
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in metal-free catalytic enantioselective higher-order cycloadditions
Bei
Zhang
b and
Jian
Wang
 *ac
*ac
aCollege of Chemistry and Materials Science, Anhui Normal University, Wuhu 241002, China. E-mail: wangjian2012@tsinghua.edu.cn
bSchool of Chemistry and Materials Science, Institute of Advanced Materials and Flexible Electronics (IAMFE), Nanjing University of Information Science and Technology, 219 Ningliu Road, Nanjing, 210044, China
cSchool of Pharmaceutical Sciences, Tsinghua University, Beijing 100084, China
First published on 1st February 2024
Abstract
Higher-order cycloadditions (HOCs) present notable benefits for the synthesis of complex molecules, particularly those featuring medium size and multiple rings. The research on HOCs has experienced remarkable advancements since the influential exchange between Woodward and Hoffmann. In recent decades, significant progress has been achieved in enantioselective metal-free catalytic HOCs. These methodologies have facilitated the regulation of peri- and enantioselectivity during ring formation. With the diversification of activation modes, the variety of intermediates used in HOCs has also been greatly expanded. This review focuses on strategies for metal-free catalytic construction of chiral frameworks. To our knowledge, there are still many unknowns to be discovered in enantioselective HOCs.
 Bei Zhang | Dr Bei Zhang obtained her BSc from Dalian University of Technology in 2017. She completed her PhD study under the guidance of Professor Jian Wang at Tsinghua University in 2023. Her research interest mainly focuses on the NHC-catalyzed radical reaction and the merger of photocatalysis and organocatalysis. She is now an assistant professor at the School of Chemistry and Materials Science, Nanjing University of Information Science and Technology. |
 Jian Wang | Jian Wang obtained his BSc from An Hui Normal University in China and completed his PhD studies at the University of New Mexico, USA. He did postdoctoral research at the Scripps Research Institute, then moved to the National University of Singapore (NUS) as an assistant professor, and finally joined Tsinghua University full-time, where he is now a tenured full professor. His research focuses on asymmetric carbene organocatalysis, novel synthesis methods and natural product synthesis, as well as innovative drugs in pain and neurodegenerative disorder diseases. |
1. From classical cycloadditions to higher-order cycloadditions
Cycloaddition reactions are a class of powerful organic transformations.1 These reactions involve the formation of cyclic compounds from two or more reactants, often accompanied by the generation of multiple new carbon–carbon bonds.Classical cycloadditions, usually referring to cyclisations with electrons less than or equal to 6π, have been widely utilized to form cyclic products.2 These reactions occur via the simultaneous formation of two or more new bonds, typically with the concomitant loss of one or more π-bonds. The most well-known classical cycloaddition is the Diels–Alder reaction,3,4 which involves the reaction of a conjugated diene with an alkene or alkyne to build a six-membered ring. Other important examples include 1,3-dipolar cycloaddition (e.g., diazo compounds react with alkenes or alkynes) and [2 + 2] cycloaddition (e.g., two alkenes or alkynes react with each other to form a quaternary ring).5
In 1965, Hoffman and Woodward conducted an important academic exchange, after which Houk experimentally verified the feasibility of the [6 + 4] cycloaddition reaction.6,7 Almost simultaneously, Inoue, Morrison, and their colleagues independently demonstrated the viability of this new class of cyclic reactions.8,9 Thus, the concept of higher-order cycloadditions was established and subsequently developed rapidly. Higher-order cycloadditions (HOCs)10 generally refer to reactions in which more than six π-electrons participate, such as [6 + 2], [6 + 4], [8 + 2], [8 + 4], [10 + 2], [12 + 2] cycloaddition, etc. These reactions have attracted much attention due to their ability to produce complex polycyclic structures (e.g. natural products, pharmaceuticals, or valuable intermediates)11–55 (Scheme 1).
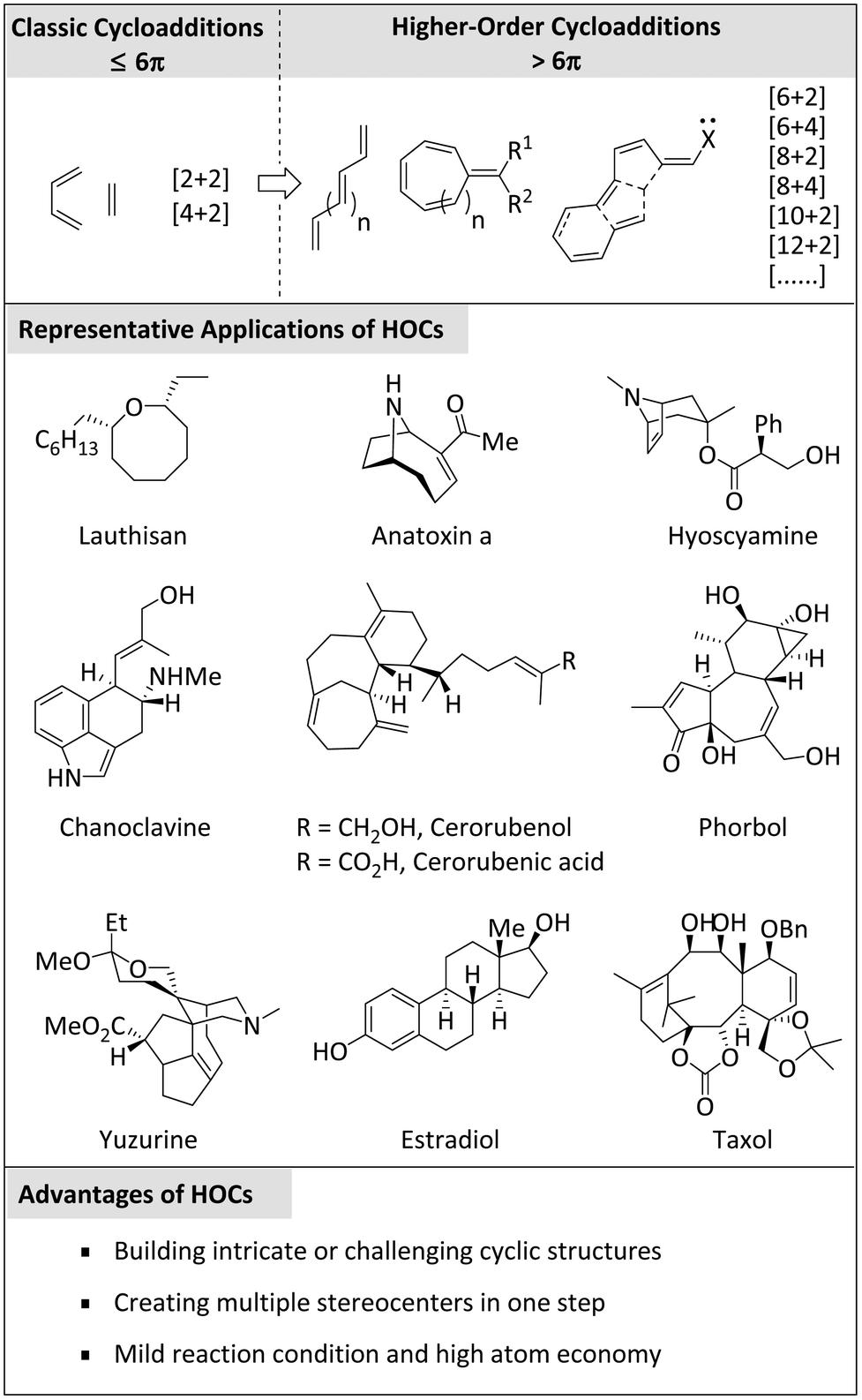 | ||
| Scheme 1 Brief introduction of higher-order cycloadditions. | ||
While HOCs offer many advantages, many issues, such as steric hindrance, mechanical complexity, and selective control, are often encountered in their investigation. To overcome these problems, researchers continue to uncover new solutions to further promote the field of HOCs.
2. From racemic HOCs to asymmetric HOCs
The development of asymmetric HOCs has always been an important topic in synthetic chemistry. Racemic HOCs typically rely on heat to drive and enable reactions. Thermal HOCs involve the reaction of three or more molecules without the use of a catalyst. While these reactions can be used to synthesize complex molecules, there are some important questions. In fact, one of the main problems with thermal HOCs is that they are often difficult to be controlled. Since reactions usually proceed at high temperatures, side reactions, decomposition, or incomplete transformations are prone to occur. Furthermore, thermal HOCs often require long reaction times or high pressures to achieve reasonable yields.9 Another significant challenge for thermal HOCs is regioselective control.8 In short, considering the high temperature, long reaction time and purification problem, the application of HOCs in industry is extremely challenging.56 In 2002, Rigby and coworkers reported an intramolecular [6 + 4] cycloaddition of Ti(IV)-assisted diene-tethered tropones for the construction of spirocyclic oxindoles,57 indicating both high peri- and diastereoselectivity. Subsequently, the same team successfully synthesized the ingenane sesquiterpene core with high peri- and diastereoselectivities as well as moderate enantioselectivities via the use of a chiral Lewis acid catalyst (Scheme 2).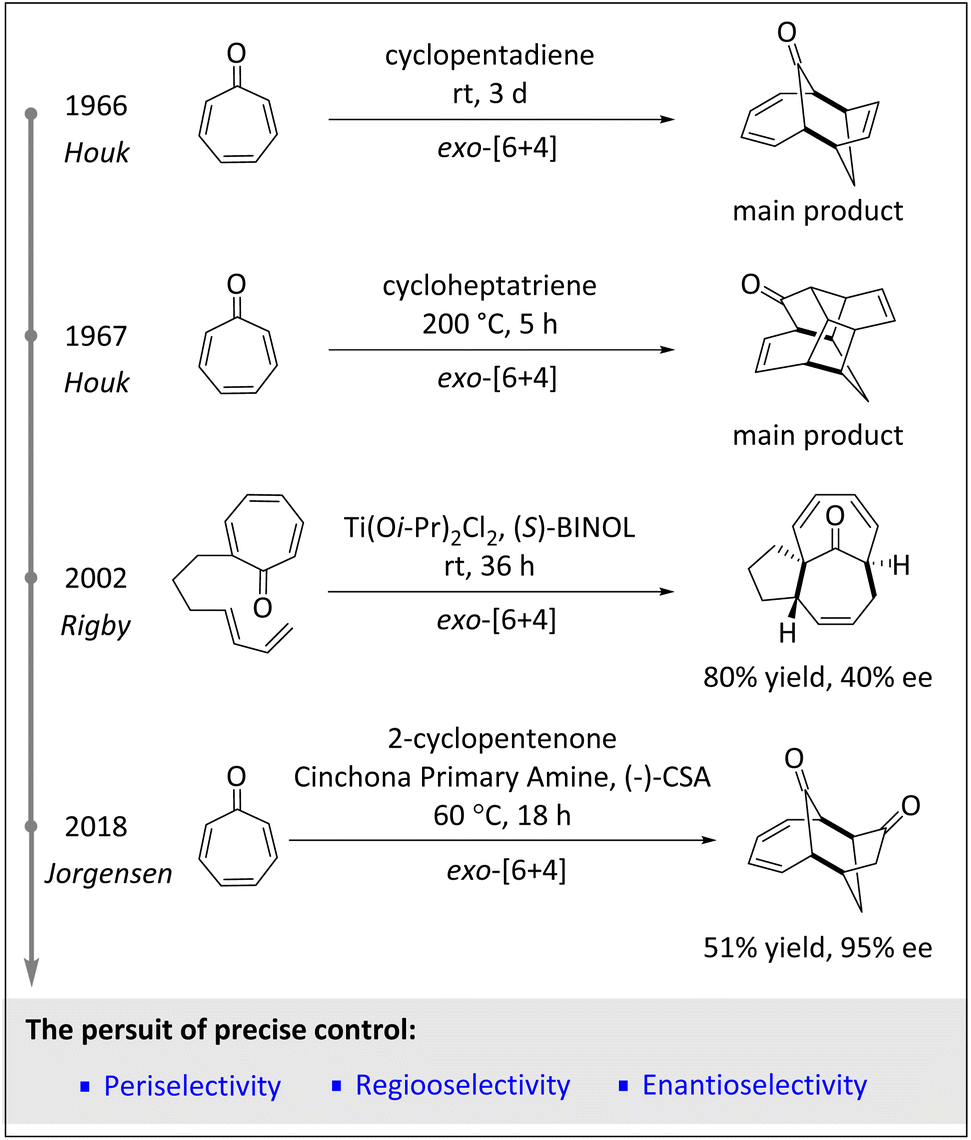 | ||
| Scheme 2 The brief history of higher-order cycloadditions. | ||
The emergence of organocatalysis has paved an alternative way for obtaining new intermediates and achieving excellent stereoselectivities. Before chiral amino catalysts were applied to this field, enantioselective HOCs were very rare.57,58 The Hayashi group first employed diphenylprolinol silyl ether as the organocatalyst to accomplish the asymmetric intramolecular [6 + 2] cycloaddition of fulvenes, resulting in the formation of valuable linear triquinane derivatives with high yields and exceptional enantioselectivities.59 In 2018, the Jørgensen group60 utilized cinchona alkaloid-derived amino catalysts to efficiently promote [6 + 4] and [8 + 2] cycloadditions, exhibiting excellent diastereo- and enantioselective control (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, ≥95% ee). Additionally, the periselectivities are well regulated by the combination of the ring size of the 2-cycloalkenone and the substituents of the heptafulvenoid.
:1 dr, ≥95% ee). Additionally, the periselectivities are well regulated by the combination of the ring size of the 2-cycloalkenone and the substituents of the heptafulvenoid.
Since then, organocatalytic HOCs have entered a new stage of rapid development. With the support of catalytic modes of enamine, imine, enolate and ion-pair catalysis, high efficiency and selectivity are regularly observed. In addition, computational chemistry interventions help researchers more accurately predict and design new asymmetric HOC reactions.
This review provides an overview of asymmetric organocatalytic HOCs. We summarize and classify HOCs according to the activation mode and discuss the fundamentals of reaction control. It also includes key advances in understanding and application, as well as challenges and opportunities for further development. Specifically, we focus on the design and formation of new synthons, the understanding of reaction pathways, and their application in natural product and drug synthesis. Overall, this review aims to provide a perspective on the current state of the art in asymmetric HOCs and highlights the exciting potential for future developments in this field.
3. Organocatalytic activation modes
The development of asymmetric HOCs is generally classified according to the types of intermediates formed by different catalysts.3.1 Enamine catalysis
Hayashi and co-workers reported an effective intramolecular [6 + 2] cycloaddition reaction for the synthesis of linear triquinane derivatives by employing diphenylprolinol silyl ether as the chiral organocatalyst (Scheme 3).59 The combination of linear aldehyde 1 and chiral aminocatalyst C1 offers A1, a HOMO-raising 2π component. The electron rich 2π addend then reacts with a 6π fulvene core viaA2 to generate a triquinane structure A3. A systematic theoretical study of the reaction mechanism was carried out for a deeper understanding. DFT calculations show that the intermolecular [6 + 2] cycloaddition between the fulvene and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of enamine is formed by the stepwise addition of the zwitterionic intermediate. In contrast, the intramolecular [6 + 2] cycloaddition to form a cis-fused triquinone backbone is carried out through a highly asynchronous transition state and a synergistic mechanism. The 6π fulvene functional group and 2π enamine moiety adapt the gauche–syn conformation during ring generation. The energy profiles also provide a plausible explanation for the exclusive formation of the cis-fused triquinane skeleton. A detailed discussion on enantioselectivity control of this [6 + 2] process is also included.
C bond of enamine is formed by the stepwise addition of the zwitterionic intermediate. In contrast, the intramolecular [6 + 2] cycloaddition to form a cis-fused triquinone backbone is carried out through a highly asynchronous transition state and a synergistic mechanism. The 6π fulvene functional group and 2π enamine moiety adapt the gauche–syn conformation during ring generation. The energy profiles also provide a plausible explanation for the exclusive formation of the cis-fused triquinane skeleton. A detailed discussion on enantioselectivity control of this [6 + 2] process is also included.
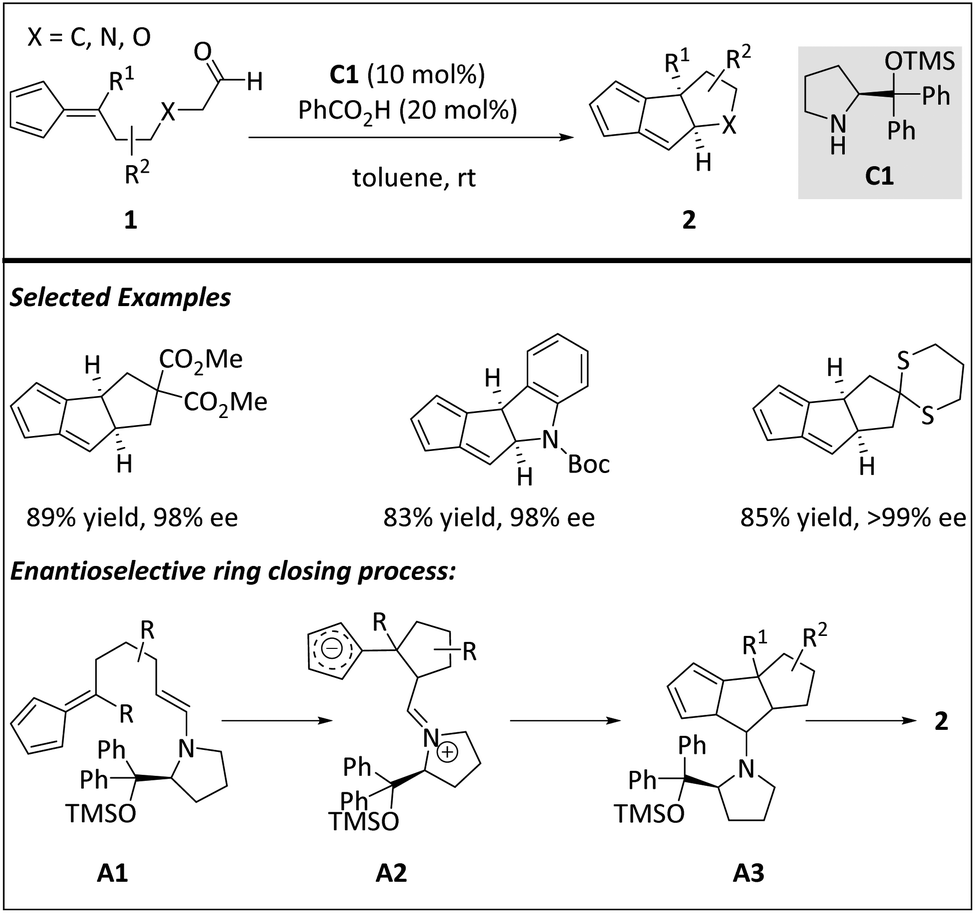 | ||
| Scheme 3 Enantioselective synthesis of tricyclopentanoids via an intramolecular [6 + 2] cycloaddition reaction. | ||
In 2017, Zhou and Chen et al. reported a series of [n + 2] (n = 2, 4, 6) intermolecular cycloadditions of alkylidene-2-cyclopentenones (Scheme 4).61 When 3-olefin(7-aza)oxindole is used as a raw material, the primary amine catalysts C2–C4 derived from cinchona alkaloids work with different Brønsted acids to facilitate the occurrence of switchable [6 + 2] HOC and [4 + 2] cyclization. When 3 reacts with maleimide 7, an intriguing, γ-regioselective [2 + 2] cycloaddition process produces fused cyclobutanes. As mentioned before, the peri- and stereoselectivities are difficult to control in HOCs. The switching of different ring sizes is an absolute challenge.62–65
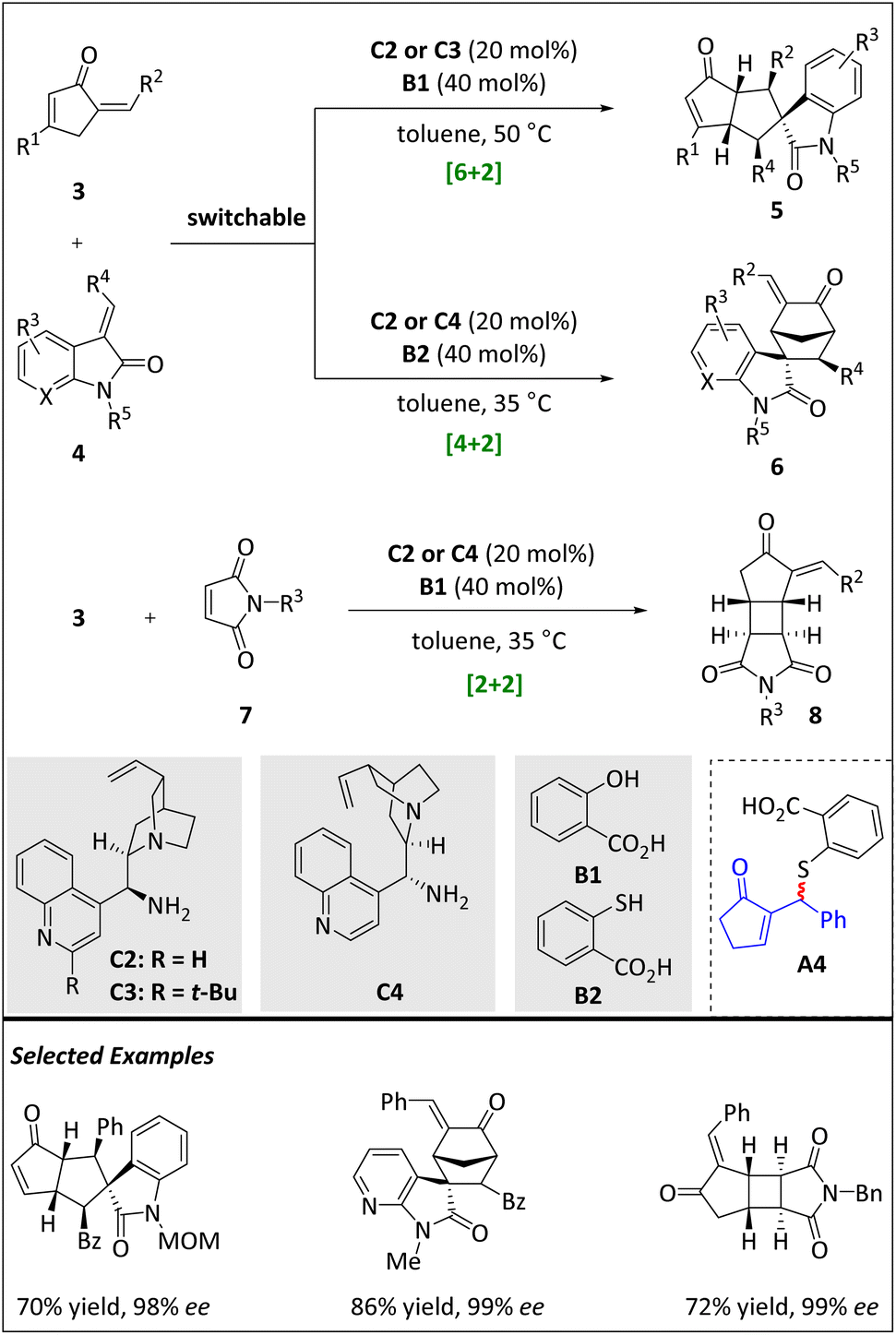 | ||
| Scheme 4 Switchable asymmetric [6 + 2], [4 + 2] and [2 + 2] cycloadditions. | ||
The generated highly reactive 6π component, 4-amino-fulvene intermediate, then reacts with activated 2π alkene 4 to efficiently accomplish γ,β′-regioselective [6 + 2] HOCs. An array of bicyclic skeletons with five consecutive stereogenic centers is obtained through this pathway. Under the same reaction conditions, HOMO-raising trienamines react with suitable N-benzyl maleimide 7 to afford a number of tricyclic cyclobutanes in moderate yields but with excellent diastereo- and enantioselectivities. DFT calculations reveal that the previous regioselective [6 + 2] cycloaddition may occur through a stepwise Michael–Michael reaction mechanism, resulting in the formation of more thermodynamically stable [6 + 2] products. On the other hand, the [2 + 2] reaction is promoted through a kinetically favourable pathway by means of ipso, γ-regioselective [4 + 2] cycloaddition between maleimides and the HOMO-raised 4-aminofulvenes.
When acid B1 is replaced by thiol acid B2, amine C2 or C4 successfully catalyzes the α,γ-regioselective [4 + 2] cycloaddition, affording a bridged structure 6 in high yields with excellent diastereo- and enantioselectivities (Scheme 4). A brief mechanistic study reveals that acid B2 can add directly to 3 in an α,β′-regioselective manner, leading to the formation of enone with a racemic sulfide moiety A4. Upon isomerization of the CC bond, the [4 + 2] cycloaddition reaction mediated by dienamine is effectively promoted by the addition of olefin 7 and catalyst C4.
In the same year, the Jørgensen group developed a series of stereoselective cycloadditions (e.g. [6 + 4], [8 + 2] and formal [4 + 2]) through two types of dienamine intermediates (Scheme 5).60 When the catalyst cinchona alkaloid-derived primary amine C5 activates 2-cyclic enone 9, a mixture of dienamine species, including cross dienamine, endocyclic linear dienamine, and exocyclic linear dienamine, is formed. The kinetically generated cross-dienamine intermediates drive the occurrence of [6 + 4] HOC and exhibit excellent stereoselectivities and moderate yields. Meanwhile, the carbonyl group of tropone 10 has proven to be a necessary functionality. By replacing 2-cyclopentenone with 2-cyclohexaenone, an inverse electron demand [4 + 2] cycloadduct is obtained. Notably, catalytic γ-functionalization by endocyclic linear dienylamines has not been reported. The authors hypothesize that [4 + 2] cycloadducts are made by an initial [8 + 2] cycloaddition followed by a rearrangement.
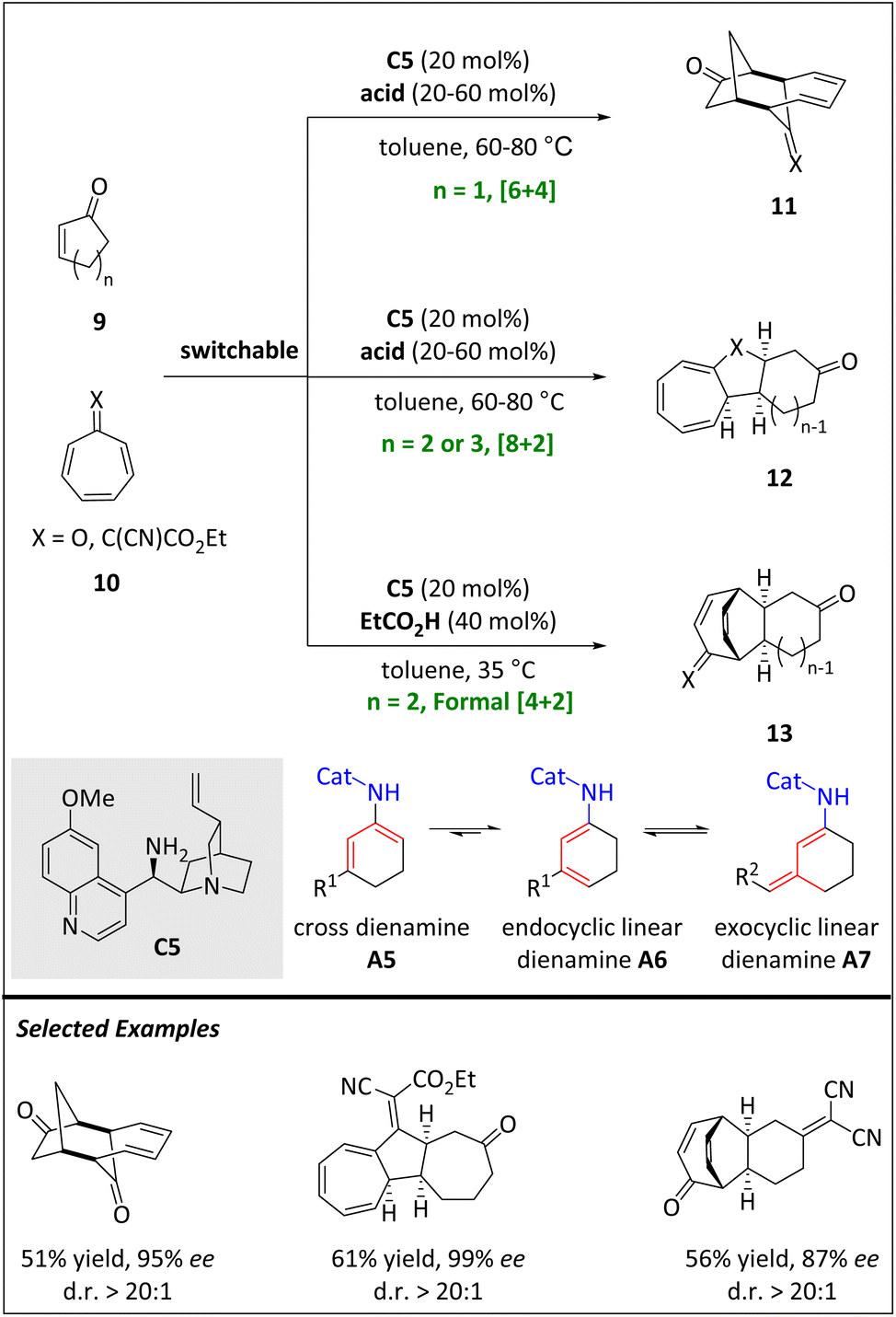 | ||
| Scheme 5 Stereoselective intermolecular [6 + 4], [8 + 2] and formal [4 + 2] via aminocatalysis. | ||
The thermodynamic distribution favours the transformation of 3-alkyl-2-cycloalkenones into endo- or exocyclic linear dienamines, while 2-cycloalkenones form endocyclic linear dienamines.66 However, the cross dienamine is usually not observed in small amounts. Interestingly, the thermodynamic distribution of the dienamine mixtures is not reflected in the aminocatalytic reactions. Instead, 3-alkyl-2-cycloalkenones react through cross-dienamines or exocyclic linear-dienamines, while 2-cycloalkenones react through cross-dienamines. The control of periselectivities in cycloadditions is determined both by the ring size of 2-cycloalkenones and the substitution patterns of heptafulvenes.
In 2019, Houk and Jørgensen et al. presented an elegant work on asymmetric hetero-[6 + 4] and [6 + 2] HOCs.67
Specifically, formyl aldehydes derived from pyrroles, imidazoles, and pyrazoles can react with amino-catalyst C6 or C7 to generate electron-rich hetero-6π aza- and diazafulvenes. This component then undergoes chemo-, regio-, and stereoselective cyclization of electron-deficient diene 15 and nitroalkene 17 (Scheme 6). The hetero-[6 + 4] cycloaddition of pyrrole systems with dienes allows for a wide variation of both reaction partners, ultimately yielding pyrrolo-azepine products with high yields and excellent enantioselectivities. Importantly, the authors also extend this hetero-[6 + 4] cycloaddition protocol to imidazoles and pyrazoles, resulting in the production of imidazolo- and pyrazolo-azepines. This activation mode also achieves success in hetero-[6 + 2] cycloadditions of pyrroles with nitroolefins, synthesizing pyrrolizidine alkaloid scaffolds. DFT calculations showcase that the stereoselectivity is dominated by the stepwise addition and ring closing step. Various substrates and catalysts have an effect on enantioselectivity.
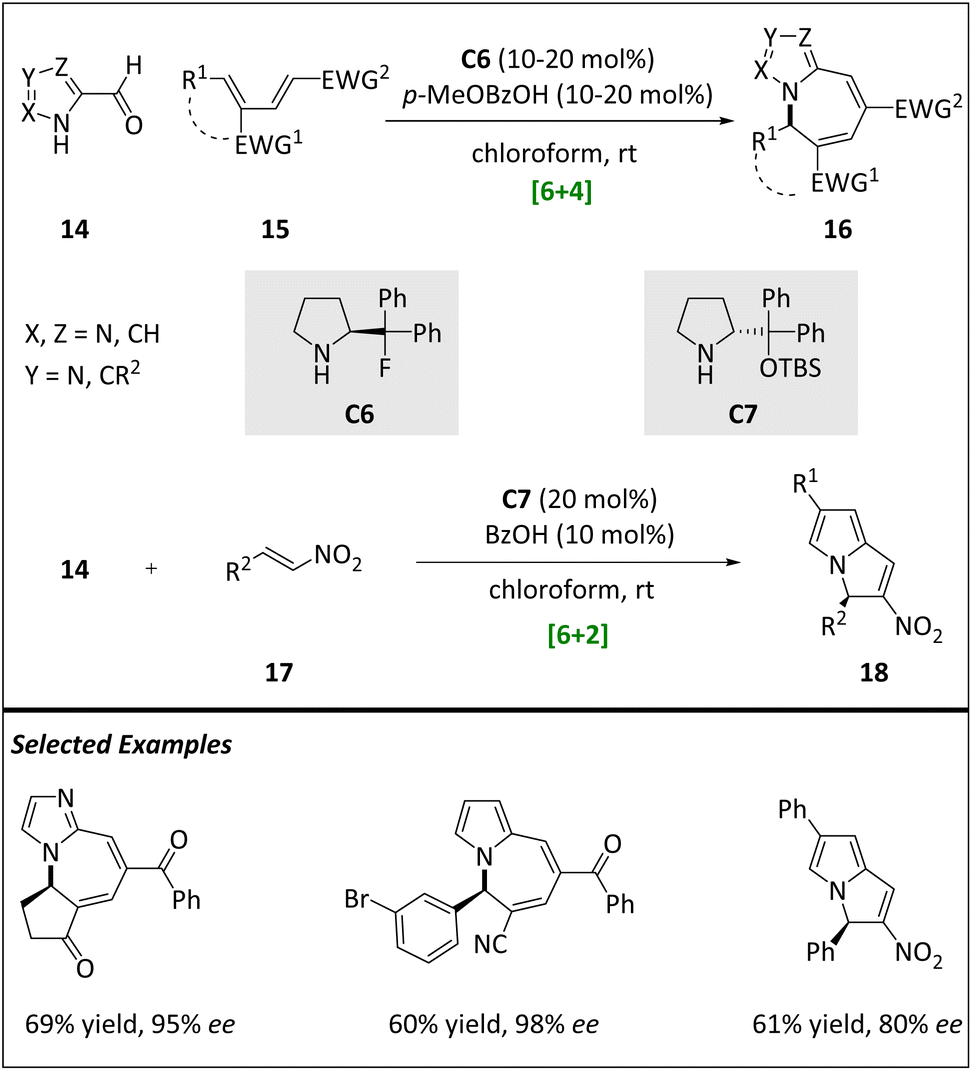 | ||
| Scheme 6 Stereoselective intermolecular hetero-[6 + 4] and [6 + 2] cycloadditions. | ||
Due to high ring-strain and entropy, the formation of an eight-membered ring presents a challenging task. Although cyclooctanoid terpenes are an important class of structurally diverse natural products, the total synthesis of cyclooctane scaffolds lacks general synthetic methods. McLeod and Jørgensen et al. reported the first enantioselective 1,3-dipolar [6 + 4] cycloaddition of pyrylium ions A8 with fulvenes 19via primary amine organocatalysis (Scheme 7).68 This method enables the rapid and stereoselective production of cyclooctane rings 21. The diverse functional groups and transannular bridges facilitate the transformation of multiple stereoselective structures into complex structures, demonstrating the versatility of the core for a range of cyclooctanoid natural products. Computational studies of both catalytic and non-catalytic reactions indicate a stepwise mechanism, which agrees with the experimental results regarding regio- and stereochemical outcomes. The reaction exhibits excellent diastereoselectivity and facilitates a set of transformations, allowing for the efficient assembly of complex scaffolds.
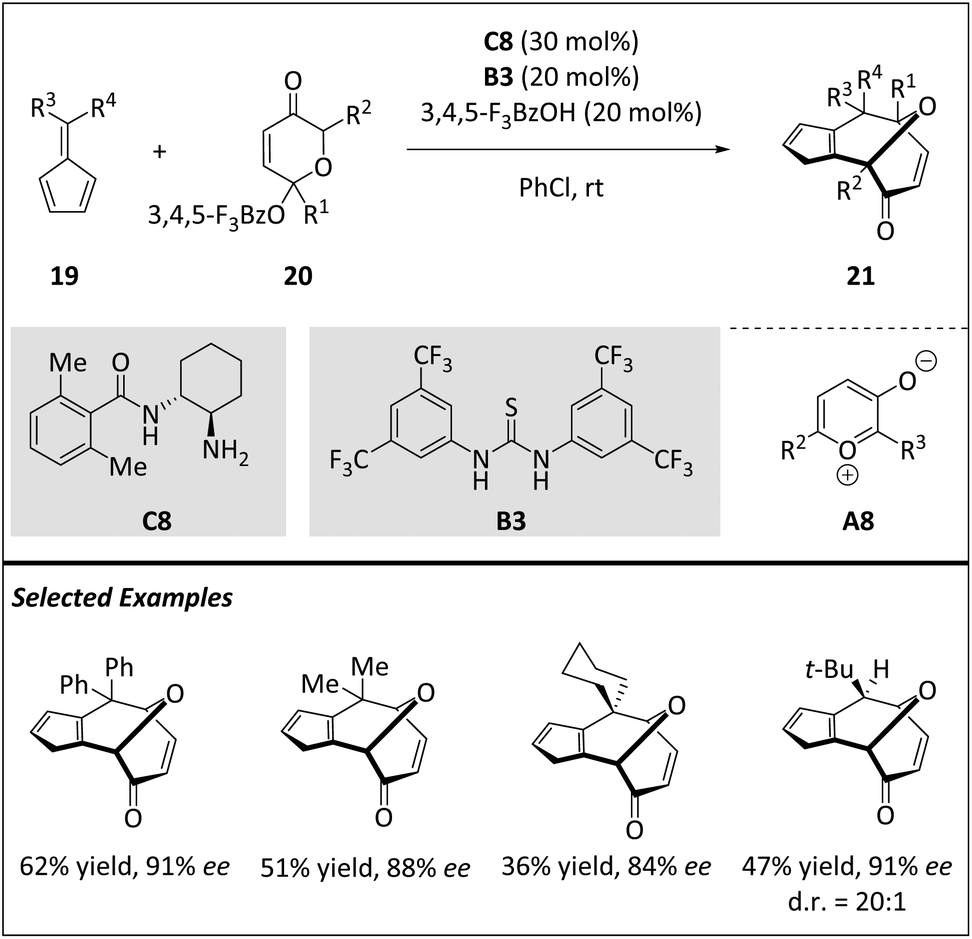 | ||
| Scheme 7 Asymmetric intermolecular 1,3-dipolar [6 + 4] cycloadditions via pyrylium ions. | ||
Jørgensen and co-workers also disclosed a unique [8 + 2] cycloaddition of indene-2-carbaldehydes 22 and olefins 17 or 23 through an unprecedented catalytic amino isobenzofulvene intermediate A10 (Scheme 8).69 In 1968, Hafner and Bauer first used stoichiometric isobenzofulvenes in cycloaddition.70 By introducing an amino moiety into exocyclic carbon, isobenzofulvene becomes more stable. It was found that the depicted amino isobenzofulvenes A9 produced a blend of [10 + 2] cycloadducts when they reacted with N-phenylmaleimides. In the presence of chiral A10, a number of enantioenriched benzonorbornenes, which serve as common scaffolds in commercial pesticides and neurotransmitter analogs, are generated by [8 + 2] cycloadditions. In such a highly peri-, diastereo-, and enantioselective cyclization process, the C2-symmetric catalyst C9 is the key factor determining the stereochemistry of five contiguous centers. DFT analysis showed that an energetically feasible [10 + 4] cycloaddition pathway is plausible and the proposed zwitterionic structure can ultimately result in the [8 + 2] cycloaddition product.
 | ||
| Scheme 8 Peri- and stereoselective intermolecular [8 + 2] cycloadditions via an isobenzofulvene intermediate. | ||
According to the computational results of [8 + 2] cycloaddition, the [10 + 4] cycloadduct has the potential to be prepared through the kinetically preferred intermediate A13. The Jørgensen group realized this hypothesis by choosing electron-deficient diene 26 as the 4π component (Scheme 9).69 Initially, the diphenylproline methylsilyl ether C7 catalyzes the reaction between indane-2-carbaldehyde 25 and diene 26, resulting in a mixture consisting of a small amount of presumed [8 + 2] cycloadducts and a separable quantity of [10 + 4] cycloadducts. However, by blocking the 3-position of indene-2-carbaldehydes, a single diastereoisomer 27 is obtained with a higher yield and high enantioselectivity. Both experimental and computational data show that the observed stereoselectivity is dominated by the formation of aminoisobenzofulvenes under kinetic control. The high yields, along with significant peri-, diastereo-, and enantioselectivities, and a broad scope of substrates make this technique extremely useful in the construction of intricate enantioenriched frameworks.
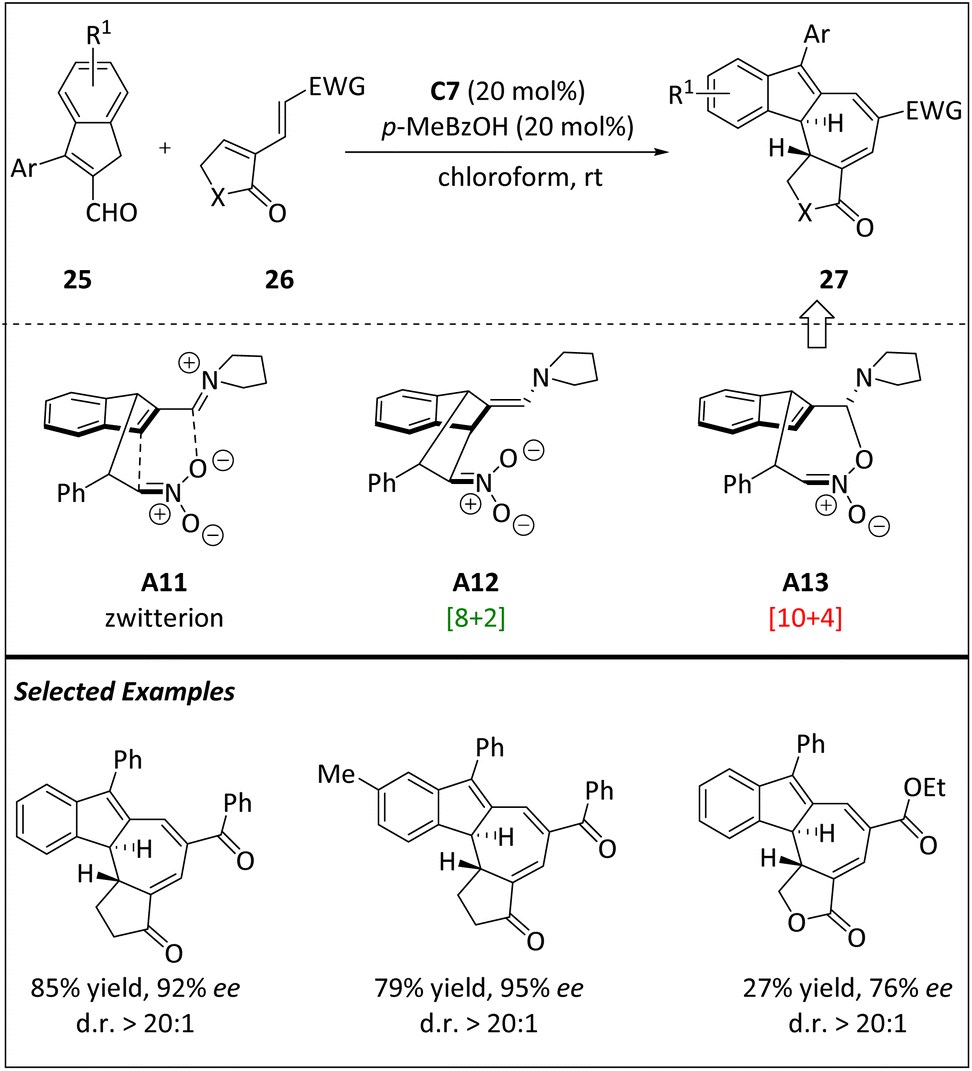 | ||
| Scheme 9 Asymmetric intermolecular [10 + 4] cycloadditions via the isobenzofulvene intermediate. | ||
Although the asymmetric [8 + 4] cycloaddition of isobenzofurane can potentially be used for the direct construction of chiral bicyclo[4.2.1]nonane, the asymmetric version has not been achieved. Indeed, when Jørgensen et al. accomplished the asymmetric [8 + 4] cycloaddition reaction between indenhydrin-2-carboxaldehyde and methylene oxalate, they encountered several challenges in achieving isobenzofurans.71 For example, (1) how to achieve periselectivity between 8π and 10π-components of isobenzofulvene? (2) how to ensure chemoselectivity among Michael addition and [8 + 2] and [8 + 4] cycloadditions? and (3) how to control the stereochemistry?
Later on, Zhao and Deng et al. developed a new method for asymmetric [8 + 4] cycloaddition of isobenzofuvene A14 and electron-deficient dienes (IQDMs) with the assistance of secondary amine catalysts (Scheme 10).72 This strategy efficiently offers indole cyclic bicyclo[4.2.1]nonane 32 with good yields, good enantioselectivities and moderate diastereoselectivities. In addition, the pyrrolidone-3,4-diene 29 is also well tolerated, yielding cyclic bicyclo[4.2.1]nonane 31 with good yields, high enantioselectivities and excellent diastereoselectivities.
 | ||
| Scheme 10 Asymmetric intermolecular [8 + 4] cycloadditions via the amino isobenzofulvene intermediate. | ||
Jørgensen and coworkers reported a novel stereoselective [10 + 2] cycloaddition between homologated indenecarbaldehydes 33 and α,β-unsaturated aldehydes 34, resulting in the assembly of tetrahydro-cyclopenta[a]indenes (Scheme 11).73 The process involves the organocatalytic generation of homologated amino isobenzofulvene intermediate A15. Kinetic studies, DFT calculations, and isolation of intermediates suggest that a stepwise cycloaddition mechanism involving the Curtin–Hammett scenario is plausible. The stereo-determining step was the irreversible ring closure process, and the initial formation of precyclization intermediates is rapid and reversible. The reaction scope is thoroughly investigated, and the observed selectivity supports the mechanistic conclusions. This study contributes to the further development of cycloadditions with more than 6π-electrons, suggesting that these reactions tend to react selectively with specific partners based on electron effects and substrates.
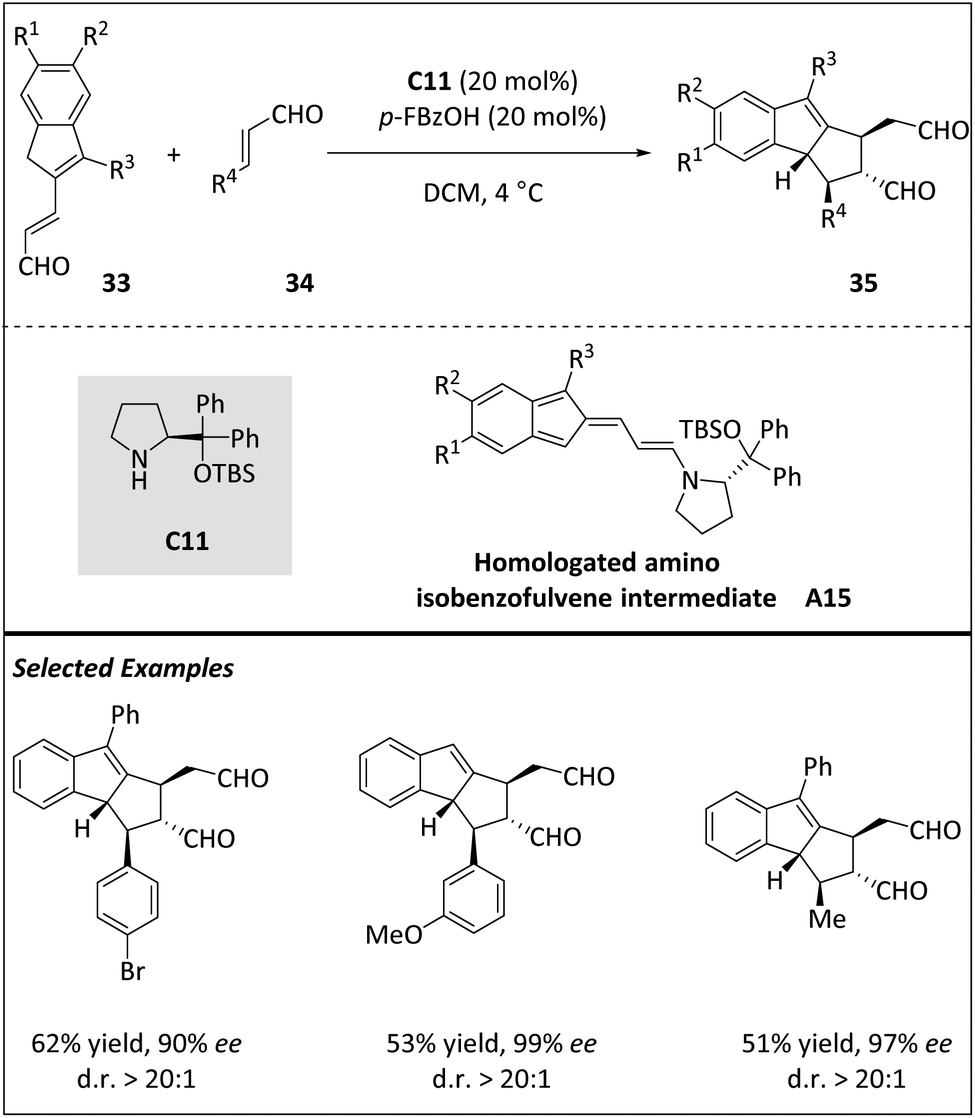 | ||
| Scheme 11 Asymmetric intermolecular [10 + 2] cycloadditions via the homologated amino isobenzofulvene intermediate. | ||
A one-step and direct organocatalytic process has been developed by Jessen and Jørgensen et al. for the enantioselective construction of a chiral cycl[3.2.2]azine core (Scheme 12).74 This is the first enantioselective example of [12 + 2] cycloaddition of a tricyclic skeleton with a central ring-junction nitrogen atom. The electron-rich 12π component intermediate A16 can be obtained from 5H-benzo[a]pyrrolizine-3-carbaldehyde 36 in the presence of aminocatalyst C12. Intermediate A16 acts as a nucleophile and subsequently as an electrophile through the exocyclic position, enabling higher-order cycloadditions with electron-poor 2π-components to construct the cycl[3.2.2]azine core. It is worth noting that even-membered ring syntheses based on higher-order cycloadditions are uncommon but can be achieved in this case.
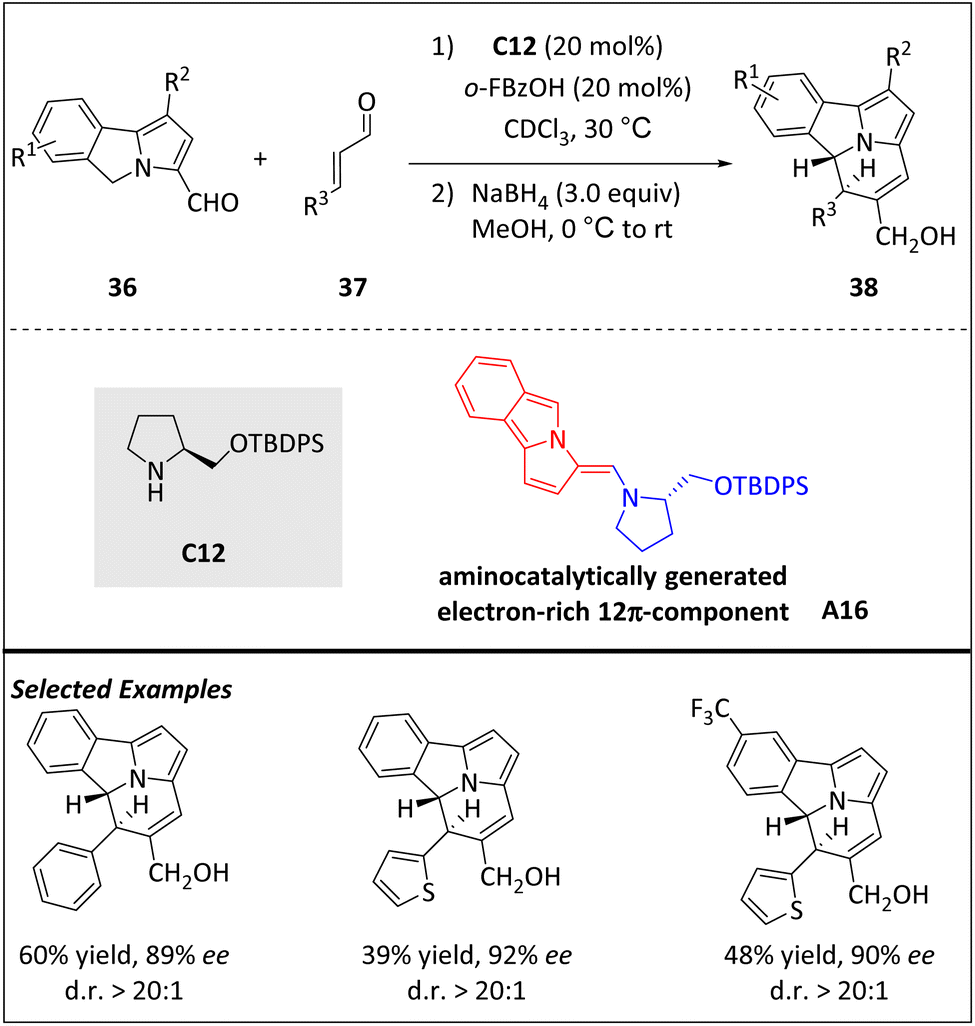 | ||
| Scheme 12 Asymmetric intermolecular [12 + 2] cycloadditions via the aminocatalytical 12π intermediate. | ||
Although partial erosion of the diastereomeric ratio occurred during purification, one-pot reduction of the formyl group of cycloadducts preserves the stereochemical information. DFT calculations reveal that the observed high stereoselectivity relies on the energetic barrier of ring closure. Minor modifications to the reaction conditions allow for the introduction of trans-benzo[a]cycl[3.2.2]azines from α,β-unsaturated ketoesters and nitroolefins. Fully unsaturated and photoluminescent benzo[a]cycl[3.2.2]azines are also obtained in the presence of vinyl sulfones.
The higher-order cycloaddition reaction is not only useful in constructing multiple asymmetric centers in complex ring systems but also enables the atroposelective establishment of C(sp2)–C(sp3). Most recently, the Jørgensen group developed a novel [12 + 2] HOC to access enantioenriched atropisomeric scaffolds that exhibit a stable C(sp2)–C(sp3) stereogenic axis along with two chiral centers (Scheme 13).75 Electron-rich aminocatalytic 12π intermediates A17, which were generated from 5H-benzo[a]pyrrolizine-3-carbaldehydes 36 with C12 or C13, react with electron-poor olefins 39, resulting in the formation of cycl[3.2.2]azine-based frameworks 40. This approach is highly versatile, accommodating a wide range of substrates and producing various chiral products. The rotational barrier of parent compounds has been determined through experimentation, and both atropisomers have been separated and identified following thermal equilibration. The conformational stability of compounds with different substitution patterns is also evaluated, with a half-life of over 640 years. Additionally, the isolation of these unique compounds provides empirical evidence of the principle behind central-to-axial chirality conversions through a simple oxidative transformation. DFT calculations and the isolation of a reactive intermediate demonstrate that the reaction's stereoselectivity is controlled by the Curtin–Hammett principle, with a single transition state (the elimination step) governing both the atropo- and enantio-induction.
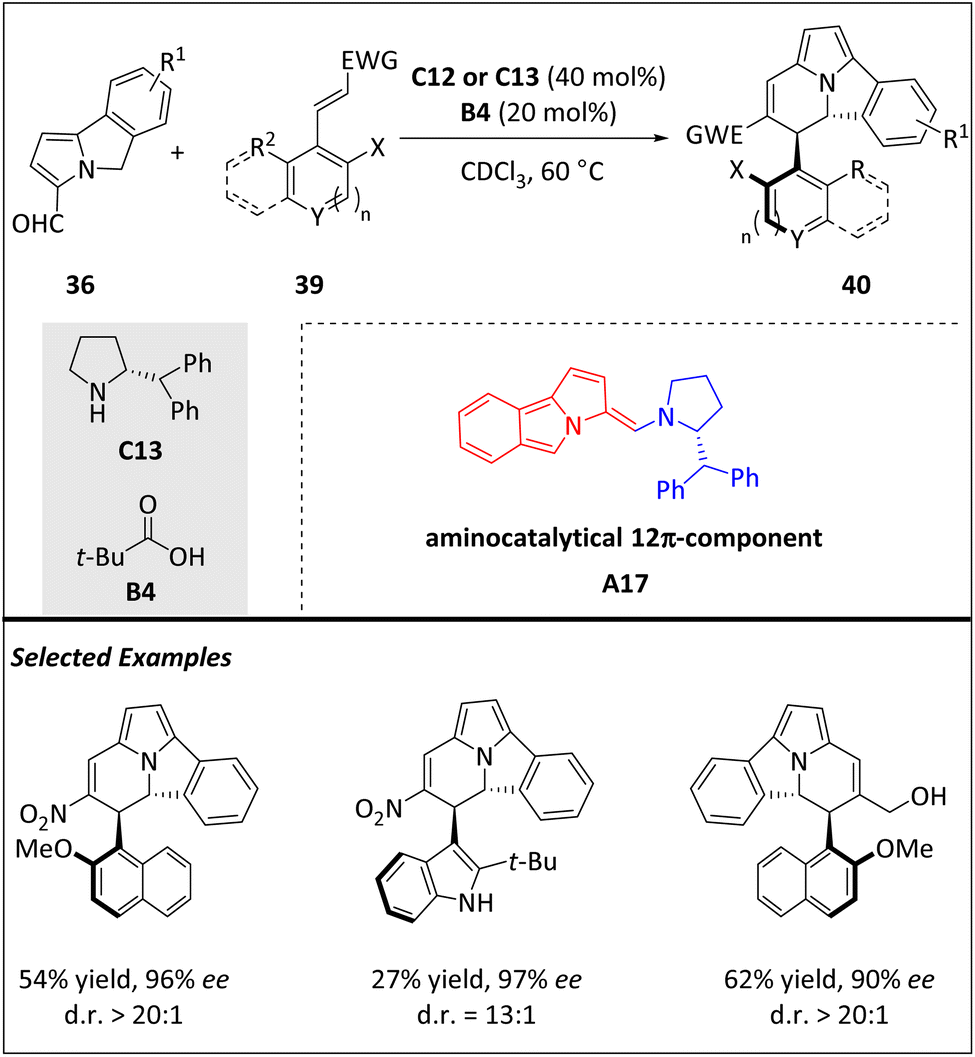 | ||
| Scheme 13 Enantioselective intermolecular [12 + 2] cycloadditions for the construction of C(sp2)–C(sp3) atropisomers. | ||
3.2 Iminium catalysis
Lately, there has been growing interest in utilizing higher-order cycloadditions as a versatile platform to develop novel asymmetric transformations. Notably, organocatalytic activation strategies have gained attention in this regard. Among these, chiral amines have shown remarkable efficacies when utilizing a HOMO-raising activation pattern. The strategy of LUMO-lowering through iminium ion intermediates has greatly contributed to the development of asymmetric HOC as well.In 2019, Albrecht and coworkers successfully utilized LUMO-activated iminium ions to react with tropothione 41 through an efficient [8 + 2] cycloaddition (Scheme 14).76 In this finding, 41 has proven to be a highly stable and valuable reactant that readily engages in enantioselective higher-order cycloadditions. Unlike tropone and derivatives, which behaved as 8π or 6π-components and participate through their LUMOs as electron-poor systems, tropothiones act as electron-rich 8π-components based on their HOMOs. The stereochemistry is controlled by aminocatalytic LUMO-activation of the carbonyl reactant.
 | ||
| Scheme 14 Asymmetric intermolecular [8 + 2] cycloadditions via the LUMO-lowering strategy. | ||
A rational design utilizing novel (E)-3-benzylidene-3H-pyrrolizines in iminium-ion-catalyzed [8 + 2] cycloaddition reactions proposed by Jessen and Jørgensen has facilitated the efficient and highly stereoselective synthesis of chiral cycl[3.2.2]azines (Scheme 15).77 The protocol is capable of diverse enals, enolizable aldehydes, and substrates with extended conjugation. The resulting product has an electron-rich alkenylpyrrole and an electron-deficient carbonaldehyde moiety, both of which can be selectively modified without altering the stereochemical information in the [8 + 2] cycloaddition. The addition of electron-rich substrate 44 at its 5-position to the nonshielded face of iminium-ion substrates results in the formation of the first two stereocenters (A19). The tetrahydrocycl[3.2.2]azine scaffold, which contains four consecutive stereocenters (A20), is then completed by ring closure through nucleophilic addition to the electrophilic pyrrolizine moiety. The aminocatalyst is then liberated through hydrolysis. The investigation of the acid-catalyzed isomerization pathway of cycloadducts emphasizes the importance of maintaining the balance between the loading of amino catalysts and acid additives.
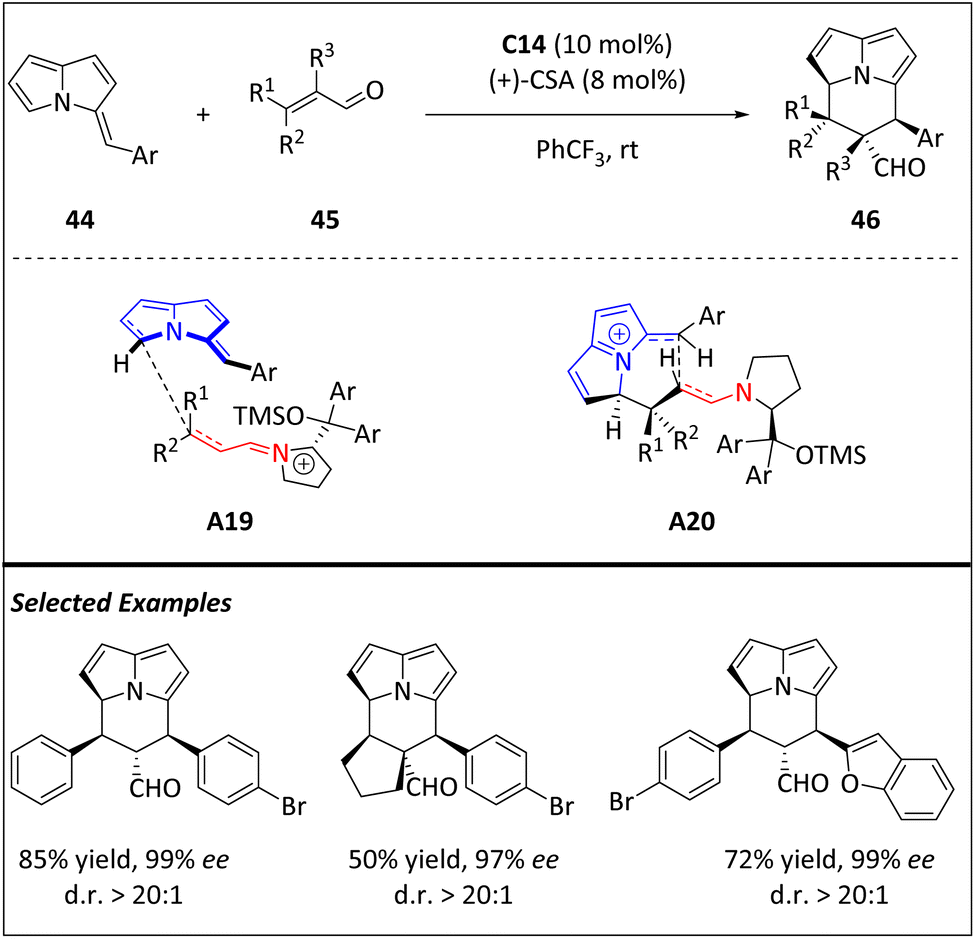 | ||
| Scheme 15 Asymmetric intermolecular [8 + 2] cycloadditions via iminium-ion catalysis. | ||
Yang and Wang et al. uncovered a new atroposelective [8 + 2] cycloaddition between pyridinium/isoquinolinium ylides and ynals (Scheme 16).78 Significantly, this approach demonstrates a fresh illustration of the organocatalyzed atropoenantioselective higher-order cycloaddition, furnishing diverse axially chiral 3-arylindolizines 49 with exceptional yields and enantioselectivities. Due to their valuable photochemical properties and diverse therapeutic indications, 3-arylindolizines are recognized as one of the most significant heterocyclics. It is proposed that the chiral amine catalyst C15 first reacts with alkynaldehyde 47 to the alkynylamine cationic intermediate A21 through dehydration. Subsequently, the Michael addition of A21 with 48 builds axially chiral allenamine A22. Next, intramolecular cyclization of A22 leads to the formation of axial chiral heterodiaryl intermediates, which are then hydrolyzed into product 49 and release catalyst C15 for the next catalytic cycle. The axially chiral 49 derivatives obtained in this reaction offer a wide range of functional groups, providing numerous opportunities for chemical transformations and drug discovery.
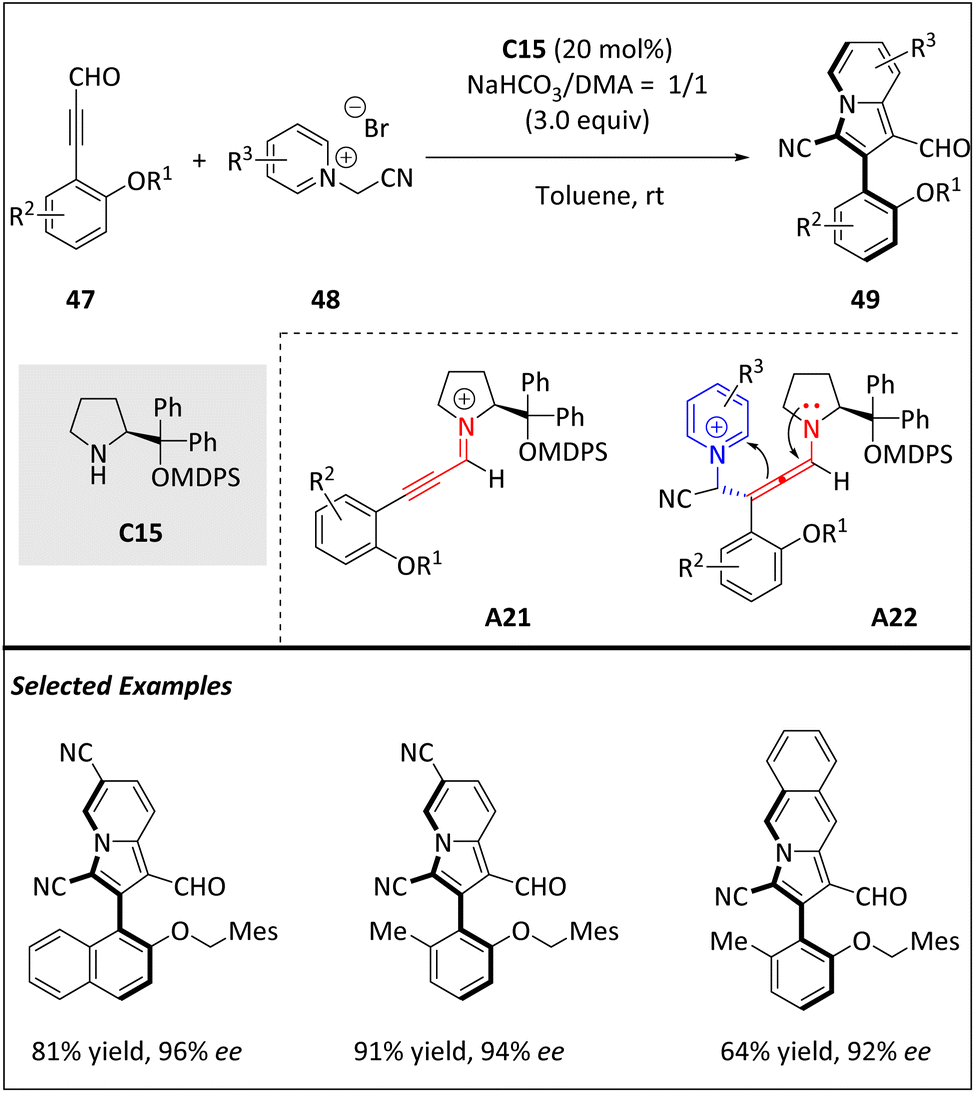 | ||
| Scheme 16 Atroposelective intermolecular [8 + 2] cycloadditions via iminium-ion catalysis. | ||
3.3 Enolate catalysis
In 2017, Pericàs and colleagues presented a nice approach to produce cycloheptatriene fused pyrrolidone rings 50 with high enantioselectivities (Scheme 17).79 It involves an isothiourea-catalyzed periselective [8 + 2] cycloaddition of chiral ammonium enolates A23, which are in situ generated from carboxylic acids 50 and azaheptafulvenes 51. This straightforward process yields architecturally polycyclic structures.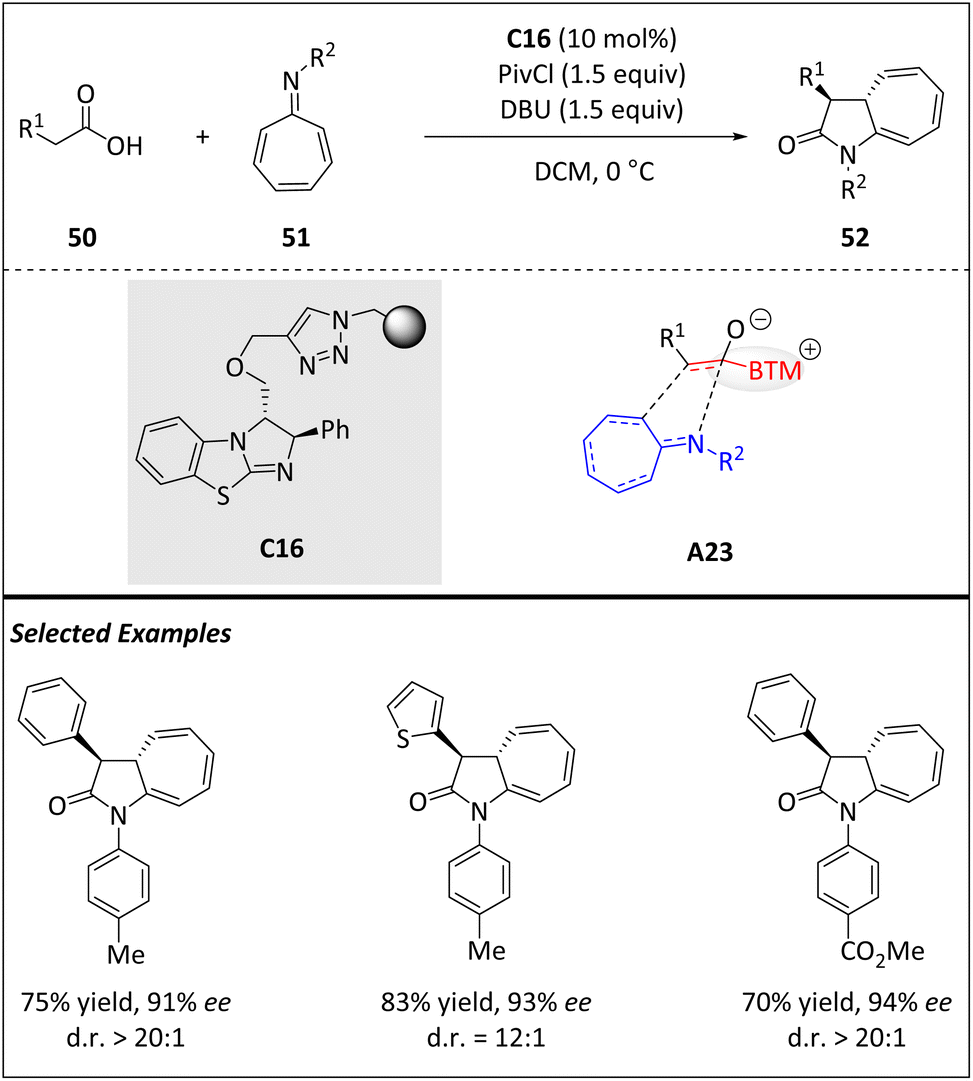 | ||
| Scheme 17 Stereoselective intermolecular [8 + 2] cycloadditions via isothiourea catalysis. | ||
In terms of practicality, the ability to recycle the immobilized isothiourea catalyst C16 is highly desirable as it reduces costs and improves overall efficiency. In order to achieve this goal, catalyst C16 is recovered through simple filtration and reused by adding fresh reactants after every single run. Surprisingly, no significant decrease in stereoselectivity is observed and only a slight yield decreases over the first four runs. The recycling experiments showcase an accumulated turnover number (TON = 44.7). Mechanistically, the mixed anhydride is produced in situ by the reaction of 50 with pivaloyl chloride. The corresponding acyl ammonium species is generated from the anhydride and isothiourea C16.
In 2019, a similar asymmetric [8 + 2] NHC-catalyzed cycloaddition reaction of azaheptafulvenes 51 with α-chloro aliphatic aldehydes 53 has been developed by Xu and coworkers (Scheme 18).80 This reaction yields cis-cycloheptatriene-fused γ-lactams 54 with excellent enantioselectivities, moderate to good diastereoselectivities, and good yields. The resulting cycloadducts can easily transform into valuable structures under mild reaction conditions. In contrast to Pericas's method, the α-alkyl substituted reactants are well tolerated and a series of diastereo-cycloadducts are obtained. The proposed mechanism involves the formation of the Breslow intermediate A24via the addition of NHC catalyst C17 to aldehyde 53. Intermediate A24 is then dechlorinated and deprotonated to produce NHC-bound enolate intermediate A25. Next, Mg(OTf)2 coordinates with the oxygen atom of intermediate A25 and the nitrogen atom of azaheptafulvene 51 to form a transition state. Finally, intramolecular lactamization produces cycloheptatriene-fused γ-lactam 54 and regenerates catalyst C17.
 | ||
| Scheme 18 Stereoselective intermolecular [8 + 2] cycloadditions via NHC catalysis. | ||
The development of chiral catalysts has led to an expanding toolbox capable of covering various chemical spaces through numerous transformations. However, the emphasis is usually placed on selectivity rather than versatility. The Pericas group utilized the NHC catalyst to exploit the distinctive reactivity of enals compared to tropones, resulting in high enantiomeric purity of cyclohepta[b]furan-2-ones via higher order cycloadditions (Scheme 19).81 In fact, the same group has recently published findings that demonstrate the ability of isothiourea catalysts to facilitate highly stereo- and periselective [8 + 2] annulations of azaheptafulvenes, producing [5.3.0] bicyclic products. However, a certain number of restrictions remained, such as the ineffectiveness of tropones and the tolerance solely of arylacetic acids as the 2π component. Consequently, they began to explore whether NHC catalysts could also facilitate these higher order cycloadditions. In this case, the Breslow intermediate A26 smoothly yields azolium enolate A27, which acts as the 2π component in a redox-neutral process. Moreover, the reaction can be directed towards different diastereomers by modifying the reaction conditions, resulting in a shift from kinetic to thermodynamic control. The process is well tolerated with substituted tropones and the resulting cycloaddition products can be modified via hydrogenation or methanolysis. DFT computational kinetic modelling has provided strong support for the experimental results in terms of periselectivity and stereoselectivity.
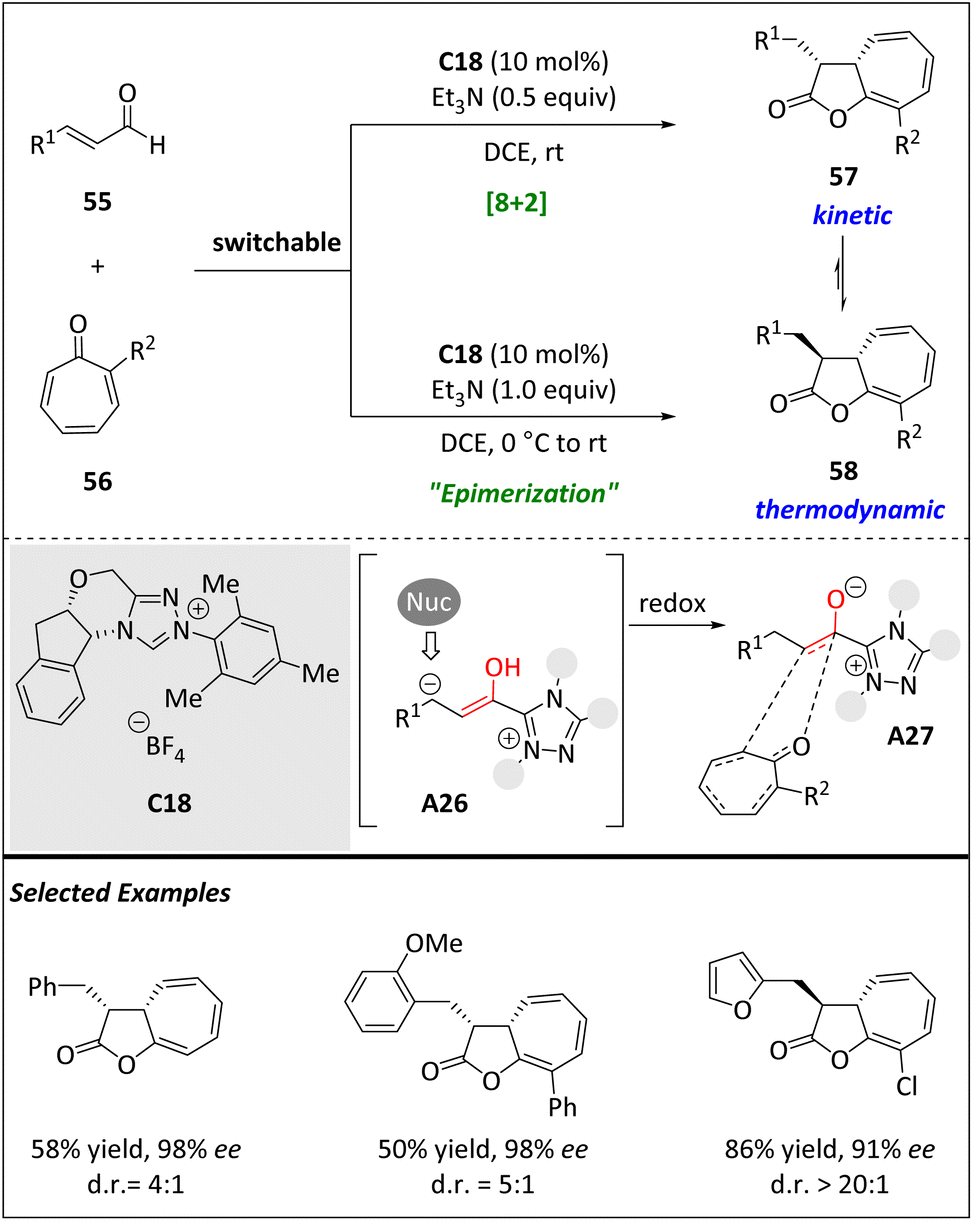 | ||
| Scheme 19 Diastereodivergent asymmetric intermolecular [8 + 2] cycloadditions via NHC catalysis. | ||
In 2019, Chi and co-workers disclosed an enantioselective [10 + 2] cycloaddition by employing indole-7-carbaldehyde as the 10π electron component with the aid of chiral NHC catalysts (Scheme 20).82 The process involves the formation of an acyl azolium ester intermediate, which then transforms into an aza-o-quinone methides (aza-o-QMs)-containing intermediate A28 through proton transfer. The subsequent procedure entails a [10 + 2] annulation between A28 and a reactive ketone substrate 58, yielding acetal product 59 with good yield and favourable enantiomeric ratios. The products obtained through this method contain pyrroloquinazoline or oxazinoindole scaffolds that are commonly found in bioactive molecules. The method demonstrates the ability of NHCs to activate the aldehyde group at a remote site of the indole molecule and control the stereoselectivity. Unlike previous studies where nitrogen or oxygen atoms are attached to benzene as reaction sites, the reactive nitrogen atom is located within the aldehyde substrate's aromatic ring in this method. To expand the versatility and practicality of this protocol, isatin derivatives for the assembling of spiro-cyclics are disclosed. The asymmetric aminals and N,O-acetal units in the cycloadducts hold significant synthetic values.
 | ||
| Scheme 20 Enantioselective intermolecular [10 + 2] cycloadditions via the 10π aza-o-QM intermediate. | ||
Zhang and Wang carried out a theoretical investigation on such an asymmetric [10 + 2] HOC process.83 According to the computational results, the stereoselectivity is primarily influenced by the ring-forming step between the aza-o-QMs intermediate A28 and ketone 58, resulting in a predominantly R-configured product. The researchers identify a weak interaction, which is analyzed qualitatively and quantitatively as LP⋯π, C–H⋯F, C–F⋯O, and C–F⋯F interactions in terms of NCI and AIM analyses.
Simultaneously, Chi and coworkers uncovered a novel method to activate nitrogen atoms in heteroarenes for achieving enantioselective [10 + 2] HOCs (Scheme 21).84 The acylazolium intermediate A29 is generated through the carbene-addition step under an oxidant. This oxidation step eliminates the electron density from the indole (or pyrrole), resulting in a less nucleophilic overall π-system in contrast to the arylaldehyde substrate. For example, the carbon, located in the 2- or 3-position, is a possible nucleophilic center. Conversely, the nitrogen atom is selectively activated and serves as the reactive center through A29 deprotonation, resulting in the formation of the azafulvene intermediate A30. The activation achieved via carbene catalysis leads to the creation of novel aza-fulvene acylazolium intermediates that selectively react with ketones, forming N,O-acetals with exceptional yields and enantiomeric ratios. The chiral N,O-acetal products display promising antibacterial properties against Ralstonia solanacearum and hold significance in the advancement of novel chiral agrichemicals for safeguarding plants. This study marks the initial NHC-catalyzed activation of nitrogen atoms incorporated into aromatic π-rings, thereby expanding the potential of carbene catalysis in accomplishing asymmetric reactions associated with aromatic skeletons. The [10 + 2] cycloaddition of 60 with indoline-2,3-dione 61 was also reported by the Hui group.85
 | ||
| Scheme 21 Enantioselective intermolecular [10 + 2] cycloadditions via the 10π aza-fulvene intermediate. | ||
Peng and Wang et al. independently reported a novel catalytic and enantioselective [10 + 2] higher-order cycloaddition of trifluoromethyl ketone derivatives with indole-2-carbaldehydes (Scheme 22), highlighting the potential of synthesized molecules due to the significance of polycyclic structures and the incorporation of “F”-containing fragments.86
 | ||
| Scheme 22 Asymmetric intermolecular [12 + 2] cycloadditions via the aminocatalytical 12π intermediate. | ||
The Wang group efficiently constructed enantioenriched polycycles with fluorinated substituents via an NHC-bound aza-benzofulvene intermediate A30, marking the first use of NHC-bound aza-arylfulvene intermediates in catalytic and enantioselective [10 + 2] or [14 + 2] reactions. Kinetic studies reveal that the reaction shows nearly first-order depending on catalyst C22 and zero-order relying on substrates 67, 68, and 3,3′,5,5′-tetra-tert-butyl diphenoquinone (DQ). An intrinsic reaction coordinate calculation (IRC) of the transition state is conducted, indicating a concerted asynchronous process where the covalent bond between the C1 of trifluoroacetophenone and the N1 of indole is formed first. Additionally, DFT calculations enable accurate prediction of and a thorough understanding of enantio- and stereo-control. Similar HOC reactions were also discovered by the Biju group.87
In 2021, Yang and Chi et al. reported a process that utilizes carbene catalysis to activate nitrogen atoms through covalent interactions (Scheme 23).88 This approach enables the asymmetric synthesis of heterocycles, (benz)imidazole-derived imines 70 and activated ketones 61 or 68via a highly enantioselective HOC manner. The process requires the use of the chiral C23 or C24 catalyst and an oxidant (DQ or MnO2) to obtain the desired skeletons. At the onset of the reaction, catalyst C23 or C24 is introduced to an aldimine substrate derived from (benz)imidazole. The following steps involve oxidation and proton transfer, forming triaza-diene intermediate A31. In a previous report, the reactive nucleophilic heteroatoms are situated at the γ-position relative to the carbonyl groups of the substrates. These intermediates (e.g. o-QMs,89,90 aza-o-QMs A2891,92 and aza-fulvene A30) are comparable to vinyl enolate intermediates, bearing a heteroatom at the γ-position. This is the first success in applying a triaza-diene intermediate for asymmetric HOCs. Building upon a relaxed 2D potential energy surface (PES) scan, the concerted synchronous [10 + 2] cycloaddition pathway is ruled out. Alternatively, a highly asynchronous concerted mechanism is identified with the aid of DFT calculations.
 | ||
| Scheme 23 Enantioselective hetero-[10 + 2] cycloadditions via the triaza-diene intermediate. | ||
Most recently, the enantio- and diastereoselective [12 + 2] higher-order cycloaddition between 5H-benzo[a]pyrrolizine-3-carbaldehydes 73 and isatine-derivatives 61 has been developed by Chi et al. via NHC-catalyzed remote C(sp3)–H activation (Scheme 24).93 The reaction allows the rapid construction of a sophisticated tricyclic core bearing a morpholine moiety under mild conditions and easily accessible starting materials. Specifically, aldehydes 73 react with carbene catalyst C23 to generate the Breslow intermediate. Then, the NHC-bound acyl azolium intermediate A32 is obtained from the Breslow intermediate via oxidation. Deprotonation of intermediate A32 gives rise to a carbon anion, which then undergoes tautomerization to form 12π species A33. Upon addition of A33 to 2π electrophile 61, the target product is formed.
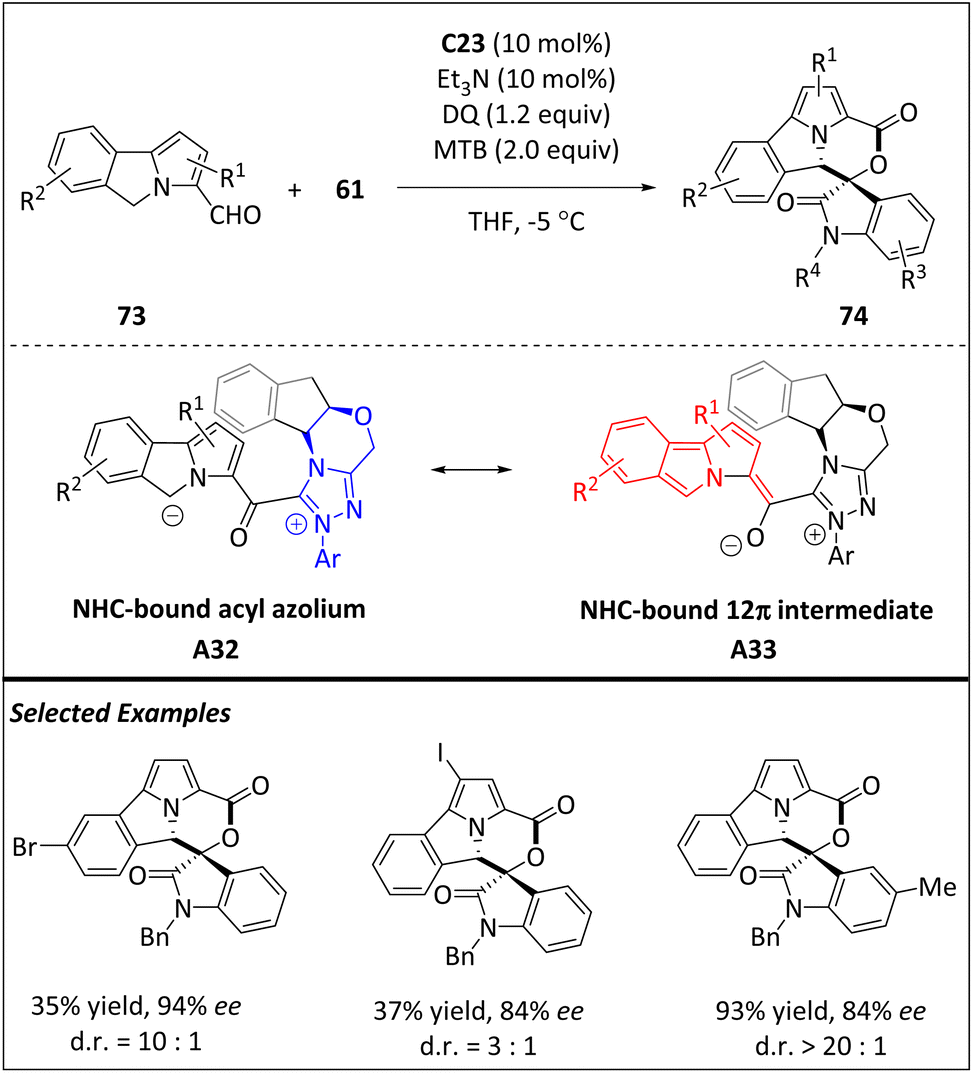 | ||
| Scheme 24 Enantioselective [12 + 2] cycloadditions via the NHC-bound 12π intermediate. | ||
3.4 Ion-pairing catalysis
Gao and Guo et al. reported the phosphine-catalyzed [8 + 2]-annulation of heptafulvene 75 with allenoates 76, yielding bicyclo[5.3.0]decane-fused spirobarbiturates 77 in moderate to excellent yields (Scheme 25).94 By using chiral Kwon's phosphine catalyst C25, enantioenriched products are obtained in moderate to high yields and with excellent enantioselectivities. The reaction involves the formation of the β-phosphonium dienolate intermediate A34, which is generated from the conjugate addition of phosphine to allenoate 76. This intermediate then attacks heptafulvene-barbiturate 75, which has never been used as an 8π component in an [8 + 2] cycloaddition before. A subsequent intramolecular Michael addition leads to product 77. In addition, the β-phosphonium dienolate intermediate A34 undergoes Si-face attack to form 75, ultimately generating (S)-tricyclic products.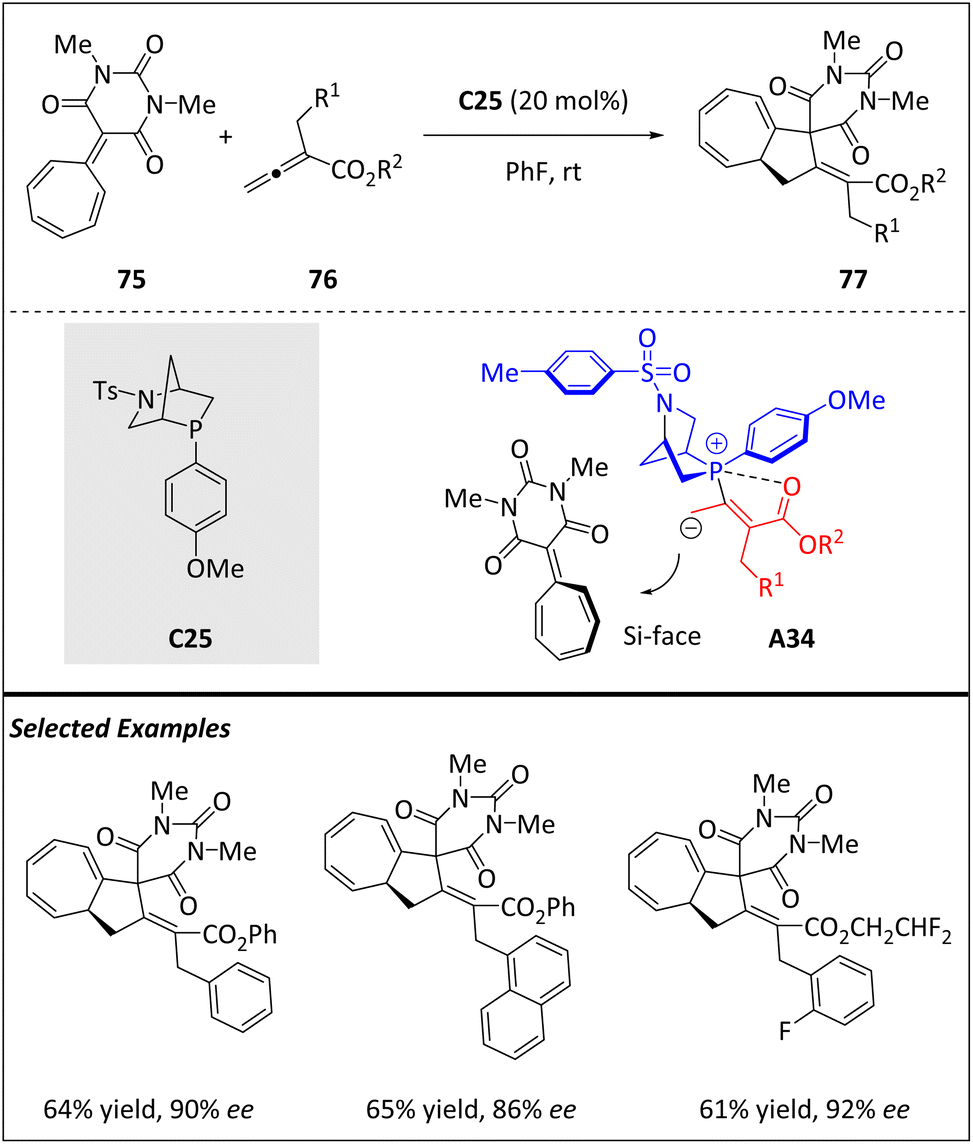 | ||
| Scheme 25 Enantioselective [8 + 2] cycloadditions via the phosphine-bound 2π intermediate. | ||
Kallweit and Schneider have developed a one-step process (Scheme 26) for synthesizing densely substituted 2,3-dihydro-1H-pyrrolizines 80, which are often found in many biologically active natural products.95 The process involves an organocatalytic [6 + 2]-cycloaddition between 1H-pyrrole-2-yl carbinols 78 and 2-vinylindoles 79, catalyzed by a BINOL-derived phosphoric acid C26. The reaction proceeds via the formation of chiral hydrogen-bonded 2-methide-2H-pyrroles A35 and results in the formation of single diastereomers with excellent enantioselectivity and good yields. The resulting 2,3-dihydro-1H-pyrrolizines bear three contiguous stereogenic centers, making them highly valuable synthetic targets for developing new biologically active compounds.
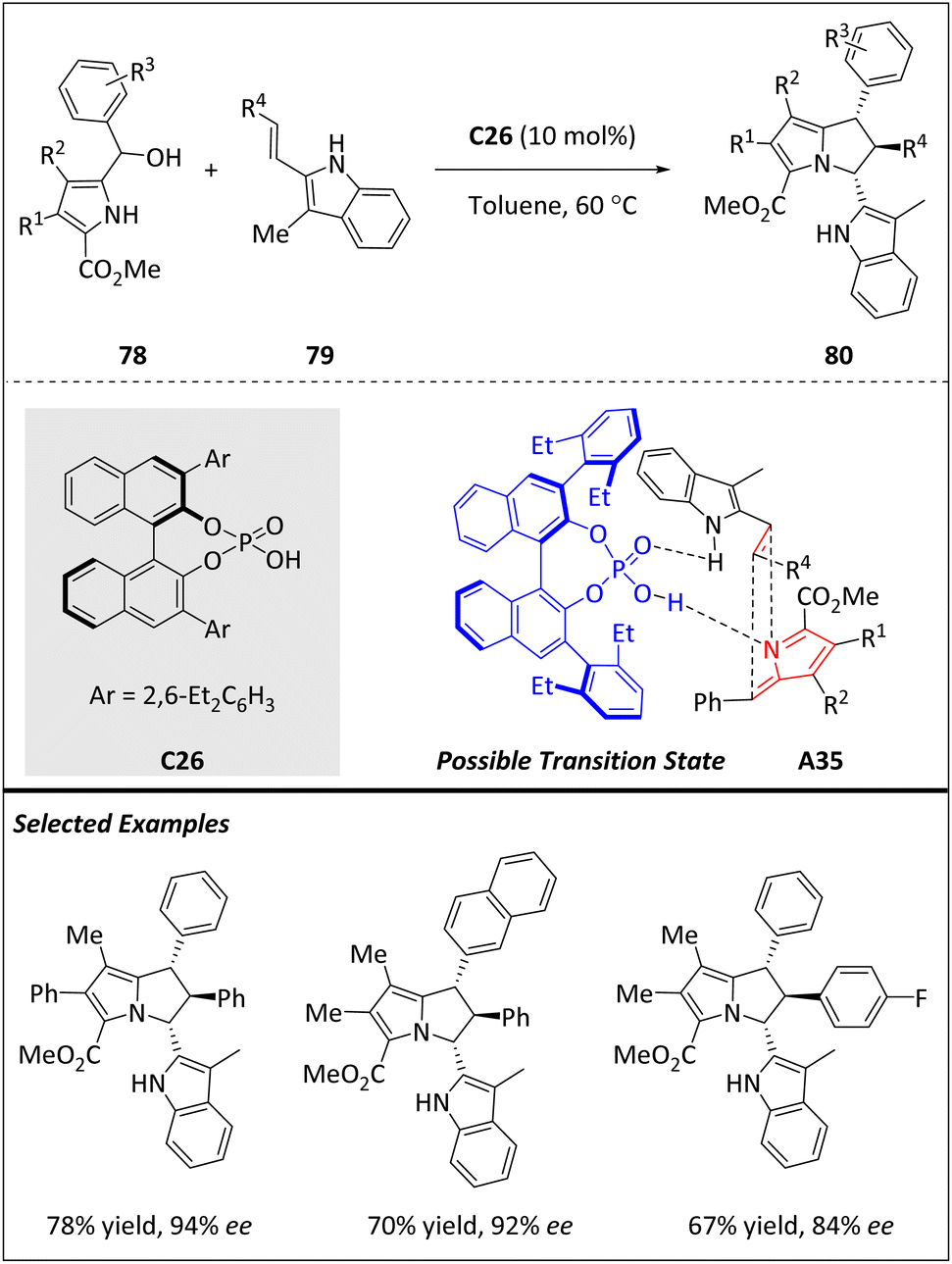 | ||
| Scheme 26 Enantioselective [6 + 2] cycloadditions catalyzed by phosphoric acid via 2-methide-2H-pyrroles. | ||
In the presence of chiral C26, carbinols 78 can be successfully dehydrated to form chiral hydrogen-bonded 2-methide-1H-indoles, which subsequently react with various nucleophiles to produce both open-chain and annulated indole-based heterocycles. The intermediate 2-methide-2H-pyrroles A35 plays a vital role in the biosynthesis of porphyrines, myrmicarin alkaloids and (+)-roseophilin. However, their applications in synthetic chemistry have been restricted due to the instability of pyrrol-2-yl-carbinols and the high reactivity of the pyrrole nucleus towards electrophilic attack. To address these challenges, the authors hypothesize that introducing an electron-withdrawing group at the C5 position of the pyrrole nucleus would effectively block this site and prevent oligomerization through electronic deactivation. By attenuating the reactivity of pyrrole, the full synthetic potential of 2-methide-2H-pyrroles can be realized.
In the control experiment, no conversion is observed when subjected to the reaction with 78, indicating the crucial role of the free N–H moiety for double hydrogen bonding as described previously. A plausible transition state is also proposed to explain the observed diastereo- and enantioselectivity (Scheme 26). In fact, the lower 3-aryl group in the BINOL backbone shields the bottom side of the A35 effectively and the incoming dienophile is guided to the top face.
As mentioned above, the ability of tropone 56 to serve as a 2π, 4π, 6π, or 8π component makes it a fascinating and challenging cyclic polyene.96 By reacting with 1,3-dipoles, a variety of fused or bridged polycycles can be prepared smoothly. Enantioselective transition metal catalysis has been reported to facilitate stereoselective 1,3-dipolar higher-order cycloadditions of tropone (or heptafulvene derivatives).97–102 However, there is a noticeable lack of organocatalytic 1,3-dipolar higher-order cycloadditions of tropones in the literature. Using chiral phosphoric acids as catalysts for 1,3-dipolar cycloadditions of azomethine ylides A36 with electron-deficient 2π components provides a reliable protocol for the preparation of enantioenriched pyrrolidines. In 2020, Jørgensen and coworkers reported the first example of chiral phosphonic acid (CPA)-catalyzed 1,3-dipolar [6 + 4] cycloaddition of 56 with A36 (Scheme 27).103 This approach relies on the distinctive reactivity of iminomalonate esters, which are obtained through the condensation of aldehydes 81 with 2-aminomalonates 82. The desired dipoles A36 are produced through a CPA-facilitated prototropic shift. The resulting structural motif forms the central core of bioactive Lycopodium and Daphniphyllum alkaloids. A proposed transition state explained the relative and absolute configuration of compound 83. The CPA catalyst C27 plays multiple roles in the reaction.
 | ||
| Scheme 27 Enantioselective 1,3-dipolar [6 + 4] cycloadditions catalyzed by phosphoric acid. | ||
In 2020, Vicario and Manzano et al. reported a phosphine-catalyzed enantioselective [8 + 2] HOC reaction using racemic γ-substituted allenes 84 and azaheptafulvenes 51 (Scheme 28).104 This reaction offers a direct and straightforward approach for synthesizing aza-azulenoid scaffolds. By optimizing the reaction conditions, a wide range of mono- and disubstituted cycloadducts with good yields and precise control over peri-, regio-, diastereo-, and enantioselectivity were successfully afforded. In contrast to previous reports regarding 84 as the C4-component, in this study, they demonstrated the exceptional role of these allenes as excellent 2π components. The suggested mechanism entails a nucleophilic assault by catalyst C26 on allenic amides 84, leading to the generation of an intermediate known as an allyl phosphonium ylide. This intermediate undergoes a selective reaction guided by a three-dimensional distribution, facilitated by an H-bond association, ultimately determining the stereochemical outcome.
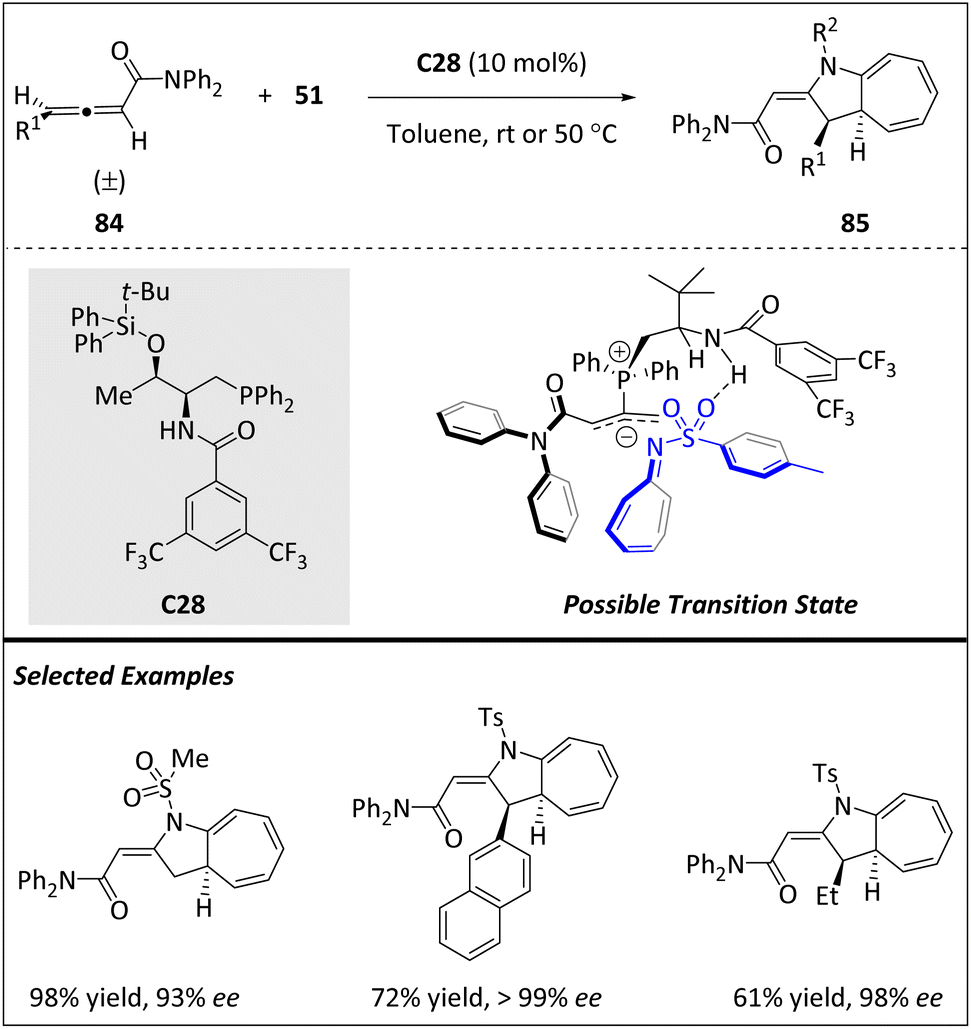 | ||
| Scheme 28 Enantioselective [8 + 2] cycloadditions catalyzed by phosphine. | ||
The Chen group has developed an asymmetric higher order [10 + 2] cycloaddition by employing 2-alkyl-idene-1-indanones and electron-deficient alkenes (Scheme 29). Previously, 2-benzylidene-1-indanone could transform into a 1-hydroxyl isobenzofulvene anion intermediate, and subsequently carried out a [10 + 2] cycloaddition with alkenes. By introducing chiral phase-transfer catalysis, they successfully generated a novel 1-activated isobenzofulvene, enabling the realization of an asymmetric version of this transformation. The incorporation of various alkenes, such as 86, 2-alkyl-idene-benzofuran-3(2H)-one, 2-alkyl-idene-1,3-dione, and α,β-unsaturated ketones, significantly enhanced the structural diversity and versatility of the resulting products.105
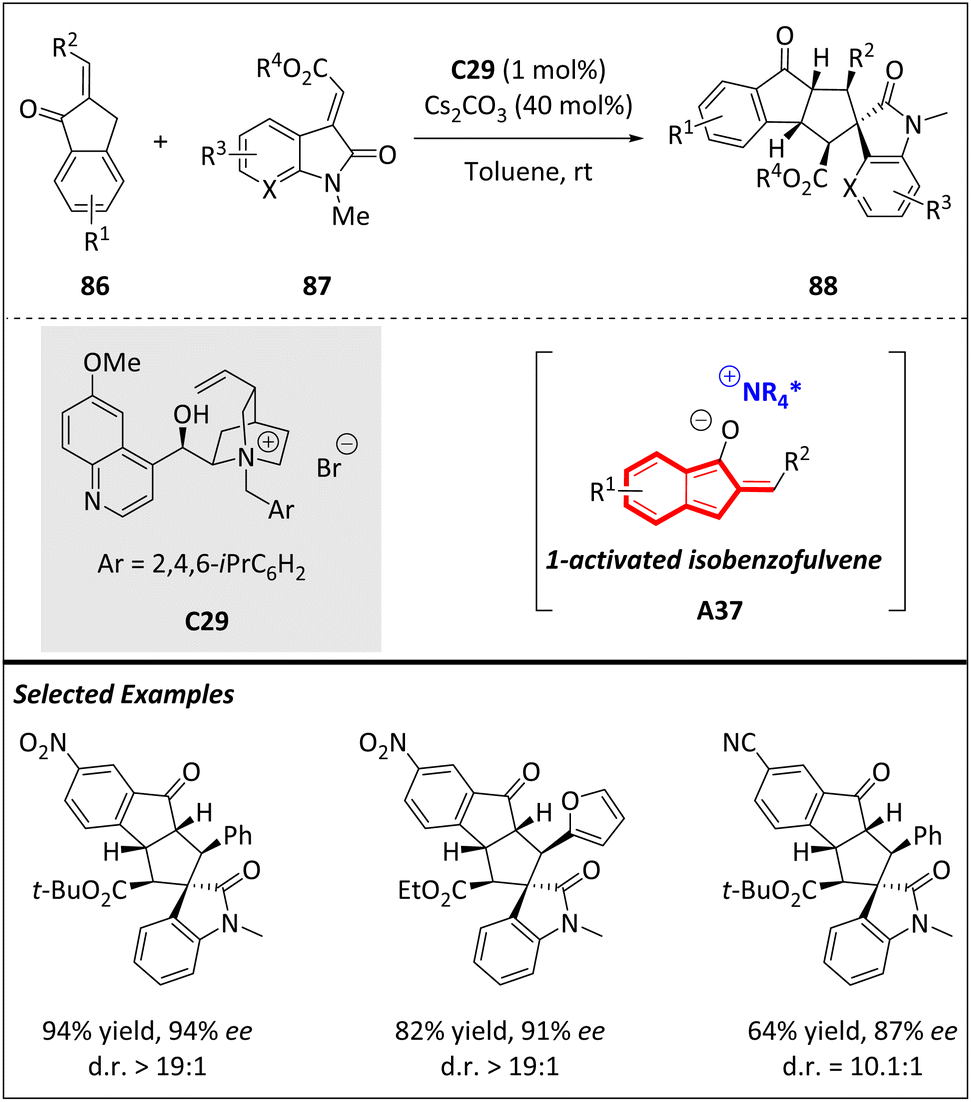 | ||
| Scheme 29 Enantioselective [8 + 2] cycloadditions catalyzed by phosphine. | ||
To address the challenges of diastereo- and enantioselectivity issues, a newly designed cinchona-based ammonium salt with a bulky substituent was disclosed. Surprisingly, this catalyst indicates high efficiency even at low loadings (1 mol%), even if the reactive site is physically distant from the chiral ion pair complex. This finding represents a rare instance of achieving asymmetric higher-order cycloaddition using an isobenzofulvene-based 10π intermediate.
In a recent study, Zhao and Yuan along with their colleagues developed a dearomative higher-order [10 + 2] cycloaddition of 2-alkylidene-1-indanones and 3-nitroindoles.106 Utilizing benzyl triethyl ammonium chloride (TEBA) as the phase-transfer catalyst under basic conditions, a broad array of polycyclic cyclopenta[b]indoline derivatives were prepared under mild conditions, achieving excellent results (up to 99% yield and >20:1 dr).
4. Summary and outlook
Higher order cycloadditions (HOCs) offer distinct advantages in the synthesis of complex molecules, particularly those with medium size and multiple rings. The field of asymmetric HOC has witnessed significant progress since the influential exchange between Woodward and Hoffmann. In the last few decades, significant advancements have been made in the field of asymmetric HOC, particularly through the utilization of organocatalysis. These approaches have facilitated the control of peri- and enantioselectivity in the ring formation process. Diverse optically pure functional structures were smoothly prepared, showcasing attractive applications in synthetic chemistry and biological studies. This review highlights various activation strategies employed in constructing chiral skeletons in the presence of organocatalysts. However, novel activation modes, including radical-type cycloaddition107 and transition metal/organocatalyst dual systems, have limited progress. Additionally, the control of axial, planar, spiro, and other forms of chirality through asymmetric HOCs remains very rare. In summary, there is ample room for future development in the field of asymmetric higher order cycloadditions. Advancements in this area would greatly enhance the value of precise control and efficient assembly in synthetic chemistry.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
Generous financial support for this work was provided by the National Natural Science Foundation of China (21871160, 21672121, and 22071130), the Bayer Investigator Fellowship, and the Fellowship of Tsinghua-Peking Center for Life Sciences (CLS).References
- W. C. Herndon, Theory of Cycloaddition Reactions, Chem. Rev., 1972, 72, 157–179 CrossRef CAS.
- N. I. Jessen, D. McLeod and K. A. Jørgensen, Higher-Order Cycloadditions in the Age of Catalysis, Chem, 2022, 8, 20–30 CAS.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, The Diels–Alder Reaction in Total Synthesis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS PubMed.
- J.-A. Funel and S. Abele, Industrial Applications of the Diels–Alder Reaction, Angew. Chem., Int. Ed., 2013, 52, 3822–3863 CrossRef CAS PubMed.
- T. Hashimoto and K. Maruoka, Recent Advances of Catalytic Asymmetric 1,3-Dipolar Cycloadditions, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed.
- K. N. Houk, L. J. Luskus and N. S. Bhacca, Novel Double [6 + 4] Cycloaddition of Tropone to Dimethylfulvene, J. Am. Chem. Soc., 1970, 92, 6392–6394 CrossRef CAS.
- K. N. Houk and R. B. Woodward, Cycloaddition Reactions of Cycloheptatriene and 2,5-Dimethyl-3,4-Diphenylcyclopentadienone, J. Am. Chem. Soc., 1970, 92, 4143–4145 CrossRef CAS.
- S. Itô, Y. Fujise, T. Okuda and Y. Inoue, Reaction of Tropone with Cyclopentadiene, Bull. Chem. Soc. Jpn., 1966, 39, 1351–1351 CrossRef.
- R. C. Cookson, B. V. Drake, J. Hudec and A. Morrison, The Adduct of Tropone and Cyclopentadiene: A New Type of Cyclic Reaction, Chem. Commun., 1966, 1, 15–16 RSC.
- D. McLeod, M. K. Thøgersen, N. I. Jessen, K. A. Jørgensen, C. S. Jamieson, X.-S. Xue, K. N. Houk, F. Liu and R. Hoffmann, Expanding the Frontiers of Higher-Order Cycloadditions, Acc. Chem. Res., 2019, 52, 3488–3501 CrossRef CAS PubMed.
- W. Oppolzer, Regio- and Stereo-Selective Syntheses of Cyclic Natural Products by Intramolecular Cycloaddition- and Ene-Reactions, Pure Appl. Chem., 1981, 53, 1181–1201 CrossRef CAS.
- H. S. Ateeq, Higher-Order Cycloaddition Reactions in Natural Product Synthesis, Pure Appl. Chem., 1994, 66, 2029–2032 CrossRef CAS.
- S. M. Sieburth and N. T. Cunard, The [4 + 4] Cycloaddition and Its Strategic Application in Natural Product Synthesis, Tetrahedron, 1996, 52, 6251–6282 CrossRef CAS.
- J. H. Rigby, N. C. Warshakoon and A. J. Payen, Studies on Chromium(0)-Promoted Higher-Order Cycloaddition-Based Benzannulation. Total Synthesis of (+)-Estradiol, J. Am. Chem. Soc., 1999, 121, 8237–8245 CrossRef CAS.
- B. Zhang, K. B. Wang, W. Wang, X. Wang, F. Liu, J. Zhu, J. Shi, L. Y. Li, H. Han, K. Xu, H. Y. Qiao, X. Zhang, R. H. Jiao, K. N. Houk, Y. Liang, R. X. Tan and H. M. Ge, Enzyme-Catalysed [6 + 4] Cycloadditions in the Biosynthesis of Natural Products, Nature, 2019, 568, 122–126 CrossRef CAS PubMed.
- Y.-Y. Cao, Z. Wang, Z.-H. Wang, X.-G. Jiang and W.-H. Lu, Inhibition of miR-155 Alleviates Sepsis-Induced Inflammation and Intestinal Barrier Dysfunction by Inactivating NF-κB Signaling, Int. Immunopharmacol., 2021, 90, 107218 CrossRef CAS PubMed.
- X. Wang, D. Zhou, W. Zhou, J. Liu, Q. Xue, Y. Huang, C. Cheng, Y. Wang, J. Chang, P. Wang and C. Miao, Clematichinenoside AR Inhibits the Pathology of Rheumatoid Arthritis by Blocking the circPTN/miR-145-5p/FZD4 Signal Axis, Int. Immunopharmacol., 2022, 113, 109376 CrossRef CAS PubMed.
- W.-G. Chen, S.-S. zhang, S. Pan, Z.-F Wang, J.-Y. Xu, X.-H Sheng, Q. Yin and Y.-J. Wu, α-Mangostin Treats Early-Stage Adjuvant-Induced Arthritis of Rat by Regulating the CAP-SIRT1 Pathway in Macrophages, Drug Des. Dev. Ther., 2022, 16, 509–520 CrossRef CAS PubMed.
- S.-P. Sun, Y.-Y. Du, Q. Sun, L.-J. He, E. Zhu and J.-R. Li, Oxidative Stress-Mediated Hepatotoxicity in Rats Induced by Ethanol Extracts of Different Parts of Chloranthus Serratus, Pharm. Biol., 2020, 58, 1286–1298 CrossRef PubMed.
- G. Wang, H. Zhang, J. Sun, Y. Zhang, F. He and J. Zou, Cyclosporin A Impairs Neurogenesis and Cognitive Abilities in Brain Development via the IFN-γ-Shh-BDNF Pathway, Int. Immunopharmacol., 2021, 96, 107744 CrossRef CAS PubMed.
- Y. Jiang, W. Li, J. Wang and G.-G. Wang, Cardiac Dysfunction Is Attenuated by Ginkgolide B via Reducing Oxidative Stress and Fibrosis in Diabetic Rats, Iran. J. Basic Med. Sci., 2020, 23, 1078–1084 Search PubMed.
- Y. Li, M. Dai, L. Wang and G. Wang, Polysaccharides and Glycosides from Aralia Echinocaulis Protect Rats from Arthritis by Modulating the Gut Microbiota Composition, J. Ethnopharmacol., 2021, 269, 113749 CrossRef CAS PubMed.
- J. Chen, D. Cao, S. Jiang, X. Liu, W. Pan, H. Cui, W. Yang, Z. Liu, J. Jin and Z. Zhao, Triterpenoid Saponins from Ilex Pubescens Promote Blood Circulation in Blood Stasis Syndrome by Regulating Sphingolipid Metabolism and the PI3K/AKT/eNOS Signaling Pathway, Phytomedicine, 2022, 104, 154242 CrossRef CAS PubMed.
- L. H. Zhou Hua and W. Haihua, Potential Protective Effects of the Water-Soluble Chinese Propolis on Experimental Ulcerative Colitis, J. Tradit. Chin. Med., 2023, 43, 925–933 Search PubMed.
- S. Wang, S. Gao, W. Ye, Y. Li, J. Luan and X. Lv, The Emerging Importance Role of m6A Modification in Liver Disease, Biomed. Pharmacother., 2023, 162, 114669 CrossRef CAS PubMed.
- H. W. Li Cheng, Z. Wang, H. Huang, D. Zhuo and J. Lin, Leflunomide Inhibits Proliferation and Induces Apoptosis via Suppressing Autophagy and PI3K/Akt Signaling Pathway in Human Bladder Cancer Cells, Drug Des. Dev. Ther., 2020, 14, 1897–1908 CrossRef PubMed.
- Z.-S. Tao, T.-L. Li and S. Wei, Silymarin Prevents Iron Overload Induced Bone Loss by Inhibiting Oxidative Stress in an Ovariectomized Animal Model, Chem.-Biol. Interact., 2022, 366, 110168 CrossRef CAS PubMed.
- X. He, G. Liu, X. Chen, Y. Wang, R. Liu, C. Wang, Y. Huang, J. Shen and Y. Jia, Pharmacokinetic and Pharmacodynamic Interactions between Henagliflozin, a Novel Selective SGLT-2 Inhibitor, and Warfarin in Healthy Chinese Subjects, Clin. Ther., 2023, 45, 655–661 CrossRef CAS PubMed.
- Y. Wang, H. Wu, Z. Han, H. Sheng, Y. Wu, Y. Wang, X. Guo, Y. Zhu, X. Li and Y. Wang, Guhong Injection Promotes Post-Stroke Functional Recovery via Attenuating Cortical Inflammation and Apoptosis in Subacute Stage of Ischemic Stroke, Phytomedicine, 2022, 99, 154034 CrossRef CAS PubMed.
- L. Wang, P. Wang, D. Wang, M. Tao, W. Xu and O. J. Olatunji, Anti-Inflammatory Activities of Kukoamine A from the Root Bark of Lycium Chinense Miller, Nat. Prod. Commun., 2020, 15, 20912088 Search PubMed.
- L. Zang, H. Xu, C. Huang, C. Wang, R. Wang, Y. Chen, L. Wang and H. Wang, A Link between Chemical Structure and Biological Activity in Triterpenoids, Recent Pat. Anticancer Drug Discov., 2022, 17, 145–161 CrossRef CAS PubMed.
- Y. Li, H. Xu, H. Wang, K. Yang, J. Luan and S. Wang, TREM2: Potential Therapeutic Targeting of Microglia for Alzheimer's Disease, Biomed. Pharmacother., 2023, 165, 115218 CrossRef CAS PubMed.
- H. Hong, Q. Zou, Y. Liu, S. Wang, G. Shen and X. Yan, Supramolecular Nanodrugs Based on Covalent Assembly of Therapeutic Peptides toward in Vitro Synergistic Anticancer Therapy, ChemMedChem, 2021, 16, 2381–2385 CrossRef CAS PubMed.
- A. Song, T. Ding, N. Wei, J. Yang, M. Ma, S. Zheng and H. Jin, Schisandrin B Induces HepG2 Cells Pyroptosis by Activating NK Cells Mediated Anti-Tumor Immunity, Toxicol. Appl. Pharmacol., 2023, 472, 116574 CrossRef CAS PubMed.
- Y. Huang, R. Liu, Y. Wang, G. Liu, C. Wang, X. Chen, Y. Jia and J. Shen, Evaluation of Pharmacokinetic Interactions between the New SGLT2 Inhibitor SHR3824 and Valsartan in Healthy Chinese Volunteers, Clin. Ther., 2022, 44, 945–956 CrossRef CAS PubMed.
- D. Ding, X. Shen, L. Yu, Y. Zheng, Y. Liu, W. Wang, L. Liu, Z. Zhao, S. Nian and L. Liu, Timosaponin BII Inhibits TGF-β Mediated Epithelial-Mesenchymal Transition through Smad-Dependent Pathway During Pulmonary Fibrosis, Phytother. Res., 2023, 37, 2787–2799 CrossRef CAS PubMed.
- J. Hao, J. Bei, Z. Li, M. Han, B. Ma, P. Ma and X. Zhou, Qing'E Pill Inhibits Osteoblast Ferroptosis via ATM Serine/Threonine Kinase (ATM) and the PI3K/AKT Pathway in Primary Osteoporosis, Front. Pharmacol., 2022, 13, 902102 CrossRef CAS PubMed.
- D. Wang, X. Li, G. Gong, Y. Lu, Z. Guo, R. Chen, H. Huang, Z. Li and J. Bian, An Updated Patent Review of Glutaminase Inhibitors (2019–2022), Expert Opin. Ther. Pat., 2023, 33, 17–28 CrossRef CAS PubMed.
- X. Wang, C. Shen, X. Wang, J. Tang, Z. Wu, Y. Huang, W. Shao, K. Geng, H. Xie and Z. Pu, Schisandrin Protects against Ulcerative Colitis by Inhibiting the SGK1/NLRP3 Signaling Pathway and Reshaping Gut Microbiota in Mice, Chinese Med., 2023, 18, 112 CrossRef CAS PubMed.
- Y.-Y. Chen, X.-Z. Wang, Q.-Y. Hu and L.Y. Zhou, Self-Emulsifying System Co-Loaded with Paclitaxel and Coix Seed Oil Deeply Penetrated to Enhance Efficacy in Cervical Cancer, Curr. Drug Deliv., 2023, 20, 919–926 CrossRef CAS PubMed.
- Z.-S. Tao, W.-S. Zhou, H.-G. Xu and M. Yang, Simvastatin Can Enhance the Osseointegration of Titanium Rods in Ovariectomized Rats Maintenance Treatment with Valproic Acid, Biomed. Pharmacother., 2020, 132, 110745 CrossRef CAS PubMed.
- T. Tang, S.-N. Wang, T.-Y. Cai, H. Tao, Q. Zhang, S.-M. Qi and Z.-L. Qi, Aloin Inhibits the Proliferation and Migration of Gastric Cancer Cells by Regulating NOX2-ROS-Mediated Pro-Survival Signal Pathways, Drug Des. Dev. Ther., 2020, 14, 145–155 CrossRef PubMed.
- Y.-J. Xing, B.-H. Liu, S.-J. Wan, Y. Cheng, S.-M. Zhou, Y. Sun, X.-M. Yao, Q. Hua, X.-J. Meng, J.-H. Cheng, M. Zhong, Y. Zhang, K. Lv and X. Kong, A SGLT2 Inhibitor Dapagliflozin Alleviates Diabetic Cardiomyopathy by Suppressing High Glucose-Induced Oxidative Stress in vivo and in vitro, Front. Pharmacol., 2021, 12, 708177 CrossRef CAS PubMed.
- S. Sun, S. Li, Y. Du, C. Wu, M. Zhang, J. Li and X. Zhang, Anti-Inflammatory Effects of the Root, Stem and Leaf Extracts of Chloranthus Serratus on Adjuvant-Induced Arthritis in Rats, Pharm. Biol., 2020, 58, 528–537 CrossRef CAS PubMed.
- J. Zhang, W. Zhou, Y. Chen, Y. Wang, Z. Guo, W. Hu, Y. Li, X. Han and S. Si, Small Molecules Targeting Pin1 as Potent Anticancer Drugs, Front. Pharmacol., 2023, 14, 1073037 CrossRef CAS PubMed.
- H. Hong, Q. Zou, Y. Liu, S. Wang, G. Shen and X. Yan, Supramolecular Nanodrugs Based on Covalent Assembly of Therapeutic Peptides toward in vitro Synergistic Anticancer Therapy, ChemMedChem, 2021, 16, 2381–2385 CrossRef CAS PubMed.
- S. Zhao, L. Ma, Z. Chu, H. Xu, W. Wu and F. Liu, Regulation of Microglial Activation in Stroke, Acta Pharm. Sin., 2017, 38, 445–458 CrossRef CAS PubMed.
- X.-F. Wang, W. Lei, C.-M. Liu, J. Yang and Y.-H. Zhu, BOLA3 is a Prognostic-Related Biomarker and Correlated with Immune Infiltrates in Lung Adenocarcinoma, Int. Immunopharmacol., 2022, 107, 108652 CrossRef CAS PubMed.
- J. Liu, X. Zhao, F. Qin, J. Zhou, F. Ding, G. Zhou, X. Zhang, Z. Zhang and Z. Li, Isoliquiritigenin Mitigates Oxidative Damage after Ssubarachnoid Hemorrhage in vivo and in vitro by Regulating Nrf2-Dependent Signaling Pathway via Targeting of SIRT1, Phytomedicine, 2022, 105, 154262 CrossRef CAS PubMed.
- X. Wang, X. Wang, H. Yao, C. Shen, K. Geng and H. Xie, A Comprehensive Review on Schisandrin and Its Pharmacological Features, Naunyn-Schmiedeberg's Arch. Pharmacol., 2024, 397, 783–794 CrossRef CAS PubMed.
- L. Zha, L. Pan, J. Guo, N. French, E.V. Villanueva and B. Tefsen, Effectiveness and Safety of High Dose Tigecycline for the Treatment of Severe Infections: A Systematic Review and Meta-Analysis, Adv. Ther., 2020, 37, 1049–1064 CrossRef CAS PubMed.
- W. Zhou, K. Yang, J. Zeng, X. Lai, X. Wang, C. Ji, Y. Li, P. Zhang and S. Li, FordNet: Recommending Traditional Chinese Medicine Formula via Deep Neural Network Integrating Phenotype and Molecule, Pharmacol. Res., 2021, 173, 105752 CrossRef CAS PubMed.
- D. Liang, J. Shen, Y. Jia, M. Dai, X. Li, L. Zhou, W. Wang, B. Yang, J. Shao, Y. Jiang, H. Xie and H. Sun, Pharmacokinetic Properties of S-oxiracetam after Single and Multiple Intravenous Infusions in Healthy Volunteers, Eur. J. Drug Metab. Pharmacokinet., 2021, 46, 793–805 CrossRef CAS PubMed.
- Y. Chen, S. Wang, Q. Hu and L. Zhou, Self-Emulsifying System Co-Loaded with Paclitaxel and Coix Seed Oil Deeply Penetrated to Enhance Efficacy in Cervical Cancer, Curr. Drug Deliv., 2023, 20, 919–926 CrossRef CAS PubMed.
- H. Wang, Y. Chen, L. Wang, Q. Liu, S. Yang and C. Wang, Advancing Herbal Medicine: Enhancing Product Quality and Safety through Robust Quality Control Practices, Front. Pharmacol., 2023, 14, 1265178 CrossRef PubMed.
- T. A. Palazzo, R. Mose and K. A. Jørgensen, Cycloaddition Reactions: Why Is It So Challenging to Move from Six to Ten Electrons?, Angew. Chem., Int. Ed., 2017, 56, 10033–10038 CrossRef CAS PubMed.
- J. H. Rigby and M. Fleming, Construction of the Ingenane Core Using an Fe(III) or Ti(IV) Lewis Acid-Catalyzed Intramolecular [6 + 4] Cycloaddition, Tetrahedron Lett., 2002, 43, 8643–8646 CrossRef CAS.
- M. Xie, X. Liu, X. Wu, Y. Cai, L. Lin and X. Feng, Catalytic Asymmetric [8 + 2] Cycloaddition: Synthesis of Cycloheptatriene-Fused Pyrrole Derivatives, Angew. Chem., Int. Ed., 2013, 52, 5604–5607 CrossRef CAS PubMed.
- Y. Hayashi, H. Gotoh, M. Honma, K. Sankar, I. Kumar, H. Ishikawa, K. Konno, H. Yui, S. Tsuzuki and T. Uchimaru, Organocatalytic, Enantioselective Intramolecular [6 + 2] Cycloaddition Reaction for the Formation of Tricyclopentanoids and Insight on Its Mechanism from a Computational Study, J. Am. Chem. Soc., 2011, 133, 20175–20185 CrossRef CAS PubMed.
- P. Yu, C. Q. He, A. Simon, W. Li, R. Mose, M. K. Thøgersen, K. A. Jørgensen and K. N. Houk, Organocatalytic [6 + 4] Cycloadditions Via Zwitterionic Intermediates: Chemo-, Regio-, and Stereoselectivities, J. Am. Chem. Soc., 2018, 140, 13726–13735 CrossRef CAS PubMed.
- Z. Zhou, Z.-X. Wang, Y.-C. Zhou, W. Xiao, Q. Ouyang, W. Du and Y.-C. Chen, Switchable Regioselectivity in Amine-Catalysed Asymmetric Cycloadditions, Nat. Chem., 2017, 9, 590–594 CrossRef CAS PubMed.
- L. Dell'Amico, G. Rassu, V. Zambrano, A. Sartori, C. Curti, L. Battistini, G. Pelosi, G. Casiraghi and F. Zanardi, Exploring the Vinylogous Reactivity of Cyclohexenylidene Malononitriles: Switchable Regioselectivity in the Organocatalytic Asymmetric Addition to Enals Giving Highly Enantioenriched Carbabicyclic Structures, J. Am. Chem. Soc., 2014, 136, 11107–11114 CrossRef PubMed.
- C. Guo, B. Sahoo, C. G. Daniliuc and F. Glorius, N-Heterocyclic Carbene Catalyzed Switchable Reactions of Enals with Azoalkenes: Formal [4 + 3] and [4 + 1] Annulations for the Synthesis of 1,2-Diazepines and Pyrazoles, J. Am. Chem. Soc., 2014, 136, 17402–17405 CrossRef CAS PubMed.
- C. Guo, M. Fleige, D. Janssen-Müller, C. G. Daniliuc and F. Glorius, Switchable Selectivity in an NHC-Catalysed Dearomatizing Annulation Reaction, Nat. Chem., 2015, 7, 842–847 CrossRef CAS PubMed.
- R. Mose, G. Preegel, J. Larsen, S. Jakobsen, E. H. Iversen and K. A. Jørgensen, Organocatalytic Stereoselective [8 + 2] and [6 + 4] Cycloadditions, Nat. Chem., 2017, 9, 487–492 CrossRef CAS PubMed.
- N. F. Firrell and P. W. Hickmott, Enamine Chemistry. Part VI. Structure and Proton Magnetic Resonance Spectra of Dienamines, J. Chem. Soc. B, 1969, 293–298, 10.1039/j29690000293.
- G. Bertuzzi, M. K. Thøgersen, M. Giardinetti, A. Vidal-Albalat, A. Simon, K. N. Houk and K. A. Jørgensen, Catalytic Enantioselective Hetero-[6 + 4] and -[6 + 2] Cycloadditions for the Construction of Condensed Polycyclic Pyrroles, Imidazoles, and Pyrazoles, J. Am. Chem. Soc., 2019, 141, 3288–3297 CrossRef CAS PubMed.
- D. McLeod, A. Cherubini-Celli, N. Sivasothirajah, C. H. McCulley, M. L. Christensen and K. A. Jørgensen, Enantioselective 1,3-Dipolar [6 + 4] Cycloaddition of Pyrylium Ions and Fulvenes Towards Cyclooctanoids, Chem. – Eur. J., 2020, 26, 11417–11422 CrossRef CAS PubMed.
- B. S. Donslund, A. Monleón, T. A. Palazzo, M. L. Christensen, A. Dahlgaard, J. D. Erickson and K. A. Jørgensen, Organocatalytic Enantioselective Higher-Order Cycloadditions of in Situ Generated Amino Isobenzofulvenes, Angew. Chem., Int. Ed., 2018, 57, 1246–1250 CrossRef CAS PubMed.
- K. Hafner and W. Bauer, Isobenzofulvenes, Angew. Chem., Int. Ed. Engl., 1968, 7, 297–299 CrossRef CAS.
- B. S. Donslund, N. I. Jessen, G. Bertuzzi, M. Giardinetti, T. A. Palazzo, M. L. Christensen and K. A. Jørgensen, Catalytic Enantioselective [10 + 4] Cycloadditions, Angew. Chem., Int. Ed., 2018, 57, 13182–13186 CrossRef CAS PubMed.
- J. Zhao, X. Zheng, Y.-S. Gao, J. Mao, S.-X. Wu, W.-L. Yang, X. Luo and W.-P. Deng, Organocatalytic Enantioselective [8 + 4] Cycloadditions of Isobenzofulvenes for the Construction of Bicyclo[4.2.1]Nonanes, Chin. J. Chem., 2021, 39, 3219–3224 CrossRef CAS.
- D. McLeod, J. A. Izzo, D. K. B. Jørgensen, R. F. Lauridsen and K. A. Jørgensen, Development and Investigation of an Organocatalytic Enantioselective [10 + 2] Cycloaddition, ACS Catal., 2020, 10, 10784–10793 CrossRef CAS.
- N. I. Jessen, G. Bertuzzi, M. Bura, M. L. Skipper and K. A. Jørgensen, Enantioselective Construction of the Cycl[3.2.2]Azine Core via Organocatalytic [12 + 2] Cycloadditions, J. Am. Chem. Soc., 2021, 143, 6140–6151 CrossRef CAS PubMed.
- G. Bertuzzi, V. Corti, J. A. Izzo, S. Ričko, N. I. Jessen and K. A. Jørgensen, Organocatalytic Enantioselective Construction of Conformationally Stable C(Sp2)–C(Sp3) Atropisomers, J. Am. Chem. Soc., 2022, 144, 1056–1065 CrossRef CAS PubMed.
- S. Frankowski, A. Skrzyńska and Ł. Albrecht, Inverting the Reactivity of Troponoid Systems in Enantioselective Higher-Order Cycloaddition, Chem. Commun., 2019, 55, 11675–11678 RSC.
- N. I. Jessen, M. Bura, G. Bertuzzi and K. A. Jørgensen, Aminocatalytic [8 + 2] Cycloaddition Reactions toward Chiral Cyclazines, Angew. Chem., Int. Ed., 2021, 60, 18527–18531 CrossRef CAS PubMed.
- G. Yang, Z. Li, Y. Liu, D. Guo, X. Sheng and J. Wang, Organocatalytic Higher-Order [8 + 2] Cycloaddition for the Assembly of Atropoenantiomeric 3-Arylindolizines, Org. Lett., 2021, 23, 8109–8113 CrossRef CAS PubMed.
- S. Wang, C. Rodríguez-Escrich and M. A. Pericàs, Catalytic Asymmetric [8 + 2] Annulation Reactions Promoted by a Recyclable Immobilized Isothiourea, Angew. Chem., Int. Ed., 2017, 56, 15068–15072 CrossRef CAS PubMed.
- C. He, Z. Li, H. Zhou and J. Xu, Stereoselective [8 + 2] Cycloaddition Reaction of Azaheptafulvenes with α-Chloro Aldehydes via N-Heterocyclic Carbene Catalysis, Org. Lett., 2019, 21, 8022–8026 CrossRef CAS PubMed.
- S. Wang, C. Rodríguez-Escrich, M. Fianchini, F. Maseras and M. A. Pericàs, Diastereodivergent Enantioselective [8 + 2] Annulation of Tropones and Enals Catalyzed by N-Heterocyclic Carbenes, Org. Lett., 2019, 21, 3187–3192 CrossRef CAS PubMed.
- X. Yang, G. Luo, L. Zhou, B. Liu, X. Zhang, H. Gao, Z. Jin and Y. R. Chi, Enantioselective Indole N–H Functionalization Enabled by Addition of Carbene Catalyst to Indole Aldehyde at Remote Site, ACS Catal., 2019, 9, 10971–10976 CrossRef CAS.
- L. Zhang and Y. Wang, NHC-Catalyzed NH Functionalization/Cycloaddition Reaction of Indole Aldehyde and Ketone: A Dft Perspective, Comput. Theor. Chem., 2023, 1220, 114007 CrossRef CAS.
- Y. Liu, G. Luo, X. Yang, S. Jiang, W. Xue, Y. R. Chi and Z. Jin, Carbene-Catalyzed Enantioselective Aromatic N-Nucleophilic Addition of Heteroarenes to Ketones, Angew. Chem., Int. Ed., 2020, 59, 442–448 CrossRef CAS PubMed.
- C. Wang, Z. Li, J. Zhang and X.-P. Hui, Asymmetric N-Alkylation of Indoles with Isatins Catalyzed by N-Heterocyclic Carbene: Efficient Synthesis of Functionalized Cyclic N,O-Aminal Indole Derivatives, Org. Chem. Front., 2020, 7, 1647–1652 RSC.
- Q. Peng, S. J. Li, B. Zhang, D. Guo, Y. Lan and J. Wang, N-Heterocyclic Carbene-Catalyzed Enantioselective Hetero-[10 + 2] Annulation, Commun. Chem., 2020, 3, 177 CrossRef CAS PubMed.
- K. Balanna, K. Madica, S. Mukherjee, A. Ghosh, T. Poisson, T. Besset, G. Jindal and A. T. Biju, N-Heterocyclic Carbene-Catalyzed Formal [6 + 2] Annulation Reaction via Cross-Conjugated Aza-Trienolate Intermediates, Chem. – Eur. J., 2020, 26, 818–822 CrossRef CAS PubMed.
- X. Yang, Y. Xie, J. Xu, S. Ren, B. Mondal, L. Zhou, W. Tian, X. Zhang, L. Hao, Z. Jin and Y. R. Chi, Carbene-Catalyzed Activation of Remote Nitrogen Atoms of (Benz)Imidazole-Derived Aldimines for Enantioselective Synthesis of Heterocycles, Angew. Chem., Int. Ed., 2021, 60, 7906–7912 CrossRef CAS PubMed.
- X. Chen, S. Yang, B.-A. Song and Y. R. Chi, Functionalization of Benzylic C(Sp3)-H Bonds of Heteroaryl Aldehydes through N-Heterocyclic Carbene Organocatalysis, Angew. Chem., Int. Ed., 2013, 52, 11134–11137 CrossRef CAS PubMed.
- D. Janssen-Müller, S. Singha, T. Olyschläger, C. G. Daniliuc and F. Glorius, Annulation of o-Quinodimethanes through N-Heterocyclic Carbene Catalysis for the Synthesis of 1-Isochromanones, Org. Lett., 2016, 18, 4444–4447 CrossRef PubMed.
- X. Chen, H. Wang, K. Doitomi, C. Y. Ooi, P. Zheng, W. Liu, H. Guo, S. Yang, B.-A. Song, H. Hirao and Y. R. Chi, A Reaction Mode of Carbene-Catalysed Aryl Aldehyde Activation and Induced Phenol OH Functionalization, Nat. Commun., 2017, 8, 15598 CrossRef PubMed.
- A. Lee, J. L. Zhu, T. Feoktistova, A. C. Brueckner, P. H. Y. Cheong and K. A. Scheidt, Carbene-Catalyzed Enantioselective Decarboxylative Annulations to Access Dihydrobenzoxazinones and Quinolones, Angew. Chem., Int. Ed., 2019, 58, 5941–5945 CrossRef CAS PubMed.
- H. Ji, C. Mou, J. Zou, Y. Liu, S.-C. Ren and Y. R. Chi, NHC-Catalyzed [12 + 2] Reaction of Polycyclic Arylaldehydes for Access to Indole Derivatives, Chem. Commun., 2023, 59, 6351–6354 RSC.
- Z.-Z. Gao, C. Wang, L.-J. Zhou, C.-H. Yuan, Y.-M. Xiao and H.-C. Guo, Allenoates and Its Asymmetric Variant: Construction of Bicyclo[5.3.0]decane Scaffold, Org. Lett., 2018, 20, 4302–4305 CrossRef CAS PubMed.
- I. Kallweit and C. Schneider, Brønsted Acid Catalyzed [6 + 2]-Cycloaddition of 2-Vinylindoles with in Situ Generated 2-Methide-2H-Pyrroles: Direct, Catalytic, and Enantioselective Synthesis of 2,3-Dihydro-1H-Pyrrolizines, Org. Lett., 2019, 21, 519–523 CrossRef CAS PubMed.
- V. Nair and K. G. Abhilash, [8 + 2] Cycloaddition Reactions in Organic Synthesis, Synlett, 2008, 301–312 CrossRef CAS.
- B. M. Trost, P. J. McDougall, O. Hartmann and P. T. Wathen, Asymmetric Synthesis of Bicyclo[4.3.1]Decadienes and Bicyclo[3.3.2]Decadienes via [6 + 3] Trimethylenemethane Cycloaddition with Tropones, J. Am. Chem. Soc., 2008, 130, 14960–14961 CrossRef CAS PubMed.
- B. M. Trost and P. J. McDougall, Access to a Welwitindolinone Core Using Sequential Cycloadditions, Org. Lett., 2009, 11, 3782–3785 CrossRef CAS PubMed.
- R. Tejero, A. Ponce, J. Adrio and J. C. Carretero, Ni-Catalyzed [8 + 3] Cycloaddition of Tropones with 1,1-Cyclopropanediesters, Chem. Commun., 2013, 49, 10406–10408 RSC.
- H. Liu, Y. Wu, Y. Zhao, Z. Li, L. Zhang, W. Yang, H. Jiang, C. Jing, H. Yu, B. Wang, Y. Xiao and H. Guo, Metal-Catalyzed [6 + 3] Cycloaddition of Tropone with Azomethine Ylides: A Practical Access to Piperidine-Fused Bicyclic Heterocycles, J. Am. Chem. Soc., 2014, 136, 2625–2629 CrossRef CAS PubMed.
- H.-L. Teng, L. Yao and C.-J. Wang, Cu(I)-Catalyzed Regio- and Stereoselective [6 + 3] Cycloaddition of Azomethine Ylides with Tropone: An Efficient Asymmetric Access to Bridged Azabicyclo[4.3.1]Decadienes, J. Am. Chem. Soc., 2014, 136, 4075–4080 CrossRef CAS PubMed.
- J. Zhang, W. Xiao, H. Hu, L. Lin, X. Liu and X. Feng, Catalytic Asymmetric [8 + 3] Annulation Reactions of Tropones or Azaheptafulvenes with Meso-Aziridines, Chem. – Eur. J., 2018, 24, 13428–13431 CrossRef CAS PubMed.
- G. Bertuzzi, D. McLeod, L.-M. Mohr and K. A. Jørgensen, Organocatalytic Enantioselective 1,3-Dipolar [6 + 4] Cycloadditions of Tropone, Chem. – Eur. J., 2020, 26, 15491–15496 CrossRef CAS PubMed.
- R. Manzano, A. Romaniega, L. Prieto, E. Díaz, E. Reyes, U. Uria, L. Carrillo and J. L. Vicario, γ-Substituted Allenic Amides in the Phosphine-Catalyzed Enantioselective Higher Order Cycloaddition with Azaheptafulvenes, Org. Lett., 2020, 22, 4721–4725 CrossRef CAS PubMed.
- Y. Yang, Y. Jiang, W. Du and Y.-C. Chen, Asymmetric Cross [10 + 2] Cycloadditions of 2-Alkylidene-1-Indanones and Activated Alkenes under Phase-Transfer Catalysis, Chem. – Eur. J., 2020, 26, 1754–1758 CrossRef CAS PubMed.
- J.-Q. Zhao, S. Zhou, H.-L. Qian, Z.-H. Wang, Y.-P. Zhang, Y. You and W.-C. Yuan, Higher-Order [10 + 2] Cycloaddition of 2-Alkylidene-1-Indanones Enables the Dearomatization of 3-Nitroindoles: Access to Polycyclic Cyclopenta[b]Indoline Derivatives, Org. Chem. Front., 2022, 9, 3322–3327 RSC.
- X.-C. Yu, C.-C. Zhang, L.-T. Wang, J.-Z. Li, T. Li and W.-T. Wei, The Synthesis of Seven- and Eight-Membered Rings by Radical Strategies, Org. Chem. Front., 2022, 9, 4757–4781 RSC.
| This journal is © the Partner Organisations 2024 |