
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced copper-catalyzed alkoxycarbonylation of alkyl fluorides†
Peng
Yang
a,
Yan-Hua
Zhao
a and
Xiao-Feng
Wu
 *ab
*ab
aLeibniz-Institut für Katalyse e.V, Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: xiao-feng.wu@catalysis.de
bDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, Liaoning, China
First published on 6th March 2024
Abstract
As the strongest σ bond in organic chemistry, the functionalization of the carbon–fluoride bond (BDE = 485 kJ mol−1) remains a significant challenge. Herein, we developed a carbonylation procedure that utilizes a copper catalyst and visible light irradiation to enable the production of esters from alkyl fluorides and phenols for the first time. From our mechanistic studies, the use of magnesium iodide as an additive was found to be crucial for the success of this protocol which drives the halide exchange and removes fluoride from the reaction.
Ester compounds are of great importance as they are found in numerous natural products, pharmaceuticals, polymers, synthetic intermediates, etc.1 Traditionally, esters are mainly synthesized from the esterification between the corresponding carboxylic acid derivatives and alcohols. However, the requirement of strongly alkaline conditions or/and a stoichiometric amount of activators restricts the application of this type of reaction.
On the other hand, using carbon monoxide as a cheap and readily available C1 source, the carbonylation reaction has become a powerful and dominant tool for producing various carbonylated compounds in a highly atom-economical manner.2 In this area, the carbonylation reaction of organic halides has received considerable attention since 1963.3 Generally, catalytic methods for carbonylation of organic halides proceed in two possible pathways:4 (i) transition metal-catalyzed carbonylation of sp2–X bonds and benzylic halides via an ionic pathway (Scheme 1a (1)); Morikawa, Fuchikami, Beller and many other research groups developed various novel protocols for the carbonylation of aryl, allyl, benzyl, and perfluoroalkyl halides with alcohols.5 The procedure applicable for alkyl halides is still very limited due to the barrier in oxidative addition and fast β-hydride elimination of the alkyl metal species.6 Recently, (ii) radical carbonylation became a new and alternative way to resolve these problems (Scheme 1a (2)).7 In 1988, Watanabe and co-workers developed a transition metal carbonyl complex-catalyzed radical carbonylation of alkyl iodide under visible light irradiation.8 Subsequently, Ryu's group reported a visible light-induced Pd-catalyzed radical carbonylation reaction of alkyl iodide with alcohols.9 In 2016, Alexanian and co-workers developed a Pd-catalyzed radical alkoxycarbonylation of alkyl bromides using the NHC ligand.10a More recently, Beller's group disclosed the Rh-catalyzed radical carbonylation of primary alkyl chlorides with sodium iodide as the additive.10b Arndtsen and co-workers achieved novel catalytic systems for the carbonylative transformations of organic halides by the combination of a palladium catalyst and visible light.11 In recent years, our group has also been interested in the metal-catalyzed carbonylation of alkyl halides under mild conditions, and various transition metal catalysts (Ru, Rh, Cu, and Fe) were studied.12 However, for the carbonylation of alkyl fluorides, which have the highest bond dissociation energy (485 kJ mol−1; Scheme 1b),13,14 no procedure yet has been reported.
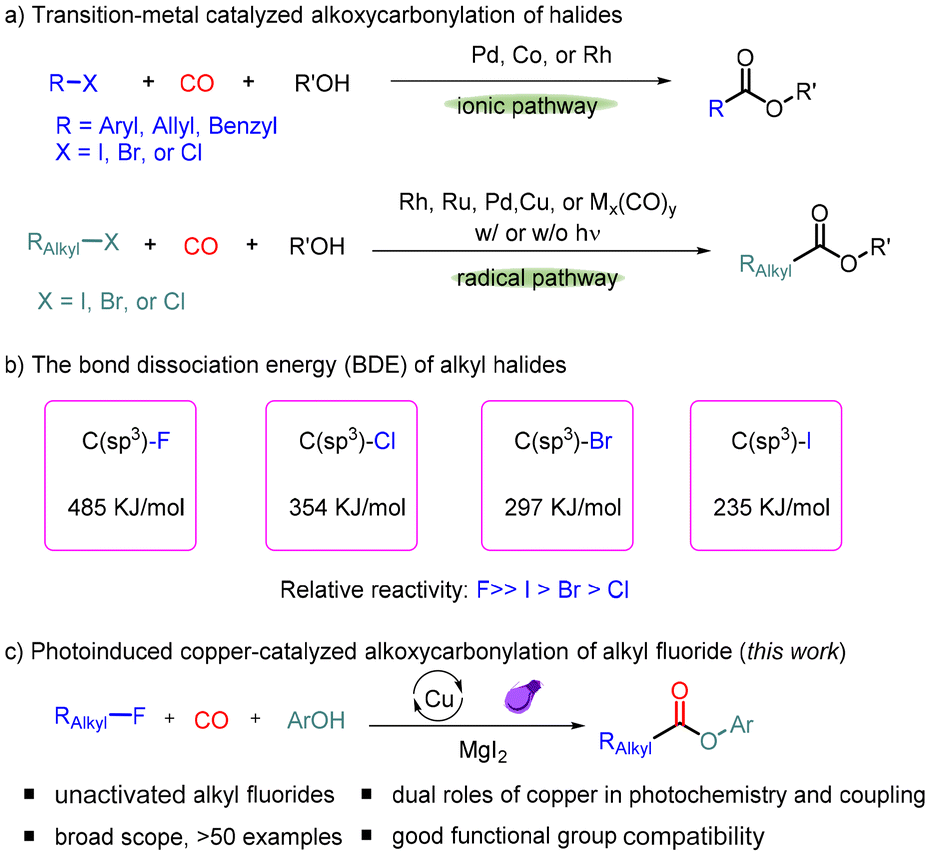 | ||
| Scheme 1 Alkoxycarbonylation of alkyl halides. | ||
Herein, we present the successful alkoxycarbonylation of alkyl fluorides with phenols under light irradiation catalyzed by a copper catalyst (Scheme 1c). The addition of magnesium iodide was found to be crucial for the success of this transformation, which not only generates more reactive species in situ via halogen exchange but also avoids the influence of fluoride ions in the reaction solution on the subsequent radical course. A wide range of valuable esters were produced in high yields with excellent functional group adaptations.
Our initial studies were carried out with 1-fluorooctane (1a) and phenol (2a) as the model substrates to examine the reaction parameters (Table 1 and also see the ESI, Tables S1–S5†). After a series of studies, we were pleased to find that the desired reaction indeed worked smoothly at 80 °C, when employing a catalytic system consisting of Cu(CH3CN)4PF6 (10 mol%) and Xantphos (11 mol%) in the presence of 2.5 equivalents of K2CO3 and 1.5 equivalents of MgI2, and the desired product 3a was formed in 95% NMR yield (Table 1, entry 1). Control experiments, without light irradiation or the use of blue light, confirmed that the reaction proceeded through a photocatalytic process (Table 1, entries 2 and 3). No reaction occurred at room temperature and the alkyl fluoride remained unreacted (Table 1, entry 4). The use of other metal iodides such as LiI and CaI2 instead of MgI2 resulted in no reaction despite the detection of 1-iodooctane (Table 1, entries 5 and 6). Notably, the valence of the copper salt is important for the onset of the reaction as well; Cu1+ could initiate the alkoxycarbonylation, whereas no product could be found when Cu2+ was used or in the absence of a copper salt (Table 1, entries 7–9). We then investigated other bidentate phosphine ligands and an obvious effect on the yield of the final product was found, with Xantphos still being the best ligand of choice (Table 1, entries 10 and 11). The effects of bases were tested, and poor yields were obtained with either other inorganic bases or organic bases (Table 1, entries 12–14). Moreover, we also found that the reaction appeared to be sensitive to the properties of solvents; a strongly polar solvent completely inhibited the formation of the targeted ester, and a non-polar or weakly polar solvent ensured the proceeding of the desired alkoxycarbonylation, with anisole being the best solvent (Table 1, entries 15 and 16).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | 3a yield (%) |
| a Standard conditions: 1a (0.2 mmol, 2.0 eq.), 2a (0.1 mmol, 1.0 eq.), MgI2 (0.15 mmol, 1.5 eq.), Cu(CH3CN)4PF6 (10 mol%), Xantphos (11 mol%), K2CO3 (0.25 mmol, 2.5 eq.), CO (40 bar), anisole (1 mL), stirred at 80 °C for 24 h under irradiation (320–400 nm); yields were determined by 1H NMR with 1,3,5-trimethoxybenzene as the internal standard. b Isolated yield. | ||
| 1 | None | 95 (92)b |
| 2 | No light | 0 |
| 3 | Blue light | 0 |
| 4 | r.t. | 0 |
| 5 | LiI instead of MgI2 | 0 |
| 6 | CaI2 instead of MgI2 | 0 |
| 7 | No Cu(CH3CN)4PF6 | 0 |
| 8 | CuI instead of Cu(CH3CN)4PF6 | 15 |
| 9 | Cu(OAc)2 instead of Cu(CH3CN)4PF6 | 0 |
| 10 | BINAP instead of Xantphos | <5 |
| 11 | Niphos instead of Xantphos | 16 |
| 12 | Na2CO3 instead of K2CO3 | <5 |
| 13 | Cs2CO3 instead of K2CO3 | 33 |
| 14 | DBU instead of K2CO3 | 0 |
| 15 | CH3CN as solvent | 0 |
| 16 | Toluene as solvent | 58 |
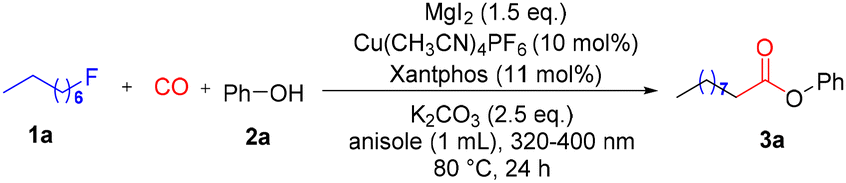
With the optimized reaction conditions in hand, we investigated the substrate scope of this reaction, and the results are shown in Scheme 2. At the first stage, a range of phenols with electron-donating groups or electron-withdrawing groups could serve as effective partners, providing the desired esters in good to excellent yields. Versatile functional groups, such as alkyl (3b–3d, 3o), phenyl (3e), halogen (3f–3i), trifluoromethyl (3j), cyano (3k), alkoxyl (3l), alkylthio (3m), alkylsulfonyl (3n), and Bpin (3p), were all tolerated well in the reaction system with high yields. Subsequently, the steric hindrance effect was tested. Phenols with different groups substituted at the meta-position were used, and the desired meta-substituted phenol esters were obtained (3q–3s). Phenols bearing ortho-substituents gave the desired esters in moderate to good yields as well irrespective of the steric hindrance effect (3t–3w). Furthermore, phenols possessing di-substituents still reacted well and gave the products in good yields (3x–3z, 4a, and 4b). The natural product sesamol can gave the corresponding product with excellent yield in this reaction (4c). Next, we investigated the reaction with naphthols, and 2-naphthol gave a better yield than 1-naphthol (4d and 4e). Aliphatic alcohol was also tested but was proven to be an unsuitable reaction partner, which gave the corresponding product in only 22% yield (4f). Remarkably, an interesting product was formed when we used 4-hydroxybenzyl alcohol as the substrate. The 4-hydroxybenzyl alcohol reacted with anisole first through electrophilic aromatic substitution,15 and then alkoxycarbonylation occurred to give the final product (4g) in 80% yield.
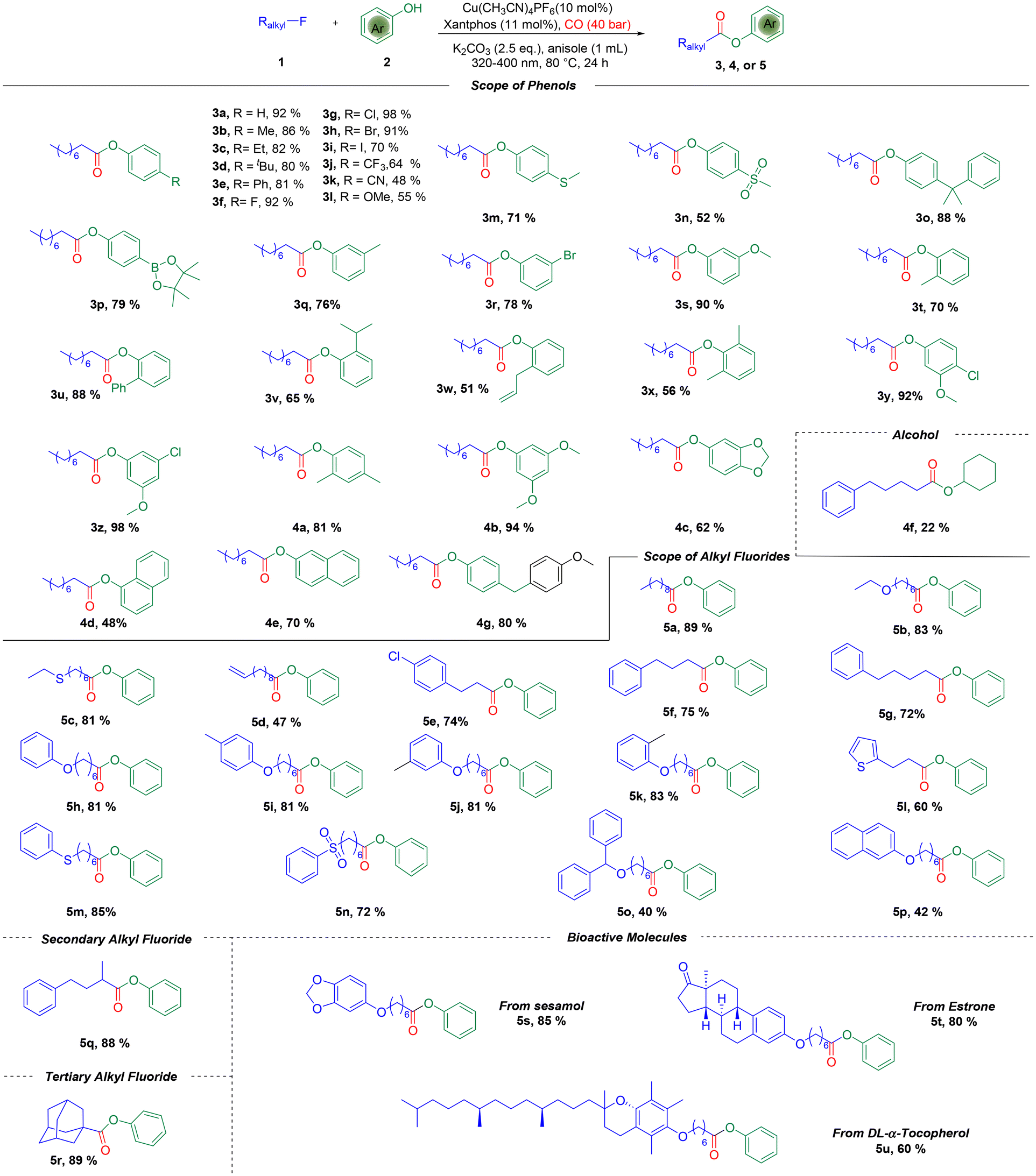 | ||
| Scheme 2 Reaction scope. Reaction conditions: alkyl fluorides 1 (0.2 mmol, 2eq.), phenols 2 (0.1 mmol, 1.0 eq.), Cu(CH3CN)4PF6 (10 mol%), Xantphos (11 mol%), MgI2 (1.5 eq.), K2CO3 (2.5 eq.), anisole (1 mL), CO (40 bar), 80 °C, irradiation (320–400 nm), 24 h. | ||
Subsequently, the scope of different types of alkyl fluorides was examined (5a–5u). The alkyl fluoride with a long carbon chain also gave an excellent yield (5a), and functional groups such as ether (5b), thioether (5c), alkenyl (5d), and alkyl groups (5e–5g) were all well tolerated. Phenoxy fluorides with different substituents also gave excellent yields (5h–5k). Meanwhile, heterocycle (5l), phenthio (5m), phensulfonyl (5n), and naphthyloxy (5p) groups were all compatible under the standard conditions to give the desired esters in moderate to good yields. It is noteworthy that the reaction was also extended to the alkoxycarbonylation of secondary and tertiary alkyl fluorides, with the corresponding esters being delivered smoothly (5q and 5r). Importantly, the alkoxycarbonylation can incorporate an ester fragment into natural products, while the alkyl fluoride derivatives from sesamol, estrone, and DL-α-tocopherol could also be converted into the corresponding alkoxycarbonylation products in 60–85% yields (5s–5u).
Control studies were also carried out to explore the reaction mechanism. At first, we studied the conversion of alkyl fluoride to alkyl iodide, and the fluoride/iodide exchange reaction proceeded with an excellent yield at 80 °C (Scheme 3a-I). To gain more insight into the mechanism and to identify the rate-determining step of the reaction, we studied the kinetic profile of the halogen exchange reaction. As shown in Fig. 1, in the first 2 h, 70% of 1-fluorooctane was converted. Then, the conversion rate gradually slowed down, and conversion was achieved in 85% yield after 24 h. The above results indicate that the rate-determining step of our reaction is not halogen exchange. Subsequently, alkyl iodide was tested in this reaction. The reaction of 1b at room temperature or under heating all gave the desired product in good yields (Scheme 3a-II), which means heating is not necessary for the proceeding of alkoxycarbonylation. For a better understanding of the role of magnesium iodide in this reaction, reactions with alkyl iodide and metal fluoride were performed under our standard conditions. The results show that magnesium fluoride has no effect on the carbonylation (Scheme 3a-III). However, when using other fluorides as additives, the reaction gave no desired ester product but converted back to alkyl fluoride (Scheme 3a-IV). More specifically, halogen exchange is a reversible reaction, and in the presence of fluoride ions, alkyl iodide will preferentially undergo halogen exchange to the more stable alkyl fluoride rather than carbonylation. Thanks to the ultra-low solubility of magnesium fluoride in anisole, our standard reaction is essentially free of fluoride ions, which is the key to the successful achievement of the reaction with good yields of the desired ester products.
 | ||
| Scheme 3 Mechanistic experiments. | ||
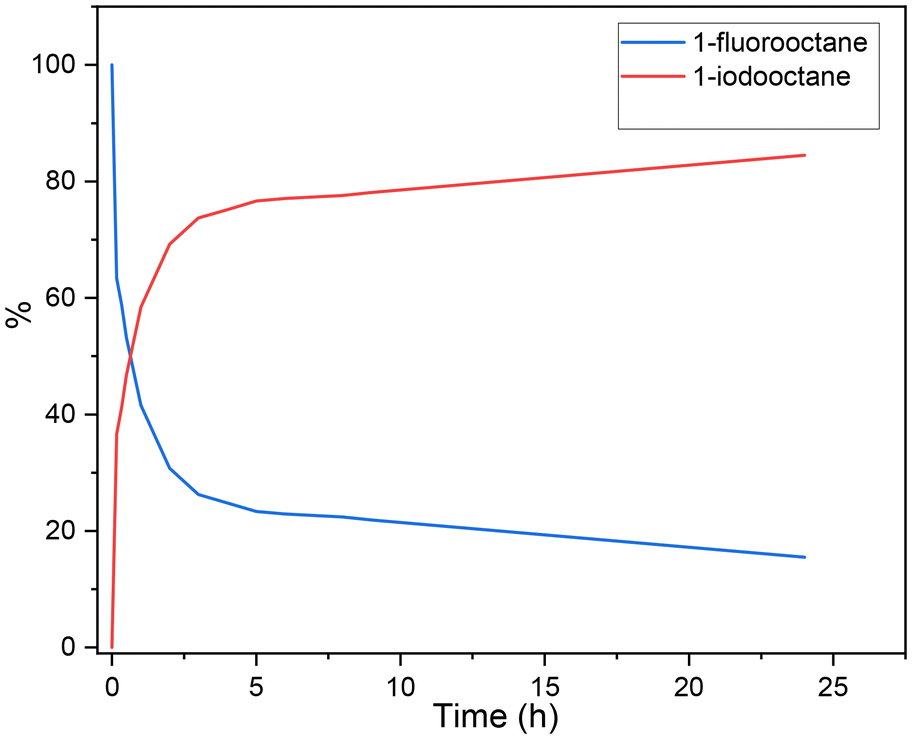 | ||
| Fig. 1 Kinetic profile of the halogen exchange reaction between 1-fluorooctane and 1-iodooctane. | ||
To further shed light on the mechanism, additional mechanistic experiments were conducted. When stoichiometric radical scavengers, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and 1,1-diphenylethene (1,1-DPE), were added to the model reaction of 1a and 2a under standard alkoxycarbonylation conditions, the formation of ester 3a was completely suppressed. The adducts of olefin with alkyl radical 6a were detected by GC-MS (Scheme 3b). In the radical inhibition experiments, the yields gradually decreased with the gradual addition of 2,6-di-tert-butyl 4-methylphenol (BHT, a radical scavenger, 0–2 equiv.) (Scheme 3c). Subsequently, in radical clock experiments, reactions using phenol and (1-fluorohex-5-en-3-yl)benzene as the substrates, the corresponding cyclization product 7 was obtained as the major ester (Scheme 3d). All these results imply the existence of radical intermediates in the reaction.
Based on the above results and literature reports,7–14 we proposed a mechanism for this reaction (Scheme 4). Initially, the LCuIOAr species (I) was generated from the copper salt through ligand exchange with phenol and the ligand.16 Then the complex I excited to its excited state adduct II under irradiation. Meanwhile, the alkyl fluoride undergoes halogen exchange to generate the corresponding alkyl iodide. Afterwards, complex II engaged in a single electron transfer reaction with alkyl iodide to form an alkyl radical and complex III. The alkyl radical was then captured by carbon monoxide to afford an acyl radical which will react with complex III to give acyl copper complex IV. Finally, the target ester can be eliminated by reductive elimination and regenerate CuI species for the next catalytic cycle.
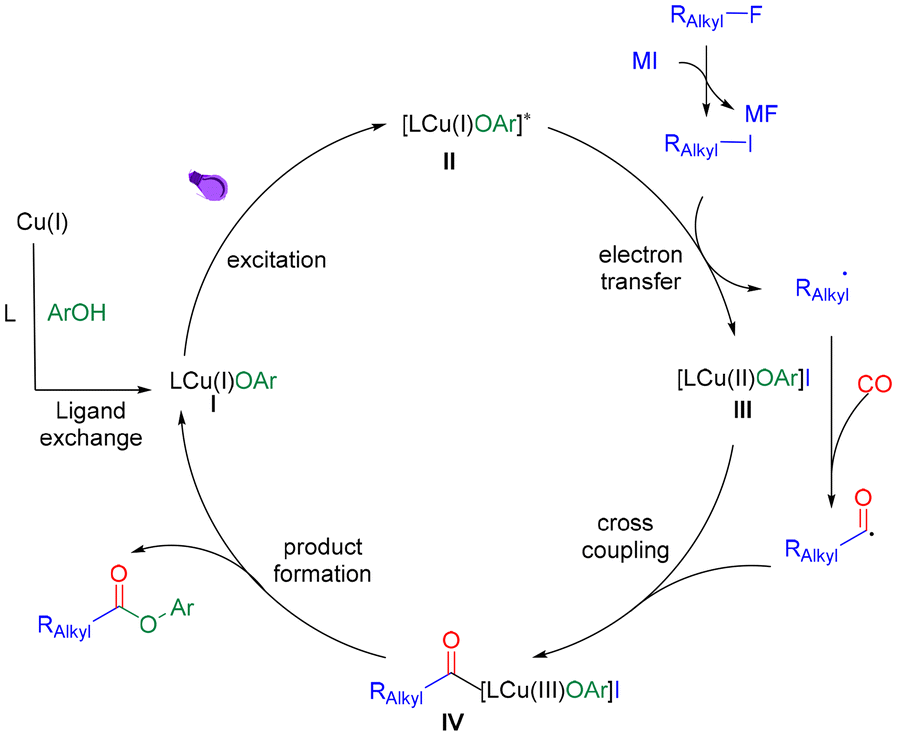 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In summary, we have developed a photoinduced copper-catalyzed alkoxycarbonylation of alkyl fluorides with phenols to synthesize various esters. The remarkable scope and functional group tolerance were evidenced by >50 examples and also the late-stage modification of natural product-related substrates. As the first example of the carbonylation of alkyl fluorides, the procedure provides a new pathway for the functionalization of alkyl fluorides.Author contributions
X.-F. W. conceived and designed the project; P. Y. and Y.-H. Z. conducted the experiments; P.Y. analysed and interpreted the experimental data; P. Y. and X.-F. W. prepared the manuscript; P. Y. and X.-F. W. prepared the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the financial support from the Chinese Scholarship Council and National Key R&D Program of China (2023YFA1507500). We thank the analytical department of Leibniz-Institute for Catalysis for their excellent analytical service. We appreciate the general support from Professor Armin Börner in LIKAT.References
- (a) R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley-VCH, 1999 Search PubMed; (b) J. Otera, Esterification: Methods, Reactions, and Applications, Wiley, 2010 Search PubMed; (c) C. Liu, S. Tang and A. Lei, Oxidant controlled Pd-catalysed selective oxidation of primary alcohols, Chem. Commun., 2013, 49, 1324–1326 RSC; (d) C. Locatelli, F. B. Filippin-Monteiro and T. B. Creczynski-Pasa, Alkyl esters of gallic acid as anticancer agents: A review, Eur. J. Med. Chem., 2013, 60, 233–239 CrossRef CAS PubMed; (e) B. Liu, F. Hu and B.-F. Shi, Recent Advances on Ester Synthesis via Transition-Metal Catalyzed C-H Functionalization, ACS Catal., 2015, 5, 1863–1881 CrossRef CAS.
- (a) B. Gabriele, Carbon Monoxide in Organic Synthesis: Carbonylation Chemistry, Wiley, 2021 CrossRef; (b) J.-B. Peng, F.-P. Wu and X.-F. Wu, First-Row Transition-Metal-Catalyzed Carbonylative Transformations of Carbon Electrophiles, Chem. Rev., 2019, 119, 2090–2127 CrossRef CAS PubMed; (c) J.-B. Peng, H.-Q. Geng and X.-F. Wu, The Chemistry of CO: Carbonylation, Chem, 2019, 5, 526–552 CrossRef CAS.
- R. F. Heck and D. S. Breslow, Carboxyalkylation reactions catalyzed by cobalt carbonylate ion, J. Am. Chem. Soc., 1963, 85, 2779–2782 CrossRef CAS.
- B. Lu, M. Xu, X. Qi, M. Jiang, W.-J. Xiao and J.-R. Chen, Switchable Radical Carbonylation by Philicity Regulation, J. Am. Chem. Soc., 2022, 144, 14923–14935 CrossRef CAS PubMed.
- (a) J. Tsuji, J. Kiji, S. Imamura and M. Morikawa, Organic Syntheses by Means of Noble Metal Compounds. VIII.1 Catalytic Carbonylation of Allylic Compounds with Palladium Chloride, J. Am. Chem. Soc., 1964, 86, 4350–4353 CrossRef CAS; (b) U. Hisao, O. Kosukegawa, Y. Ishii, H. Yugari and T. Fuchikami, Carbonylation of 1-perfluoroalkyl-substituted 2-iodoalkanes catalyzed by transition-metal complexes, Tetrahedron Lett., 1989, 30, 4403–4406 CrossRef; (c) W. Mägerlein, M. Beller and A. F. Indolese, Palladium-catalyzed carbonylation of aryl halides - a detailed investigation of the alkoxycarbonylation of 4-bromoacetophenone, J. Mol. Catal. A: Chem., 2000, 156, 213–221 CrossRef; (d) A. M. Trzeciak, W. Wojtków, Z. Ciunik and J. J. Ziółkowski, Low-Pressure Carbonylation of Benzyl Bromide with Palladium Complexes Modified with PNS or P(OPh)3. Structural Identification of Palladium-Catalyst Intermediate, Catal. Lett., 2001, 77, 245–249 CrossRef CAS.
- (a) A. C. Bissember, A. Levina and G. C. Fu, A Mild, Palladium-Catalyzed Method for the Dehydrohalogenation of Alkyl Bromides: Synthetic and Mechanistic Studies, J. Am. Chem. Soc., 2012, 134, 14232–14237 CrossRef CAS PubMed; (b) L. Wu, X. Fang, Q. Liu, R. Jackstell, M. Beller and X.-F. Wu, Palladium-catalyzed carbonylative transformation of C (sp3)-X bonds, ACS Catal., 2014, 4, 2977–2989 CrossRef CAS; (c) X.-W. Gu, Y. Zhang, F. Zhao, H.-J. Ai and X.-F. Wu, Phosphine-catalyzed photo-induced alkoxycarbonylation of alkyl iodides with phenols and 1,4-dioxane through charge-transfer complex, Chin. J. Catal., 2023, 48, 214–223 CrossRef CAS.
- (a) I. Ryu and N. Sonoda, Free-Radical Carbonylations: Then and Now, Angew. Chem., Int. Ed. Engl., 1996, 35, 1050–1066 CrossRef; (b) I. Ryu, Radical carboxylations of iodoalkanes and saturated alcohols using carbon monoxide, Chem. Soc. Rev., 2001, 30, 16–25 RSC; (c) S. Zhao and N. P. Mankad, Metal-catalysed radical carbonylation reactions, Catal. Sci. Technol., 2019, 9, 3603–3613 RSC.
- (a) T. Kondo, Y. Tsuji and Y. Watanabe, Photochemical carbonylation of alkyl iodides in the presence of various metal carbonyls, Tetrahedron Lett., 1988, 29, 3833–3836 CrossRef CAS; (b) T. Kondo, Y. Sone, Y. Tsuji and Y. Watanabe, Photo-, electro-, and thermal carbonylation of alkyl iodides in the presence of group 7 and 8-10 metal carbonyl catalysts, J. Organomet. Chem., 1994, 473, 163–173 CrossRef CAS.
- (a) A. Fusano, S. Sumino, S. Nishitani, T. Inouye, K. Morimoto, T. Fukuyama and I. Ryu, Pd/Light-Accelerated Atom-Transfer Carbonylation of Alkyl Iodides: Applications in Multicomponent Coupling Processes Leading to Functionalized Carboxylic Acid Derivatives, Chem. – Eur. J., 2012, 18, 9415–9422 CrossRef CAS PubMed; (b) S. Sumino, A. Fusano, T. Fukuyama and I. Ryu, Carbonylation Reactions of Alkyl Iodides through the Interplay of Carbon Radicals and Pd Catalysts, Acc. Chem. Res., 2014, 47, 1563–1574 CrossRef CAS PubMed.
- (a) B. T. Sargent and E. J. Alexanian, Palladium-Catalyzed Alkoxycarbonylation of Unactivated Secondary Alkyl Bromides at Low Pressure, J. Am. Chem. Soc., 2016, 138, 7520–7523 CrossRef CAS PubMed; (b) P. Wang, Y. Wang, H. Neumann and M. Beller, Rh-catalyzed alkoxycarbonylation of unactivated alkyl chlorides, Chem. Sci., 2022, 13, 13459–13465 RSC.
- (a) G. M. Torres, Y. Liu and B. A. Arndtsen, A dual light-driven palladium catalyst: Breaking the barriers in carbonylation reactions, Science, 2020, 368, 318–323 CrossRef CAS PubMed; (b) Y. Liu, C. Zhou, M. Jiang and B. A. Arndtsen, Versatile Palladium-Catalyzed Approach to Acyl Fluorides and Carbonylations by Combining Visible Light- and Ligand-Driven Operations, J. Am. Chem. Soc., 2022, 144, 9413–9420 CrossRef CAS PubMed; (c) K. E. Chami, Y. Liu, M. A. Belahouane, Y. Ma, P.-L. Lagueux-Tremblay and B. A. Arndtsen, A Visible Light Driven Nickel Carbonylation Catalyst: The Synthesis of Acid Chlorides from Alkyl Halides, Angew. Chem., Int. Ed., 2023, 62, e202213297 CrossRef PubMed.
- (a) H.-J. Ai, Y. Yuan and X.-F. Wu, Ruthenium pincer complex-catalyzed heterocycle compatible alkoxycarbonylation of alkyl iodides: substrate keeps the catalyst active, Chem. Sci., 2022, 13, 2481–2486 RSC; (b) X. Qi, R. Zhou, H.-J. Ai and X.-F. Wu, HMF and furfural: Promising platform molecules in rhodium-catalyzed carbonylation reactions for the synthesis of furfuryl esters and tertiary amides, J. Catal., 2020, 381, 215–221 CrossRef CAS; (c) Y. Li and X.-F. Wu, Copper/iron co-catalyzed alkoxycarbonylation of unactivated alkyl bromides, Commun. Chem., 2018, 1, 39 CrossRef; (d) F. Zhao, P. Russo, R. Mancuso, B. Gabriele and X.-F. Wu, Copper-catalyzed carbonylative coupling of alkyl iodides with phenols for the synthesis of esters, J. Catal., 2022, 413, 907–912 CrossRef CAS.
- (a) H. Yamamoto, T. Hiyama, K. Kanie, T. Kusumoto, Y. Morizawa and M. Shimzu, Organofluorine Compounds: Chemistry and Applications, Springer Berlin Heidelberg, 2000 Search PubMed; (b) H. Amii and K. Uneyama, C-F Bond Activation in Organic Synthesis, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed; (c) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Functionalization of Fluorinated Molecules by Transition-Metal-Mediated C-F Bond Activation To Access Fluorinated Building Blocks, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed; (d) T. Iwasaki and N. Kambe, Cross- and Multi-Coupling Reactions Using Monofluoroalkanes, Chem. Rec., 2023, 23, e202300033 CrossRef CAS PubMed.
- (a) J. Choi, D. Y. Wang, S. Kundu, Y. Choliy, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Net Oxidative Addition of C(sp3)-F Bonds to Iridium via Initial C-H Bond Activation, Science, 2011, 332, 1545–1548 CrossRef CAS PubMed; (b) C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Lewis Acidity of Organofluorophosphonium Salts: Hydrodefluorination by a Saturated Acceptor, Science, 2013, 341, 1374–1377 CrossRef CAS PubMed; (c) W. Gu, M. R. Haneline, C. Douvris and O. V. Ozerov, Carbon−Carbon Coupling of C(sp3)−F Bonds Using Alumenium Catalysis, J. Am. Chem. Soc., 2009, 131, 11203–11212 CrossRef CAS PubMed; (d) M. Ahrens, G. Scholz, T. Braun and E. Kemnitz, Catalytic Hydrodefluorination of Fluoromethanes at Room Temperature by Silylium-ion-like Surface Species, Angew. Chem., Int. Ed., 2013, 52, 5328–5332 CrossRef CAS PubMed; (e) C. Douvris and O. V. Ozerov, Hydrodefluorination of Perfluoroalkyl Groups Using Silylium-Carborane Catalysts, Science, 2008, 321, 1188–1190 CrossRef CAS PubMed; (f) C. Douvris, C. M. Nagaraja, C.-H. Chen, B. M. Foxman and O. V. Ozerov, Hydrodefluorination and Other Hydrodehalogenation of Aliphatic Carbon-Halogen Bonds Using Silylium Catalysis, J. Am. Chem. Soc., 2010, 132, 4946–4953 CrossRef CAS PubMed.
- (a) S. F. R. Taylor, J. Sá and C. Hardacre, Friedel-Crafts Alkylation of Aromatics with Benzyl Alcohol over Gold-Modified Silica, ChemCatChem, 2011, 3, 119–121 CrossRef CAS; (b) M. Rueping and B. J. Nachtsheim, A review of new developments in the Friedel-Crafts alkylation – From green chemistry to asymmetric catalysis, Beilstein J. Org. Chem., 2010, 6, 6, DOI:10.3762/bjoc.6.6.
- (a) Y. Jia, Z. Zhang, G.-M. Yu, X. Jiang, L.-Q. Lu and W.-J. Xiao, Visible Light Induced Copper-Catalyzed Enantioselective Deaminative Arylation of Amino Acid Derivatives Assisted by Phenol, Angew. Chem., Int. Ed., 2023, 62, e202312102 CrossRef CAS PubMed; (b) J.-P. Yue, J.-C. Xu, H.-T. Luo, X.-W. Chen, H.-X. Song, Y. Deng, L. Yuan, J.-H. Ye and D.-G. Yu, Metallaphotoredox-enabled aminocarboxylation of alkenes with CO2, Nat. Catal., 2023, 6, 959–968 CrossRef CAS; (c) C. Chen, J. C. Peters and G. C. Fu, Photoinduced copper-catalysed asymmetric amidation via ligand cooperativity, Nature, 2021, 596, 250–256 CrossRef CAS PubMed; (d) C. Chen and G. C. Fu, Copper-catalysed enantioconvergent alkylation of oxygen nucleophiles, Nature, 2023, 618, 301–307 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General procedure, analytical data, and NMR spectra. See DOI: https://doi.org/10.1039/d4qo00041b |
| This journal is © the Partner Organisations 2024 |