
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of the pendant substituent at the C1 position of a spirobifluorene scaffold on the electronic properties†
Lambert
Sicard
a,
Clément
Brouillac
 a,
Nicolas
Leclerc
b,
Sadiara
Fall
c,
Nicolas
Zimmerman
c,
Olivier
Jeannin
a,
Joëlle
Rault-Berthelot
a,
Cassandre
Quinton
a and
Cyril
Poriel
*a
a,
Nicolas
Leclerc
b,
Sadiara
Fall
c,
Nicolas
Zimmerman
c,
Olivier
Jeannin
a,
Joëlle
Rault-Berthelot
a,
Cassandre
Quinton
a and
Cyril
Poriel
*a
aUniv Rennes, CNRS, ISCR-UMR 6226, F-35000 Rennes, France. E-mail: cyril.poriel@univ-rennes1.fr
bInstitut de Chimie et Procédés pour l’Énergie, l’Environnement et la Santé (ICPEES), UMR CNRS 7515, 67087 Strasbourg, France
cLaboratoire ICube, Université de Strasbourg, UMR CNRS 7357, 67087 Strasbourg, France
First published on 1st January 2024
Abstract
1-Substituted fluorenes have emerged in recent years as being among the most efficient functional materials for phosphorescence and thermally activated delayed fluorescence OLEDs. The position C1 of the fluorene is indeed highly interesting to design functional materials with specific properties. This scaffold displays two main characteristics. First, there is a strong π-conjugation disruption between the pendant substituent attached at C1 and the substituted fluorene. Second, the specific geometry of the C1-fluorene allows through-space interactions between the substituent attached at C1 and the cofacial building unit attached at C9. However, to date, this family of organic-semi-conductors has not been intensively studied yet. In this work, we report the synthesis and the structural, electrochemical and photophysical properties of four 1-substituted spirobifluorene derivatives, constructed with widely known extended π-conjugated systems: naphthalene, anthracene, phenanthrene and pyrene at the C1 position of spirobifluorene. Using a structure–property relationship approach, we show how the nature of the substituent grafted at C1 significantly modifies the electronic properties (HOMO/LUMO energy levels, triplet state energy levels, etc.). As 1-substituted spirobifluorenes are undoubtedly very promising organic semi-conductors, such a study provides fundamental knowledge to construct future efficient organic materials for specific applications.
Introduction
9,9′-Spirobifluorene (SBF) is one of the most important building blocks used in the synthesis of organic semi-conductors (OSCs) for organic electronics (OEs).1–5 The most studied position of substitution is the C2 position, known to significantly increase the conjugation length between the fluorene and its substituent.1,2,6 However, in recent years, manipulating the substitution pattern of SBF (positions of substitution C1, C3 or C4) has appeared as a very efficient tool to further tune the electronic properties of SBF-based materials.1 The particularity of the substitution at C1,7–9 C3,8,10–16 or C48,17–21 is the restriction of the electronic coupling between the fluorene and the attached substituent. This key characteristic has been advantageously used to design high triplet energy materials (ET > 2.7 eV), which are particularly attractive for hosting phosphors in phosphorescence OLEDs (PhOLEDs).22–24 Poly-substituted organic materials with specific properties have also been investigated, taking advantage from the different SBF positions (C125–28 and C329–31).In a C4-substituted SBF, constructed on a twisted ortho linkage, the π-conjugation restriction is induced by strong steric hindrance between the fluorene and its substituent.1,17 In a C3-substituted SBF, the π-conjugation restriction is caused by the electronic decoupling induced by the presence of a meta linkage. The C1 position combines these two features, meta linkage and strong steric hindrance, between the fluorene and the substituent. Thanks to the strong electronic decoupling induced by these two features, a C1-substituted SBF scaffold has appeared as the most efficient regioisomer to construct high ET materials, recently providing important advances in the field of PhOLEDs with the most efficient blue and white PhOLEDs ever reported in the literature.23 The substitution at the C1-position of SBF displays another interesting characteristic that is the intramolecular interaction occurring between the pendant substituent at C1 and the cofacial fluorene. This characteristic has been efficiently used in thermally activated delayed fluorescence (TADF) based OLEDs, providing cofacial donor/acceptor systems.32–36
Thus, the substitution at C1 has many advantages, which have allowed designing high efficiency materials for electronics. However, as almost all the C1-based materials reported to date were designed to host phosphors in PhOLEDs, only small molecular units with short π-conjugation pathways were attached such as phenyl,7 biphenyl,23 carbazole,7 and m-terphenyl.23 Thus, in order to increase the molecular diversity of the emerging family of 1-substituted SBFs and to well apprehend their electronic properties, we wish to report herein a series of 1-substituted SBFs incorporating various π-conjugated molecular fragments possessing different conjugation lengths, namely naphthyl, phenanthrenyl, anthracenyl or pyrenyl. The present work reports a detailed structure–property relationship study of 1-naphthyl-, 1-phenanthrenyl-, 1-anthracenyl- and 1-pyrenyl-spirobifluorenes (1-Napht-SBF, 1-Phen-SBF, 1-Anth-SBF and 1-Pyr-SBF, respectively, Chart 1) with their constituting building blocks (naphthalene, phenanthrene, anthracene and pyrene) and the structurally related analogue spirobifluorene (SBF). We show the importance of the substituent in the modulation of the electronic properties (HOMO/LUMO energy levels, optical transitions, triplet state energy ET, charge transport, etc.). This feature provides an interesting degree of tuning for this emerging family of OSCs.
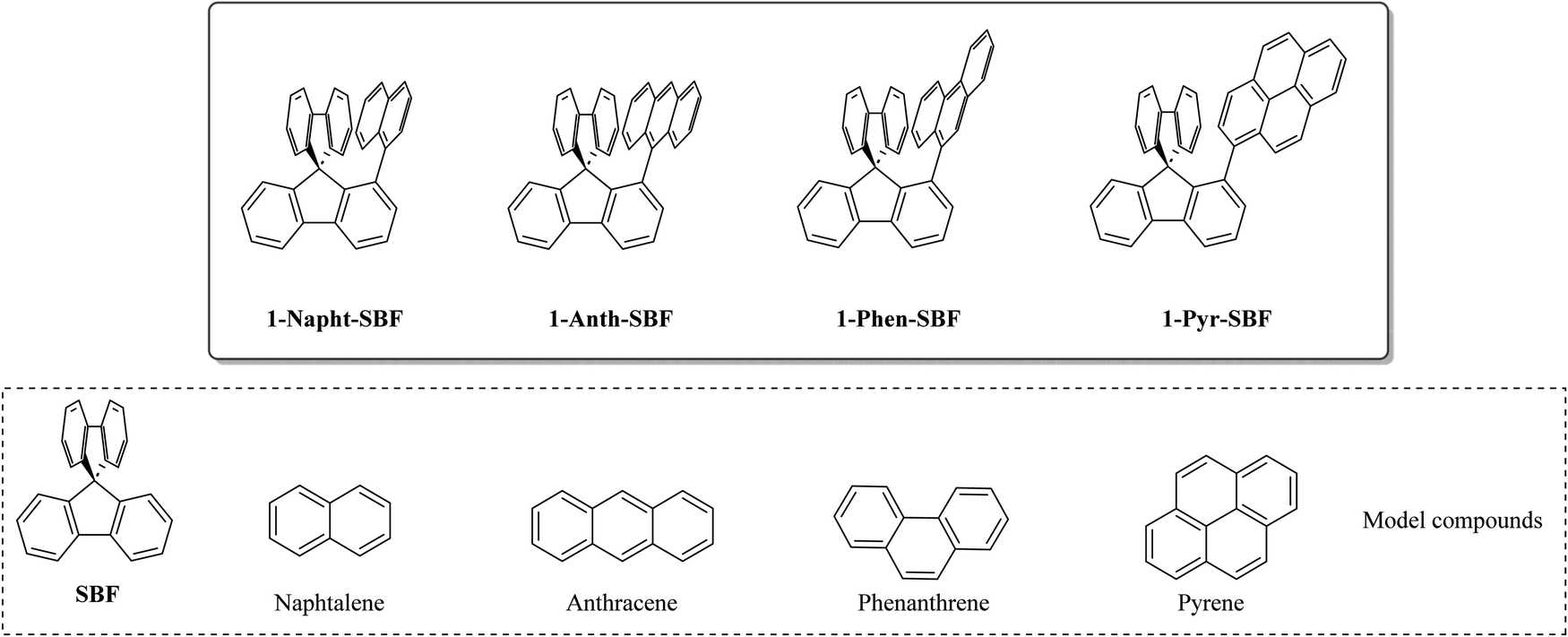 | ||
| Chart 1 Molecular structures of 1-Napht-SBF, 1-Phen-SBF, 1-Anth-SBF and 1-Pyr-SBF and constituting building blocks (spirobifluorene, naphthalene, phenanthrene, anthracene and pyrene). | ||
Results and discussion
Synthesis
Since 1-bromo-fluorenone (1-Br-FO) can be obtained at the multigram scale,7 the synthesis of 1-Napht-SBF, 1-Phen-SBF, 1-Anth-SBF and 1-Pyr-SBF is straightforward with high yield (Scheme 1). 1-Br-FO was obtained as previously reported using a one pot efficient synthesis method reported by Sorensen and coworkers.37 The aryl fragments were then attached via Suzuki–Miyaura cross-coupling to give the corresponding 1-aryl-fluorenones with yields varying from 43 to 78%. The synthesis of the targeted 1-aryl-spirobifluorenes was then carried out using a classical two-step procedure: the lithium-iodine exchange of 2-iodobiphenyl with n-butyllithium at low temperatures, followed by addition of 1-Br-FO affording the corresponding fluorenol (not isolated), which was further involved in an intramolecular cyclisation reaction (AcOH/HCl) to provide the corresponding 1-aryl-spirobifluorenes with overall yields in the range of 61 to 82% from 1-aryl-fluorenones.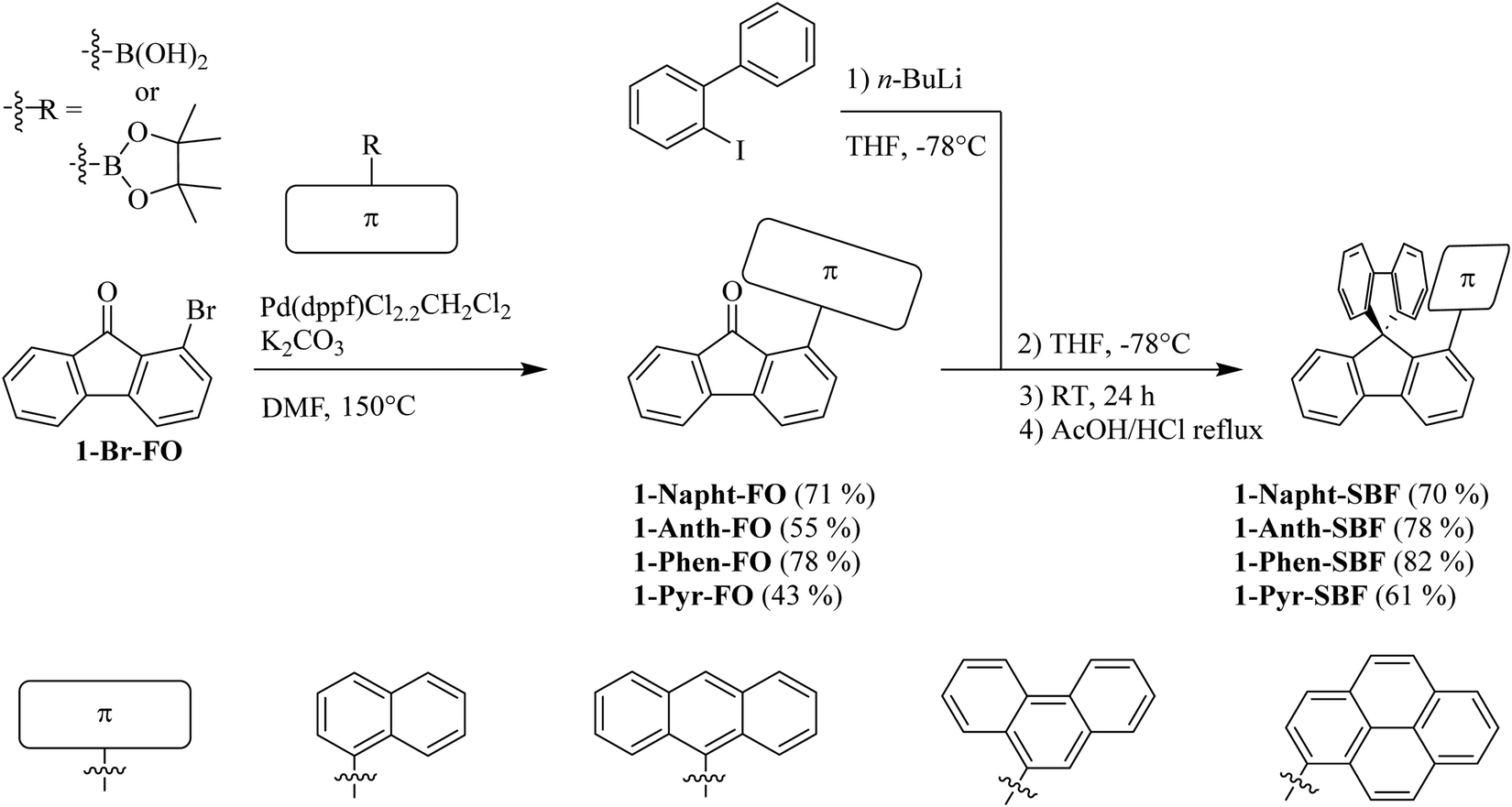 | ||
| Scheme 1 Synthesis of 1-Napht-SBF, 1-Phen-SBF, 1-Anth-SBF and 1-Pyr-SBF. | ||
Structural properties
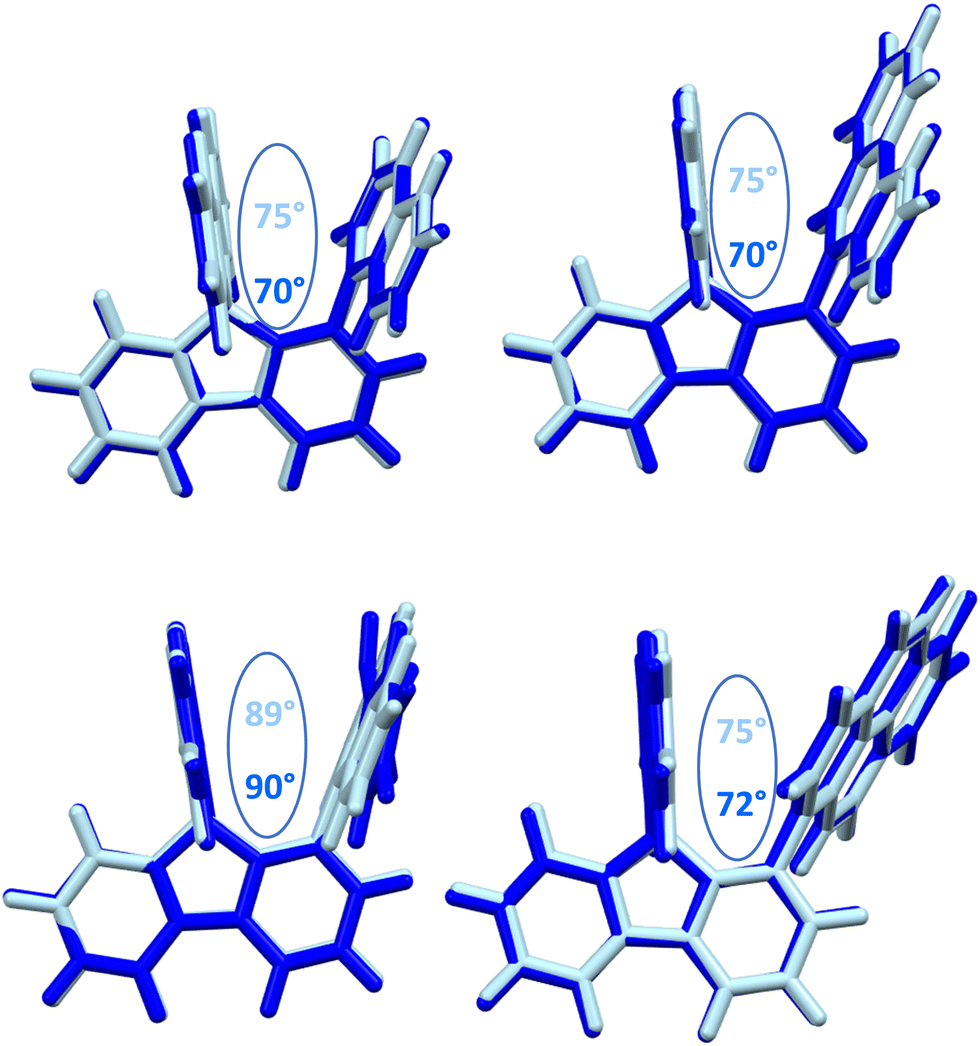
The structural arrangement of the four derivatives obtained by X-ray diffraction are depicted in Fig. 1. The most important structural feature is the relative position of the pendant substituent with respect to the phenyl ring of the substituted fluorene. For the four molecules, the angle between the substituted phenyl ring and the grafted phenyl of the substituent is very high: 75° for 1-Napht-SBF, 79° and 86° for 1-Anth-SBF (four molecules are indeed present in the asymmetric unit), 81° for 1-Pyr-SBF and 89° for 1-Phen-SBF. This feature indicates a significant conjugation breaking between the fluorene and its substituent at C1. The model compound 1-Phenyl-SBF, previously reported in the literature, displays an angle of 75°.8 Thus, from phenyl to naphthalene, the dihedral angle remains identical, showing that the steric hindrance is not increased when an additional phenyl ring is fused. However, the fusion of an additional phenyl ring in anthracene increases the dihedral angle (from 80° to 86°). The highest value is obtained for 1-Phen-SBF, 89°, in which the fused phenyl rings are in ortho and meta positions of the 1-SBF linkage.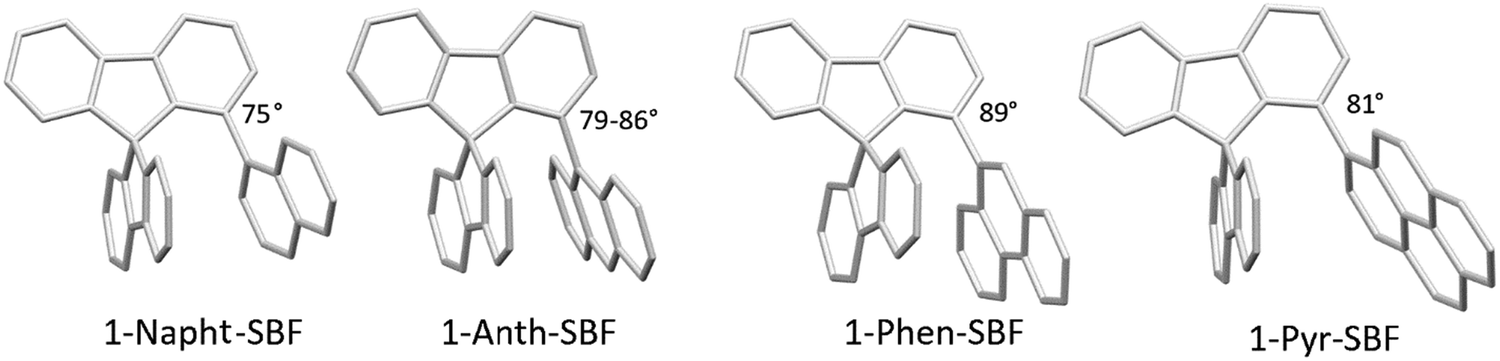 | ||
| Fig. 1 Molecular structures from X-ray diffraction of 1-Napht-SBF, 1-Anth-SBF, 1-Phen-SBF and 1-Pyr-SBF and the angles between the substituted phenyl ring and the phenyl of the substituent. | ||
Due to the particular geometry of the 1-SBF scaffold, the molecular structures of the four compounds display many short intramolecular C–C distances between the two cofacial fragments (see the ESI†). These C–C distances are shorter than the sum of their van der Waals radii (3.4 Å)38 and translate a sterically hindered environment. In order to evaluate the strength of these interactions between the pendant substituent and the cofacial fluorene, three structural parameters have been evaluated for the cofacial phenyl rings, namely ring-centroid/ring-centroid distance dC–C, vertical displacements d1 and d2 and slippage angles θ1 and θ2 (Fig. 2, see the ESI† for methods). As discussed by Janiak and coworkers, these parameters can reflect the strength of the interactions between two phenyl rings.39–43 Indeed, ring-centroid/ring-centroid distances below 3.8 Å, vertical displacements below 1.5 Å, and ring slippage angles below 25° indicate strong interactions between two interacting phenyl rings. In the present case, the central cyclopentadienyl ring of the fluorene will also be considered.
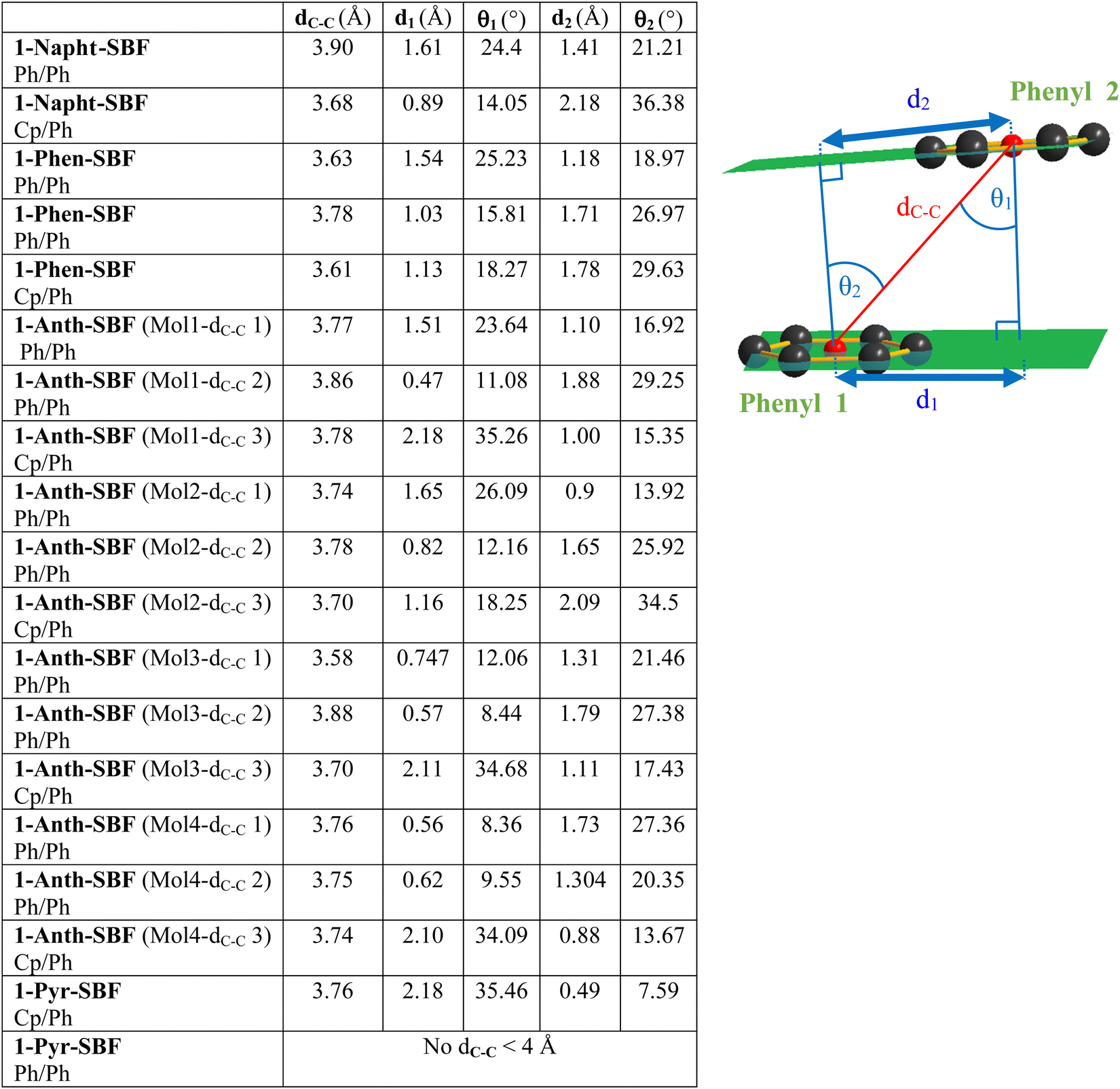 | ||
| Fig. 2 Left: selected crystallographic data of 1-Napht-SBF, 1-Anth-SBF, 1-Phen-SBF and 1-Pyr-SBF. Right: reprinted (adapted) with permission from J. Phys. Chem. C 2019, 123, 19094–19104. Copyright 2023 American Chemical Society. | ||
All molecules display short ring-centroid/ring-centroid distances between two cofacial phenyl or cyclopentadienyl rings (below 3.9 Å). Very small distances in the series are detected, notably for 1-Phen-SBF and 1-Anth-SBF with short dC–C of 3.61 and 3.58 Å, respectively. Many phenyl/phenyl and cyclopentadienyl/phenyl couples also display very short vertical displacements (below 1.5 Å) and ring slippage angles (below 25°) satisfying the above-mentioned criteria. Three of the four molecules present in the asymmetric unit of 1-Anth-SBF fully satisfy the three Janiak's criteria, indicating strong intramolecular interactions between the facing anthracene and fluorene units. Compounds 1-Phen-SBF and 1-Napht-SBF also present several phenyl/phenyl and cyclopentadienyl/phenyl interactions. These interactions partially satisfy all the above-mentioned criteria and translate therefore moderately to strong π–π interactions between the cofacial systems. In contrast, 1-Pyr-SBF only displays one short dC–C and the pyrene unit is therefore less influenced by the facing fluorene. This shows that the C1 grafting allows not only modulation of the fluorene–substituent direct conjugation, but also fine tuning of the interactions between the second SBF-included fluorene and the grafted substituent at C1. Especially, these data show that the substitution at C1 provides a sterically hindered environment which seems to govern the π–π interactions between the cofacial systems, and these interactions are modulated by the size of the grafted substituent.
Photophysical properties
UV-Vis absorption and emission spectra of the four compounds, 1-Napht-SBF, 1-Anth-SBF, 1-Phen-SBF and 1-Pyr-SBF, were recorded in dilute cyclohexane solution at room temperature (Fig. 3) in addition to their corresponding building units, either naphthalene for 1-Napht-SBF, anthracene for 1-Anth-SBF, phenanthrene for 1-Phen-SBF and pyrene for 1-Pyr-SBF and SBF. The photophysical properties are collected in Table 1 (and in Table S1, ESI† for their building units).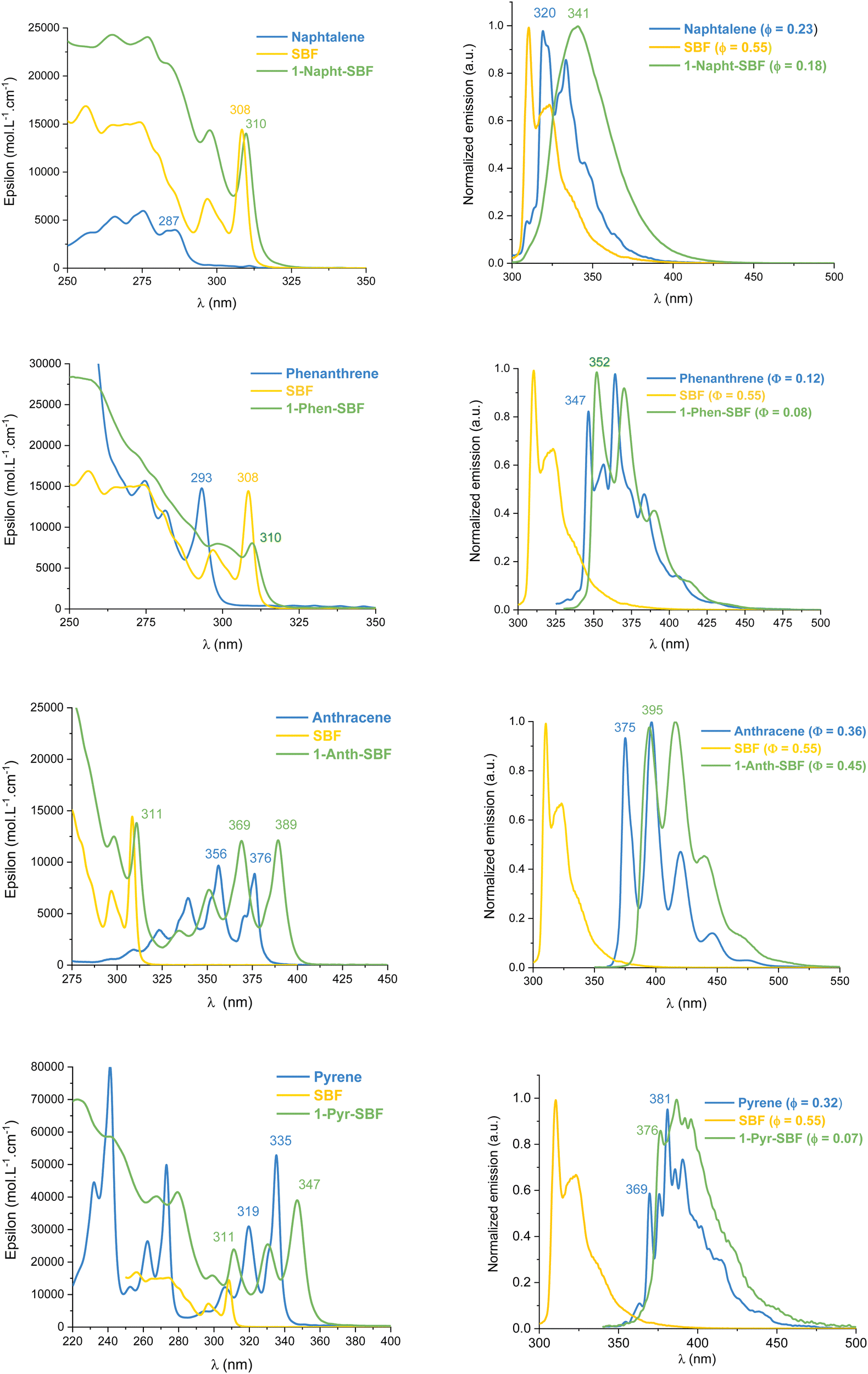 | ||
| Fig. 3 Absorption (left) and emission (right) at room temperature in cyclohexane of 1-aryl-SBFs (green), SBF (yellow) and the substituents (blue). | ||
| 1-Napht-SBF | Naphtalene | 1-Phen-SBF | Phenanthrene | 1-Pyr-SBF | Pyrene | 1-Anth-SBF | Anthracene | SBF 45 | |
|---|---|---|---|---|---|---|---|---|---|
| a In cyclohexane. b Quantum yield θ measured with quinine sulphate as the reference. c λ exc = 310 nm. d From the onset of fluorescence spectra in cyclohexane. e From the pic maximum in 2-MeTHF at 77 K. f From theoretical calculations (TD-DFT, b3lyp, 6-311+g(d,p)). g From CV in Bu4NPF6 0.2 M in DCM for oxidation. h From CV in Bu4NPF6 0.1 M in DMF. i From the onset of the absorption spectrum in cyclohexane. | |||||||||
| λ abs (ε) [nm] (×104 L mol−1 cm−1) | 265 (2.4) | 257 (0.37) | 298 (0.79) | 274 (1.5) | 221 (7.0) | 232 (4.4) | 298 (1.24) | 309 (0.15) | 297 (0.72) |
| 277 (2.4) | 265 (0.53) | 310 (0.8) | 281 (1.2) | 243 (5.8) | 241 (8.0) | 311 (1.38) | 324 (0.34) | 308 (1.45) | |
| 283 (2.1) | 275 (0.59) | 293 (1.47) | 267 (4.0) | 252 (1.24) | 335 (0.33) | 339 (0.66) | |||
| 298 (1.4) | 283 (0.39) | 279 (4.1) | 262 (2.62) | 351 (0.73) | 352 (0.35) | ||||
| 310 (1.4) | 286 (0.39) | 299 (1.5) | 273 (4.98) | 369 (1.2) | 356 (0.97) | ||||
| 311 (2.3) | 294 (0.47) | 389 (1.2) | 371 (0.48) | ||||||
| 331 (2.5) | 306 (1.21) | 376 (0.88) | |||||||
| 347 (3.9) | 319 (3.09) | ||||||||
| 335 (5.3) | |||||||||
| λ em [nm] | 341 | 320, 333 | 352, 370, 390 | 347, 356, 364, 373, 383, 406, 431 | 376, 388, 397, 423 | 370, 376, 381, 386, 390, 399, 413, 427, 439, 468 | 395, 416, 439, 467 | 375, 396, 420, 446, 477 | 310, 323 |
| 413, 438 | |||||||||
| θ | 0.18 | 0.23 | 0.08 | 0.12 | 0.07 | 0.32 | 0.45 | 0.36 | 0.55 |
| τ S [ns]c | 4.8 | 16.4 | 15 | 14.3 | 1.9 | 17.2 | 4.8 | 4.2 | 4.6 |
| k r [×107 s−1] | 3.8 | 14 | 0.5 | 0.8 | 3.7 | 1.9 | 9.4 | 8.6 | 12 |
| k nr [×107 s−1] | 17 | 47 | 6.1 | 6.1 | 49 | 4.0 | 11 | 15 | 10 |
| E S [eV]d | 3.94 | 4.03 | 3.58 | 3.63 | 3.34 | 3.52 | 3.23 | 3.36 | 4.07 |
| E T [eV] expe | 2.61 | 2.65 | 2.64 | 2.71 | 2.07 | 2.29 | — | — | 2.89 |
| E T [eV] calcf | 2.31 | 2.36 | 2.38 | 2.45 | 1.80 | 1.83 | — | — | |
| τ T [s]e | 2.7 | 2.8 | 3.6 | 3.4 | 0.6 | — | — | — | 5.4 |
| Ox potentials [V vs. SCE]g | 1.58, 1.77(sh), 1.92 | 1.75, 1.99 (sh) | 1.55, 1.88 (sh), 2.02 | 1.69, 2.1 | 1.22, 1.82 | 1.22(sh),1.29, 1.44, >2.0 | 1.12, 1.82, 2.17 | 1.23, 1.92 | 1.67, 1.86 |
| HOMO [eV] | |||||||||
| Elg | −5.90 | −5.94 | −5.85 | −5.89 | −5.52 | −5.54 | −5.43 | −5.46 | −5.95 |
| Calf | −5.91 | −6.14 | −5.88 | −6.08 | −5.52 | −5.67 | −5.40 | −5.57 | −5.99 |
| LUMO [eV] | |||||||||
| Elh | −1.97 | −1.87 | −2.00 | −1.95 | −2.29 | −2.31 | −2.36 | −2.42 | −1.74 |
| Calf | −1.36 | −1.41 | −1.41 | −1.42 | −1.81 | −1.87 | −1.95 | −2.04 | −1.26 |
| ΔE [eV] | |||||||||
| Opti | 3.94 | 4.25 | 3.93 | 4.16 | 3.47 | 3.64 | 3.10 | 3.26 | 3.97 |
| Elg | 3.93 | 4.07 | 3.85 | 3.94 | 3.23 | 3.23 | 3.07 | 3.04 | 4.21 |
| Calf | 4.55 | 4.73 | 4.47 | 4.66 | 3.71 | 3.80 | 3.45 | 3.53 | 4.73 |
| μ h (cm2 V−1 s−1) | 1.1 × 10−6 | — | 3.5 × 10−6 | — | 0.15 × 10−6 | — | 3.2 × 10−6 | — | — |
For the four 1-SBF derivatives, the absorption ranges from 250 to 400 nm (Fig. 3-left). First, it should be noted that unsubstituted SBF exhibits two thin absorption bands at ca. 297 and 308 nm (π–π* transitions). Moreover, the aromatic substituents display absorption bands, which can be classified in three categories:44 the first bands are weak (ε = 102–103 mol L−1 cm−1) and may be hidden by the others (they can be seen above 300 nm for naphthalene), and the second bands are moderately intense (ε = 104 mol L−1 cm−1) with a vibrational structure displaying peaks spaced by 1400 cm−1 and the third bands are very intense (ε = 105 mol L−1 cm−1) and unstructured. The bands corresponding to each fragment (both SBF and the substituents) can be found in the 1-substituted SBF. It should be noted that the bands corresponding to anthracene and pyrene are clearly red-shifted in 1-Ant-SBF and 1-Pyr-SBF in comparison to the model compounds (by 13 and 12 nm, respectively) while the other bands corresponding to the SBF seem relatively unchanged. There is indeed a slight red shift of 2 nm between the SBF building block and the corresponding band of the 1-substituted SBFs. This shows that the substituent is not completely isolated from the SBF backbone (through bonds and/or through-space interactions). As the dihedral angle between the C1-substituted fluorene and the substituent attached is very high in these molecules, π-conjugation extension by through-bond interactions should be very weak. However, as shown above, through-space interactions occur and these interactions might be responsible for the red shift observed in the absorption spectra. This hypothesis will be verified below by cyclic voltammetry and discussed throughout this work.
Theoretical calculations were performed using DFT and TD-DFT (Fig. 4). For both 1-Napht-SBF and 1-Phen-SBF, HOMO/HOMO−1 and LUMO/LUMO+1 levels are approximately <0.1 eV. The HOMO of 1-Napht-SBF is spread out on a naphthyl unit and, to a lesser extent, substituted fluorene. Interestingly, HOMO−1, which is very close in energy to the HOMO, is spread out on both the naphthyl unit and its cofacial fluorene, translating the potential cofacial interaction. For 1-Napht-SBF, the TD-DFT calculations indicate that the band experimentally found at 310 nm is due to two transitions (corresponding to transitions from the ground to the second and the third excited states). In addition to the HOMO−1 → LUMO+1 transition localized on the SBF core (found in SBF), the HOMO →LUMO+1 transition occurs from the naphthyl unit to the substituted fluorene. By comparison, 1-Phenyl-SBF displays a very intense HOMO/LUMO transition8 with both molecular orbitals centred on the fluorene, showing how the substituent can significantly modify the nature of the transitions.
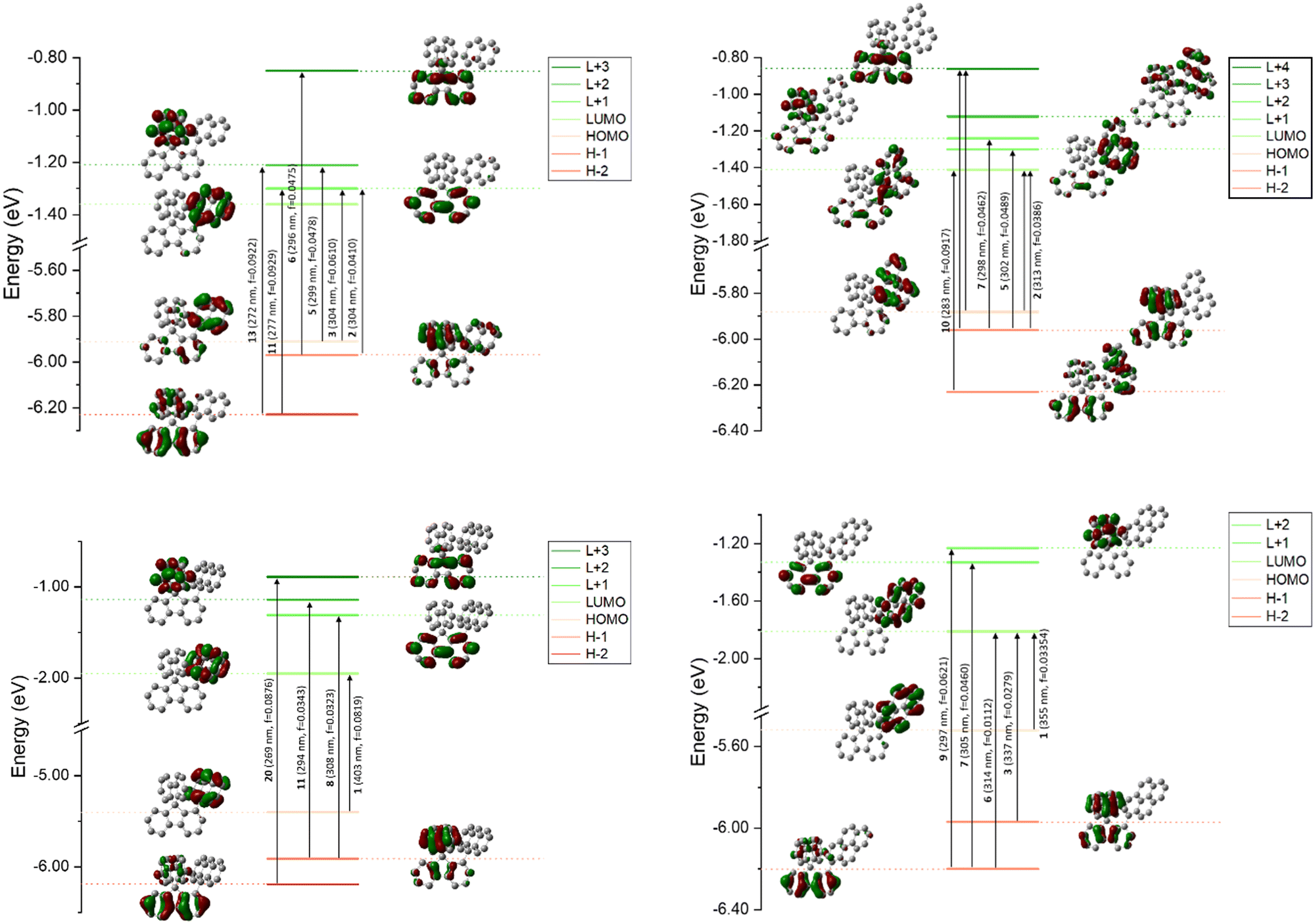 | ||
| Fig. 4 Representation of the energy levels and the molecular orbitals involved in the main electronic transitions of 1-Napht-SBF (top-left), 1-Phen-SBF (top-right), 1-Anth-SBF (bottom-left) and 1-Pyr-SBF (bottom-right) as obtained by TD-DFT, B3LYP/6-311 + G(d,p) shown with an isovalue of 0.04 (e Bohr−3)1/2. | ||
With the phenanthrene attached, the HOMO/LUMO transition of 1-Phen-SBF is detected with both the HOMO and the LUMO mainly spread out on the phenanthrene core. However, the LUMO of 1-Phen-SBF displays more density on the fluorene than the LUMO of 1-Napht-SBF whereas the opposite is observed for the HOMO. This is due to the different π-conjugation pathway observed in phenanthrene vs. naphthalene as phenanthrene can be seen as a naphthalene unit with an additional phenyl core fused in C3 and C4 positions. This results in the modification of different transitions in 1-Phen-SBF.
Both the HOMO and the LUMO of 1-Anth-SBF and 1-Pyr-SBF are exclusively spread out on the pendant substituent with no participation of the fluorenyl units. The HOMO/LUMO transitions in these two compounds fully involve the anthracene or pyrene units (a high oscillator strength of 0.082 is noted for this transition in the case of 1-Anth-SBF). Moreover, both the HOMO and LUMO energies are calculated with a large energy difference from their respective HOMO−1 and LUMO+1 orbital levels. This is a different behaviour from what was observed above for 1-Napht-SBF and 1-Phen-SBF. It should be noted that the substitution of anthracene with SBF increases both the HOMO and LUMO levels, to a lesser extent for the LUMO, which induces a red shift for 1-Anth-SBF compared to anthracene (15 nm according to the calculations in agreement with experimental data, 13 nm). Similar observations can explain the red shift of 1-Pyr-SBF compared to that of pyrene (theoretical calculations: 15 nm vs. experimental data: 12 nm).
From the onset absorption wavelength, the optical energy gaps were calculated as 3.94 eV for 1-Napht-SBF and 3.93 eV for 1-Phen-SBF; both are significantly lower than those of their corresponding building block (4.25 eV for naphthalene and 4.16 eV for phenanthrene) and both are slightly lower than that of SBF (3.97 eV). This indicates that (i) the SBF unit mainly drives the optical gap due to the short π-conjugation pathways of both naphthalene and phenanthrene and (ii) there is a small but not null impact of the substituent. The optical gaps of 1-Anth-SBF (3.10 eV) and 1-Pyr-SBF (3.47 eV) are significantly contracted and are both lower than that of SBF, due to the extended π-system of the substituent. These gaps are also contracted compared to those of their building units anthracene (3.26 eV) and pyrene (3.64 eV). The optical gaps are then measured as follows: naphthalene > phenanthrene > pyrene > anthracene (4.25, 4.16, 3.64, and 3.26 eV, respectively) showing the different π-conjugation pathways in accordance with the above-stated conclusions. Hence, for all compounds, one can note that the C1-substituted compounds display a shorter optical gap than their constituting building units with however a different magnitude. Through-space interactions between the substituent and its cofacial fluorene are at the origin of this behaviour. Thus, the optical gap is driven by the most conjugated compound and tuned by the strength of the intramolecular interactions.
The emission spectra recorded in cyclohexane are presented in Fig. 3-right. Compared to the fluorescence of their related aryl units, 1-aryl-SBFs display emission spectra red shifted and roughly less defined. Superposition of optimized geometries of the ground state (S0) and the first singlet excited state (S1) indicates, except for 1-Anth-SBF, a significant decrease (ca 20/25°) of the dihedral angle between the fluorene and its substituent (Fig. 5). Therefore, this planification provides an increase of the π-conjugation extension between the two fragments at the excited state. Thus, in fluorescence, both through-space and through-bond interactions seem to be involved. For 1-Anth-SBF, the dihedral angle remains very high and, in this case, through-space interaction seems to be involved in the red shift observed compared to anthracene fluorescence. We can finally note that the quantum yields of the substituents are not significantly affected, except in the case of pyrene, by their incorporation at C1 of the SBF.
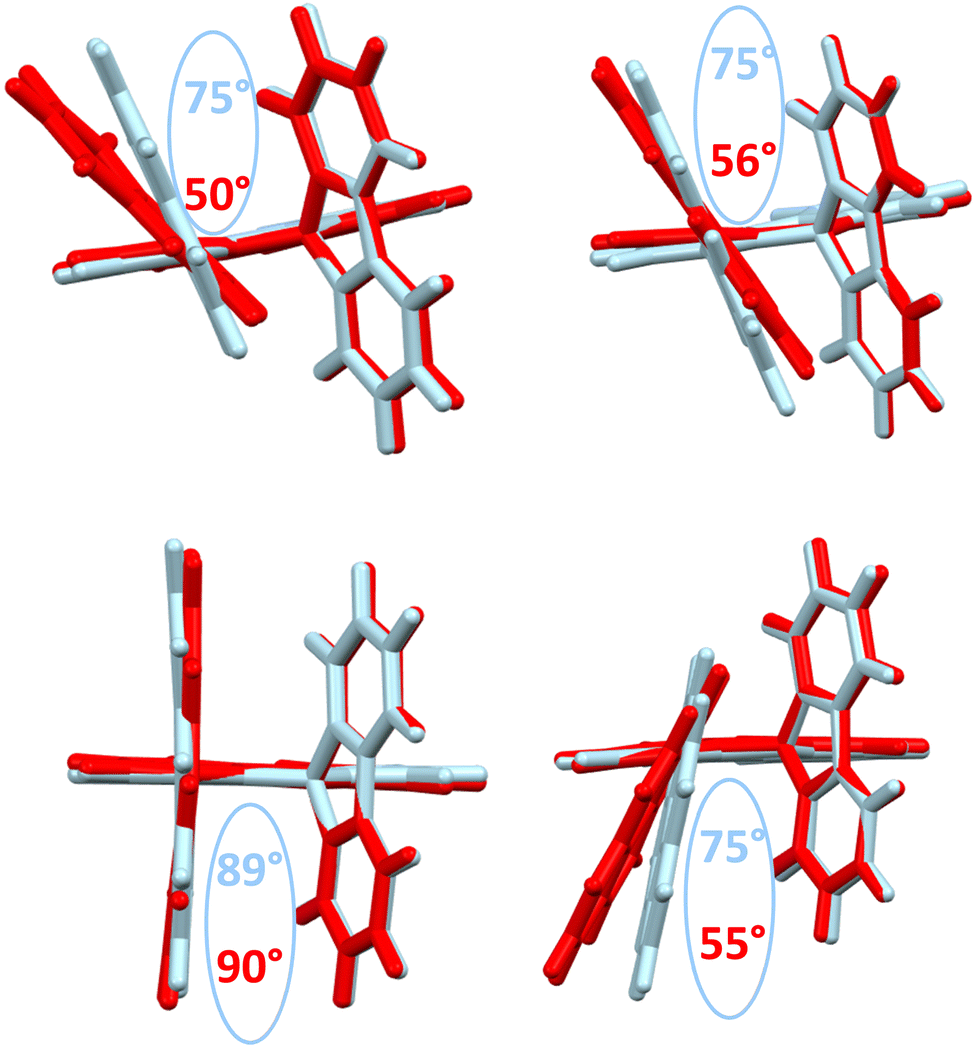 | ||
| Fig. 5 Superposition of optimized geometries: S0 (ground state, light blue) and S1 (first singlet excited state, red). Top: 1-Napht-SBF (left) and 1-Phen-SBF (right) and bottom: 1-Anth-SBF (left) and 1-Pyr-SBF (right). On each structure, the modelized dihedral angle between the substituted fluorene and the substituent in the S0 and S1 states is presented. | ||
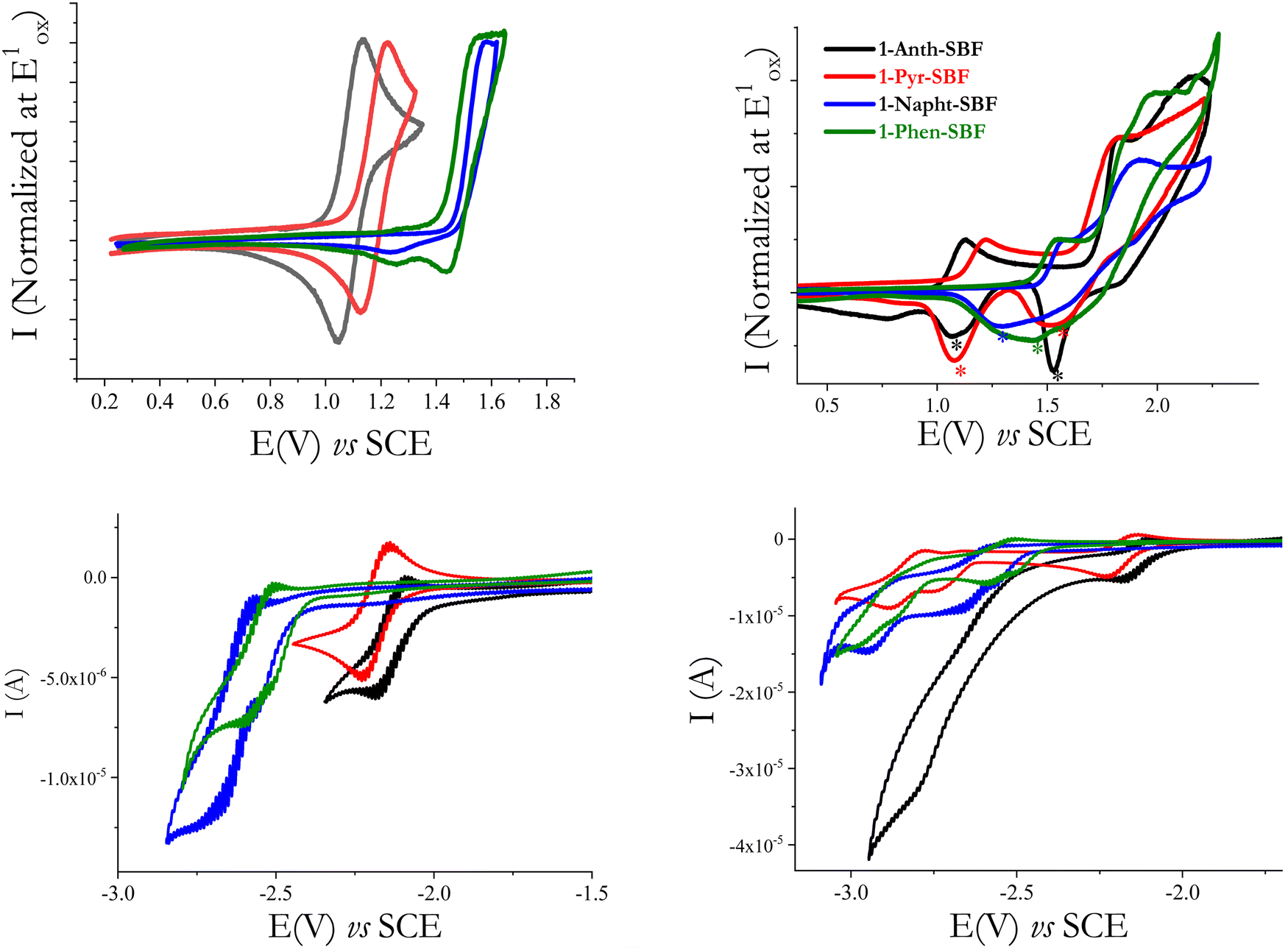 | ||
| Fig. 6 Cyclic voltammograms of 1-Napht-SBF (blue line), 1-Phen-SBF (green line), 1-Anth-SBF (black line) and 1-Pyr-SBF (red line): (top-left) up to their first oxidation wave or (top-right) between 0.5 and 2.3/2.4 V in CH2Cl2 0.2 M + Bu4NPF6 and (bottom-left) to their first reduction wave or (bottom-right) up to their highest reduction step in DMF 0.1 M + Bu4NPF6 sweep-rate: 100 mV s−1 and a platinum disk working electrode. | ||
The first oxidation wave is reversible for 1-Anth-SBF and 1-Pyr-SBF, while it is not reversible for 1-Napht-SBF and 1-Phen-SBF. When oxidized at higher potential values (Fig. 6-top-right), the four compounds present other oxidation waves and are involved in electrodeposition processes which are indicated by the appearance of new redox systems (marked with a * in Fig. 6-top right), by their growth along recurrent sweeps and by the coverage of the electrode surface by an insoluble and electroactive deposit (see for each compound the detailed study of the electrodeposition process in the ESI†). Such an electrodeposition process is classically observed with fluorene or SBF based compounds46–48 and more generally with bridged oligophenylenes (indenofluorene49 or carbazole50). The HOMO levels, determined from the onset of the first oxidation potential, decrease from −5.43/−5.52/−5.85 to −5.90 eV from 1-Anth-SBF/1-Pyr-SBF/1-Phen-SBF to 1-Napht-SBF. The same trend is observed by theoretical calculations from −5.40/−5.52/−5.88 to −5.91 eV from 1-Anth-SBF/1-Pyr-SBF/1-Phen-SBF to 1-Napht-SBF (Table 1). In order to check the exact influence of the pendant substituent, the HOMO energy levels of the constituting building blocks have also been measured and a similar trend was observed: −5.46 eV for anthracene, −5.54 eV for pyrene, −5.89 eV for phenanthrene and −5.94 eV for naphthalene. For the four compounds, the HOMO is slightly higher (by 0.02–0.04 eV) than the one of the corresponding substituents. This can be, in principle, assigned to two features, which have the same effect on the HOMO energy level. First, the electronic coupling between the fluorene and its substituent increases the conjugation length and in turn the HOMO energy. Second, the cofacial arrangement found in 1-aryl SBF induces through-space π–π interactions between the fluorene and its facing pendant substituent, which are known to decrease the oxidation potential, increasing in turn the HOMO energy.51–53 As the extension of conjugation is weak at the C1 position due to the high dihedral angle (especially in the HOMO as previously reported8,22,45), the cofacial arrangement and the resulting interactions are surely the main parameters involved in the increase of the HOMO energy. This was confirmed by plotting the electrostatic potential surfaces, which show an electron dense region between the face of the unsubstituted fluorene and the substituent (see the ESI†).
In reduction, recorded in DMF (see Fig. 6-Bottom), the first reduction wave of both 1-Napht-SBF and 1-Phen-SBF is irreversible and occurs at −2.72 V for 1-Napht-SBF and −2.60 V for 1-Phen-SBF. The two other C1-substituted SBFs, 1-Pyr-SBF and 1-Anth-SBF, are reduced in a first reversible process peaking at higher potential values, −2.22 and −2.18 V, respectively. The LUMO energy levels calculated from the onset reduction potentials decrease from −1.97 eV for 1-Napht-SBF to −2.0 eV for 1-Phen-SBF, −2.29 eV for 1-Pyr-SBF and −2.36 eV for 1-Anth-SBF following the same trend as their the respective aryl units (−1.87/−1.95/−2.31 and −2.42 eV for naphthalene/phenanthrene/pyrene and anthracene). The same conclusions as those for the HOMO can be then drawn. This is in accordance with the electron density of the LUMO, which is mainly distributed over the substituent. Thus, one can note that, compared to SBF (LUMO: −1.74 eV), the LUMO energy levels of the four C1-substituted SBFs are significantly more modified than their HOMO energy levels. Similar effects have been observed for C1-based dimers.45 The electrochemical gaps calculated from the HOMO (in DCM) and the LUMO (in DMF) following the trend of the substituent (3.93/3.85/3.23/3.07 eV for 1-Napht-SBF/1-Phen-SBF/1-Pyr-SBF/1-Anth-SBF) are in accordance with those obtained by theoretical calculations (Table 1) and confirm the above-mentioned conclusions on the impact of the substituent.
The influence of the nature of the substituent on the triplet state energy level (ET) is also of chief importance in the design of organic materials when hosting a phosphor in PhOLEDs is the goal.45 For the last 20 years, the field of host materials for PhOLEDs has been driven by this feature. Since the first report, in 2017, of a C1-substituted SBF as the host material in a blue PhOLED,8 the C1-substituted scaffold has become one of the most efficient molecular platforms to reach high performance devices.1,22,23,54
At 77 K, the phosphorescence contributions of the emission spectra of both 1-Napht-SBF and 1-Phen-SBF are similar with a first band centred at 475/470 nm leading to ET values of 2.61 and 2.64 eV, respectively (Fig. 7). The ET values are slightly lower than for their corresponding substituents, 2.65 and 2.71 eV respectively, in accordance with a triplet exciton spread out exclusively on the substituent (Fig. 7, inset). The small decrease can be assigned to the through-space interaction as detailed above. Interestingly, the ET value is significantly decreased compared to that of SBF, evaluated at ET = 2.89 eV, highlighting the significant influence of the substituent on the T1 state. This is a different result to what was reported for structurally related C1-substituted SBFs previously reported in the literature such as 1-Phenyl-SBF for which the ET is maintained very high, 2.86 eV, as a very small π-conjugated unit, i.e. the phenyl unit, is attached. Thus, the ET can be easily tuned by the grafted substituent. As the π-conjugation of the attached substituent increases, there is a drastic change in the corresponding phosphorescence contributions. Indeed, in 1-Pyr-SBF, the ET is significantly decreased to 2.07 eV, with a shift of the first emission band at 597 nm (the SDD of the triplet is exclusively centred on the pyrene unit). The ET of pyrene, measured 2.29 eV, is higher than that 1-Pyr-SBF in accordance with the above-mentioned conclusion. Finally, with anthracene grafted in 1-Anth-SBF, there is no phosphorescence contribution detected in the range as anthracene with its three fused phenyl ring provides a highly conjugated system. Radiative deactivation of the triplet state has also been measured and appears to be very slow under these experimental conditions: the phosphorescence decay was measured and the lifetimes of the T1 state were found to be 2.7 and 3.6 s for 1-Napht-SBF and 1-Phen-SBF, respectively. In 1-Pyr-SBF, the lifetime is significantly shorter, 0.6 s, highlighting the more important contribution of the pyrene fragment.
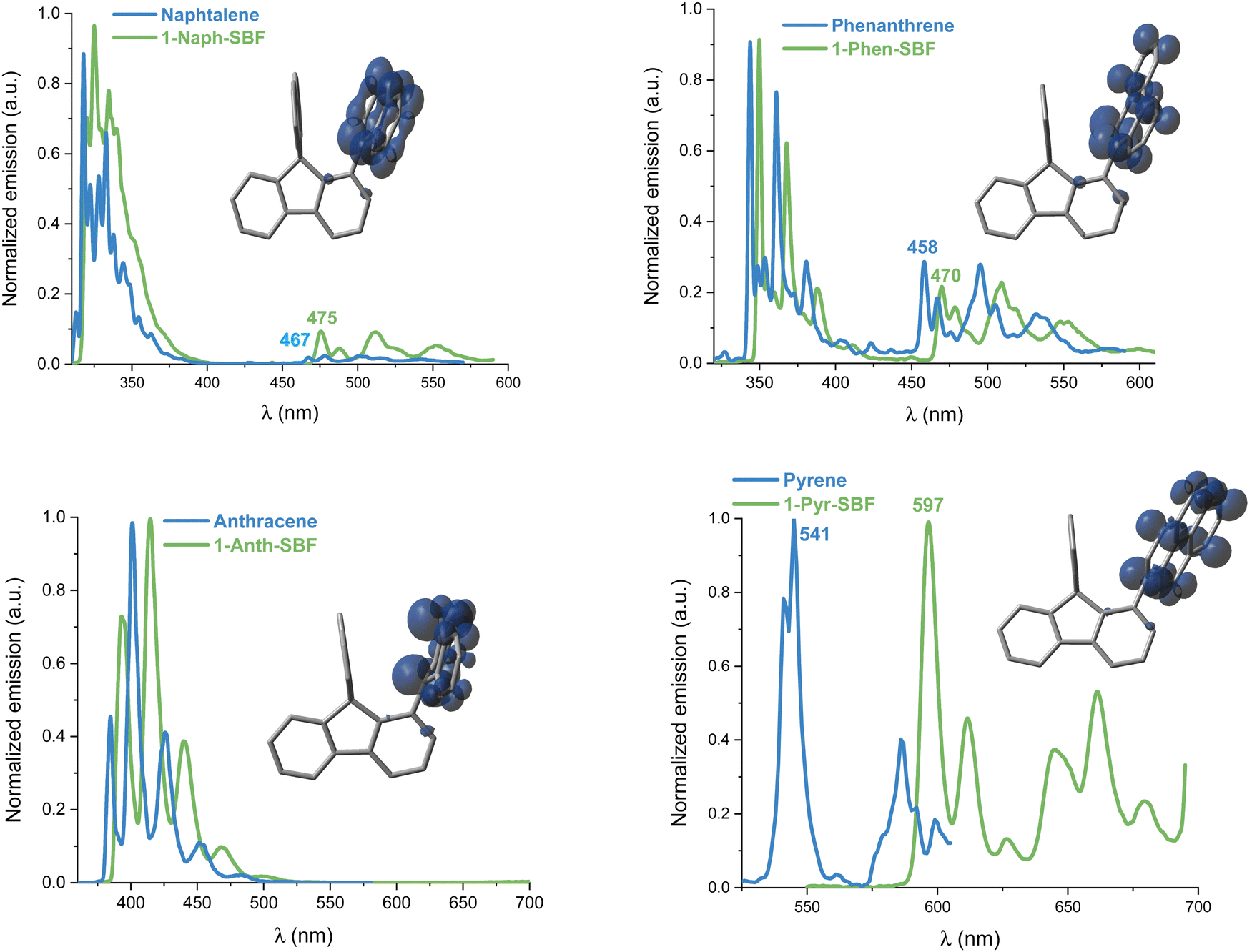 | ||
| Fig. 7 Emission spectra of 1-Napht-SBF/Naphtalene (top-left, λex = 300 nm), 1-Phen-SBF/Phenanthrene (top-right, λex = 310 nm), 1-Anth-SBF/Anthracene (bottom-left, λex = 350 nm), and 1-Pyr-SBF/Pyrene (bottom-right, λex = 310/340 nm) at 77 K (2-Me-THF). Inset: spin density distribution (SDD) of the triplet of 1-aryl-SBF (isovalue = 0.004). | ||
Superposition of optimized geometries of the ground state (S0) and the first triplet excited state (T1) shows that the high rigidity and the particular molecular arrangement of the 1-SBF scaffold strongly prevents reorganization at the first triplet excited state oppositely to what was observed for S1 (see above) (Fig. 8). For the ground state, the substituted-fluorene dihedral angle is very high maintaining the π-conjugation breaking between the two fragments (Table 1). The ET decrease of 1-SBFs compared to their building units can then not be assigned to an increase of the π-conjugation between the substituent and the fluorene but more to the cofacial arrangement.
 | ||
| Fig. 8 Superposition of optimized geometries: S0 (ground state, light blue) and T1 (first triplet excited state) blue of 1-Napht-SBF (top-left), 1-Phen-SBF (top-right), 1-Anth-SBF (bottom-left) and 1-Pyr-SBF (bottom-right). On each structure, the modelized dihedral angle between the substituted fluorene and the substituent in the S0 and T1 states is presented. | ||
Charge transport properties
Finally, as the C1 position of SBF and related fluorene compounds has been successfully used in the OLED field with very high efficiencies either in phosphorescence or in TADF,23,33 the charge transport properties were investigated using space charge limited current (SCLC) devices (see the device architecture in Fig. 9) to measure the out-of-plane hole mobilities. The whole series of C1-substituted SBF compounds exhibit a hole mobility of ca 10−6 cm2 V−1 s−1, which are intrinsically low values but in line with other reports for materials constructed on SBF scaffolds (3D shape). In fact, compared with very high performance host materials for PhOLEDs recently reported, the present mobilities are in the same range.23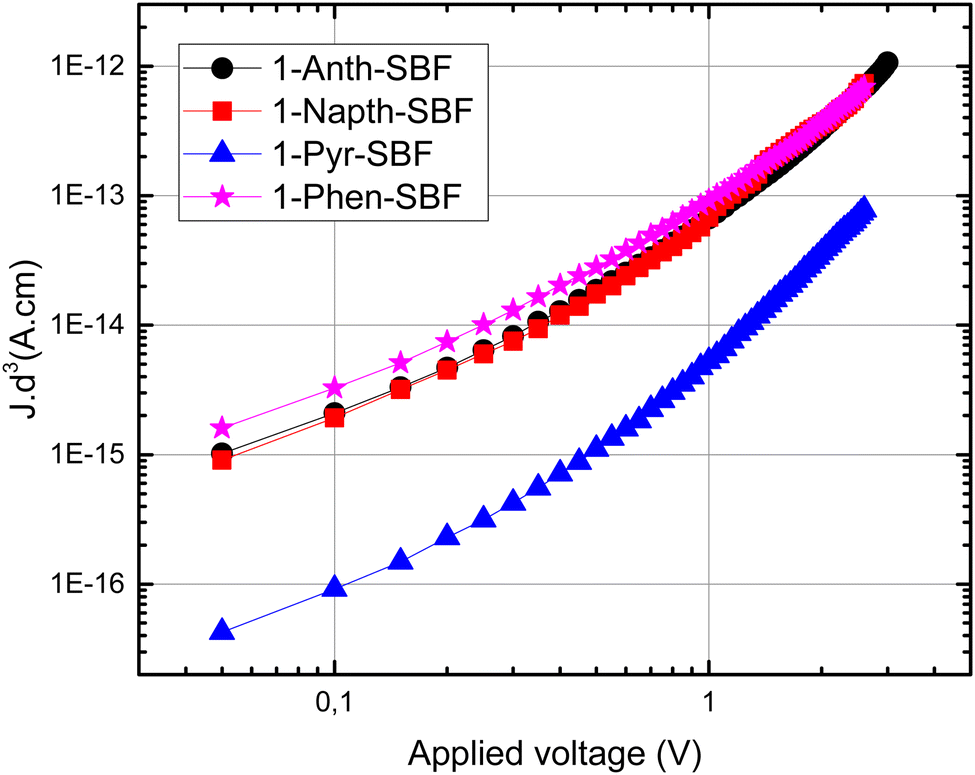 | ||
| Fig. 9 SCLC device (left): thickness-scaled current–voltage characteristics of 1-Napht-SBF, 1-Phen-SBF, 1-Anth-SBF and 1-Pyr-SBF hole-only SCLC devices (right): The dotted lines indicate the SCLC regime and the continuous ones indicate the Ohmic regime. | ||
The small differences observed within the C1-substituted SBF molecules suggest that the nature of the substituents does not significantly influence the π-overlaps between neighbouring molecules in the solid state, up to a certain level of steric hindrance. Indeed, one can nevertheless note that 1-Pyr-SBF displays a mobility of one order of magnitude lower than its analogues, despite the pyrene flat and extended π-system which can act as the π-stacking platform. This probably results from the high steric hindrance imposed by the pyrene-substituent whose conjugation plane appears to be slightly more tilted as regards to the cofacial fluorene (high slippage angle).
Conclusions
The present work reports the synthesis and the study of a series of organic semi-conductors constructed on an emerging molecular fragment, namely the C1-substituted SBF. This scaffold is nowadays one of the most efficient materials for phosphorescence and TADF OLEDs and deserves particular attention. However, this fragment has been, to date, used only with small substituents introduced at the C1 position. In the present work, extended π-conjugated systems, namely naphthalene, anthracene, phenanthrene and pyrene, were introduced and their effects on the electronic properties were investigated. We show that the substituent attached mainly drives the optical and electrochemical properties, which are nevertheless modulated by the π–π intramolecular interactions occurring with the cofacial fluorene. Thanks to the structural study based on the work of Janiak and coworkers, we demonstrate that the particular arrangement of the C1 SBF scaffold induces moderate to strong cofacial interactions with the pendant substituent attached. The impact of this cofacial interaction appears to be different as a function of the substituent and of the property investigated: HOMO/LUMO energy levels, absorption, fluorescence, and phosphorescence. As tuning intramolecular interactions is of great interest notably to control the ΔES–T in TADF OLEDs, this study shows how the fragment grafted at C1 has a key role in this interaction. This study will help to further design organic semi-conductors based on this more and more used molecular scaffold.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been financially supported by the ANR (N°22-CE07-0024-Evolution Project). CB and LS thank the ADEME (Ecoelec Project, Dr Bruno Lafitte) and the ANR (N°14-CE05-0024-Men In Blue Project) for the PhD grant. We also thank the CRMPO (Rennes) for mass analyses and GENCI (Project N°AD010814136) for computing time.References
- C. Poriel, L. Sicard and J. Rault-Berthelot, New generations of spirobifluorene regioisomers for organic electronics: tuning electronic properties with the substitution pattern, Chem. Commun., 2019, 55, 14238 RSC.
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Spiro Compounds for Organic Optoelectronics, Chem. Rev., 2007, 107, 1011 CrossRef CAS PubMed.
- Y.-K. Qu, Q. Zheng, J. Fan, L.-S. Liao and Z.-Q. Jiang, Spiro Compounds for Organic Light-Emitting Diodes, Acc. Mater. Res., 2021, 2, 1261 CrossRef CAS.
- T. Fuhrmann and J. Salbeck, Functional Molecular Glasses: Building Blocks for Future Optoelectronics, Adv. Photochem., 2002, 27, 83 CAS.
- S. Liu, D. Xia and M. Baumgarten, Rigidly-Fused Spiro-Conjugated π-Systems, ChemPlusChem, 2021, 21, 36 CrossRef PubMed.
- M. Zhu and C. Yang, Blue fluorescent emitters: design tactics and applications in organic light-emitting diodes, Chem. Soc. Rev., 2013, 42, 4963 RSC.
- L. Sicard, C. Quinton, F. Lucas, O. Jeannin, J. Rault-Berthelot and C. Poriel, 1-Carbazolyl Spirobifluorene: Synthesis, Structural, Electrochemical, and Photophysical Properties, J. Phys. Chem. C, 2019, 123, 19094 CrossRef CAS.
- L. Sicard, C. Quinton, J.-D. Peltier, D. Tondelier, B. Geffroy, U. Biapo, R. Métivier, O. Jeannin, J. Rault-Berthelot and C. Poriel, Spirobifluorene Regioisomerism: A Structure–Property Relationship Study, Chem. – Eur. J., 2017, 23, 7719 CrossRef CAS PubMed.
- Y. Luo, Z. Liu, G. Yang, T. Wang, Z. Bin, J. Lan, D. Wu and J. You, Iridium(III)-Catalyzed Diarylation/Annulation of Benzoic Acids: Facile Access to Multi-Aryl Spirobifluorenes as Pure Hydrocarbon Hosts for High-Performance OLEDs, Angew. Chem., Int. Ed., 2021, 60, 18852 CrossRef CAS PubMed.
- L.-S. Cui, S.-C. Dong, Y. Liu, M.-F. Xu, Q. Li, Z.-Q. Jiang and L.-S. Liao, meta-Linked spirobifluorene/phosphine oxide hybrids as host materials for deep blue phosphorescent organic light-emitting diodes, Org. Electron., 2013, 14, 1924 Search PubMed.
- L.-S. Cui, Y.-M. Xie, Y.-K. Wang, C. Zhong, Y.-L. Deng, X.-Y. Liu, Z.-Q. Jiang and L.-S. Liao, Pure Hydrocarbon Hosts for ≈100% Exciton Harvesting in Both Phosphorescent and Fluorescent Light-Emitting Devices, Adv. Mater., 2015, 27, 4213 Search PubMed.
- J.-J. Liang, Y. Li, Y. Yuan, S.-H. Li, X.-D. Zhu, S. Barlow, M.-K. Fung, Z.-Q. Jiang, S. R. Marder and L.-S. Liao, A blue thermally activated delayed fluorescence emitter developed by appending a fluorene moiety to a carbazole donor with meta-linkage for high-efficiency OLEDs, Mater. Chem. Front., 2018, 2, 917 RSC.
- A. Eckstein-Andicsová, Z. Tokárová, E. Kozma, R. Balogh, A. Vykydalová, W. Mróz and K. Tokár, Thiazolo[5,4-d]thiazoles with a spirobifluorene moiety as novel D–π–A type organic hosts: design, synthesis, structure–property relationship and applications in electroluminescent devices, New J. Chem., 2023, 47, 11165–11175 Search PubMed.
- C. Du, T. Lu, Z. Cheng, Y. Chang, H. Liu, J. Wang, L. Wan, Y. Lv and P. Lu, Rational molecular design of phenanthroimidazole-based fluorescent materials towards high-efficiency non-doped deep blue OLEDs, J. Mater. Chem. C, 2022, 10, 14186 Search PubMed.
- C. Zhang, Y. Lu, Z. Liu, Y. Zhang, X. Wang, D. Zhang and L. Duan, A π–D and π–A Exciplex-Forming Host for High-Efficiency and Long-Lifetime Single-Emissive-Layer Fluorescent White Organic Light-Emitting Diodes, Adv. Mater., 2020, 32, 2004040 CrossRef CAS PubMed.
- X.-Y. Liu, F. Liang, L. Ding, S.-C. Dong, Q. Li, L.-S. Cui, Z.-Q. Jiang, H. Chen and L.-S. Liao, The study on two kinds of spiro systems for improving the performance of host materials in blue phosphorescent organic light-emitting diodes, J. Mater. Chem. C, 2015, 3, 9053 RSC.
- S. Thiery, C. Declairieux, D. Tondelier, G. Seo, B. Geffroy, O. Jeannin, R. Métivier, J. Rault-Berthelot and C. Poriel, 2-Substituted vs. 4-substituted-9,9′-spirobifluorene host materials for green and blue phosphorescent OLEDs: a structure–property relationship study, Tetrahedron, 2014, 70, 6337 CrossRef CAS.
- C. Quinton, S. Thiery, O. Jeannin, D. Tondelier, B. Geffroy, E. Jacques, J. Rault-Berthelot and C. Poriel, Electron-Rich 4-Substituted Spirobifluorenes: Toward a New Family of High Triplet Energy Host Materials for High-Efficiency Green and Sky Blue Phosphorescent OLEDs, ACS Appl. Mater. Interfaces, 2017, 9, 6194 CrossRef CAS PubMed.
- Z.-Y. Wang, J.-W. Zhao, B. Liu, C. Cao, P. Li, Q.-X. Tong and S.-L. Tao, Universal materials for high performance violet-blue OLEDs (CIEy < 0.06) and PhOLEDs, Dyes Pigm., 2019, 163, 213 CrossRef CAS.
- G. Tian, Y. Jiang, P. Wu, J. Huang, Q. Zou, Q. Wang, H. Mu and J. Su, Pure hydrocarbon host materials based on spirofluorene with excellent performances for green phosphorescent light-emitting devices, New J. Chem., 2016, 40, 9500 RSC.
- C. Fan, Y. Chen, P. Gan, C. Yang, C. Zhong, J. Qin and D. Ma, Tri-, Tetra- and Pentamers of 9,9′-Spirobifluorenes through Full ortho-Linkage: High Triplet-Energy Pure Hydrocarbon Host for Blue Phosphorescent Emitter, Org. Lett., 2010, 12, 5648 CrossRef CAS PubMed.
- L. J. Sicard, H.-C. Li, Q. Wang, X.-Y. Liu, O. Jeannin, J. Rault-Berthelot, L.-S. Liao, Z.-Q. Jiang and C. Poriel, C1-Linked Spirobifluorene Dimers: Pure Hydrocarbon Hosts for High-Performance Blue Phosphorescent OLEDs, Angew. Chem., Int. Ed., 2019, 58, 3848 CrossRef CAS PubMed.
- F. C. Kong, Y. L. Zhang, C. Quinton, N. McIntosh, S. Y. Yang, J. Rault-Berthelot, F. Lucas, C. Brouillac, O. Jeannin, J. Cornil, Z. Q. Jiang, L. S. Liao and C. Poriel, Pure Hydrocarbon Materials as Highly Efficient Host for White Phosphorescent Organic Light-Emitting Diodes: A New Molecular Design Approach, Angew. Chem., Int. Ed., 2022, 61, e202207204 Search PubMed.
- Y. Wang, J. H. Yun, L. Wang and J. Y. Lee, High Triplet Energy Hosts for Blue Organic Light-Emitting Diodes, Adv. Funct. Mater., 2020, 31, 2008332 CrossRef.
- H. Zhang, Q. Guo, H. Cheng, C. Ran, D. Wu and J. Lan, An umpolung strategy for rapid access to thermally activated delayed fluorescence (TADF) materials based on phenazine, Chem. Commun., 2022, 58, 1581 RSC.
- X. Lv, Y. Wang, N. Li, X. Cao, G. Xie, H. Huang, C. Zhong, L. Wang and C. Yang, Regulating the photophysical properties of highly twisted TADF emitters by concurrent through-space/-bond charge transfer, Chem. Eng. J., 2020, 402, 126173 Search PubMed.
- X. Lv, W. Zhang, D. Ding, C. Han, Z. Huang, S. Xiang, Q. Zhang, H. Xu and L. Wang, Integrating the Emitter and Host Characteristics of Donor–Acceptor Systems through Edge-Spiro Effect Toward 100% Exciton Harvesting in Blue and White Fluorescence Diodes, Adv. Opt. Mater., 2018, 6, 1800165 CrossRef.
- B. Kobin, J. Schwarz, B. Braun-Cula, M. Eyer, A. Zykov, S. Kowarik, S. Blumstengel and S. Hecht, Spiro-Bridged Ladder-Type Oligo(para-phenylene)s: Fine Tuning Solid State Structure and Optical Properties, Adv. Funct. Mater., 2017, 27, 1704077 CrossRef.
- H. Lee, H. Jung, S. Kang, J. H. Heo, S. H. Im and J. Park, Three-Dimensional Structures Based on the Fusion of Chrysene and Spirobifluorene Chromophores for the Development of Blue OLEDs- high performance in non doped blue oled, J. Org. Chem., 2018, 83, 2640 CrossRef CAS PubMed.
- D. Xia, X. Guo, M. Wagner, M. Baumgarten, D. Schollmeyer and K. Müllen, Cruciform Electron Acceptors Based on Tetraindeno-Fused Spirofluorene, Cryst. Growth Des., 2017, 17, 2816 Search PubMed.
- D. Xia, D. Gehrig, X. Guo, M. Baumgarten, F. Laquai and K. Mullen, A spiro-bifluorene based 3D electron acceptor with dicyanovinylene substitution for solution-processed non-fullerene organic solar cells, J. Mater. Chem. A, 2015, 3, 11086 Search PubMed.
- C. C. Peng, S. Y. Yang, H. C. Li, G. H. Xie, L. S. Cui, S. N. Zou, C. Poriel, Z. Q. Jiang and L. S. Liao, Highly Efficient Thermally Activated Delayed Fluorescence via an Unconjugated Donor-Acceptor System Realizing EQE of Over 30%, Adv. Mater., 2020, 32, 2003885 CrossRef CAS PubMed.
- S. Y. Yang, Z. Q. Feng, Z. Fu, K. Zhang, S. Chen, Y. J. Yu, B. Zou, K. Wang, L. S. Liao and Z. Q. Jiang, Highly Efficient Sky-Blue pi-Stacked Thermally Activated Delayed Fluorescence Emitter with Multi-Stimulus Response Properties, Angew. Chem., Int. Ed., 2022, e202206861 CAS.
- S.-Y. Yang, Q.-S. Tian, Y.-J. Yu, S.-N. Zou, H.-C. Li, A. Khan, Q.-H. Wu, Z.-Q. Jiang and L.-S. Liao, Sky-Blue Thermally Activated Delayed Fluorescence with Intramolecular Spatial Charge Transfer Based on a Dibenzothiophene Sulfone Emitter, J. Org. Chem., 2020, 85, 10628 CrossRef CAS PubMed.
- S.-Y. Yang, Y.-K. Wang, C.-C. Peng, Z.-G. Wu, S. Yuan, Y.-J. Yu, H. Li, T.-T. Wang, H.-C. Li, Y.-X. Zheng, Z.-Q. Jiang and L.-S. Liao, Circularly Polarized Thermally Activated Delayed Fluorescence Emitters in Through-Space Charge Transfer on Asymmetric Spiro Skeletons, J. Am. Chem. Soc., 2020, 142, 17756 CrossRef CAS PubMed.
- S.-Y. Yang, Y.-K. Qu, L.-S. Liao, Z.-Q. Jiang and S.-T. Lee, Research Progress of Intramolecular π-Stacked Small Molecules for Device Applications, Adv. Mater., 2022, 34, 2104125 Search PubMed.
- X.-Y. Chen, S. Ozturk and E. J. Sorensen, Synthesis of Fluorenones from Benzaldehydes and Aryl Iodides: Dual C–H Functionalizations Using a Transient Directing Group, Org. Lett., 2017, 19, 1140 Search PubMed.
- A. Bondi, Rayon de VDW Bible a utiliser, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- X.-J. Yang, F. Drepper, B. Wu, W.-H. Sun, W. Haehnel and C. Janiak, From model compounds to protein binding: syntheses, characterizations and fluorescence studies of [RuII(bipy)(terpy)L]2+ complexes (bipy = 2,2′-bipyridine; terpy = 2,2′:6′,2′′-terpyridine; L = imidazole, pyrazole and derivatives, cytochrome c), Dalton Trans., 2005, 256 RSC.
- E. Craven, C. Zhang, C. Janiak, G. Rheinwald and H. Lang, Z. Anorg. Allg. Chem., 2003, 629, 2282 CrossRef CAS.
- C. Janiak, A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- S. Banerjee, A. Ghosh, B. Wu, P.-G. Lassahn and C. Janiak, Polymethylene spacer regulated structural divergence in cadmium complexes: Unusual trigonal prismatic and severely distorted octahedral coordination, Polyhedron, 2005, 24, 593 CrossRef CAS.
- T. Dorn, C. Janiak and K. Abu-Shandi, CrystEngComm, 2005, 7, 633 Search PubMed.
- J. Delaire, J. Piard, R. Méallet-Renault and G. Clavier, Photophysique et photochimie: Des fondements aux applications, EDP Sciences, 2017 Search PubMed.
- C. Poriel, C. Quinton, F. Lucas, J. Rault-Berthelot, Z. Q. Jiang and O. Jeannin, Spirobifluorene Dimers: Understanding How The Molecular Assemblies Drive The Electronic Properties, Adv. Funct. Mater., 2021, 2104980 CrossRef CAS.
- C. Poriel, Y. Ferrand, P. Le Maux, J. Rault-Berthelot and G. Simonneaux, Organic Cross-Linked Electropolymers as Supported Oxidation Catalysts:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Poly((tetrakis(9,9′-spirobifluorenyl)porphyrin)manganese) Films, Inorg. Chem., 2004, 43, 5086 CrossRef CAS PubMed.
Poly((tetrakis(9,9′-spirobifluorenyl)porphyrin)manganese) Films, Inorg. Chem., 2004, 43, 5086 CrossRef CAS PubMed. - C. Poriel, Y. Ferrand, P. Le Maux, C. Paul-Roth, G. Simonneaux and J. Rault-Berthelot, Anodic oxidation and physicochemical properties of various porphyrin-fluorenes or -spirobifluorenes: Synthesis of new polymers for heterogeneous catalytic reactions, J. Electroanal. Chem., 2005, 583, 92 Search PubMed.
- C. Poriel, Y. Ferrand, P. Le Maux, J. Rault-Berthelot and G. Simonneaux, Syntheses of manganese and iron tetraspirobifluorene porphyrins as new catalysts for oxidation of alkenes by hydrogen peroxide and iodosylbenzene, Tetrahedron Lett., 2003, 44, 1759 CrossRef CAS.
- J. Rault-Berthelot, C. Poriel, F. Justaud and F. Barrière, Anodic oxidation of indenofluorene. Electrodeposition of electroactive poly(indenofluorene), New J. Chem., 2008, 32, 1259 RSC.
- F. Lucas, L. Sicard, O. Jeannin, J. Rault-Berthelot, E. Jacques, C. Quinton and C. Poriel, [4]Cyclo-N-ethyl-2,7-carbazole: Synthesis, Structural, Electronic and Charge Transport Properties, Chem. – Eur. J., 2019, 25, 7740 CrossRef CAS PubMed.
- D. Thirion, C. Poriel, F. Barrière, R. Métivier, O. Jeannin and J. Rault-Berthelot, Tuning the Optical Properties of Aryl-Substituted Dispirofluorene-Indenofluorene Isomers through Intramolecular Excimer Formation, Org. Lett., 2009, 11, 4794 CrossRef CAS PubMed.
- R. Rathore, S. H. Abdelwahed and I. A. Guzei, Synthesis, structure, and evaluation of the effect of multiple stacking on the electron-donor properties of π-stacked polyfluorenes, J. Am. Chem. Soc., 2003, 125, 8712 CrossRef CAS PubMed.
- V. J. Chebny, R. Shukla, S. V. Lindeman and R. Rathore, Molecular Actuator: Redox-Controlled Clam-Like Motion in a Bichromophoric Electron Donor, Org. Lett., 2009, 11, 1939 CrossRef CAS PubMed.
- C. Poriel, J. Rault-Berthelot and Z.-Q. Jiang, Are pure hydrocarbons the future of host materials for blue phosphorescent organic light-emitting diodes?, Mater. Chem. Front., 2022, 6, 1246 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2297372–2297374, 2297377. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qm01106b |
| This journal is © the Partner Organisations 2024 |