
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA convergent paired electrochemical strategy for decarboxylative C(sp2)–C(sp3) bond formation†
Lingjiao
Li‡
ab,
Zijian
Li‡
 bc,
Wenxuan
Sun
ab and
Chao
Li
*ab
bc,
Wenxuan
Sun
ab and
Chao
Li
*ab
aTsinghua Institute of Multidisciplinary Biomedical Research, Tsinghua University, Beijing, 100084, China. E-mail: lichao@nibs.ac.cn
bNational Institute of Biological Sciences, Beijing, 102206, China
cAcademy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China
First published on 6th February 2024
Abstract
In this report, we describe a convergent paired electrochemical decarboxylative C(sp2)–C(sp3) cross-coupling reaction that employs readily available alkyl carboxylic acids and C(sp2)–iodides as direct coupling partners. This easy-to-operate electrochemical process occurs under mild conditions, uses inexpensive materials and reagents, requires no prefunctionalization of the native acids, and operates broadly across diverse aryl, heteroaryl, and alkenyl iodides, thereby providing powerful and flexible access to the construction of C(sp2)–C(sp3) bonds. The utility and practicality of this convergent paired electrolysis is demonstrated in the syntheses of high-value and synthetically challenging unnatural amino acids and terpenoid natural products. Extensive mechanistic studies shed light on the anodic PPh3/NaI-mediated generation of N-hydroxyphthalimide esters and the cathodic reductive cross-coupling processes facilitated by NiI species.
Introduction
Over the last decade, decarboxylative C(sp2)–C(sp3) cross-coupling reactions that directly employ readily available aryl/vinyl halides as one of the coupling partners have been propelled to the forefront for C(sp2)–C(sp3) bond formation.1 These appealing synthetic methods can be classified into 2 types of reactions (Fig. 1A): (1) metallaphotoredox cross-coupling, wherein an unmodified carboxylic acid is used directly and for which the homolytic cleavage of a C–CO2H bond is achieved by photomediated single-electron oxidation;2 (2) cross-electrophilic coupling (XEC), wherein a carboxylic acid is initially converted to a redox-active ester (RAE) that can abstract an electron from the low valent Ni catalyst generated in reductive conditions or from the reductants directly, driving decarboxylation of the RAE.3–6 Recently, Fu's group reported innovative electrophotochemical Ce/Ni dual transition metal-catalyzed decarboxylative arylation and alkenylation reactions by integrating the photooxidation of carboxylic acids by anodic generated CeIV with cathodic nickel-catalyzed reductive cross-coupling.7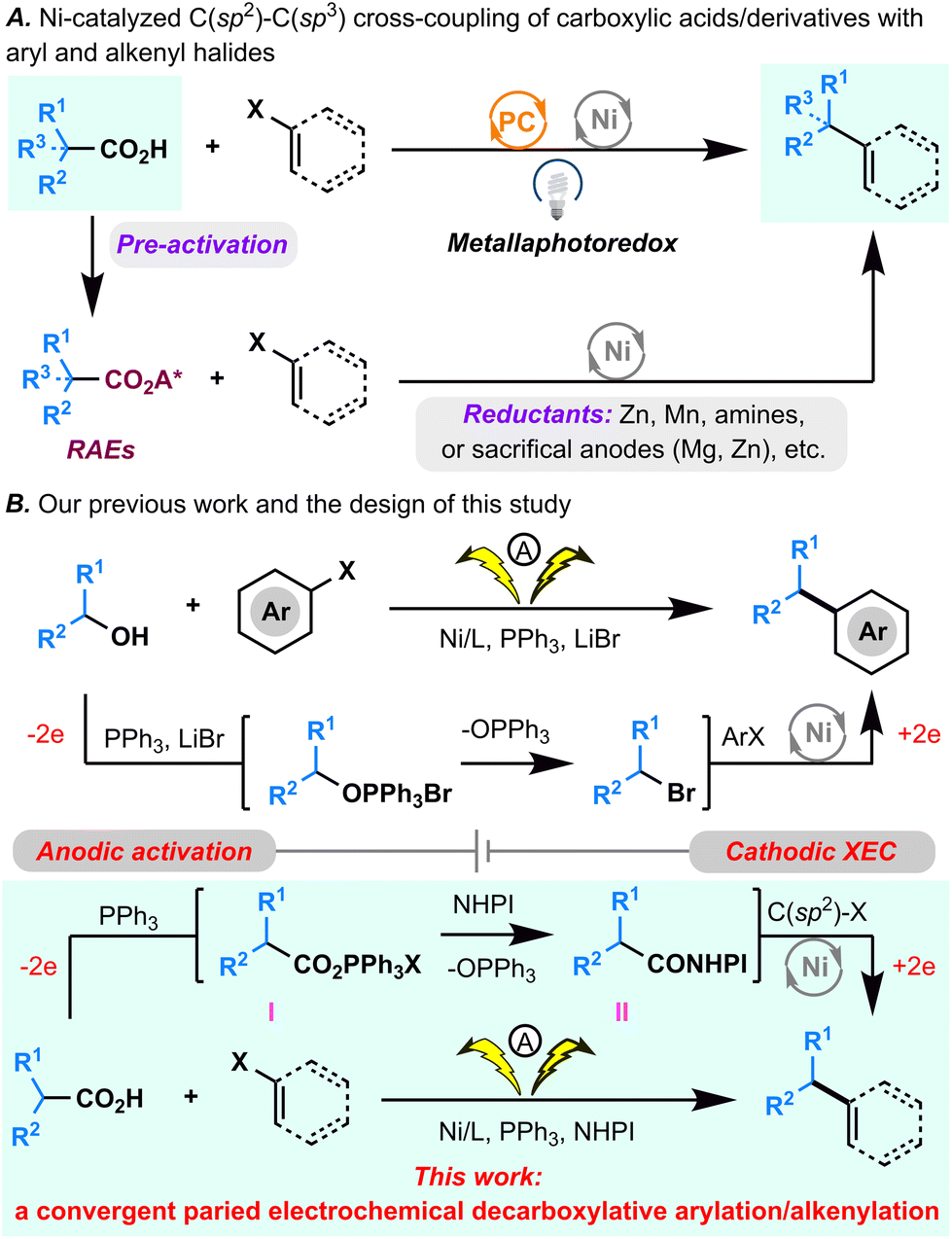 | ||
| Fig. 1 (A) Established decarboxylative arylation/alkenylation strategies. (B) Our previous study on electrochemical dehydroxylative arylation and the envisioned convergent paired electrolysis for the decarboxylative arylation and alkenylation. | ||
Despite significant advancements, several limitations persist in many reported decarboxylative C(sp2)–C(sp3) cross-coupling reactions. Notably, the high oxidative potential of carboxylic acids poses constraints on oxidative decarboxylative cross-couplings, limiting their applicability in the presence of certain oxidation-sensitive functional groups when exposed to photoexcited catalysts. Additionally, reductive approaches often necessitate the preactivation of carboxylic acids (e.g., separate preparation of RAEs) and the use of superstoichiometric reductants (e.g., Zn, Mn, or sacrificial anodes). To address these challenges, we pursued a convergent paired electrochemical decarboxylative C(sp2)–C(sp3) cross-coupling reaction by seamlessly merging the anodic generation of RAEs with cathodic nickel-catalyzed reductive cross-coupling.
Previously, we devised a convergent paired electrochemical strategy for the dehydroxylative C(sp2)–C(sp3) bond formation (Fig. 1B).8 In that approach, an unmodified alkyl alcohol underwent conversion to an alkyl bromide through an Appel reaction with anodic generated PPh3·Br2, subsequently paired with Ni-catalyzed cathodic reductive cross-coupling to achieve formal dehydroxylative C(sp2)–C(sp3) bond formation. Building on this foundation, the current study explores the potential of anodically generated PPh3·Br2 to activate carboxylic acids,9,10 yielding acyloxytriphenylphosphonium ions. In the presence of NHPI, these ions are envisioned to be subsequently transformed into NHPI esters.11 The integration of this process with cathodic decarboxylative cross-electrophilic coupling (XEC) aims to achieve formal direct decarboxylative C(sp2)–C(sp3) cross-coupling (Fig. 1B). However, given the inherent challenges associated with transition metal-catalyzed convergent paired electrolysis,12 two critical considerations emerge: ensuring the harmonization of the intrinsic redox properties of all reactants and coordinating the reaction rates of the anodic PPh3·Br2 generation, the acyloxytriphenylphosphonium ion and the NHPI ester formation, and the Ni catalytic cycle.
Results and discussion
We implemented this idea in practice by evaluating a series of electrochemical conditions using with 4-phenylbutyric acid (1) and iodobenzene (2) as model substrates (Table 1). After extensive optimization, the desired product 3 was obtained in 87% yield by running the electrolysis in a simple undivided cell under a constant current of 3 mA and using NiBr2·glyme as the precatalyst, 2,2′-bipyridine as the ligand, PPh3, DMAP, and NHPI as activating agents, graphite as the anode, Ni foam as the cathode, NaI as the electrolyte, and NMP as the solvent, each of which was found to be required to obtain 3 in high yield (entries 1–7). Notably, all reagents and materials employed in this electrolysis are cost-effective and readily available. It is noteworthy that, interestingly, even in the absence of NaI, coupling products were obtained with a commendable 79% yield, without encountering high electrical resistance. This observation suggests that the salt generated from the carboxylic acid and DMAP effectively served as the electrolyte.13 Beyond its role as a base, DMAP may also function as an acyl transfer reagent in the NHPI ester formation. This is evidenced by lower yields when replaced with Et3N or pyridine (entries 12 and 13). Furthermore, the utilization of aryl iodide is imperative; substituting it with bromobenzene led to a reduced yield of 33% (entry 14).|
|
|||||
|---|---|---|---|---|---|
| Entry | Deviation from above | Yieldb (%) | Entry | Deviation from above | Yieldb (%) |
| a Reactions were conducted on a 0.2 mmol scale in NMP (4 mL). b Yields were determined by GC/MS. c Yield of the isolated product. | |||||
| 1 | No electricity | 0 | 8 | LiCO4 instead of NaI | 78 |
| 2 | w/o NiBr2·glyme | 0 | 9 | LiBr instead of NaI | 74 |
| 3 | w/o L1 | 5 | 10 | L2 instead of L1 | 74 |
| 4 | w/o NHPI | 0 | 11 | L3 instead of L1 | 55 |
| 5 | w/o Ph3P | 0 | 12 | Et3N instead of DMAP | 61 |
| 6 | w/o DMAP | 9 | 13 | Pyridine instead of DMAP | 30 |
| 7 | w/o NaI | 79 | 14 | PhBr instead of PhI | 33 |
|
|||||

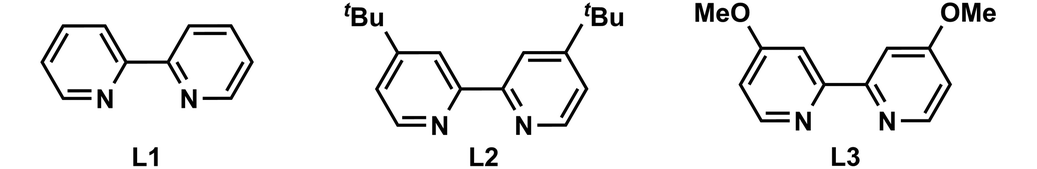
With the optimized conditions in hand, we first examined the scope of aryl iodides (Fig. 2). Gratifyingly, we found that aryl iodides containing various functional groups at the para, meta, and ortho positions (4–14) were generally well accommodated in this convergent paired electrochemical decarboxylative arylation. Electron-poor (4, 5, 9, 12, 19), electron-neutral (10, 13, 20), and electron-rich (6–8, 11, 14, 21) aryl iodides were all coupled with equal levels of efficiency, affording the desired products in good-to-high yields. Moreover, this reaction showed notable chemoselectivity toward aryl–I bonds, and a range of bromides and chlorides containing aryl fragments (15, 17, 18, 24, 29, 30) were tolerated in this convergent paired electrolysis, allowing for further downstream diversification. Notably, a series of medicinally relevant and structurally distinct heterocycles including a quinoline (22), an unprotected indole (23), pyridines with various substitution patterns (24–31), a dibenzofuran (32), a carbazole (33), and a thiophene (34), were also investigated and the desired coupling products were successfully afforded in good-to-high yields.
 | ||
Fig. 2 Scope of the convergent paired electrolysis for decarboxylative arylation. Reactions were conducted on a 0.2 mmol scale; indicated yields are for isolated products. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The reactions were carried out with L2. The reactions were carried out with L2. | ||
We subsequently explored the substrate scope for carboxylic acid coupling partners (Fig. 2A and B). Overall, a range of primary and secondary carboxylic acids bearing a variety of functionalities, including ethers (36, 41, 43, 50), a sulfone (37), an alkene (38), an ester (39), carbamates (44, 46, 47), a ketal (49), and a ketone (52) were well accommodated in this convergent paired electrolysis. Moreover, various densely functionalized bioactive molecules such as 2,4-D (53), chlorambucil (54), indometacin (55), isoxepac (56), and acetyllithocholic acid (57) could participate in this convergent paired electrolysis with high efficiency (Fig. 2C), supporting the applicability of our method for medicinal chemistry campaigns. Notably, the direct preparation of unnatural amino acids 58 (49%) and 59 (56%) from a protected glutamic acid showed comparable efficiency observed in Baran's Ag–Ni electrocatalysis (59% NMR yield for 58, 63% isolated yield for 59; note that both were prepared from NHPI esters using a sacrificial Mg anode, 20 mol% Ni catalyst, and 50 mol% AgNO3 in Baran's conditions),5e thereby demonstrating a novel approach to these synthetically challenging and highly valuable targets from simple starting materials. Note that: (i) the enantiomeric excess values of 58 and 59 were completely retained in our reaction conditions; (ii) this reaction could be performed on a gram scale (44).
In spite of the extensive success in coupling various aryl iodides and carboxylic acids, the existing reaction conditions face limitations in accommodating sterically hindered tertiary carboxylic acids, carboxylic acids with alkyne groups, or N-Boc-pyrrolidine (as illustrated in Fig. 2D). Additionally, it is noteworthy that a competition experiment, investigating diverse substituted carboxylic acids, revealed that primary carboxylic acids with comparatively lower steric hindrance exhibited higher reactivity than their more sterically hindered secondary counterparts (Fig. S4†).
Recently, Baran and co-workers developed a nickel-catalyzed electrochemical decarboxylative alkenylation of RAEs with vinyl iodides using Mg as the sacrificial anode and silver-nanoparticle-modified RVC as a cathode; this elegant electrolysis was used successfully in the preparation of a series of bioactive terpenes.14 We also evaluated the ability of this paired electrolysis for decarboxylative alkenylation. To our delight, by simply switching the ligand from bpy L1 to dtbpy L2, our protocol also showed comparable efficiency with Baran's method (Fig. 3), delivering natural products (R)-E-nerolidol (62), (E)-α-bisabolene (63), and a TBS-protected E,E-homofarnesol (64) with moderate-to-high yields directly from the corresponding acids.
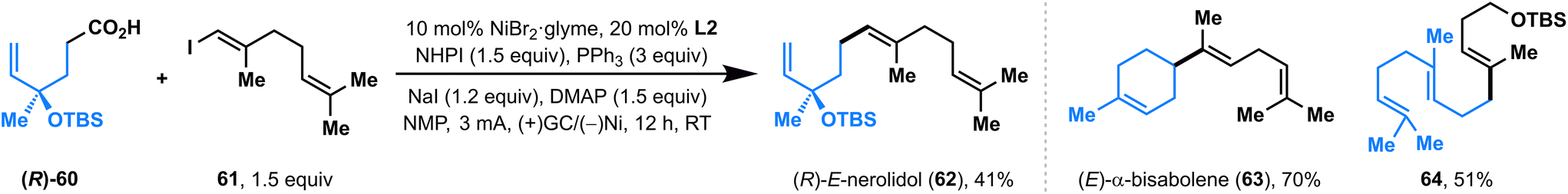 | ||
| Fig. 3 The convergent paired electrolysis for the decarboxylative alkenylation. | ||
Mechanistic studies
To unravel the intricate processes involved in this paired electrolysis, a comprehensive series of experiments was conducted to illuminate the entirety of the reaction mechanism. Initially, cyclic voltammetry (CV) experiments were undertaken, revealing that the electrolyte NaI exhibited the most facile oxidation (0.19 V versus Fc/Fc+, in CH3CN, Fig. 4B) within the santandard conditions. Notably, its oxidation potential was markedly lower than that of PPh3, DMAP, NHPI, and carboxylic acid 1 (with oxidative potentials of 0.75, 0.95, 1.69, and 1.85 V versus Fc/Fc+, respectively, in CH3CN). This discrepancy strongly suggested that I2 would be initially generated through anodic oxidation. Drawing parallels with the Appel reaction mechanism,15 it was deduced that I2 could rapidly react with PPh3, yielding PPh3·I2. This deduction was substantiated by the observable significant increase in the oxidation current of NaI in the presence of PPh3 (red curve, Fig. 4B).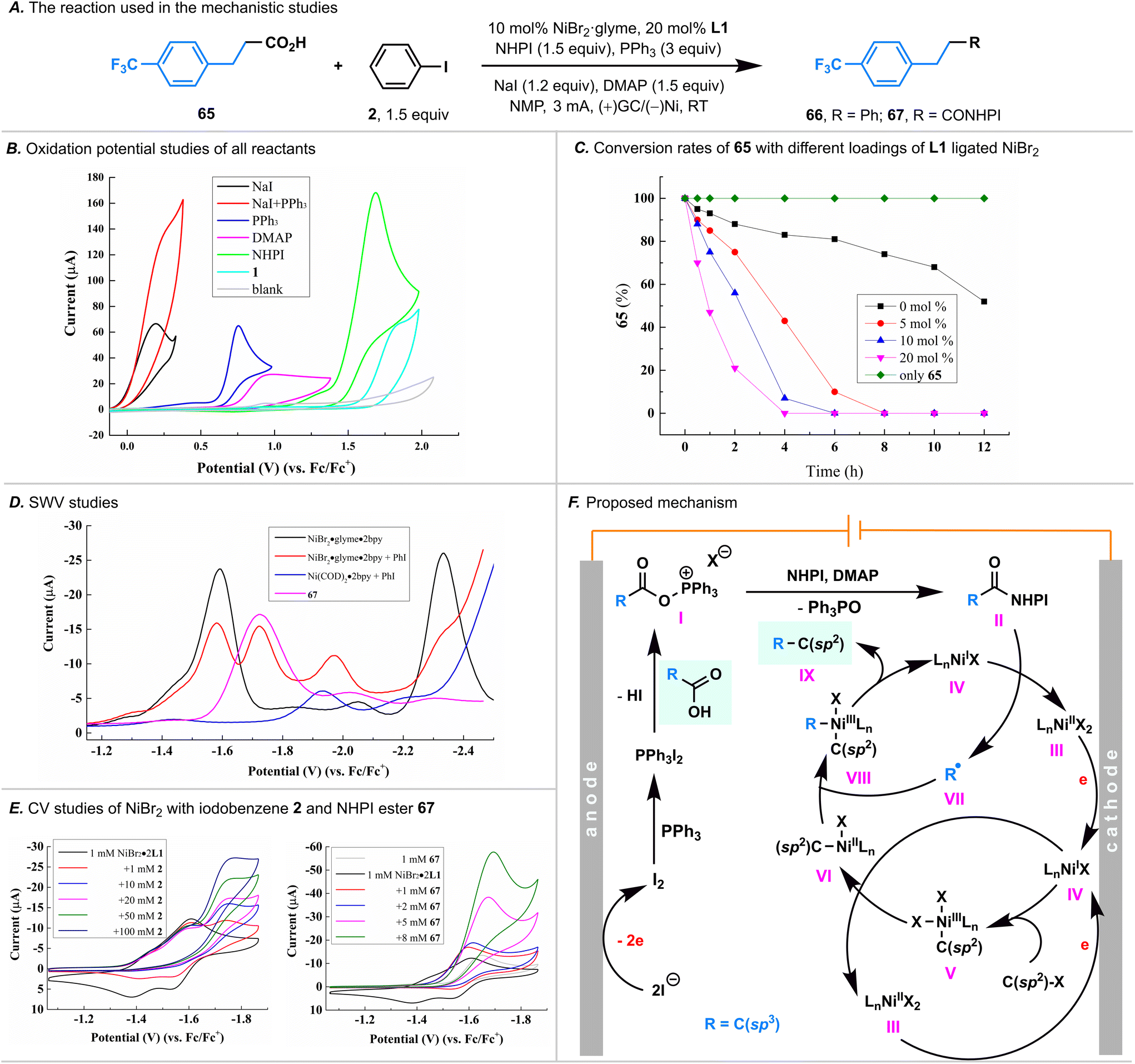 | ||
| Fig. 4 (A) The reaction used in the mechanistic investigations. (B) Cyclic voltammetry (CV) studies for the anodic oxidation processes (1 mM with 0.1 M LiClO4 in CH3CN, ν = 100 mV s−1, versus Fc/Fc+). (C) Impact of the catalyst loading on the consumption rate of carboxylic acids 65. Reactions were conducted on a 0.2 mmol scale; indicated yields were determined by 19F NMR in CDCl3 using PhCF3 as the internal standard. (D) Squarewave voltammetry (SWV) studies of various L1 ligated Ni species and 67 (1 mM with 0.1 M LiClO4 in NMP, versus Fc/Fc+) with an amplitude of 25 mV, frequency of 40 Hz, and a step potential of 5 mV. (E) CV investigation of L1 ligated NiBr2·glyme (1 mM with 0.1 M LiClO4 in NMP, ν = 100 mV s−1, versus Fc/Fc+) with 2 or 67. (F) Proposed mechanism for this convergent paired electrochemical decarboxylative arylation/alkenylation reaction. | ||
Given that the carboxylic acid 1 could be easily activated by the PPh3·I2 to form an acyloxyphosphonium iodide, we postulated that this intermediate could react with NHPI in the presence of DMAP, yielding a NHPI ester.9,11 Despite our efforts to isolate the NHPI ester of 1 by terminating the standard reaction prior to the complete consumption of 1, our attempts proved unfruitful. We also employed 19F NMR to detect the generation of the NHPI ester, utilizing carboxylic acid 65; however, the corresponding NHPI ester 67 as not detected. We attribute this outcome to the low equilibrium concentration of the NHPI ester; the in situ generated NHPI ester is rapidly consumed under the net-redox-neutral conditions characteristic of the paired electrolysis. Recognizing the rapid consumption of the NHPI ester, we performed the electrolysis of carboxylic acid 66 in the presence of PPh3, DMAP, and NaI first, followed by adding NHPI (without electric current at this stage), we successfully detected the NHPI ester 67 by 19F NMR (Fig. S5†), thereby demonstrating its generation.
Having confirmed the NHPI ester was generated through an anodic oxidation, we next turned our attention to the cathodic reduction process. Notably, by using carboxylic acid 65, we found that the rate of the consumption of carboxylic acid was highly dependent on the Ni catalyst loading (Fig. 4C). Specifically, the rate of carboxylic acid consumption accelerated proportionally with the increase in Ni catalyst loading. In the absence of the Ni catalyst, carboxylic acid exhibited a slow consumption rate, suggesting that the reduction of the in situ generated NHPI ester was predominantly catalyzed by Ni.
Our subsequent focus centered on the examination of nickel valence changes, and squarewave voltammetry (SWV, Fig. 4D) was used first to illustrate the reductive potentials of various Ni species. We found that: (i) the reductive potentials of L1 ligated NiBr2 and NiBr species were −1.59 V and −2.34 V, respectively (versus Fc/Fc+ in NMP, represented by the black curve); (ii) the reductive potentials of the L1 ligated NiII–aryl complex, generated in situ by oxidative addition of L1 ligated Ni(COD)2 to iodobenzene 2, is −1.94 V (versus Fc/Fc+ in NMP, represented by the blue curve); (iii) upon adding iodobenzene 2 to L1 ligated NiBr2, two additional peaks were detected (−1.72 V and −1.97 V, respectively, versus Fc/Fc+ in NMP, represented by the red curve) before the reductive potential of L1 ligated NiBr. Notably, the second peak (−1.97 V) being comparable to the reductive potential of the L1 ligated NiII–aryl complex (−1.94 V) led us to hypothesize that the oxidative addition process of L1 ligated NiBr to iodobenzene 2 occurred before the reduction of L1 ligated NiBr to Ni0, and the first peak (−1.72 V) is attributed to the reduction of the NiIII–aryl complex to the NiII–aryl complex.
Subsequently, we employed CV experiments to substantiate our hypothesis (Fig. 4E). Firstly, the CV experiments showed that the reduction of L1 ligated NiBr2 to NiBr is reversible (represented by the black curve). Secondly, upon introducing iodobenzene 2 to the L1 ligated NiBr2, the reductive current peak of L1 ligated NiBr2 to NiBr is observed (−1.59 V versus Fc/Fc+ in NMP), but the reversible peak disappeared (−1.54 V), indicating the consumption of L1 ligated NiBr by iodobenzene 2 through an oxidative addition process. Thirdly, a reductive peak at −1.72 V was observed, consistent with the signal observed in SWV experiments. Notably, this peak exhibited concentration-dependent increases in the reduction current, confirming the occurrence of the oxidative addition of L1 ligated NiBr to iodobenzene. Furthermore, upon introducing the NHPI ester 67 to the L1 ligated NiBr2, the reoxidative current peak of L1 ligated NiBr to NiBr2 also disappeared, suggesting that the L1 ligated NiBr could reduce the NHPI ester as well.
While further mechanistic investigations are warranted, our studies provide support for the proposed cathode-mediated Ni catalytic cycle (Fig. 4F). Firstly, NiI complex IV are preferentially generated from NiII species IIIvia a cathodic reduction. Secondly, the oxidative addition of IV to iodobenzene generates NiIII–aryl complex V, which undergoes reduction by NiI species IV through comproportionation, giving NiII–aryl complex VI and NiII species III.16III can obtain an electron from the cathode to reform NiI complex IV. Thirdly, by trapping an alkyl radical generated through the single electron reduction of NHPI ester by NiI species IV, NiII–aryl complex VI is converted to NiIII adduct VIII, which, after reductive elimination, completes the cycle by producing the desired product IX and NiI complex IV. It is worth noting that while we cannot completely rule out the possibility that NiIII–aryl complex V is directly reduced by the cathode to NiII–aryl complex VI, the similarity in reductive potentials between NiIII–aryl complex V and NHPI ester (see Fig. 4D, −1.72 V versus Fc/Fc+, in NMP) and the occurrence of homogeneous comproportionation between NiIII and NiI favor the notion that NiIII–aryl complex V is reduced by NiI species IV.
Conclusions
In conclusion, we have developed a convergent paired electrochemical decarboxylative arylation/alkenylation reaction by merging the anodic generation of RAEs and cathodic nickel-catalyzed reductive cross-coupling in an undivided electrochemical cell. This electrochemical cross-coupling reaction between alkyl carboxylic acids and C(sp2)-iodides features exceptional substrate generality, high functional group compatibility, and mild reaction conditions, which have been well demonstrated with the successful preparation of synthetically challenging unnatural amino acids and terpenoid natural products. Detailed mechanistic studies demonstrated the in situ generation of NHPI esters via an anodic generated acyloxytriphenyl-phosphonium ion intermediate and the reductive cross-coupling mediated by the cathodic generated NiI species. Moreover, we anticipate that this convergent paired electrochemical strategy will serve as a powerful and flexible method for C(sp2)–C(sp3) bond formation and is likely to find broad applications on account of its step and material economy.Author contributions
C. Li, L. Li, and Z. Li conceived and designed the experiments. L. Li, Z. Li and W. Sun performed the experiments. The manuscript was written through contributions of all authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support for this research was provided by the MOST of China, and the Tsinghua Institute of Multidisciplinary Biomedical Research, Tsinghua University.References
- (a) G. Laudadio, M. D. Palkowitz, T. E.-H. Ewing and P. S. Baran, Decarboxylative Cross-Coupling: A Radical Tool in Medicinal Chemistry, ACS Med. Chem. Lett., 2022, 13, 1413–1420 CrossRef CAS PubMed; (b) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed; (c) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Visible-Light-Induced Decarboxylative Functionalization of Carboxylic Acids and Their Derivatives, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed; (d) S. Murarka, N-(Acyloxy)phthalimides as Redox-Active Esters in Cross-Coupling Reactions, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS; (e) T. Patra and D. Maiti, Decarboxylation as the Key Step in C–C Bond-Forming Reactions, Chem. – Eur. J., 2017, 23, 7382–7401 CrossRef CAS PubMed; (f) S. K. Parida, T. Mandal, S. Das, S. K. Hota, S. D. Sarkar and S. Murarka, Single Electron Transfer-Induced Redox Processes Involving N-(Acyloxy)phthalimides, ACS Catal., 2021, 11, 1640–1683 CrossRef CAS; (g) S. Karmakar, A. Silamkoti, N. A. Meanwell, A. Mathur and A. K. Gupta, Utilization of C(sp3)-Carboxylic Acids and Their Redox-Active Esters in Decarboxylative Carbon−Carbon Bond Formation, Adv. Synth. Catal., 2021, 363, 3693–3736 CrossRef CAS; (h) Y. Jin and H. Fu, Visible-Light Photoredox Decarboxylative Couplings, Asian J. Org. Chem., 2017, 6, 368–385 CrossRef CAS; (i) H. Huang, K. Jia and Y. Chen, Radical Decarboxylative Functionalizations Enabled by Dual Photoredox Catalysis, ACS Catal., 2016, 6, 4983–4988 CrossRef CAS.
- (a) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Merging Photoredox with Nickel Catalysis: Coupling of α-Carboxyl sp3-Carbons with Aryl Halides, Science, 2014, 345, 437–440 CrossRef CAS PubMed; (b) A. Noble, S. J. McCarver and D. W. C. MacMillan, Merging Photoredox and Nickel Catalysis: Decarboxylative Cross-Coupling of Carboxylic Acids with Vinyl Halides, J. Am. Chem. Soc., 2015, 137, 624–627 CrossRef CAS PubMed.
- For selected examples using metal powders, see: (a) K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, Decarboxylative Cross-Electrophile Coupling of N-Hydroxyphthalimide Esters with Aryl Iodides, J. Am. Chem. Soc., 2016, 138, 5016–5019 CrossRef CAS PubMed; (b) D. C. Salgueiro;, B. K. Chi, I. A. Guzei, P. García-Reynaga and D. J. Weix, Control of Redox-Active Ester Reactivity Enables a General Cross-Electrophile Approach to Access Arylated Strained Rings, Angew. Chem. Int. Ed., 2022, 61, e202205673 CrossRef PubMed.
- For selected examples using organic reducing agents, see: N. Suzuki, J. L. Hofstra, K. E. Poremba and S. E. Reisman, Nickel-Catalyzed Enantioselective Cross-Coupling of N-Hydroxyphthalimide Esters with Vinyl Bromides, Org. Lett., 2017, 19, 2150–2153 CrossRef CAS PubMed.
- For selected examples using electrochemical methods, see: (a) H. Li, C. P. Breen, H. Seo, T. F. Jamison, Y.-Q. Fang and M. M. Bio, Ni-Catalyzed Electrochemical Decarboxylative C–C Couplings in Batch and Continuous Flow, Org. Lett., 2018, 20, 1338–1341 CrossRef CAS PubMed; (b) T. Koyanagi, A. Herath, A. Chong, M. Ratnikov, A. Valiere, J. Chang, V. Molteni and J. Loren, One-Pot Electrochemical Nickel-Catalyzed Decarboxylative Sp2−Sp3 Cross-Coupling, Org. Lett., 2019, 21, 816–820 CrossRef CAS PubMed; (c) F. Lian, K. Xu, W. Meng, H. Zhang, Z. Tan and C. Zeng, Nickel-Catalyzed Electrochemical Reductive Decarboxylative Coupling of N-Hydroxyphthalimide Esters with Quinoxalinones, Chem. Commun., 2019, 55, 14685–14688 RSC; (d) Y. Liu, L. Xue, B. Shi, F. Bu, D. Wang, L. Lu, R. Shi and A. Lei, Catalyst-Free Electrochemical Decarboxylative Cross-Coupling of N-Hydroxyphthalimide Esters and N-Heteroarenes towards C(sp3)–C(sp2) Bond Formation, Chem. Commun., 2019, 55, 14922–14925 RSC; (e) M. D. Palkowitz, G. Laudadio, S. Kolb, J. Choi, M. S. Oderinde, T. E.-H. Ewing, P. N. Bolduc, T. Chen, H. Zhang, P. T. W. Cheng, B. Zhang, M. D. Mandler, V. D. Blasczak, J. M. Richter, M. R. Collins, R. L. Schioldager, M. Bravo, T. G. M. Dhar, B. Vokits, Y. Zhu, P.-G. Echeverria, M. A. Poss, S. A. Shaw, S. Clementson, N. N. Petersen, P. K. Mykhailiuk and P. S. Baran, Overcoming Limitations in Decarboxylative Arylation via Ag–Ni Electrocatalysis, J. Am. Chem. Soc., 2022, 144, 17709–17720 CrossRef CAS PubMed.
- For selected examples using photoreductant, see: L. M. Kammer, S. O. Badir, R.-M. Hu and G. A. Molander, Photoactive Electron Donor–Acceptor Complex Platform for Ni-Mediated C(sp3)–C(sp2) bond formation, Chem. Sci., 2021, 12, 5450–5457 RSC.
- (a) K. Yang, J. Lu, L. Li, S. Luo and N. Fu, Electrophotochemical Metal-Catalyzed Decarboxylative Coupling of Aliphatic Carboxylic Acids, Chem. – Eur. J., 2022, 28, e202202370 CrossRef CAS PubMed; (b) J. Lu, Y. Yao, L. Li and N. Fu, Dual Transition Metal Electrocatalysis: Direct Decarboxylative Alkenylation of Aliphatic Carboxylic Acids, J. Am. Chem. Soc., 2023, 145, 26774–26782 CrossRef CAS PubMed.
- Z. Li, W. Sun, X. Wang, L. Li, Y. Zhang and C. Li, Electrochemically Enabled, Nickel-Catalyzed Dehydroxylative Cross-Coupling of Alcohols with Aryl Halides, J. Am. Chem. Soc., 2021, 143, 3536–3543 CrossRef CAS PubMed.
- H. Ohmori, H. Maeda, M. Kikuoka, T. Maki and M. Masui, Electrochemical Generation and Reactions of Acyloxytriphenylphosphonium ions, Tetrahedron, 1991, 47, 767–776 CrossRef CAS.
- For a recent review, see: Y. Wang, J. Xu, Y. Pan and Y. Wang, Recent Advances in Electrochemical Deoxygenation Reactions of Organic Compounds, Org. Biomol. Chem., 2023, 21, 1121–1133 RSC.
- For selected examples, see: (a) A. Kumar, H. K. Akula and M. K. Lakshman, Simple Synthesis of Amides and Weinreb Amides Using PPh3 or Polymer-Supported PPh3 and Iodine, Eur. J. Org. Chem., 2010, 2709–2715 CrossRef CAS; (b) C. Duangkamol, S. Wangngae, M. Pattarawarapan and W. Phakhodee, Acyloxyphosphonium versus Aminophosphonium Intermediates: Application to the Synthesis of N-Acylbenzotriazoles, Eur. J. Org. Chem., 2014, 7109–7112 CrossRef CAS; (c) S. P. Morcillo, L. Á. de Cienfuegos, A. J. Mota, J. Justicia and R. Robles, Mild Method for the Selective Esterification of Carboxylic Acids Based on the Garegg-Samuelsson Reaction, J. Org. Chem., 2011, 76, 2277–2281 CrossRef CAS PubMed; (d) N. Mamidi and D. Manna, Zn(OTf)2-P romoted Chemoselective Esterification of Hydroxyl Group Bearing Carboxylic Acids, J. Org. Chem., 2013, 78, 2386–2396 CrossRef CAS PubMed; (e) S.-P. Wang, C. W. Cheung and J.-A. Ma, Direct Amidation of Carboxylic Acids with Nitroarenes, J. Org. Chem., 2019, 84, 13922–13934 CrossRef CAS PubMed.
- For selected reviews on paired electrolysis, see: (a) N. Sbei, T. Hardwick and N. Ahmed, Green Chemistry: Electrochemical Organic Transformations via Paired Electrolysis, ACS Sustainable Chem. Eng., 2021, 9, 6148–6169 CrossRef CAS; (b) J. C. Vantourout, From Bench to Plant: An Opportunity for Transition Metal Paired Electrocatalysis, Org. Process Res. Dev., 2021, 25, 2581–2586 CrossRef CAS; (c) Y. Zhang, W. Sun and C. Li, Nickel-Catalyzed Paired Electrochemical Cross-Coupling of Aryl Halides with Nucleophiles, Synthesis, 2022, 281–294 Search PubMed; (d) K.-J. Jiao, X.-T. Gao, C. Ma, P. Fang and T.-S. Mei, Recent Applications of Paired Electrolysis in Organic Synthesis, Isr. J. Chem., 2023, e202300085 CrossRef; (e) R. Zhang, L. Li, K. Zhou and N. Fu, Radical-Based Convergent Paired Electrolysis, Chem. – Eur. J., 2023, 29, e202301034 CrossRef CAS PubMed.
- Building upon our prior investigation (ref. 8), it was established that a catalytic quantity of bromide ions, derived from NiBr2, could serve as the initiator for the reaction, leading to the formation of PPh3·Br2. In the present study, we posit that a catalytic quantity of bromide ion, originating from NiBr2, also instigated the reaction. Furthermore, we propose that the liberation of the iodide ion from iodobenzene played a pivotal role in promoting the reaction.
- S. J. Harwood, M. D. Palkowitz, C. N. Gannett, P. Perez, Z. Yao, L. Sun, H. D. Abruña, S. L. Anderson and P. S. Baran, Modular Terpene Synthesis enabled by Mild Electrochemical Couplings, Science, 2022, 375, 745–752 CrossRef CAS PubMed.
- R. Appel, Tertiary Phosphane/Tetrachloromethane, a Versatile Reagent for Chlorination, Dehydration, and P–N Linkage, Angew. Chem., Int. Ed. Engl., 1975, 14, 801–811 CrossRef.
- Y. Kawamata, J. C. Vantourout, D. P. Hickey, P. Bai, L. Chen, Q. Hou, W. Qiao, K. Barman, M. A. Edwards, A. F. Garrido-Castro, J. N. deGruyter, H. Nakamura, K. Knouse, C. Qin, K. J. Clay, D. Bao, C. Li, J. T. Starr, C. Garcia-Irizarry, N. Sach, H. S. White, M. Neurock, S. D. Minteer and P. S. Baran, Electrochemically Driven, Ni Catalyzed Aryl Amination: Scope, Mechanism, and Applications, J. Am. Chem. Soc., 2019, 141, 6392–6402 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qo02141f |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2024 |