singapore 637459, Singapore. E-mail: jmlee@ntu.edu.sg
cSchool of Energy and Power Engineering, Nanjing University of Science and Technology, 200 Xiaolingwei Street, Jiangsu Province 210094, China. E-mail: jzhou@njust.edu.cn
dJiangsu Key Laboratory of New Power Batteries, Jiangsu Collaborative Innovation Centre of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing 210023, China
First published on 1st November 2023
Abstract
The rational structural and compositional design of Pt-based electrocatalysts is effective in synergizing the physicochemical and electrochemical properties of Pt for developing high-performance hydrogen evolution reaction (HER) electrocatalysts. In this study, we present the synthesis of one-dimensional (1D) Au@PtPd@Pt nanowires using a template-induced method. The nanowires are composed of ultrathin PtPd alloy layers coated on the surface of Au nanowires (NWs) and ultrafine island-like Pt nanoparticles grown on the surface of a PtPd alloy shell. The resulting Au@PtPd@Pt nanowires exhibit exceptional electrocatalytic activity and excellent long-term electrochemical stability toward the alkaline HER, which is attributed to the synergistic effect of the anisotropic ultrathin 1D nanowires with a prominent core–shell–satellite structure and the trimetallic elemental compositions. The nanowires demonstrate a low overpotential of 23 mV at 10 mA cm−2, outperforming commercial Pt/C, and bimetallic counterparts. Additionally, the integrated Au@PtPd@Pt//RuO2 electrolyzer achieves a lower operating potential of 1.52 V to achieve 10 mA cm−2, surpassing the performance of the commercial Pt/C//RuO2 configuration. This highlights its great potential for practical water electrolysis. This work showcases an effective design approach that comprehensively and rationally considers both the designed nanostructure and multiple compositions to achieve high-performance electrocatalysts for various electrochemical reactions.
Introduction
The hydrogen evolution reaction (HER) resulting from the electrochemical water splitting process, driven by green and renewable energy, offers a promising way to reduce carbon emissions and realize carbon neutralization due to its high utilization efficiency and energy density as well as zero carbon emissions.1–3 However, despite extensive efforts dedicated to the rational design and preparation of efficient HER catalysts, the bottleneck in hydrogen generation through water splitting is closely related to the limitations of the oxygen evolution reaction (OER). In particular, the poor stability of OER catalysts under acidic conditions necessitates the development of novel engineering strategies to create multifunctional electrode materials suitable for the HER in neutral or alkaline electrolytes.4–8 Although extensive research has been focused on Pt-free catalysts to reduce costs, while ensuring reasonable efficiency, a discernible performance disparity between Pt-free and Pt-based counterparts remains evident. Pt-based materials currently continue to demonstrate state-of-the-art catalytic activity for the HER due to the rapid recombination of hydrogen intermediates (Tafel step, 2H* → H2), but are largely restricted by the poor activation for the dissociation of water (Volmer step, 2H2O + 2e−→ H2 + 2HO−), and the prohibitive costs and scarce reserves. Meanwhile, the kinetic limitation drastically decreases the rate of the HER process by 2 to 3 orders of magnitude under neutral and alkaline conditions, when compared to acidic conditions.9,10The construction of Pt-based catalysts with a core–shell structure is a powerful and promising strategy to accelerate both H2O dissociation and H* recombination, leading to the development of highly effective Pt-based HER catalysts. Recent advancements have also unveiled the potential of core–shell nanostructures in facilitating the systematic design and synthesis of high-performance Pt-based catalysts with tailored morphologies and compositions.11–14 For instance, Zeng and co-workers have successfully synthesized ultrathin Pt nanoshells at the subnanoscale on finely AgPd alloy cores, which enhances both the activity and stability of the HER by dually optimizing the lattice strain and electron effects.14 However, the shell regions in the core–shell structure usually exhibit smooth and dense surfaces, which could pose challenges for guest species to access the inner cores, thereby diminishing the benefits of the core–shell structure.15,16 In contrast, the core–satellite nanostructure possesses abundant surface satellite particles, exhibits an open outer surface, and combines the advantages of a core–shell structure. The distinctive structure endows the catalysts with short proton/electron transport distances, low material density, and large volume gaps that are available for reactants.17–20 Ultrathin anisotropic nanowires (NWs) can serve as ideal templates for the construction of one-dimensional (1D) core–shell electrocatalysts. This is due to their high aspect ratio and superb resistance to dissolution and aggregation, which can suppress physical Ostwald ripening, increase atomic utilization, and facilitate mass and electron transfer during electrocatalysis.15,21–24 The ultrathin Au nanowires, known for their excellent chemical stability and corrosion resistance under electrochemical conditions, are especially beneficial for fabricating desired core–shell electrocatalysts and enhancing the structural stability of the target catalysts.25–29
Recognizing the significance of architectural superiority and compositional synergy, a facile template method is presented in this study to fabricate Au@PtPd@Pt NWs with a core–shell–satellite configuration. The nanowires comprise a core of Au NWs, with an ultrathin Pd-rich PtPd alloy layer coated on the surface through the layer growth mode, and ultrafine Pt nanoparticles as satellites formed on the surface of the PtPd layer via the island growth mode. The nanowires consist of Au NWs as the core, an ultrathin Pd-rich PtPd alloy layer coated on the surface of the Au core, and ultrafine island-like Pt satellite nanoparticles grown on the surface of the PtPd alloy shell. The ultrathin 1D nanostructure and the ultrafine Pt satellite nanoparticles surrounded by the ultrathin PtPd alloy layer facilitate highly exposed surface areas, maximizing the atomic utilization of Pt atoms and promoting efficient reactant transport. Additionally, the strong interactions between the surface PtPd shell and Pt satellite and the inner Au core modify the electronic structure of Pt, resulting in a downshift of the d-band center of Pt atoms. As a result, the adsorption energy of hydrogen on the surfaces of the PtPd shell and Pt satellites is weakened, enhancing the kinetics of the HER. The unique configuration of Au@PtPd@Pt nanowires provides superior HER activity and stability compared to that of the commercial Pt/C catalysts.
Results and discussion
The fabrication process of 1D ultrathin core–shell–satellite structured Au@PtPd@Pt NWs using a template-induced method under water-bath conditions is illustrated in Fig. 1a. Detailed descriptions can be found in the experiment section. Initially, Au NWs were synthesized by aging a mixture of HAuCl4 aqueous solution and 1-naphthol ethanol solution. 1-Naphthol acts as both a reductant and structure-directing agent due to its abundant hydroxyl groups and aromatic rings.30 Transmission electron microscopy (TEM) images clearly depict the uniform 1D ultrathin nanowire structure of the synthesized Au NWs, with no other shapes observed (Fig. S1, ESI†). The Au NWs have a length of several hundred nanometers, a mean diameter of 4.87 nm, and a lattice spacing of 0.235 nm, corresponding to the (111) lattice plane of the fcc Au (JCPDS no. 65-2870).31 Subsequently, the Au NWs serve as a hard template and core for the reaction with PdCl2 and H2PtCl6, which act as metal precursors for the shell, resulting in the fabrication of Au@PtPd@Pt NWs through reduction with ascorbic acid (AA) in the subsequent step. The metal-stable properties of Au NWs make them suitable templates for the in situ growth of PtPd alloy layers on their surface, while the ultrathin 1D nanostructure imparts the acquired Au@PtPd@Pt sample with the same structural and morphological characteristics as the Au NWs. Additionally, numerous ultrafine Pt nanoparticles, resembling satellites, are distinctly observed randomly distributed on the surface of the product (Fig. 1b and Fig. S2, ESI†). The core–shell–satellite configuration provides a considerably increased exposed active surface area, enhanced electrical conductivity, and improved structural stability for catalytic reactions.32 As observed in the high-resolution TEM (HRTEM) image, continuous lattice fringes extend from the inner Au core to the outer shell (Fig. 1c), indicating an in situ epitaxial deposition of Pt/Pd atoms with high crystallinity on the surface of Au NWs, leading to the formation of the core–shell–satellite structure. The Fourier-filtered lattice fringe images and the corresponding integrated pixel intensities transformed from the three representative areas are shown in Fig. 1d and e. The average lattice spacings of Au(111), PtPd(111), and Pt(111) are measured to be 0.235 nm (orange rectangular region), 0.230 nm (green rectangular region), and 0.230 nm (yellow rectangular region), respectively. These measurements indicate the alloy nature of the PtPd layer coating the surface of Au NWs and the presence of Pt nanoparticle satellites on the alloy surface. The energy dispersive X-ray (EDX) line-scanning profiles recorded across two nanowires provide strong evidence that the signal-intensity fitting curve of Au exhibits a “volcano” shape (Fig. 1f and g), whereas the curves of Pd and Pt show a distinct “valley” shape, indicating the presence of a PdPt alloy layer on the surface of Au NWs. Impressively, the Pt signals exhibit obviously higher intensities in the 0–5 nm area (blue shadow area in Fig. 1g), providing strong evidence for the “Au core–PtPd shell–Pt satellite” configuration of the Au@PtPd@Pt NWs, which is consistent with the EDX element mapping images (Fig. 1h–l). The presence of numerous Pt islands on the alloy surface is further elucidated through the enhanced EDX line-scan profile and corresponding element mapping images (Fig. S3, ESI†). As illustrated in Fig. S3a–e (ESI†), abundant Pt islands (marked by yellow dotted circles) flourish across the surface of the PtPd alloy layer, in harmony with the elemental mapping results. Significantly, the Pt signals distinctly intensify within the 0–5 nm, 12–16 nm, and 23–25 nm intervals (indicated by the blue shaded area in Fig. S3f, ESI†), providing compelling evidence of multiple Pt islands serving as satellites on the PtPd layer's surface, further affirming the “Au core–PtPd shell–Pt satellite” configuration of the Au@PtPd@Pt NWs. The exact atomic ratio of the metals in Au@PdPt@Pt NWs, as determined from the EDX spectrum, is 71.01![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15.86:13.12, closely resembling the feeding ratio of the metal precursors and ICP-OES(Fig. S4 and Table S1, ESI†). Notably, the presence of pure Pt nanoparticles on the surface indicates a Pd-rich composition in the PtPd shell, which is supported by the similar atomic ratios of Pt and Pd. This unique 1D wire-like core–shell–satellite structure likely originates from the differing growth modes of Pd and Pt atoms on Au NWs.28,33
:15.86:13.12, closely resembling the feeding ratio of the metal precursors and ICP-OES(Fig. S4 and Table S1, ESI†). Notably, the presence of pure Pt nanoparticles on the surface indicates a Pd-rich composition in the PtPd shell, which is supported by the similar atomic ratios of Pt and Pd. This unique 1D wire-like core–shell–satellite structure likely originates from the differing growth modes of Pd and Pt atoms on Au NWs.28,33
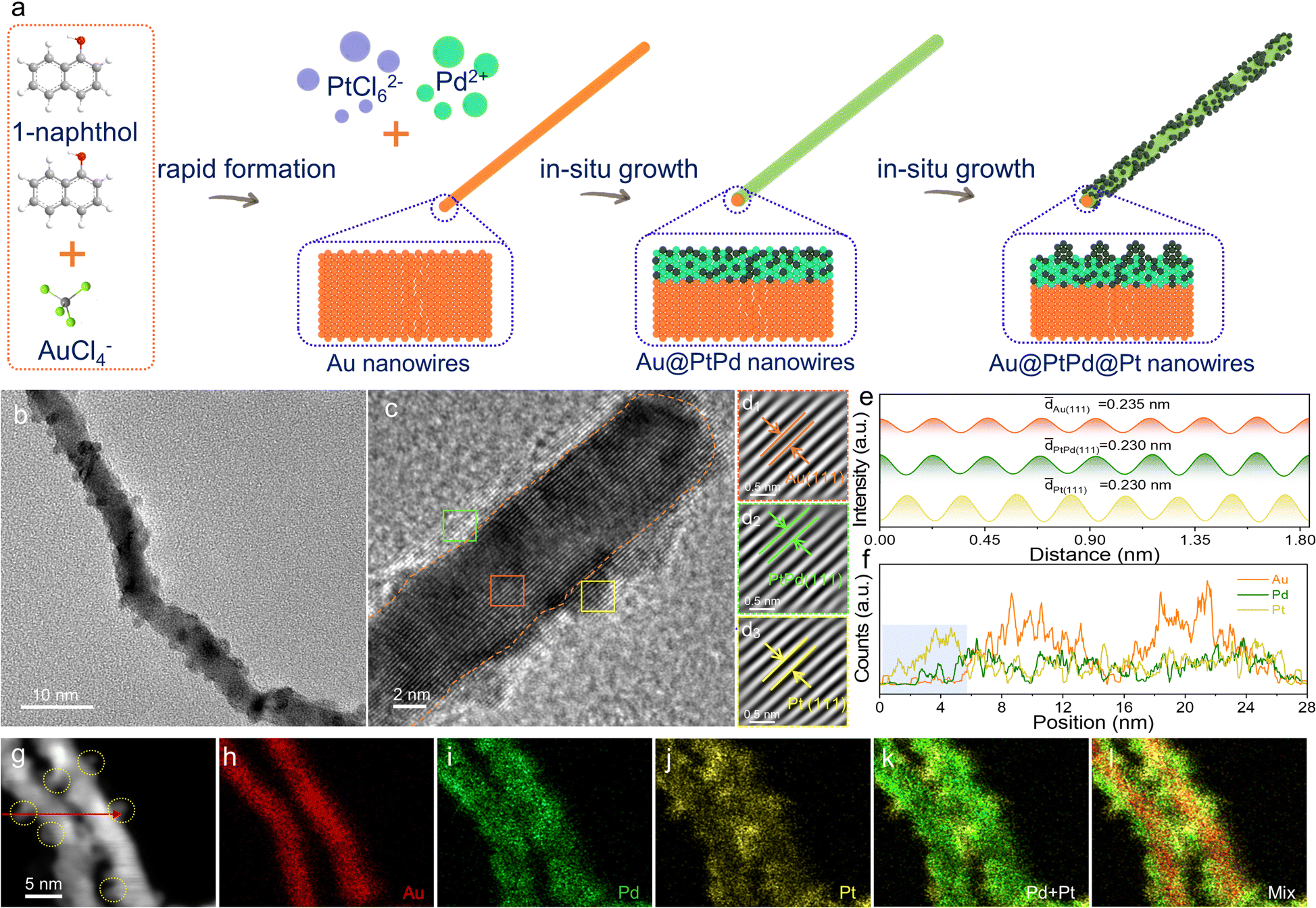 | ||
| Fig. 1 Preparation and structural characterization of the Au@PtPd@Pt NWs. (a) Scheme of the preparation of Au@PtPd@Pt NWs. (b) TEM and (c) HRTEM images. (d) Fourier-filtered lattice fringe images taken from the rectangular regions in c, and (e) corresponding integrated pixel intensities. (f) EDX line scan profile, and (g)–(l) STEM image and the corresponding EDX elemental mapping images. | ||
To investigate the formation process of Au@PtPd@Pt NWs, bimetallic Au@Pt and Au@Pd NWs were also synthesized using a similar method. Fig. 2a and Fig. S5 (ESI†) reveal the growth of Pt nanoparticles as islands on Au NWs, suggesting a potential core–satellite structure. EDX mapping images and line-scan profiles (Fig. 2b and Fig. S6, ESI†) confirm this structure, with Au predominantly located in the center and small Pt satellite particles randomly dispersed on the outer surface. Indeed, the previous report has demonstrated that the lattice mismatch of 4.08% between Pt and Au can induce a notable interface energy between the deposited Pt atoms and the surface of Au NWs, prompting island formation and contributing significantly to the emergence of core–satellite structures.34 This core–satellite nanostructure showcases an abundance of surface satellite particles and an exposed outer surface, synergizing the advantages inherent in a core–shell configuration, which endows the catalysts with ample proton/electron transfer pathways, reduced material density, and generous volume gaps available to reactants.17–20 Furthermore, the EDX spectrum (Fig. S7, ESI†) shows that the element composition of Au@Pt NWs is Au68.37Pt31.63, which closely matches the total feeding ratio of Au and Pt in Au@PtPd@Pt NWs. HRTEM images provide further insights into the core–satellite nanowire architecture (Fig. 2c). Fourier-filtered lattice fringe images and their corresponding integrated pixel intensities reveal lattice spacing distances of 0.235 nm and 0.230 nm, corresponding to the Au (111) and Pt (111) crystal planes, respectively. This provides additional evidence that the Pt nanoparticles grow as island-like structures on Au NWs (Fig. 2d and e). Importantly, the island growth mode of Pt is significantly distinct from the layer growth mode of Pd on Au NWs. TEM images (Fig. S8a and S9, ESI†) depict a rough shell surface and larger particle size (∼7.36 nm) compared to Au NWs. Observations from the EDX element mapping images and corresponding line-scan profiles (Fig. S8b and S10, ESI†) show that the Au element is primarily distributed in the inner core area, while the Pd element is dispersed around the Au core in the outer shell area. The molar ratio of Au/Pd is estimated to be approximately 70.79:29.21, as confirmed by the EDX spectrum (Fig. S11, ESI†), which aligns with the Au/Pt feeding ratio in Au@Pt NWs. HRTEM images demonstrate the consistent orientation of the lattice fringe across the core nanowire and layer, indicating the epitaxial growth of the Pd layer on Au NWs (Fig. S8c, ESI†). Fourier-filtered lattice fringe images in Fig. S8d (ESI†) and their corresponding integrated pixel intensities in Fig. S8e (ESI†) reveal lattice spacing distances of 0.235 nm and 0.230 nm, respectively, corresponding to the Au (111) and Pd (111) crystal planes. This further supports the notion of layer growth of Pd on Au NWs (Fig. S8d and e, ESI†). These investigations clearly demonstrate the distinct growth modes of Pd and Pt atoms on Au NWs.
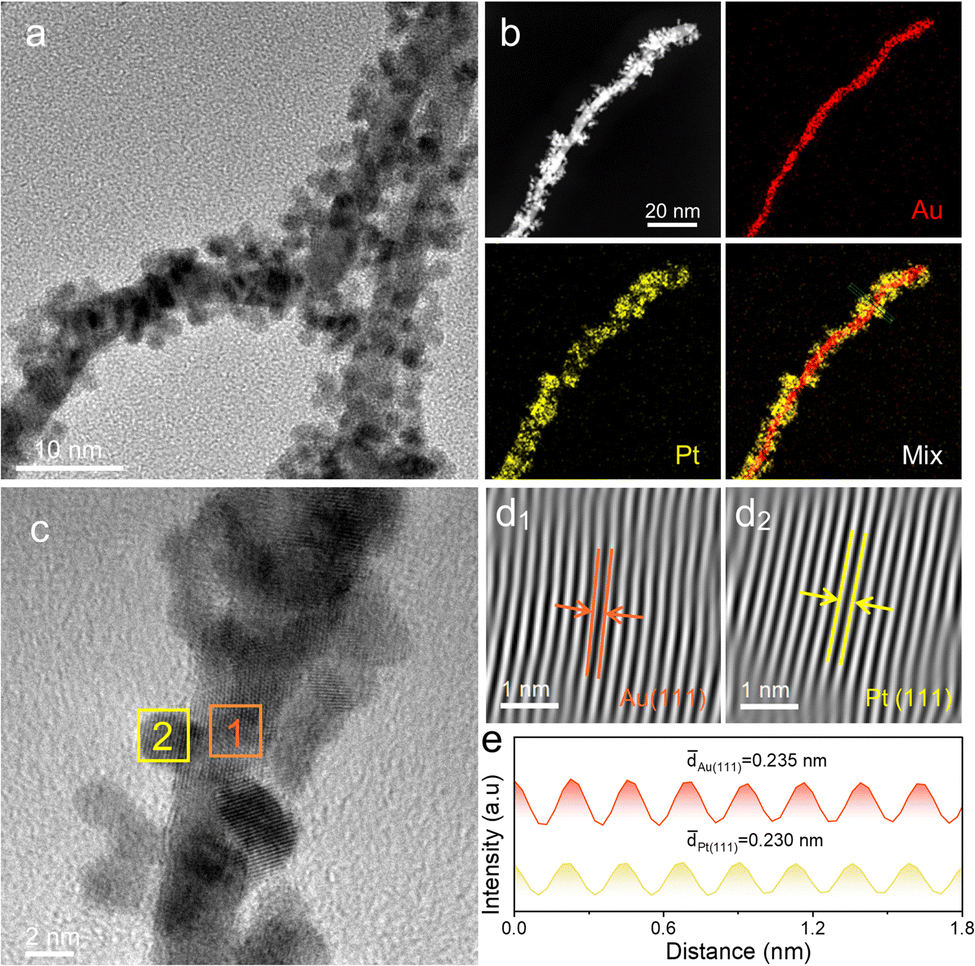 | ||
| Fig. 2 Structural characterizations of core–satellite Au@Pt NWs. (a) TEM image. (b) EDX elemental mapping images. (c) HRTEM image. (d) Fourier-filtered lattice fringe images taken from the rectangular regions in (c), and (e) corresponding integrated pixel intensities. | ||
To gain a better understanding of the growth process of Au@PtPd@Pt NWs, we monitored the time-dependent growth using TEM. It was observed that the layer growth of the Pd-rich PtPd alloy shell initially follows a layer growth mode with fewer Pt nanoparticles on the nanowire surface within the first 40 minutes (Fig. S12a, ESI†). This suggests that the Pd precursor is more easily deposited through epitaxial growth on the outer surface of Au NWs compared to the Pt precursor. As the reaction time increased, more Pt nanoparticles accumulated on the nanowires. The continued growth of these islands led to the formation of small Pt columns with similar diameters to the islands, extending from the PtPd alloy layer. Further growth, as depicted in Fig. S12b and c (ESI†), resulted in a dense “forest” of single-crystal Pt columns. After 120 minutes, Au@PtPd@Pt NWs were formed via the layer-island growth mode, with ultrathin PtPd alloy layers coating the surface of Au NWs and ultrafine island-like Pt nanoparticles grown on the surface of the PtPd alloy shell. The structure exhibits open channels between the columns, promoting the exposure of active sites in the inner PtPd layer. To comprehensively validate that the growth mechanism of Au@PtPd@Pt is predicated on distinct growth modes of Pt and Pd on Au seeds, we also monitored the time-dependent structural evolution of Au@Pt NWs and Au@Pd NWs using TEM. We can observe that Au@Pt NWs undergo growth characterized by an initial island Pt shell and adhere to an island growth mode, as demonstrated by the presence of fewer Pt islands on the surface of the Au NWs during the initial 40 minutes (Fig. S13, ESI†). Conversely, Au@Pd NWs follow a layer growth mode, leading to the deposition of thin Pd layers on the surface of Au NWs within the first 40 minutes (Fig. S14, ESI†). With the progression of the reaction time, there was an increased aggregation of Pt nanoparticles into islands, accompanied by the deposition of Pd nanoparticles onto the substrate in a layer-by-layer manner. Following 120 minutes, the successful formation of Au@Pt NWs displaying abundant and uniformly distributed Pt islands, as well as the creation of Au@Pd NWs with uniform Pd layers, further underscores the distinct growth modes of Pt and Pd on Au seeds.
X-Ray diffraction (XRD) was performed to determine the crystalline structure of the prepared samples. As shown in Fig. 3a, all the nanowires exhibit a series of diffraction peaks corresponding to the face-centered cubic (fcc) Au phase (PDF# 65-2870), which is consistent with the HRTEM results (Fig. 1, 2 and Fig. S8, ESI†). Additionally, Au@Pt, Au@Pd, and Au@PtPd@Pt samples display weak shoulder peaks, marked by arrows in Fig. 3a and Fig. S15 (ESI†). These weak signals from the outer layers can be attributed to the presence of Pd and Pt, which are partially obscured by the strong Au crystalline signals. This observation further confirms the presence of ultrathin shells in the core–shell nanowires. Furthermore, the valence states and surface compositions of the samples were investigated using X-ray photoelectron spectroscopy (XPS). As shown in Fig. 3b, all the nanowires exhibit characteristic doublets in the Au 4f XPS spectrum, corresponding to the Au 4f5/2 and Au 4f7/2 peaks. Notably, the Au 4f peaks in Au@Pt, Au@Pd, and Au@PtPd@Pt NWs are shifted towards lower binding energies compared to that of Au NWs, indicating a strong interaction between Au and the other metallic atoms. In the Pd 3d region, the peaks in the Au@PtPd@Pt NWs can be deconvoluted into two pairs of spin–orbit separations. The stronger pair, located at binding energies of 340.50 eV and 335.18 eV, is attributed to metallic Pd atoms, while the weaker pair at 336.85 eV and 341.61 eV indicates the presence of a small percentage of Pd2+ species (Fig. 3c). Notably, the observed metallic Pd 3d5/2 peak in Au@PtPd@Pt is shifted to a higher binding energy by 0.16 eV compared to Au@Pd, suggesting the formation of PtPd alloy and lower electron density of Pd due to the modified electronic structure. In the Pt 4f XPS spectrum, the two prominent peaks at 74.30 eV and 70.88 eV correspond to the Pt 4f5/2 and Pt 4f7/2 orbitals of metallic Pt, respectively. Additionally, a weaker pair of peaks at 75.90 eV and 71.84 eV indicates the presence of Pt2+ species (Fig. 3d). It is worth noting that Au@PtPd@Pt NWs display a subtle positive shift in the metallic Pd 3d5/2 peak, whereas the Pt 4f7/2 peaks exhibit an opposite negative shift of 0.20 eV. This observation implies a charge transfer from Pd to Pt due to their disparate electronegativities, which further support the formation of a PtPd alloy and the electronic interactions between the shell and satellite materials.35 The electronic interaction leads to the coupling of d-orbitals between Pd (with fully occupied d-orbitals) and Pt (with unsaturated d-orbitals), thereby inducing a shift in the Pt work function.36 Consequently, the d-band centers of the shell and satellite metals shift towards a more negative position in relation to the reference Fermi level, which ultimately diminishes the adsorption strength of hydrogen on the surfaces of both the PtPd shell and the Pt satellite.37,38
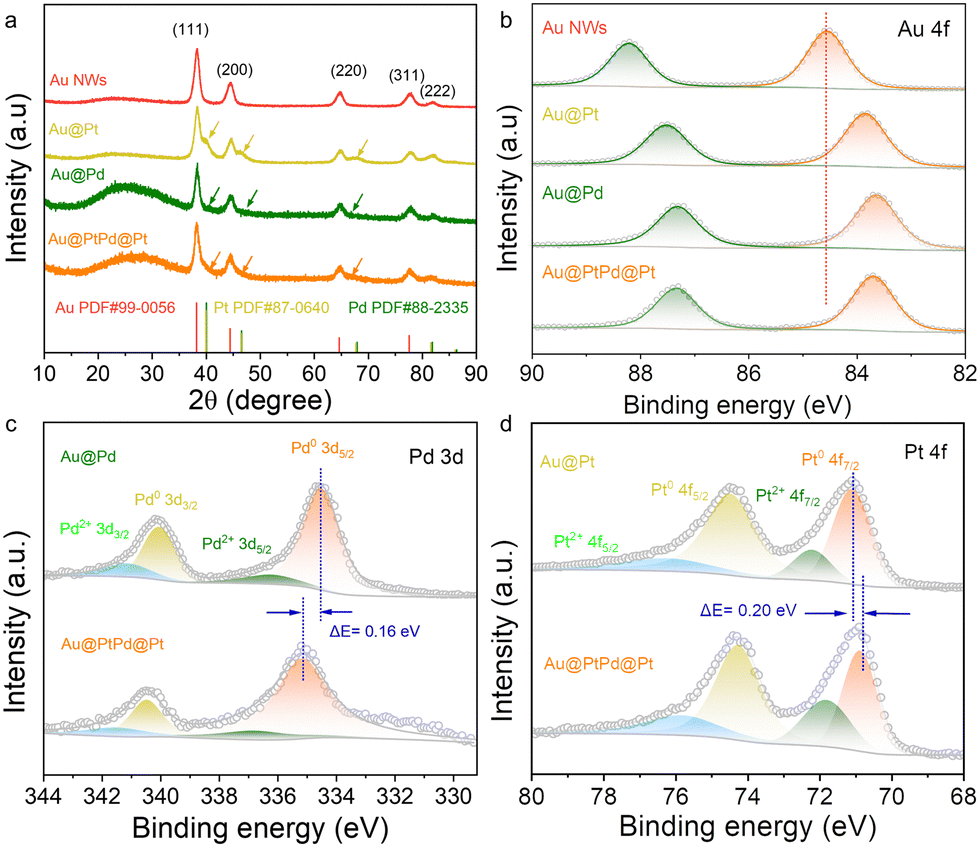 | ||
| Fig. 3 Composition and surface analyses of the obtained catalysts. (a) XRD pattern. High-resolution XPS spectrum in (b) Au 4f, (c) Pd 3d, and (d) Pt 4f regions. | ||
The unique core–shell–satellite composition, combined with the ultrathin 1D nanowire structure and the electronic interactions between trimetallic compositions, positions Au@PtPd@Pt NWs as a potential highly efficient catalyst for the HER. The HER performance of the catalysts was evaluated in N2-saturated 1.0 M KOH electrolyte using a typical three-electrode system with a scan rate of 5 mV s−1. Benchmark catalysts, including core–shell Au@Pd NWs, core–satellite Au@Pt NWs, and commercial Pt/C, were employed as reference catalysts. As depicted in Fig. 4a and c, Au@PtPd@Pt NWs exhibited superior HER activity, manifesting the lowest overpotentials at current densities of 10 mA cm−2 (η10) and 50 mA cm−2 (η50), ranking as follows: Au@PtPd@Pt (η10: 23 mV; η50: 61 mV) > Au@Pt (η10: 35 mV; η50: 74 mV) > commercial Pt/C (η10: 38 mV; η50: 80 mV) > Au@Pd (η10: 41 mV; η50: 91 mV). To gain a fundamental insight into the inherent properties of HER catalysts, Tafel plots were calculated based on HER polarization curves, as depicted in Fig. 4b. Typically, the Tafel slope serves as an indicator of the rate-determining step in the HER process, which can be categorized into either the Volmer–Tafel process or the Volmer–Heyrovsky pathway under alkaline conditions.39,40 In accordance with theoretical calculations, Tafel slopes of approximately 120 mV dec−1, 40 mV dec−1, and 30 mV dec−1 are attributed to the Volmer reaction, the Heyrovsky reaction, and the Tafel reaction, respectively, as the rate-determining steps (RDS).41,42 Note that the Tafel slope for Au@PtPd@Pt NWs is measured at 32.12 mV dec−1, which is lower than that of their counterparts, which implies that the HER process exhibits faster kinetics through the Volmer–Tafel mechanism, alongside electrochemical hydrogen desorption as the rate-determining step in HER electrocatalysis.40 The notably enhanced HER kinetics for Au@PtPd@Pt NWs can be attributed to the synergistic integration of the core–shell–satellite configuration and the 1D ultrathin nanowire structure, which provides an abundance of active sites, rapid electron transport, and minimal charge-transfer resistance.30,43–45
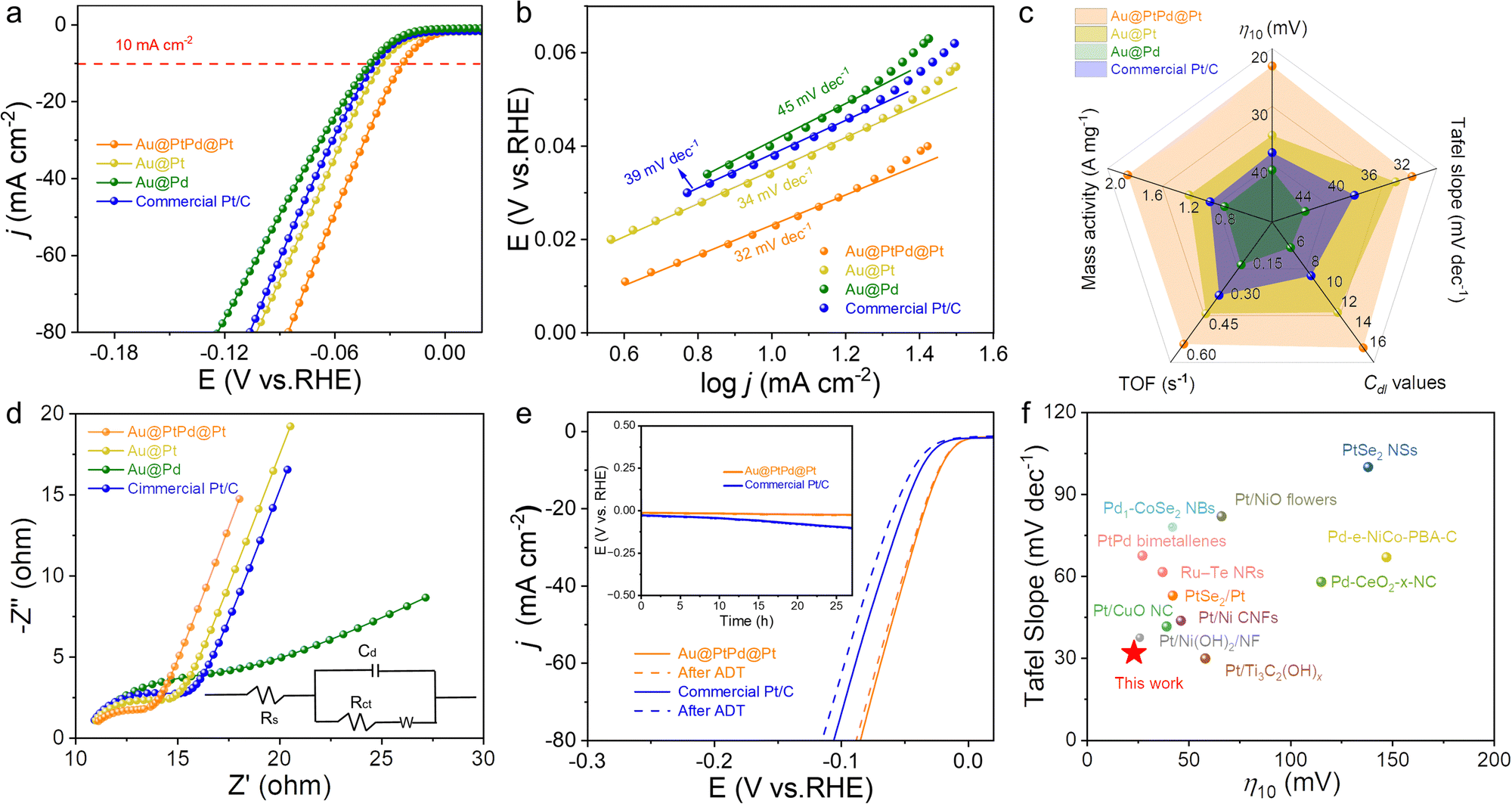 | ||
| Fig. 4 Compare the electrocatalytic alkaline HER performances of the obtained Au@PtPd@Pt and other catalysts. (a) LSV curves in N2-saturated 1.0 M KOH solution. (b) Corresponding Tafel plots. (c) Comparison of the HER performance metrics of different electrocatalysts. (d) Nyquist plots. (e) LSV polarization curves before and after the ADT test for Au@PtPd@Pt NWs and commercial Pt/C. The inset in (e) shows corresponding chronopotentiometry tests at 10 mA cm−2. (f) Comparison of η10 and the Tafel slope for reported noble metal-based catalysts in the recent literature. | ||
To clearly demonstrate this, we systematically compared the HER performance metrics of the catalysts, including η10, Tafel slope, double-layer capacitance (Cdl) values, mass activity (MA) and turnover frequency (TOF) at the 50 mV overpotential. As shown in Fig. 4c, all these merits of core–shell–satellite Au@PtPd@Pt NWs are superior with respect to bimetallic nanowires, demonstrating the key role of core–shell–satellite configuration and PtPd alloy layer composition in boosting HER performance. In detail, the Au@PtPd@Pt NWs demonstrated the lowest TOF value of 0.60 s−1 and the highest MA value of 1.88 A mg−1 at 50 mV, outperforming the other catalysts (TOFAu@Pt: 0.45 s−1; TOFPt/C: 0.36 s−1; TOFAu@Pd: 0.21 s−1; MAAu@Pt: 1.21 A mg−1; MAPt/C: 0.98 A mg−1; MAAu@Pd: 0.82 A mg−1). These results emphasize the favorable impact of the PtPd alloy shell formation and the presence of Pt satellites on the surface of Au NWs in reducing charge transfer resistance and facilitating electrochemical and mass transfer processes. To thoroughly investigate the intrinsic activity, the electrochemical surface areas (ECSA) of the studied catalysts were estimated based on their Cdl, obtained from cyclic voltammogram curves (CVs) at different scan rates (Fig. S16, ESI†).46,47 As depicted in Fig. S17 (ESI†), the Cdl value of Au@PtPd@Pt NWs was determined to be 16.54 mF cm−2, which is significantly larger than that of Au@Pt (13.01 mF cm−2), Au@Pd (6.54 mF cm−2), and commercial Pt/C (9.36 mF cm−2). Consistent with the expectations, the Au@PtPd@Pt NWs exhibit higher ECSA-normalized current densities at low potentials (Fig. S18, ESI†). This result unequivocally reveals that Au@PtPd@Pt NWs possess a higher ECSA due to the presence of an ample number of catalytically active sites on their external surface. Furthermore, it indicates that the ultrathin 1D wire-like core–shell–satellite configuration is advantageous for enhancing the number of accessible active sites, thereby improving the electrochemical activity. Additionally, electrochemical impedance spectroscopy (EIS) measurements were performed to gain further insights into the kinetics of the electrocatalytic interfacial reactions involved in the HER. As depicted in Fig. 4d, the Au@PtPd@Pt NWs had the smallest impedance arc diameter, indicating the lowest charge transfer resistance and the fastest HER kinetics among all tested electrodes. When compared to recently reported noble metal-based catalysts, the Au@PtPd@Pt NWs exhibit highly competitive electrochemical HER performance in alkaline electrolytes, as summarized in Fig. 4f and Table S2 (ESI†).
In addition to the catalytic activity, the electrocatalytic stability serves as another informative indicator for assessing the practicality of a catalyst. The stability of Au@PtPd@Pt NWs was assessed through an accelerating durability test (ADT). Fig. 4e illustrates that Au@PtPd@Pt NWs exhibit only a slight decline in performance after undergoing 10000 cycles of ADT, whereas commercial Pt/C experiences an irreversible loss in performance. Furthermore, the exceptional stability of Au@PtPd@Pt NWs was confirmed by conducting chronopotentiometry measurements. The overpotentials of Au@PtPd@Pt remained nearly constant even after continuous operation at a current density of 10 mA cm−2 for over 24 hours, demonstrating superior stability when compared to commercial Pt/C (inset of Fig. 4e). Faradaic efficiency is also a crucial parameter for quantifying the effectiveness of the HER catalysts. The H2 gas generated on the surface of Au@PtPd@Pt was collected using a straightforward drainage method within an H-type electrolytic cell with an anion-exchange membrane as a separator to calculate the Faraday efficiency of the HER (Fig. 5a and Fig. S19, ESI†). As referred to Fig. 5b, the experimental H2 volumes demonstrate a strong time-based correlation and closely correspond to the theoretical volumes, indicating that the Faraday efficiency of Au@PtPd@Pt NWs remains near 100% throughout the entire HER process. Additionally, the Au@PtPd@Pt NWs were utilized as the anode in a two-electrode electrolysis setup to achieve overall water splitting in a 1.0 M KOH electrolyte to showcase their practical applicability. As indicated in Fig. 5c, the Au@PtPd@Pt//RuO2 configuration exhibits a lower cell voltage of only 1.52 V, enabling an electrolytic current density of 10 mA cm−2, which outperforms the commercial Pt/C//RuO2 configuration (1.58 V). Importantly, the water splitting chronopotentiometry curve in Fig. 5d at operating voltage shows that the Au@PtPd@Pt configuration exhibits negligible attenuation of potential even after a long period of 50 h, indicating its extraordinary electrochemical stability in the alkaline medium.
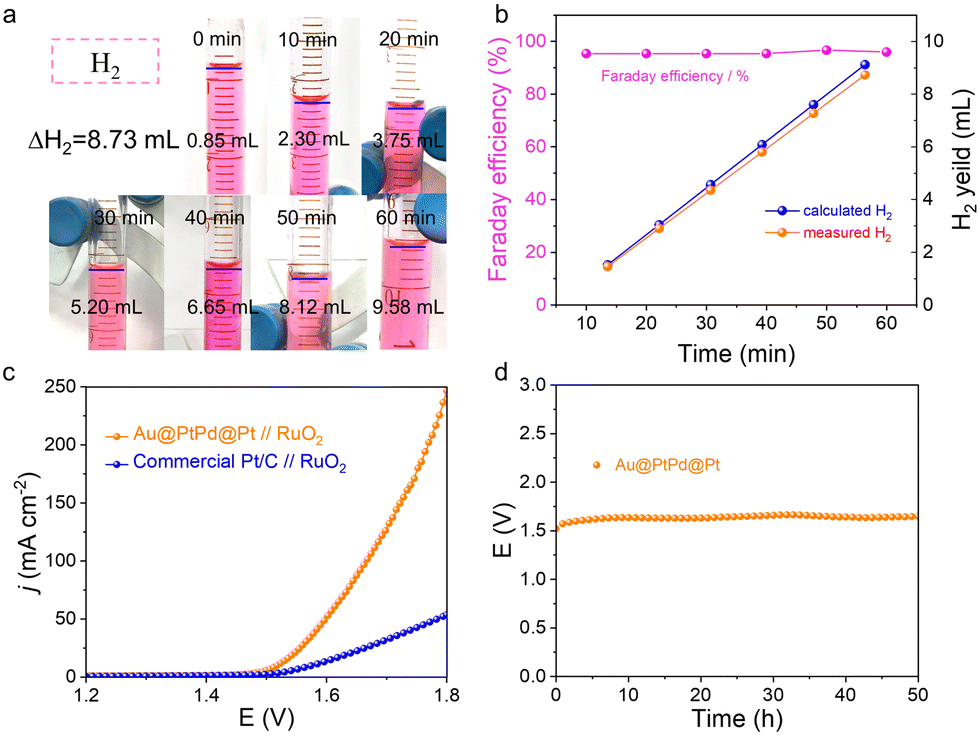 | ||
| Fig. 5 Comparison of the overall water-splitting performance. (a) Digital photographs of the volume changes of H2via the drainage method. (b) Experimental and theoretical volumes of produced H2 during water splitting and Faraday efficiency of Au@PtPd@Pt catalysts. (c) LSV curves of different two-electrode HER/OER electrolyzers in 1.0 M KOH media; the inset shows the overall water-splitting photograph of the electrode. (d) Water splitting chronopotentiometry curve of the Au@PtPd@Pt-based electrolyze at operating voltage. | ||
As supported by experimental results and related discussions, the outstanding HER performance of the core–shell–satellite Au@PtPd@Pt NWs can be attributed to the following synergies aspects: (i) the introduction of 1D ultrathin Au NWs as a growth template allows for the secure growth of the active PtPd shell@satellite Pt on the Au core due to its superior chemical stability. Importantly, the inertness of Au NWs during the electrocatalytic process can inhibit undesirable substrate corrosion and structure collapse at high potentials.48–50 As a result, this phenomenon significantly mitigates the undesirable dissolution, agglomeration, and Ostwald ripening of Au@PtPd@Pt NWs, rendering excellent long-term stability for the HER. (ii) The core–shell–satellite configuration with massive volume gaps and active Pt nanoislands could render the reactive sites more accessible and promote mass transport, electrolyte permeation, and gas release, enormously accelerating the reaction kinetics.17–20,32 (iii) In addition to the unique configuration, the synergistic effect between Pt and Pd plays a crucial role in enhancing the performance of Au@PtPd@Pt NWs. XPS analysis reveals that the modification of the electronic structure between the PtPd shell and the Pt satellites significantly reduces the d-band center of the Pt atoms (or upshifts the Fermi level), thereby weakening the adsorption energy of hydrogen on the surfaces of the PtPd shell and Pt satellites.51,52 This modification not only endows Au@PtPd@Pt NWs with highly catalytic HER performance but also confers excellent electrocatalytic stability upon them.
Conclusions
In summary, we have developed a novel and facile synthesis of 1D ultrathin Au@PtPd@Pt nanowires by utilizing Au nanowires (NWs) as a growth template for a highly active HER. The Au core surface was coated with ultrathin PtPd alloy layers through the layer growth mode, while ultrafine Pt nanoparticles were deposited on the PtPd shell surface using the island growth mode. XPS analysis demonstrates that the formation of a PdPd@Pt shell–satellite structure can modify the electronic structure of Pt, leading to a lower d-band center of Pt atoms. As a result, the adsorption energy of hydrogen on both the PtPd shell and Pt satellites is weakened, thereby enhancing the kinetics of the HER. The anisotropic structure of the 1D nanowires, along with their unique composition of Au core–PtPd shell–Pt satellites, contributes to their exceptional HER activity and remarkable stability, surpassing that of commercial Pt/C and other bimetallic catalysts. Furthermore, Au@PtPd@Pt NWs also exhibit significant potential in practical catalysis for water splitting. Our work provides valuable insights into the rational design of Pt-based catalysts through compositional regulation and nanoarchitecture engineering for the alkaline HER.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22232004, 22279062 and 22202104), the Natural Science Foundation of Jiangsu Province (BK20220933), and the ShuangChuang Doctor Plan of Jiangsu Province, Jiangsu Specially Appointed Professor Plan. The authors are also grateful for the support from the National and Local Joint Engineering Research Center of Biomedical Functional Materials and a project sponsored by the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- D. Wang, Q. Li, C. Han, Q. Lu, Z. Xing and X. Yang, Atomic and electronic modulation of self-supported nickel-vanadium layered double hydroxide to accelerate water splitting kinetics, Nat. Commun., 2019, 10, 3899 CrossRef PubMed.
- Y. Xu, S. Yu, T. Ren, S. Liu, Z. Wang, X. Li, L. Wang and H. Wang, Hydrophilic/Aerophobic Hydrogen-Evolving Electrode: NiRu-Based Metal-Organic Framework Nanosheets In Situ Grown on Conductive Substrates, ACS Appl. Mater. Interfaces, 2020, 12, 34728–34735 CrossRef CAS.
- K. L. Zhou, Z. Wang, C. B. Han, X. Ke, C. Wang, Y. Jin, Q. Zhang, J. Liu, H. Wang and H. Yan, Platinum single-atom catalyst coupled with transition metal/metal oxide heterostructure for accelerating alkaline hydrogen evolution reaction, Nat. Commun., 2021, 12, 3783 CrossRef CAS.
- J. Guo, Y. Zhang, A. Zavabeti, K. Chen, Y. Guo, G. Hu, X. Fan and G. K. Li, Hydrogen production from the air, Nat. Commun., 2022, 13, 5046 CrossRef CAS PubMed.
- K. Sun, X. Wu, Z. Zhuang, L. Liu, J. Fang, L. Zeng, J. Ma, S. Liu, J. Li, R. Dai, X. Tan, K. Yu, D. Liu, W.-C. Cheong, A. Huang, Y. Liu, Y. Pan, H. Xiao and C. Chen, Interfacial water engineering boosts neutral water reduction, Nat. Commun., 2022, 13, 6260 CrossRef CAS PubMed.
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Electrolysis of low-grade and saline surface water, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- L. Godeffroy, P. Ciocci, N. Ortiz Peña, D. Alloyeau, J.-M. Noël, J.-F. Lemineur and F. Kanoufi, Imaging the Footprint of Nanoscale Electrochemical Reactions for Assessing Synergistic Hydrogen Evolution, Angew. Chem., Int. Ed., 2023, 135, e202304950 CrossRef.
- M. Chen, Z. Lin, Y. Ren, X. Wang, M. Li, D. Sun, Y. Tang and G. Fu, Universal synthesis of rare earth-doped FeP nanorod arrays for the hydrogen evolution reaction, Mater. Chem. Front., 2023, 7, 4132–4141 RSC.
- X.-L. Liu, Y.-C. Jiang, J.-T. Huang, W. Zhong, B. He, P.-J. Jin and Y. Chen, Bifunctional PdPt bimetallenes for formate oxidation-boosted water electrolysis, Carbon Energy, 2023, e367 CrossRef.
- Z. Wu, P. Yang, Q. Li, W. Xiao, Z. Li, G. Xu, F. Liu, B. Jia, T. Ma, S. Feng and L. Wang, Microwave Synthesis of Pt Clusters on Black TiO2 with Abundant Oxygen Vacancies for Efficient Acidic Electrocatalytic Hydrogen Evolution, Angew. Chem., Int. Ed., 2023, 62, e202300406 CrossRef CAS PubMed.
- X. Zhang, Z. Sun, R. Jin, C. Zhu, C. Zhao, Y. Lin, Q. Guan, L. Cao, H. Wang, S. Li, H. Yu, X. Liu, L. Wang, S. Wei, W.-X. Li and J. Lu, Conjugated dual size effect of core-shell particles synergizes bimetallic catalysis, Nat. Commun., 2023, 14, 530 CrossRef CAS PubMed.
- Q. Lu, A.-L. Wang, Y. Gong, W. Hao, H. Cheng, J. Chen, B. Li, N. Yang, W. Niu, J. Wang, Y. Yu, X. Zhang, Y. Chen, Z. Fan, X.-J. Wu, J. Chen, J. Luo, S. Li, L. Gu and H. Zhang, Crystal phase-based epitaxial growth of hybrid noble metal nanostructures on 4H/fcc Au nanowires, Nat. Chem., 2018, 10, 456–461 CrossRef CAS PubMed.
- H. Zhang, J. Diao, M. Ouyang, H. Yadegari, M. Mao, M. Wang, G. Henkelman, F. Xie and D. J. Riley, Heterostructured Core-Shell Ni-Co@Fe-Co Nanoboxes of Prussian Blue Analogues for Efficient Electrocatalytic Hydrogen Evolution from Alkaline Seawater, ACS Catal., 2023, 13, 1349–1358 CrossRef CAS PubMed.
- Q. Zeng, J. Song, P. Cui, H. Liu, L. Tian, D. Chen and J. Yang, Optimizing Lattice Strain and Electron Effect of Ultrathin Platinum Nanoshells through Core-Shell Construction toward Superior Electrocatalytic Hydrogen Evolution, Ind. Eng. Chem. Res., 2022, 61, 7529–7536 CrossRef CAS.
- Q. Xue, H.-Y. Sun, Y.-N. Li, M.-J. Zhong, F.-M. Li, X. Tian, P. Chen, S.-B. Yin and Y. Chen, Au@Ir core-shell nanowires towards oxygen reduction reaction, Chem. Eng. J., 2021, 421, 129760 CrossRef CAS.
- X. Yue, X. Zhang, M. Zhang, W. Du and H. Xia, The enhancement in the performance of ultra-small core-shell Au@AuPt nanoparticles toward HER and ORR by surface engineering, Nanoscale, 2023, 15, 4378–4387 RSC.
- D. Han, D.-L. Zhou, Q.-Y. Guo, X. Lin, Q. Zhang and Q. Fu, Engineering the Surface Pattern of Microparticles: From Raspberry-like to Golf Ball-like, ACS Appl. Mater. Interfaces, 2021, 13, 31215–31225 CrossRef CAS.
- H. Li, L. Jian, Y. Chen, G. Wang, J. Lyu, X. Dong, X. Liu and H. Ma, Fabricating Bi2MoO6@Co3O4 core-shell heterogeneous architectures with Z-scheme for superior photoelectrocatalytic water purification, Chem. Eng. J., 2022, 427, 131716 CrossRef CAS.
- B. T. Sneed, C.-H. Kuo, C. N. Brodsky and C.-K. Tsung, Iodide-Mediated Control of Rhodium Epitaxial Growth on Well-Defined Noble Metal Nanocrystals: Synthesis, Characterization, and Structure-Dependent Catalytic Properties, J. Am. Chem. Soc., 2012, 134, 18417–18426 CrossRef CAS PubMed.
- L. Wang and Y. Yamauchi, Metallic nanocages: synthesis of bimetallic Pt-Pd hollow nanoparticles with dendritic shells by selective chemical etching, J. Am. Chem. Soc., 2013, 135, 16762–16765 CrossRef CAS.
- Q. Xue, H. Huang, J.-Y. Zhu, Y. Zhao, F.-M. Li, P. Chen and Y. Chen, Au@Rh core-shell nanowires for hydrazine electrooxidation, Appl. Catal., B, 2020, 278, 119269 CrossRef CAS.
- X. Wang, J. Qi, X. Luo, Z. Yang, Y. Fan, Z. Jiang, C. Liu, J. Yang and W. Chen, Core-shell Au@PtIr nanowires with dendritic alloy shells as efficient bifunctional catalysts toward methanol oxidation and hydrogen evolution reactions, Int. J. Hydrogen Energy, 2021, 46, 36771–36780 CrossRef CAS.
- F. Li, Y. Ding, X. Xiao, S. Yin, M. Hu, S. Li and Y. Chen, From monometallic Au nanowires to trimetallic AuPtRh nanowires: interface control for the formic acid electrooxidation, J. Mater. Chem. A, 2018, 6, 17164–17170 RSC.
- C. Wang, X. Jiang, Y. Wang, Y. Tang, J. Zhou and G. Fu, Recent advances in nonmetallic modulation of palladium-based electrocatalysts, Chem. Synth., 2023, 3, 8 CrossRef CAS.
- X. Jiang, Y. Xiong, R. Zhao, J. Zhou, J.-M. Lee and Y. Tang, Trimetallic Au@PdPb nanowires for oxygen reduction reaction, Nano Res., 2020, 13, 2691–2696 CrossRef CAS.
- H. Wang, D. Yang, S. Liu, S. Yin, Y. Xu, X. Li, Z. Wang and L. Wang, Metal-Nonmetal One-Dimensional Electrocatalyst: AuPdP Nanowires for Ambient Nitrogen Reduction to Ammonia, ACS Sustainable Chem. Eng., 2019, 7, 15772–15777 CrossRef CAS.
- S. Liu, S. Yin, H. Zhang, S. Jiao, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, Trimetallic Au@PdPt porous core-shell structured nanowires for oxygen reduction electrocatalysis, Chem. Eng. J., 2022, 428, 131070 CrossRef CAS.
- Q. Xue, J. Bai, C. Han, P. Chen, J.-X. Jiang and Y. Chen, Au Nanowires@Pd-Polyethylenimine Nanohybrids as Highly Active and Methanol-Tolerant Electrocatalysts toward Oxygen Reduction Reaction in Alkaline Media, ACS Catal., 2018, 8, 11287–11295 CrossRef CAS.
- Q. Xue, X.-Y. Bai, Y. Zhao, Y.-N. Li, T.-J. Wang, H.-Y. Sun, F.-M. Li, P. Chen, P. Jin, S.-B. Yin and Y. Chen, Au core-PtAu alloy shell nanowires for formic acid electrolysis, J. Energy Chem., 2022, 65, 94–102 CrossRef CAS.
- X. Jiang, X. Qiu, G. Fu, J. Sun, Z. Huang, D. Sun, L. Xu, J. Zhou and Y. Tang, Highly simple and rapid synthesis of ultrathin gold nanowires with (111)-dominant facets and enhanced electrocatalytic properties, J. Mater. Chem. A, 2018, 6, 17682–17687 RSC.
- Y. He, Q. Wang, Z. Wei, L. Xu, Z. Wang, H. Huang and H. Jiao, Soft Template-Directed Reactions: One-Pot Synthetic Route for Bimetallic Core-Satellite-Shell Structured Electrocatalytic Nanospheres, ChemCatChem, 2018, 10, 2546–2550 CrossRef CAS.
- H. Yang, Platinum-Based Electrocatalysts with Core-Shell Nanostructures, Angew. Chem., Int. Ed., 2011, 50, 2674–2676 CrossRef CAS PubMed.
- S. I. Lim, M. Varon, I. Ojea-Jiménez, J. Arbiol and V. Puntes, Pt nanocrystal evolution in the presence of Au(III)-salts at room temperature: spontaneous formation of AuPt heterodimers, J. Mater. Chem., 2011, 21, 11518–11523 RSC.
- J. Feng, D. Xu, F. Yang, J. Chen, C. Wu and Y. Yin, Surface Engineering and Controlled Ripening for Seed-Mediated Growth of Au Islands on Au Nanocrystals, Angew. Chem., Int. Ed., 2021, 60, 16958–16964 CrossRef CAS PubMed.
- X. Yang, Q. Wang, S. Qing, Z. Gao, X. Tong and N. Yang, Modulating Electronic Structure of an Au-Nanorod-Core-PdPt-Alloy-Shell Catalyst for Efficient Alcohol Electro-Oxidation, Adv. Energy Mater., 2021, 11, 2100812 CrossRef CAS.
- Y.-C. Jiang, Y. Ding, F.-M. Li, S.-B. Yin, D.-S. Li, X.-F. Li and Y. Chen, Ultrafine Pd@PdPt nanowires with a single-atom alloy shell for efficient formate oxidation, J. Mater. Chem. A, 2022, 10, 24701–24707 RSC.
- B. S. Gu, S. Dutta, Y.-R. Hong, O. F. Ngome Okello, H. Im, S. Ahn, S.-Y. Choi, J. Woo Han, S. Ryu and I. S. Lee, Harmonious Heterointerfaces Formed on 2D-Pt Nanodendrites by Facet-Respective Stepwise Metal Deposition for Enhanced Hydrogen Evolution Reaction, Angew. Chem., Int. Ed., 2023, 62, e202307816 CrossRef PubMed.
- F. Li, G.-F. Han, H.-J. Noh, J.-P. Jeon, I. Ahmad, S. Chen, C. Yang, Y. Bu, Z. Fu, Y. Lu and J.-B. Baek, Balancing hydrogen adsorption/desorption by orbital modulation for efficient hydrogen evolution catalysis, Nat. Commun., 2019, 10, 4060 CrossRef.
- J. Mahmood, F. Li, S.-M. Jung, M. S. Okyay, I. Ahmad, S.-J. Kim, N. Park, H. Y. Jeong and J.-B. Baek, An efficient and pH-universal ruthenium-based catalyst for the hydrogen evolution reaction, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS.
- M. Gong, W. Zhou, M.-C. Tsai, J. Zhou, M. Guan, M.-C. Lin, B. Zhang, Y. Hu, D.-Y. Wang, J. Yang, S. J. Pennycook, B.-J. Hwang and H. Dai, Nanoscale nickel oxide/nickel heterostructures for active hydrogen evolution electrocatalysis, Nat. Commun., 2014, 5, 4695 CrossRef CAS PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Recent Advances in Electrocatalytic Hydrogen Evolution Using Nanoparticles, Chem. Rev., 2020, 120, 851–918 CrossRef CAS.
- C. G. Morales-Guio, L.-A. Stern and X. Hu, Nanostructured hydrotreating catalysts for electrochemical hydrogen evolution, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- X. Jiang, G. Fu, X. Wu, Y. Liu, M. Zhang, D. Sun, L. Xu and Y. Tang, Ultrathin AgPt alloy nanowires as a high-performance electrocatalyst for formic acid oxidation, Nano Res., 2018, 11, 499–510 CrossRef CAS.
- H. Yin, S. Zhao, K. Zhao, A. Muqsit, H. Tang, L. Chang, H. Zhao, Y. Gao and Z. Tang, Ultrathin platinum nanowires grown on single-layered nickel hydroxide with high hydrogen evolution activity, Nat. Commun., 2015, 6, 6430 CrossRef CAS PubMed.
- H. Wang, S. Jiao, S. Liu, S. Wang, T. Zhou, Y. Xu, X. Li, Z. Wang and L. Wang, Mesoporous Bimetallic Au@Rh Core-Shell Nanowires as Efficient Electrocatalysts for pH-Universal Hydrogen Evolution, ACS Appl. Mater. Interfaces, 2021, 13, 30479–30485 CrossRef CAS PubMed.
- K. Deng, T. Ren, Y. Xu, S. Liu, Z. Dai, Z. Wang, X. Li, L. Wang and H. Wang, Crystalline core-amorphous shell heterostructures: epitaxial assembly of NiB nanosheets onto PtPd mesoporous hollow nanopolyhedra for enhanced hydrogen evolution electrocatalysis, J. Mater. Chem. A, 2020, 8, 8927–8933 RSC.
- Y. Zhao, J. Xu, K. Huang, W. Ge, Z. Liu, C. Lian, H. Liu, H. Jiang and C. Li, Dopant- and Surfactant-Tuned Electrode-Electrolyte Interface Enabling Efficient Alkynol Semi-Hydrogenation, J. Am. Chem. Soc., 2023, 145, 6516–6525 CrossRef CAS PubMed.
- X. Zhou, Y. Ma, Y. Ge, S. Zhu, Y. Cui, B. Chen, L. Liao, Q. Yun, Z. He, H. Long, L. Li, B. Huang, Q. Luo, L. Zhai, X. Wang, L. Bai, G. Wang, Z. Guan, Y. Chen, C.-S. Lee, J. Wang, C. Ling, M. Shao, Z. Fan and H. Zhang, Preparation of Au@Pd Core–Shell Nanorods with fcc-2H-fcc Heterophase for Highly Efficient Electrocatalytic Alcohol Oxidation, J. Am. Chem. Soc., 2022, 144, 547–555 CrossRef CAS PubMed.
- J. Xia, L. Dong, X. Liu, R. Chernikov, M. Shakouri, Y. Hu, Y. Guo, J. Wang, K. Song, P. Hu, Y. Wang and H. Wang, Identifying the Activity Origin of a Single-Atom Au1/Nb2O5 Catalyst for Hydrodeoxygenation of Methylcatechol: A Stable Substitutional Au+ Site, ACS Catal., 2023, 13, 6093–6103 CrossRef CAS.
- X.-P. Yu, C. Yang, P. Song and J. Peng, Self-assembly of Au/MoS2 quantum dots core-satellite hybrid as efficient electrocatalyst for hydrogen production, Tungsten, 2020, 2, 194–202 CrossRef.
- L. Wang and Y. Yamauchi, Strategic Synthesis of Trimetallic Au@Pd@Pt Core-Shell Nanoparticles from Poly(vinylpyrrolidone)-Based Aqueous Solution toward Highly Active Electrocatalysts, Chem. Mater., 2011, 23, 2457–2465 CrossRef CAS.
- H. Lv, X. Chen, D. Xu, Y. Hu, H. Zheng, S. L. Suib and B. Liu, Ultrathin PdPt bimetallic nanowires with enhanced electrocatalytic performance for hydrogen evolution reaction, Appl. Catal., B, 2018, 238, 525–532 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qm00618b |
| ‡ These authors contributed equally to the paper. |
| This journal is © the Partner Organisations 2024 |