
Iron-catalyzed asymmetric Csp3–H/Csp3–H coupling: improving the chirality induction by mechanochemical liquid-assisted grinding†
Ping
Ying‡
ab,
Tao
Ying‡
a,
Hong
Chen
a,
Keyu
Xiang
 a,
Weike
Su
a,
Haijiao
Xie
c and
Jingbo
Yu
*a
a,
Weike
Su
a,
Haijiao
Xie
c and
Jingbo
Yu
*a
aLaboratory of Pharmaceutical Engineering of Zhejiang Province, Key Laboratory for Green Pharmaceutical Technologies and Related Equipment of Ministry of Education, Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals. Zhejiang University of Technology, Hangzhou 310014, P.R. China. E-mail: yjb@zjut.edu.cn
bCollege of Ecology. Lishui University, Lishui, 323000, P.R. China
cHangzhou Yanqu Information Technology Co., Ltd, Y2, 2nd Floor, Building 2, Xixi Legu Creative Pioneering Park, No. 712 Wen'er West Road, Xihu District, Hangzhou, Zhejiang Province 310003, P.R. China
First published on 10th November 2023
Abstract
The iron-catalyzed asymmetric oxidative coupling is a challenging transformation that is typically restricted to naphthol substrates (Csp2–H) with carefully designed chiral ligands. Herein, we established a mechanochemical protocol for iron-catalyzed asymmetric Csp3–H/Csp3–H coupling between glycines and β-ketoesters. By using size-tunable liquid additives via non-covalent bond interactions with simply designed chiral salen ligands and substrates under mechanochemical treatment, it is possible to improve the asymmetric induction and offer a variety of structurally diverse α-amino acid derivatives in high enantiopurity. Mechanistic studies revealed that the iminium ion derived from acid-assisted aerobic oxidation of glycine ester was the key intermediate of the reaction, and the liquid additive t-BuOH acted both as a stabilizer for the iminium ion via N–H⋯O interaction and as an assistant for enantio-control. Moreover, a safer, cleaner, and more energy-conserving route via mechanochemically accelerated aging was first disclosed for this asymmetric Csp3–H/Csp3–H coupling reaction.
Introduction
The development of the transition-metal catalyzed asymmetric cross-dehydrogenative coupling (CDC) reaction1 is highly important because it offers a sustainable alternative to traditional cross-coupling reactions, which heavily rely on prefunctionalization of the coupling partners (Fig. 1A). Although significant contributions on metal-catalyzed CDC reactions2 have been made by the Li group and others, their catalytic asymmetric variants3 are much less operational. Even more frustrating is the lack of Csp3–H/Csp3–H coupling reactions and only a few metal-catalyzed studies have been reported using Cu,4 Ni5 and cooperative bimetallic6 catalyst systems. In recent years, iron catalysis has aroused much attention due to its advantages of earth abundance, eco-friendliness, and low toxicity.7 However, sporadic examples of iron catalyzed CDCs have been reported, which are limited to the oxidative cross couplings between naphthols8 or with azonaphthalenes9 which are induced by chiral redox iron complexes (Fig. 1B). The main issues resulting in the absence of enantioselective versions potentially arise from two aspects: (1) the wide array of formal oxidation states and thus the multifarious chemical nature of the iron metal, particularly under strong oxidative conditions, made the stereo-control of iron-catalyzed CDC reactions highly challenging;10 (2) reversible single electron transfer (SET) and delocalization of the Fe–substrate complex often led to depreciation of the enantiopurity (Fig. 1B).8c,d Therefore, the development of a reliable chiral iron catalytic system to access the asymmetric CDC reaction with alternative hybrid carbon under facile oxidative conditions is a highly important objective.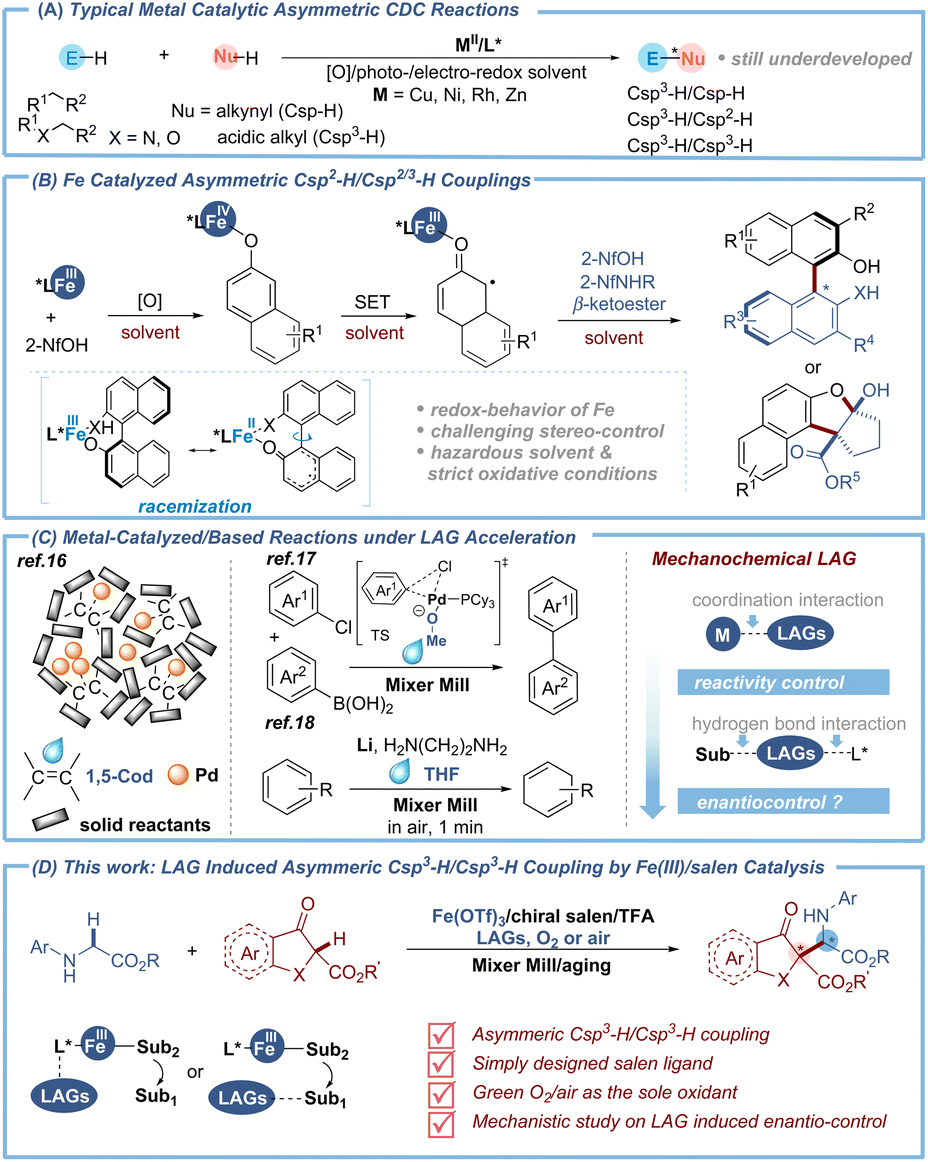 | ||
| Fig. 1 Background studies and this work. | ||
Mechanochemistry11 provides exciting synthetic opportunities with no dissolution concerns to generate new chemical reactivity12 and selectivity.13 However, the utilization of ball milling in asymmetric organic synthesis has not been adequately investigated which concentrated on the condensation, addition and substitution reactions.14 Recently, our group focused on asymmetric oxidative coupling reactions using the liquid-assisted grinding (LAG)15 strategy, a useful technique derived from mechanochemistry, by which tiny liquid additives could participate in the catalytic cycle to endow high reactivity and enantioselectivity.15b Besides, LAG plays an important role in accelerating some metal-catalyzed/based reactions via coordination interactions (Fig. 1C). For example, Ito and co-workers’ recent elegant studies showed that the coordinative 1,5-cyclooctadiene (1,5-cod) acts as both a dispersant and stabilizer for a palladium catalyst to facilitate solid-state cross-coupling.16 Meanwhile methanol (MeOH) was proved by our group to accelerate the palladium-catalyzed oxidative addition of carbon–chloride bonds via ligand exchange.17 Besides, tetrahydrofuran (THF) has been used as an alternative ligand to suppress the side reaction of lithium-based Birch reduction and to improve the reactivity of magnesium-based carbon nucleophiles, respectively.18 Inspired by our previous studies on mechano-synthesis and enantioselective reactions,19 we envisioned whether the non-covalent bond interactions between size-tunable liquid additives and chiral ligands/substrates promoted by mechanochemical LAG could be employed to create a suitable spatial environment that facilitates asymmetric induction for iron-catalyzed oxidative cross-coupling. This strategy, if successful, would provide an alternative expedient to metal catalytic chirality induction.
Herein, we demonstrated the realization of this strategy for iron-catalyzed asymmetric oxidative Csp3–H/Csp3–H coupling of glycines and β-ketoesters using a salen-type ligand (Fig. 1D). Under mechanochemical LAG, the liquid additive t-BuOH of high steric hindrance could significantly improve the enantioselectivity and stabilize the iminium intermediate via a N–H⋯O interaction as evidenced by experimental and computational studies. This reaction provides an expedient way to access enantioenriched unnatural α-amino acid derivatives,20 with salient features that include (1) the first iron catalyzed asymmetric coupling reaction, (2) green oxygen or air as the sole oxidant, (3) excellent chirality control with a simply designed salen ligand, and (4) mechanistic insights into LAG induced enantiocontrol.
Results and discussion
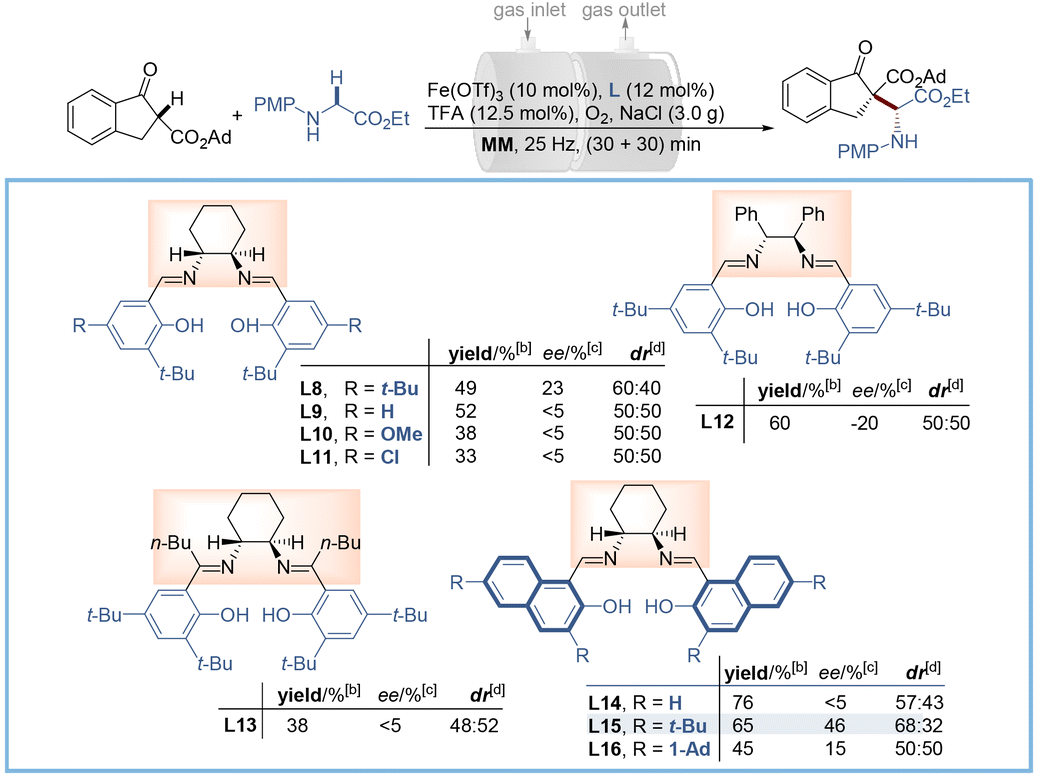
We commenced our study by examining the asymmetric coupling reaction between β-indanone ester 1a and N-p-methoxy phenylglycine ethyl ester 2a using Fe(OTf)3 as the catalyst. Preliminary screening of the ligand scaffold and oxidant showed that the salen-type ligand gave superior stereoselectivity and yield, and oxygen was necessary and optimal for this transformation (for details, see Table S1†). Considering that a Brønsted acid is beneficial for driving the aerobic oxidation of glycine derivatives,21 several acidic additives were then tested (Table S1,† entries 10–14) and trifluoroacetic acid (TFA) gave the best result (49% yield with 23% ee and 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 dr). Further optimization of iron salts revealed Fe(OTf)3 to be the best choice (Table S1,† entries 15–19). To improve the enantio- and diastereo-selectivity, a series of salen ligands with different steric hindrance and electronic properties were examined (Table 1, L8–L13, and Table S2†). The results indicated that the steric hindrance from the phenol ring rather than the amine sources (Table 1, L12–L13) was effective for the stereo-control of this transformation, while electronic properties proved to be futile (Table 1, L9–L11). Encouraged by these results, we replaced the phenol moiety with bulky naphthol (Table 1, L14) and incorporated tertiary butyl (Table 1, t-Bu, L15) and adamantyl (Table 1, Ad, L16) groups at both C3 and C6 positions of the naphthol ring. Pleasingly, L15 was found to be optimal to give 3aa in 65% yield with 46% ee and 68:32 dr, while the over-congested Ad group led to depreciation in both yield and enantioselectivity. Nevertheless, there is still plenty of room for improvement of selectivity.
:40 dr). Further optimization of iron salts revealed Fe(OTf)3 to be the best choice (Table S1,† entries 15–19). To improve the enantio- and diastereo-selectivity, a series of salen ligands with different steric hindrance and electronic properties were examined (Table 1, L8–L13, and Table S2†). The results indicated that the steric hindrance from the phenol ring rather than the amine sources (Table 1, L12–L13) was effective for the stereo-control of this transformation, while electronic properties proved to be futile (Table 1, L9–L11). Encouraged by these results, we replaced the phenol moiety with bulky naphthol (Table 1, L14) and incorporated tertiary butyl (Table 1, t-Bu, L15) and adamantyl (Table 1, Ad, L16) groups at both C3 and C6 positions of the naphthol ring. Pleasingly, L15 was found to be optimal to give 3aa in 65% yield with 46% ee and 68:32 dr, while the over-congested Ad group led to depreciation in both yield and enantioselectivity. Nevertheless, there is still plenty of room for improvement of selectivity.
| a Reaction conditions: Fe(OTf)3 (10 mol%), L (12 mol%), TFA (12.5 mol%), and NaCl (3.0 g) were pre-milled at 25 Hz for 30 min, using two stainless-steel balls (dMB = 1.0 cm) in a 25 mL stainless-steel vial, then, 1a (0.2 mmol), 2a (0.2 mmol) and O2 were added in through a gas inlet valve and milled for another 30 min. b Yield of the isolated product. c Determined by HPLC analysis. d Determined by 1H NMR analysis. n.d. = not detected. MM = mixer mill. |
|---|
|
To avoid elaborate ligand design, we then took advantage of the LAG strategy by exploring suitable liquid additives (LAGs) with good hydrogen bonding interaction with the ligand and/or substrates to further improve the stereo-control under ball-milling conditions. The results depicted in Fig. 2A and Table S3† show that the reaction enantioselectivity and diastereoselectivity are highly sensitive to the steric hindrance of these liquid additives, and among them t-BuOH (η = 1.16) of high steric hindrance gave the best performance (75% yield, 87% ee and 85:15 dr). Further experimental insights into the influence of mechanochemical parameters on the asymmetric oxidative reaction are also presented (Table S4†). A medium frequency (20 Hz) ball-milling for 55 min using two stainless-steel balls (14 diameter) and 2.0 g of NaCl was finally chosen to give the best yield (88%), ee (95%) and dr (90:10) values. In this case, the amount of t-BuOH could be reduced to 75 μL (η = 0.58) (Table S4,† entry 10). Comparisons to solution-stirred and neat-stirred approaches were then carried out, demonstrating that the solution analogue (in 4 mL t-BuOH) is slower than the ball-milling process and poorer enantio-control was imparted, as only 7% yield and 46% ee of the product were observed after 30 min (Fig. 2B), which implies the importance of reaction acceleration by mechanochemical LAG. Likewise, the dry-stirring analogue with t-BuOH (150 μL, 4 equiv.) as an additive was much inferior to the ball-milling approach, providing only 7% yield of 3aa with 16% ee after 24 h (Fig. 2C). Besides, under Wang's solution conditions3a using a copper/L1 catalyst and DDQ oxidant, the reaction showed little activity (Fig. 2D). In so doing has highlighted the importance of the iron catalytic system and the mechanical treatment for the enantiocontrol of the current transformation.
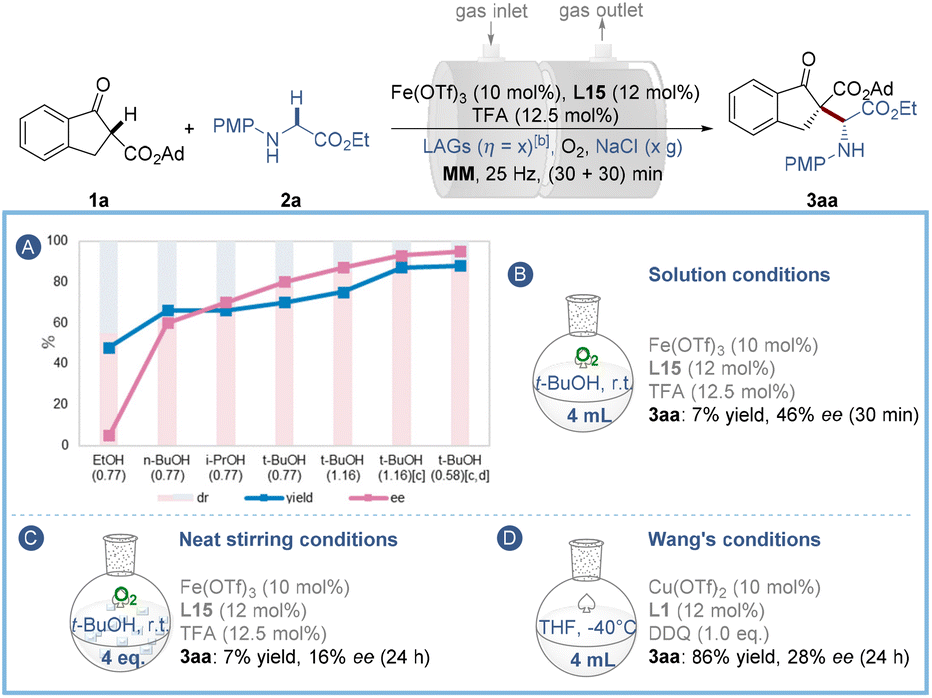 | ||
| Fig. 2 Screening of LAGsa,b (A) and comparative experiments (B–D). aFor reaction conditions see Table 1. bLAGs [η = V (liquid; μL)/m (reagents; mg)] were added in the pre-milling process. cTwo stainless-steel balls (dMB = 1.4 cm) were used and the mixtures were milled at 20 Hz for (30 + 25) min. dNaCl (2.0 g) and t-BuOH (η = 0.58) were used. MM = mixer mill. | ||
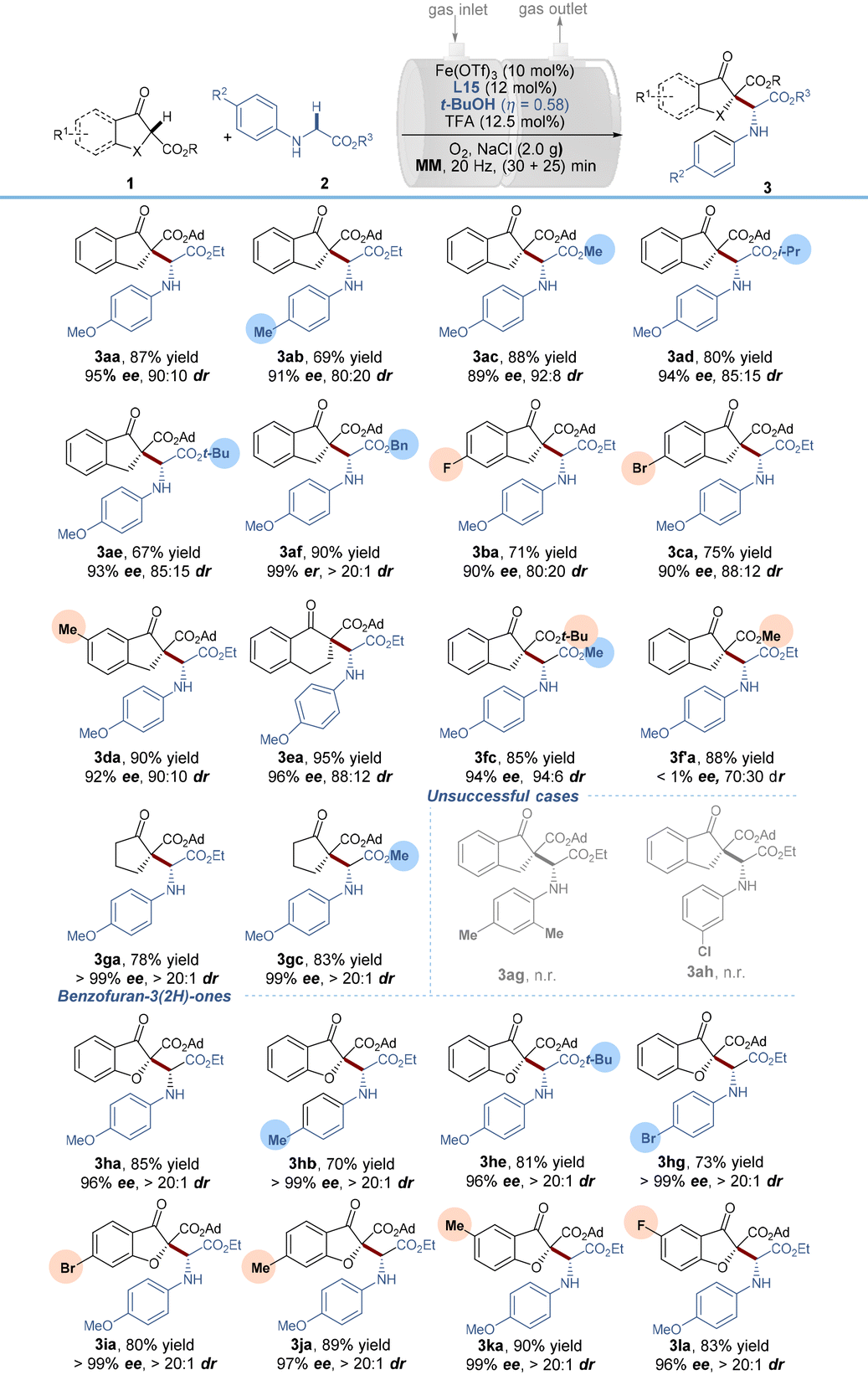
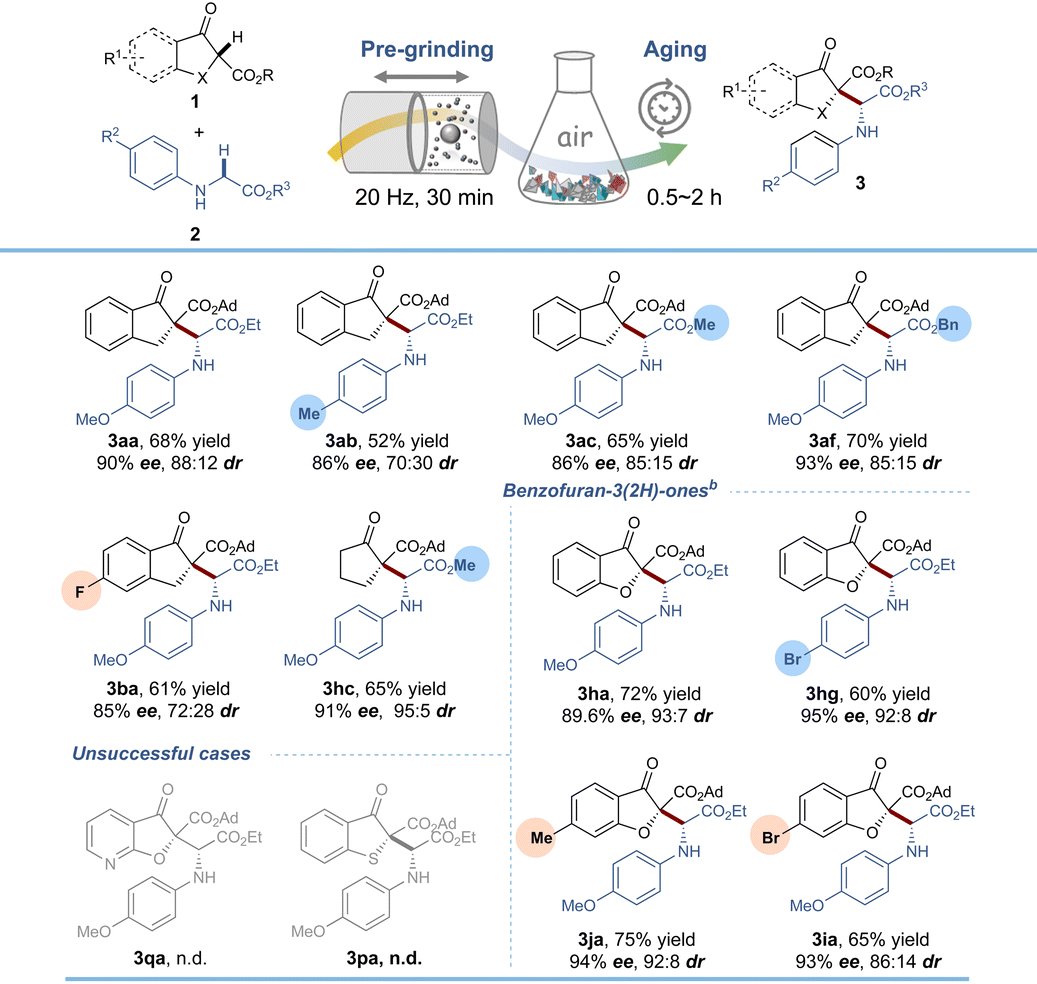
With the optimized reaction conditions in hand, we began to explore the reaction scope by varying the glycine component 2 in combination with β-indanone esters (Table 2). The reaction is amenable for a range of glycine esters that possess different ester groups (OEt, OMe, Oi-Pr, Ot-Bu and OBn), resulting in moderate to good product yields (67–90%), excellent enantioselectivities (89–99%) and satisfactory diastereoselectivities (85:15 to >20:1 dr). However, the substituents on benzene rings were limited to electron-donating groups (Me and OMe) in the para position, while a 2,6-dimethyl substituent (3ag) and an m-chloro substituent (3ah) failed to react. It was probably due to the beneficial effects of para electron-donating substituents for aerobic α-C–H oxidation. With respect to β-indanone esters, both electron-rich and electron-deficient substrates were found to efficiently react with N-PMP glycine ethyl ester 2a, as demonstrated by the good to excellent yields and ee values of the coupling products 3ba–3da. The tert-butyl ester substituent in 1f was compatible with the reaction conditions, maintaining high enantioselectivity and diastereoselectivity (3fc). However, a small-sized methyl ester 1f′ completely suppressed the reaction enantioselectivity (3f′a, no ee value), indicating the vital importance of the steric hindrance of the ester groups. As extensions of this reaction, tetralone β-keto ester and aliphatic cyclic β-keto ester derivatives were tolerated as well in the current system, generating the related products 3ea, 3ga and 3gc in good to excellent yields (78–95%) and high enantio- and diastereoselectivities (96–>99% ee and 88:12–>20:1 dr). It is worth noting that switching the carbocyclic β-keto ester to biologically active benzofuran-3(2H)-ones gave rise to enantioenriched α-alkyl α-amino acid derivatives in 70–90% isolated yields with 96–99% ee values and >20:1 dr values. In these cases, regardless of the electronic properties and/or positions of the substituents on the benzene rings of benzofuran-3(2H)-ones, all of the reactions ran well, constituting an efficient and unprecedented method for accessing benzofuranone derived α-amino acid derivatives. The relative configuration of minor diastereomers [(RR)-form and (SS)-form] of 3aa was confirmed by X-ray crystallography analysis22 and HPLC analysis (for details see the ESI, section 5†) and the absolute configuration of the major stereoisomer was further supported by DFT calculations (vide infra).
| a Reaction conditions: Fe(OTf)3 (10 mol%), L15 (12 mol%), TFA (12.5 mol%), t-BuOH (η = 0.58) and NaCl (2.0 g) were pre-milled at 20 Hz for 30 min, using two stainless-steel balls (dMB = 1.4 cm) in a 25 mL stainless-steel vial, then, 1a (0.2 mmol) and 2a (0.2 mmol) were added and O2 was supplied through a gas inlet valve and milled for another 30 min. MM = mixer mill. |
|---|
|
Our subsequent efforts were directed toward further increasing the greenness of the reaction through the accelerated aging (AA) technique, which possesses inherently simple, safe, low-cost, and energy-saving properties.23 Despite being rarely studied by organic chemists, it has been practically applied to the C–H functionalization of glycines by our group very recently.24 After optimization of the aging conditions (Table S5†), 3aa could be formed in 68% yield and 90% ee by 30 min of pre-grinding and 2 h of aging without any additional oxidant. Substrate adaptability under AA conditions was also tested and the results are shown in Table 3. Generally, regardless of the type of β-keto ester and glycine ester, all of the reactions ran smoothly under open air, furnishing the corresponding coupling products in 52–75% isolated yields with 85% to 95% ee values and 70:30 to 93:7 dr values, which were lower than those obtained by a continuous LAG strategy. Among them, benzofuran-3(2H)-ones (aging for 0.5 h) gave better performance than β-indanone esters in terms of enantioselectivity and diastereoselectivity. Nevertheless, pyridinofuran-3(2H)-one 1q and benzothiophen-3(2H)-one 1p could not furnish any products. Additionally, a series of green chemistry metrics including the environmental impact factor (E-factor), solvent intensity (SI) and mass intensity (MI) were quantified which showed this methodology to be sustainable and cost-effective (see Table S6 and Scheme S6†).
| a Reaction conditions: 1a (0.2 mmol), 2a (0.2 mmol), Fe(OTf)3 (10 mol%), L15 (12 mol%), TFA (12.5 mol%), t-BuOH (η = 0.58) and NaCl (2.0 g) were pre-milled at 20 Hz for 30 min, using two stainless-steel balls (dMB = 1.4 cm) in a 25 mL stainless-steel vial, and then aged in an open flask (50 mL) for 2 h. MM = mixer mill. b Aging for 0.5 h. |
|---|
|
To gain insights into the mechanism of the reaction, control experiments were conducted with the reaction between 1a and 2a (Fig. 3). In sharp contrast to the high efficiency exhibited by the reaction carried out under the standard LAG conditions (87% yield and 95% ee), no reaction occurred for the model substrates under inert gases (Fig. 3A), even in the presence of a stoichiometric Fe(III) catalyst (Fig. 3B). In addition, the reaction was completely inhibited in the presence of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or 2,6-di-t-butyl-4-methylphenol (BHT), and the latter could trap an amine radical to afford 2a-BHT in 30% isolated yield (Fig. 3C and Scheme S4†). These results revealed that oxygen is essential and it is the only oxidant for this mechanochemical asymmetric Csp3–H/Csp3–H coupling reaction, which is possibly involved in the radical oxidation of the glycine esters.
 | ||
| Fig. 3 Control experiments. Mechanochemical reactions of 1a and 2a under nitrogen atmospheres (A and B). Mechanochemical reaction of 1a and imine 2–2′ (C). Radical trapping experiments (D). Solution reactions of 1a and 2a in THF with or without t-BuOH (4 equiv., 75 μL) as the additive (E). Iron-complex capturing experiments (F). | ||
When replacing 2a with imine 2–2′, it can be easily captured by 1a even without the assistance of a chiral catalyst, leading to a strong racemic background reaction. In this case, 3aa was obtained in 88% yield with only <5% ee value (Fig. 3D and Scheme S3†). Besides, solution reactions in both t-BuOH and THF exhibited poor performance, and very low transformations were observed during the first 30–60 min, while prolonging the reaction to 240 min gave rise to obvious racemization25 (Fig. 3E). It should be noted that an obvious enhancement of the ee value was observed by adding small drops of t-BuOH (4 eq.) to the THF solution, indicating that the coordination effect was not exclusive to mechanochemistry. However, this positive effect was weakened by thermodynamically controlled racemization, highlighting the importance of reaction acceleration by mechanochemistry to ensure the stereoselectivity.
The dramatic enhancement of enantioselectivities under LAG conditions prompted us to probe the iron-complexes in the LAG system. ESI-MS analysis19c was employed to detect the mixtures after ball-milling of Fe(OTf)3, L15 and t-BuOH (Fig. 3F). Two peaks attributed to Fe-complexes [L15-Fe]+ (m/z 700.4) and [L15-Fe-t-BuOH + MeCN]+ (m/z 815.5) were found. In stark contrast, neat grinding of Fe(OTf)3 and L15 followed by physical mixing with t-BuOH offered only [L15-Fe]+ (m/z 700.4) and [L15 + H]+ (m/z 647.5) species, implying that the mechanical impact was essential for accessing the L-Fe-butanol complex, which is likely an active catalytic species, and the interference caused by ionization during the MS detection could also be excluded (for details see Scheme S5 and Fig. S1 in the ESI†). However, when 2a was added to the mixtures, no Fe-complexes containing 2a were detected after ball-milling which means that the β-keto ester might fail to coordinate with the iron center by direct ligand exchange (vide infra).
Collectively, the overall catalytic cycle was proposed through the combination of density functional theory (DFT) calculations using PBE0 methods26 (for details see the ESI, section 9†). As illustrated in Fig. 4B, the catalytic cycle started with the LFe complex INT 1 (although the dissociation of INT 1 into [LFe]+ had a high free energy barrier of 99.5 kcal mol−1, OTf− could be lost during the MS detection27). Direct ligand exchange of OTf− with t-BuOH or 1f-enol was energetically disfavoured (Fig. 4A), while an associative pathway involving INT 1, 1f-enolate and t-BuOH was workable to afford INT 3 bearing t-BuOH on the naphthol-O of L15 (−24.3 kcal mol−1). In this context, 1f-enolate was obtained from the deprotonation of 1f by CF3CO2− (20.5 kcal mol−1) rather than by OTf− (43.1 kcal mol−1), while the former was derived from the TFA assisted two-step single-electron oxidation of 2c. Next, Si- or Re-face coordination of INT 3 to two faces of the iminium 2c-2 led to two potential enantiomeric intermediates INT 4SR and INT 4RS, respectively (here only the major diastereomers were considered), where the iminium ions were stabilized by t-BuOH via a N–H⋯O interaction. Afterwards, nucleophilic addition occurred in transition states (TSSR and TSRS) to yield the LFe-containing adducts (INT 5SR and INT 5RS) and then the catalyst and t-BuOH were released to close the catalytic cycle and afford the product 3fc in two enantiomeric forms. As showcased in Fig. 4C, the favourable transition state TSSR leading to the desired (SR)-form final product PSR was computed to be lower in free energy than TSRS forming the (RS)-form product PRS by about 4.1 kcal mol−1 in the vacuum phase, indicating the (SR)-form to be the major enantiomer. Notably, a comparison of free energy barriers of INT 4SR and INT 4SR-1 suggests that t-BuOH plays a major role in stabilizing the iminium ion (Fig. S3†). The enantioselectivity seems to be provided by the steric interaction between the salen ligand, ketoester and t-BuOH. As seen from the bottom view of the enantioselectivity models (Fig. S3†), the tert-butyl group of ketoester in TSSR is bent significantly to be away from the bulky naphthol of the ligand (Fe–OA as 97.3°), which might contribute to the lower free energy of TSSR and is consistent with the experimental results (3aavs.3f′a) that a smaller-sized ester group of 1 was detrimental for the enantiocontrol. In addition, the H–H repulsion in TSRS between the butyl group of t-BuOH and the cyclohexyl group on the ligand also contributes to the enantiocontrol which results in higher free energy (Fig. 4C). Above all, given the X-ray crystallography analysis and DFT calculations, the absolute configuration of the product was speculated as the (SR)-form.
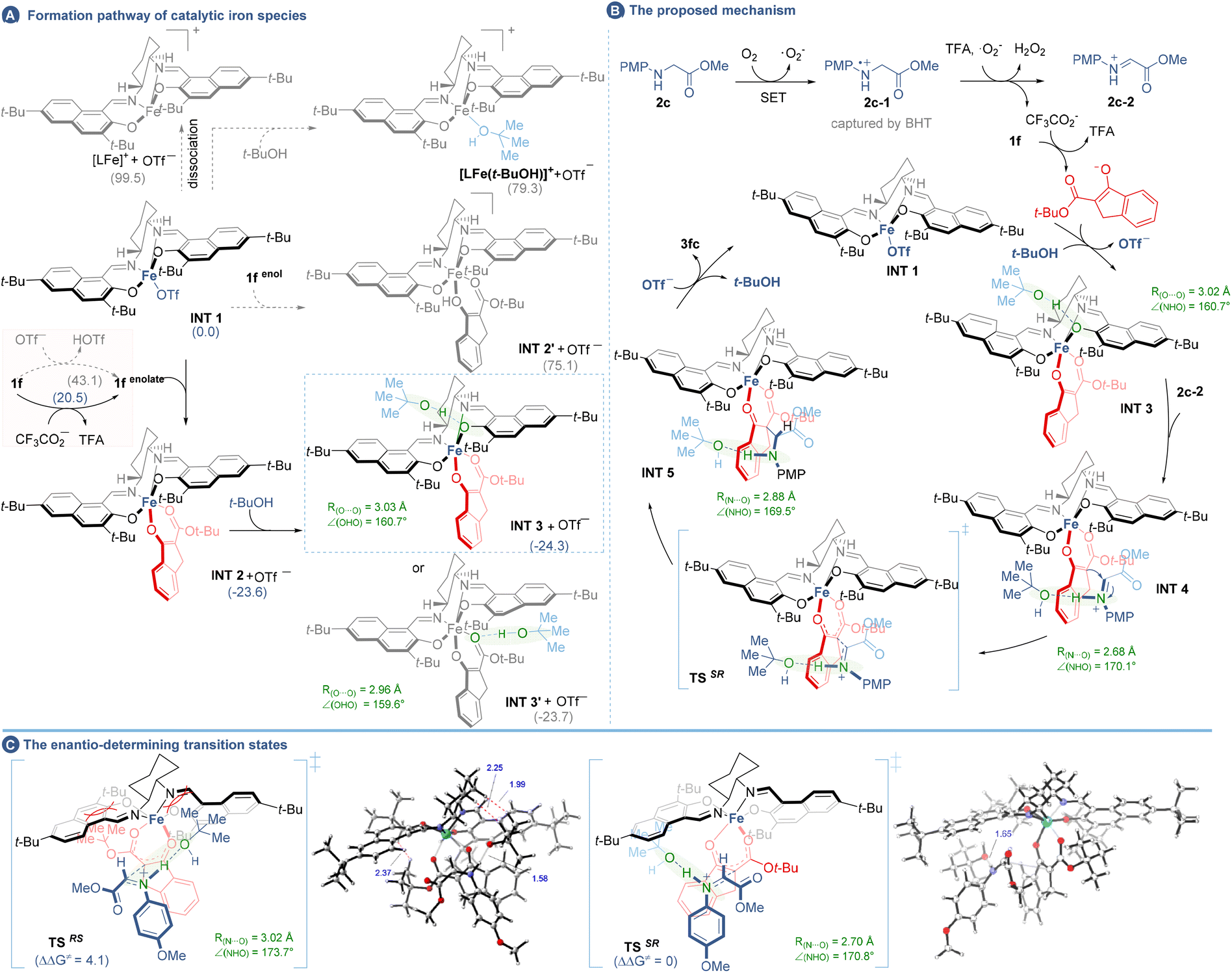 | ||
| Fig. 4 DFT studies and the proposed mechanism. (A) Formation pathway of the catalytic iron species. (B) The proposed mechanism. (C) The enantio-determining transition states. The DFT-calculated free energies, in kcal mol−1, are shown in parentheses. All of the Fe-complexes are in the sextet spin state. | ||
Conclusions
In summary, a chiral Fe/salen (L15)/O2 system was used to drive the mechanochemical asymmetric oxidative coupling of glycines and β-ketoesters in a highly stereoselective manner with the assistance of t-BuOH as a liquid additive. This method provides a new mode of chiral control which is barely effective in solution chemistry. Notably, various amino acid derivatives bearing β-indanone esters, aliphatic cyclic β-ketoesters, tetralone β-ketoesters and benzofuran-3(2H)-ones were produced in good yields with high enantiopurity, demonstrating the capability of this mechanochemical asymmetric protocol. The mechanistic study elucidated that the mechanical impact was essential for accessing the active catalytic species (L-Fe-butanol complex), and t-BuOH played a crucial role in stabilizing the iminium intermediate and boosting enantiocontrol via non-covalent interactions in the enantio-determining step. In addition, a mechanochemical accelerated aging (AA) strategy with short-lasting low-energy impetus was applied to this asymmetric Csp3–H/Csp3–H coupling, which will open a new avenue for iron catalyzed asymmetric reactions.Author contributions
P. and J. B. suggested the idea; P., T. and H. performed the experiments. K. Y. conducted catalyst recovery experiments. W. K. and J. B. supervised the project. Writing – review and editing were conducted by P. and J. B. All authors have read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21978270), the Zhejiang Provincial Natural Science Foundation of China (LY23B060005), and the National Key R&D Program of China (2021YFC2101000) for financial support.References
- A. M. Faisca Phillips, M. d. F. C. Guedes da Silva and A. J. L. Pombeiro, New Trends in Enantioselective Cross-Dehydrogenative Coupling, Catalysts, 2020, 10, 529–564 CrossRef.
- For selected reviews, see: (a) C.-Y. Huang, H. Kang, J. Li and C.-J. Li, En Route to Intermolecular Cross-Dehydrogenative Coupling Reactions, J. Org. Chem., 2019, 84, 12705–12721 CrossRef CAS; (b) T. Tian, Z. Li and C.-J. Li, Cross-Dehydrogenative Coupling: A Sustainable Reaction for C–C Bond Formations, Green Chem., 2021, 23, 6789–6862 RSC; (c) H. Wang, P. Ying, J. Yu and W. Su, Alternative Strategies Enabling Cross-Dehydrogenative Coupling: Access to Carbon-Carbon Bonds, Chin. J. Org. Chem., 2021, 41, 1897–1924 CrossRef CAS; (d) Y. L. Tsang, P. Y. Choy, M. P. Leung, X. He and F. Y. Kwong, Recent Advances in Rhodium-Catalysed Cross-Dehydrogenative-Coupling between Two C(sp2)–H Bonds, Org. Chem. Front., 2022, 9, 1992–2012 RSC; (e) I. N. Egorov, A. Mukherjee, S. Santra, D. S. Kopchuk, I. S. Kovalev, Y. Liu, G. V. Zyryanov, A. Majee, O. N. Chupakhin and B. C. Ranu, Mechanochemically Induced Cross Dehydrogenative Coupling Reactions under Ball Milling, Adv. Synth. Catal., 2022, 364, 2462–2478 CrossRef CAS; (f) J. Li, C.-Y. Huang and C.-J. Li, Cross-Dehydrogenative Coupling of Unactivated Alkanes, Trends Chem., 2022, 4, 479–494 CrossRef CAS.
- (a) Y.-L. Zhao, Y. Wang, Y.-C. Luo, X.-Z. Fu and P.-F. Xu, Asymmetric C–H Functionalization Involving Organocatalysis, Tetrahedron Lett., 2015, 56, 3703–3714 CrossRef CAS; (b) S. Gandhi, Catalytic Enantioselective Cross Dehydrogenative Coupling of sp3 C-H of Heterocycles, Org. Biomol. Chem., 2019, 17, 9683–9692 RSC.
- (a) G. Zhang, Y. Zhang and R. Wang, Catalytic Asymmetric Activation of a Csp3-H Bond Adjacent to a Nitrogen Atom: A Versatile Approach to Optically Active α-Alkyl α-Amino Acids and C1-Alkylated Tetrahydroisoquinoline Derivatives, Angew. Chem., Int. Ed., 2011, 50, 10429–10432 CrossRef CAS; (b) C. Guo, J. Song, S.-W. Luo and L.-Z. Gong, Enantioselective Oxidative Cross-Coupling Reaction of 3-Indolylmethyl C-H Bonds with 1,3-Dicarbonyls Using a Chiral Lewis Acid-Bonded Nucleophile to Control Stereochemistry, Angew. Chem., Int. Ed., 2010, 49, 5558–5562 CrossRef CAS; (c) A. Lee, R. Betori, E. Crane and K. A. Scheidt, An Enantioselective Cross Dehydrogenative Coupling Catalysis Approach to Substituted Tetrahydropyrans, J. Am. Chem. Soc., 2018, 140(20), 6212–6216 CrossRef CAS PubMed; (d) K. Zhou, Y. Yu, Y.-M. Lin, Y. Li and L. Gong, Copper-Catalyzed Aerobic Asymmetric Cross-Dehydrogenative Coupling of C(sp3)-H Bonds Driven by Visible Light, Green Chem., 2020, 22, 4597–4603 RSC.
- G. Wang, X. Xin, Z. Wang, G. Lu, Y. Ma and L. Liu, Catalytic Enantioselective Oxidative Coupling of Saturated Ethers with Carboxylic Acid Derivatives, Nat. Commun., 2019, 10, 559–567 CrossRef.
- W. Cao, X. Liu, R. Peng, P. He, L. Lin and X. Feng, Catalytic Asymmetric Cross-Dehydrogenative Coupling: Activation of C–H Bonds by A Cooperative Bimetallic Catalyst System, Chem. Commun., 2013, 49, 3470–3472 RSC.
- (a) K. S. Egorova and V. P. Ananikov, Which Metals are Green for Catalysis? Comparison of the Toxicities of Ni, Cu, Fe, Pd, Pt, Rh, and Au Salts, Angew. Chem., Int. Ed., 2016, 55, 12150–12162 CrossRef CAS; (b) A. Piontek, E. Bisz and M. Szostak, Iron–Catalyzed Cross–Couplings in the Synthesis of Pharmaceuticals: In Pursuit of Sustainability, Angew. Chem., Int. Ed., 2018, 57, 11116–11128 CrossRef CAS; (c) P. S. Steinlandt, X. Xie, S. Ivlev and E. Meggers, Stereogenic-at-Iron Catalysts with a Chiral Tripodal Pentadentate Ligand, ACS Catal., 2021, 11, 7467–7476 CrossRef CAS.
- (a) H. Egami and T. Katsuki, Iron-Catalyzed Asymmetric Aerobic Oxidation: Oxidative Coupling of 2-Naphthols, J. Am. Chem. Soc., 2009, 131, 6082–6083 CrossRef CAS; (b) H. Egami, K. Matsumoto, T. Oguma, T. Kunisu and T. Katsuki, Enantioenriched Synthesis of C1-Symmetric BINOLs: Iron-Catalyzed Cross-Coupling of 2-Naphthols and Some Mechanistic Insight, J. Am. Chem. Soc., 2010, 132, 13633–13635 CrossRef CAS PubMed; (c) S. Narute, R. Parnes, D. Toste and D. Pappo, Enantioselective Oxidative Homocoupling and Cross-Coupling of 2-Naphthols Catalyzed by Chiral Iron Phosphate Complexes, J. Am. Chem. Soc., 2016, 138, 16553–16560 CrossRef CAS PubMed; (d) S. Narute and D. Pappo, Iron Phosphate Catalyzed Asymmetric Cross-Dehydrogenative Coupling of 2-naphthols with β-ketoesters, Org. Lett., 2017, 19, 2917–2920 CrossRef CAS; (e) T. Horibe, K. Nakagawa, T. Hazeyama, K. Takeda and K. Ishihara, An Enantioselective Oxidative Coupling Reaction of 2-Naphthol Derivatives Catalyzed by Chiral Diphosphine Oxide–Iron(II) Complexes, Chem. Commun., 2019, 55, 13677–13680 RSC.
- A. Dyadyuk, V. Vershinin, H. Shalit, H. Shalev, N. Y. More and D. Pappo, A Chiral Iron Disulfonate Catalyst for the Enantioselective Synthesis of 2-Amino-2′-Hydroxy-1, 1′-Binaphthyls (NOBINs), J. Am. Chem. Soc., 2022, 144, 3676–3684 CrossRef CAS PubMed.
- (a) A. Fürstner, Iron Catalysis in Organic Synthesis: a Critical Assessment of What it Takes to Make This Base Metal a Multitasking Champion, ACS Cent. Sci., 2016, 2, 778–789 CrossRef; (b) S. Rana, J. P. Biswas, S. Paul, A. Paik and D. Maiti, Organic Synthesis with the Most Abundant Transition Metal–Iron: from Rust to Multitasking Catalysts, Chem. Soc. Rev., 2021, 50, 243–472 RSC.
- For selected reviews, see: (a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Mechanochemistry: Opportunities for New and Cleaner Synthesis, Chem. Soc. Rev., 2012, 41, 413–447 RSC; (b) J. G. Hernández, C–H Bond Functionalization by Mechanochemistry, Chem. – Eur. J., 2017, 23, 17157–17165 CrossRef; (c) J. L. Howard, Q. Cao and D. L. Browne, Mechanochemistry as An Emerging Tool for Molecular Synthesis: What Can It Offer?, Chem. Sci., 2018, 9, 3080–3094 RSC; (d) T. Friščić, C. Mottillo and H. M. Titi, Mechanochemistry for Synthesis, Angew. Chem., Int. Ed., 2020, 59, 1018–1041 CrossRef; (e) X.-J. Yang, C.-Y. Wu, W.-K. Su and J.-B. Yu, Mechanochemical C–X/C–H Functionalization: An Alternative Strategy Access to Pharmaceuticals, Eur. J. Org. Chem., 2022, e202101440 CrossRef CAS.
- (a) J.-L. Do, C. Mottillo, D. Tan, V. Štrukil and T. Friščić, Mechanochemical Ruthenium-Catalyzed Olefin Metathesis, J. Am. Chem. Soc., 2015, 137, 2476–2479 CrossRef CAS PubMed; (b) R. Takahashi, T. Seo, K. Kubota and H. Ito, Palladium-Catalyzed Solid-State Polyfluoroarylation of Aryl Halides Using Mechanochemistry, ACS Catal., 2021, 11, 14803–14810 CrossRef CAS; (c) P. Gao, J. Jiang, S. Maeda, K. Kubota and H. Ito, Mechanochemically Generated Calcium–Based Heavy Grignard Reagents and Their Application to Carbon–Carbon Bond–Forming Reactions, Angew. Chem., Int. Ed., 2022, 61, e202207118 CrossRef CAS.
- (a) J. G. Hernández and C. Bolm, Altering Product Selectivity by Mechanochemistry, J. Org. Chem., 2017, 82, 4007–4019 CrossRef; (b) J. L. Howard, M. C. Brand and D. L. Browne, Switching Chemoselectivity: Using Mechanochemistry to Alter Reaction Kinetics, Angew. Chem., Int. Ed., 2018, 57, 16104–16108 CrossRef CAS.
- I. N. Egorov, S. Santra, D. S. Kopchuk, I. S. Kovalev, G. V. Zyryanov, A. Majee, B. C. Ranu, V. L. Rusinov and O. N. Chupakhin, Ball Milling: An Efficient and Green Approach for Asymmetric Organic Syntheses, Green Chem., 2020, 22, 302–315 RSC.
- (a) G. A. Bowmaker, Solvent-Assisted Mechanochemistry, Chem. Commun., 2013, 49, 334 RSC; (b) P. Ying, J.-B. Yu and W.-K. Su, Liquid–Assisted Grinding Mechanochemistry in the Synthesis of Pharmaceuticals, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef CAS.
- (a) K. Kubota, T. Seo, K. Koide, Y. Hasegawa and H. Ito, Olefin-Accelerated Solid-State C–N Cross-Coupling Reactions Using Mechanochemistry, Nat. Commun., 2019, 10, 111 CrossRef; (b) T. Seo, T. Ishiyama, K. Kubota and H. Ito, Solid-State Suzuki–Miyaura Cross-Coupling Reactions: Olefin-Accelerated C–C Coupling Using Mechanochemistry, Chem. Sci., 2019, 10, 8202–8210 RSC.
- Z.-J. Jiang, Z.-H. Li, J.-B. Yu and W.-K. Su, Liquid-Assisted Grinding Accelerating: Suzuki–Miyaura Reaction of Aryl Chlorides under High-Speed Ball-Milling Conditions, J. Org. Chem., 2016, 81, 10049–10055 CrossRef CAS PubMed.
- (a) Y. Gao, K. Kubota and H. Ito, Mechanochemical Approach for Air–Tolerant and Extremely Fast Lithium–Based Birch Reductions in Minutes, Angew. Chem., Int. Ed., 2023, 62, e202217723 CrossRef CAS; (b) R. Takahashi, A. Hu, P. Gao, Y. Gao, Y. Pang, T. Seo, J. Jiang, S. Maeda, H. Takaya, K. Kubota and H. Ito, Mechanochemical Synthesis of Magnesium-Based Carbon Nucleophiles in Air and Their Use in Organic Synthesis, Nat. Commun., 2021, 12, 6691 CrossRef CAS PubMed.
- (a) J.-B. Yu, Z.-H. Li, K.-Y. Jia, Z.-J. Jiang, M.-L. Liu and W.-K. Su, Fast, Solvent-Free Asymmetric Alkynylation of Prochiral sp3 C–H Bonds in A Ball Mill for the Preparation of Optically Active Tetrahydroisoquinoline Derivatives, Tetrahedron Lett., 2013, 54, 2006–2009 CrossRef CAS; (b) Z. Li, Z. Jiang and W. Su, Fast, Solvent-Free, Highly Enantioselective Three-Component Coupling of Aldehydes, Alkynes, and Amines Catalysed by the Copper(II) Pybox Complex under High-Vibration Ball-Milling, Green Chem., 2015, 17, 2330–2334 RSC; (c) K.-Y. Jia, J.-B. Yu, Z.-Z. Jiang and W.-K. Su, Mechanochemically Activated Oxidative Coupling of Indoles with Acrylates through C–H Activation: Synthesis of 3-Vinylindoles and β,β-Diindolyl Propionates and Study of the Mechanism, J. Org. Chem., 2016, 81, 6049–6055 CrossRef CAS; (d) J.-B. Yu, H.-W. Shou, W.-Y. Yu, H.-D. Chen and W.-K. Su, Mechanochemical Oxidative Heck Coupling of Activated and Unactivated Alkenes: A Chemo–, Regio–and Stereo–Controlled Synthesis of Alkenylbenzenes, Adv. Synth. Catal., 2019, 361, 5133–5139 CrossRef CAS; (e) J.-B. Yu, P. Ying, H. Wang, K.-Y. Xiang and W.-K. Su, Mechanochemical Asymmetric Cross–Dehydrogenative Coupling Reaction: Liquid–Assisted Grinding Enables Reaction Acceleration and Enantioselectivity Control, Adv. Synth. Catal., 2020, 362, 893–902 CrossRef CAS; (f) C.-Y. Wu, T. Ying, X.-J. Yang, W.-K. Su, A. V. Dushkin and J.-B. Yu, Mechanochemical Magnesium-Mediated Minisci C–H Alkylation of Pyrimidines with Alkyl Bromides and Chlorides, Org. Lett., 2021, 23, 6423–6428 CrossRef CAS PubMed; (g) X.-J. Yang, H. Wang, Y.-H. Zhang, W.-K. Su and J.-B. Yu, Generation of Aryl Radicals from in situ Activated Homolytic Scission: Driving Radical Reactions by Ball Milling, Green Chem., 2022, 24, 4557–4565 RSC; (h) C.-Y. Wu, T. Ying, H.-Q. Fan, C.-H. Hu, W.-K. Su and J.-B. Yu, C-4 Regioselective Alkylation of Pyridines Driven by Mechanochemically Activated Magnesium Metal, Org. Lett., 2023, 25, 2531–2536 CrossRef CAS PubMed.
- (a) A. B. Hughes, Amino Acids, Peptides and Proteins in Organic Chemistry. Vol. 1: Origins and Synthesis of Amino Acids, Wiley-VCH, Weinheim, Germany, 2009 Search PubMed; (b) L. Pollegioni and S. Servi, Unnatural Amino Acids, Springer, New York, 2012 CrossRef.
- (a) C. Huo, Y. Yuan, M. Wu, X. Jia, X. Wang, F. Chen and J. Tang, Auto-Oxidative Coupling of Glycine Derivatives, Angew. Chem., Int. Ed., 2014, 53, 13544–13547 CrossRef CAS; (b) Z. Q. Zhu, P. Bai and Z. Z. Huang, Dehydrogenative Cross-Coupling Reaction by Cooperative Transition-Metal and Brønsted Acid Catalysis for the Synthesis of β-Quinolinyl α-Amino Acid Esters, Org. Lett., 2014, 16, 4881–4883 CrossRef CAS; (c) Y. Wei, J. Wang, Y. Wang, X. Yao, C. Yang and C. Huo, Auto-Oxidation Promoted sp3 C–H Arylation of Glycine Derivatives, Org. Biomol. Chem., 2018, 16, 4985–4989 RSC.
- CCDC 2271241† (minor diastereomers of 3aa) contains the supplementary crystallographic data for this paper.
- I. Huskić, C. B. Lennox and T. Friščić, Accelerated Ageing Reactions: Towards Simpler, Solvent-Sree, Low Energy Chemistry, Green Chem., 2020, 22, 5881–5901 RSC.
- K.-Y. Xiang, P. Ying, T. Ying, W.-K. Su and J.-B. Yu, α-C–H Functionalization of Glycine Derivatives under Mechanochemical Accelerated Aging en Route the Synthesis of 1,4-Dihydropyridines and α-Substituted Glycine Esters, Green Chem., 2023, 25, 2853–2862 RSC.
- O. Takahashi, K. Kobayashi and A. Oda, Computational Modeling of the Enolization in a Direct Mechanism of Racemization of the Aspartic Acid Residue, Chem. Biodivers., 2010, 7, 1630–1633 CrossRef CAS.
- C. Adamo and V. Barone, Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- C. Zang, Y. Liu, Z.-J. Xu, C.-W. Tse, X. Guan, J. Wei, J.-S. Huang and C.-M. Che, Highly Enantioselective Iron-Catalyzed cis-Dihydroxylation of Alkenes with Hydrogen Peroxide Oxidant via an FeIII-OOH Reactive Intermediate, Angew. Chem., Int. Ed., 2016, 55, 10253–10257 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC: 2271241. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01467c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |