
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bio-based palladium catalyst in cryogel for cross-coupling reactions†
Elisabetta Grazia
Tomarchio
ab,
Chiara
Zagni
 *a,
Vincenzo
Paratore
c,
Guglielmo Guido
Condorelli
c,
Sabrina Carola
Carroccio
d and
Antonio
Rescifina
a
*a,
Vincenzo
Paratore
c,
Guglielmo Guido
Condorelli
c,
Sabrina Carola
Carroccio
d and
Antonio
Rescifina
a
aDepartment of Drug and Health Sciences, University of Catania, V.le A. Doria 6, 95125, Catania, Italy. E-mail: chiara.zagni@unict.it
bDepartment of Biomedical and Biotechnological Sciences, University of Catania, Via Santa Sofia 97, 95123 Catania, Italy
cDepartment of Chemical Science, Università degli Studi di Catania, Viale Andrea Doria 6, 95125, Catania, Italy
dInstitute for Polymers, Composites, and Biomaterials CNR-IPCB, Via Paolo Gaifami 18, 95126, Catania, Italy
First published on 26th September 2024
Abstract
Biobased catalysts play a crucial role in sustainable chemistry, using natural resources to support eco-friendly processes. While palladium catalysts are essential for various industrial applications, they often pose environmental challenges due to their non-reusability and tendency to degrade. To address these issues, we developed an innovative phenylalanine-based catalyst containing palladium (C-PhebPd) designed for the Suzuki–Miyaura reaction. The natural amino acids, used as monomers, chelate palladium, preventing leaching, unlike other heterogeneous catalysts that use palladium nanoparticles, which can be released over time, leading to catalyst degradation. Such catalyst exhibits outstanding performance in aqueous media at moderate temperatures, facilitating cross-coupling reactions between various aryl halides and arylboronic acids with high yields of up to 99%. The affordable synthetic procedure and C-PhebPd's stability make it potentially scalable for industrial applications. The robustness of this catalyst was also proved by recyclability tests up to seven cycles. Further investigation into its capabilities could unlock additional insights for various catalytic transformations.
Introduction
The Suzuki–Miyaura cross-coupling reaction of arylboronic acids and aryl halides, catalyzed by palladium complexes, is a precious method in organic synthesis for creating biaryls by forming new carbon–carbon bonds.1 This reaction is notable for its mild conditions, which can accommodate various functional groups,2–4 making it especially useful for the convergent syntheses of pharmaceutical intermediates and fine chemicals. In pursuit of enhanced reactivity and selectivity in this cross-coupling reaction, researchers have explored various homogeneous catalytic systems featuring diverse ligands or ligand-less approaches.5 Recent advancements in homogeneous Pd catalysts have revolutionized catalytic processes by significantly reducing catalyst loading. Homogeneous Pd catalysts have been extensively explored to enhance reactivity and selectivity, ranging from simple Pd(0) complexes, like Pd acetate and Pd tetrakis, to more complex forms, such as Pd precatalysts with phosphine ligands or fully formed (pre)catalysts, often formulated as air-stable complexes, offering enhanced convenience for bench chemists during handling and experimentation.6 Nonetheless, the environmental impact of homogeneous catalysts is significant due to challenges in separation processes and the potential contamination of products from palladium leaching.7Efficient separation and recycling of catalyst systems remain ongoing challenges crucial for economic and ecological reasons. From a pharmaceutical perspective, ease of product isolation and minimizing residual impurities are essential factors. Moreover, homogeneous catalysts often rely on harmful organic solvents for optimal reactivity. This leads to diminished performance in the “Do Not Significantly Harm” (DNSH) principle and recycling difficulties due to palladium leaching.8
Given the challenges associated with homogeneous catalytic systems, such as palladium leaching and the reliance on toxic organic solvents, there is significant interest in developing efficient heterogeneous catalysts.9,10 These catalysts are actively being researched for their potential to offer robust reusability and enhance environmental sustainability.11 The goal is to create cleaner and more efficient catalytic systems that eliminate the need for harmful solvents and improve overall environmental impact. For this purpose, several heterogeneous catalysts have been developed, which show prominent advantages such as ease of handling, recyclability, and broad substrate scope.12 Most of the previous heterogeneous catalysis involving palladium metal is transitioning into nanoparticle exploitation.13–17 However, significant drawbacks constrain a wide application due to their high costs, elevated temperatures and pressure required for the process, generation of numerous side products, and management of the catalyst in leaching and recovery. In recent decades, Suzuki–Miyaura coupling reactions carried out on supported Pd nanoparticles provided outstanding ability in converting the reactants. Various porous materials like silica-supported salts,18–20 zeolites,21 carbon-based materials,22 and porous organic polymers (POPs)23 have been utilized as efficient supports.24,25 However, mechanistic studies have shown that using supported Pd nanoparticles as catalysts causes the release of Pd atoms from the catalyst surface during the oxidative addition of Pd(0) species with aryl halides. This leads to the formation of soluble Pd(II) complexes that promote the reaction through a (quasi)homogeneous mechanism. Therefore, preventing catalytic deactivation and product contamination from Pd leaching is difficult.26 Unlike supported Pd nanoparticles, anchoring Pd complexes onto solid supports via well-defined coordination sites is advantageous for producing highly dispersed and stabilized Pd species. This approach helps preserve catalytic stability throughout the reaction and mitigates leaching phenomena. Therefore, in light of these considerations and given a transition towards greener chemistry practices, we synthesized a novel macroporous biopolymeric material derived from phenylalanine complexed with palladium ions as a heterogeneous catalyst. The new material was synthesized by radical polymerization at subzero temperatures in the presence of water.27,28 This technique allows the production of a sponge-like material with high porosity, stability, and good absorption capacity.29 Within the pores of these cryogels, organic molecules become concentrated, facilitating efficient reactions. This innovative approach, which utilizes a Palladium complex, offers promising applications in various catalysis fields and provides several benefits, including affordable natural feedstocks, high product yields, short reaction times, excellent atom economy, and recyclability.
Experimental
Materials
The starting materials L-phenylalanine, 4-vinylbenzyl chloride, 2-hydroxyethyl methacrylate (HEMA), N,N′-methylenebisacrylamide (MBAA), palladium(II) acetate, tetramethyl-ethylene-diamine (TEMED), ammonium persulfate (APS), absolute ethanol (EtOH), deuterated water (D2O), deuterated sodium hydroxide (NaOD), dimethyl sulfoxide-d6 (DMSO-d6) were purchased from Sigma-Aldrich. A Milli-Q water purification system produced deionized water. Precoated aluminum sheets (silica gel 60 F254, Merck) were used for thin-layer chromatography (TLC). All NMR experiments, including 2D spectra (g-COSY, g-HSQCAD, and g-HMBCAD), were software supplied by the manufacturer and acquired at 300 K on Varian UNITY Inova at 500 MHz.Synthetic procedure
1H NMR (500 MHz, DMSO-d6) δ 7.35 (d, J = 7.8 Hz, 2H), 7.20 (ddd, J = 22.4, 14.9, 7.9 Hz, 7H), 6.68 (dd, J = 17.6, 10.9 Hz, 1H), 5.77 (d, J = 17.7 Hz, 1H), 5.20 (d, J = 11.0 Hz, 1H), 3.73 (d, J = 13.7 Hz, 1H), 3.55 (d, J = 13.7 Hz, 1H), 3.22 (d, J = 6.6 Hz, 1H), 2.92 (dd, J = 13.6, 5.9 Hz, 1H), 2.80 (dd, J = 13.6, 7.4 Hz, 1H).
13C-NMR (126 MHz, DMSO-d6): δ = 174.67, 139.63, 138.73, 136.95, 129.81, 128.87, 128.51, 126.47, 114,37, 62.59, 50.86, 40.08.
1H NMR (500 MHz, DMSO-d6) δ 7.55–7.41 (m, 4H), 7.28–7.22 (m, 5H), 6.75 (dd, J = 17.6, 10.9 Hz, 1H), 6.12 (m, J = 4.7 Hz, 1H), 5.88 (d, J = 17.7 Hz, 1H), 5.31 (d, J = 11.1 Hz, 1H), 3.79 (dd, J = 13.4, 5.0 Hz, 1H), 3.47 (dd, J = 13.4, 3.5 Hz, 1H), 3.29 (d, J = 6.0 Hz, 1H), 3.14 (dd, J = 14.2, 6.0 Hz, 1H), 3.02 (dd, J = 14.2, 6.4 Hz, 1H). 13C-NMR (125 MHz, DMSO-d6): δ = 180.01, 137.33, 137.02, 136.14, 133.64, 133.64, 130.73, 129.40, 128.33, 126.59, 126.25, 114.87, 63.56, 53.54, 37.71.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio) was dissolved in water. The crosslinker agent MBAA was added at a monomers/crosslinker molar ratio of 6:1 and mixed until complete dissolution. The mixture was then cooled, and the water content was adjusted to achieve a total concentration of polymerizable compounds of 10% w/v.32
:1 weight ratio) was dissolved in water. The crosslinker agent MBAA was added at a monomers/crosslinker molar ratio of 6:1 and mixed until complete dissolution. The mixture was then cooled, and the water content was adjusted to achieve a total concentration of polymerizable compounds of 10% w/v.32
Subsequently, 1.5% v/v of 10% APS and TEMED ready solutions were added to the mixture. After stirring for 1 minute, the solution was transferred into a 10 mL capped syringe and placed in a cryostatic bath at −15 °C for 24 hours. The resulting cryogel was then thawed, rinsed with water and ethanol, and dried, yielding an 84% polymerization efficiency.
Catalyst characterization
Transmission FT-IR measurements of samples in KBr pellets were recorded using a JASCO FTIR 4600LE spectrometer (Easton, MD, USA) in the spectral range 560–4000 cm−1 (resolution 4 cm−1). All the samples were characterized by preparing a KBr pill with a ratio of 1:100 (i.e., sample: KBr).
The catalyst's thermal stability was assessed using a PerkinElmer thermogravimetric apparatus under a nitrogen atmosphere (flow rate 60 mL min−1), employing a heating rate of 10 °C min−1 from 90 °C to 700 °C. The TGA sensitivity was 0.1 μg, with a weighting precision of ± 0.01%.
The macroporous morphology of the synthesized material was confirmed by scanning electron microscopy (SEM) with a Phenomenex microscope. Before testing, samples were coated with gold to enhance conductivity. Images were captured to assess the cryogel morphology, with data acquisition and processing conducted using Phenom Porometric 1.1.2.0 software by Phenom-World BV, Eindhoven, The Netherlands. Furthermore, the elemental composition of the catalyst was measured by energy-dispersive X-ray spectroscopy (EDX).
Swelling tests were carried out on dried bare cryogels, measuring mass before and after water uptake. Total uptake was determined by observing cumulative mass increase over fixed intervals.33 Equilibrium swelling was assessed by weighing wet samples immersed for 30 minutes. Adsorption kinetics were analyzed by exposing samples to excess water for specific durations and rapidly removing unabsorbed water. The adsorbed water was quantified by weighing samples over time and normalizing the data. Three parallel samples were tested, and the average result was computed. The standard deviation was found to be less than 5%.
X-ray photoelectron spectroscopy (XPS) was carried out with a PHI 5000 Versa Probe Instrument (Chanhassen, MN, USA) using a monochromatic Al Kα X-ray source excited with a micro-focused electron beam. All the analyses were performed with a photoelectron take-off angle of 45° (relative to the sample Surface). The XPS binding energy (B.E.) scale was calibrated on the C1s peak of adventitious carbon at 285.0 eV.
General procedure of Suzuki coupling reactions
In an appropriate volume vial, aromatic halide (0.3 mmol), phenylboronic acid (0.36 mmol), alkali (0.6 mmol), solvent (2 mL), and wet cryogel catalyst (9 mg) were mixed. The reaction mixture was stirred at 80 °C until consumption of the starting material, monitored by TLC. The organic layer was extracted with DCM, dried with anhydrous Na2SO4, and concentrated under reduced pressure. The product was purified by silica column chromatography with cyclohexane/ethyl acetate as the eluent when necessary.Reusability test
To assess the durability and reusability of the produced catalyst, the reaction mixture was separated from the catalyst. The catalyst itself was washed and employed for other Suzuki reactions.Heterogeneity test for C-PhebPd catalyst
The Sheldon's test was carried out to ensure the heterogeneous nature of the synthesized material and whether any Pd species leached out in the filtrate solution. Reactions between iodobenzene and phenylboronic acid were initiated in the presence of the catalyst at 80 °C for varying durations: 5, 10, 15, and 20 minutes. Subsequently, the catalyst was removed, and the reaction was allowed for the remaining time to ensure a total reaction time of 30 minutes. The resulting products were quantitatively analyzed.ICP/MS
Metal ions in solution were quantitatively analyzed using inductively coupled plasma–mass spectrometry (ICP/MS). The Nexion 300X, a device developed by PerkinElmer Inc. (Waltham, Massachusetts, USA), was used, incorporating kinetic energy discrimination (KED) to minimize interference. The accuracy of the analytical procedure was verified by testing a standard reference material, NIST 1640a (trace elements in natural water), with no significant deviations observed. Batch equilibrium experiments were conducted to determine the percentage of metal ion removal.34,35A sample of 5 mg of C-PhebPd was poured into a vial containing 5 ml of water to determine the possible Pd leaching. After 24 h, 100 μL of the sample was taken for analysis to allow it to reach the equilibrium.36
Results and discussion
Synthesis of the catalyst
Macroporous materials are highly suitable for catalytic support due to their characteristic structure, allowing for intimate molecular contact and confined reaction spaces.37 Their versatility is further advantaged by rapid mass diffusion properties and the ability to accommodate volume changes, making them valuable tools in various catalytic applications.38
L-Phenylalanine (L-Phe) contains a nonpolar phenyl group and is a prevalent aromatic amino acid in living organisms.39 Owing to their oxygen and nitrogen atoms, two L-Phe molecules can interact with palladium, forming a stable complex.40 In the pursuit of synthesizing biobased materials, L-Phe underwent functionalization through a reaction with 4-vinylbenzyl chloride in the presence of a base at room temperature (Fig. 1). Initially, the reaction was carried out in methanol, which produced the desired product within two days but also led to the formation of side products. To overcome this issue, the solvent was switched to water. While this change extended the reaction time to three days, it enabled the isolation of pure compound (Pheb) through straightforward filtration. Additionally, this approach aligns with the DNSH principle, providing further environmental advantages. The obtained monomer was mixed with palladium acetate in a 1:1 water/acetone mixture to form the final complex PhebPd 4.
 | ||
| Fig. 1 (a) Synthetic route of monomer PhebPd. (b) Illustration of PhebPd structure. | ||
PhebPd 4 underwent radical polymerization in the presence of MBAA at subzero temperature, achieving a grey-coloured macroporous biopolymer-based PhebPd cryogel (Fig. 2).28 The hydrophilic material obtained possesses high swelling and absorption capacity, providing an ideal space to confine organic molecules that favour interactions after solution uptake. Consequently, it is reasonable to suppose this could facilitate intimate contact between the metal and reactants, enhancing the catalytic efficiency.41
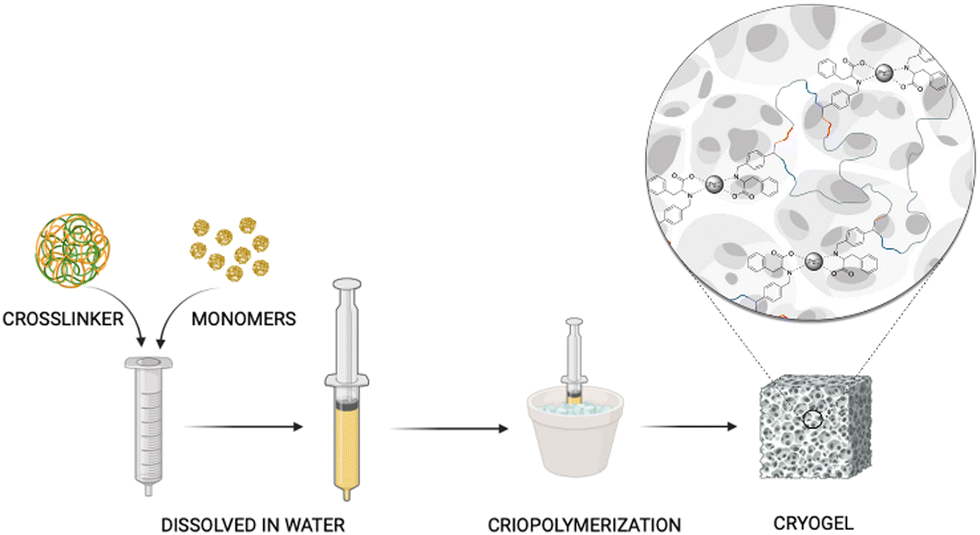 | ||
| Fig. 2 Schematic representation of cryogel preparation. | ||
Characterization of the catalyst
The monomer and complex were characterized using a combination of 1H and 13C NMR analyses and two-dimensional techniques such as COSY, HSQC, and HMBC. These methods were employed to confirm the formation of compound 3 (Pheb) and the Pd-complex 4 (PhebPd) (Fig. S1–S8, ESI†). The successful synthesis of complex 4 was corroborated by the observed shifts in proton signals in the 1H NMR spectrum. In monomer 3, benzylic hydrogens showed peaks at 2.79–2.92 ppm, and signals from the –CH2 group linked to the styrenyl moiety appeared at 3.57–3.72 ppm. Upon formation of complex 4, these peaks shifted to 3.02–3.14 and 3.47–3.79 ppm, respectively. The 13C NMR spectra also displayed a notable shift, particularly the carbonyl signal, which shifted from 174 ppm to 180 ppm upon complex formation.FTIR spectra of monomer and polymer were carried out to confirm further the formation of the palladium complex and the polymerization that occurred (Fig. 3(a)). The spectrum of Pheb 3 (black curve) shows an intense peak at 1572 cm−1 related to the asymmetric stretch of the carboxylate group of the amino acid derivative in zwitterionic form, with the amine exhibiting a weaker signal around 1497 cm−1. In the PhebPd spectrum (red curve), a shift of the two signals to 1685 cm−1 and 1560 cm−1 can be observed, corresponding to the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and NH deformation, respectively, indicating coordination with Pd. This is further corroborated by the shift in the asymmetric stretching of NH at 3185 cm−1. Additionally, the difference between the symmetric and asymmetric stretching of the carboxylate group (C–O–Pd and CO at 1658 cm−1 and 1352 cm−1, respectively) can confirm the formation of a monodentate bond with Pd.42 In the blue curve, representing the FT-IR analysis of C-PhebPd, a strong signal at 1700 cm−1 is observed, related to the stretching of CO from the carboxylic groups present in the polymer. Peaks corresponding to C–O–Pd, NH–Pd, and NH stretching, similar to the spectrum of PhebPd, are also observed. These peaks in C-PhebPd exhibit no significant shifts but have lower intensities due to the mixture in the polymer matrix. Across all curves, a signal around 3400 cm−1, related to OH stretching from water absorption, is present.
O and NH deformation, respectively, indicating coordination with Pd. This is further corroborated by the shift in the asymmetric stretching of NH at 3185 cm−1. Additionally, the difference between the symmetric and asymmetric stretching of the carboxylate group (C–O–Pd and CO at 1658 cm−1 and 1352 cm−1, respectively) can confirm the formation of a monodentate bond with Pd.42 In the blue curve, representing the FT-IR analysis of C-PhebPd, a strong signal at 1700 cm−1 is observed, related to the stretching of CO from the carboxylic groups present in the polymer. Peaks corresponding to C–O–Pd, NH–Pd, and NH stretching, similar to the spectrum of PhebPd, are also observed. These peaks in C-PhebPd exhibit no significant shifts but have lower intensities due to the mixture in the polymer matrix. Across all curves, a signal around 3400 cm−1, related to OH stretching from water absorption, is present.
 | ||
| Fig. 3 (a) FTIR spectra of Pheb 3, C-Pheb, and PhebPd; (b) thermogravimetric analysis (TGA) of C-Pheb and C-PhebPd. | ||
Thermogravimetric analyses (TGA) were conducted to examine the synthesized cryogels’ thermal stability and the residue's Pd content (Fig. 3(b)). The TG curve of C-PhebPd exhibits two distinct weight loss stages. The first stage in the temperature range of 220–240 °C shows a sharp weight loss of 20%, corresponding to the Phe degradation.43 As the temperature rises, a series of reactions occur, causing dehydration, chain cleavage, and depolymerization, with the most significant loss of weight observed. Notably, the residual part obtained at 600 °C is significantly higher than C-Pheb (10%) due to the presence of the metal residue.44
The surface morphology of the new material was investigated using SEM and XPS. Scanning electron microscopy image illustrating the C-PhebPd catalyst revealed a morphology characterized by a macroporous structure with pore diameter ranging from 10 to 60 μm (Fig. 4 and Fig. S10, ESI†). Additionally, the elemental compositions of C-PhebPd material were ascertained using energy-dispersive X-ray (EDX) analysis. As expected, the peak around 3 keV (Fig. S11, ESI†) confirms the presence of Pd, which is also visible to the necked eye, as seen in the grey-coloured cryogel in Fig. 4(b). From the EDX analysis, the weight concentration of Pd content in the polymer was determined to be 9.6%, which agrees with the TGA data.
 | ||
| Fig. 4 (a) SEM image of C-PhebPd; (b) representative image of C-PhebPd; (c) table with element composition of C-PhebPd. | ||
The pore properties obtained through SEM image analysis are reported in Table S1 (ESI†).
The XPS analysis was performed to support FT-IR observations regarding the formation of the complex and the complementary NMR investigation. The study shows the formation of the palladium complex with PheB (Fig. 5). As depicted in Fig. 5(a), the C1s signal shows three components: a main component at 285 eV due to the typical carbon atoms in hydrocarbon configuration (C–C/C–H), a second component at 288,1 eV due to the presence of carboxylate groups and another component assigned to carbon atoms bonded to one oxygen atom or nitrogen (C–O/C–N) at 286.6 eV. The O1s region shows a main peak at 531.0 eV assigned to the oxygen of COO− groups (Fig. 5(b)). Additionally, the N1s signal (Fig. 5(c)) exhibits a narrow peak at 401.4 eV, associated with –N+–H groups, confirming the presence of derived amino acid molecules. In Fig. 5(d)–(g), the signals related to C1s, O1s, N1s, and Pd3d are reported, confirming the formation of the metal–organic palladium complex. The C1s signal in Fig. 5(d) shows, besides the component due to the C–C/C–H atoms, a C–O/C–N peak at 285.9 eV and a peak at 289.0 eV, assigned to the carbon of the OC–O–Pd group. The O1s signal in Fig. 5(e) shows a component assigned to the CO/C–O–Pd, a peak assigned to C–OH, and another peak related to physisorbed H2O at 532.0 eV, 533.3 eV, and 535.4 eV respectively.45 Additionally, a peak at 400.8 eV in the N1s signal (Fig. 5(f)) and a peak at 338.8 eV related to the presence of ionic Pd (Fig. 5(d)) are consistent with the predicted structure. Table 1 reports a quantitative analysis of XPS outputs. This data show an N:Pd2+ ratio of about 2:1 and a CO/C–O–Pd:Pd2+ ratio of approximately 4:1, confirming the coordination of Pd2+ with the NH and COO− groups of Pheb molecules.
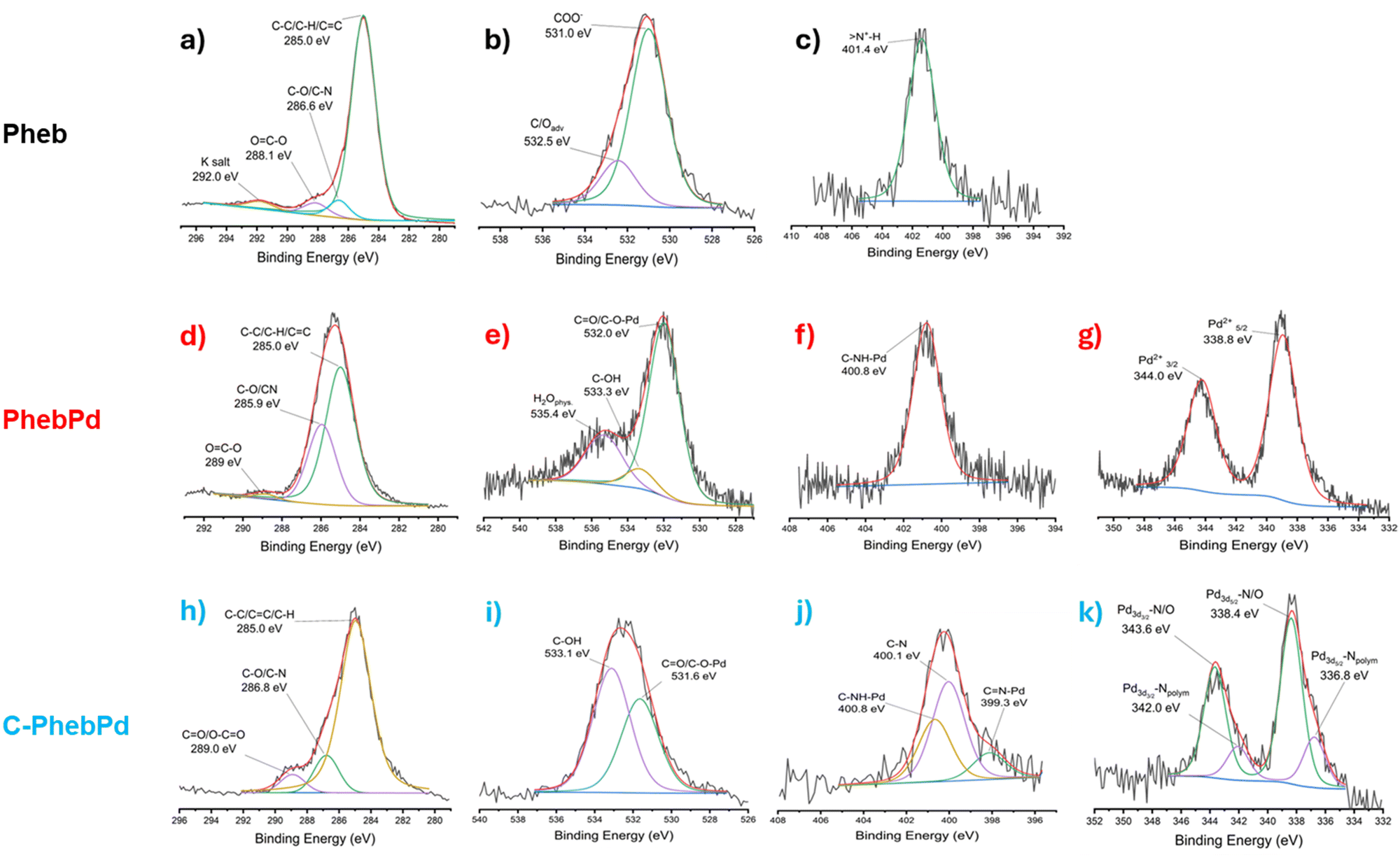 | ||
| Fig. 5 XPS spectra of (a)–(c) Pheb, (d)–(g) PhebPd and (h)–(m) C-PhebPd. | ||
| Element | Atomic concentration |
|---|---|
| O–Pd | 3.74 |
| N–Pd | 4.73 |
| Pd2+ | 2.45 |
| C1s | 78.06 |
The presence of the Pheb complex in the organic matrix was also confirmed by XPS analysis (Fig. 5(h) and (k)). In the O1s and N1s signals, the peaks at 531.6 eV and 400.8 eV, respectively, indicate the presence of the CO/C–O–Pd and C–NH–Pd bonding in the complex. This assumption is confirmed in the Pd3d peak at 338.4 eV related to ionic palladium, which is related to the Pd–O/N signals. Moreover, a second peak in the N1S and Pd3d peaks related to the CN–Pd and Pd–N at 399.3 eV and 336.8 eV, respectively, can be associated with the bonding between the polymeric matrix and the ionic Pd.
The swelling properties of C-PhebPd were evaluated using a standard gravimetric procedure and compared to a cryogel formed by sola HEMA.46 The newly synthesized macroporous material showed significant swelling capacity. In two seconds, up to 8 times its original weight is achieved (Fig. S12, ESI†). This can be attributed to the synergistic effect of the cryogels’ interconnected porous structure and the pronounced hydrophilicity of the functional groups within the polymer network. Data indicate that palladium complexation did not alter the water uptake capacity of the sponge. This feature is relevant to allowing a fast diffusion of reactant mixture within the catalyst, thus favouring the coupling reaction in a confined area.
Suzuki–Miyaura coupling reactions catalyzed by C-PhebPd catalyst
In the initial phase of our catalytic studies, we investigated the coupling reaction between iodobenzene and phenylboronic acid due to its rapid kinetics and excellent yields, with the added benefit of spontaneous product crystallization, simplifying isolation and ensuring purity. Unsurprisingly, conducting the reaction under base-free conditions failed to yield the desired product, highlighting the importance of the base in facilitating the coupling process. Through systematic exploration of various reaction parameters, including the choice of base, solvent, and reaction temperature, as summarized in Table 2, we optimized the reaction conditions. While employing NaOH as a base resulted in unsatisfactory coupling yields, the use of K2CO3 proved to be significantly more effective. Notably, employing K2CO3 improved both the yields and reaction kinetics, achieving a 99% conversion of reagents within just 30 minutes in an H2O/MeOH mixture at 80 °C (entry 2). The minimal effect on yield when substituting methanol with ethanol further highlights the robustness of the reaction conditions (entry 3). Moreover, it was observed that reducing the reaction temperature to 60 °C lowered the yield to 75%, emphasizing the critical role of temperature optimization, with 80 °C identified as the optimal temperature for maximizing efficiency.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Base | Solvent | Catalyst mol% | Time/min | T (°C) | Yield % |
| 1 | K2CO3 | H2O/MeOH | — | 30 | 80 | 0 |
| 2 | K2CO3 | H2O/MeOH | 2.5 | 30 | 80 | 99 |
| 3 | K2CO3 | H2O/EtOH | 2.5 | 30 | 80 | 99 |
| 4 | K2CO3 | H2O/MeOH | 2.5 | 30 | 60 | 75 |
| 5 | Na2CO3 | H2O/MeOH | 2.5 | 30 | 80 | 78 |
| 6 | K2CO3 | MeOH | 2.5 | 30 | 80 | 15 |
| 7 | K2CO3 | EtOH | 2.5 | 30 | 80 | 9 |
| 8 | NaOH | H2O/MeOH | 2.5 | 30 | 80 | 60 |
| 9 | NaOH | H2O | 2.5 | 30 | 80 | 13 |
| 10 | TEA | H2O/MeOH | 2.5 | 30 | 80 | 55 |

Further investigations showed that replacing K2CO3 with Na2CO3 (entry 5) led to a slight decrease in product formation. Using organic bases such as TEA (entry 10) caused a significant drop in yields and promoted the formation of undesired side products. Moreover, omitting water and using solely alcohol as the solvent yielded poor product formation, highlighting the importance of solvent composition in this catalytic system. This phenomenon can be elucidated by the swelling behaviour exhibited by our catalyst in aqueous environments. Such swelling facilitates the penetration of reagents into the catalyst's pores, thereby promoting their proximity to each other and enhancing interaction with the catalytic metal species.
The optimal reaction conditions were achieved using K2CO3 as the base and H2O/MeOH (1:1) as the solvent. Under these conditions, the desired product was typically obtained neatly and isolated through a simple extraction process without further purification. All yields were calculated based on the clean, isolated product.
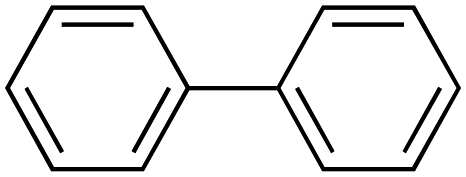
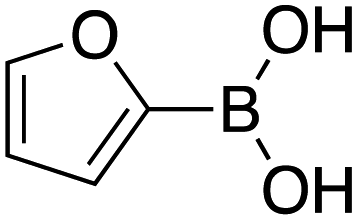
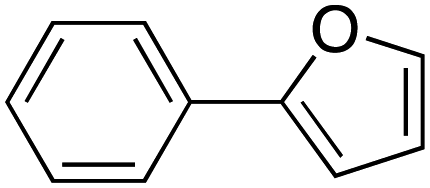
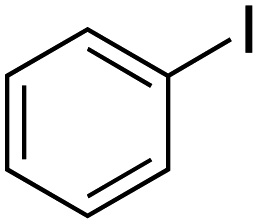
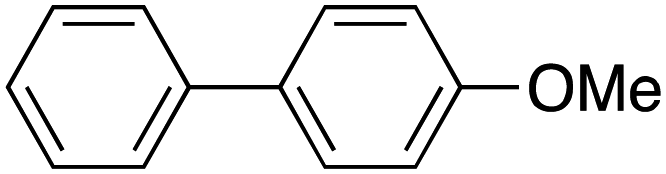
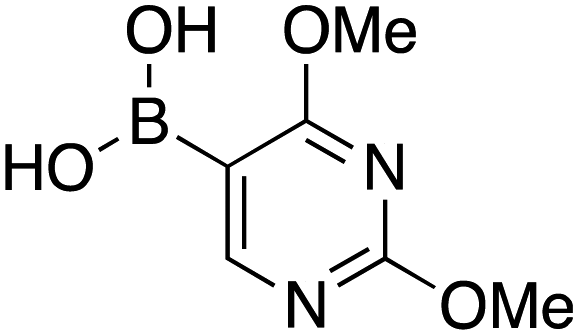
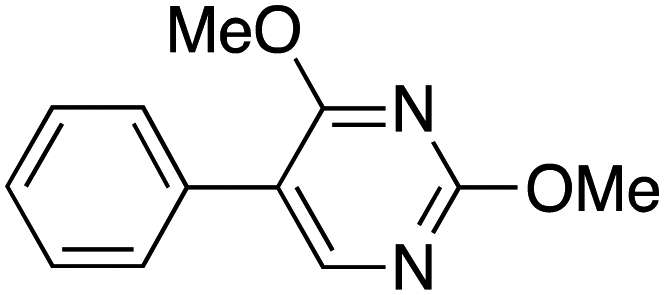
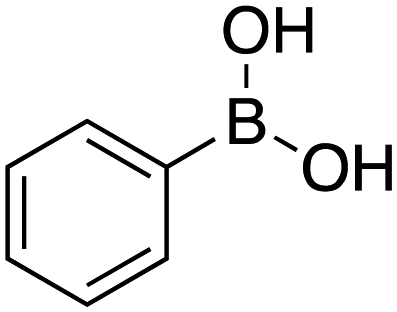
Therefore, the C-PhebPd catalyst application was extended using various aryl halides and boronic acids (Table 3). Starting with standard iodobenzene, we performed the coupling reaction with a range of boronic acids, achieving good to excellent yields for most products (Table 3, entries 1–4, 6–12, and 14). However, the reactions with pyrimidine boronic acid and 2-bromofuran resulted in poor yields (Table 3, entries 5 and 13). In detail, introducing electron-donating substituents on the aryl halide also enhances product yields (Table 3, entries 6–8). Specifically, the synthesis of 4-methoxy-1,1′-biphenyl via the coupling of iodobenzene with (4-methoxyphenyl)boronic acid (Table 3, entry 4) outperforms the reaction between 1-iodo-4-methoxybenzene with phenylboronic acid (Table 3, entry 6), yielding 87% compared to 74%. Similarly, optimal yields for forming 2-phenylfuran were achieved when iodobenzene reacted with furan-2-ylboronic acid (Table 3, entry 2), whereas reversing the reactants (Table 3, entry 13) resulted in minimal product formation. Even in cases where aryls bear electron-withdrawing substituents, the reaction proceeded rapidly, and yields were consistently high (Table 3, entry 14). In most instances, the product quickly precipitated and spontaneously crystallized, simplifying the isolation process and ensuring the purity of the final product. To measure the efficiency of the catalyst, the turnover frequency number (TOF) was calculated for all reactions following eqn (1):
 | (1) |
| Entry | Aryl halides | Arylboronic acid | Time (min) | Productsa | Yieldb (%) | TOF (h−1) |
|---|---|---|---|---|---|---|
|
a Reaction conditions: aryl halide (0.3 mmol), arylboronic acid (0.36 mmol), K2CO3 (0.6 mmol), MeOH/H2O (1:1, 2 mL), C-Pheb-Pd (9 mg, wet), 80 °C.
b Yield of isolated product.
c Arylboronic acid (2 eq.).
|
||||||
| 1 |
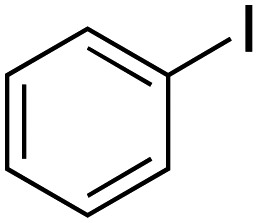
|
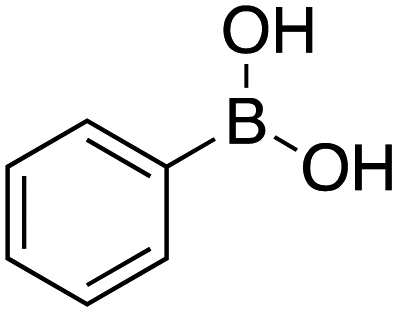
|
25 |
|
≥99 | 95.58 |
| 2 |

|
|
30 |
|
89 | 70.27 |
| 3 |
|
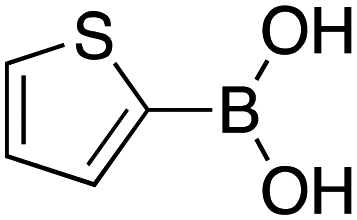
|
30 |
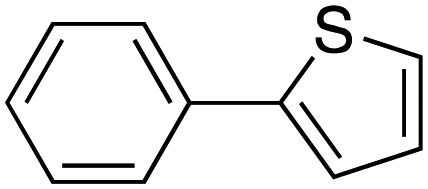
|
90 | 71.35 |
| 4 |
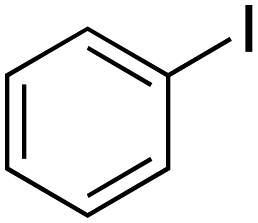
|
|
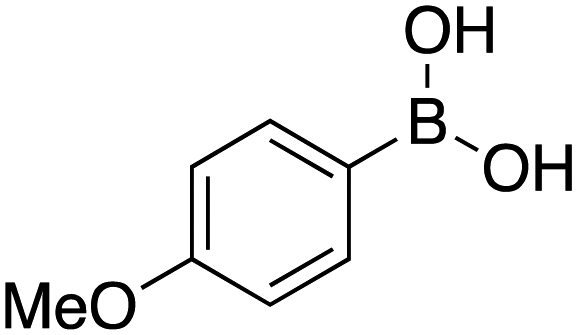
25 |
|
87 | 83.71 |
| 5 |

|
|
30 |
|
14 | 10.81 |
| 6 |

|
|
25 |
|
74 | 62.42 |
| 7 |
|
|
20 |

|
89 | 93.28 |
| 8 |
|

|
15 |
|
58 | 80.21 |
| 9 |

|
|
40 |

|
84 | 55.69 |
| 10 |

|
|
30 |
|
75c | 65.40 |
| 11 |

|
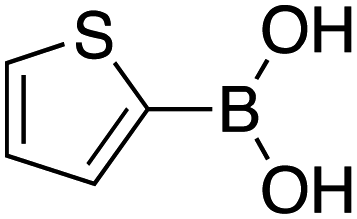
|
15 |

|
45 | 77.83 |
| 12 |

|
|
30 |

|
44 | 32.43 |
| 13 |
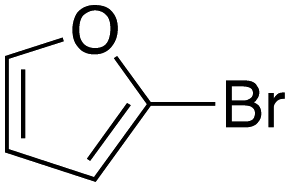
|
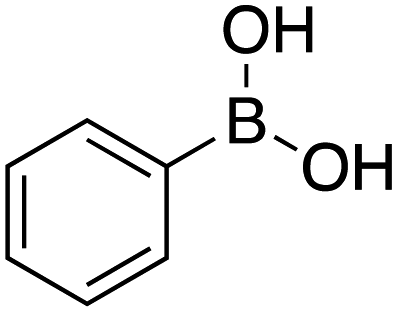
|
120 |

|
Trace | 0.54 |
| 14 |
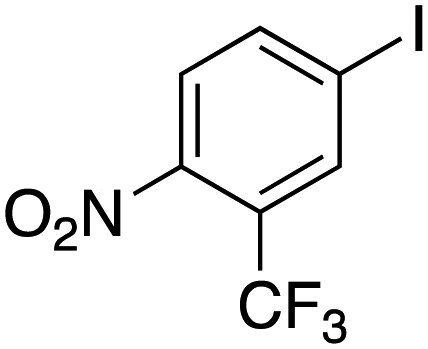
|

|
30 |

|
94 | 48.08 |
Table 4 compares our catalyst's performance with other heterogeneous catalysts reported in the literature. The table summarizes key metrics such as solvent, time, temperature, and TOF under similar reaction conditions (Table 3, entry 1). This comparison highlights our catalyst's advantages and potential limitations relative to existing alternatives, providing a comprehensive overview of its performance and applicability.
| Catalyst | Solvent | Temperature (°C) | Time (h) | TOF (h−1) | Ref. |
|---|---|---|---|---|---|
| C-PhebPd | H2O/MeOH (1:1) |
80 | 0.41 | 95 | This work |
| G-COOH-Pd-10 | H2O/DMF | 70 | 3.00 | 52 | 47 |
| PPI-1-NPy-Pd | H2O | 100 | 24.00 | 462 | 48 |
| Pd-PEPPSI | H2O/i-PrOH (3:1) |
r.t. | 1.00 | 1000 | 49 |
| PPI-2-NPy-Pd | H2O | 100 | 24.00 | 685 | 48 |
| Pd/MPA | Toluene | 80 | 0.03 | 980 | 50 |
| Pd/N-C-300 | n-Butanol | 60 | 24.00 | 198 | 51 |
It is important to highlight that some systems discussed rely on non-sustainable chemicals and demanding conditions, while others utilize more environmentally friendly aromatic catalysts. For example, the catalyst based on Pd immobilized on polyamide (Pd/MPA) demonstrates good catalytic activity at a relatively low cost.50 However, the use of melamine, classified as “carcinogenic to humans”52 and a potential groundwater contaminant, raises serious environmental and health concerns. Moreover, the reaction requires toluene as a solvent and must be performed under a nitrogen atmosphere, limiting its sustainability and safety.
Similarly, while Pd-PEPPSI complexes offer impressive stability in air and water and facilitate reactions at lower temperatures, their synthesis involves harmful solvents like pyridine and dichloromethane, compromising their overall environmental compatibility. Even systems such as PPI-1-NPy-Pd, which allow reactions in water, suffer from lower yields and slower reaction times than our catalyst.49
In contrast, our system addresses these sustainability challenges by avoiding toxic solvents and harmful conditions while delivering high performance with fast reaction times and high yields. This combination of environmental responsibility and catalytic efficiency underscores the potential of our catalyst to drive advancements in green chemistry.
Reusability and Pd leaching tests
Following the urge to avoid waste for green and sustainable development, catalysts should have a long lifetime and be easily recyclable. To this end, we performed a reusability assay to verify the heterogeneity and recyclability of the C-PhebPd catalyst. Catalyst recycling experiments were conducted by washing the post used catalyst with a methanol/water mixture and drying it (Fig. 6(a)). It was observed that the weight of the catalyst remained stable, and the reaction yields were consistently maintained even after five cycles, with values ranging from 99% to 91%. However, in the seventh cycle, a slight reduction in catalytic activity is observed, resulting in a 70% product yield.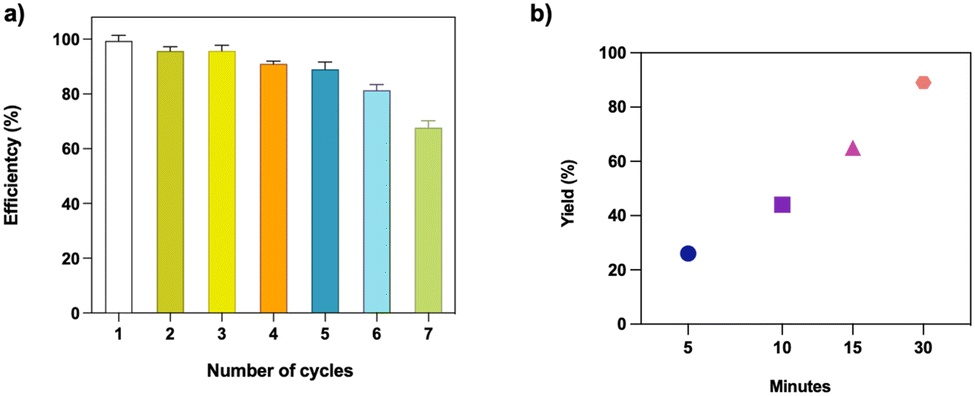 | ||
| Fig. 6 (a) Recycling test of catalyst in Suzuki–Miyaura reaction between iodobenzene and phenylboronic acid. (b) Yield of standard Suzuki–Miyaura reaction as a function of catalyst contact time. | ||
A prevalent concern associated with heterogeneous catalysts is the leaching of the metal salt, which poses dual drawbacks: (i) a decline in the catalyst's activity over successive cycles and (ii) potential environmental implications. A test was conducted to confirm the heterogeneity of the C-PhebPd catalyst and assess the potential for metal species leaching. The reaction was initiated and allowed to proceed for a specific duration at 80 °C, varying the contact duration with the catalyst. After removing the catalyst, the reaction continued at 80 °C for 30 minutes. Fig. 6(b) vividly illustrates a decline in catalytic activity upon removing the catalyst from the reaction mixture. These findings unequivocally suggest negligible leaching of palladium ions under the current reaction conditions (Fig. 6(b)).
Mass spectrometry (ICP-MS) was used to further prove the absence of Pd release in water. After the reaction, the Pd content in solution (leaching) was 185 ± 7 ppb.
Conclusions
Our research has successfully developed a novel bio-based heterogenous palladium catalyst with remarkable efficacy in Suzuki–Miyaura cross-coupling. This hybrid system improves palladium-catalysed reactions since they are conducted in aqueous media at moderate temperatures, giving products with high yields. Notably, it offers several benefits, including its increased sustainability owing to the phenylalanine synthon, resistance to palladium leaching, and exceptional reusability without significant loss of catalytic activity and multiple cycles. C-PhebPd catalyst, thanks to its robustness, could be ad hoc designed for industrial processes, specifically in pharmaceutical production. Its synthetic chemistry versatility opens a new perspective for advancements in other cross-coupling reactions.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results received funding from the MIUR-PNRR project SAMOTHRACE – Sicilian MicronanoTech Research and Innovation Center (ECS00000022, CUP B63C2 2000620005). The views and opinions expressed are those of the authors only and do not necessarily reflect those of the European Union or the European Commission. Neither the European Union nor the European Commission can be held responsible for them. The authors thank the Bio-nanotech Research and Innovation Tower (BRIT) laboratory of the University of Catania.References
- J. P. Wolfe, R. A. Singer, B. H. Yang and S. L. Buchwald, Highly Active Palladium Catalysts for Suzuki Coupling Reactions, J. Am. Chem. Soc., 1999, 121, 9550–9561 CrossRef CAS.
- L. Yin and J. Liebscher, Carbon−Carbon Coupling Reactions Catalyzed by Heterogeneous Palladium Catalysts, Chem. Rev., 2007, 107, 133–173 CrossRef CAS.
- N. Miyaura and A. Suzuki, Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. Suzuki, Cross-Coupling Reactions Of Organoboranes: An Easy Way To Construct C–C Bonds (Nobel Lecture), Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- N. Oger and F.-X. Felpin, Heterogeneous Palladium Catalysts for Suzuki–Miyaura Coupling Reactions Involving Aryl Diazonium Salts, ChemCatChem, 2016, 8, 1998–2009 CrossRef CAS.
- Y. Zhang and S. N. Riduan, Functional porous organic polymers for heterogeneous catalysis, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- E. O. Pentsak, L. U. Dzhemileva, V. A. D’yakonov, R. R. Shaydullin, A. S. Galushko, K. S. Egorova and V. P. Ananikov, Comparative assessment of heterogeneous and homogeneous Suzuki–Miyaura catalytic reactions using bio-Profiles and bio-Factors, J. Organomet. Chem., 2022, 965–966, 122319 CrossRef CAS.
- S.-Y. Zhang, K. Yu, Y.-S. Guo, R.-Q. Mou, X.-F. Lu and D.-S. Guo, Preparation and Reactivation of Heterogeneous Palladium Catalysts and Applications in Sonogashira, Suzuki, and Heck Reactions in Aqueous Media, ChemistryOpen, 2018, 7, 803–813 CrossRef CAS PubMed.
- Z. Chen, E. Vorobyeva, S. Mitchell, E. Fako, M. A. Ortuño, N. López, S. M. Collins, P. A. Midgley, S. Richard, G. Vilé and J. Pérez-Ramírez, A heterogeneous single-atom palladium catalyst surpassing homogeneous systems for Suzuki coupling, Nat. Nanotechnol., 2018, 13, 702–707 CrossRef CAS PubMed.
- M. Ashraf, M. S. Ahmad, Y. Inomata, N. Ullah, M. N. Tahir and T. Kida, Transition metal nanoparticles as nanocatalysts for Suzuki, Heck and Sonogashira cross-coupling reactions, Coord. Chem. Rev., 2023, 476, 214928 CrossRef CAS.
- A. Srivastava, H. Kaur, H. Pahuja, T. M. Rangarajan, R. S. Varma and S. Pasricha, Optimal exploitation of supported heterogenized Pd nanoparticles for C–C cross-coupling reactions, Coord. Chem. Rev., 2024, 507, 215763 CrossRef CAS.
- Q. Zhang, Z. Mao, K. Wang, N. T. S. Phan and F. Zhang, Microwave-assisted aqueous carbon–carbon cross-coupling reactions of aryl chlorides catalysed by reduced graphene oxide supported palladium nanoparticles, Green Chem., 2020, 22, 3239–3247 RSC.
- R. Tao, X. Ma, X. Wei, Y. Jin, L. Qiu and W. Zhang, Porous organic polymer material supported palladium nanoparticles, J. Mater. Chem. A, 2020, 8, 17360–17391 RSC.
- Z. Li, B. Yao, C. Cheng, M. Song, Y. Qin, Y. Wan, J. Du, C. Zheng, L. Xiao, S. Li, P.-F. Yin, J. Guo, Z. Liu, M. Zhao and W. Huang, Versatile Structural Engineering of Metal–Organic Frameworks Enabling Switchable Catalytic Selectivity, Adv. Mater., 2024, 36, 2308427 CrossRef CAS.
- Z. Song, M. Wang, Y. Qin, J. Du, Z. Li, M. Song, Y. Wan, S. Li, J. Guo and M. Zhao, Thioether functionalized vinylene-linked covalent organic frameworks boost the semi-hydrogenation selectivity of alkynes, Mater. Chem. Front., 2024, 8, 2610–2618 RSC.
- L. Xiao, C. Cheng, Z. Li, C. Zheng, J. Du, M. Song, Y. Wan, S. Li, G. Jun and M. Zhao, Dynamically modulated synthesis of hollow metal-organic frameworks for selective hydrogenation reactions, Nano Res., 2023, 16, 11334–11341 CrossRef CAS.
- T. Das, H. Uyama and M. Nandi, Pronounced effect of pore dimension of silica support on Pd-catalyzed Suzuki coupling reaction under ambient conditions, New J. Chem., 2018, 42, 6416–6426 RSC.
- D. Sahu and P. Das, Phosphine-stabilized Pd nanoparticles supported on silica as a highly active catalyst for the Suzuki–Miyaura cross-coupling reaction, RSC Adv., 2014, 5, 3512–3520 RSC.
- A. Aschenaki, F. Ren, J. Liu, W. Zheng, Q. Song, W. Jia, J. J. Bao and Y. Li, Preparation of a magnetic and recyclable superparamagnetic silica support with a boronic acid group for immobilizing Pd catalysts and its applications in Suzuki reactions, RSC Adv., 2021, 11, 33692–33702 RSC.
- M. Gao, J. Wang, W. Shang, Y. Chai, W. Dai, G. Wu, N. Guan and L. Li, Zeolite-encaged palladium catalysts for heterogeneous Suzuki–Miyaura cross-coupling reactions, Catal. Today, 2023, 410, 237–246 CrossRef CAS.
- E. Pérez-Mayoral, V. Calvino-Casilda and E. Soriano, Metal-supported carbon-based materials: opportunities and challenges in the synthesis of valuable products, Catal. Sci. Technol., 2016, 6, 1265–1291 RSC.
- R. Tao, X. Ma, X. Wei, Y. Jin, L. Qiu and W. Zhang, Porous organic polymer material supported palladium nanoparticles, J. Mater. Chem. A, 2020, 8, 17360–17391 RSC.
- S. E. Hooshmand, B. Heidari, R. Sedghi and R. S. Varma, Recent advances in the Suzuki–Miyaura cross-coupling reaction using efficient catalysts in eco-friendly media, Green Chem., 2019, 21, 381–405 RSC.
- H. Salemi, M. Debruyne, V. V. Speybroeck, P. V. D. Voort, M. D’hooghe and C. V. Stevens, Covalent organic framework supported palladium catalysts, J. Mater. Chem. A, 2022, 10, 20707–20729 RSC.
- Z. Zhang and Y. M. A. Yamada, Recent Advancements in Continuous-Flow Suzuki–Miyaura Coupling Utilizing Immobilized Molecular Palladium Complexes, Chem. – Eur. J., 2024, 30, e202304335 CrossRef CAS PubMed.
- P. A. Shiekh, S. M. Andrabi, A. Singh, S. Majumder and A. Kumar, Designing cryogels through cryostructuring of polymeric matrices for biomedical applications, Eur. Polym. J., 2021, 144, 110234 CrossRef CAS.
- C. Zagni, A. Coco, T. Mecca, G. Curcuruto, V. Patamia, K. Mangano, A. Rescifina and S. C. Carroccio, Sponge-like macroporous cyclodextrin-based cryogels for controlled drug delivery, Mater. Chem. Front., 2023, 7, 2693–2705 RSC.
- C. Zagni, V. Patamia, S. Dattilo, V. Fuochi, S. Furnari, P. M. Furneri, S. C. Carroccio, G. Floresta and A. Rescifina, Supramolecular biomaterials as drug nanocontainers with iron depletion properties for antimicrobial applications, Mater. Adv., 2024, 5, 3675–3682 RSC.
- H.-W. Shao, Y. Wu and R. Li, A Highly Practical Method for Monobenzylation of Amino Acids, Synth. Commun., 2000, 30, 1911–1915 CrossRef CAS.
- D. B. Hobart, J. S. Merola, H. M. Rogers, S. Sahgal, J. Mitchell, J. Florio and J. W. Merola, Synthesis, Structure, and Catalytic Reactivity of Pd(II) Complexes of Proline and Proline Homologs, Catalysts, 2019, 9, 515 CrossRef CAS.
- C. Zagni, A. A. Scamporrino, P. M. Riccobene, G. Floresta, V. Patamia, A. Rescifina and S. C. Carroccio, Portable Nanocomposite System for Wound Healing in Space, Nanomaterials, 2023, 13, 741 CrossRef CAS PubMed.
- C. Zagni, Single and dual polymeric sponges for emerging pollutants removal, Eur. Polym. J., 2022, 179(5), 111556 CrossRef CAS.
- S. Scurti, S. Dattilo, D. Gintsburg, L. Vigliotti, A. Winkler, S. C. Carroccio and D. Caretti, Superparamagnetic Iron Oxide Nanoparticle Nanodevices Based on Fe3O4 Coated by Megluminic Ligands for the Adsorption of Metal Anions from Water, ACS Omega, 2022, 7, 10775–10788 CrossRef CAS.
- M. V. Di Natale, S. C. Carroccio, S. Dattilo, M. Cocca, A. Nicosia, M. Torri, C. D. Bennici, M. Musco, T. Masullo, S. Russo, A. Mazzola and A. Cuttitta, Polymer aging affects the bioavailability of microplastics-associated contaminants in sea urchin embryos, Chemosphere, 2022, 309, 136720 CrossRef CAS PubMed.
- E. Conti, S. Dattilo, G. Costa and C. Puglisi, Bioaccumulation of trace elements in the sandhopper Talitrus saltator (Montagu) from the Ionian sandy coasts of Sicily, Ecotoxicol. Environ. Saf., 2016, 129, 57–65 CrossRef CAS PubMed.
- J. Qi, W. Zhang and R. Cao, Porous Materials as Highly Efficient Electrocatalysts for the Oxygen Evolution Reaction, ChemCatChem, 2018, 10, 1206–1220 CrossRef CAS.
- S. Dattilo, C. Zagni, T. Mecca, V. Patamia, G. Floresta, P. Nicotra, S. C. Carroccio and A. Rescifina, Solvent-free conversion of CO2 in carbonates through a sustainable macroporous catalyst, Giant, 2024, 18, 100258 CrossRef CAS.
- M. E. Hossain, M. M. Hasan, M. E. Halim, M. Q. Ehsan and M. A. Halim, Interaction between transition metals and phenylalanine: A combined experimental and computational study, Spectrochim. Acta, Part A, 2015, 138, 499–508 CrossRef.
- D. B. Hobart, M. A. G. Berg, H. M. Rogers and J. S. Merola, Synthesis, Characterization, and Non-Covalent Interactions of Palladium(II)–Amino Acid Complexes, Molecules, 2021, 26, 4331 CrossRef CAS.
- V. Giglio, C. Zagni, E. T. A. Spina, F. Cunsolo and S. C. Carroccio, Polyvinylimidazole-Based Cryogel as an Efficient Tool for the Capture and Release of Oleuropein in Aqueous Media, Polymers, 2024, 16, 2339 CrossRef CAS.
- K. Nakamoto and R. S. Czernuszewicz, Methods in Enzymology, Academic Press, 1993, vol. 226, pp. 259–289 Search PubMed.
- J. Lu, Characterization and pseudopolymorphism of l-phenylalanine anhydrous and monohydrate forms, Afr. J. Pharm. Pharmacol., 2012, 6, 269–277 CrossRef CAS.
- S. Scurti, G. P. Salanitri, T. Mecca, E. Rodríguez-Aguado, J. A. Cecilia, G. Curcuruto, S. C. Carroccio, D. Caretti and N. Dimitratos, pH-dependent catalytic activity of Au and Pd-based hybrid cryogels by investigating the acid/base nature of the polymeric phase, Mater. Today Chem., 2024, 38, 102046 CrossRef CAS.
- J.-H. Jhang, J. A. Boscoboinik and E. I. Altman, Ambient pressure x-ray photoelectron spectroscopy study of water formation and adsorption under two-dimensional silica and aluminosilicate layers on Pd(111), J. Chem. Phys., 2020, 152, 084705 CrossRef CAS.
- C. Zagni, A. Coco, S. Dattilo, V. Patamia, G. Floresta, R. Fiorenza, G. Curcuruto, T. Mecca and A. Rescifina, HEMA-based macro and microporous materials for CO2 capture, Mater. Today Chem., 2023, 33, 101715 CrossRef CAS.
- R. S. Erami, D. Díaz-García, S. Prashar, A. Rodríguez-Diéguez, M. Fajardo, M. Amirnasr and S. Gómez-Ruiz, Suzuki–Miyaura C–C Coupling Reactions Catalyzed by Supported Pd Nanoparticles for the Preparation of Fluorinated Biphenyl Derivatives, Catalysts, 2017, 7, 76 CrossRef.
- E. R. Rangel, E. M. Maya, F. Sánchez, J. G. de la Campa and M. Iglesias, Palladium-heterogenized porous polyimide materials as effective and recyclable catalysts for reactions in water, Green Chem., 2014, 17, 466–473 RSC.
- S. Alnasser, N. Touj, S. Alomar, L. Mansour, M. Sauthier, N. Gurbuz, I. Özdemir and N. Hamdi, Pd–PEPPSI-type expanded ring N-heterocyclic carbene complexes: synthesis, characterization, and catalytic activity in Suzuki–Miyaura cross coupling, Green Chem. Lett. Rev., 2024, 17, 2370268 CrossRef.
- J. Chen, J. Zhang, W. Sun, K. Song, D. Zhu and T. Li, Pd immobilized on polyamide based on melamine and terephalic acid as an efficient and recyclable catalyst for Suzuki–Miyaura coupling reaction, Appl. Organomet. Chem., 2018, 32, e4135 CrossRef.
- L. Wang, S. Lyu, P. Zhang, X. Tian, D. Wang, W. Huang and Z. Liu, Nitrogen-bonded ultrasmall palladium clusters over the nitrogen-doped carbon for promoting Suzuki cross-coupling reactions, Adv. Compos. Hybrid Mater., 2022, 5, 1396–1403 CrossRef CAS.
- ECHA RAC: New hazard classification melamine and BPS – Food Packaging Forum, https://www.foodpackagingforum.org/news/echa-rac-new-hazard-classification-melamine-and-bps, (accessed 10 September 2024).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qm00800f |
| This journal is © the Partner Organisations 2024 |