
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNADH-mediated primordial synthesis of amino acids†
Noemí
Nogal‡
 a,
Javier
Luis-Barrera‡
a,
Sonia
Vela-Gallego
a,
Fernando
Aguilar-Galindo
bc and
Andrés
de la Escosura
*ac
a,
Javier
Luis-Barrera‡
a,
Sonia
Vela-Gallego
a,
Fernando
Aguilar-Galindo
bc and
Andrés
de la Escosura
*ac
aDepartment of Organic Chemistry, Universidad Autónoma de Madrid, Campus de Cantoblanco, 28049 Madrid, Spain. E-mail: andres.delaescosura@uam.es
bDepartment of Chemistry, Universidad Autónoma de Madrid, Campus de Cantoblanco, 28049 Madrid, Spain
cInstitute for Advanced Research in Chemistry (IAdChem), Cantoblanco, 28049 Madrid, Spain
First published on 14th February 2024
Abstract
The pathways that could explain the abiotic synthesis of amino acids are different from those observed in living organisms, which leaves a gap of knowledge between bottom-up approaches towards the understanding of abiogenesis and studies on the emergence and evolution of metabolism. Finding non-enzymatic versions of current key metabolic reactions represents a powerful strategy to fill such a gap, and the use of natural cofactors can be useful to aid that chemistry in the absence of enzymes. In this direction, herein we describe how NADH mediates the reductive amination of α-ketoacids in the presence of ammonia, allowing the production of various amino acids. The reaction occurs at room temperature in ammonium/ammonia solutions at pH > 7, conditions that are compatible with some plausible scenarios on the prebiotic Earth. A combination of NMR and mass spectrometry data with computational calculations supports that the reaction proceeds through a hemiaminal intermediate, which subsequently gets dehydrated into the iminium species. This allows for the overcoming of the low reactivity of NADH as a hydride donor to the carbonyl group of α-ketoacids, while the cofactor loses the nicotinamide ring during the process. Overall, these results demonstrate that NADH can assist in the production of amino acids through a route that was postulated as primordial by the chemoautotrophic theory, thus providing a link of continuity with the chemistry of these important building blocks in ancient protometabolism.
Introduction
Amino acids play a fundamental role in the chemistry sustaining life, not only as the building blocks of peptides and proteins but also as important metabolic intermediates.1 In the context of the origin of life, it is therefore essential to investigate plausible primitive sources of these biomolecules. The relevance of amino acids in prebiotic chemistry dates back to the seminal Miller–Urey experiment,2,3 and they have been found in different Miller-type spark discharge conditions,4 meteorites,5,6 tails of comets7 and in the interstellar medium.8 In parallel, various synthetic routes have been investigated to explain their abiotic emergence, including the hydrolysis of α-aminonitriles produced by the Strecker reaction or that of hydantoins obtained through the Bücherer–Bergs process,9 as well as the chemistry behind the cyanosulfidic scenario.10,11 Whether these transformations represented the main source of amino acids for the first protocellular systems is, however, an open question, as there are no traces of such synthetic pathways in the biochemistry of any extant living organism. In contrast, amino acids are normally synthesized from α-ketoacids, either through reductive amination for glutamate or transamination for the rest of the amino acids (with glutamate as the source of nitrogen).12,13 Chemoautotrophic theory postulates, indeed, that reductive amination would have been the main prebiotic route.14In the search for non-enzymatic versions of the reactions that lead to central metabolites,12,15 it is common to use cofactors in order to aid that chemistry.16 It has been proposed that some cofactors, especially those containing phosphate and ribonucleotide moieties (i.e., NADH/NAD+, FADH/FAD+ and CoA), are widespread across the tree of life and so they could be ancient.17 Moreover, they are involved in different steps of the TCA/rTCA (tricarboxylic acids) cycle, also called the (reductive) Krebs cycle,18 which, independently of whether it is run in the catabolic or anabolic direction, supplies the five universal metabolic precursors: acetate, succinate, pyruvate, oxaloacetate and α-ketoglutarate.19 The latter three are α-ketoacids and can be subjected to reductive amination with the help of a reducing agent.20 Various iron-based mineral catalysts and nucleophilic hydride species have been employed for this purpose,21–25 but they seem disconnected from the extant metabolic routes. NAD(P)H is normally the hydride donor when such a reaction occurs in current biochemistry. Consequently, herein we have searched for prebiotic conditions that allow NADH to mediate the reductive amination of α-ketoacids (Scheme 1), as a primordial synthesis of amino acids that connects prebiotic chemistry with the metabolic pathways that led to these biomolecules in the first autotrophic protocells.
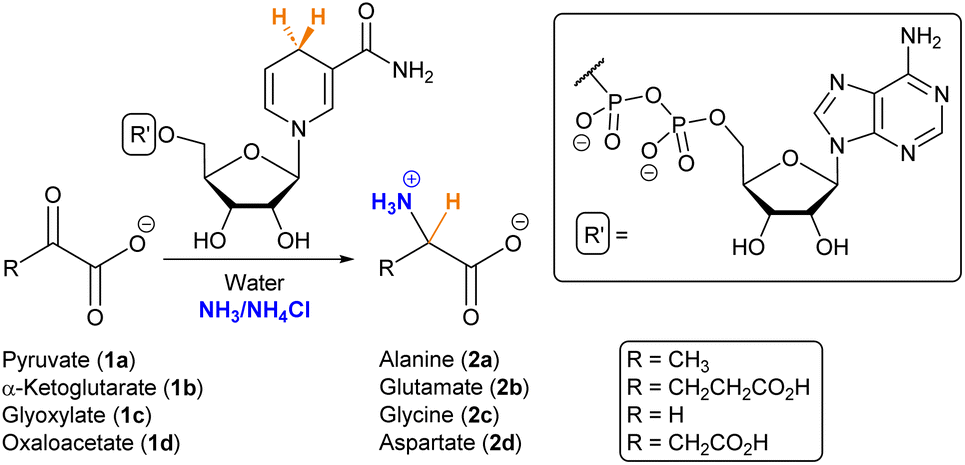 | ||
| Scheme 1 Reductive amination of α-ketoacids in the presence of NADH. | ||
As a redox-active species, NAD+ has been utilized for various oxidation processes, in combination with iron–sulfur peptides,26 organometallic rhodium complexes27 or titanium oxide nanoparticles.28 Moreover, NAD+ has shown the capacity to react with pyruvate at a basic pH in an oxidative decarboxylation that leads to acetate, with this reaction being applicable to other α-ketoacids.29 This relevant finding raises the question of whether there was a primordial NAD+/NADH-dependent protometabolism. One could, for example, question if NADH can also act as a reducing agent for the same substrates. In a recent paper by Moran and coworkers, experiments carried out in phosphate buffer, at pH between 5 and 7, suggested that NADH is not a feasible hydride donor for that purpose,30 probably due to its hydrolysis and low reactivity under the mildly acidic conditions required for carbonyl group activation. Such a limitation in reactivity, however, is only problematic when the medium does not permit the formation of more electrophilic carbonyl derivatives,31,32 for instance, the iminium ion, which is the most likely pathway to take place in an ammonium/ammonia solution. Hence, in the work below, we explore the use of ammonia to drive a NADH-mediated reductive amination of α-ketoacids.
Results and discussion
Formation of amino acids
For gaining insight into the reactivity between α-ketoacids and NADH, the reaction was tested with 30 mM pyruvate (1a) as the substrate and one equivalent of NADH at 25 °C under a nitrogen atmosphere, either in phosphate or ammonia (200 mM each) solutions, with the pH being adjusted to 8 in both cases.33 In phosphate buffer, the NMR spectra did not show any amino acid formation over 72 h (Fig. S28B†). In ammonia solution, in contrast, while the single components did not yield any significant reaction (Fig. S10 and S13†), clear spectral changes were observed in the reaction mixture (Fig. 1A and S14†), indicative of the appearance of a new species with a quartet (J = 7.2 Hz) at 3.80 ppm and a partially overlapped doublet (J = 7.2 Hz) at 1.49 ppm (blue dots). The 1H-NMR spectra were calibrated with respect to the internal standard 3-(trimethylsilyl)-2,2,3,3-tetradeuteropropionic acid (TMSP-d4), and then compared with that of an alanine (2a) authentic sample under the same conditions, revealing a perfect overlap of the two mentioned signals. The product identity was further corroborated through GC-MS: both the reaction mixture and the alanine pure sample were subjected to an established derivatization protocol19 with ethyl chloroformate and subsequently analysed (see the protocol in the ESI†). The reaction mixture chromatogram presents a peak whose retention time and mass spectrum match those for the main peak of pure alanine (Fig. 1B), confirming the amino acid formation.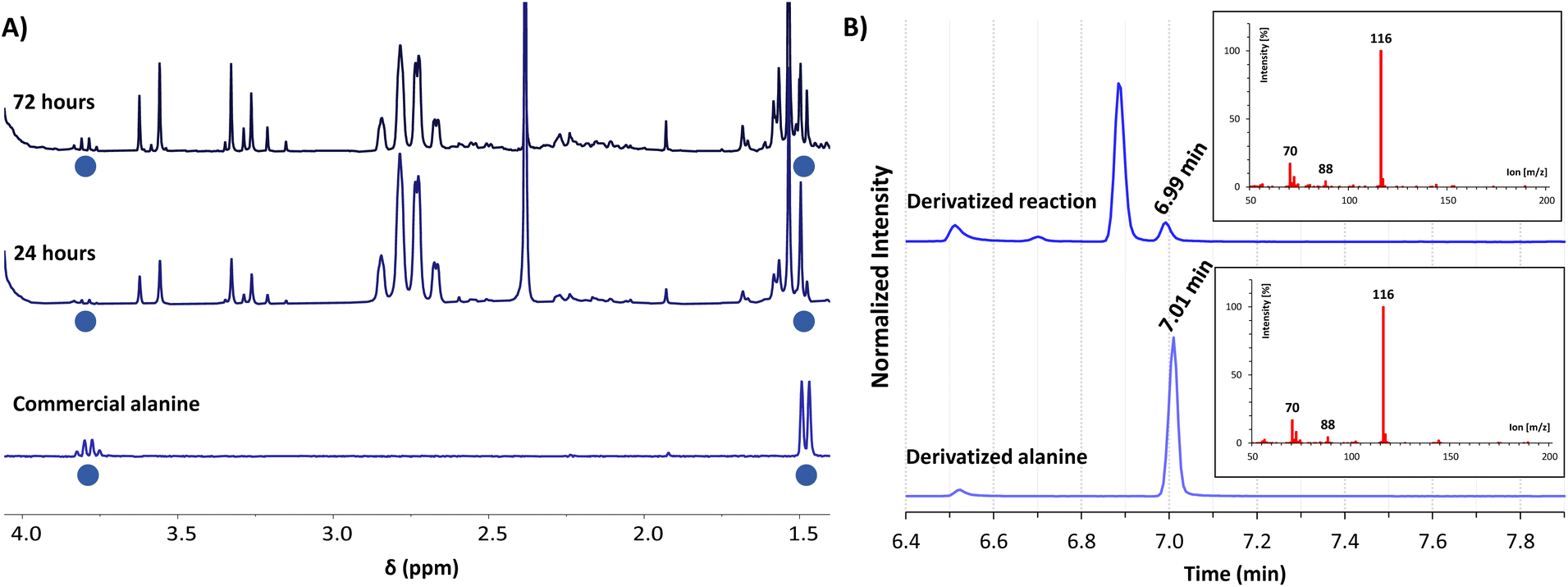 | ||
Fig. 1 (A) Selected region of the 1H-NMR spectra in H2O/D2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, with water signal suppression) of the reaction between pyruvate (1a, 30 mM) and NADH (30 mM) in ammonia solution (200 mM) at pH 8, after 24 h (middle spectrum) and 72 h (top) of reaction, compared with an authentic sample of alanine (2a, 3 mM) in the same solution (bottom). Chemical shifts are given using TMSP-d4 as a reference (0.00 ppm). (B) GC-MS chromatograms of the derivatized reaction mixture (top) and a derivatized authentic sample of alanine (2a, bottom), showing in both cases the mass spectrum associated with the peak at 7.0 min. Derivatizations made with ethyl chloroformate (Scheme S2†). :1, with water signal suppression) of the reaction between pyruvate (1a, 30 mM) and NADH (30 mM) in ammonia solution (200 mM) at pH 8, after 24 h (middle spectrum) and 72 h (top) of reaction, compared with an authentic sample of alanine (2a, 3 mM) in the same solution (bottom). Chemical shifts are given using TMSP-d4 as a reference (0.00 ppm). (B) GC-MS chromatograms of the derivatized reaction mixture (top) and a derivatized authentic sample of alanine (2a, bottom), showing in both cases the mass spectrum associated with the peak at 7.0 min. Derivatizations made with ethyl chloroformate (Scheme S2†). | ||
The achieved conversion can be determined through 1H-NMR in experiments where the internal standard TMSP-d4 was added to the NMR tubes at a known concentration, allowing us to relate the integral of its reference peak with those arising from the amino acid (e.g., the quartet at 3.80 ppm for alanine).34 With this approach, it was possible to compare the 1H-NMR yields obtained at different times, pH conditions, NH3/NH4Cl concentrations, and stoichiometric ratios (see section 3 of the ESI† for details about calibrations and 1H-NMR yield calculations). As shown in Fig. 2A, a slow but constant reaction is observed at pH 7 and 8, reaching a conversion of 11% after 72 h of reaction.35 In contrast, a drastic drop in efficacy occurs in more alkaline media, with lower conversions at pH 9 (e.g., 3% at 72 h) and no reaction at pH 10. Considering that the process proceeds through a pyruvate iminium ion as the species accepting the cofactor hydride donation (see Scheme 4, blue path, and the related DFT calculations below), this result is in good agreement with previous studies pointing to a maximum stationary concentration of iminium ion between pH 4 and 8, while it decreases at higher pH values.30
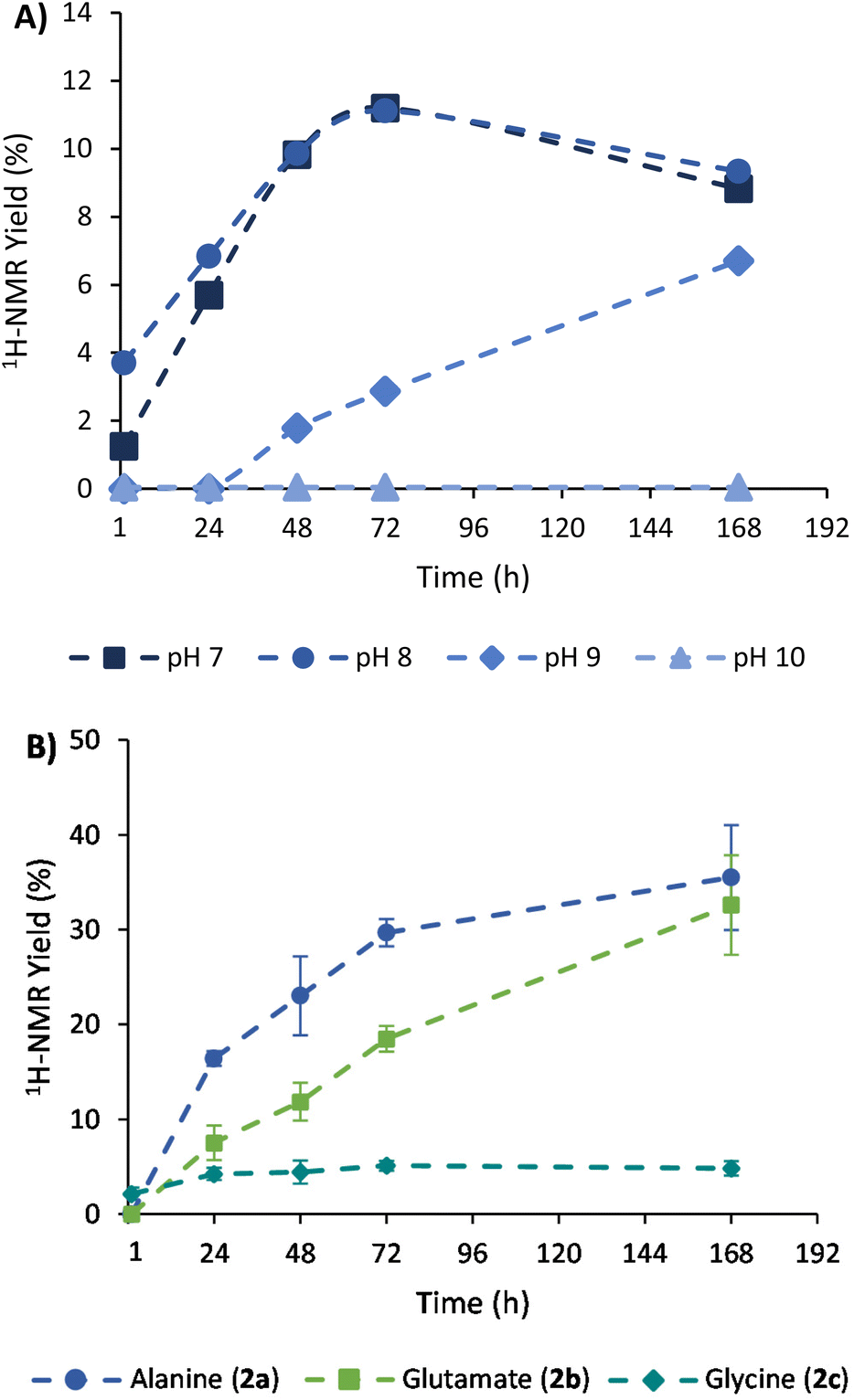 | ||
| Fig. 2 Plots of 1H-NMR yield (%) relative to TMSP-d4 at different times of: (A) the reaction of pyruvate (1a, 30 mM) and NADH (30 mM) in ammonia solution (200 mM) to produce alanine (2a) at different pH values; (B) reaction of the different α-ketoacids (1a, 1b or 1c, 30 mM) with NADH (90 mM) at pH 8 in ammonia solution (1 M), yielding the corresponding amino acids (2). Under these optimal conditions, yields were calculated as an average from three independent experiments, and so the error bars are shown. | ||
In order to increase the substrate conversion into alanine (2a), the most favourable pH (8) was fixed36 while other variables were evaluated. For example, the 1H-NMR yield significantly improved when the NH3/NH4Cl concentration was increased to 600 mM or 1 M (Table 1, entries 2 and 3). The cofactor/substrate ratio was also addressed, obtaining better yields when three equivalents of NADH were employed (entries 4–6). Overall, the highest yield was obtained with three equivalents of NADH (90 mM) in 1 M NH3/NH4Cl at pH 8 (entry 6).
| Entry | KAa:NADH |
NH3/NH4Cl (mM) |
2ab (%) |
2bb (%) |
2cb (%) |
|---|---|---|---|---|---|
|
a KA: α-ketoacid.
b 1H-NMR yields determined at 168 h of reaction, using TMSP-d4 as an internal standard.
c In the parenthesis, the yield at 72 h of reaction is given. See Tables S8–S10 and Fig. S7–S9† for yields at different reaction times.
|
|||||
| 1 | 1:1 |
200 | 9 | 11 | 2 (2)c |
| 2 | 1:1 |
600 | 16 | 24 | 3 |
| 3 | 1:1 |
1000 | 21 | 13 | 5 |
| 4 | 1:3 |
200 | 19 | 15 | 1 |
| 5 | 1:3 |
600 | 28 | 18 | 4 |
| 6 | 1:3 |
1000 | 36 | 33 | 5 |
Once the conditions had been adjusted, we proceeded to study the reaction scope, employing α-ketoglutarate (1b), glyoxylate (1c) and oxaloacetate (1d) as alternative substrates (structures in Scheme 1). In control experiments, none of these ketoacids gave rise to the corresponding amino acids when the cofactor was not present in the medium (Fig. S17, S20 and S25†). In the presence of NADH, Table 1 shows the 1H-NMR yields under different conditions, and Fig. 2B represents a comparison of the reaction progression under the optimal conditions for the three substrates that reacted through the expected pathway (i.e., pyruvate, α-ketoglutarate and glyoxylate). The identity of glutamate and glycine was also confirmed by GC-MS analysis (Fig. S33 and S34†). It is noteworthy that alanine (2a) and glutamate (2b) were obtained in 36% and 33% yield, respectively (entry 6), while a significant yield reduction occurred in the formation of glycine (2c). Such a decrease in efficiency happens for all the conditions tested in Table 1 (last column) and has led us to investigate, in the next section, the possible decomposition paths that are detrimental to the reaction yield. The formation of the corresponding α-hydroxyacids, on the other hand, was not detected in any case (Fig. S14–16, S31–S32 and S34–35†), ruling out the possibility that NADH could reduce the ketoacids directly without incorporating nitrogen from ammonia.
An interesting, different case is the evolution of oxaloacetate (1d) under the studied conditions (Scheme 2). The amino acid aspartate (2d) was not detected by 1H-NMR. However, careful analysis of the spectra revealed that, due to its 1,3-ketoacid nature, this species undergoes decarboxylation into pyruvate (1a), which subsequently evolves into alanine (2a) (Fig. S25–27†). The decarboxylation process also occurs in a control experiment that lacks the cofactor (Fig. S25†), yet in that case without the last reductive amination step.
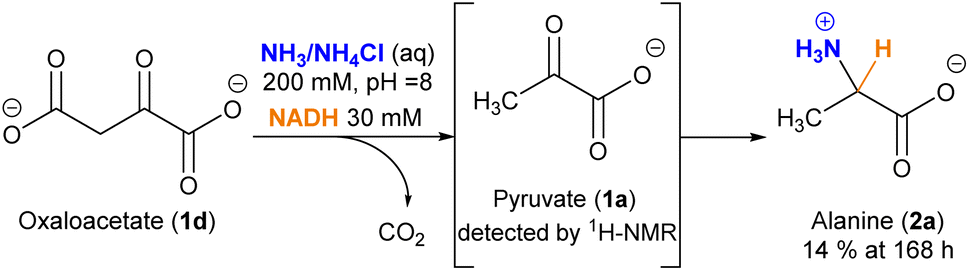 | ||
| Scheme 2 Evolution of oxaloacetate (1d) under the conditions explored for reductive amination. | ||
Monitoring of cofactor byproducts
NAD+ is the direct product obtained when NADH transfers a hydride (Scheme 3). It is known that both NAD+ and NADH can suffer hydrolysis, losing the nicotinamide moiety and giving adenosine diphosphate ribose (ADPR).37 During the reductive amination of all ketoacids (1), small amounts of NAD+ appeared as the amino acid (2) was formed. The formation of ADPR was in turn observed in all cases, in proportions that depend strongly on the substrate.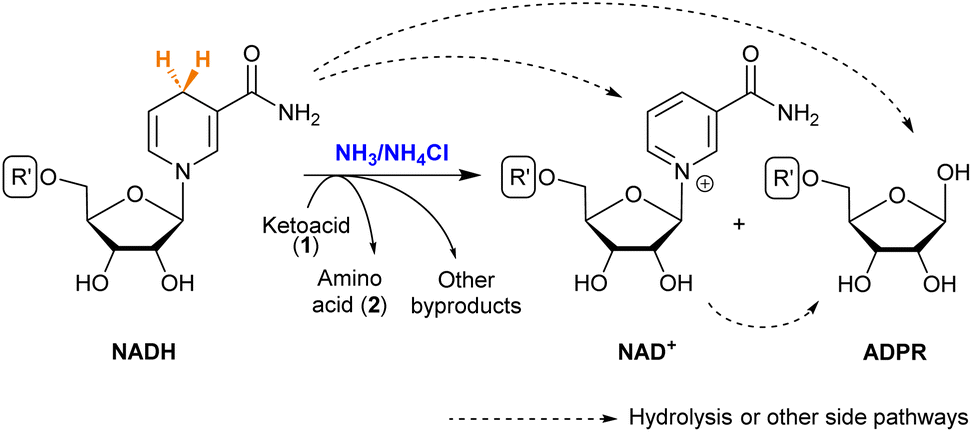 | ||
| Scheme 3 Possible products of NADH in the reductive amination of ketoacids. See the R′ structure in Scheme 1. | ||
This phenomenon is clearly more significant in the reductive amination of glyoxylate (1c). Hence, 1H-NMR monitoring of the reaction of this precursor with one equivalent of NADH in 200 mM NH3/NH4Cl solution (Table 1, entry 1) is shown in Fig. 3A as a representative example. During this process, the disappearance of signals from NADH (orange circles) is accompanied by the onset and increase in the intensity of a new set of signals (brown squares) matching the ADPR spectroscopic data.38 Signals corresponding to NAD+ are also detected, although they are really minor (red dots). Remarkably, no free nicotinamide was observed, and complex mixtures appeared in some regions of the 1H-NMR spectrum (Fig. S21†). The glyoxylate (1c) signal, on the other hand, is not always appreciable because a mixture of species in equilibrium can be formed from this compound in ammonia solution.39
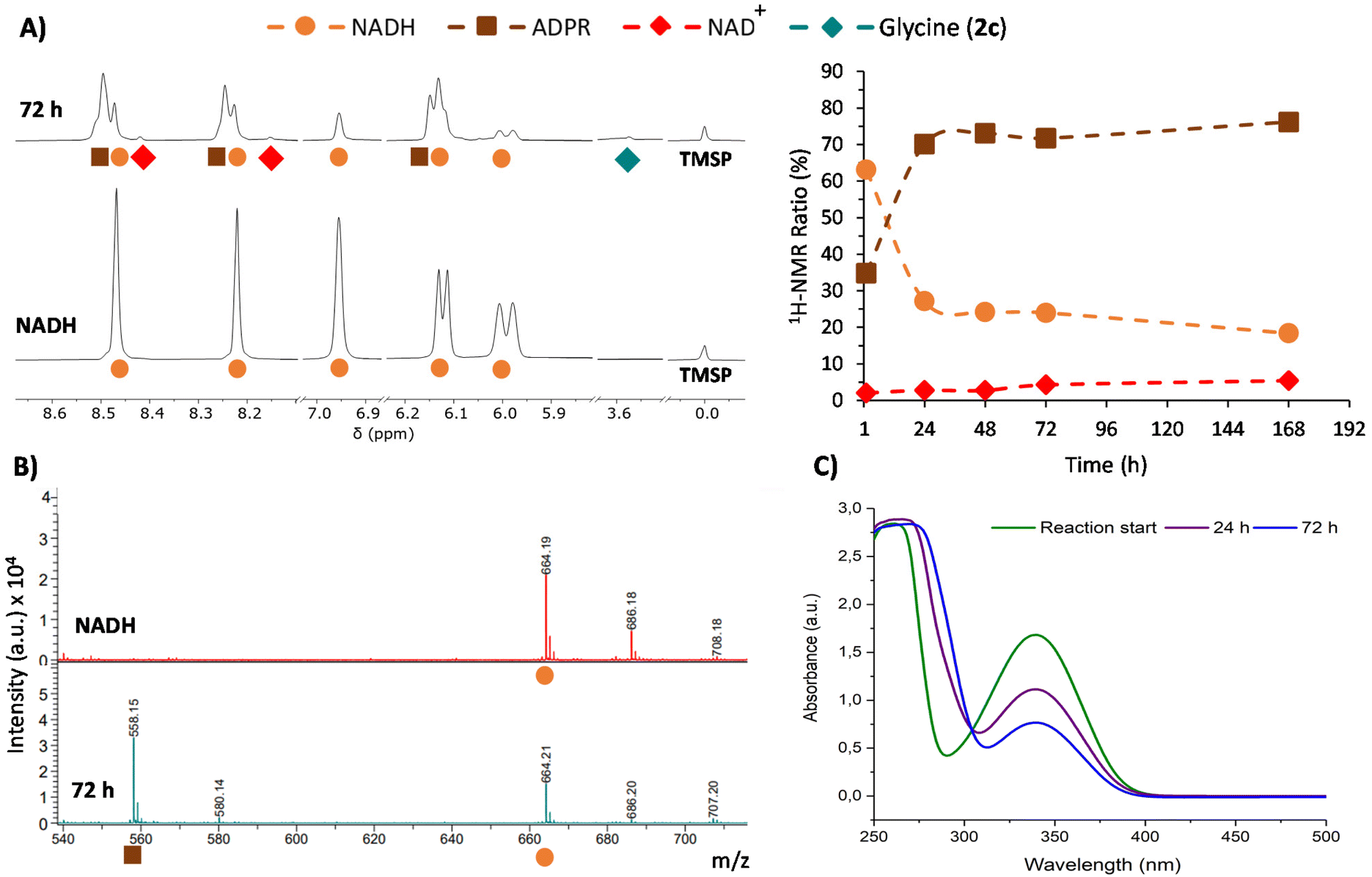 | ||
| Fig. 3 Monitoring of the conversion of NADH into NAD+ and ADPR in the reaction between glyoxylate (1c, 30 mM) and NADH (30 mM) in ammonia solution (200 mM) at pH 8. (A) Left: selected regions of the 1H-NMR spectra in H2O/D2O (9:1, with water signal suppression) of the reaction (top spectrum) and a pure sample NADH (30 mM) in the same ammonia solution (bottom spectrum). Chemical shifts are given using TMSP-d4 as a reference (0.00 ppm). Right: relative conversion (%) of NADH into ADPR and NAD+ in the reaction. (B) Selected region of the MALDI-TOF MS spectra from NADH and the reaction mixture. (C) Cofactor UV-Vis absorption decay during the reaction. | ||
Further insights were obtained through MALDI-TOF MS, which was recorded in negative mode from aliquots taken at different reaction times. While in the spectrum of NADH, a major peak corresponding to [M − H]− was observed at m/z 664.2 (Fig. 3B), the spectra of the reaction mixture showed a new peak at m/z 558.1 ([M − 123 + 17]−). This peak is associated with the loss of the dihydronicotinamide ring (123 Da) and the gain of an OH (17 Da) group (Fig. 3B, bottom), thus confirming the presence of ADPR. Importantly, the ratio of intensities of the peaks at m/z 558.1 and 664.2 keeps increasing over time (Fig. S38†). The spectra also show a gradual reduction of the relative intensity of the peak for the glyoxylate (1c) molecular ion at m/z 241.0 ([Mglyo + (2,3,4-THAP) − H]−) (Fig. S39†), corroborating its implication in the cofactor transformation. Along with these NMR and MS changes, UV-Vis spectroscopy revealed a gradual decay of NADH absorption (λmax = 340 nm) over time (Fig. 3C and Fig. S41†).
Once all the NADH byproducts were identified, the cofactor relative conversion into NAD+ and ADPR over time could be assessed by 1H-NMR analysis (Fig. 3A, right). At 72 h, for instance, the conversion values for NAD+ and ADPR were 4% and 72%, respectively, while only 2% of glycine (2c) was reached at that point (Table 1, entry 1). Different control experiments indicate that when glyoxylate (1c) is dissolved together with NADH in the absence of ammonia, similar amounts of ADPR and degradation mixtures are obtained (see Tables S14–S15 and Fig. S28–S29†). This set of evidence suggests that there are two processes competing. First, a slow process of transformation of glyoxylate (1c) with ammonia and the subsequent hydride transfer from NADH give rise to glycine (2c) and NAD+.40 Second, a faster reaction of glyoxylate (1c) directly with the reduced nicotinamide ring of NADH releases the rest of the molecules as ADPR and forms a complex mixture of degradation products. The prevalence of the latter process drastically decreases the yield of glycine (2c).41
The analysis of NAD+ and ADPR formation was also performed for the reductive amination of pyruvate (1a) and α-ketoglutarate (1b) under identical conditions (1 eq. of NADH and 200 mM ammonia solution at pH 8; see Tables S8 and S9†), revealing substantially different results. For 1a, 11% of alanine (2a) and 19% of ADPR were observed after 72 h. The same reaction with 1b yielded 8% of glutamate (2b) and 17% of ADPR at 72 h. Thus, the side reaction by which ADPR is overproduced is much less prominent in these examples than from glyoxylate (1c), allowing the ketoacids 1a and 1b to evolve into the corresponding amino acids with better yields.
Computational DFT studies
NADH can act via two different mechanisms:42 a one-step hydride anion transfer or a multistep hydride transfer, initiated by a single electron transfer. The latter possibility implies the formation of radical intermediates. While radical NADH˙+ (or radical derivatives) has usually been detected in CH3CN solutions,42,43 hydride transfer from NADH to positively charged organic molecules in aqueous solution usually follows kinetics that are typical of a one-step nucleophilic attack.44 Based on this, we carried out DFT calculations to check the viability of the “hydride anion transfer” mechanism under the studied conditions. In this hypothetical scenario, NADH would transfer the hydride anion to any sufficiently electrophilic species that is present in the equilibrium mixture of the ketoacid (1) with ammonia, such as the iminium ion.DFT calculations were performed with the M06-2X functional,45 which is one of the best-performing global hybrids,46 and the triple-ζ cc-pVTZ basis set. To reduce the computational effort, only the active part of the cofactor, i.e., the nicotinamide ring with an adjacent simplified ribose unit (Fig. S42†), was considered in the calculations. The solvent (water) was simulated using the SMD implicit model47 and, in order to accurately describe the protonation steps in this medium, the energy of a proton was computed as the energy difference between NH4+ and NH3, given that ammonium is the main proton source under the studied conditions. For reaction steps that imply a proton exchange (i.e., hemiaminal formation or its dehydration into the imine/iminium species), an explicit water or ammonium molecule was also considered. The calculations were performed for the different steps of our model reaction, i.e., the synthesis of alanine (2a) from pyruvate (1a) (Scheme 4). On this basis, the free energies and structures of all intermediates and transition states (TS) through the two studied pathways are displayed in Fig. 4. The zero energy value has been established in the reactants.
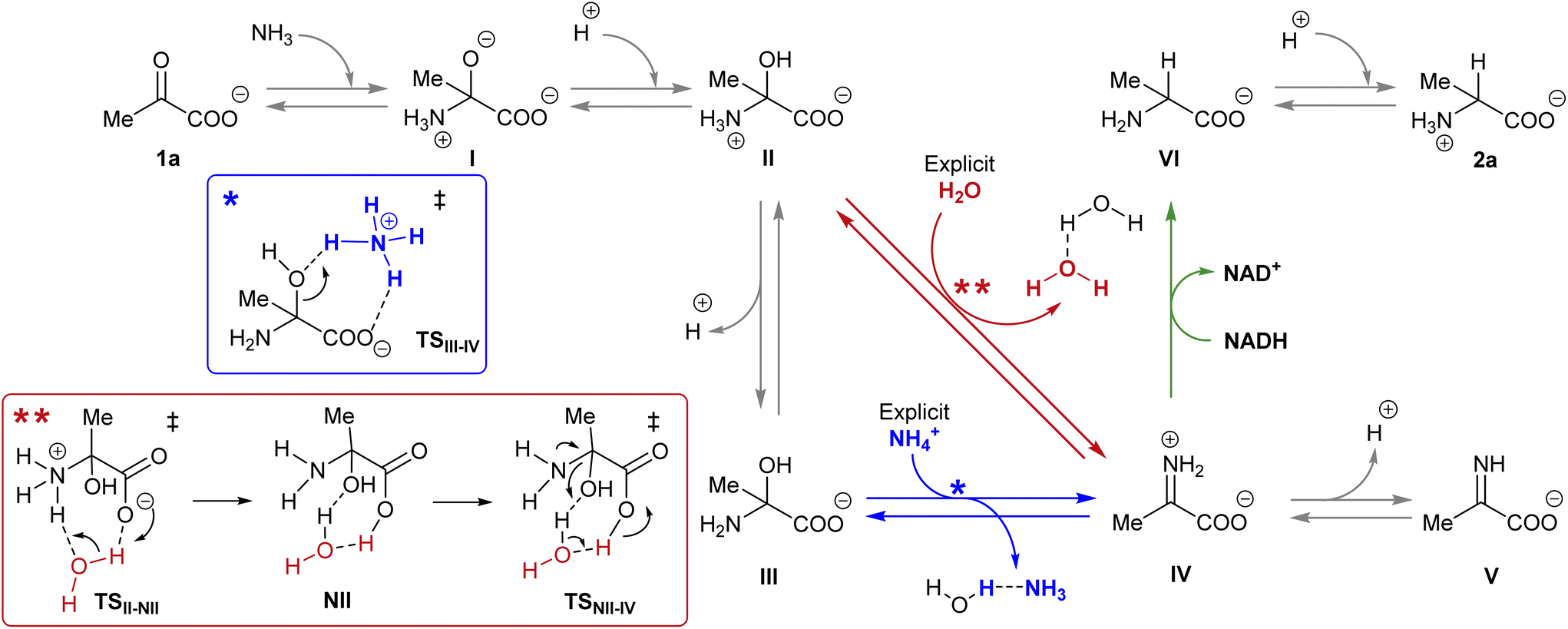 | ||
| Scheme 4 Reaction pathways that could lead to the synthesis of amino acid 2a from pyruvate 1a in the presence of NADH. The different colour arrows indicate different possible pathways for the formation of iminium ion IV (red or blue) and the hydride transfer from NADH (green) and match the calculated ones in Fig. 4. Inset: Action of the explicit ammonium ion (in blue) or the explicit water (in red) in assisting the dehydration of hemiaminal (II or III) into IV. | ||
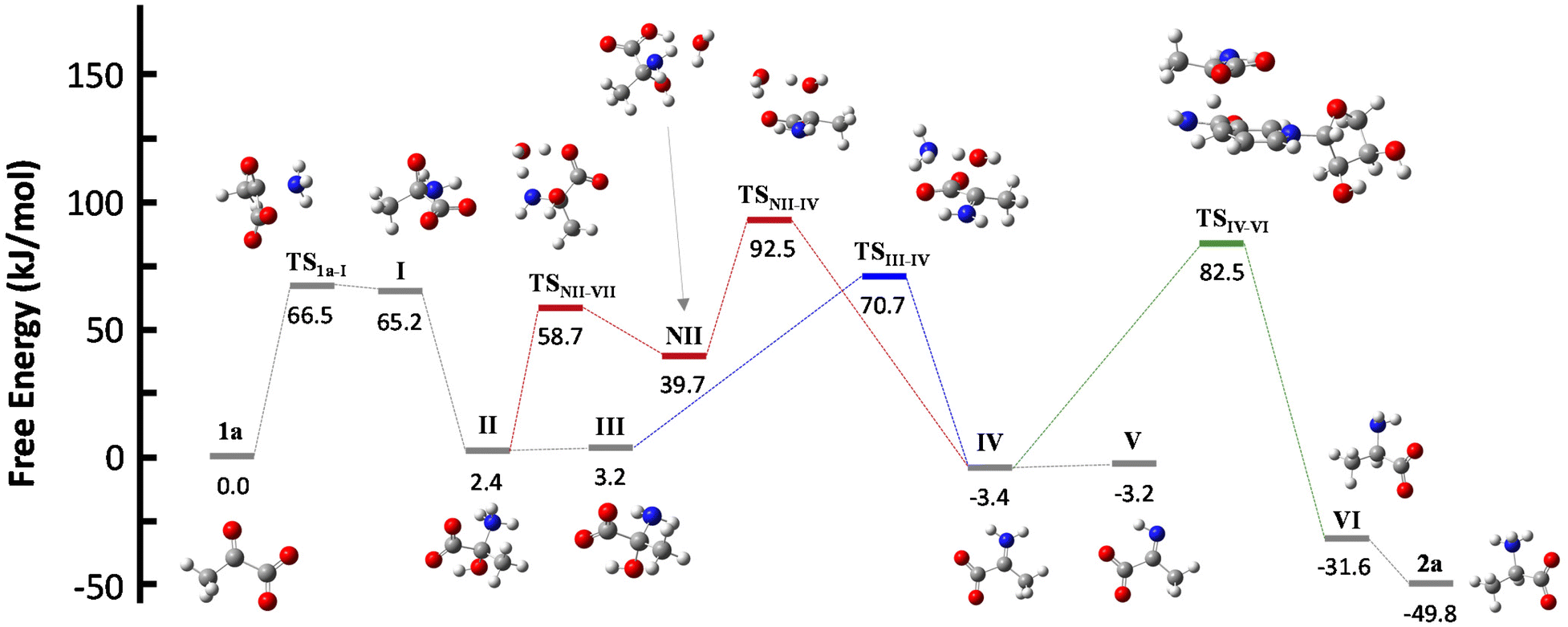 | ||
| Fig. 4 Structures and relative free energies of the different intermediates and transition states for the studied hypothetical mechanisms, applied to the case of pyruvate (1a) as the substrate, pointing to the initial formation of hemiaminal species (II and III). Dehydration of II or III to form an iminium derivative IV, and subsequent reaction with the NADH active part leads to alanine (2a) through accessible energy barriers. Carbon: grey, hydrogen: white, nitrogen: blue, oxygen: red. | ||
It is known that imine species are generated when α-ketoacids are dissolved in aqueous ammonia/ammonium solution via a hemiaminal intermediate.48 Accordingly, the calculated process begins with the nucleophilic attack of an ammonia molecule on the carbonyl group of the ketoacid 1a, affording intermediate I with a barrier of 66.5 kJ mol−1 (TS1a-I). Protonation of I leads to the more thermodynamically favored hemiaminal II,49 with the whole process being slightly endergonic (ΔG1a-II = 2.4 kJ mol−1). Intermediate II can lose a proton from its amine group and get converted into hemiaminal III, having similar free energies.
Dehydration of this hemiaminal would lead to the formation of the imine derivative. However, the implicit solvent model was not enough to describe this step accurately (Fig. S44†), since there must necessarily be explicit molecules of the medium capable of exchanging protons with the intermediate to release a water molecule.50,51 In particular, an explicit ammonium ion, which is abundant in the reaction medium, can in principle form hydrogen bonds with the hydroxyl and carboxylate groups of hemiaminal III. Considering this interaction, the ammonium ion could catalyze the hydroxyl cleavage (first inset of Scheme 4), releasing it in the form of water and ammonia, in an asynchronous one-step process that occurs through a transition state at 70.7 kJ mol−1 (blue pathway, TSIII–IV). In a similar way, the solvent has been evaluated as a possible mediator of this step (second inset of Scheme 4): an explicit water molecule could assist in a double proton transfer of II into non-zwitterionic hemiaminal NII and subsequently promote the dehydration into IV across a transition state at 92.5 kJ mol−1 (red pathway, TSNII–IV). Therefore, although ammonium provides a dehydration mechanism with a lower activation energy, water cannot be discarded to mediate this process.
In any case, dehydration leads to the iminium ion IV, which is moderately more thermodynamically stable than the hemiaminal or pyruvate (ΔG1a-IV = −3.4 kJ mol−1) and constitutes a conjugate acid–base pair with imine V, with both species having similar energy. On these bases, it seems that the equilibrium between pyruvate, its hemiaminal and the imine/iminium species is established through transition states that can be easily overcome at room temperature.
For the hydride transfer step, in turn, two possible pathways were considered. The direct reaction between the hemiaminal II and the active part of NADH has a huge energy barrier of more than 300 kJ mol−1 (TSII–VI, Fig. S45†). Hence, this SN2-type process is not expected to play a significant role in the mechanism. On the other hand, hydride donation from the active part of NADH to iminium ion IV gives rise to amino acid VI with an energy barrier of 85.9 kJ mol−1 with respect to IV (TSIV–VI, green pathway in Scheme 4 and Fig. 4), making the hydride transfer to the iminium ion much more plausible. From there, product VI is finally protonated to the most stable zwitterionic form of 2a (majoritarian species at pH 8). The overall process has a reaction energy of ΔG1a–2a = −49.8 kJ mol−1, therefore being exergonic, as was previously determined experimentally.52
Conclusions
In summary, in this article we have demonstrated how amino acids can be produced through a reaction that is compatible with the chemoautotrophic theory.14 The process occurs under the action of a cofactor, NADH, but in the absence of enzymes, thus providing a link of continuity between prebiotic chemistries leading to the origin of life and the metabolic pathways observed in present-day organisms. Our results show how the reductive amination of α-ketoacids could occur in slightly basic aqueous solutions where ammonia/ammonium is present, mostly through the conversion of their hemiaminal derivative into the iminium species and subsequent hydride donation by the cofactor. Under the studied conditions, the most effective ammonia concentration is 1 M, although the reaction also takes place at lower concentrations, which could be a more plausible scenario on the ancient Earth.53 Concerning the comparison of reactivity between the different ketoacids, glyoxylate (1c) would not be a suitable substrate for the synthesis of glycine through this pathway because, despite its higher reactivity, it tends to degrade the cofactor. Oxaloacetate (1d) is not an adequate substrate either because it undergoes decarboxylation. This leaves pyruvate (1a) and α-ketoglutarate (1b) as the two possible core precursors of amino acids in the first autotrophic protocells, while the rest could be synthesized by transamination using glutamate as the nitrogen source.13Overall, these results provide support for the idea of a primordial NAD+/NADH-dependent protometabolism facilitating certain abiotic chemistries.28 Of course, the existence of this cofactor in prebiotic times cannot be taken for granted, but there exist other dihydronicotinamide hydride donor analogues with much simpler structures that could have played a role in establishing a sequence of stages with increasing efficiencies of the cofactor activity. Another possible limitation of the described reaction, its slow kinetics, could be overcome by using catalysts that operate in conjunction with the dihydronicotinamide derivative, based on either metal ions or small organic molecules. Finally, it may be feasible to turn the process stereoselective, as it has been recently demonstrated for the reduction of α-ketoacids into the corresponding hydroxyacids.54 These and other aspects will be investigated in the near future in order to address the potential prebiotic relevance of this cofactor-mediated synthesis of amino acids.
Abbreviations
| ADPR | Adenosine diphosphate ribose |
| DFT | Density functional theory |
| GC | Gas chromatography |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADH | Reduced nicotinamide adenine dinucleotide |
| NMR | Nuclear magnetic resonance |
| MALDI-TOF | Matrix-assisted laser desorption/ionization – time-of-flight |
| MS | Mass spectrometry |
| TCA | Tricarboxylic acids |
| TMSP-d4 | 3-(Trimethylsilyl)-2,2,3,3-tetradeuteropropionic acid |
| TS | Transition state |
| UV-Vis | Ultraviolet-visible |
Author contributions
N. N. and J. L.-B. performed the experiments, F. A.-G. performed the DFT calculations, A. d. l. E. conceived the project, N. N., J. L.-B., S. V.-G and A. d. l. E. designed and analyzed the experiments, N. N., J. L.-B. and A. d. l. E. wrote and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Spanish Agencia Estatal de Investigación (AEI, projects PID2020-119306GB-100 and PID2022-138470NB-I00) and the European Union (the CLASSY project, the Horizon 2020 research and innovation program, Grant Agreement No. 862081; and the CORENET project, supported by the HorizonEurope research and innovation program and the Swiss State Secretariat for Education, Research and Innovation (SERI), Grant Agreement No. 101046294).References
- M. Frenkel-Pinter, M. Samanta, G. Ashkenasy and L. J. Leman, Prebiotic Peptides: Molecular Hubs in the Origin of Life, Chem. Rev., 2020, 120, 4707–4765 CrossRef CAS PubMed.
- S. L. Miller, A Production of Amino Acids Under Possible Primitive Earth Conditions, Science, 1953, 117, 528–529 CrossRef CAS PubMed.
- S. L. Miller and H. C. Urey, Organic Compound Synthesis on the Primitive Earth, Science, 1959, 130, 245–251 CrossRef CAS PubMed.
- A. P. Johnson, H. J. Cleaves, J. P. Dworkin, D. P. Glavin, A. Lazcano and J. L. Bada, The Miller Volcanic Spark Discharge Experiment, Science, 2008, 322, 404 CrossRef CAS PubMed.
- K. Kvenvolden, J. Lawless, K. Pering, E. Peterson, J. Flores, C. Ponnamperuma, I. R. Kaplan and C. Moore, Evidence for Extraterrestrial Amino-Acids and Hydrocarbons in the Murchison Meteorite, Nature, 1970, 228, 923–926 CrossRef CAS PubMed.
- S. Pizzarello, The Chemistry of Life's Origins: A Carbonaceous Meteorite Perspective, Acc. Chem. Res., 2006, 39, 231–237 CrossRef CAS PubMed.
- K. Altwegg, H. Balsiger, A. Bar-Nun, J. J. Berthelier, A. Bieler, P. Bochsler, C. Briois, U. Calmonte, M. Combi and H. Cottin, et al., Prebiotic Chemicals – Amino acid and Phosphorus – in the Coma of Comet 67P/Churyumov-Gerasimenko, Sci. Adv., 2016, 2, e1600285 CrossRef PubMed.
- P. Ehrenfreund and S. B. Charnley, Organic Molecules in the Interstellar Medium, Comets, and Meteorites: A Voyage from Dark Clouds to the Early Earth, Annu. Rev. Astron. Astrophys., 2000, 38, 427–483 CrossRef CAS.
- K. Ruiz-Mirazo, C. Briones and A. de la Escosura, Prebiotic Systems Chemistry: New Perspectives for the Origins of Life, Chem. Rev., 2014, 114, 285–366 CrossRef CAS PubMed.
- B. H. Patel, C. Percivalle, D. J. Ritson, C. D. Duffy and J. D. Sutherland, Common Origins of RNA, Protein and Lipid Precursors in a Cyano-sulfidic Protometabolism, Nat. Chem., 2015, 7, 301–307 CrossRef CAS PubMed.
- S. Islam and M. W. Powner, Prebiotic Systems Chemistry: Complexity Overcoming Clutter, Chem, 2017, 2, 470–501 CAS.
- K. B. Muchowska, S. J. Varma and J. Moran, Nonenzymatic Metabolic Reactions and Life's Origins, Chem. Rev., 2020, 120, 7708–7744 CrossRef CAS PubMed.
- R. J. Mayer, H. Kaur, S. A. Rauscher and J. Moran, Mechanistic Insight into Metal Ion-Catalyzed Transamination, J. Am. Chem. Soc., 2021, 143, 19099–19111 CrossRef CAS PubMed.
- G. Wächtershäuser, Groundworks for an Evolutionary Biochemistry: The Iron-Sulphur World, Prog. Biophys. Mol. Biol., 1992, 58, 85–201 CrossRef PubMed.
- M. Ralser, An Appeal to Magic? The Discovery of a Non-Enzymatic Metabolism and Its Role in the Origins of Life, Biochem. J., 2018, 475, 2577–2592 CrossRef CAS PubMed.
- Q. Dherbassy, R. J. Mayer, K. B. Muchowska and J. Moran, Metal-Pyridoxal Cooperativity in Nonenzymatic Transamination, J. Am. Chem. Soc., 2023, 145, 13357–13370 CrossRef CAS PubMed.
- O. Khersonsky, S. Malitsky, I. Rogachev and D. S. Tawfik, Role of Chemistry versus Substrate Binding in Recruiting Promiscuous Enzyme Functions, Biochemistry, 2011, 50, 2683–2690 CrossRef CAS PubMed.
- K. B. Muchowska, E. Chevallot-Beroux and J. Moran, Recreating Ancient Metabolic Pathways before Enzymes, Bioorg. Med. Chem., 2019, 27, 2292–2297 CrossRef CAS PubMed.
- K. B. Muchowska, S. J. Varma and J. Moran, Synthesis and Breakdown of Universal Metabolic Precursors Promoted by Iron, Nature, 2019, 569, 104–107 CrossRef CAS PubMed.
- S. Pulletikurti, M. Yadav, G. Springsteen and R. Krishnamurthy, Prebiotic Synthesis of α-Amino Acids and Orotate from α-Ketoacids Potentiates Transition to Extant Metabolic Pathways, Nat. Chem., 2022, 14, 1142–1150 CrossRef CAS PubMed.
- R. F. Borch, M. D. Bernstein and H. D. Durst, The Cyanohydroborate Anion as a Selective Reducing Agent, J. Am. Chem. Soc., 1971, 93, 2897–2904 CrossRef CAS.
- S. Ogo, K. Uehara, T. Abura and S. Fukuzumi, pH-Dependent Chemoselective Synthesis of α-Amino Acids. Reductive Amination of α-Ketoacids with Ammonia Catalyzed by Acid-Stable Iridium Hydride Complexes in Water, J. Am. Chem. Soc., 2004, 126, 3020–3021 CrossRef CAS PubMed.
- C. Huber and G. Wächtershäuser, Primordial Reductive Amination Revisited, Tetrahedron Lett., 2003, 44, 1695–1697 CrossRef CAS.
- W. Wang, Q. Li, B. Yang, X. Liu, Y. Yang and W. Su, Photocatalytic Reversible Amination of α-Ketoacids on a Zn-S surface: Implications for the Prebiotic Metabolism, Chem. Commun., 2012, 48, 2146–2148 RSC.
- L. M. Barge, E. Flores, M. M. Baum, D. G. Van der Velde and M. J. Russell, Redox and pH Gradients Drive Amino Acid Synthesis in Iron Oxyhydroxy Mineral Systems, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4828–4833 CrossRef CAS PubMed.
- C. Bonfio, E. Godino, M. Corsini, F. Fabrizi de Biani, G. Guella and S. S. Mansy, Prebiotic Iron-Sulfur Peptide Catalysts Generate a pH Gradient Across Model Membranes of Late Protocells, Nat. Catal., 2018, 1, 616–623 CrossRef CAS.
- P. Dalai and N. Sahai, A Model Protometabolic Pathway across Protocell Membranes Assisted by Photocatalytic Minerals, J. Phys. Chem. C, 2020, 124, 1469–1477 CrossRef CAS.
- D. P. Summers and D. Rodoni, Vesicle Encapsulation of a Nonbiological Photochemical System Capable of Reducing NAD+ to NADH, Langmuir, 2015, 31, 10633–10637 CrossRef CAS PubMed.
- S. Basak, S. Nader and S. S. Mansy, Protometabolic Reduction of NAD+ with α-Ketoacids, J. Am. Chem. Soc. Au, 2021, 1, 371–374 CAS.
- R. J. Mayer and J. Moran, Quantifying Reductive Amination in Nonenzymatic Amino Acid Synthesis, Angew. Chem., Int. Ed., 2022, 61, e202212237 CrossRef CAS PubMed.
- J. J. Steffens and D. M. Chipman, Reactions of Dihydronicotinamides. I. Evidence for an Intermediate in the Reduction of Trifluoroacetophenone by 1-Substituted Dihy-dronicotinamides, J. Am. Chem. Soc., 1971, 93, 6694–6696 CrossRef CAS PubMed.
- S. Tsukiji, S. B. Pattnaik and H. Suga, Reduction of an Aldehyde by a NADH/Zn2+-Dependent Redox Active Ribozyme, J. Am. Chem. Soc., 2004, 126, 5044–5045 CrossRef CAS PubMed.
- In this text, aqueous mixtures of NH3 and NH4Cl have been generically named “ammonia solution”, although solutions at pH 7, 8 or 9 contain more NH4Cl than NH3 (pKa NH4+ = 9.3) (Table S1†).
- Pyruvate could not be monitored because it is present in the aqueous medium as a mixture of different species (e.g., as pyruvate hydrate) in equilibrium (Fig. S13†).
- A slight (2%) decay of 1H-NMR yield was observed at 168 h for the curves recorded at pH 7 and 8 (see Fig. 2A), which does not happen under other conditions or for glutamate formation (Fig. S7 and S8†). This may be due to a slow consumption of alanine by a side process or just due to the experimental error normally associated with quantitative NMR: see. S. K. Bharti and R. Roy, Quantitative 1H NMR spectroscopy, Trends Anal. Chem., 2012, 35, 5–26 CrossRef CAS.
- pH measurements indicate that pH only undergoes small variations (±0.15 at most) throughout the reaction progression (see Table S12†).
- J. Feldmann, Y. Li and Y. Tor, Emissive Synthetic Cofactors: A Highly Responsive NAD+ Analogue Reveals Biomolecular Recognition Features, Chem. – Eur. J., 2019, 25, 4379–4389 CrossRef CAS PubMed.
- F. Ravalico, I. Messina, M. V. Berberian, S. L. James, M. E. Migaud and J. S. Vyle, Rapid Synthesis of Nucleotide Pyrophosphate Linkages in a Ball Mill, Org. Biomol. Chem., 2011, 9, 6496–6497 RSC.
- A. J. Hoefnagel, H. van Bekkum and J. A. Peters, The Reaction of Glyoxylic Acid with Ammonia Revisited, J. Org. Chem., 1992, 57, 3916–3921 CrossRef CAS.
- Part of the NAD+ originated in this first process can also be hydrolysed into ADPR (see Fig. S12†).
- Control experiments of NADH in which the ketoacids (1) are not present show that the ammonia solution (or other aqueous basic buffers) also promotes the oxidation of NADH into NAD+ and the hydrolysis of NADH and NAD+ into ADPR, albeit to a lesser extent (see Tables S13–S15†). These are minor background processes in comparison with the processes described in the main text.
- J. GÈ©bicki, A. Marcinek and J. Zielonka, Transient Species in the Stepwise Interconversion of NADH and NAD+, Acc. Chem. Res., 2004, 37, 379–386 CrossRef PubMed.
- S. Fukuzaki, O. Inada and T. Suenobu, Mechanisms of Electron-Transfer Oxidation of NADH Analogues and Chemiluminescence. Detection of the Keto and Enol Radical Cations, J. Am. Chem. Soc., 2003, 125, 4808–4816 CrossRef PubMed.
- R. J. Mayer and J. Moran, Quantification of the Hydride Donor Abilities of NADH, NADPH, and BH3CN− in Water, Org. Biomol. Chem., 2023, 21, 85–88 RSC.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermo-chemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. Bursch, J.-M. Mewes, A. Hansen and S. Grimme, Best-Practice DFT Protocols for Basic Molecular Computational Chemistry, Angew. Chem., Int. Ed., 2022, 61, e202205735 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- P. Zuman, Die Reaktion der Karbonylverbindungen mit primären Aminen, Collect. Czech. Chem. Commun., 1950, 15, 839–873 CrossRef.
- This step was also calculated considering explicitly the protonation source during the addition of ammonia, with the results not differing significantly (Fig. S43†).
- S. J. Oliphant and R. H. Morris, Density Functional Theory Study on the Selective Reductive Amination of Aldehydes and Ketones over Their Reductions to Alcohols Using Sodium Triacetoxyborohydride, ACS Omega, 2022, 7, 30554–30564 CrossRef CAS PubMed.
- C. Solís-Calero, J. Ortega-Castro, A. Hernández-Laguna and F. Muñoz, A Comparative DFT Study of the Schiff Base Formation from Acetaldehyde and Butylamine, Glycine and Phosphatidylethanolamine, Theor. Chem. Acc., 2012, 131, 1263 Search PubMed.
- C. E. Grimshaw and W. W. Cleland, Kinetic Mechanism of Bacillus subtilis L-Alanine Dehydrogenase, Biochemistry, 1981, 20, 5650–5655 CrossRef CAS PubMed.
- E. E. Stüeken, Nitrogen in Ancient Mud: A Biosignature?, Astrobiology, 2016, 16, 730–735 CrossRef PubMed.
- R. Mayer and J. Moran, Metal Ions Turn on a Stereoselective Nonenzymatic Reduction of Keto Acids by the Coenzyme NADH, ChemRxiv, 2023 DOI:10.26434/chemrxiv-2023-jdhxk.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures for the studied reaction and its analysis, calibration, tables indicating the quantified conversions under different conditions, 1H-NMR spectra, GC-MS data, MALDI-TOF MS spectra, UV-vis spectra and specific DFT calculated data. See DOI: https://doi.org/10.1039/d4qo00050a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |