
NHC–amine Ru complex catalyzed selective mono-N-methylation of amines with methanol: understanding the effect of amine ligands†
Xiuju
Cai‡
 a,
Xiaoyu
Zhou‡
b and
Ming
Huang
*a
a,
Xiaoyu
Zhou‡
b and
Ming
Huang
*a
aSchool of Pharmacy, School of Clinical Pharmacy, Guangdong Pharmaceutical University, Guangzhou 510006, P. R. China. E-mail: 1529236883@qq.com
bSchool of Materials Science & Engineering, PCFM Lab, School of Chemistry, Sun Yat-sen University, Guangzhou 510275, P. R. China
First published on 14th November 2023
Abstract
The selective mono-N-methylation of amines with methanol remains challenging, particularly for aliphatic amines. Inspired by metal–ligand cooperation, a series of N-heterocyclic carbene (NHC)–amine Ru complexes (Ru1–Ru4) with different amine ligands were developed for this transformation. Among them, complex Ru4 bearing an NHC–aniline ligand with a 7-membered chelate ring displayed the highest activity for the selective mono-N-methylation of amines with methanol. Various substrates, including aromatics, sulfonamides, and aliphatic amines, can be transformed into the desired mono-N-methylation products in excellent yields. Moreover, a series of sensitive groups such as nitro, carbonyl, hydroxyl, trifluoromethyl, and internal vinyl groups were well tolerated. Experimental and theoretical studies revealed that the catalytic mechanism involved an inner-sphere mechanism promoted by an Ru complex with NH2 as the labile ligand.
Introduction
N-Methylated amines are one of the essential motifs in numerous bioactive molecules and natural products and serve as key intermediates in synthetic chemistry.1,2 The synthesis of N-methylated amines is mainly dependent on using toxic and waste-generating alkylating agents such as formaldehyde, methyl iodide, and dimethyl sulphate.3 Leveraging methanol as a sustainable and green C1 source, the modern hydrogen autotransfer/borrowing hydrogen (HA/BH) methodology offers an attractive alternative.4,5Generally, the N-methylation of amines with methanol is difficult because the methanol dehydrogenation step has a higher activation barrier compared with heavier alcohols. To date, several metal catalysts (Scheme 1a), such as Ir,6–8 Ru,9–12 Fe,13,14 Mn,15–17 and others,18,19 have been developed in this field. However, most catalytic systems are only suitable for mono-N-methylation of anilines, and nucleophilic aliphatic amines remain challenging substrates or suffer from overmethylation. In 2018, Hong et al. successfully employed an Ru-MACHO-BH complex (Scheme 1b) for the mono-N-methylation of aliphatic amines with methanol.20 However, hydrogen gas (4 MPa) is essential for this transformation. In 2021, Natte et al. used an RuCl3·nH2O catalytic system (Scheme 1b) for the N-methylation of aliphatic amines with methanol; nonetheless, this requires an excessively strong base (2.5 equiv. KOtBu).21 Furthermore, in 2022, Rit et al. reported that an orthometalated Ru–N-heterocyclic carbene (NHC) complex (Scheme 1b) catalysed this transformation, supported by limited examples of aliphatic amines in GC yields.22 A catalytic system with high efficiency needs to be developed for the selective mono-N-methylation of aliphatic amines with methanol.
 | ||
| Scheme 1 N-Methylation of amines with methanol. | ||
Transition-metal complexes bearing amine donor ligands have increasingly attracted attention for catalytic applications.23–25 These complexes display robust amine–amide metal ligand cooperation that has been widely used in numerous (de)hydrogenation reactions. In addition, NHC ligands with strong σ-donating properties are beneficial in (de)hydrogenation reactions.17,26–28 For example, in 2015, Huynh et al. developed a mixed NHC/phosphine Ru complex that could catalyse tunable amidation and amination of alcohols with amines.26 In 2021, Gülcemal et al. reported a series of NHC–Ir complexes for the α-alkylation of arylacetonitriles with primary alcohols.29 Recently, our groups have demonstrated that bis-NHC–TM (TM = Mn, Fe or Mo) complexes are highly effective catalysts that enable the selective N-alkylation of anilines with alcohols.17,28,30 In addition, Glorius, Neugebauer, and Li et al. developed a well-defined Ru(II)–NHC diamine complex that exhibited superior activity in asymmetric hydrogenation.27
The aforementioned studies and findings stimulated our interest in designing Ru complexes with NHC–amine ligands for the mono-N-methylation of amines using methanol. A series of NHC–amine Ru complexes (Ru1–Ru4, Scheme 3) with different amine ligands have been developed. Notably, Ru4, containing an NHC–aniline ligand with a 7-membered chelate ring, showed high catalytic performance for the selective mono-N-methylation of amines with methanol, including aromatics, sulfonamides, and aliphatic amines.
Results and discussion
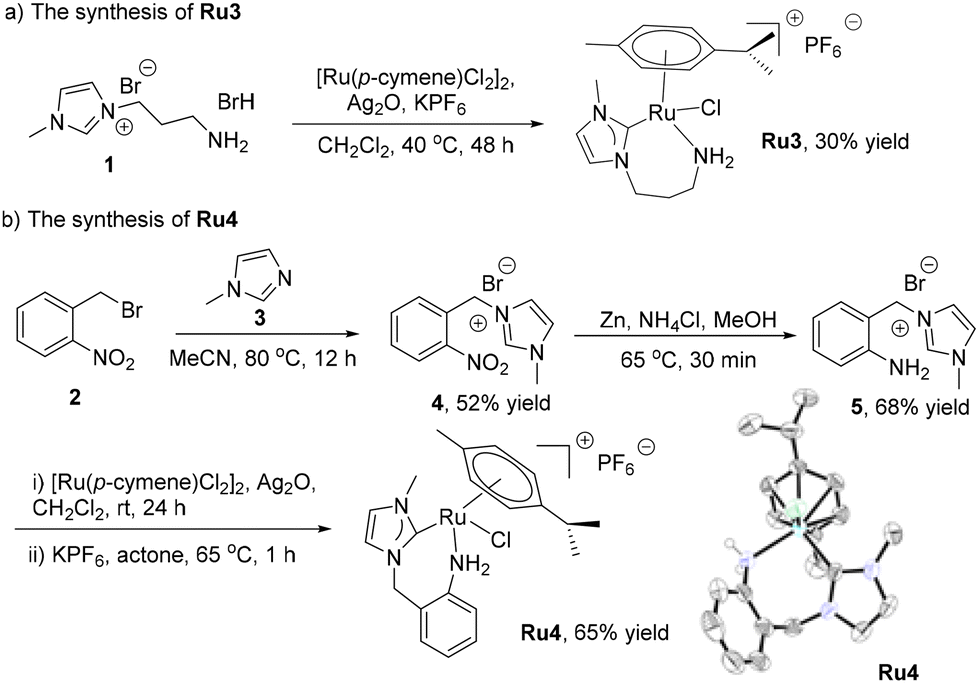
The reported NHC–Ru complexes (Scheme 3) Ru1,31Ru2,32Ru5,33Ru6,34 and Ru7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35 were synthesized according to the literature. Ru3, the new NHC–Ru complex, was prepared via a transmetalation procedure involving the reaction of ligand precursor 1,36 Ag2O, [Ru(p-cymene)Cl2]2 and KPF6 (Scheme 2a). The new NHC–Ru complex Ru4 was prepared according to the procedure presented in Scheme 2b. Intermediate 4 was obtained from the reaction of 1-methylimidazole (3) and 2-nitrobenzyl bromide (2). Following the reduction of the nitro group in 4, the ligand precursor 5 was obtained. The treatment of 5 with Ag2O, [Ru(p-cymene)Cl2]2 and KPF6 afforded the new complex Ru4. All the NHC–Ru complexes were highly stable and could be stored in air for months without decomposition. The structure of complex Ru4 was confirmed by X-ray diffraction and a typical half-sandwich complex was revealed.
35 were synthesized according to the literature. Ru3, the new NHC–Ru complex, was prepared via a transmetalation procedure involving the reaction of ligand precursor 1,36 Ag2O, [Ru(p-cymene)Cl2]2 and KPF6 (Scheme 2a). The new NHC–Ru complex Ru4 was prepared according to the procedure presented in Scheme 2b. Intermediate 4 was obtained from the reaction of 1-methylimidazole (3) and 2-nitrobenzyl bromide (2). Following the reduction of the nitro group in 4, the ligand precursor 5 was obtained. The treatment of 5 with Ag2O, [Ru(p-cymene)Cl2]2 and KPF6 afforded the new complex Ru4. All the NHC–Ru complexes were highly stable and could be stored in air for months without decomposition. The structure of complex Ru4 was confirmed by X-ray diffraction and a typical half-sandwich complex was revealed.
 | ||
| Scheme 2 Synthesis of Ru3 and Ru4. | ||
 | ||
| Scheme 3 N-Methylation of amines with methanol with different NHC–Ru complexes. | ||
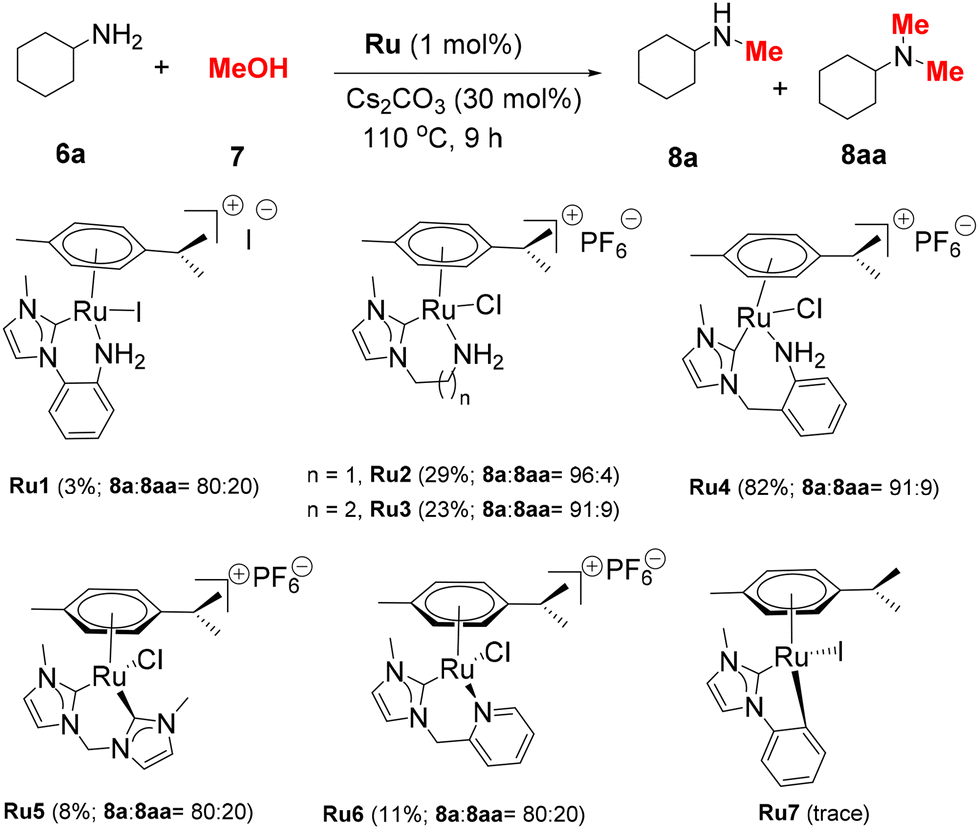
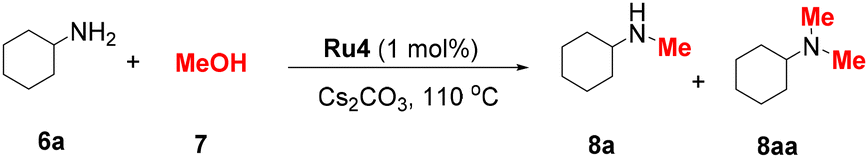
The initial test reactions using cyclohexylamine (6a) and methanol were performed at 110 °C for 9 h with a 1 mol% Ru complex and 0.3 equiv. of Cs2CO3. When Ru1–Ru3 bearing NHC–NH2 ligands were used, low yields (3–29%) of the desired product 8a were obtained. Ru4, which possesses an NHC–aniline ligand with a 7-membered chelate ring, demonstrated the highest catalytic activity, affording 8a in 82% yield. Ru5–Ru7, the complexes proven to be effective catalysts for (de)hydrogenation reactions, provided inferior yields of 8a.34,35,37 These results indicate that both the type of the NH2 ligand and the chelate ring size of NHC–Ru complexes are vital for their catalytic performance.
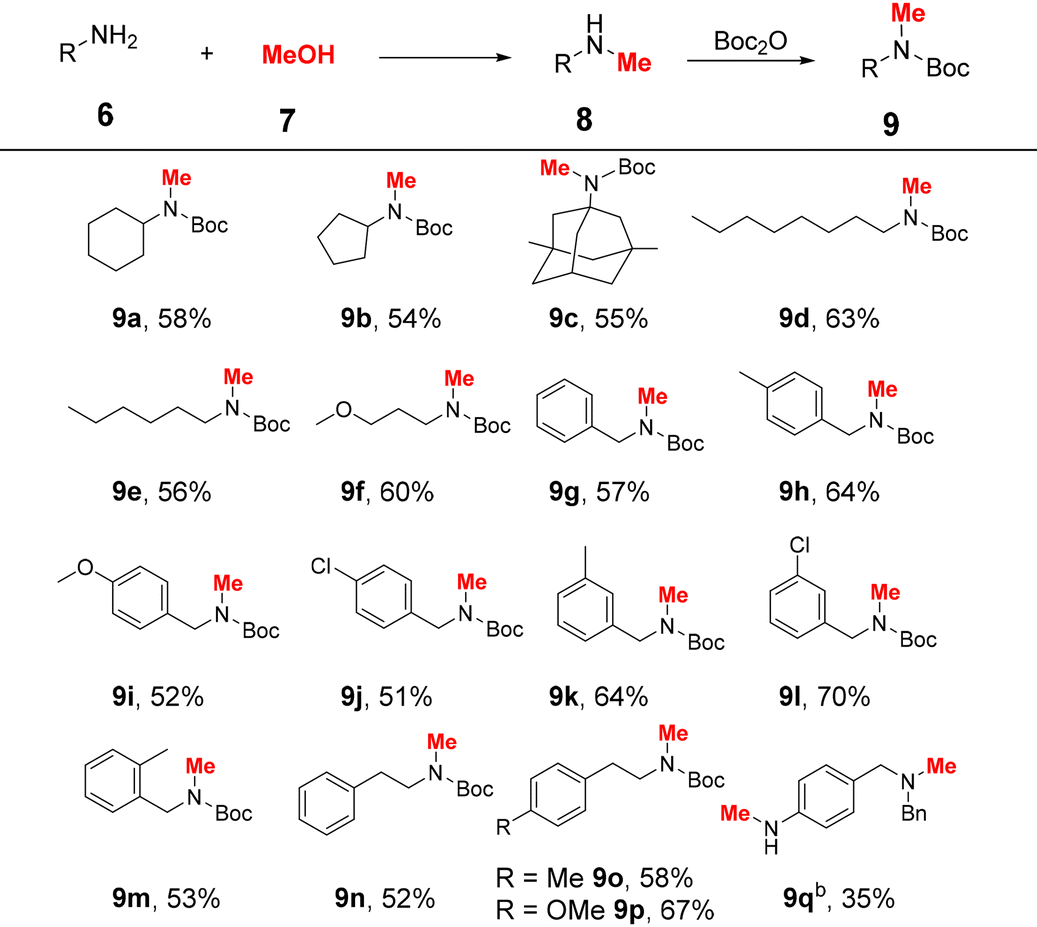
Inspired by the promising performance of the Ru4 complex, the reaction conditions were optimised (Table 1). First, various inorganic bases were investigated. However, replacing Cs2CO3 with other bases such as KOtBu, KOH, NaOtBu, NaOH, etc. resulted in decreased yields and provided products in 0–64% yields (entries 1–9). Subsequently, the amount of Cs2CO3 was tested (entries 10 and 11), and 0.2 equiv. of Cs2CO3 was sufficient to furnish the product in 85% yield. When the loading of Ru4 was reduced to 0.5 mol%, the yield of 8a decreased significantly (entry 12). Prolonging the reaction time to 12 h resulted in a slight decrease in the yield of 8a and an increase in the yield of 8aa (entry 13). Control experiments revealed that no product was formed when the reaction was performed without a catalyst or a base (entries 14 and 15). No product was observed when [((p-cymene)RuCl2)]2 was used as the catalyst (entry 16), indicating the excellent catalytic performance of our catalyst. The yield of product 8a decreased along with the decrease in the reaction temperature (Table S2,† entries 1 and 2). In addition, the influence of the amount of MeOH on the reaction was investigated (Table S2,† entries 3–7). When the amount of methanol was low, the yields of 8a were sharply decreased (Table S2,† entries 3–5). Inferior selectivity was observed when the amount of methanol was increased to 1 mL (Table S2,† entry 7). The optimal reaction conditions were demonstrated to be 20 mol% Cs2CO3 and 1 mol% Ru4 at 110 °C for 9 h (entry 10). The scope of aliphatic amines was explored under the optimal reaction conditions (Table 2). The reaction time was extended to 24 h to obtain sufficient yields during substrate screening. In general, aliphatic amines are more prone to the generation of N,N-dimethylated products because of their high nucleophilicity.6,10,38 In this catalytic system, aliphatic amines were selectively converted to the corresponding monomethylated products. To facilitate product isolation, the resulting reaction mixtures were directly subjected to Boc protection. Both cyclic and acyclic aliphatic primary amines were smoothly mono-methylated, and the desired products 9a–f were obtained in 54–63% yields. Notably, memantine (6c),39 a commercially available Alzheimer's drug, was converted to the corresponding methylated product, 9c, in 55% yield. Benzylamines with either electron-donating or electron-withdrawing groups afforded the corresponding products (9g–m) in 51–70% yields. Notably, sterically hindered 2-methylbenzylamine also provided the product 9m in 53% yield. When phenylethylamines were used as substrates, the corresponding products 9n–p were isolated in 52–67% yields. In the case of 4-aminobenzylamine (6q), the corresponding Bn group protected product 9q was obtained in 35% yield.
|
|
||||
|---|---|---|---|---|
| Entry | Base (loading, equiv.) | Time (h) | Yield of 8a (%) | Ratio (8a:8aa) |
| a Reaction conditions: 0.5 mmol 6a, 0.5 mL MeOH, 1.0 mol% Ru4, base, 110 °C, 9 h. b GC yields with mesitylene as the internal standard. c 0.5 mol% Ru4. d Without Cs2CO3. e Without Ru4. f [((p-Cymene)RuCl2)]2 as the catalyst. | ||||
| 1 | KOtBu (0.3) | 9 | 64 | 91:9 |
| 2 | KOH (0.3) | 9 | 39 | 97:3 |
| 3 | NaOtBu (0.3) | 9 | 37 | 87:13 |
| 4 | NaOH (0.3) | 9 | 23 | 91:9 |
| 5 | K2CO3 (0.3) | 9 | 64 | 94:6 |
| 6 | Na2CO3 (0.3) | 9 | 11 | 97:3 |
| 7 | KHCO3 (0.3) | 9 | 6 | 98:2 |
| 8 | NaHCO3 (0.3) | 9 | — | — |
| 9 | Cs2CO3 (0.3) | 9 | 82 | 91:9 |
| 10 | Cs2CO3 (0.2) | 9 | 85 | 90:10 |
| 11 | Cs2CO3 (0.1) | 9 | 67 | 87:13 |
| 12c | Cs2CO3 (0.3) | 9 | 49 | 96:4 |
| 13 | Cs2CO3 (0.3) | 12 | 81 | 89:11 |
| 14d | — | 9 | — | — |
| 15e | Cs2CO3 (0.3) | 9 | — | — |
| 16f | Cs2CO3 (0.3) | 9 | — | — |
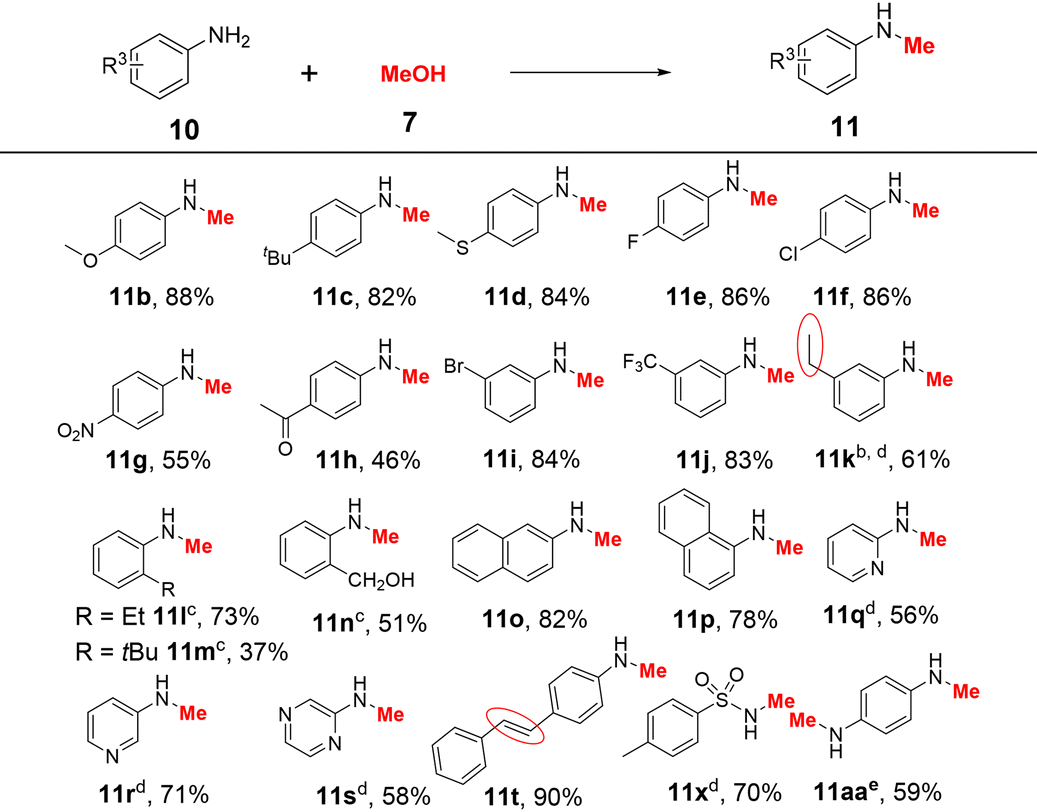
Next, we explored the variability of aromatic amines. Notably, the reactions were performed successfully using 10 mol% K2CO3 at 120 °C for 24 h (Table 3). The para- and meta-substituted aromatic amine derivatives containing various electron-withdrawing and electron-donating substituents were effectively converted to the corresponding monomethylated products in acceptable to excellent yields (46–88% for 11a–k). The steric hindrance of the substrate has a significant impact on the reaction activity (Scheme S6†). For example, when 2-ethylaniline, 2-tert-butyl aniline, and 2-aminobenzyl alcohol were used as substrates, the monomethylated products 11l–n were obtained in 37–73% yields at 130 °C using KOtBu as a base. Halogen substituents (F, Cl, and Br), functional groups (CH3O, CH3S, CF3, and CH2OH), and several reducible groups (NO2 and CH3CO) were well tolerated. In the case of 3-aminostyrene, the vinyl group was reduced and 11k was obtained in 61% yield. 2-Naphthylamine and 1-naphthylamine were also tested, and the corresponding products 11o and 11p were isolated in 82% and 78% yields, respectively. Heteroaromatic amines such as 2-aminopyridine, 3-aminopyridine, and aminopyrazine were successfully converted to the corresponding products 11q–s in 56–71% at 130 °C. Furthermore, this system was applied to 4-aminostilbene and p-methyl-benzenesulfonamide, affording the desired products 11t and 11x in 90% and 70% yields, respectively. p-Phenylenediamine (10aa) could react with methanol to afford product 11aa in 59% yield.
Several control experiments were performed to elucidate the reaction mechanism. First, we prepared the N-Me analogue (Ru4–Me) of complex Ru4 that showed decreased activity (Scheme 4a; 47% in comparison with 85% with Ru4) for the methylation reaction. This indicates the significant contribution of the N–H group of the ligand for the (de)hydrogenation reaction. The reaction was performed in the presence of 1 equiv. of mercury (Scheme 4b and S8†) and the yield was maintained. This result confirms the homogeneity of the catalytic reaction.40–42 Subsequently, two parallel reactions with CH3OH or CD3OD as methylating agents were conducted (Table S5†). A low kinetic isotope effect (KIE) (KH/KD = 1.3) was observed (Scheme 4c and S9†), indicating that the dehydrogenation of methanol might not be the rate-limiting step. Moreover, the N-methylation of 4-methylaniline proceeded with different deuterated methanol derivatives (CD3OD and CH3OD), and the deuteration pattern of the resulting products was investigated (Scheme 4d and S10†).9,12 When CD3OD was used as the starting material, a fully deuterated methyl group was found in the product (>95%), whereas CH3OD produced minor deuteration of the product's methyl group (13%). No deuterated NH groups were observed in either reaction. These results indicated that the methanol dehydrogenation step was reversible. Furthermore, several hydro signals ranging from −8 to −19 ppm were observed in the 1H NMR spectrum, indicating the presence of a [RuH] species (Fig. S5†).
 | ||
| Scheme 4 Preliminary mechanistic studies. | ||
To better understand the reaction mechanism, DFT calculations were performed (Scheme 5).43,44 The catalytic cycle involved three key steps: (1) dehydrogenation of methanol to formaldehyde, (2) in situ condensation of formaldehyde and amines to imines, and (3) hydrogenation of imines. Firstly, the active species IM0 (0.0 kcal mol−1) was produced by the activation of Ru4 with the assistance of a base and heat.32 From IM0, the dehydrogenation of methanol would occur via an inner- or outer-sphere mechanism. The inner-sphere pathway begins from IM1 (14.0 kcal mol−1), which is formed by the coordination of methanol to IM0. The proton of the methanol in IM1 is subsequently transferred to the NH ligand to provide IM2 (12.9 kcal mol−1) viaTS1 (18.5 kcal mol−1). Dissociation of the NH2 ligand affords IM3 (13.2 kcal mol−1). From IM3, β-hydride elimination (TS2, 20.3 kcal mol−1) provides IM4 (19.2 kcal mol−1). The overall barrier for the inner-sphere dehydrogenation of methanol is 20.3 kcal mol−1 relative to IM0. In the outer-sphere steps, the α-hydrogen and proton of methanol transfer synchronously to the Ru center and NH ligand, respectively, via the transition state TS5 (26.2 kcal mol−1). The overall barrier for the outer-sphere dehydrogenation is 26.2 kcal mol−1 relative to IM0, which is 5.9 kcal mol−1 higher than that of the inner-sphere pathway.
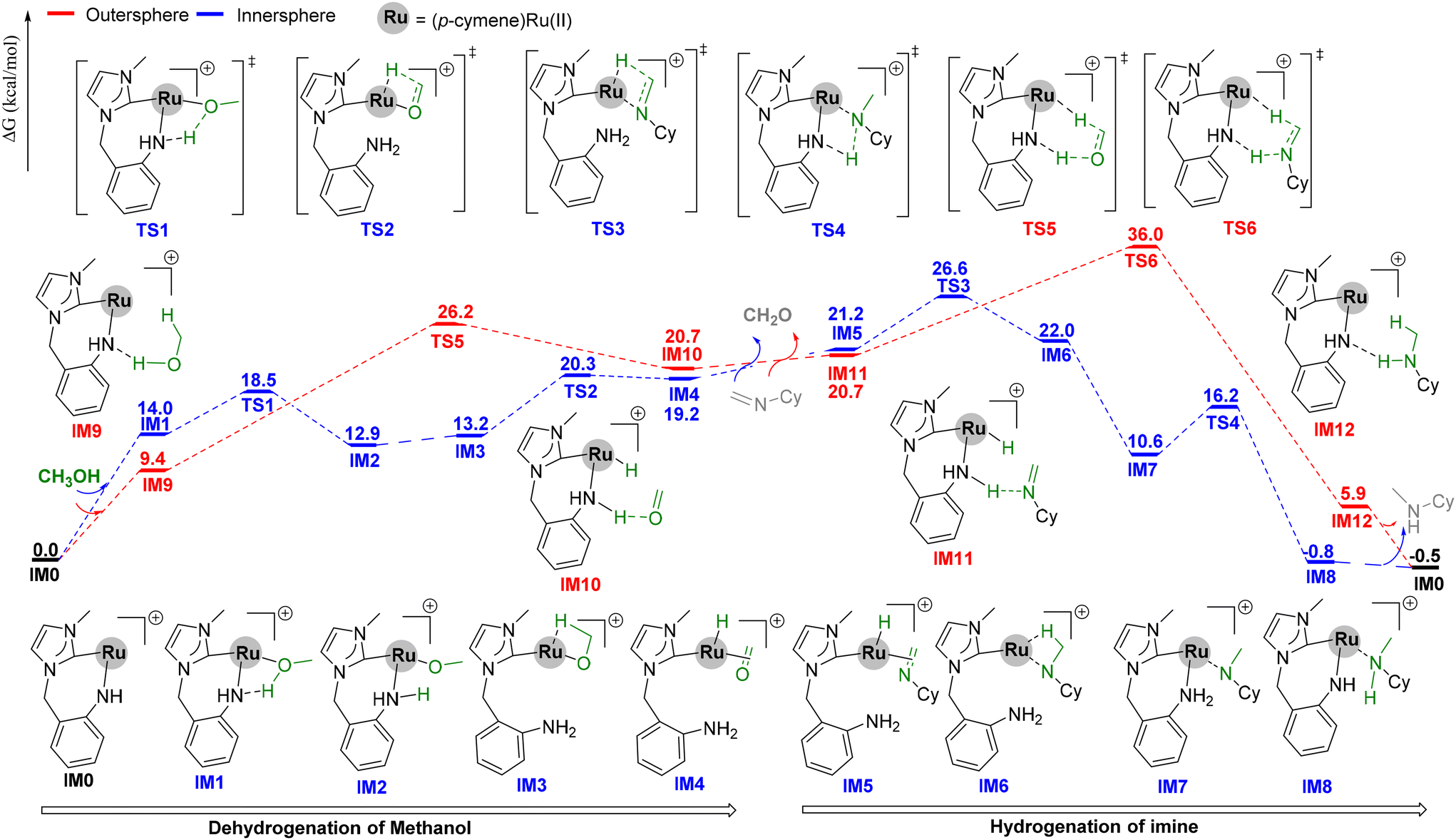 | ||
| Scheme 5 Free energy profile for Ru4-catalyzed N-methylation of amines with methanol. Free energies are given in kcal mol−1. | ||
The hydrogenation of the imine occurs in a manner similar to the reverse of alcohol oxidation. The inner-sphere pathway starts with the dissociation of the NH2 ligand and the coordination of imine to IM4, forming IM5 (21.2 kcal mol−1). From IM5, the hydrogenation of imine proceeds through transition states TS3 (26.6 kcal mol−1) and TS4 (16.2 kcal mol−1), leading to IM8 (−0.8 kcal mol−1). Finally, the product is released, and the catalytic species IM0 is regenerated. The overall barrier for the inner-sphere hydrogenation of imines is 26.6 kcal mol−1. In the outer-sphere pathway, the proton and hydride are transferred synchronously through TS6 (36.0 kcal mol−1). The overall energy barrier is 36.0 kcal mol−1, which is 9.4 kcal mol−1 higher than that of the inner-sphere pathway. Therefore, both the dehydrogenation and hydrogenation steps are favoured via the inner-sphere mechanism, and RDS is involved in the hydrogenation step, which is consistent with the experimental KIE results.
Conclusions
We developed an NHC–NH2 Ru system for the selective mono-N-methylation of amines with methanol using a carbonate salt as a base. This transformation proceeded successfully to afford various aromatic amines, sulphonamides, and aliphatic amines in high yields. Moreover, a wide range of sensitive groups such as nitro, carbonyl, hydroxyl, trifluoromethyl, and internal vinyl groups were well tolerated. A systematic study of these NHC–Ru complexes revealed that the aniline ligand and chelate ring size played crucial roles in enhancing the yield of the desired product. Experiments and DFT calculations revealed an inner-sphere mechanism promoted by the Ru complex with NH2 as the labile ligand, providing guidelines for the development of new NHC–transition metal complexes based on metal–ligand cooperation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Science Foundation of China (22002023).References
- J. Chatterjee, C. Gilon, A. Hoffman and H. Kessler, N-Methylation of Peptides: A New Perspective in Medicinal Chemistry, Acc. Chem. Res., 2008, 41, 1331–1342 CrossRef CAS PubMed.
- J. Chatterjee, F. Rechenmacher and H. Kessler, N-Methylation of Peptides and Proteins: An Important Element for Modulating Biological Functions, Angew. Chem., Int. Ed., 2013, 52, 254–269 CrossRef CAS PubMed.
- R. N. Salvatore, C. H. Yoon and K. W. Jung, Synthesis of secondary amines, Tetrahedron, 2001, 57, 7785–7811 CrossRef CAS.
- A. Corma, J. Navas and M. J. Sabater, Advances in One-Pot Synthesis through Borrowing Hydrogen Catalysis, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- T. Irrgang and R. Kempe, 3d-Metal Catalyzed N- and C-Alkylation Reactions via Borrowing Hydrogen or Hydrogen Autotransfer, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
- R. Liang, S. Li, R. Wang, L. Lu and F. Li, N-Methylation of Amines with Methanol Catalyzed by a Cp*Ir Complex Bearing a Functional 2,2′-Bibenzimidazole Ligand, Org. Lett., 2017, 19, 5790–5793 CrossRef CAS PubMed.
- D. Deng, B. Hu, M. Yang and D. Chen, Methylation of Amines and Ketones with Methanol Catalyzed by an Iridium Complex Bearing a 2-Hydroxypyridylmethylene Fragment, Organometallics, 2018, 37, 3353–3359 CrossRef CAS.
- X. Feng and M. Huang, Effect of the ancillary ligand in N-heterocyclic carbene iridium(III) catalyzed N-alkylation of amines with alcohols, Polyhedron, 2021, 205, 115289 CrossRef CAS.
- O. Ogata, H. Nara, M. Fujiwhara, K. Matsumura and Y. Kayaki, N-Monomethylation of Aromatic Amines with Methanol via PN(H)P-Pincer Ru Catalysts, Org. Lett., 2018, 20, 3866–3870 CrossRef CAS PubMed.
- P. Liu, N. T. Tung, X. Xu, J. Yang and F. Li, N-Methylation of Amines with Methanol in the Presence of Carbonate Salt Catalyzed by a Metal-Ligand Bifunctional Ruthenium Catalyst [(p-cymene)Ru(2,2′-bpyO)(H2O)], J. Org. Chem., 2021, 86, 2621–2631 CrossRef CAS PubMed.
- M. Huang, Y. Li, X. B. Lan, J. Liu, C. Zhao, Y. Liu and Z. Ke, Ruthenium(II) complexes with N-heterocyclic carbene-phosphine ligands for the N-alkylation of amines with alcohols, Org. Biomol. Chem., 2021, 19, 3451–3461 RSC.
- P. Piehl, R. Amuso, A. Spannenberg, B. Gabriele, H. Neumann and M. Beller, Efficient methylation of anilines with methanol catalysed by cyclometalated ruthenium complexes, Catal. Sci. Technol., 2021, 11, 2512–2517 RSC.
- A. Lator, S. Gaillard, A. Poater and J.-L. Renaud, Well-Defined Phosphine-Free Iron-Catalyzed N-Ethylation and N-Methylation of Amines with Ethanol and Methanol, Org. Lett., 2018, 20, 5985–5990 CrossRef CAS PubMed.
- K. Polidano, B. D. W. Allen, J. M. J. Williams and L. C. Morrill, Iron-Catalyzed Methylation Using the Borrowing Hydrogen Approach, ACS Catal., 2018, 8, 6440–6445 CrossRef CAS.
- S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Efficient and selective N-alkylation of amines with alcohols catalysed by manganese pincer complexes, Nat. Commun., 2016, 7, 12641 CrossRef PubMed.
- A. Bruneau-Voisine, D. Wang, V. Dorcet, T. Roisnel, C. Darcel and J.-B. Sortais, Mono-N-methylation of anilines with methanol catalyzed by a manganese pincer-complex, J. Catal., 2017, 347, 57–62 CrossRef CAS.
- M. Huang, Y. Li, Y. Li, J. Liu, S. Shu, Y. Liu and Z. Ke, Room temperature N-heterocyclic carbene manganese catalyzed selective N-alkylation of anilines with alcohols, Chem. Commun., 2019, 55, 6213–6216 RSC.
- D. Wei, O. Sadek, V. Dorcet, T. Roisnel, C. Darcel, E. Gras, E. Clot and J.-B. Sortais, Selective mono N-methylation of anilines with methanol catalyzed by rhenium complexes: An experimental and theoretical study, J. Catal., 2018, 366, 300–309 CrossRef CAS.
- V. Goyal, J. Gahtori, A. Narani, P. Gupta, A. Bordoloi and K. Natte, Commercial Pd/C-Catalyzed N-Methylation of Nitroarenes and Amines Using Methanol as Both C1 and H2 Source, J. Org. Chem., 2019, 84, 15389–15398 CrossRef CAS PubMed.
- G. Choi and S. H. Hong, Selective Monomethylation of Amines with Methanol as the C1 Source, Angew. Chem., Int. Ed., 2018, 57, 6166–6170 CrossRef CAS PubMed.
- N. Sarki, V. Goyal, N. K. Tyagi, Puttaswamy, A. Narani, A. Ray and K. Natte, Simple RuCl3-catalyzed N-Methylation of Amines and Transfer Hydrogenation of Nitroarenes using Methanol, ChemCatChem, 2021, 13, 1722–1729 CrossRef CAS.
- P. M. Illam and A. Rit, Electronically tuneable orthometalated RuII–NHC complexes as efficient catalysts for C–C and C–N bond formations via borrowing hydrogen strategy, Catal. Sci. Technol., 2022, 12, 67–74 RSC.
- P. A. Dub, B. L. Scott and J. C. Gordon, Why Does Alkylation of the N–H Functionality within M/NH Bifunctional Noyori-Type Catalysts Lead to Turnover?, J. Am. Chem. Soc., 2017, 139, 1245–1260 CrossRef CAS PubMed.
- P. A. Dub and J. C. Gordon, Metal–Ligand Bifunctional Catalysis: The “Accepted” Mechanism, the Issue of Concertedness, and the Function of the Ligand in Catalytic Cycles Involving Hydrogen Atoms, ACS Catal., 2017, 7, 6635–6655 CrossRef CAS.
- J. R. Khusnutdinova and D. Milstein, Metal–Ligand Cooperation, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- X. Xie and H. V. Huynh, Tunable Dehydrogenative Amidation versus Amination Using a Single Ruthenium-NHC Catalyst, ACS Catal., 2015, 5, 4143–4151 CrossRef CAS.
- W. Li, T. Wagener, L. Hellmann, C. G. Daniliuc, C. Mück-Lichtenfeld, J. Neugebauer and F. Glorius, Design of Ru(II)-NHC-Diamine Precatalysts Directed by Ligand Cooperation: Applications and Mechanistic Investigations for Asymmetric Hydrogenation, J. Am. Chem. Soc., 2020, 142, 7100–7107 CrossRef CAS PubMed.
- W. Li, M. Huang, J. Liu, Y.-L. Huang, X.-B. Lan, Z. Ye, C. Zhao, Y. Liu and Z. Ke, Enhanced Hydride Donation Achieved Molybdenum Catalyzed Direct N-Alkylation of Anilines or Nitroarenes with Alcohols: From Computational Design to Experiment, ACS Catal., 2021, 11, 10377–10382 CrossRef CAS.
- B. Arslan and S. Gülcemal, α-Alkylation of arylacetonitriles with primary alcohols catalyzed by backbone modified N-heterocyclic carbene iridium(I) complexes, Dalton Trans., 2021, 50, 1788–1796 RSC.
- Z. Ye, Z. Yang, C. Yang, M. Huang, X. Xu and Z. Ke, Disarming the alkoxide trap to access a practical FeCl3 system for borrowing-hydrogen N-alkylation, Org. Chem. Front., 2022, 9, 4803–4817 RSC.
- E. Jansen, L. S. Jongbloed, D. S. Tromp, M. Lutz, B. de Bruin and C. J. Elsevier, Ligand Effects on the Hydrogenation of Biomass-Inspired Substrates with Bifunctional Ru, Ir, and Rh Complexes, ChemSusChem, 2013, 6, 1737–1744 CrossRef CAS PubMed.
- H. Ohara, W. N. O. Wylie, A. J. Lough and R. H. Morris, Effect of chelating ring size in catalytic ketone hydrogenation: facile synthesis of ruthenium(II) precatalysts containing an N-heterocyclic carbene with a primary amine donor for ketone hydrogenation and a DFT study of mechanisms, Dalton Trans., 2012, 41, 8797–8808 RSC.
- B. Y. Tay, C. Wang, P. H. Phua, L. P. Stubbs and H. V. Huynh, Selective hydrogenation of levulinic acid to γ-valerolactone using in situ generated ruthenium nanoparticles derived from Ru–NHC complexes, Dalton Trans., 2016, 45, 3558–3563 RSC.
- F. E. Fernández, M. C. Puerta and P. Valerga, Ruthenium(II) Picolyl-NHC Complexes: Synthesis, Characterization, and Catalytic Activity in Amine N-alkylation and Transfer Hydrogenation Reactions, Organometallics, 2012, 31, 6868–6879 CrossRef.
- Z.-Q. Wang, X.-S. Tang, Z.-Q. Yang, B.-Y. Yu, H.-J. Wang, W. Sang, Y. Yuan, C. Chen and F. Verpoort, Highly active bidentate N-heterocyclic carbene/ruthenium complexes performing dehydrogenative coupling of alcohols and hydroxides in open air, Chem. Commun., 2019, 55, 8591–8594 RSC.
- P. Bhunia, E. Hwang, M. Min, J. Lee, S. Seo, S. Some and H. Lee, A non-volatile memory device consisting of graphene oxide covalently functionalized with ionic liquid, Chem. Commun., 2012, 48, 913–915 RSC.
- S. N. R. Donthireddy, P. M. Illam and A. Rit, Ruthenium(II) Complexes of Heteroditopic N-Heterocyclic Carbene Ligands: Efficient Catalysts for C–N Bond Formation via a Hydrogen-Borrowing Strategy under Solvent-Free Conditions, Inorg. Chem., 2020, 59, 1835–1847 CrossRef CAS PubMed.
- K.-i. Fujita, G. Toyooka and A. Tuji, Efficient and Versatile Catalytic Systems for the N-Methylation of Primary Amines with Methanol Catalyzed by N-Heterocyclic Carbene Complexes of Iridium, Synthesis, 2018, 4617–4626 CrossRef.
- C. Mount and C. Downton, Alzheimer disease: progress or profit?, Nat. Med., 2006, 12, 780–784 CrossRef CAS PubMed.
- H. Narjinari, N. Tanwar, L. Kathuria, R. V. Jasra and A. Kumar, Guerbet-type β-alkylation of secondary alcohols catalyzed by chromium chloride and its corresponding NNN pincer complex, Catal. Sci. Technol., 2022, 12, 4753–4762 RSC.
- J. A. Widegren and R. G. Finke, A review of the problem of distinguishing true homogeneous catalysis from soluble or other metal-particle heterogeneous catalysis under reducing conditions, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- K. Chakrabarti, B. Paul, M. Maji, B. C. Roy, S. Shee and S. Kundu, Bifunctional Ru(ii) complex catalysed carbon–carbon bond formation: an eco-friendly hydrogen borrowing strategy, Org. Biomol. Chem., 2016, 14, 10988–10997 RSC.
- A. Bartoszewicz, G. G. Miera, R. Marcos, P.-O. Norrby and B. Martín-Matute, Mechanistic Studies on the Alkylation of Amines with Alcohols Catalyzed by a Bifunctional Iridium Complex, ACS Catal., 2015, 5, 3704–3716 CrossRef CAS.
- M. Huang, Y. Li, J. Liu, X.-B. Lan, Y. Liu, C. Zhao and Z. Ke, A bifunctional strategy for N-heterocyclic carbene-stabilized iridium complex-catalyzed N-alkylation of amines with alcohols in aqueous media, Green Chem., 2019, 21, 219–224 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2281550. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01097j |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |