
Rapid modular synthesis of indole ethers via dehydrogenative cross-coupling reaction of indoles and alcohols†
Mao-Gui
Huang
,
Xiao-Hong
Chen
,
Hai-Bing
Xu
and
Yue-Jin
Liu
 *
*
Collaborative Innovation Center for Advanced Organic Chemical Materials Co-constructed by the Province and Ministry, Ministry of Education Key Laboratory for the Synthesis and Application of Organic Functional Molecules, and College of Chemistry and Chemical Engineering, Hubei University, Wuhan, 430062, China. E-mail: liuyuejin@hubu.edu.cn
First published on 14th November 2023
Abstract
Indole ethers are commonly found in pharmaceuticals, enzymes, and many bioactive compounds. The cross-coupling of indoles and alcohols by activation of the indole C–H bond would provide a direct and economical way of making these medicinally active products, but this remains an elusive transformation. Here we describe a straightforward and economical protocol for the synthesis of indole ethers by indole C–H alkoxylation with cobalt catalysis. A variety of functionalized indole ethers were modularly prepared from readily accessible indoles and alcohols. The scope of the transformation was expanded to a challenging intermolecular type reaction for the one-step construction of the core scaffold of piboserod (a 5-HT4 receptor antagonist). The synthetic applications were further demonstrated by the diversification of products and synthesis of many medical agent analogues. Preliminary mechanistic investigations indicated that the salicylaldehyde-cobalt catalyst played a key role in promoting this transformation.
Indoles are the most common organic molecular frameworks in organic chemistry,1 materials,2 pharmaceuticals,3 and life sciences.4 The C–H functionalization of indoles provides an efficient approach to the synthesis of these significant molecules, and thus has attracted widespread interest from academia to industry.5 In the past decade, significant progress has been made in the functionalization of indoles (Scheme 1B), which commonly focuses on C–C bond formation reactions, such as arylation,6 alkylation,7 and alkenylation,8 and alkynylation.9 In contrast, the construction of carbon-heteroatom bonds on the electron-rich indole motif,10 especially for heteroatoms with high electronegativity, is more difficult and less developed (Scheme 1A). Glorius11 and Ackermann12 respectively developed the C–H thiolation of indoles with Co and Cu catalysts. Recently, Shi et al. achieved indole C–H boronation without transition metal catalysts.13 Despite these elegant and significant advances, the construction of C–O bonds on indoles remains a critical challenge and is underdeveloped.
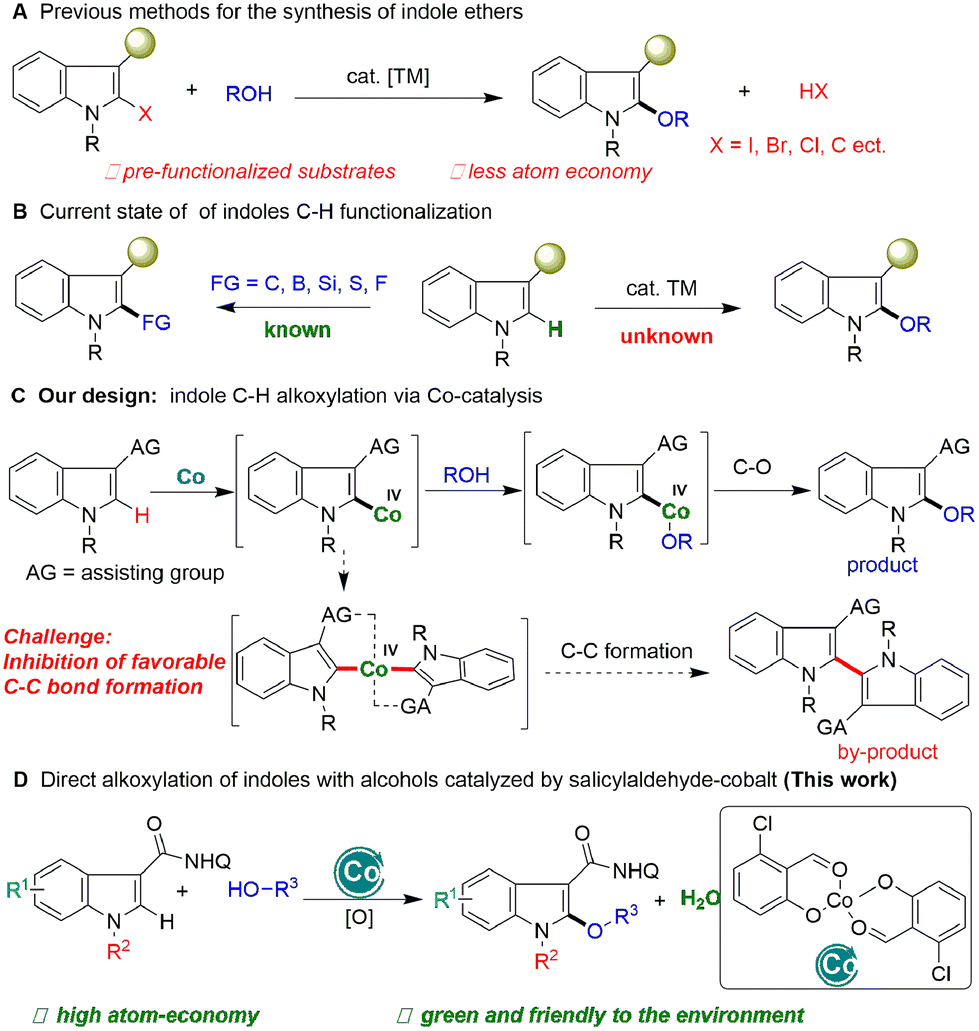 | ||
| Scheme 1 Synthesis of indole ethers via the C–H functionalization of indole. | ||
Indole ethers are an important class of structural motif, and are widely found in enzymes (tryprostatin A),14 and marketable drugs, such as indomethacin,15 an effective nonsteroidal anti-inflammatory drug which has been widely used to treat analgesia and rheumatoid arthritis (Fig. 1).16 At present, transition metal-catalyzed coupling reactions of aromatic halides with alcohols are the classic methods to achieve indole ethers (Scheme 1A).17 Notably, Zeng recently reported an elegant method for assembing 2-alkoxylindole skeletons via Rh(III)-catalyzed alkoxylation of 2-indolylalcohols with alcohols through C–C bond activation.18 Although these approaches are very efficient, there are still some shortcomings, such as the requirement of the use of pre-functionalized substrates and equivalent halogenated salt and other by-products, resulting in low atom economy and high synthetic costs. Therefore, it is still important to develop an economical and efficient method to synthesize indole ethers from simple raw materials with high atom economy.
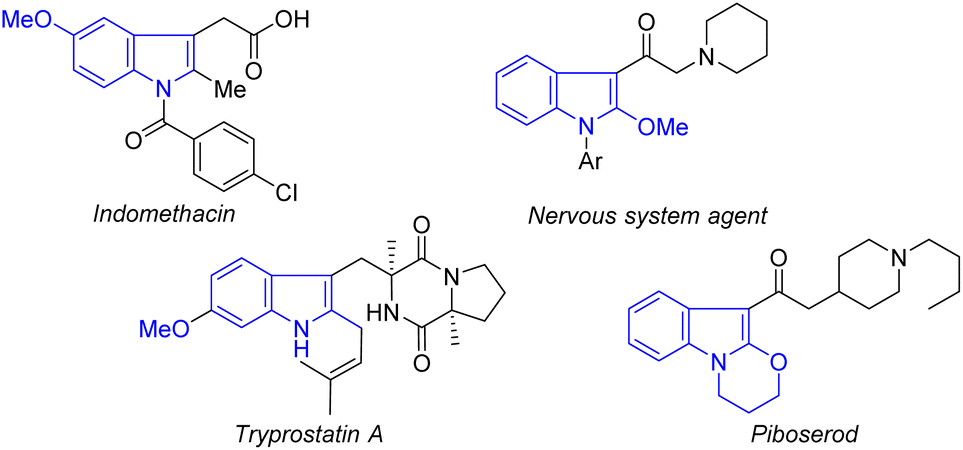 | ||
| Fig. 1 Selected bioactive molecules bearing the indole-ether motif. | ||
Cobalt is a cheap and low toxic metal element widely used in the field of C–H bond activation,19 especially in the construction of C–O bonds.20 Unfortunately, cobalt catalytic systems are mainly used for alkoxylation reactions of benzene substrates, which are difficult to apply to indole compounds. It is possible that indolyl-Co–O intermediates have good thermodynamic stability due to both the unique electron-rich characteristics of the indole motif and the high electronegativity of the O atom, which makes it difficult to form C(indolyl)–O bonds by reductive elimination. Recently, Shi's21 and other groups22 found that hydroxyoxazoline ligands can enhance cobalt catalytic activity and achieve the asymmetric functionalization of C–H bonds. Inspired by this elegant work and our recent research on salicylaldehyde-promoted cobalt-catalyzed indole C–H activation and annulation with alkynes,23 we speculated that by tuning the structure of the salicylaldehyde ligand to change the electronic and spatial properties of the cobalt catalyst, the thermodynamically stable indolyl-Co(III)–O intermediate could be oxidized to a high-valent and thermodynamically unstable salicylaldehyde-based Co(IV) species with the use of an appropriate directing group and oxidant, which can then undergo alkoxyl transfer and subsequent reductive elimination of the C–O bond, and a direct alkoxylation of indoles would be achieved. Notably, during this process the Co(IV) species may undergo C–C bond reductive elimination to form a dimerization product. How to inhibit the kinetically favorable reductive elimination of the C–C bond to form a dimerization product is a key challenge for this design (Scheme 1C). Thus, whether a direct catalytic indole C–H alkoxylation could be realized with high chemo-selectivity with a cobalt catalyst is still undetermined.
Herein, we report the unique salicylaldehyde-Co(II) promoted C–H alkoxylation of indoles with alcohols. This approach avoids the use of pre-functionalized indole substrates and excess of alcohols, and realizes a step-economical and atom-economical synthesis of indole esters (Scheme 1D).
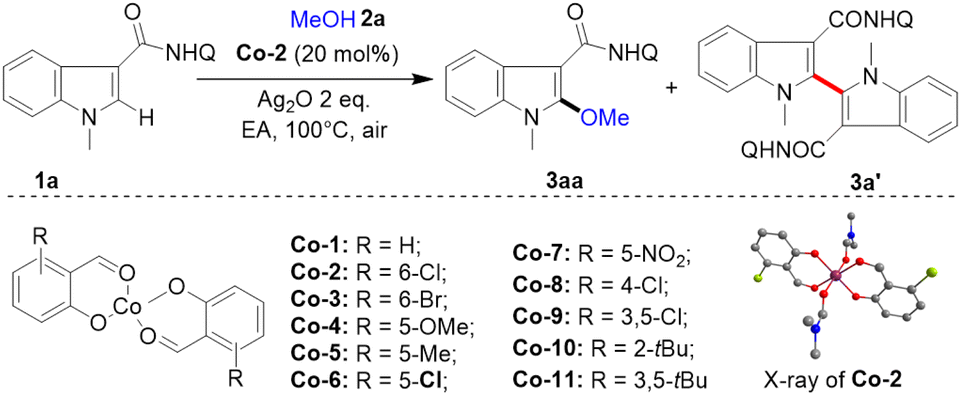
First, a series of electronically and sterically different salicylaldehyde-cobalt(II) complexes (Co-1–Co-11) were efficiently and conveniently prepared with 1 equivalent of Co(OAc)2 and 2 equivalents of salicylaldehyde under reflux in ethanol (Table 1). The divalent planar structure of the cobalt catalyst (Co-2) was determined through single crystal X-ray diffraction. With these structurally rich cobalt catalysts, the alkoxylation reaction was studied using indole-2-amide 1a and methanol 2a as raw materials. After extensive investigations (Table 1), the optimal conditions were determined as follows: in the presence of Co-2 (20 mol%), 1a (0.10 mmol, 1.0 equiv.), methanol 2a (10.0 equiv.), silver oxide (2.0 equiv.), and ethyl acetate (1.0 mL), at 100 °C for 12 h under air, the desired 2-methoxylated product 3aa was obtained in 85% yield with high selectivity (entry 1). Notably, the C–C formation reaction of indole was well inhibited, generating only a trace amount of self-coupling by-product 3a′ (<5%) with salicylaldehyde-cobalt (Co-2) catalyst. The Co(OAc)2 without salicylaldehyde ligand had low activity for this transformation (entry 2). CoNO3·6H2O failed to promote indole alkoxylation (entry 3). Instead of Co-2, with other salicylaldehyde based cobalt(II) catalysts such as Co-1, Co-3, Co-4, Co-5, Co-6, Co-7, Co-8 and Co-9 bearing both electron-rich and electron-deficient groups at the C2, C3, C4, C5 and C6 positions, high yields and chemoselectivity were obtained (entries 4–11). Notably, Co-catalysts (Co-10 and Co-11) with tert-butyl substituents at the C6 position had low activity and selectivity (entries 12 and 13), probably due to their large steric hindrances for alkoxyl transfer. No reaction could take place without the addition of cobalt-catalyst and silver oxide, indicating the importance of the Co-catalyst and silver oxidant in this conversion (entries 14 and 15). When base was added to the system, the amount of dimerization product increased (3a′, 32%), but the alkoxylation reaction was inhibited (3aa <5%) (entry 16). It may be that the addition of base accelerated the two C–H activation processes, which is conducive to the occurrence of the self-coupling reaction.
|
|
|||
|---|---|---|---|
| Entry | Variation from the optimal conditions none | Yield of 3aa (%) | Yield of 3a′ (%) |
| a Reaction conditions: 1a (0.10 mmol), 2a (10 equiv.), catalyst (20 mol%), Ag2O (2 equiv.), 1 mL EA, 12 h, under air. Yields were determined by 1H NMR. b Isolated yield. NHQ = 8-aminoquinolinyl. | |||
| 1 | None | 85 (79)b | <5 |
| 2 | Co(OAc)2 instead of Co-2 | 5 | 5 |
| 3 | Co(NO)3·6H2O instead of Co-2 | 0 | 0 |
| 4 | Co-1 instead of Co-2 | 53 | <5 |
| 5 | Co-3 instead of Co-2 | 79 | <5 |
| 6 | Co-4 instead of Co-2 | 56 | <5 |
| 7 | Co-5 instead of Co-2 | 66 | <5 |
| 8 | Co-6 instead of Co-2 | 70 | <5 |
| 9 | Co-7 instead of Co-2 | 68 | <5 |
| 10 | Co-8 instead of Co-2 | 72 | <5 |
| 11 | Co-9 instead of Co-2 | 73 | <5 |
| 12 | Co-10 instead of Co-2 | 15 | 8 |
| 13 | Co-11 instead of Co-2 | 17 | 10 |
| 14 | No Co-2 | 0 | 0 |
| 15 | No Ag2O | 0 | 0 |
| 16 | With Cs2CO3 | <5 | 32 |
With the optimized conditions in hand, the scope of indoles was investigated first (Scheme 2). Indoles bearing various common functional groups such as OMe, Me, F, and Cl could couple with ethanol, giving the corresponding C2-alkoxylated products (3ba–3ma) in moderate to good yields (56%–86%) with high chemoselectivity (3/3a′ > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Notably, the active Br group, which is a useful handle for further diversification, also tolerated the reaction conditions, generating the desired products in good yields (3ha, 76% and 3ja, 71%). We found no systematic influence of the substituents at the C4, C5, C6 and C7 positions. The reaction was sensitive to N-substituents. Electron-donating groups such as alkyl and benzyl groups with different substituents on the N atom, such as n-propyl and benzyl, could undergo C2-alkoxylation reactions smoothly, but acyl substituted substrates did not work. Moreover, the reaction could extend to the C3-alkoxylation of indole (3ta, 80%) by tuning the position of the assisting group. Furthermore, C5 and C6 alkoxylation at the phenyl ring of indoles (3ua, 74% and 3va, 52%) was also suitable for this protocol, affording a series of structurally diverse indole ethers.
:1). Notably, the active Br group, which is a useful handle for further diversification, also tolerated the reaction conditions, generating the desired products in good yields (3ha, 76% and 3ja, 71%). We found no systematic influence of the substituents at the C4, C5, C6 and C7 positions. The reaction was sensitive to N-substituents. Electron-donating groups such as alkyl and benzyl groups with different substituents on the N atom, such as n-propyl and benzyl, could undergo C2-alkoxylation reactions smoothly, but acyl substituted substrates did not work. Moreover, the reaction could extend to the C3-alkoxylation of indole (3ta, 80%) by tuning the position of the assisting group. Furthermore, C5 and C6 alkoxylation at the phenyl ring of indoles (3ua, 74% and 3va, 52%) was also suitable for this protocol, affording a series of structurally diverse indole ethers.
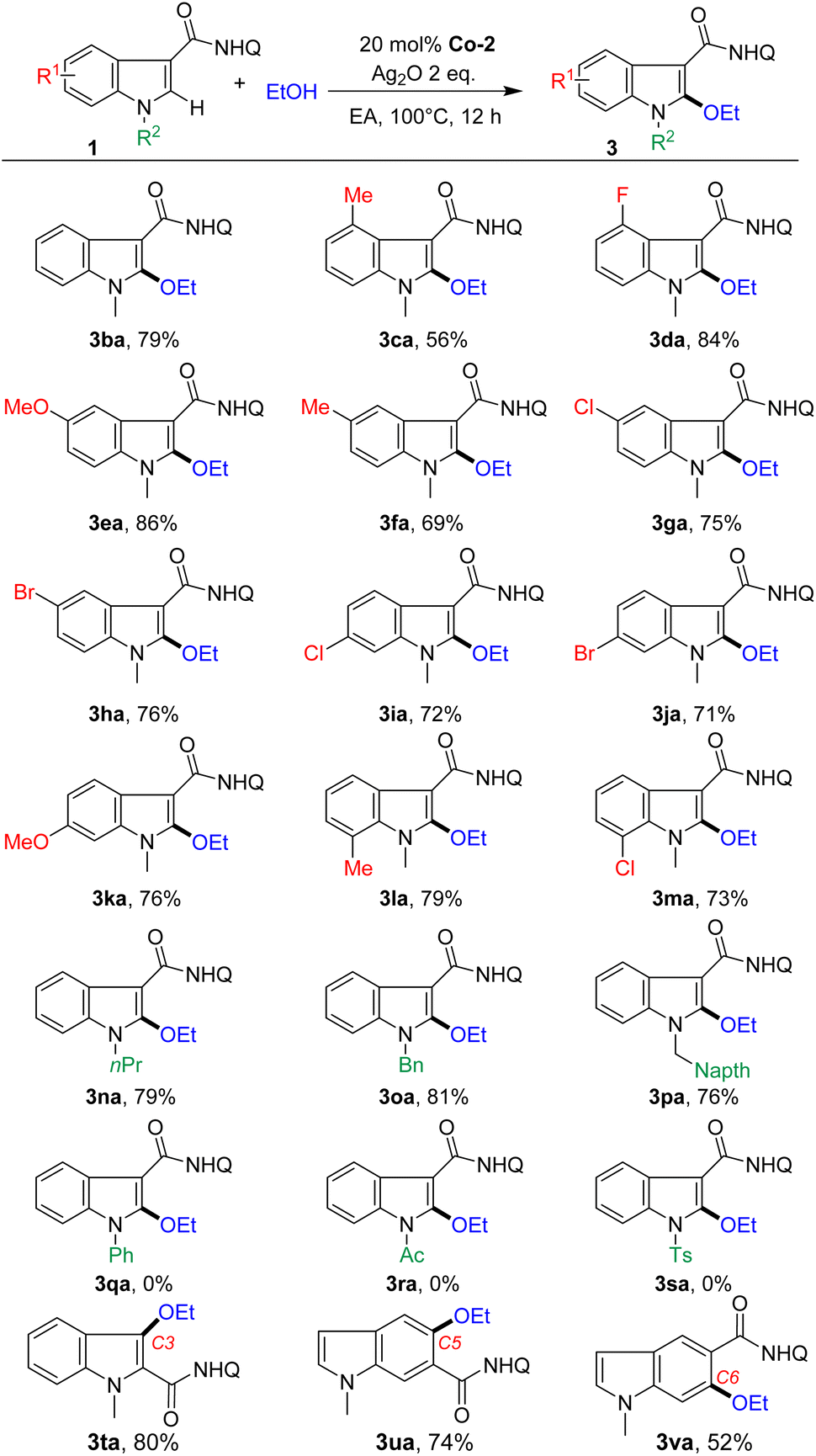 | ||
| Scheme 2 Scope of indole substrates. Reaction conditions: 1 (0.1 mmol), EtOH (1.0 mmol), Co-2 (20 mol%), Ag2O (2.0 equiv.), EA (1.0 mL), under air at 100 °C for 12 h. Isolated yields. Q = 8-aminoquinoline. | ||
Next, we studied the scope of alcohols for indole alkoxylation (Scheme 3). Diverse alcohols with various functional groups, such as Ph, OMe, OH, Cl, CCl3, Ac and CF2 groups, were all active for indole alkoxylation, affording the corresponding product in moderate to good yields. Notably, highly active alkyl bromide, which can be used as a handle for further diversification via nucleophilic substitution reactions, could also be compatible under these conditions, affording the alkoxylated product (4aj, 53%). Notably, 1,3-butanediol could undergo a cross-coupling reaction with indole, in which the alkoxylation reaction selectively occurred at the primary OH group (4an, 42%), while the secondary alcohol was completely retained. These results demonstrate the good substrate universality and wide functional group compatibility of this method.
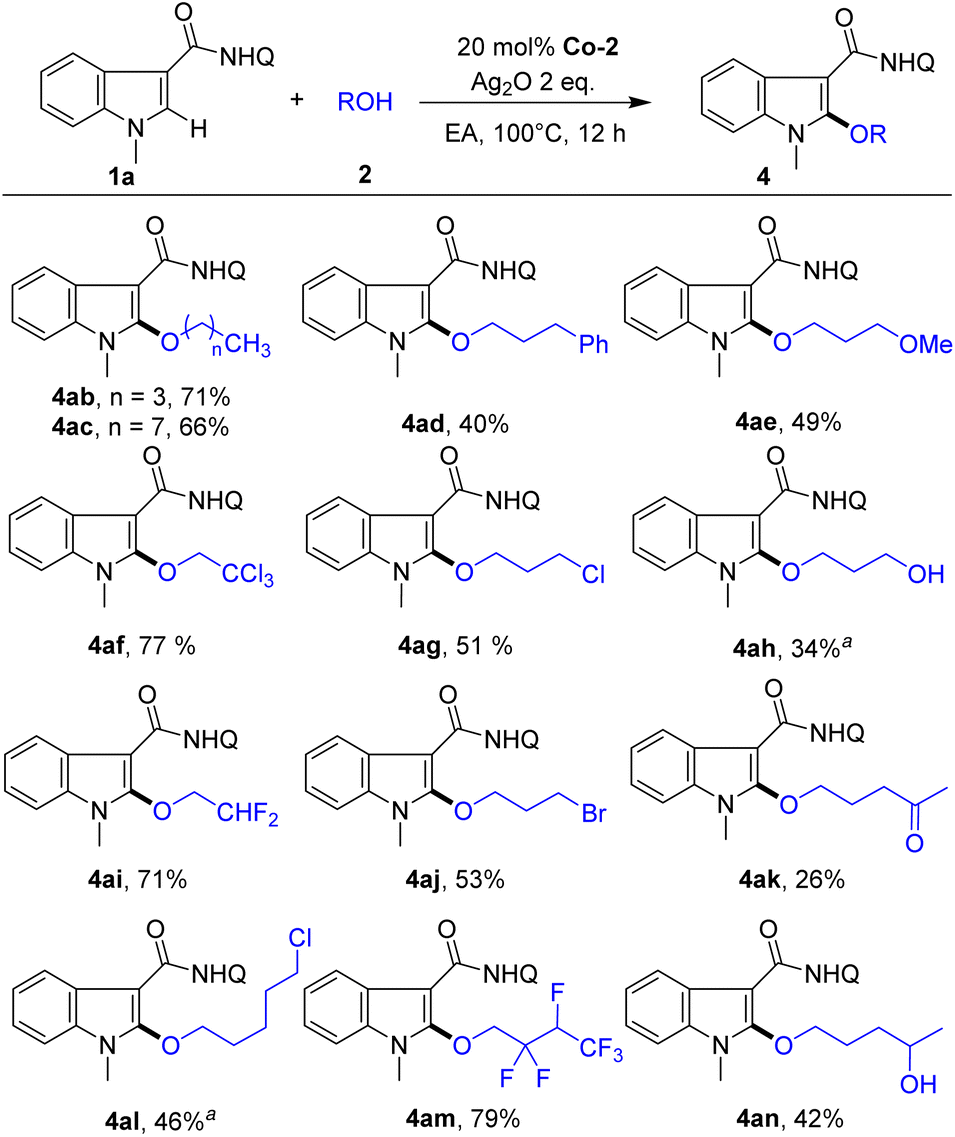 | ||
| Scheme 3 Scope of alcohols. Reaction conditions: 1a (0.1 mmol), 2 (1.0 mmol), Co-2 (20 mol%), Ag2O (2.0 equiv.), EA (1.0 mL), under air at 100 °C for 12 h. a2 (5 equiv.). Isolated yields. Q = 8-aminoquinoline. | ||
To prove the usefulness of this protocol, gram-scale reaction, removal of the assisting group, diversification of the product, derivatization of the natural product, and synthesis of medical agent analogues were conducted (Scheme 4). A high yield (0.9 g, 78%) of product 3aa was obtained with high chemoselectivity on the 3.5 mmol scale (Scheme 4a). The assisting group of 8-aminoquinoline could be removed according to previously reported methods (Scheme 4b). Alternatively, 3aa could convert methoxy groups into active hydroxyl groups for later synthesis conversion (Scheme 4c). Furthermore, the product could be transformed into oxindole (Scheme 4c), which overrides the inherent C3-selection in the indole oxidation reaction. To our delight, this method can not only efficiently synthesize anti-inflammatory agent analogue 6, but also be applied to modify a natural product (citronellol) (Scheme 4d). Finally, this protocol was further expanded to intramolecular reaction for the rapid synthesis of piboserod (a 5-HT4 receptor antagonist) analogue 8 (Scheme 4e), which further demonstrates its enormous potential in drug synthesis applications.
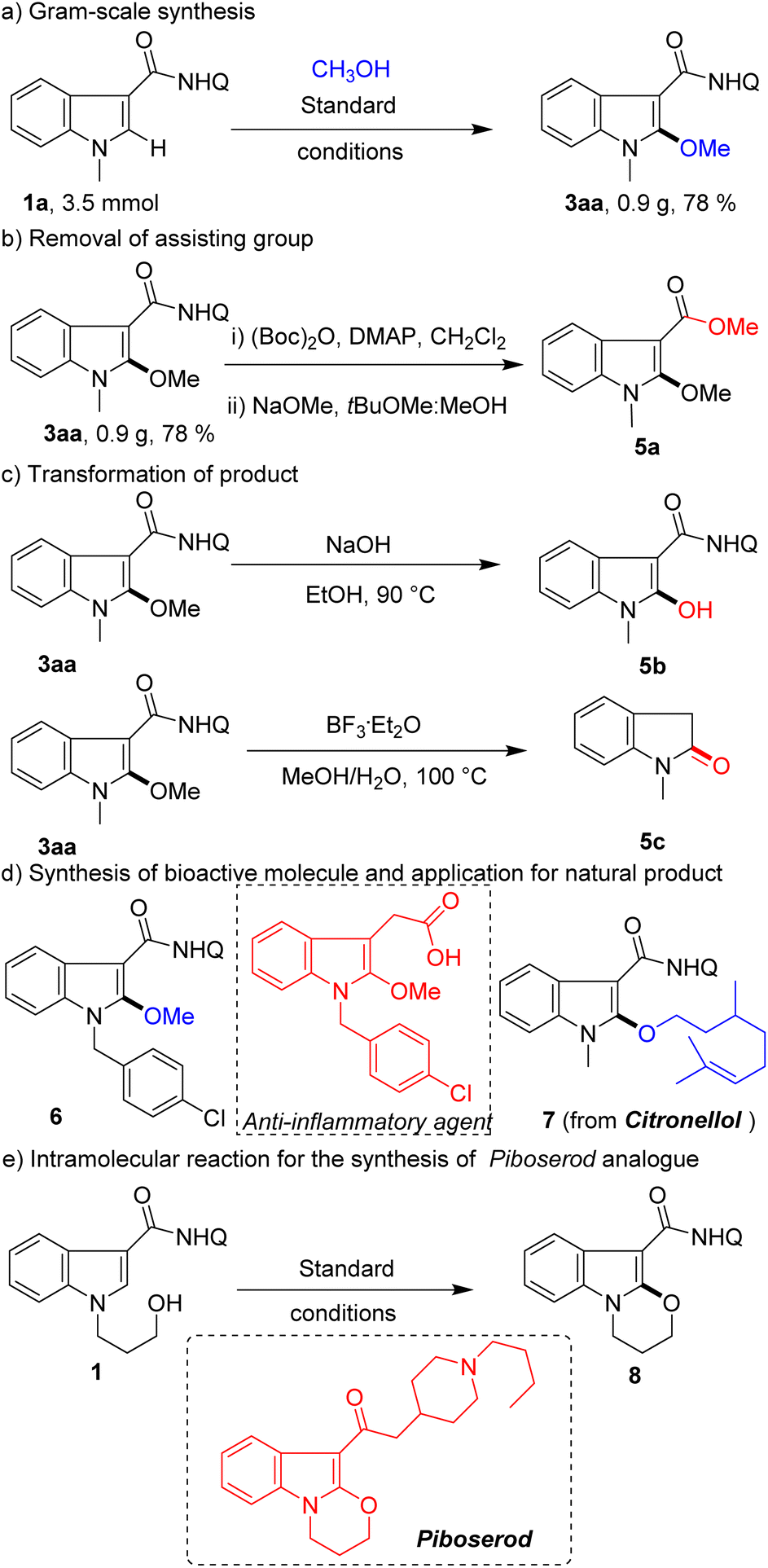 | ||
| Scheme 4 Synthetic applications. | ||
To gain insight into the mechanism of this transformation, a series of mechanistic experiments were conducted (Scheme 5). First, deuteration experiments were carried out with deuterated acetic acid and 65% deuterization was detected at the C2 position of substrate 1a (Scheme 5a), indicating that C–H activation by the Co-catalyst is a reversible process. Next, we conducted kinetic isotopic effect (KIE) studies on the indole and alcohol, respectively. The results showed that the KIE of C–H was 1.0 and the KIE of O–H was 0.94 (Scheme 5b), showing that neither C–H bond nor O–H bond cleavages were involved in the rate-limiting step. To further understand the process of the reaction, we used high-resolution mass spectrometry (HR-MS) to track and study the reaction intermediates. Among them, intermediate A detected by HR-MS (for details seeing SI†) coordinated with substrate 1a, intermediate B formed by C–H bond activation, and intermediate C generated by alkoxy group transfer were detected in the system (Scheme 5c). Unfortunately, we were not able to separate them.
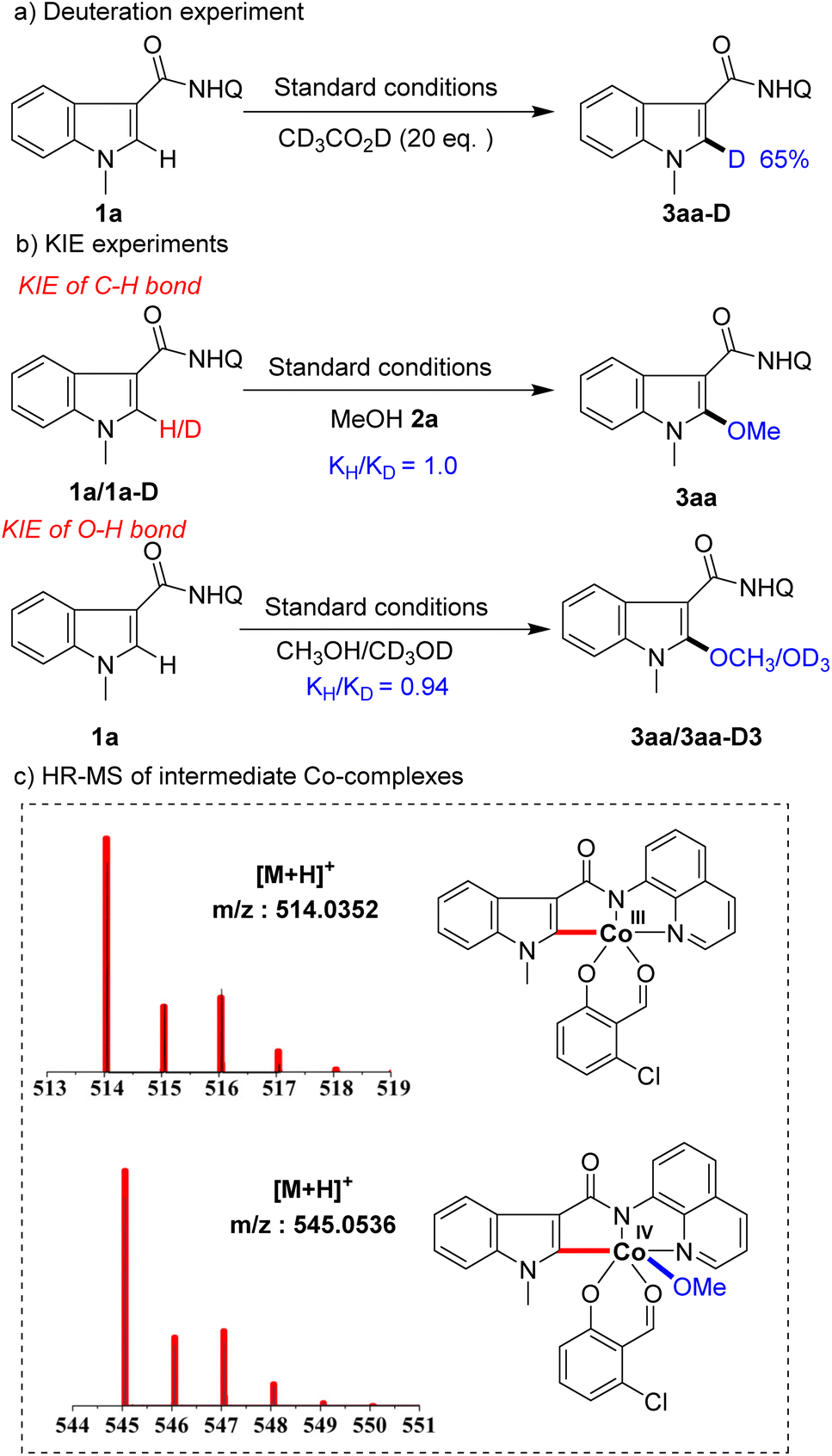 | ||
| Scheme 5 Mechanistic studies. | ||
Based on the above results and previous literature, a plausible catalytic cycle is proposed (Scheme 6). First, the indole substrate 1a coordinates with the catalyst Co-2 to form intermediate A (detected by HR-MS). Then, the five-membered cyclometalated intermediate B (detected by HR-MS) is generated from intermediate Avia reversible C–H activation. Next, complex B undergoes a methoxyl transfer with methanol to produce intermediate C (detected by HR-MS), and reductive elimination occurs to deliver the product and regenerate the Co-catalyst. On the other hand, complex B could coordinate with another indole substrate 1a to form intermediate D. Subsequently, intermediate D undergoes secondary C–H activation to generate intermediate E, which affords the by-product 3a′via C–C bond formation through reductive elimination.
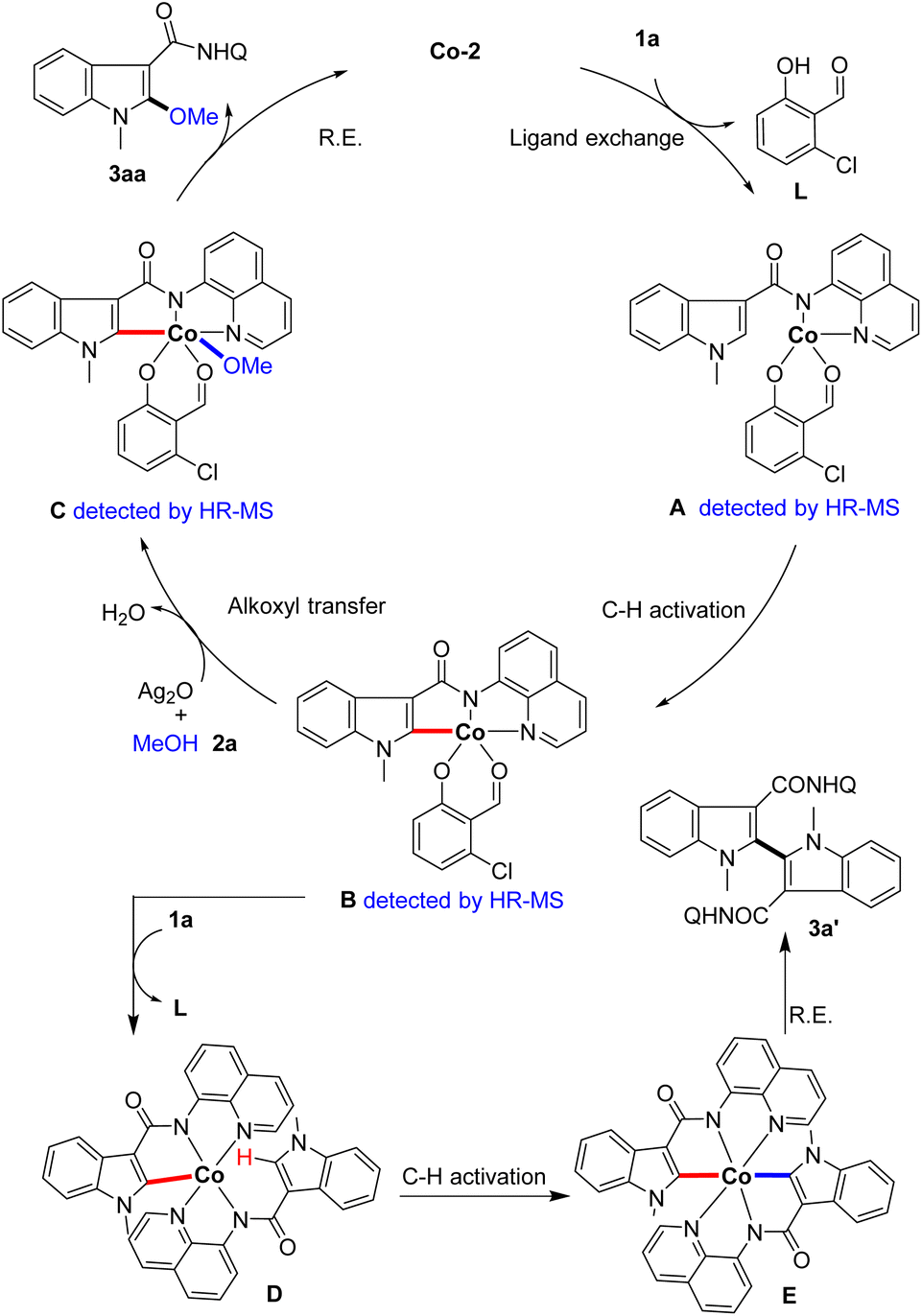 | ||
| Scheme 6 Proposed catalytic cycle. | ||
In summary, the first C–H alkoxylation reaction of indole was developed with a cobalt catalyst, which overrides the traditional favorable C–C bond formation, enabling a straightforward and economical access to indole ethers, from easily accessible and commercially available primary and secondary alcohols and various indole substrates.24 The designed Co-salicylaldehyde catalyst is the key to achieve this transformation with high chemical selectivity. This protocol can extend to a more challenging intramolecular alkoxylation reaction for the construction of indole cycloethers. The synthetic application of this transformation is demonstrated by the rapid synthesis of a series of medical agent analogues. The KIE experiments indicate that the C–H activation is not involved in the rate-determining step, and the HR-MS analyses suggest that the reaction goes through a process of oxidation, C–H activation, alkoxyl transfer and reductive elimination. This work not only fills the gap in the construction of C–O bonds in the field of indole C–H activation, but provides a novel and general strategy for the synthesis of indole ethers, which is expected to have significant applications in the discovery of new drugs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Distinguished Professor program of Hubei University.References
- (a) J. E. Saxton, Recent Progress in the Chemistry of lndole Alkaloids and Mould Metabolites, Nat. Prod. Rep., 1989, 6, 1–54 RSC; (b) L. Zeng, Y. Lin and S. Cui, Indole-N-Carboxylic Acids and Indole-N-Carboxamides in Organic Synthesis, Chem. – Asian J., 2020, 15, 973–985 CrossRef CAS PubMed; (c) M. Bandini and A. Eichholzer, Catalytic Functionalization of Indoles in a New Dimension, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 CrossRef CAS PubMed; (d) A. Alparslan and P. Ulf, Chemistry and Biology of New Marine Alkaloids from the Indole and Annelated Indole Series, Curr. Med. Chem., 2003, 10, 1113–1127 CrossRef PubMed.
- (a) D. Paul and J. John, Recent Advances towards the Synthesis and Material Applications of Indoloindoles, Chem. – Asian J., 2022, 17, e202200460 CrossRef CAS PubMed; (b) P. R. Nitha, S. Soman and J. John, Indole fused heterocycles as sensitizers in dye-sensitized solar cells: an overview, Mater. Adv., 2021, 2, 6136–6168 RSC.
- (a) P. T. Singh and M. O. Singh, Recent Progress in Biological Activities of Indole and Indole Alkaloids, Mini-Rev. Med. Chem., 2018, 18, 9–25 CrossRef PubMed; (b) F. Song, Z. Li, X. Huo, J. Fang, L. Shao and M. Zhou, Indole/isatin-containing hybrids as potential antibacterial agents, Arch. Pharm., 2020, 353, e2000143 CrossRef PubMed; (c) R. H. Mir, R. Mohi-ud-din, U. T. Wani, O. M. Dar, J. A. Shah, B. Lone, C. Pooja and H. M. Masoodi, Indole: A Privileged Heterocyclic Moiety in the Management of Cancer, Curr. Org. Chem., 2021, 25, 724–736 CrossRef CAS.
- (a) T. V. Sravanthi and S. L. Manju, Indoles - A promising scaffold for drug development, Eur. J. Pharm. Sci., 2016, 91, 1–10 CrossRef CAS PubMed; (b) V. M. Norwood and R. W. Huigens, Harnessing the Chemistry of the Indole Heterocycle to Drive Discoveries in Biology and Medicine, ChemBioChem, 2019, 20, 2273–2297 CrossRef CAS PubMed.
- (a) P. Kumar, P. J. Nagtilak and M. Kapur, Transition metal-catalyzed C-H functionalizations of indole, New J. Chem., 2021, 45, 13692–13746 RSC; (b) K. Urbina, D. Tresp, K. Sipps and M. Szostak, Recent Advances in Metal-Catalyzed Functionalization of Indoles, Adv. Synth. Catal., 2021, 363, 2723 CrossRef CAS; (c) B. Prabagar, Y.-Q. Yang and Z.-Z. Shi, Site-selective C-H functionalization to accessthe arene backbone of indoles and quinolines, Chem. Soc. Rev., 2021, 50, 11249–11269 RSC; (d) J. Wen and Z.-Z. Shi, From C4 to C7: Innovative Strategies for Site-Selective Functionalization of Indole C-H Bonds, Acc. Chem. Res., 2021, 54, 1723–1736 CrossRef CAS PubMed; (e) J. A. Leitch, Y. Bhonoah and C. J. Frost, Beyond C2 and C3: Transition-Metal-Catalyzed C-H Functionalization of Indole, ACS Catal., 2017, 7, 5618–5627 CrossRef CAS.
- (a) N. Jacob, Y. Zaid, J. C. A. Oliveira, L. Ackermann and J. Wencel-Delord, Cobalt-Catalyzed Enantioselective C-H Arylation of Indoles, J. Am. Chem. Soc., 2022, 144, 798–806 CrossRef CAS PubMed; (b) Y. Yang, P. Gao, Y. Zhao and Z.-Z. Shi, Regiocontrolled Direct C-H Arylation of Indoles at the C4 and C5 Positions, Angew. Chem., Int. Ed., 2017, 56, 3966–3971 CrossRef CAS PubMed; (c) J. Ma, R. Feng and Z.-B. Dong, Recent Advances in Indole Synthesis and the Related Alkylation, Asian J. Org. Chem., 2023, 12, e202300092 CrossRef CAS.
- (a) L.-H. Kong, X. Han, H.-H. Chen, H.-M. Sun, Y. Lan and X.-W. Li, Rhodium(II)-Catalyzed Regioselective Remote C-H Alkylation of Protic Indoles, ACS Catal., 2021, 11, 4929–4935 CrossRef CAS; (b) J. A. Leitch, C. L. McMullin, M. F. Mahon, Y. Bhonoah and C. G. Frost, Remote C6-Selective Ruthenium-Catalyzed C-H Alkylation of Indole Derivatives via σ-Activation, ACS Catal., 2017, 7, 2616–2623 CrossRef CAS; (c) v. soni, R. A. Jagtap, R. G. Gonnade and B. Punji, A Unified Strategy for Nickel-Catalyzed C-2 Alkylation of Indoles through Chelation Assistance, ACS Catal., 2016, 6, 5666–5672 CrossRef CAS.
- (a) I. Choi, A. M. Messinis and L. Ackermann, C7-Indole Amidations and Alkenylations by Ruthenium(II) Catalysis, Angew. Chem., Int. Ed., 2020, 59, 12534–12540 CrossRef CAS PubMed; (b) R. A. Jagtap, C. P. Vinod and B. Punji, Nickel-Catalyzed Straightforward and Regioselective C-H Alkenylation of Indoles with Alkenyl Bromides: Scope and Mechanistic Aspect, ACS Catal., 2019, 9, 431–441 CrossRef CAS; (c) Z. Ding and N. Yoshikai, Mild and Efficient C2-Alkenylation of Indoles with Alkynes Catalyzed by a Cobalt Complex, Angew. Chem., Int. Ed., 2012, 51, 4698–4701 CrossRef CAS PubMed.
- (a) C. N. Kona, Y. Nishii and M. Miura, Iridium-Catalyzed Direct C4- and C7-Selective Alkynylation of Indoles Using Sulfur-Directing Groups, Angew. Chem., Int. Ed., 2019, 58, 9856–9860 CrossRef CAS PubMed; (b) Z.-Z. Zhang, B. Liu, C.-Y. Wang and B.-F. Shi, Cobalt(III)-Catalyzed C2-Selective C-H Alkynylation of Indoles, Org. Lett., 2015, 17, 4094–4097 CrossRef CAS PubMed.
- (a) K. Mertins, A. Zapf and M. Beller, Catalytic borylation of o-xylene and heteroarenes via C-H activation, J. Mol. Catal. A: Chem., 2004, 207, 21–25 CrossRef CAS; (b) Z.-J. Liang, J.-L. Zhao and Y.-H. Zhang, Palladium-Catalyzed Regioselective Oxidative Coupling of Indoles and One-Pot Synthesis of Acetoxylated Biindolyls, J. Org. Chem., 2010, 75, 170–177 CrossRef CAS PubMed; (c) Y.-X. Tong, Z.-W. Wang, B. Liu, Y.-W. Xu, S. Gao, X.-B. Tang and X.-H. Zhang, Recent Advances in Synthesis of 3-Sulfenylated Indoles, Chin. J. Org. Chem., 2023, 43, 1310–1324 CrossRef; (d) X.-L. Shi, Z.-M. Wang, Y.-X. Li, X.-W. Li, X.-Q. Li and D.-Y. Shi, Palladium-Catalyzed Remote C-H Phosphonylation of Indoles at the C4 and C6 Positions by a Radical Approach, Angew. Chem., 2021, 133, 13990–13995 CrossRef.
- T. Gensch, F. J. R. Klauck and F. Glorius, Cobalt-Catalyzed C-H Thiolation through Dehydrogenative Cross-Coupling, Angew. Chem., Int. Ed., 2016, 55, 11287–11291 CrossRef CAS PubMed.
- P. Gandeepan, J. Mo and L. Ackermann, Photo-induced Copper-Catalyzed C-H Chalcogenation of Azoles at Room Temperature, Chem. Commun., 2017, 53, 5906–5909 RSC.
- J. Lv, X. Chen, X.-S. Xue, B. Zhao, Y. Liang, M. Wang, L. Jin, Y. Yuan, Y. Han, Y. Zhao, Y. Lu, J. Zhao, W.- Y. Sun, K. N. Houk and Z.-Z. Shi, Metal-free directed sp2-C-H borylation, Nature, 2019, 575, 336–340 CrossRef CAS PubMed.
- D. H. Jain, C. Zhang, S. Zhou, H. Zhou, J. Ma, X. Liu, X. Liao, A. M. Deveau, C. M. Jdieckhaus, M. A. ohnson, K. S. Smith, T. L. Macdonald, H. Kakeya, H. Osada and J. M. Cook, Synthesis and structure-activity relationship studies on tryprostatin A, an inhibitor of breast cancer resistance protein, Bioorg. Med. Chem., 2008, 16, 4626–4651 CrossRef PubMed.
- M. A. J. Duncton, R. B. Murray, G. Park and R. Singh, Tetrazolone as an acid bioisostere: application to marketed drugs containing a carboxylic acid, Org. Biomol. Chem., 2016, 14, 9343–9347 RSC.
- M. Fedouloff, F. Hossner, M. Voyle, J. Ranson, J. Powles, G. Riley and G. Sanger, Synthesis and pharmacological activity of metabolites of the 5-HT4 receptor antagonist SB-207266, Bioorg. Med. Chem., 2001, 9, 2119–2128 CrossRef CAS PubMed.
- (a) T. Ruhland, H. Pedersen and K. Andersen, Application of Polystyrene Bound Ethoxymethyl (PEOM) Chloride in the Solid-Phase Synthesis of Substituted 1-Methyl-1H-Indoles, Synthesis, 2003, 2236–2240 CrossRef CAS; (b) D. M. Ketcha and G. W. Gribble, A convenient synthesis of 3-acylindoles via Friedel Crafts acylation of 1-(phenylsulfonyl)indole. A new route to pyridocarbazole-5,11-quinones and ellipticine, J. Org. Chem., 1985, 50, 5451–5457 CrossRef CAS; (c) M. Tayu, K. Nomura, K. Kawachi, K. Higuchi, N. Saito and T. Kawasaki, Direct C2-Functionalization of Indoles Triggered by the Generation of Iminium Species from Indole and Sulfonium Salt, Chem. – Eur. J., 2017, 23, 10925–10930 CrossRef CAS PubMed; (d) T. Cao, J. Deitch, E. C. Linton and M. C. Kozlowski, Asymmetric synthesis of allenyl oxindoles and spirooxindoles by a catalytic enantioselective Saucy-Marbet Claisen rearrangement, Angew. Chem., Int. Ed., 2012, 51, 2448–2451 CrossRef CAS PubMed; (e) E. C. Linton and M. C. Kozlowski, Catalytic enantioselective Meerwein-Eschenmoser Claisen rearrangement: asymmetric synthesis of allyl oxindoles, J. Am. Chem. Soc., 2008, 130, 16162–16163 CrossRef CAS PubMed.
- C. Yang, Z. Liu, X. Hu, H. Xie, H. Jiang and W. Zeng, Rh(iii)-Catalyzed Csp2-Csp3 bond alkoxylation of α-indolyl alcohols via C-C σ bond cleavage, Org. Chem. Front., 2021, 8, 2949–2954 RSC.
- (a) M. Moselage, J. Li and L. Ackermann, Cobalt-Catalyzed C-H Activation, ACS Catal., 2016, 6, 498–525 CrossRef CAS; (b) Y. Kommagalla and N. Chatani, Cobalt(II)-catalyzed C-H functionalization using an N,N′-bidentate directing group, Coord. Chem. Rev., 2017, 350, 117–135 CrossRef CAS; (c) K. Gao and N. Yoshikai, Low-Valent Cobalt Catalysis: New Opportunities for C-H Functionalization, Acc. Chem. Res., 2014, 47, 1208–1219 CrossRef CAS PubMed; (d) Y. Zheng, C. Zheng, C. Gu and S.-L. You, Enantioselective C-H functionalization reactions enabled by cobalt catalysi, Chem Catal., 2022, 2, 2965–2985 CrossRef CAS; (e) R. Mei, U. Dhawa, R. C. Samanta, W. Ma, J. Wencel-Delord and L. Ackermann, Cobalt-Catalyzed Oxidative C-H Activation: Strategies and Concepts, ChemSusChem, 2020, 13, 3306–3356 CrossRef CAS PubMed; (f) S. Wang, S.-Y. Chen and X.-Q. Yu, C-H Functionalizations by High-valent Cp*Co(III) Catalysis, Chem. Commun., 2017, 53, 3165–3180 RSC; (g) A. Baccalini, S. Vergura, P. Dolui, G. Zanoni and D. Maiti, Recent advances in cobalt-catalysed C-H functionalizations, Org. Biomol. Chem., 2019, 17, 10119–10141 RSC.
- (a) Z.-Z. Zhang, J. Cheng, Q.-J. Yao, Q. Yue and G. Zhou, Cobalt-Catalyzed C-H Alkoxylation of Ferrocenes with Alcohols under Mild Conditions, Adv. Synth. Catal., 2021, 363, 3946 CrossRef CAS; (b) L.-B. Zhang, X.-Q. Hao, S.-K. Zhang, Z.-J. Liu, X.-X. Zheng, J.-F. Gong, J.-L. Niu and M.-P. Song, Cobalt-Catalyzed C(sp2)-H Alkoxylation of Aromatic and Olefinic Carboxamides, Angew. Chem., Int. Ed., 2015, 54, 272–275 CrossRef CAS PubMed; (c) X.-L. Li, K. Zhang, S.-Y. Chen, C. Li, F. Li, H.-J. Xu and Y. Fu, Cobalt catalyst for reductive etherification of 5-hydroxymethylfurfural to 2,5-bis(methoxymethyl)furan under mild conditions, Green Chem., 2018, 20, 1095–1105 RSC; (d) L. Rocard, D. Chen, A. Stadler, H. Zhang, R. Gil, S. Bezzenine and J. Hannedouche, Earth-Abundant 3d Transition Metal Catalysts for Hydroalkoxylation and Hydroamination of Unactivated Alkenes, Catalysts, 2021, 11, 674 CrossRef CAS.
- (a) G. Zhou, J.-H. Chen, Q.-J. Yao, F.-R. Huang, Z.-K. Wang and B.-F. Shi, Base-Promoted Electrochemical CoII-catalyzed Enantioselective C-H Oxygenation, Angew. Chem., 2023, 135, e202302964 CrossRef; (b) Q.-J. Yao, F.-R. Huang, J.-H. Chen, M.-Y. Zhong and B.-F. Shi, Enantio- and Regioselective Electrooxidative Cobalt-Catalyzed C-H/N-H Annulation with Alkenes, Angew. Chem., Int. Ed., 2023, 62, e202218533 CrossRef CAS PubMed; (c) Z.-K. Wang, Y.-J. Wu, Q.-J. Yao and B.-F. Shi, Synthesis of Chiral Diarylmethylamines by Cobalt-Catalyzed Enantioselective C-H Alkoxylation, Angew. Chem., Int. Ed., 2023, 62, e202304706 CrossRef CAS PubMed; (d) Q.-J. Yao, J.-H. Chen, H. Song, F.-R. Huang and B.-F. Shi, Cobalt/Salox-Catalyzed Enantioselective C-H Functionalization of Arylphosphinamides, Angew. Chem., Int. Ed., 2022, 61, e202202892 CrossRef CAS PubMed; (e) J.-H. Chen, M.-Y. Teng, F.-R. Huang, H. Song, Z.-K. Wang, H.-L. Zhuang, Y.-J. Wu, X. Wu, Q.-J. Yao and B.-F. Shi, Cobalt/Salox-Catalyzed Enantioselective Dehydrogenative C-H Alkoxylation and Amination, Angew. Chem., Int. Ed., 2022, 61, e202210106 CrossRef CAS PubMed; (f) Y.-J. Wu, Z.-K. Wang, Z.-S. Jia, J.-H. Chen, F.-R. Huang, B.-B. Zhan, Q.-J. Yao and B.-F. Shi, Synthesis of Axially Chiral Biaryls through Cobalt(II)-Catalyzed Atroposelective C-H Arylation, Angew. Chem., Int. Ed., 2023, 62, e202310004 CrossRef CAS PubMed.
- (a) L. Gong, S. P. Mulcahy, K. Harms and E. Meggers, Chiral-Auxiliary-Mediated Asymmetric Synthesis of Tris-Heteroleptic Ruthenium Polypyridyl Complexes, J. Am. Chem. Soc., 2009, 131, 9602–9603 CrossRef CAS PubMed; (b) X.-J. Si, D. Yang, M.-C. Sun, D.-H. Wei, M.-P. Song and J.-L. Niu, Atroposelective isoquinolinone synthesis through cobalt-catalysed C-H activation and annulation, Nat. Synth., 2022, 1, 709–718 CrossRef.
- M.-G. Huang, S. Shi, M. Li, Y.-J. Liu and M.-H. Zeng, Salicylaldehyde-Promoted Cobalt-Catalyzed C-H/N-H Annulation of Indolyl Amides with Alkynes: Direct Synthesis of a 5-HT3 Receptor Antagonist Analogue, Org. Lett., 2021, 23, 7094–7099 CrossRef CAS PubMed.
- P. Wei, Y. Zhu, J. Ying and X.-F. Wu, Cobalt-Catalyzed Direct Alkoxylation of Indoles, J. Org. Chem., 2023, 88, 6274–6280 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2291138 and 2291139. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01501g |
| This journal is © the Partner Organisations 2024 |