
Photoredox Ni-catalyzed decarboxylative arylation of oxamic acids for amide synthesis†
Depeng
Duan
and
Lu
Song
 *
*
Key Laboratory of Bio-Inspired Smart Interfacial Science and Technology of Ministry of Education, School of Chemistry, Beihang University, Beijing, China. E-mail: songlu@buaa.edu.cn
First published on 7th November 2023
Abstract
Decarboxylative cross-coupling of oxamic acids with (hetero)aryl halides has been accomplished through the synergistic merger of organic photoredox with nickel catalysis. Readily accessible oxamic acids serve as convenient starting materials that generate the key carbamoyl radicals via organic photoredox-catalyzed decarboxylation. This new, mild, and operationally simple amide synthesis protocol transforms a wide range of oxamic acids, along with (hetero)aryl halides, into the corresponding carbamides in good to excellent yields. This method is easily scalable and applicable to late-stage modification of complex molecules. Mechanistic data are consistent with the proposed reaction mechanism, in which the capture of carbamoyl radicals by ligated Ni(0) species occurs prior to the oxidative addition to an aryl electrophile.
Introduction
Amides are ubiquitous structural motifs commonly found in proteins, pharmaceuticals, agrochemicals, medicinally relevant natural products, and functional materials.1 In fact, the amide bond-forming reactions are among the most executed transformations in synthetic chemistry.2 Despite their significant importance, amide bond synthesis relies largely on the coupling reactions of amines and carboxylic acid derivatives by the use of stoichiometric amounts of dehydrative coupling reagents, which are usually expensive, toxic, and will generate stoichiometric quantities of waste by-products. In the context of practical applications, these synthetic methods are often hampered by the cost associated with the high molecular weights of coupling reagents along with the environmental impact of waste streams. Thus, the development of more efficient and greener approaches for the synthesis of the amide functionality, in a catalytic and waste-free fashion, is still in great demand in the synthetic chemistry community.3Motivated by the carbonylation process using carbon monoxide (CO) as the carbonyl source, transition metal-catalyzed carbonylative amidation of organohalides under a CO atmosphere becomes the key advance in catalytic amidation (Scheme 1A).4 However, mainly due to the strong coordination ability of CO to the metal center, the oxidative addition step of organohalide to the metal center will be confronted with a high energy barrier in order to overcome metal catalyst deactivation by CO coordination, thus resulting in high reaction temperature or high CO pressure conditions. Although it is an attractive strategic innovation, the necessity for the use of toxic CO gas under forcing conditions, together with precious transition metal catalysts has compromised wider adoption of this method.5
 | ||
| Scheme 1 Background information and working hypothesis of catalytic amide synthesis with oxamic acids as carbamoyl radical precursors. | ||
Recently, the synergistic merger of single-electron activation strategies with transition metal catalysis has empowered synthetic chemists with new and powerful tools for addressing modern synthetic problems.6 As an elegant example, photoredox catalysis has been well recognized as an effective tool in energy conversion transformations and has become a pillar of modern synthetic chemistry in the past decade.7 In particular, highly reactive radical species generated in a mild and selective manner could be involved in transition metal-catalyzed processes to access novel reactivities that are otherwise challenging or impossible in traditional chemistry.8 Related to our present work, Melchiorre and co-workers reported that redox-active 4-carbamoyl-1,4-dihydropyridine derivatives could be employed as carbamoyl radical sources and applied in photoredox nickel-catalyzed carbamidation of aryl bromides with good reaction efficiency (Scheme 1B, eqn (1)). However, the lengthy preparation steps of this kind of carbamoyl radical precursor make this method less practical with low atom economy.9 Quite recently, the Maiti group devised a silyl radical-mediated protocol for the generation of carbamoyl radicals from widely available carbamoyl chlorides and successfully demonstrated its application in photoredox nickel-catalyzed carbamidation of aryl bromides (Scheme 1B, eqn (2)). The cross-electrophile coupling design of this work is efficient, but this approach is limited by the need for expensive stoichiometric tris(trimethylsilyl)silane, which is critical to the formation of carbamoyl radicals via halogen atom transfer from carbamoyl chloride.10
Oxamic acids are bench stable and can be easily prepared by reaction of amines with commercially available oxalic acid monoester derivatives. Furthermore, single-electron oxidation-mediated decarboxylation of oxamic acids for the generation and application of carbamoyl radicals has been well established in the literature.11 In contrast to this background information, however, decarboxylative arylation of oxamic acid derivatives for the synthesis of amides remains rare.12 Herein, we describe a photoredox Ni-catalyzed protocol for the synthesis of aryl amides by reaction of readily accessible oxamic acids with aryl halides (Scheme 1C). Specifically, we anticipated that single-electron oxidation of oxamic acids would produce the required carbamoyl radical species Avia radical decarboxylation. Concurrently, we hoped that the nickel-mediated activation of aryl halides via oxidative addition to the low-valent nickel center would generate the aryl nickel(II) intermediate B, which is capable of trapping the carbon-center radical A to yield the high-valent nickel(III) adduct C. Upon feasible reductive elimination, the desired amide coupling product will be delivered along with the generation of nickel(I) species. An organic photoredox catalyst is designed to act as an electron shuttle to enable the single-electron transfer events in the reaction.13
Results and discussion
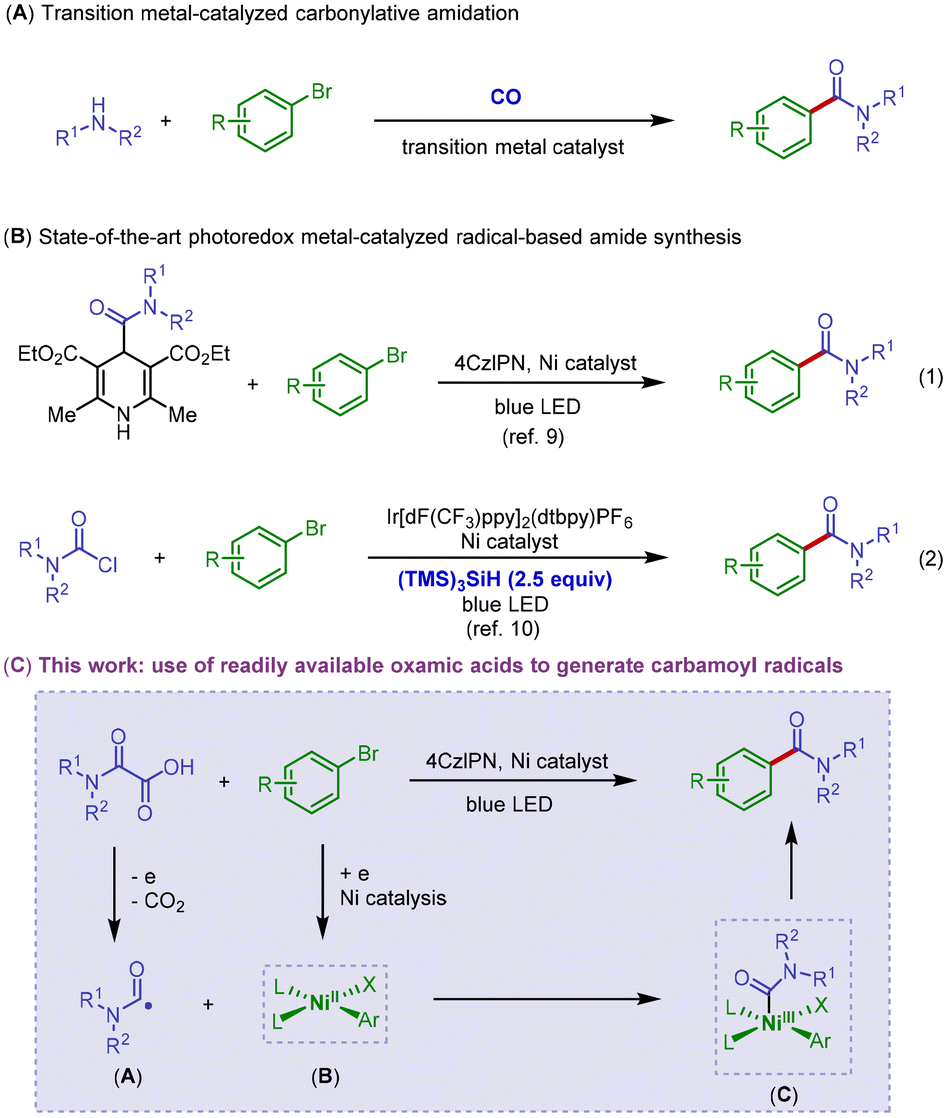
Our initial investigations into the proposed decarboxylative carbamidation reaction focused on the reaction of commercially available N,N-diethyloxamic acid 1 with 1-bromo-4-fluorobenzene 2 (Table 1). We were delighted to find that the irradiation of oxamic acid and aryl bromide in the presence of organic photocatalyst 4CzIPN (2.5 mol%), NiBr2·glyme (10 mol%), dtbbpy L2 (15 mol%), and Na2CO3 in dioxane provided the desired amide product 3 in 30% yield (entry 1). Screening of reaction media resulted in no improvement in the reaction efficiency (entry 2 and see the ESI† for more information).14 However, after surveying several cosolvents, we discovered that the solvent system of dioxane/acetonitrile (MeCN) led to a notable increase in reaction efficiency (entries 3–5). Ligand optimization for the nickel center indicated that electron-deficient bipyridine showed essentially no catalytic activity (entry 8), likely implying that the activation of the aryl electrophile via a nickel-catalyzed oxidative addition step was unfavorable. This is consistent with the observation that the use of sterically congested ligand L5 also showed no catalytic efficiency (entry 9). Ultimately, using electron-rich ligand L3 and increasing the photocatalyst loading to 3.0 mol% provided the best reaction conditions, delivering the desired coupling product in an excellent yield (entry 12, in 80% isolated yield).|
|
|||
|---|---|---|---|
| Entry | Solvent | Ligand | Yieldb (%) |
| a Conditions: reactions were performed on a 0.2 mmol scale at ambient temperature for 12 hours under illumination of blue LEDs (440 nm, 10 W) with 1 (0.3 mmol, 1.5 equiv.), 2 (0.2 mmol, 1.0 equiv.), and Na2CO3 (1.5 equiv.) in 2.0 mL of solvent. b Yield was determined by 19F NMR analysis of the crude mixture using 1-bromo-2-fluorobenzene as the internal standard. c 4CzIPN loading was 1.0 mol%. d 4CzIPN loading was 3.0 mol%. e Yield of the isolated product is given in parentheses. | |||
| 1 | Dioxane | L2 | 30 |
| 2 | MeCN | L2 | 23 |
| 3 | MeCN/dioxane (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 1:3) :v = 1:3) |
L2 | 62 |
| 4 | MeCN/dioxane (v:v = 1:1) |
L2 | 56 |
| 5 | MeCN/dioxane (v:v = 3:1) |
L2 | 37 |
| 6 | MeCN/dioxane (v:v = 1:3) |
L1 | 44 |
| 7 | MeCN/dioxane (v:v = 1:3) |
L3 | 63 |
| 8 | MeCN/dioxane (v:v = 1:3) |
L4 | <5 |
| 9 | MeCN/dioxane (v:v = 1:3) |
L5 | <5 |
| 10 | MeCN/dioxane (v:v = 1:3) |
L6 | 19 |
| 11c | MeCN/dioxane (v:v = 1:3) |
L3 | 46 |
| 12d | MeCN/dioxane (v:v = 1:3) |
L3 | 81(80)e |
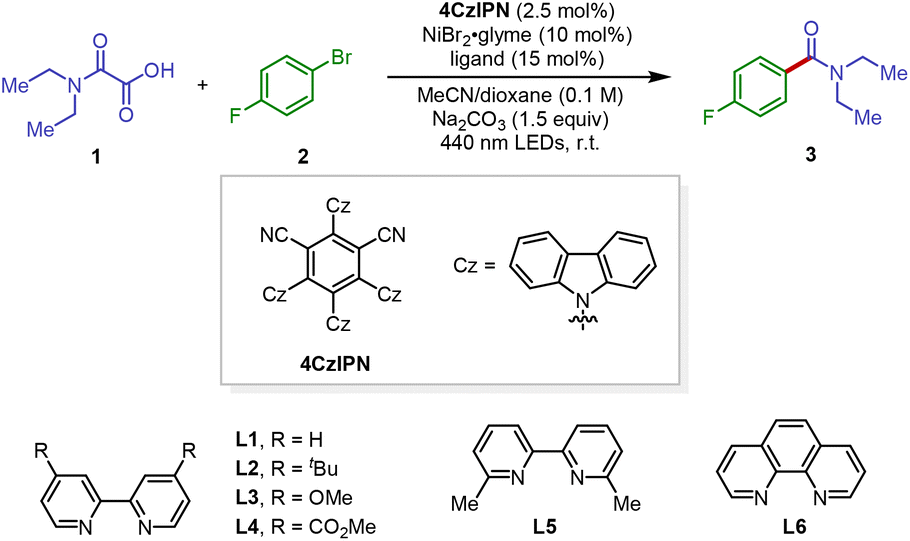
With the optimized reaction conditions in hand, we subsequently sought to determine the substrate scope and functional group compatibility of our decarboxylative carbamidation protocol (Scheme 2). In general, a diverse array of electron-deficient aryl bromides underwent the reaction well to deliver the corresponding amide products in good yields (3–10). Specifically, aryl bromides that contain a wide variety of synthetically versatile substituents including fluorine (3 and 8), trifluoromethyl (4), ester (5), aldehyde (6), ketone (9) and nitrile (7) groups were found to be excellent coupling substrates, affording the corresponding decarboxylative amidation products in good yields (3–9, 69–86% yields). However, presumably due to the steric sensitivity in the oxidative addition step, the reaction efficiency was impeded by an ortho substituent on the aromatic ring (10).
 | ||
| Scheme 2 Aryl halide scope of photoredox Ni-catalyzed decarboxylative arylation with oxamic acids: electronic properties and substitution patterns. | ||
In addition to electron-deficient electrophiles, we were pleased to find that both electron-neutral and electron-rich aryl bromides were also effective electrophiles, providing the desired coupling products with generally good reaction yields (11–16). Lastly, synthetically useful handles such as N-methoxy amide (17) and Bpin (18), and medicinally important sulfonamides (19 and 20) could be well tolerated in the reaction.
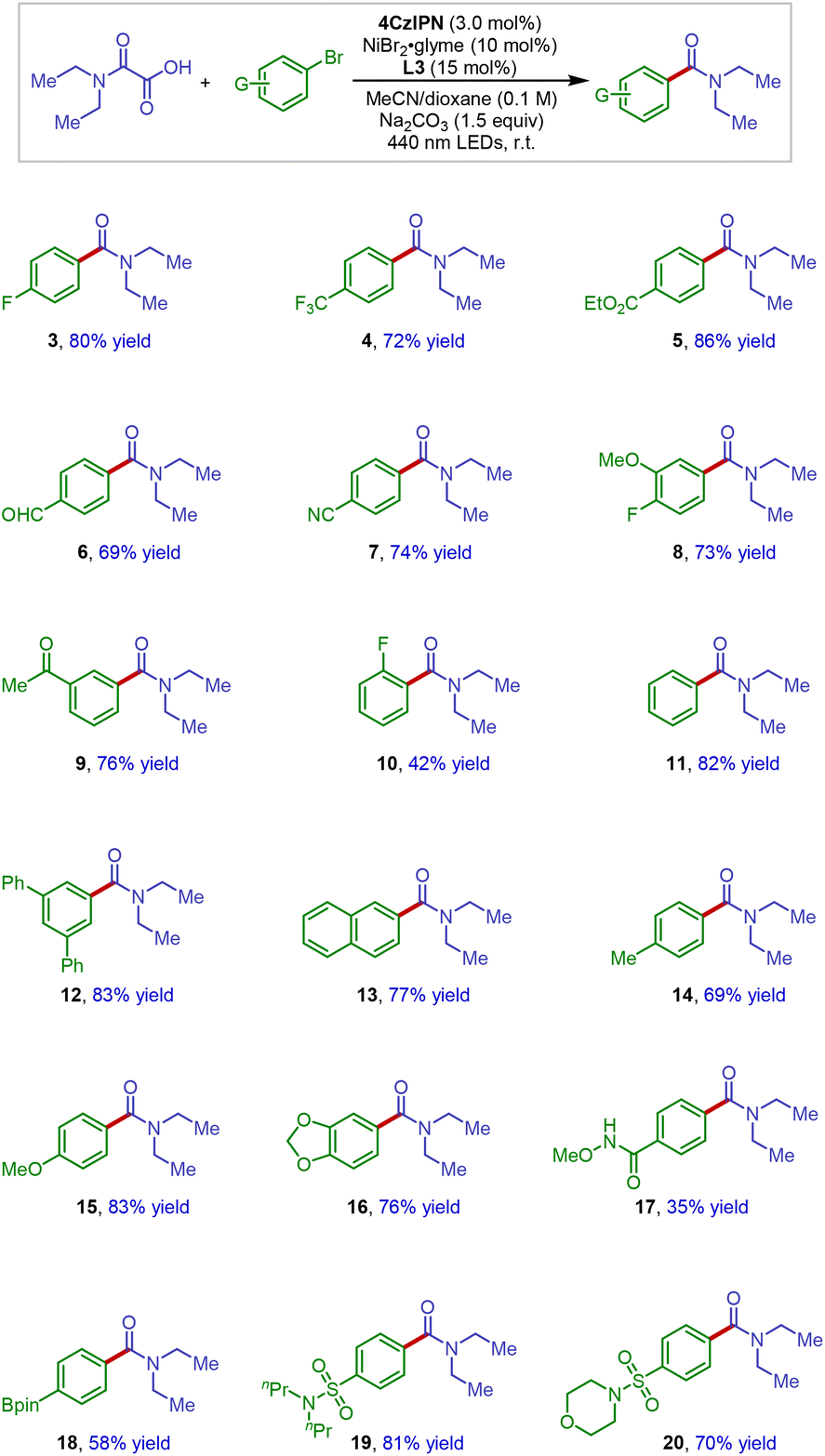
To demonstrate the applicability of this new amide synthesis protocol in medicinal chemistry, the scope of hetero aryl bromides was next investigated (Scheme 3). It has been reported that heterocycles bearing coordination sites are challenging substrates in photoredox nickel catalysis.15,16 Thus, it is noteworthy that a wide variety of medicinally attractive motifs such as pyridine (21), quinoline (22), thiophenes (23 and 24), dibenzothiophenes (24 and 25), benzothiophene (27), dibenzofuran (29), pyrazol (31), and even benzothiazole (28) could be successfully incorporated under our reaction conditions, giving the cross-coupling products with high reaction efficiency. These results clearly demonstrate the synthetic utility of this method in medicinal chemistry and also suggest potential application in late-stage modification of complex molecular structures. Indeed, an amide product derived from diacetonefructose (32) could be smoothly obtained in 90% isolated yield.
 | ||
| Scheme 3 Aryl halide scope of photoredox Ni-catalyzed decarboxylative arylation with oxamic acids: heteroaromatic coupling partners and complex molecules. | ||
We next examined the scope of the oxamic acid component in this new decarboxylative amidation protocol. As shown in Scheme 4, when using aryl bromide 33 as the representative electrophile in the reaction, a series of structurally diverse oxamic acid derivatives underwent the efficient cross-coupling reaction to afford the amide products in synthetically useful yields (34–41). In the case of the synthesis of secondary amide products, the reaction efficiency appeared to be influenced by the free N–H group, with more sterically hindered amides resulting in better yields (38–41).
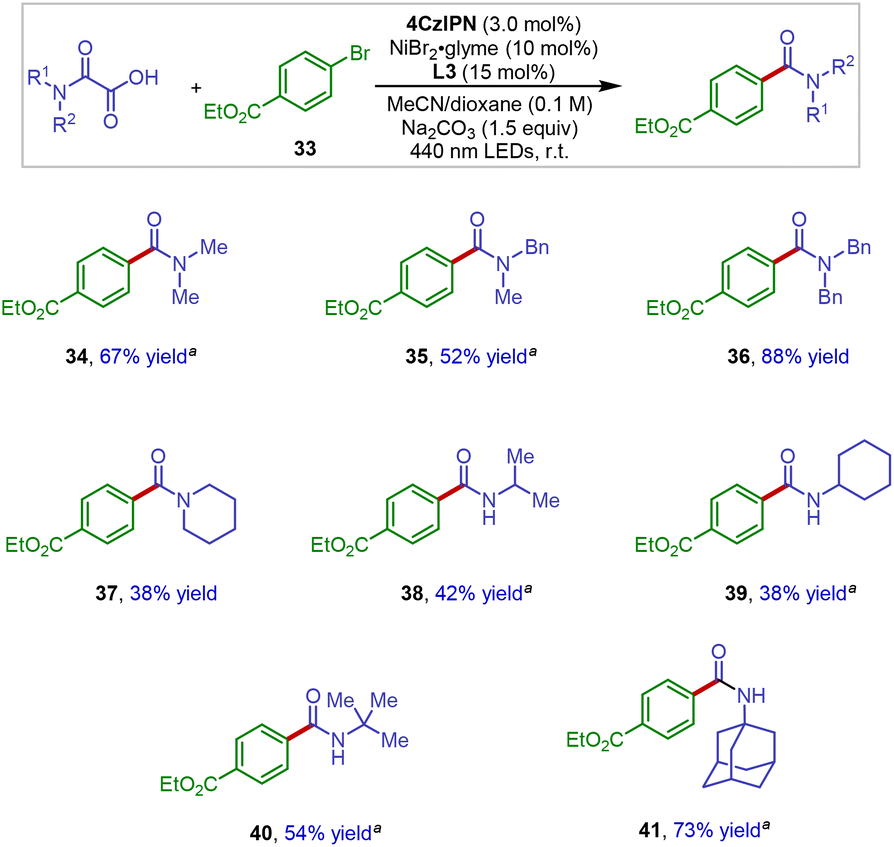 | ||
| Scheme 4 Oxamic acid scope in photoredox Ni-catalyzed decarboxylative arylation. aTMG (1,1,3,3-tetramethylguanidine) was used instead of Na2CO3 as the base additive. | ||
Finally, for practical reactions, we attempted to augment the synthetic value of this new decarboxylative amidation reaction by developing a gram-scale procedure. As shown in Scheme 5, this protocol could be straightforwardly scaled up to 6.0 mmol without any modification to the reaction conditions (Scheme 5, eqn (1)). The reaction between N,N-diethyloxamic acid 1 and ethyl 4-bromobenzoate 33 produced the corresponding amide product 5 in 79% isolated yield. More importantly, under the standard conditions, this photoredox catalytic system could be successfully applied to decarboxylative cross-coupling of oxamic acid 1 with vinyl bromide 42, giving α,β-unsaturated amide product 43 in 69% yield (Scheme 5, eqn (2)). Our reaction thus provides a new and efficient means to access α,β-unsaturated amides from readily available oxamic acids and vinyl halides.
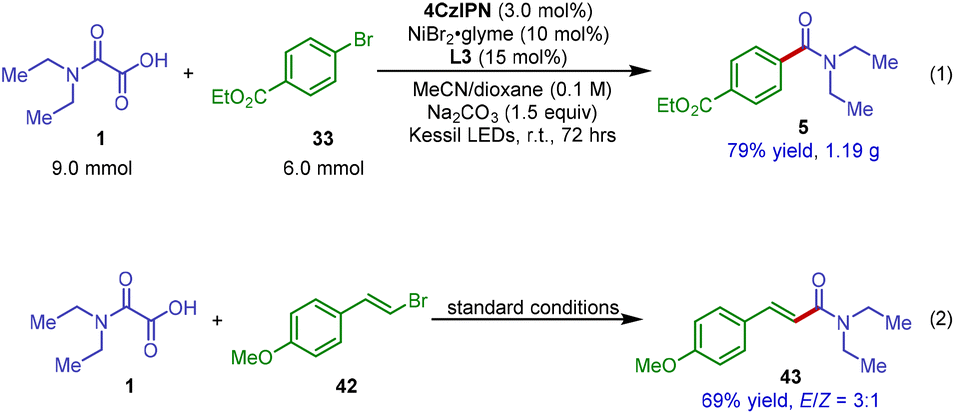 | ||
| Scheme 5 Gram-scale synthesis and strategy expansion to decarboxylative coupling with vinyl bromide for the synthesis of unsaturated amide. | ||
A series of control experiments were conducted to gain insights into the reaction mechanism of this new photoredox nickel-catalyzed decarboxylative amidation method. As expected, no desired amide product could be observed by omitting each individual component from the reaction system, thus highlighting the crucial roles played by the photoredox catalyst, nickel catalyst, ligand, light, and base additive in promoting the designed transformation.17 In addition, cyclic voltammetry studies of oxamic acid 1 in the presence of a base exhibited an irreversible oxidation wave with Ep = 1.05 V vs. SCE.18 It has been reported that the photoexcitation of the photoredox catalyst 4CzIPN produces a highly oxidizing species (4CzIPN*/4CzIPN˙− = 1.43 V vs. SCE in acetonitrile).19 This finding thus provides strong support for the involvement of an electron transfer event between the excited photoredox catalyst 4CzIPN* and the oxamic acid substrate, which triggers the radical decarboxylation process to generate the key carbamoyl radical A for the reaction.
To further confirm the radical nature of the reaction, we performed the reaction in the presence of radical scavengers (Fig. 1A). For example, by adding TEMPO to the reaction system, the reaction was fully inhibited and no desired product could be detected (Fig. 1A, eqn (1)). Moreover, when the reaction was carried out with the addition of 45, the radical addition product 46 was isolated in 30% yield (Fig. 1A, eqn (2)).20 These mechanistic data clearly demonstrated the formation of carbamoyl radical species in the reaction.
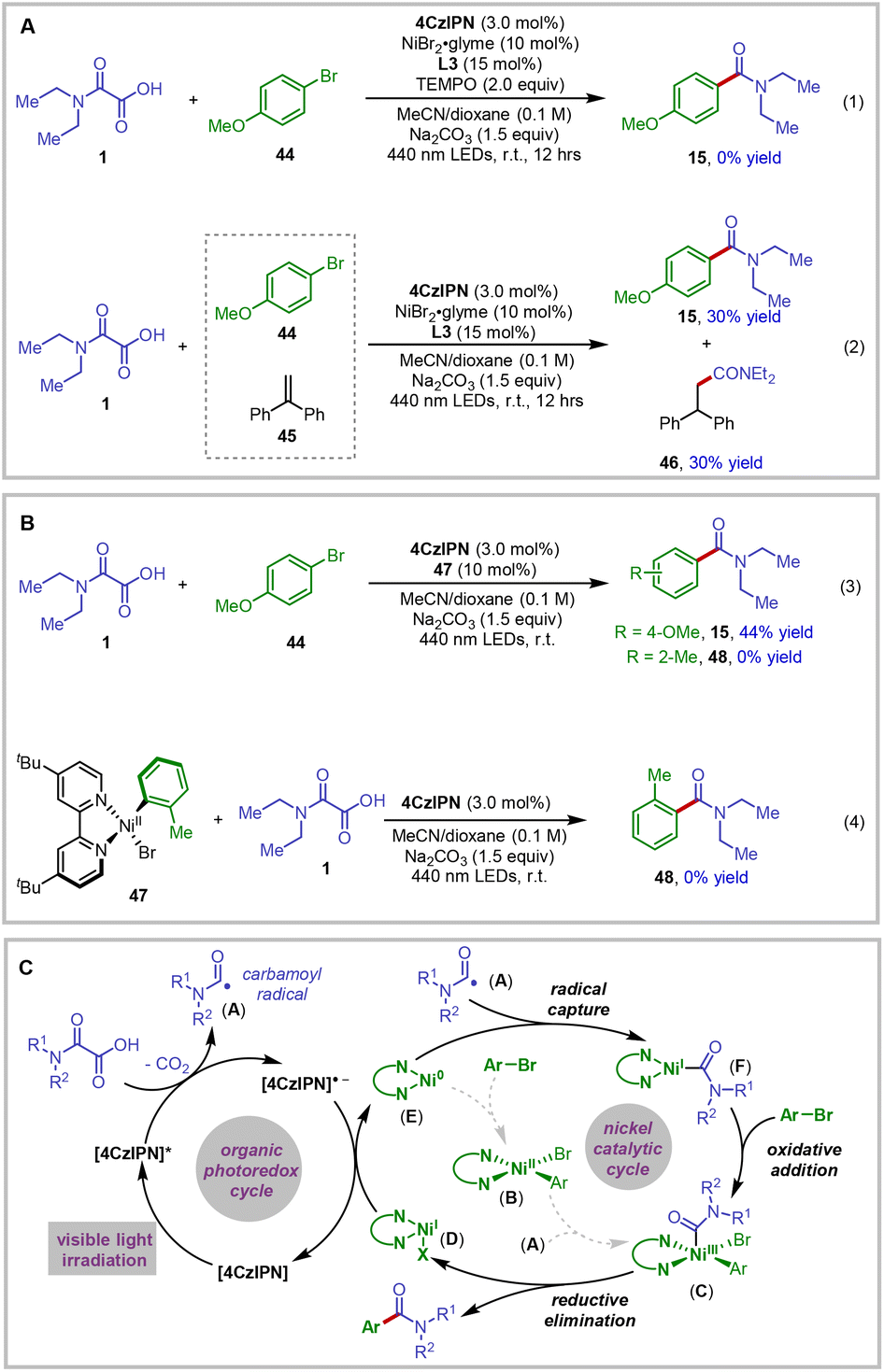 | ||
| Fig. 1 Mechanistic studies and the proposed reaction mechanism. (A) Radical trapping experiments; (B) experiments with nickel complex 47; (C) proposed reaction mechanism. | ||
Mechanistically, two alternative reaction pathways are in theory possible for the generation of carbamoyl aryl Ni(III) intermediates en route to the product formation via reductive elimination from the nickel center.21 Aryl nickel(II) complex 47 was thus synthesized and subjected to the standard conditions to further probe the reaction mechanism.22 As shown in Fig. 1B, the use of nickel complex 47 as a precatalyst delivered the decarboxylative coupling product 15 in 44% yield from the reaction of 44 and 1. However, the amide product 48, formed through the addition of the carbamoyl radical to the aryl nickel(II) complex 47, could not be detected (eqn (3)). Meanwhile, the experiment using stoichiometric aryl nickel(II) complex 47 failed to give the amide product 48 (eqn (4)). These reaction results led us to the conclusion that the aryl nickel(II) could not be the intermediate in the productive reaction pathway.22b,d
On the basis of our experimental observations, a plausible catalytic mechanism of the reaction is proposed in Fig. 1C. It is well established that the organic photoredox catalyst 4CzIPN readily absorbs photons upon visible-light irradiation to generate the highly oxidizing excited state.19 Single electron transfer from the oxamic acid to the excited photoredox catalyst 4CzIPN*, probably promoted by the base in the reaction system, could be thermodynamically feasible. This results in the formation of the corresponding carboxyl radical, which readily undergoes CO2 extrusion to produce the key carbamoyl radical A. This racial could be captured by ligated Ni(0) species E to afford Ni(I) intermediate F.23 Subsequent oxidative addition of aryl bromide to this nickel species should deliver carbamoyl aryl Ni(III) C. Thereafter, reductive elimination would afford the desired amide product and Ni(I) species D. After that, single electron transfer from the reduced photocatalyst 4CzIPN˙− (4CzIPN/4CzIPN˙− = −1.21 V vs. SCE in acetonitrile)19 to the Ni(I) species D would simultaneously turn over the nickel and photoredox catalytic cycles.24
Conclusions
To summarize, we have developed a novel, practical, and widely applicable protocol for decarboxylative coupling of oxamic acids with (hetero)aryl halides. This method provides an alternative means for amide synthesis. Notably, this new synthetic strategy relies on the use of more accessible and bench-stable oxamic acid derivatives to generate carbamoyl radicals under mild conditions and exhibits broad substrate scope and wide functional group compatibility with respect to both coupling partners. The potential synthetic utility has been demonstrated by gram-scale and complex organic molecule synthesis. Future efforts by our group are directed toward expanding the new radical-based application of oxamic acids to other types of synthetically valuable transformations.Author contributions
L.S. designed the project. D.D. performed the experiments and collected the data. Both authors analysed the data and contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (No. 22101014) and the Beihang University Young Talent Support Program for financial support.References
- G. Arthur, The Amide Linkage: Selected Structural Aspects in Chemistry, Biochemistry, and Materials Science, Wiley-Interscience, 2000 Search PubMed.
- G. Benz, Synthesis of Amides and Related Compounds: Comprehensive Organic Synthesis, Pergamon, 1991 Search PubMed.
- (a) V. R. Pattabiraman and J. W. Bode, Rethinking amide bond synthesis, Nature, 2011, 480, 471–479 CrossRef CAS; (b) E. Massolo, M. Pirola and M. Benaglia, Amide Bond Formation Strategies: Latest Advances on a Dateless Transformation, Eur. J. Org. Chem., 2020, 4641–4651 CrossRef CAS.
- (a) A. Brennführer, H. Neumann and M. Beller, Palladium-Catalyzed Carbonylation Reactions of Aryl Halides and Related Compounds, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef; (b) C. Shen and X.-F. Wu, Palladium-Catalyzed Carbonylative Multicomponent Reactions, Chem. – Eur. J., 2017, 23, 2973–2987 CrossRef.
- (a) R. Grigg and S. P. Mutton, Pd-catalysed carbonylations: versatile technology for discovery and process chemists, Tetrahedron, 2010, 66, 5515–5548 CrossRef CAS; (b) J. R. Martinelli, T. P. Clark, D. A. Watson, R. H. Munday and S. L. Buchwald, Palladium-Catalyzed Aminocarbonylation of Aryl Chlorides at Atmospheric Pressure: The Dual Role of Sodium Phenoxide, Angew. Chem., Int. Ed., 2007, 46, 8460–8463 CrossRef CAS; (c) P. G. Alsabeh, M. Stradiotto, H. Neumann and M. Beller, Aminocarbonylation of (Hetero)aryl Bromides with Ammonia and Amines using a Palladium/DalPhos Catalyst System, Adv. Synth. Catal., 2012, 354, 3065–3070 CrossRef CAS.
- (a) L. Song and N. Fu, Single-Electron Strategies in Organometallic Methods: Photoredox, Electrocatalysis, Radical Relay, and Beyond, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O'Hare, Elsevier, 2022, 13, 339–403 Search PubMed; (b) M. D. Levin, S. Kim and F. D. Toste, Photoredox Catalysis Unlocks Single-Electron Elementary Steps in Transition Metal Catalyzed Cross-Coupling, ACS Cent. Sci., 2016, 2, 293–301 CrossRef CAS; (c) G. C. Fu, Transition-Metal Catalysis of Nucleophilic Substitution Reactions: A Radical Alternative to SN1 and SN2 Processes, ACS Cent. Sci., 2017, 3, 692–700 CrossRef CAS PubMed; (d) F. Wang, P. Chen and G. Liu, Copper-Catalyzed Radical Relay for Asymmetric Radical Transformations, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS; (e) Z. L. Li, G. C. Fang, Q. S. Gu and X. Y. Liu, Recent advances in copper-catalysed radical-involved asymmetric 1,2-difunctionalization of alkenes, Chem. Soc. Rev., 2020, 49, 32–48 RSC; (f) J. Liu, L. Lu, D. Wood and S. Lin, New Redox Strategies in Organic Synthesis by Means of Electrochemistry and Photochemistry, ACS Cent. Sci., 2020, 6, 1317–1340 CrossRef CAS.
- (a) L. Candish, K. D. Collins, G. C. Cook, J. J. Douglas, A. Gómez-Suárez, A. Jolit and S. Keess, Photocatalysis in the Life Science Industry, Chem. Rev., 2022, 122, 2907–2980 CrossRef CAS PubMed; (b) D. A. Corbin and G. M. Miyake, Photoinduced Organocatalyzed Atom Transfer Radical Polymerization (O-ATRP): Precision Polymer Synthesis Using Organic Photoredox Catalysis, Chem. Rev., 2022, 122, 1830–1874 CrossRef CAS; (c) N. Holmberg-Douglas and D. A. Nicewicz, Photoredox-Catalyzed C–H Functionalization Reactions, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS; (d) L. Capaldo, D. Ravelli and M. Fagnoni, Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C–H Bonds Elaboration, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed; (e) L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Alkoxy Radicals See the Light: New Paradigms of Photochemical Synthesis, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed; (f) S. P. Pitre and L. E. Overman, Strategic Use of Visible-Light Photoredox Catalysis in Natural Product Synthesis, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS.
- (a) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Ed-ward, L. I. Frye, O. L. Garry, M. N. Lavagnino, B.-X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed; (b) K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Visible Light-Induced Transition Metal Catalysis, Chem. Rev., 2022, 122, 1543–1625 CrossRef.
- N. Alandini, L. Buzzetti, G. Favi, T. Schulte, L. Candish, K. D. Collins and P. Melchiorre, Amide Synthesis by Nickel/Photoredox-Catalyzed Direct Carbamoylation of (Hetero)Aryl Bromides, Angew. Chem., Int. Ed., 2020, 59, 5248–5253 CrossRef CAS PubMed.
- S. Maiti, S. Roy, P. Ghosh, A. Kasera and D. Maiti, Photo-Excited Nickel-Catalyzed Silyl-Radical-Mediated Direct Activation of Carbamoyl Chlorides To Access (Hetero)aryl Carbamides, Angew. Chem., Int. Ed., 2022, 61, e202207472 CrossRef CAS PubMed.
- I. M. Ogbu, G. Kurtay, F. Robert and Y. Landais, Oxamic acids: useful precursors of carbamoyl radicals, Chem. Commun., 2022, 58, 7593–7607 RSC.
- W. M. Cheng, R. Shang, H. Z. Yu and Y. Fu, Room-Temperature Decarboxylative Couplings of α-Oxocarboxylates with Aryl Halides by Merging Photoredox with Palladium Catalysis, Chem. – Eur. J., 2015, 21, 13191–13195 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly efficient organic light-emitting diodes from delayed fluorescence, Nature, 2012, 492, 234–238 CrossRef CAS.
- Please refer to Table S1 in the ESI† for details.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Merging photoredox with nickel catalysis: Coupling ofα-carboxyl sp3-carbons with aryl halides, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- C. N. P. Kullmer, J. A. Kautzky, S. W. Krska, T. Nowak, S. D. Dreher and D. W. C. MacMillan, Accelerating reaction generality and mechanistic insight through additive mapping, Science, 2022, 376, 532–539 CrossRef CAS.
- Please refer to Table S7 in the ESI† for details.
- Please refer to Fig. S2 in the ESI† for details.
- J. Luo and J. Zhang, Donor–Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(sp3)–C(sp2) Cross-Coupling, ACS Catal., 2016, 6, 873–877 CrossRef CAS.
- L. Cardinale, M. O. Konev and A. J. von Wangelin, Photoredox-Catalyzed Addition of Carbamoyl Radicals to Olefins: A 1,4-Dihydropyridine Approach, Chem. – Eur. J., 2020, 26, 8239–8243 CrossRef CAS PubMed . For more radical trapping experiments, please see the ESI† for details.
- J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Alkyl Carbon–Carbon Bond Formation by Nickel/Photoredox Cross-Coupling, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS.
- (a) G. Li, L. Yang, J.-J. Liu, W. Zhang, R. Cao, C. Wang, Z. Zhang, J. Xiao and D. Xue, Light-Promoted C–N Coupling of Aryl Halides with Nitroarenes, Angew. Chem., Int. Ed., 2021, 60, 5230–5234 CrossRef CAS PubMed; (b) W.-D. Li, Y.-Q. Jiang, Y.-L. Li and J.-B. Xia, Photoredox Ni-Catalyzed Selective Coupling of Organic Halides and Oxalates to Esters via Alkoxycarbonyl Radical Intermediates, CCS Chem., 2022, 4, 1326–1336 CrossRef CAS; (c) P. Xue, L. Li and N. Fu, Pairing Iron and Nickel Catalysis for Electrochemical Esterification of Aryl Halides with Carbazates, Org. Lett., 2022, 24, 7595–7599 CrossRef CAS; (d) P. Fan, Y. Lan, C. Zhang and C. Wang, Nickel/Photo-Cocatalyzed Asymmetric Acyl-Carbamoylation of Alkenes, J. Am. Chem. Soc., 2020, 142, 2180–2186 CrossRef CAS PubMed.
- P. Fan, Y. Lan, C. Zhang and C. Wang, Nickel/Photo-Cocatalyzed Asymmetric Acyl-Carbamoylation of Alkenes, J. Am. Chem. Soc., 2020, 142, 2180–2186 CrossRef CAS PubMed.
- Please refer to Fig. S4 in the ESI† for cyclic voltammetry of the nickel catalyst in the reaction.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qo01370g |
| This journal is © the Partner Organisations 2024 |