
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advancements in the synthesis of polyoxygenated oxepanes and thiepanes for applications to natural products
Aditya R.
Pote†
 a,
Shayne M.
Weierbach†
b,
Mark W.
Peczuh
c and
Kyle M.
Lambert
*b
a,
Shayne M.
Weierbach†
b,
Mark W.
Peczuh
c and
Kyle M.
Lambert
*b
aAstraZeneca PLC, 35 Gatehouse Drive, Waltham, MA 02451, USA
bDepartment of Chemistry and Biochemistry, Old Dominion University, 4501 Elkhorn Ave, Norfolk, VA 23529, USA. E-mail: kmlamber@odu.edu
cDepartment of Chemistry, University of Connecticut, 55 North Eagleville Rd, Storrs, CT 06269, USA
First published on 10th May 2024
Abstract
Oxepanes are central motifs and tenants of many biologically important molecules, and their synthetic construction often presents a challenge to chemists due to consequential entropic and enthalpic barriers that have limited the synthetic toolbox to access these seven-membered oxacycles. This review covers the breadth of synthetic methods to afford the oxepane/thiepane moiety, with a focus on polyoxygenated oxepanes and includes radical cyclizations, Lewis acid-mediated cyclizations, ring closing-metathesis, Nicholas–Ferrier rearrangement, homologations, and ring-expansion strategies. Implementation of these tactics towards sugar-based and non-sugar based (de novo) approaches is presented alongside their extensive application to the total synthesis of several complex polyoxygenated oxepane-containing natural products, which are also highlighted.
 Aditya R. Pote | Dr Aditya R. Pote is currently employed at AstraZeneca Pharmaceuticals LC as a Senior Scientist within the Early Oncology Chemistry (TTD) department. Dr Pote received his Ph.D. degree (2018) in Chemistry from the University of Connecticut under the guidance of Prof. Mark W. Peczuh where his dissertation research focused on the development of new strategies for synthesizing seven-membered septanose glycosides. He worked as a postdoctoral associate in Prof. Andrew G. Myers’ laboratory at Harvard University from 2018–2020 until starting his industrial career at AstraZeneca as a medicinal chemist. His current area of research is focused on the development of Antibody Drug Conjugates (ADC) for cancer treatment. |
 Shayne M. Weierbach | Shayne M. Weierbach obtained his B.S. in Chemistry at Christopher Newport University in 2021. At CNU he participated in undergraduate research and worked on the total synthesis of the torreyunlignans A–D under Prof. Jeffrey Carney. Shayne is currently a Ph.D. candidate in Chemistry at Old Dominion University working under the supervision of Prof. Kyle Lambert and is investigating the development of new oxidative methods and other novel synthetic transformations, as well as natural product total synthesis. |
 Mark W. Peczuh | Mark W. Peczuh received his Ph.D. degree (1999) in Organic Chemistry under the mentorship of Andrew Hamilton at Yale University. A post-doctoral stint at Princeton University with Dan Kahne (1999–2001) working on the chemoenzymatic synthesis of vancomycin analogs sparked his interest in carbohydrate chemistry. Peczuh began his independent career at the University of Connecticut in 2001 where he is now Professor of Chemistry. There his research program focuses on the design, synthesis, and utilization of glycomimetics, most notably seven-membered ring septanoses as ring-expanded analogs of pyranoses. |
 Kyle M. Lambert | Dr Kyle M. Lambert is an Assistant Professor in the Department of Chemistry and Biochemistry at Old Dominion University. Dr Lambert received his PhD in Chemistry in 2017 from the University of Connecticut under the supervision of Prof. Bill Bailey where his dissertation research focused on developing selective oxidations using oxoammonium salts. He was a NIH Ruth L. Kirschstein postdoctoral fellow in Prof. John Wood's group at Baylor University from 2017–2020 until starting his independent career at Old Dominion University. His group's research is focused on the synthesis of natural products, transition metal catalysis, computational chemistry, and developing new oxidative transformations. |
1. Introduction
Oxepanes are seven-membered cyclic ethers that are important motifs found within physiologically relevant small molecules and exhibit complementary, as well as differing biological activities to their six-membered counterparts.1 Oxepanes can vary in structural or stereochemical complexity and are often found as core structures embedded within biologically active natural product targets such as hemibrevetoxin B (1), nakorone (2), gambierol (3), zoapatanol (4), as well as pharmaceuticals such as artemisinin (5) (Fig. 1).1–3 Given the prevalence of this cyclic ether motif in these synthetically challenging targets combined with the unfavorable entropic and enthalpic barriers that preclude formation of such ring-expanded oxacycles, efficient methods for their preparation are of high interest to synthetic and medicinal chemists.4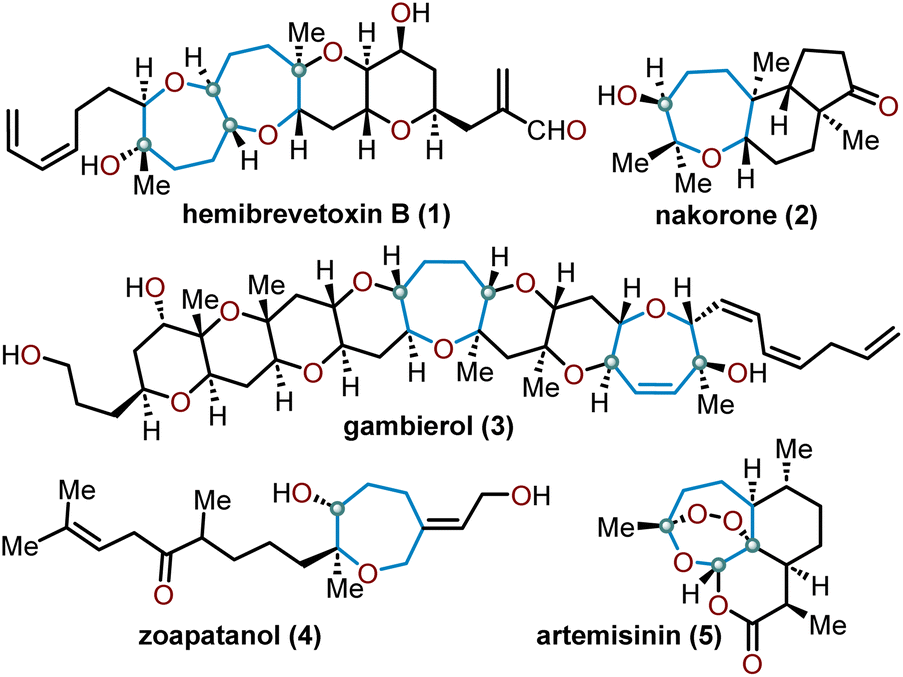 | ||
| Fig. 1 Biologically relevant natural products containing an oxepane. | ||
There is a plethora of developed methodologies that have been utilized to construct the oxepane moiety (Scheme 1). Various strategies such as radical cyclizations, Lewis acid-mediated cyclizations, and ring-closing metathesis are widely accepted cyclization strategies to access oxepanes from acyclic precursors. Additionally, ring-expansion of cyclopropanated glycals, homologations, and the Nicholas–Ferrier rearrangement are commonly used synthetic tactics to derive oxepanes from their pyranyl analogs. The routes taken to prepare these seven-membered oxacycles arise from either sugar-based or non-sugar-based starting materials. Advancements in the preparation of these seven-membered cyclic ethers have substantially impacted the synthesis of complex natural products laden with oxepane units within their scaffolds.
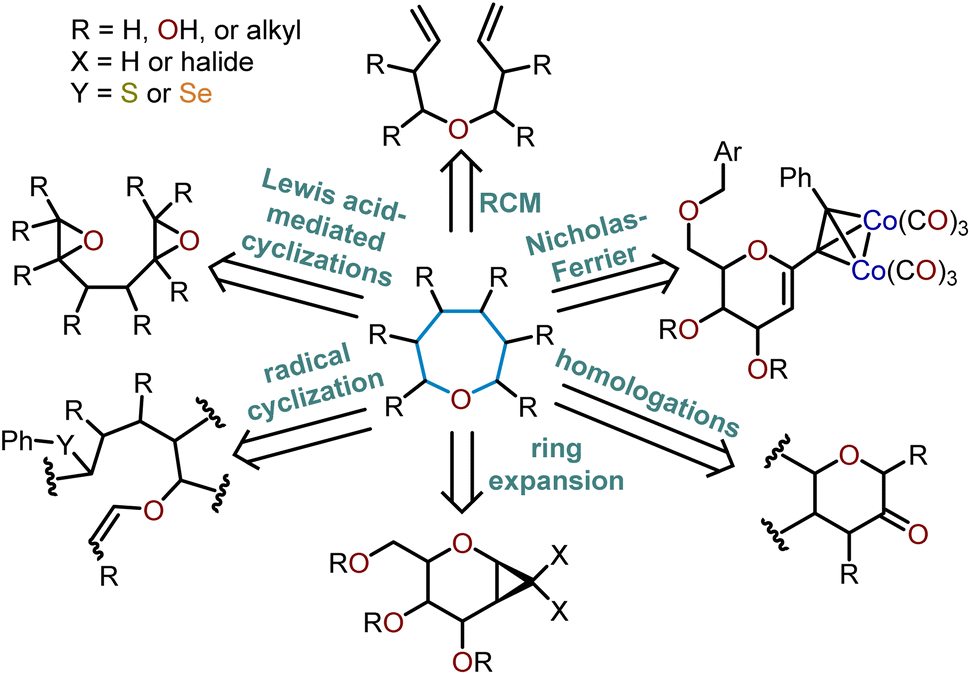 | ||
| Scheme 1 General strategies to access seven-membered oxacycles. | ||
The oxepane motif is common within the core structures of various biologically relevant molecules of marine origin. Amongst the array of oxepane-containing natural products, the biological effects range from extremely lethal to potential therapeutic agents. Additionally, the total synthesis of these target molecules has been a daunting task due to the extensive structural and stereochemical complexity of these seven-membered polycyclic ethers. To this point, the total synthesis of these complex molecules has been a highly sought-after area of research.
Other implications of deriving the seven-membered oxacycles come from their close resemblance to their six-membered analogs. In terms of physiochemical properties as well as activity towards biological targets (i.e., serving as ligands for lectins and substrates for glycosidases)5,6 the synthesis of polyoxygenated oxepanes has medicinal importance.
Current synthetic routes to access oxepanes are outlined in the present work, which covers the synthesis of functionalized oxacycles with an emphasis on methods to prepare polyoxygenated oxepanes via cyclizations, ring-closing metathesis (RCM), rearrangement, ring expansion, and homologation strategies (Scheme 1). Applications of these synthetic advancements to the construction of bioactive oxepane-containing natural products and synthetic analogs is also covered.
2. Sugar-based approaches to oxepanes and septanoses
The preparation of oxepanes from pyranose sugars or glycals is among the most commonly used synthetic strategy mainly because it incorporates nearly all of the atoms and sets the stereochemistry at the required stereocenters without any additional effort. Sugar-based approaches to synthesize the oxepanes can be divided into four different sub-categories based on the treatment of the sugar derivatives in order to obtain the appropriate starting material.2.1. Ring-expansion of cyclopropanated glycals
Ring-expansion via cyclopropanated glycals is among the most widely explored area for the synthesis of polyhydroxylated oxepanes and septanoses. Such a strategy involves the cyclopropanation of carbohydrate-based glucals or galactals followed by an acid or base-mediated ring-opening event to obtain the corresponding seven-membered oxepine (e.g., Scheme 2).7,8 The resulting oxepine provides a valuable olefin functional handle that allows for further derivatization (e.g., dihydroxylation, halogenation, arylation, etc.) to afford a variety of oxepanes and septanose carbohydrates. In the following section, strategies to construct functionalized oxepanes from commercially available glycals through ring-expansion approaches are covered.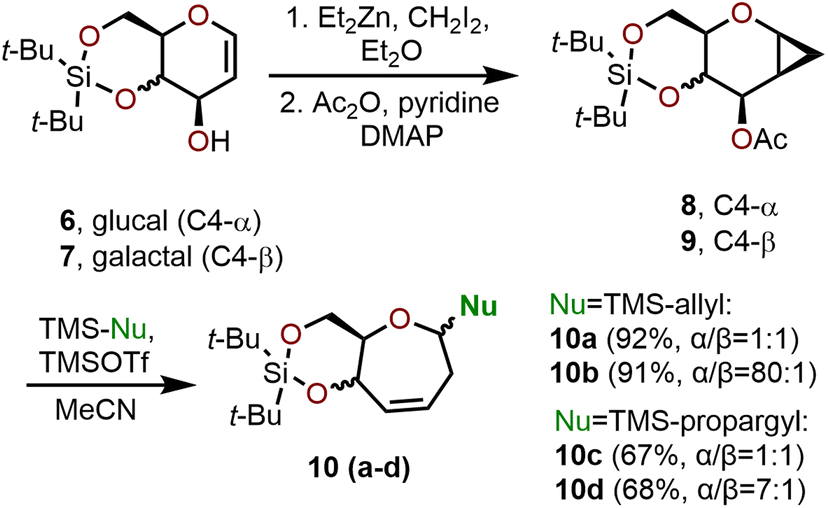 | ||
| Scheme 2 Hoberg and coworkers’ ring expansion to access oxepanes using silylated C-nucleophiles.7–11 | ||
The ring-expansion of cyclopropanated carbohydrates to afford oxepanes has been thoroughly investigated by Hoberg and coworkers, who have demonstrated that the Lewis acid-catalyzed ring expansion of glucal and galactal systems can be executed with trimethylsilyl triflate (TMSOTf) in a process that is compatible with many silylated nucleophiles.7–11 Hoberg and coworkers’ strategy begins with silyl-protected glycals (e.g., 6 or 7), which are converted to cyclopropanated sugars using the Furukawa modification of the Simmons–Smith reaction as depicted in Scheme 2. The hydroxyl-directed cyclopropanation proceeds from the β-face with a 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio (dr).8 To set the stage for the ring expansion event, the C3 hydroxyl group (pyranose numbering) of the glycal is acetylated using acetic anhydride and pyridine as a base. In the event, the acetylated glycal (e.g., 8 or 9) is treated with a catalytic amount of TMSOTf, which results in the loss of the C3 acetate and enables the ring-opening of the cyclopropane to afford a seven-membered oxonium intermediate that is then intercepted by a nucleophile at C1.7 The diastereoselectivity of the reaction was found to vary based on the nucleophile and starting material used. Cyclopropanated galactals resulted in higher diastereoselectivity (up to 80:1 dr), whereas their glucal counterparts resulted in much lower diastereoselectivity. For example, in the case of silyl-protected glucal 8, the formation of allylated oxepane 10a and allenylated oxepane 10c is observed from treatment with the corresponding TMS-allyl and TMS-propargyl nucleophiles affording the products in 92% and 67% yields, respectively, but the process is not diastereoselective resulting in 1:1 α/β selectivity in both cases.8 In contrast, when silyl-protected galactal 9 is treated with the same two nucleophiles, allylated oxepane 10b and allenylated oxepane 10d are in turn formed in 91% and 68% yields and with a high degree of α/β selectivity at C1 (10b = 80:1 and 10d = 7:1). This large difference in selectivity is explained by the planar nature of the oxocarbenium ion intermediate formed from glucals, which can be accessed from both faces by approaching nucleophiles during anomeric bond formation, whereas in galactals the β-face is hindered by the tethered silanediyl resulting in preferential approach of the nucleophile from the α-face.9,11
:1 diastereomeric ratio (dr).8 To set the stage for the ring expansion event, the C3 hydroxyl group (pyranose numbering) of the glycal is acetylated using acetic anhydride and pyridine as a base. In the event, the acetylated glycal (e.g., 8 or 9) is treated with a catalytic amount of TMSOTf, which results in the loss of the C3 acetate and enables the ring-opening of the cyclopropane to afford a seven-membered oxonium intermediate that is then intercepted by a nucleophile at C1.7 The diastereoselectivity of the reaction was found to vary based on the nucleophile and starting material used. Cyclopropanated galactals resulted in higher diastereoselectivity (up to 80:1 dr), whereas their glucal counterparts resulted in much lower diastereoselectivity. For example, in the case of silyl-protected glucal 8, the formation of allylated oxepane 10a and allenylated oxepane 10c is observed from treatment with the corresponding TMS-allyl and TMS-propargyl nucleophiles affording the products in 92% and 67% yields, respectively, but the process is not diastereoselective resulting in 1:1 α/β selectivity in both cases.8 In contrast, when silyl-protected galactal 9 is treated with the same two nucleophiles, allylated oxepane 10b and allenylated oxepane 10d are in turn formed in 91% and 68% yields and with a high degree of α/β selectivity at C1 (10b = 80:1 and 10d = 7:1). This large difference in selectivity is explained by the planar nature of the oxocarbenium ion intermediate formed from glucals, which can be accessed from both faces by approaching nucleophiles during anomeric bond formation, whereas in galactals the β-face is hindered by the tethered silanediyl resulting in preferential approach of the nucleophile from the α-face.9,11
The utility of this ring-expansion protocol toward polyoxygenated oxepanes was demonstrated by subjecting cyclopropanated galactal 9 to treatment with TMSOTf in presence of silylketene acetal 11 to afford oxepine 12 in 77% yield and 80:1 dr (Scheme 3). The olefin moiety in 12 underwent dihydroxylation from the α-face (>100:1 selectivity) under Upjohn conditions to provide 13 in a 59% yield. Other strategies have also been utilized for olefin functionalization such as epoxidation, halogenation and hydroboration–oxidation to obtain the respective oxepane systems with yields ranging from 40–90%.10
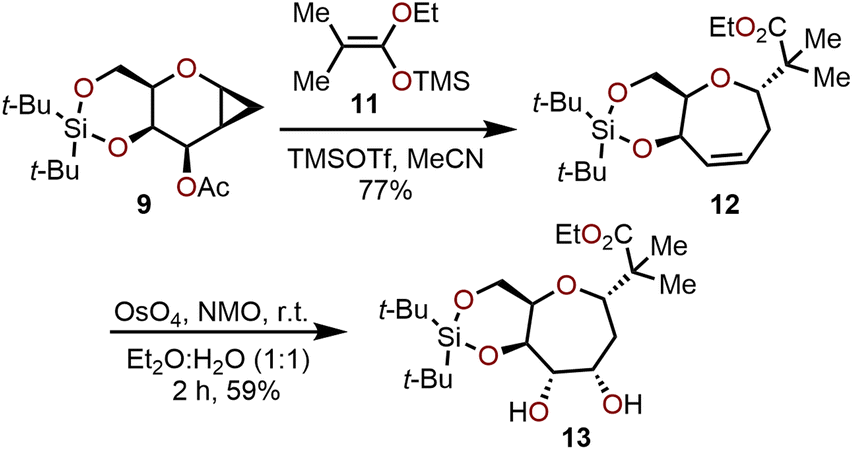 | ||
| Scheme 3 Advancement of cyclopropanated sugar 9 toward polyoxygenated oxepanes through Hoberg's ring-expansion protocol and dihydroxylation.10 | ||
Sabatino and coworkers used a similar cyclopropanation/ring-expansion protocol for the synthesis of oxepane nucleic acids (ONAs) (Scheme 4).12 ONAs are sugar phosphate oligomers in which the pentafuranose ring of DNA and RNA is replaced with a seven-membered oxepane ring. These unnatural analogs have been probed in biological studies which have provided new insights into the structure and function of natural and unnatural genetic systems. The same glycal donor 8 used by Hoberg and coworkers was treated with TMSOTf in acetonitrile solvent in presence of a persilylated thymine (Thy) or persilylated N6-benzoyladenine (AdeBz) nucleophile. The reaction affords oxepine nucleosides 14a or 14b in 40% and 45% yield respectively alongside 15–30% of diene 15. The product distribution of the ring opening reaction was found to be dependent on the potency of the nucleophile and in the case of persilylated thymine the reaction proceeded at a slower rate with an observed α:β selectivity of 1:10, whereas for persilylated N6-benzoyladenine the α:β selectivity was only 1:2.12 The oxepine products 14a (N1) and 14b (N9) were then desilylated using tetrabutylammonium fluoride (TBAF) and the olefinic moiety at C3–C4 (oxepine numbering) was reduced using standard catalytic hydrogenation conditions (Pd/C, 1 atm H2, MeOH) to obtain the saturated ONAs in 60–63% yield over these two-steps.
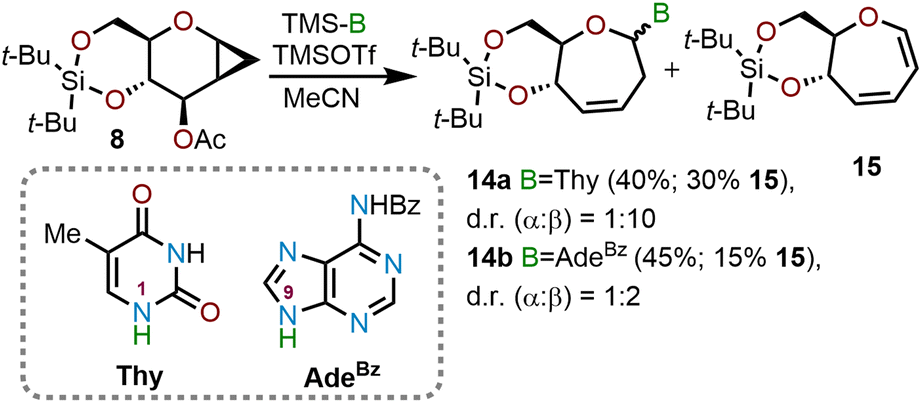 | ||
| Scheme 4 Synthesis of oxepane nucleic acids (ONAs) via cyclopropanation strategy.12 | ||
A ring-expansion strategy to access oxepanes has also been used by Dey and coworkers who investigated the expansion of dihalocyclopropane oxyglycals (Scheme 5).13,14 Their efforts began with a methylene insertion into the globally Bn-protected oxyglycal 16 using dibromocarbene generated via a haloform reaction under phase-transfer conditions using benzyltriethylammonium chloride (TEBAC).
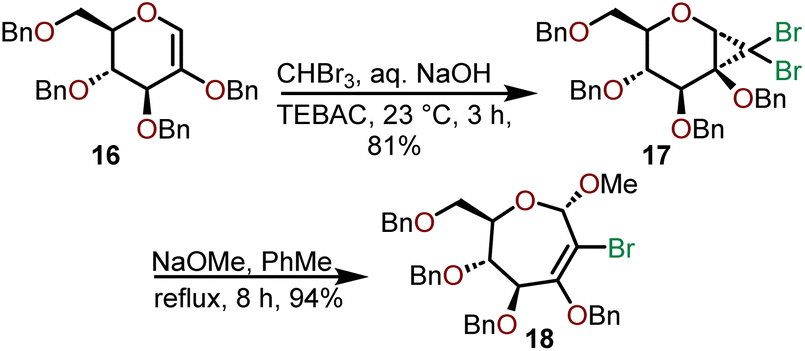 | ||
| Scheme 5 Synthesis of dibromocyclopropane oxyglycal via ring-expansion.13,14 | ||
The dihalocyclopropyl unit is then opened using sodium methoxide in refluxing toluene to afford 2-bromooxepine 18, which can serve as a versatile intermediate that can be subjected to further oxidation or used in organometallic transformations as shown in Scheme 6.13,14
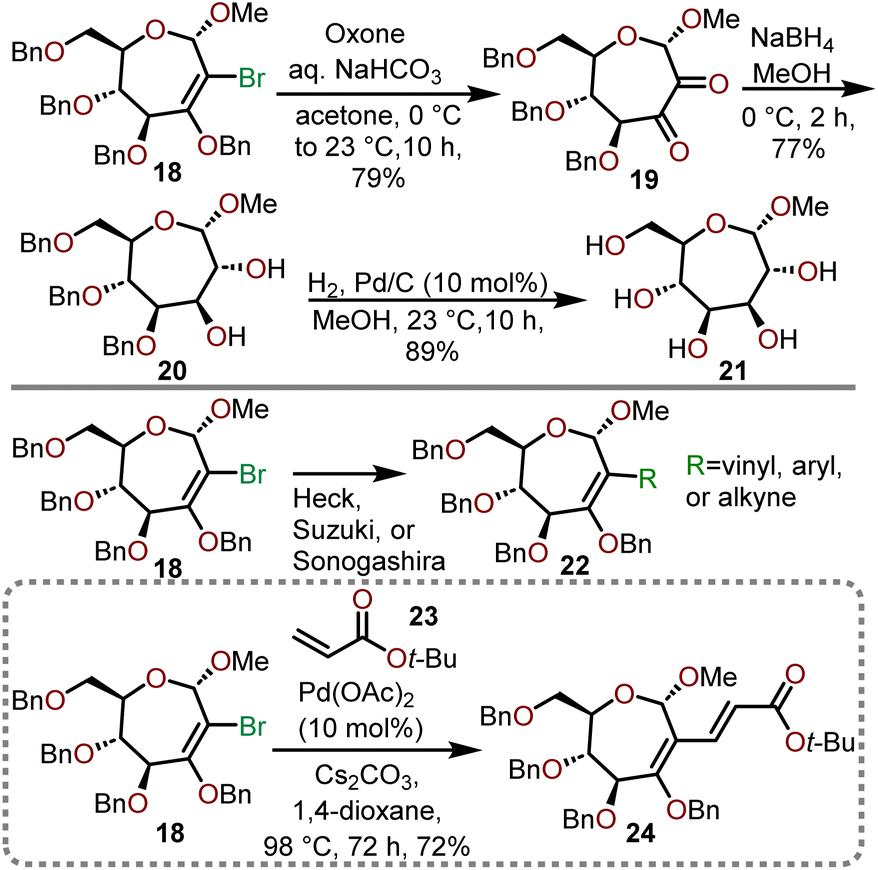 | ||
| Scheme 6 Examples of further functionalization and advancement of key intermediate 18 toward polyoxygenated oxepanes.13,14 | ||
Epoxidation of key intermediate 18 with in situ generated dimethyldioxirane (DMDO) under aqueous alkaline conditions provides dione 19, which can be subsequently reduced with sodium borohydride (NaBH4) to afford the diol 20. Catalytic hydrogenolysis with palladium on carbon was sufficient to globally deprotect the remaining benzyl ethers to yield polyhydroxylated oxepane 21 in good yield. The ability of 18 to engage in C–C bond-forming reactions mediated by transition metals further highlights its versatility as a key intermediate towards polyoxygenated oxepanes (18 → 22). Dey and coworkers demonstrate that the vinyl bromide functional handle in 18 can undergo a Heck coupling with tert-butyl acrylate (23) in the presence of Pd(OAc)2 catalyst to provide desired oxepane 24 in 72% yield (Scheme 6).13,14
2.2 Nicholas–Ferrier rearrangement of pyranosidic cations
The Ferrier rearrangement and Nicholas reaction are widely explored synthetic transformations which involve the formation of cationic intermediates.15,16 Gómez and coworkers have studied the behavior of Nicholas pyranosidic cations leading towards the formation of polyhydroxylated oxepanes from C6 O-arylated derivatives of D-glucal sugars (Scheme 7).17 The precursors to the pyranosidic cations (26) were prepared in four steps from D-glucals (25). Beginning with treatment of the Nicholas products 26 with boron trifluoride diethyl etherate (BF3·OEt2) in methylene chloride solvent, the transformation takes place through a three-step reaction sequence (Scheme 7; bottom). The process involves: (i) a 1,6-hydride transfer onto the cyclic double bond of the C3 debenzylated intermediate 29 to generate the more reactive oxocarbenium species 30, (ii) a two-step electrophilic addition (AdE) by the olefin moiety in 30 to form bicyclic system 31, and (iii) pyranosidic ring opening by way of an intermediate hemiacetal to obtain the desired oxepane system 28. The reaction can be conducted with a variety of aryl substituents on C6–OH (pyranose numbering) and through removal of adventitious water with 4 Å molecular sieves, the corresponding oxepanes were formed in moderate to high yields. The oxepane product 28 is formed preferentially over the 1,6-anhydro product 27 when molecular sieves are utilized to prevent the hydrolysis of oxocarbenium ion intermediate 30, which leads to 27 by way of 30a and 31a (Scheme 7).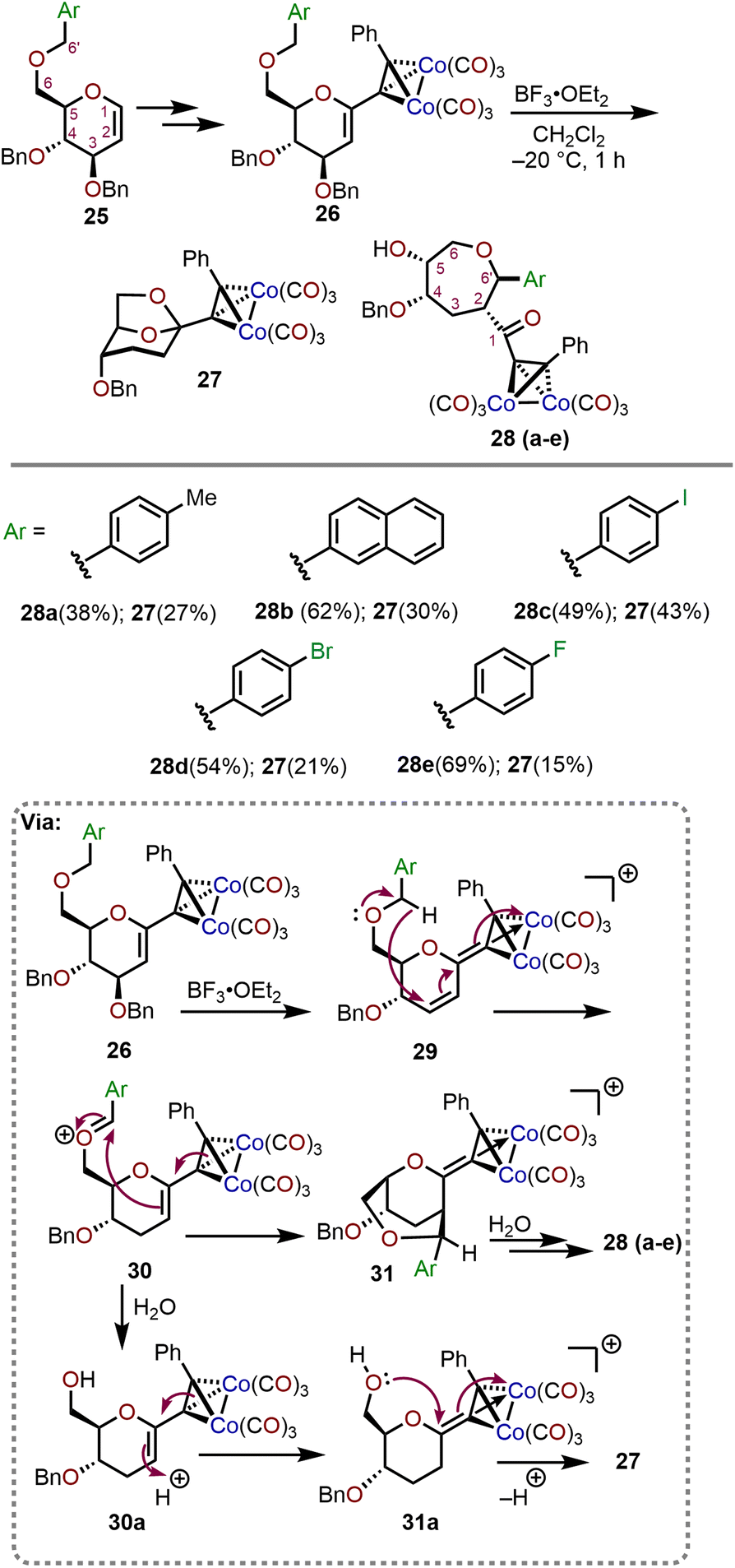 | ||
| Scheme 7 Formation of oxepanes via Nicholas–Ferrier rearrangement.17 | ||
Further studies were conducted with electronically differentiated aryl substituted glycals allowing for synthesis of the respective oxepanes (28a–e) with yields ranging from 38–69%. As observed in Scheme 7, the substrates with electron-withdrawing groups favor the formation of the oxepane system, whereas electron-rich aryl systems result in a less electrophilic oxocarbenium 30 and are prone to hydrolysis by adventitious water affording greater amounts of 27. Furthermore, two stereogenic centers are formed during the process and the stereochemical outcome at C2 (carbohydrate numbering) is governed by the geometric restrictions imposed on the approach of the of C6 substituent (carbohydrate numbering). The stereogenic center at C6′ (carbohydrate numbering) is dictated by the preferred rotamer of oxocarbenium 30 rotating the aryl group away from the bulky dicobalt hexacarbonyl moiety. The presence of a benzyl-type substituent at C6 (pyranose numbering) triggers the formation of substituted oxepanes and can be eliminated by use of a different substituent (e.g., TBS) at the same position.
2.3 Cyclization of sugar-based polyhydric alcohols
Another approach that harnesses the innate stereochemistry of sugar-based starting materials to access polyoxygenated oxepanes is the use of polyhydric alcohols. Pavlik and coworkers have demonstrated that the formation of larger cyclic ethers can be accomplished by utilizing carbohydrate-based starting materials such as mannitol or sorbitol.18 This approach is useful to generate five, seven, and eight-membered cyclic ethers without the need for protection of pendant hydroxyl units, and furthermore, the ring closure proceeds with retention of stereochemistry. The authors first observed this reactivity when D-sorbitol (32) was treated with 1 mol% of triflic acid (TfOH) in refluxing toluene, which upon careful NMR analysis was found to have afforded 1,6-anhydrosorbitol (33) as a single product, which was isolated in a 79% yield (Scheme 8).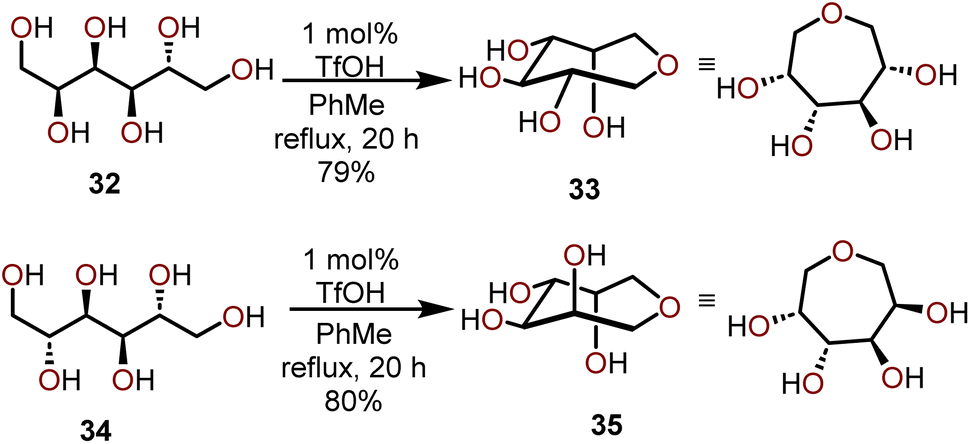 | ||
| Scheme 8 Synthesis of oxepanes via cyclization of polyhydric alcohols.18 | ||
Similarly, when D-mannitol (34) was treated with 1 mol% TfOH under the same conditions, they observed the formation of a single tetrahydroxy oxepane product 35, which was isolated in an 80% yield. The stereochemistry of the hydroxyl units in the starting materials was retained in both reactions, and the formed products were found to be stable under acidic conditions. The structure of the product obtained from the reaction of D-mannitol (34) was unambiguously determined to be 1,6-anhydromannitol (35) by spectroscopic comparison to a synthetic standard of 35 prepared in a six-step sequence from a 2,3-O-isopropylidene-D-erythronolactone commercial starting material.
An alternative strategy for utilizing chiral dianhydrosugars for regio- and stereoselective cyclizations has been demonstrated by Satoh and coworkers.19 The C2-symmetric dianhydrosugar 36 with a (2R,5R)-configuration contains two epoxy groups with similar reactivity. As depicted in Scheme 9, a (R,R)-(salen)-Co(III)Ac catalyst 37 is able to promote the cyclization of 36 at room temperature with a substrate:catalyst ratio of 200:1, producing ca. 28% of 1,6-anhydro-3,4-di-O-methyl-D-mannitol (39) along with ca. 57% of 2,5-anhydro-3,5-di-O-methyl-D-glucitol (38) and ca. 6% of a bicyclic dianhydro product 1,6:2,5-dianhydro-3,4-di-O-methyl-D-glucitol (40). The oxepane product 39 arises from the enantioselective hydrolysis of one of the epoxides by (R,R)-37, followed by cyclization of resulting diol into the other epoxide (e.g., 42; Scheme 9). First, the starting material 36 is coordinated with catalyst (R,R)-37 to allow the endo cleavage of one of the epoxide groups in presence of water. During the process, the hydroxyl group remains coordinated to the catalyst and upon coordination of the other epoxy group with another molecule of (R,R)-37 catalyst, an intramolecular 7-endo-cyclization affords oxepane 39.19–21 By-product 38 results from 5-exo-cyclization of the secondary hydroxyl (e.g., 41; Scheme 9). It is important to note that oxepane 39 is formed by a 7-endo cyclization of the initially formed epoxy alcohol, meaning that its formation is not affected by inherent stereoelectronic preference for the intramolecular exo-attack for cyclization as predicted by Baldwin's rules.21 The mechanism of formation of seven-membered ring by endo attack was confirmed by the increase in the molar fraction of the (R,R)-37 catalyst in the reaction system led to increasing yields of oxepane 39.
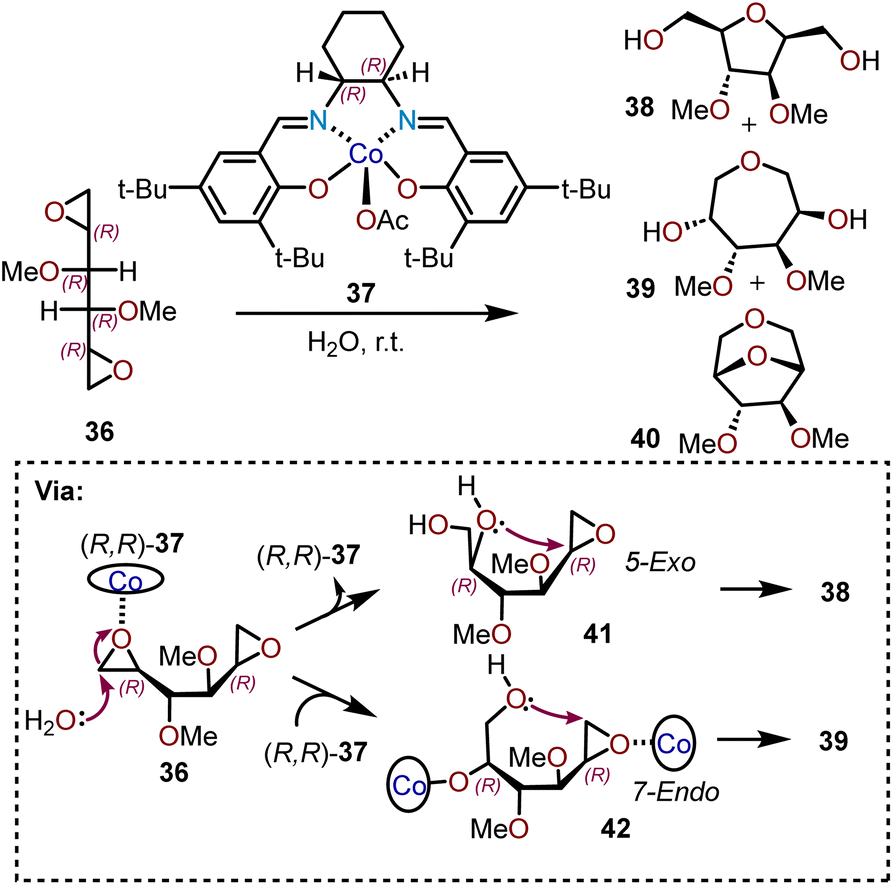 | ||
| Scheme 9 Cyclization of dianhydrosugar alcohols.19 | ||
2.4 Stepwise homologation of pyranoses and furanoses
Homologation of commercially available pyranose derivatives provides another useful strategy towards the synthesis of polyoxygenated oxepanes.22 Vannam and Peczuh have demonstrated a concise synthesis of oxepanes via electrophilic C–O cyclization of allylic alcohols prepared from pyranose lactols as illustrated in the Scheme 10.23 Their synthesis began with addition of vinylmagnesium bromide to tetra-O-benzyl-D-glucose 43 to give a 3:2 mixture of diastereomeric allylic alcohols. Treatment of the major isomer, 44, with Hg(OTFA)2 in THF initiated a diastereoselective electrophilic cyclization to an organomercuric species that was subsequently iodinated in methylene chloride to obtain compound 45 in a 53% yield. To facilitate the characterization, a three-step sequence of dehalogenation, hydrogenolysis, and acetate protection was performed to give 46 with a 92% yield. The minor isomer, epimeric at the allylic alcohol, produced 47 as a major product. When tetra-O-benzyl-D-galactose is used as the starting pyranose, the transformation yielded 49 and 50 in 84% and 92% yields, respectively.
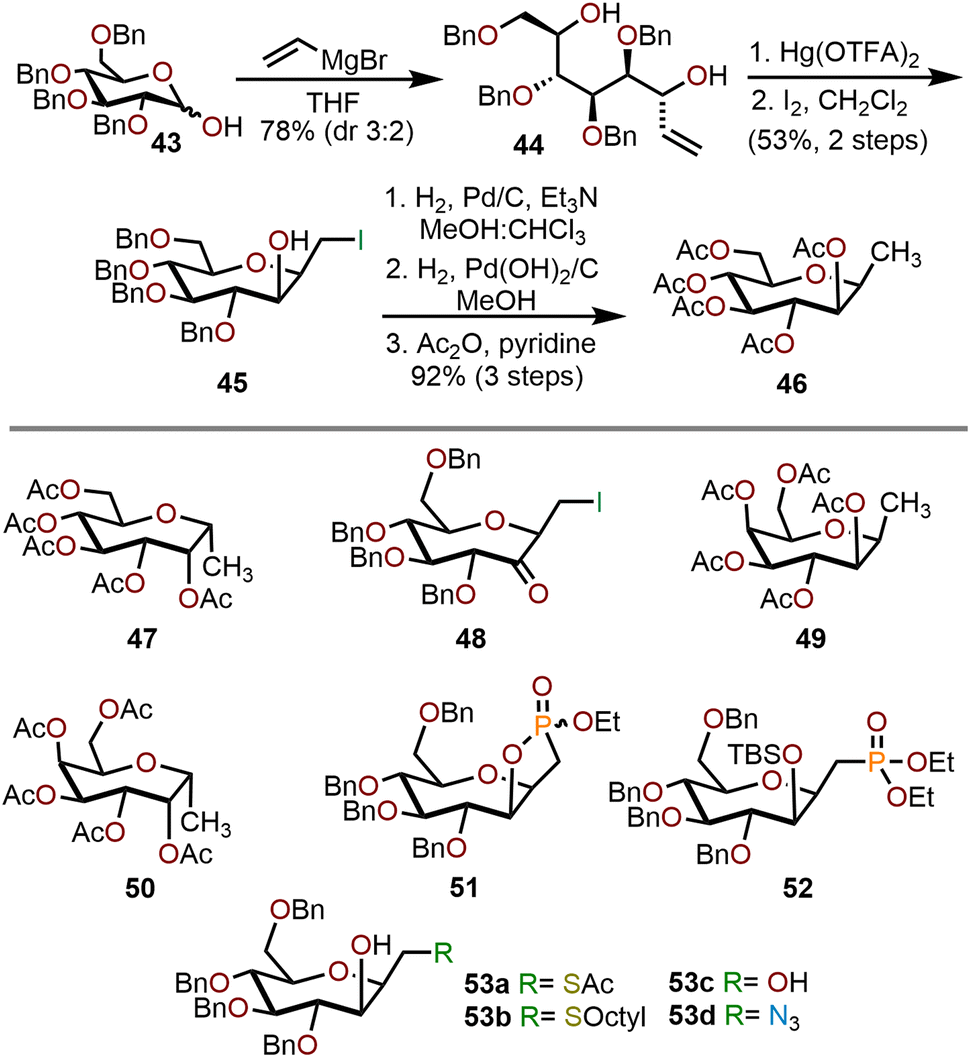 | ||
| Scheme 10 Stepwise homologation of pyranoses to synthesize polyoxygenated oxepanes.23 | ||
Compound 45 can be further derivatized using a variety of nucleophiles under known conditions to obtain fully functionalized oxepanes (53a–d) with yields ranging from 62–95%. Protection of the C2 hydroxyl group as the TBS ether followed by phosphonate formation gave 52 with a 65% yield over two steps. Alternatively, if the C2 hydroxyl group was left unprotected, attack by triethyl phosphite gave cyclic phosphonate 51 in 80% yield as a mixture of diastereomers. Finally, oxidation of the C2 hydroxyl group using pyridinium chlorochromate (PCC) provided ketone 48 in 92% yield.
A stepwise homologation approach has also been employed for the synthesis of biologically active, C1 unsubstituted oxepanes (e.g., 59 and 60) by Peczuh, Ernst, and coworkers.24 Oxepane 60 was found to serve as an excellent mannopyranoside mimic adopting the same hydrogen bond network as parent antagonists for the mannose-specific lectin FimH receptor on bacterial pili of uropathogenic E. coli; which mediates attachment of the pathogen to urothelial cells thereby playing an essential role in the first step of urinary tract infections. The synthetic route to access 59 and 60 involved the preparation of oxepine 56 from 2,3,4,5-diisopropylidene-D-mannose 54, which allowed for further functionalization of the olefin (Scheme 11).24,25 The sequence begins with subjecting 54 to a Wittig olefination followed by vinyl ether formation using a catalytic amount Pd(OAc)2 to afford diene 55. Cyclization of 55 was achieved via a ring closing metathesis (RCM) mediated by 20 mol% of Schrock's catalyst to afford acetonide protected oxepine 56 in a 89% overall yield.25,26 Next, a regio- and diastereoselective hydroboration-oxidation of 56 afforded access to oxepane 57 in 65% yield. Alternatively, O-alkylation of 57 with heptyl bromide afforded oxepane 58 in 83% yield.24 Lastly, the acetonide groups were deprotected using Amberlite IRA-120 resin to obtain polyhydroxylated oxepane 59 in a 90% yield and O-alkylated oxepane 60 in a 93% yield.
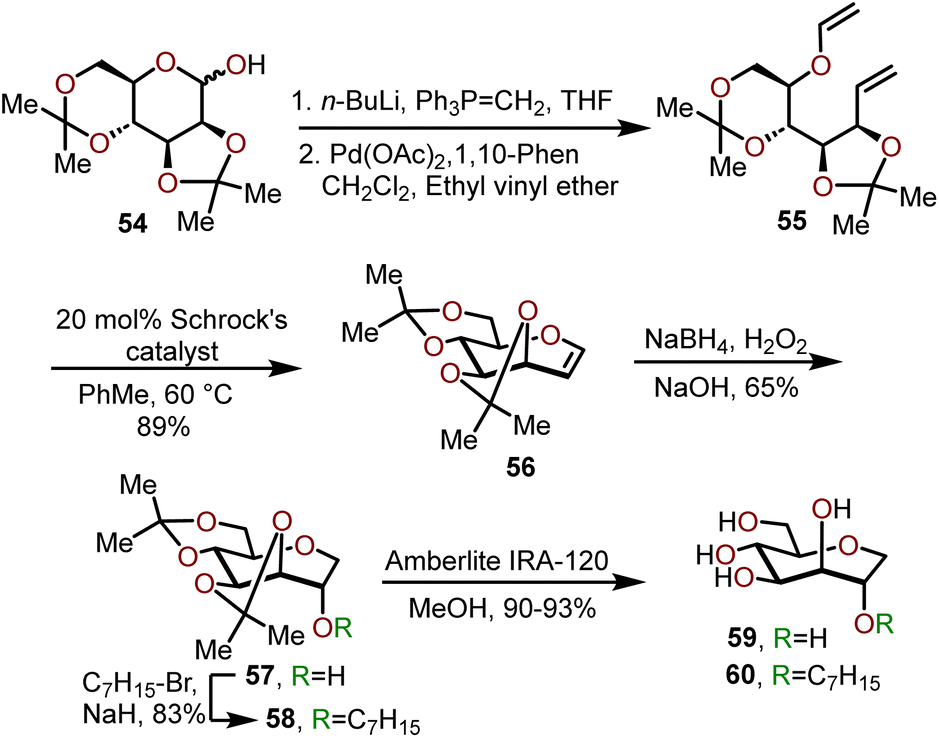 | ||
| Scheme 11 Synthesis of C2 substituted oxepanes via a RCM strategy.24 | ||
An alternative, scalable route to acetonide-protected oxepine 56 utilizes a vinylation–cyclic hemiacetal formation strategy as illustrated in Scheme 12.27 The synthesis starts with 2,3,4,6-tetra-O-benzyl-D-mannose (61), which was treated with vinylmagnesium bromide to obtain allylic alcohol 62 in 92% yield and a 3:2 diastereomeric mixture. The diastereomeric mixture of allylic alcohol 62 was directly subjected to ozonolysis conditions and trapped as the seven-membered oxacycle via acetate protection to obtain a 1,2-di-O-acetyl-3,4,5,7-tetra-O-benzyl mannoseptanoside 64 with 74% yield over the two steps. Global benzyl deprotection of 64 was achieved by hydrogenolysis, then an acid-catalyzed acetonide protection using 2,2-dimethoxypropane afforded 1,2-di-O-acetyl-3,4,5,7-diisopropylidene mannoseptanoside 65 in a 64% yield over two steps. Bromination of septanose 65 followed by a Schwartz reductive elimination with titanocene dichloride yields acetonide protected oxepine 56 in a 20% yield over the two steps.27 The above strategy was demonstrated to be a scalable synthesis for several polyoxygenated oxepanes with inexpensive starting materials and reagents, thereby it is complementary to RCM methods requiring Mo- or Ru-based catalysts.
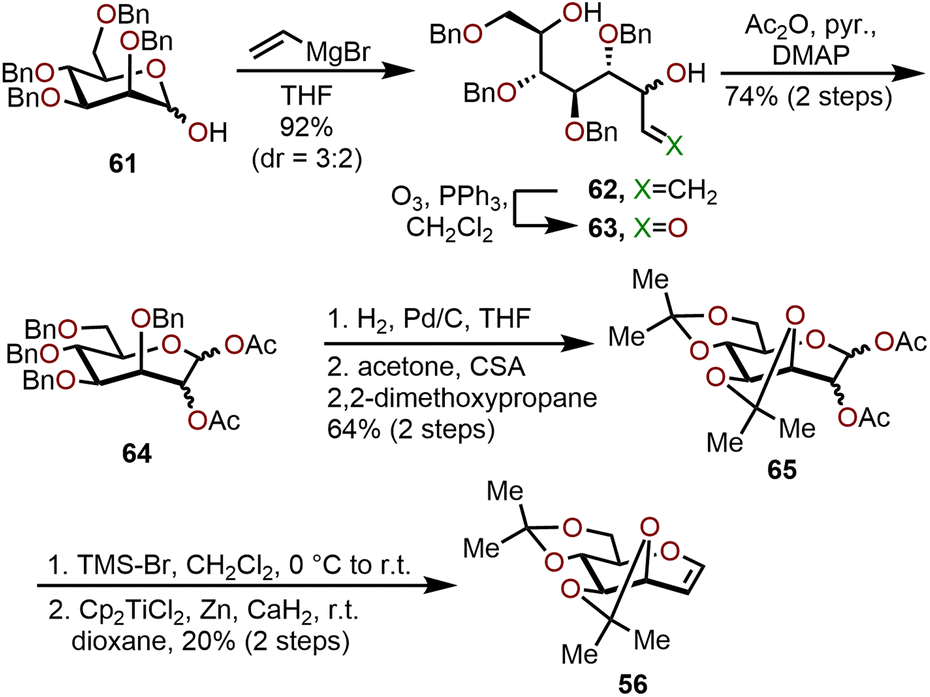 | ||
| Scheme 12 Alternative strategy for synthesis of oxepine 56 required to access polyoxygenated oxepanes 59 and 60.27 | ||
Wong and coworkers have reported the synthesis of polyoxygenated oxepanes using a RCM strategy enabled by Grubb's 1st generation catalyst; the method is compatible with substrates accessible from pentose sugars.28 An illustrative example of the protocol with diene 66, available from D-ribose is shown in the Scheme 13.28
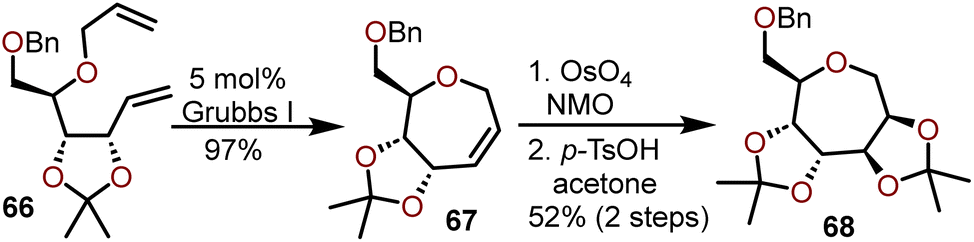 | ||
| Scheme 13 Polyoxygenated oxepanes by RCM strategy from furanoses.28 | ||
The intramolecular olefin metathesis of 66 at a concentration of 0.02 M afforded oxepine 67 in 97% yield. Advancement of 67 to afford polyoxygenated oxepane 68 in 52% yield was accomplished in a two-step process by Upjohn dihydroxylation of the olefin and acetonide protection of the resulting diol. Highly oxygenated substrates such as 66 worked well under these conditions to afford better overall yields of oxepine product in comparison to non-oxygenated substrates. While it may seem trivial, the use of a pentose versus a hexose sugar as a starting material not only alters the final position of the olefin in the generated oxepine, but it dictates which metal catalyst should be used for a successful RCM reaction. Wong and coworkers as well as Van Boom and coworkers successfully used the Grubb's 1st generation catalyst to perform a RCM with pentose-derived dienes, but when hexose-derived dienes were used by Peczuh and Snyder in a related oxepine synthesis strategy, the Grubb's 1st generation catalyst was found to be much less effective and the more reactive Schrock catalyst was required to afford the desired oxepines.25,28,29 Both RCM strategies are very efficient and provide access to C1–C2 or C2–C3 oxepine targets with high selectivity allowing advancement to a variety of polyoxygenated oxepanes.
2.5 Radical cyclizations
Radical cyclizations of advanced glucal-derived intermediates offer another avenue to quickly access complex polyoxygenated oxepane targets. Reductive couplings promoted by samarium diiodide have been widely used in natural product synthesis and have been harnessed to construct polycyclic ethers with embedded oxepanes.30,31 Nakata and coworkers were the first to explore the application of SmI2-induced reductive couplings toward the synthesis of oxepanes (Scheme 14), which was expanded upon from their method to construct polycyclic tetrahydrofurans.32,33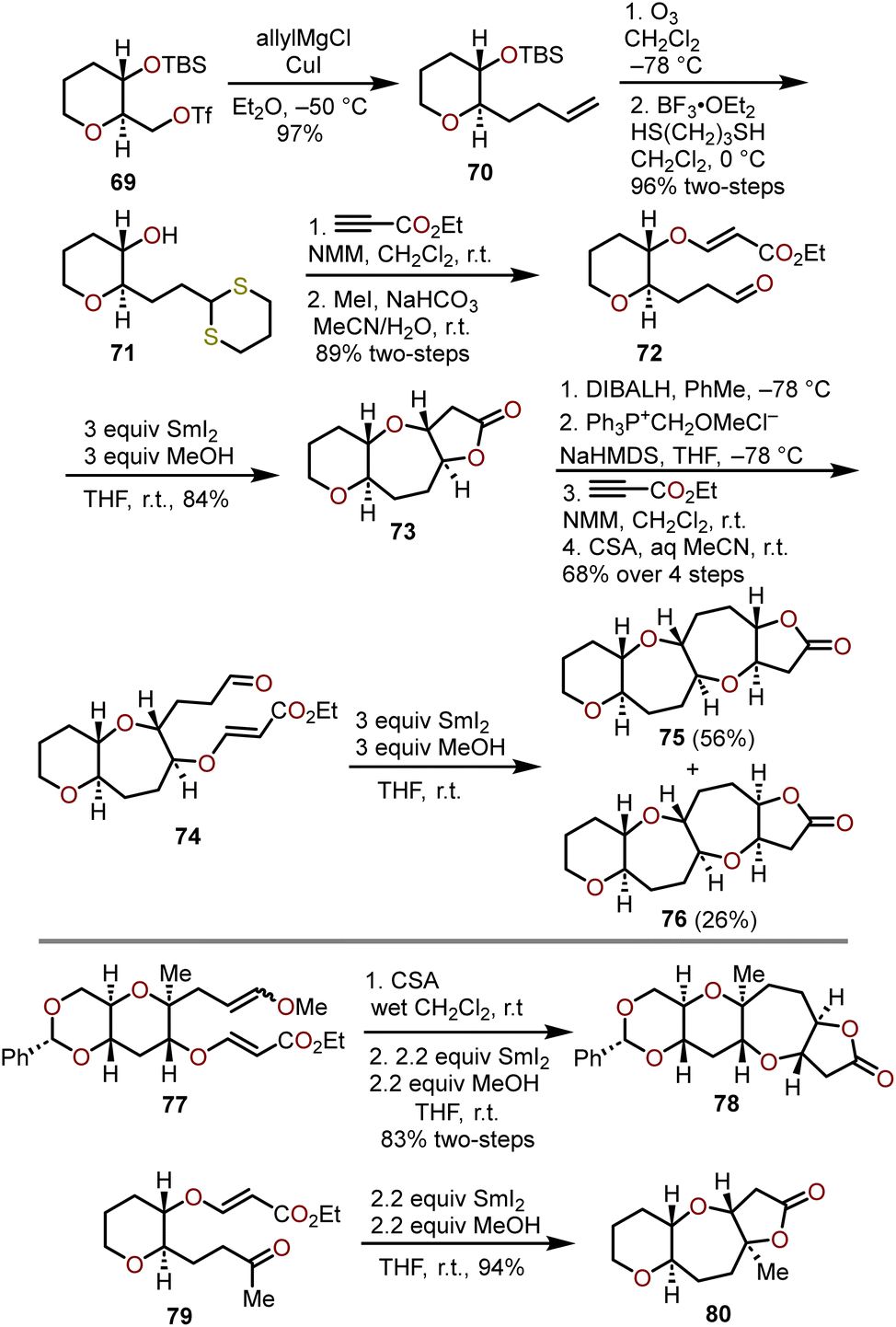 | ||
| Scheme 14 SmI2 mediated radical cyclization strategy toward fused polyoxygenated oxepanes.32,34 | ||
Beginning with optically active triflate 69, available from tri-O-acetyl-D-glucal in three steps,35,36 allylation with allylmagnesium chloride and copper(I) iodide afforded olefin 70 in high yield. Ozonolysis and dithiane protection of the resulting aldehyde proceeded efficiently with concomitant desilylation to afford alcohol 71 in a 96% yield over the two steps. Treatment of 71 with ethyl propiolate in the presence of N-methylmorpholine (NMM) base resulted in a hetero-Michael addition between the secondary alcohol and ethyl propiolate, which was followed by dethioacetalization with methyl iodide in aqueous MeCN solvent to afford aldehyde 72. Upon exposure of aldehyde 72 to three equivalents of SmI2, reductive cyclization of the in situ generated ketyl radical with the tethered α,β-unsaturated ester afforded the lactone-containing 2,7-syn-2,3-trans-oxepane 73 as the sole product in 84% yield. Further advancement of 73 was accomplished through a DIBAL-H reduction of the lactone, Wittig reaction to install the methyl enol ether, a hetero-Michael addition of the free alcohol with ethyl propiolate, and treatment with camphor sulfonic acid to afford to aldehyde 74, which allowed for the demonstration of the iterative power of the SmI2 reductive cyclization to construct fused polyoxygenated oxepanes 75 and 76 in 56% and 26% yields respectively (Scheme 14).
Nakata and coworkers followed up on their investigations and found that the reductive cyclization protocol can be conducted with 2.2 molar equivalents of SmI2 (e.g., 77 → 78) and the aldehyde can be replaced with a methyl ketone to afford fused oxepanes containing an angular methyl group (e.g., 79 → 80) in excellent overall yield.34
Sasaki and coworkers investigated the use of monoselenoacetals (e.g., 87 in Scheme 15) as precursors to α-alkoxyalkyl radicals and demonstrated that an efficient radical cyclization with a tethered β-alkoxyacrylate provides access to O-linked oxepane ring systems such as 88 in good yield.37 To access the requisite monoselenoacetal 87, diol 81 and aldehyde 82, both available from tri-O-acetyl-D-glucal,35,36 were treated with camphorsulfonic acid (CSA) to afford acetal 83. Regioselective cleavage of the acetal was accomplished with diisobutylaluminum phenylselenide at low temperature to afford a single diastereomer of monoselenoacetal 84.
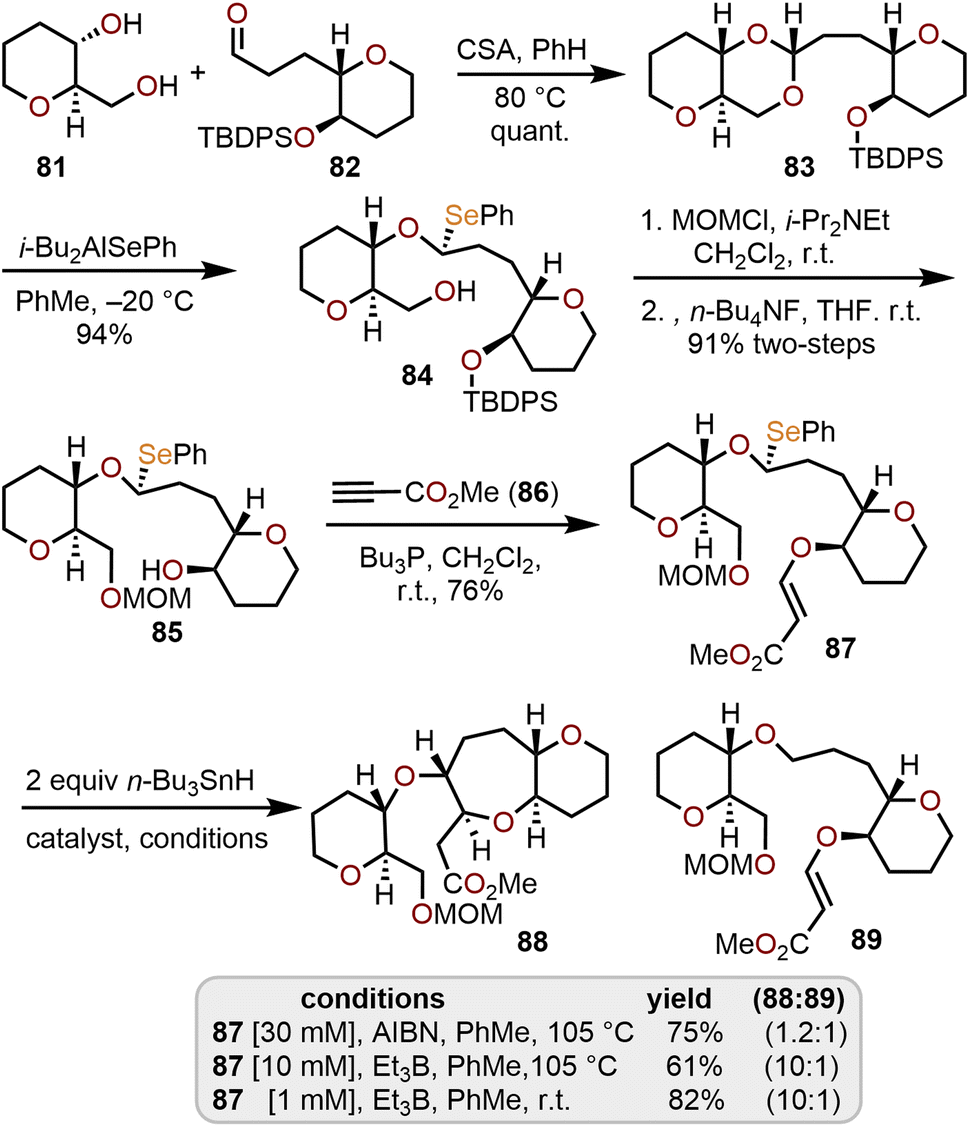 | ||
| Scheme 15 Radical cyclization strategy using monoselenoacetals to access O-linked oxepanes.37 | ||
This selectivity is presumed to arise from a tight-ion paired SN1-type mechanism involving regioselective coordination of the bulky i-Bu2AlSePh reagent with the less sterically hindered oxygen of the acetal followed by intramolecular attack of the phenylselenide anion syn to the cleaved C–O bond.37 Further advancement to 87 required manipulation of the hydroxyl protecting groups to give alcohol 85 followed by hetero-Michael addition to methyl propiolate.
A handful of conditions were screened to achieve the key intramolecular radical cyclization and it was found that both 2,2′-azobisisobutyronitrile (AIBN) and triethylborane (Et3B) in the presence of tributyltin hydride (n-Bu3SnH) were effective radical initiators. To prevent formation of the reduction product 89 room temperature and high dilution conditions (1 mM) with Et3B as the initiator were necessary to provide good yields of 88 (Scheme 15). This protocol has been demonstrated and adapted for the construction of key fragments of ciguatoxin, a complex polyether natural product vide infra.38,39
A radical cyclization of bridged dithionolides was developed by Nicolaou and coworkers to provide entry into tetracyclic ring systems containing polyoxygenated oxepanes (Scheme 16).36,40 Beginning with benzylated hydroxy acid derivatives 90 and 91 that are derived from tri-O-acetyl-D-glucal,36,40–42 sequential esterification through DCC coupling of the two fragments, debenzylation, and Corey–Nicolaou macrolactonization afforded diolide 92 in 64% yield over the three steps. Lawesson's reagent was used to access bridged dithionolide 93, which upon exposure to sodium naphthalide in THF solvent at −78 °C then quenching with methyl iodide afforded the cis-bridged tetracycle 94 in 80% yield. Removal of the methylsulfides was accomplished using n-Bu3SnH with azobisisobutyronitrile (AIBN) as an initiator afforded olefin 95 in excellent yield. Catalytic hydrogenation of 95 with Pearlman's catalyst or treatment with triethylsilane (Et3SiH) under protic conditions afforded the 6/7/7/6-tetracycle 96, which possessed the cis-fused oxepane rings. Attempts were made to reduce 94 to the trans-fused oxepane ring system using Et3SiH in the presence of silver tetrafluoroborate and were initially reported as successful,40 however subsequent studies revealed these conditions resulted in rearrangement to a 6/6–6/6-system.43,44 When dithionolide 93 was exposed to UV light for a short period of time, in contrast to treatment with sodium naphthalide, it was converted to dithiatopazine 97 in a 65% yield, alongside small amounts of recovered starting material 93 and olefin 95 (Scheme 16).45 Treatment of 97 with triphenylphosphine at 25 °C resulted in extrusion of one of the sulfur atoms resulting in episulfide-containing oxepane 98 and spiro ketal-thioketone 99 in near equal quantities.
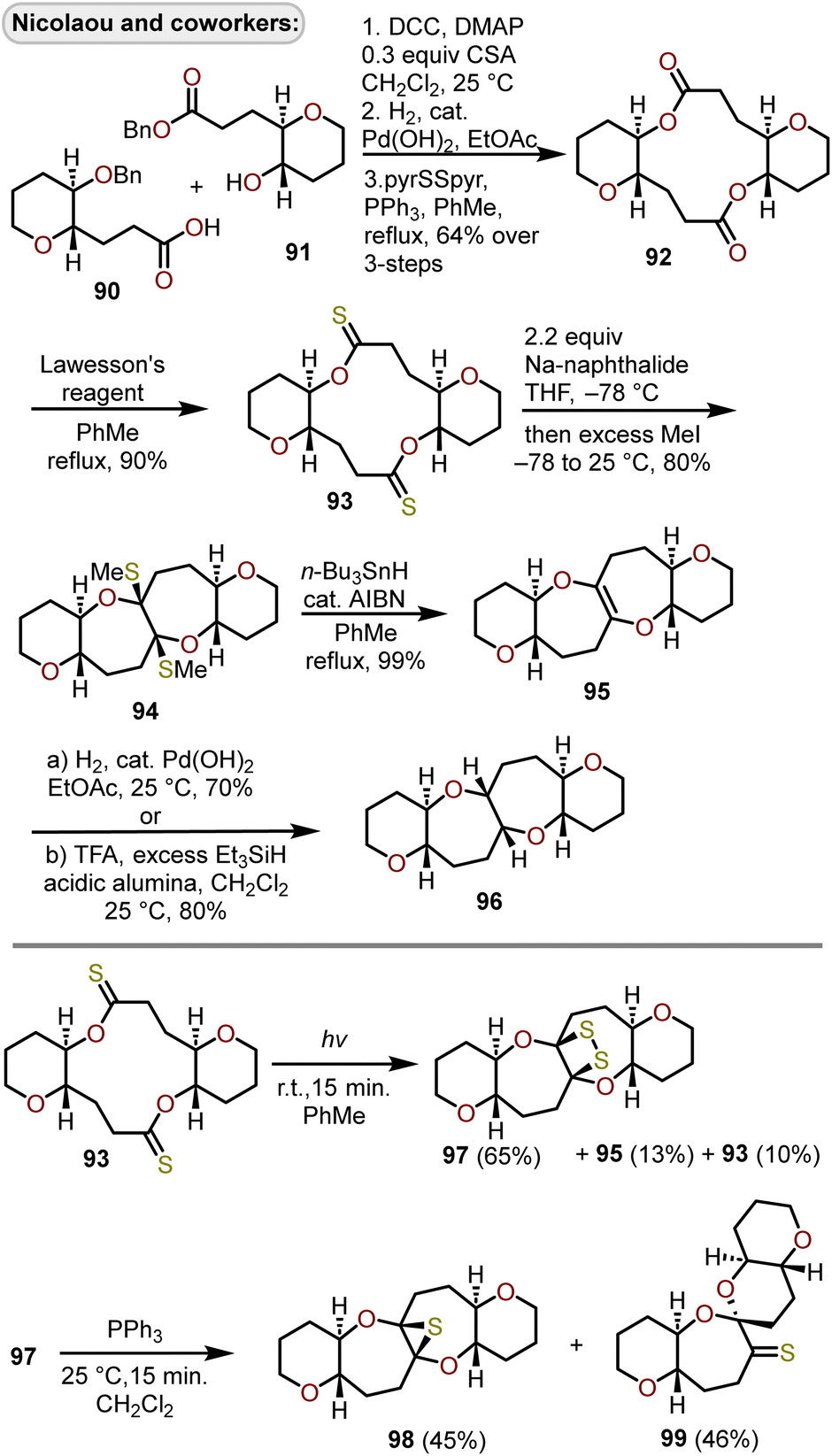 | ||
| Scheme 16 Access to 6/7/7/6-tetracyclic ring systems via radical cyclization of bridged dithionolides.36,40,45 | ||
2.6 Lewis acid-catalyzed methods
Lewis acid-catalyzed ring expansions as well as cyclizations have been developed to construct polyoxygenated oxepanes from advanced intermediates which arise from glucals or erythrose sugars and have been used to construct oxepanes within fused polycyclic ether scaffolds. Peters and coworkers in their efforts towards polycyclic ether containing natural products advanced a BF3·OEt2-mediated ring expansion to homologate 4-methoxylevoglucosenone derivative (100) with (trimethylsilyl)diazomethane (TMSCHN2)46 to afford a mixture of structural isomers 101, 102, and 103 (Scheme 17).47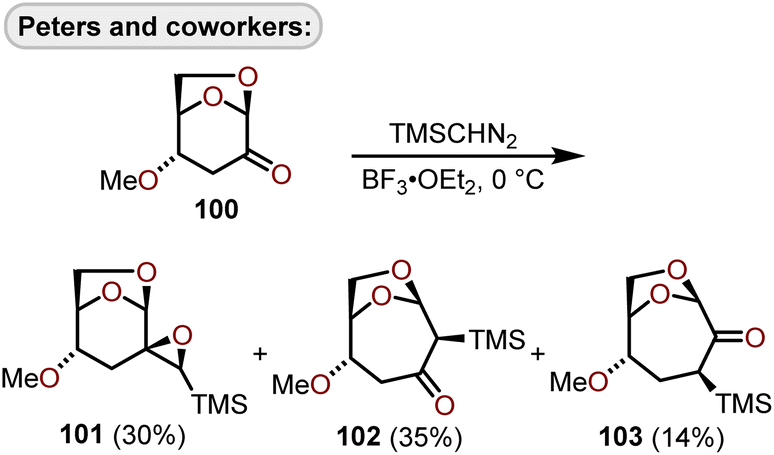 | ||
| Scheme 17 Lewis acid-catalyzed ring expansion of 1,6-anhydrohexos-2-uloses with (trimethylsilyl)diazomethane.47 | ||
The ring expansion to oxepanes 102 and 103 proceeds diastereoselectively affording products with the trimethylsilyl group on the β-face. The 4-methoxy substituent was found to be important to achieve high diastereofacial selectivity, and furthermore, unfunctionalized levoglucosenone resulted in competing 1,2- and 1,4-addition of TMSCHN2 without ring expansion to the oxepane.47
Mori and coworkers have developed conditions using TMSCHN2 to minimize the production of multiple homologation products arising from Lewis acid-catalyzed ring expansions of cyclic ketones using diazomethane (Scheme 18).35,48,49 Starting with the trans-fused 6,6-bicyclic ketone 104, which was constructed using an oxiranyl anion alkylation/6-endo cyclization strategy from 81,35,50 it was found that cryogenic temperatures using BF3·OEt2 as the Lewis acid engendered formation of 105, which after acid hydrolysis afforded 107 in 76% isolated yield over regioisomeric ketone 108. The trimethylsilyl group directs the initial formation of the less sterically hindered α-trimethylsilyl ketone 105, which quickly rearranges to silyl enol ether 106 thereby preventing the production of multiple homologation products. The selective reduction of ketone 107 with sodium borohydride (NaBH4) or diisobutylaluminum hydride (DIBAL-H) afforded cis alcohol 110b over trans alcohol 110a, with DIBAL-H providing greater selectivity over NaBH4 (Scheme 18).
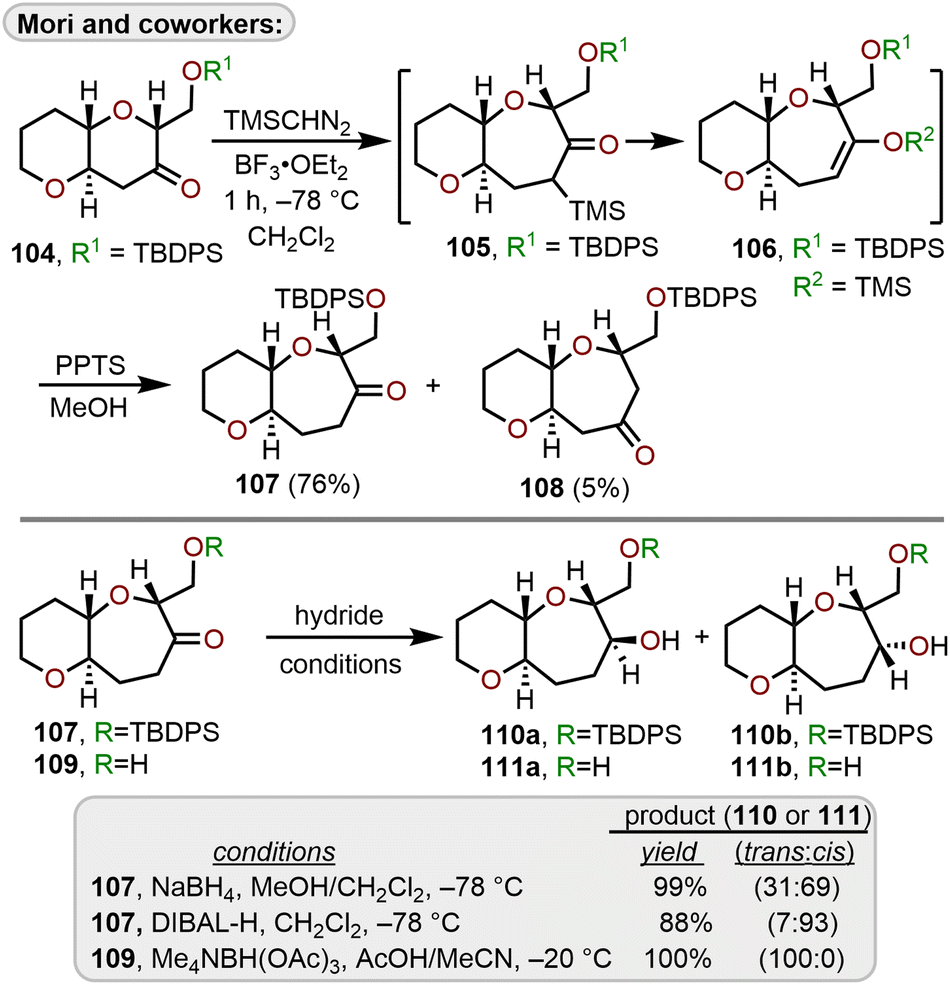 | ||
| Scheme 18 Access to 6/7-bicyclic ring systems via Lewis acid-catalyzed ring expansion of bicyclic ketones with (trimethylsilyl)diazomethane.51 | ||
By removal of the silyl protecting group on 107 to afford hydroxy ketone 109, a selective, hydroxyl-directed reduction using tetramethylammonium triacetoxyborohydride provided trans alcohol 111a quantitatively. Mori and coworkers advanced diol 111a to ketone 116 and demonstrated the utility of this ring expansion protocol in an iterative approach to construct trans-fused 6/7/7-tricyclic ketone 116 which could be reduced to trans-fused 6/7/7-tricyclic diol 117 that is primed for further iterations of this sequence to construct larger polycyclic ether systems within natural products (Scheme 19).51
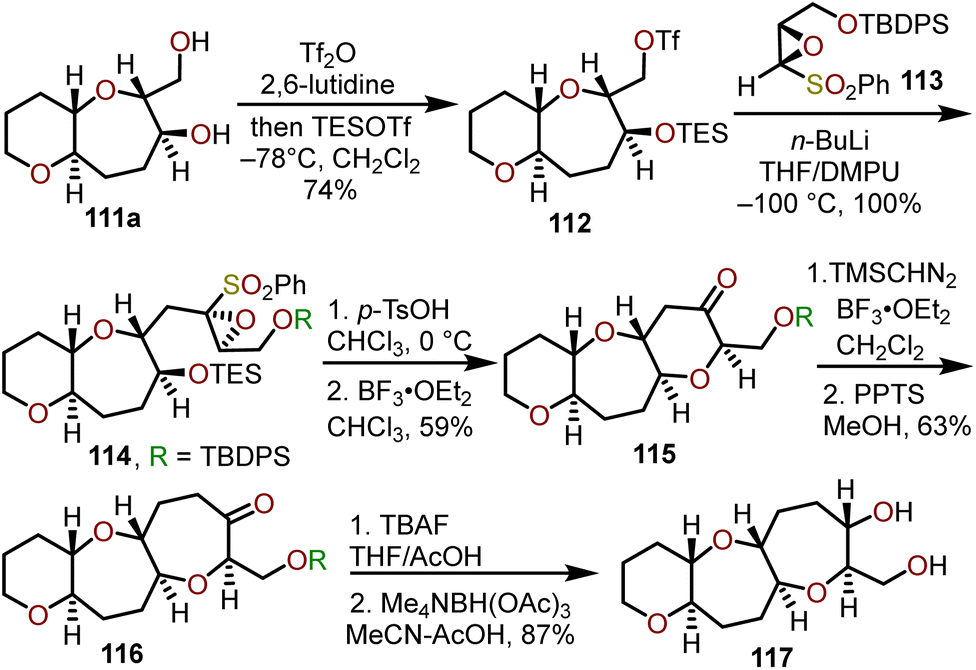 | ||
| Scheme 19 Access to trans-fused 6/7/7-tricyclic ring systems via Lewis acid-catalyzed ring expansion of bicyclic ketones with (trimethylsilyl)diazomethane.51 | ||
Complementary to the radical-based cyclizations of bridged dithionolides detailed in section 2.5 above,36,52 Nicolaou and coworkers showed that tethered dithionoesters such as 118 could be cyclized into hydroxy ketone-containing oxepanes (e.g., 121, Scheme 20).44,52 The process is proposed to proceed through a 1,2-dithietane intermediate (e.g., 119), which through expulsion of disulfur under the reaction conditions produce didehydrooxepanes (e.g., 120). Fluoride-mediated desilylation of 120 afforded hydroxy ketone-containing oxepane 121, which enabled access to trans-fused 6/7/7/6-tetracyclic ring systems, such as the CDEF-ring skeleton of brevetoxin B (122a), through a TMSOTf-catalyzed reductive cyclization in the presence of trialkylsilanes (Scheme 20).44,53,54 Of the trialkylsilanes evaluated, Et3SiH was found to be an effective reductant, but methyldiphenylsilane (Ph2MeSiH) resulted in better overall yield and selectivity (ca. 4:1) for the desired trans-fused 6,7,7,6-tetracyclic ring system 122a over 122b (Scheme 20). Reductive cyclizations of hydroxy ketones are often used to construct oxepanes within the cores of complex natural products vide infra.55,56
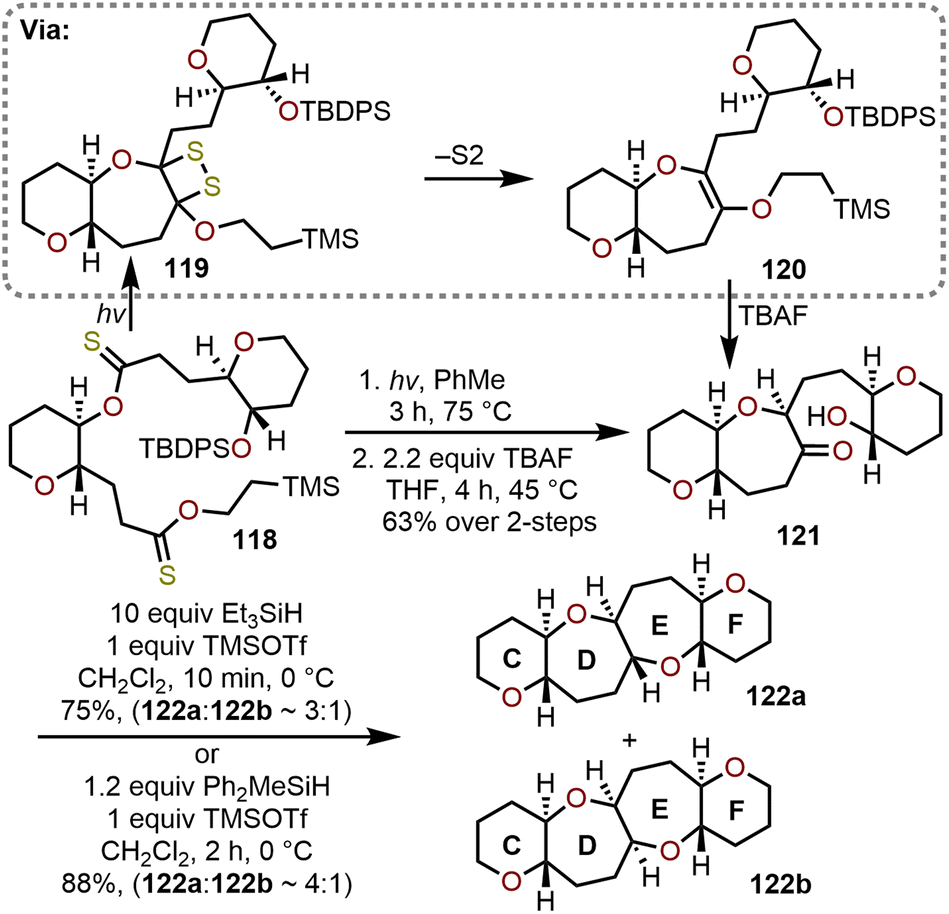 | ||
| Scheme 20 Access to trans-fused 6/7/7/6-tetracyclic ring systems via Lewis acid-catalyzed reductive cyclization of hydroxy ketones with TMSOTf and trialkylsilanes.44,52 | ||
Isobe and coworkers have leveraged the ability of dicobalt intermediates (e.g., 126) to participate in the Nicholas reaction in the presence of a Lewis or Brønsted acid to afford a stable propargylic cation and have shown that intramolecular trapping by a tethered alcohol can afford syn–trans fused 6/7-bicyclic ethers (e.g., 128; Scheme 21).57 The exemplified sequence in Scheme 21 was refined from their earlier work on Brønsted acid-catalyzed cyclizations58–60 and begins with iodide 123 prepared from glucal triacetate, which allows for conversion to acetylene 124.61,62 Addition of the lithium acetylide of 124 to cinnamaldehyde and trapping of the resulting alkoxide with pivaloyl chloride afforded the propargylic pivalate 125. Acetylene dicobalt hexacarbonyl complex 126 was generated in good yield upon exposure of 125 to dicobalt octacarbonyl and when treated with BF3·OEt2 underwent cyclization to the oxepane affording syn–trans fused 6/7-bicyclic ether 127 in 89% yield. Wilkinson's catalyst was used under high pressure hydrogenation conditions to remove the cobalt complex to afford 128.
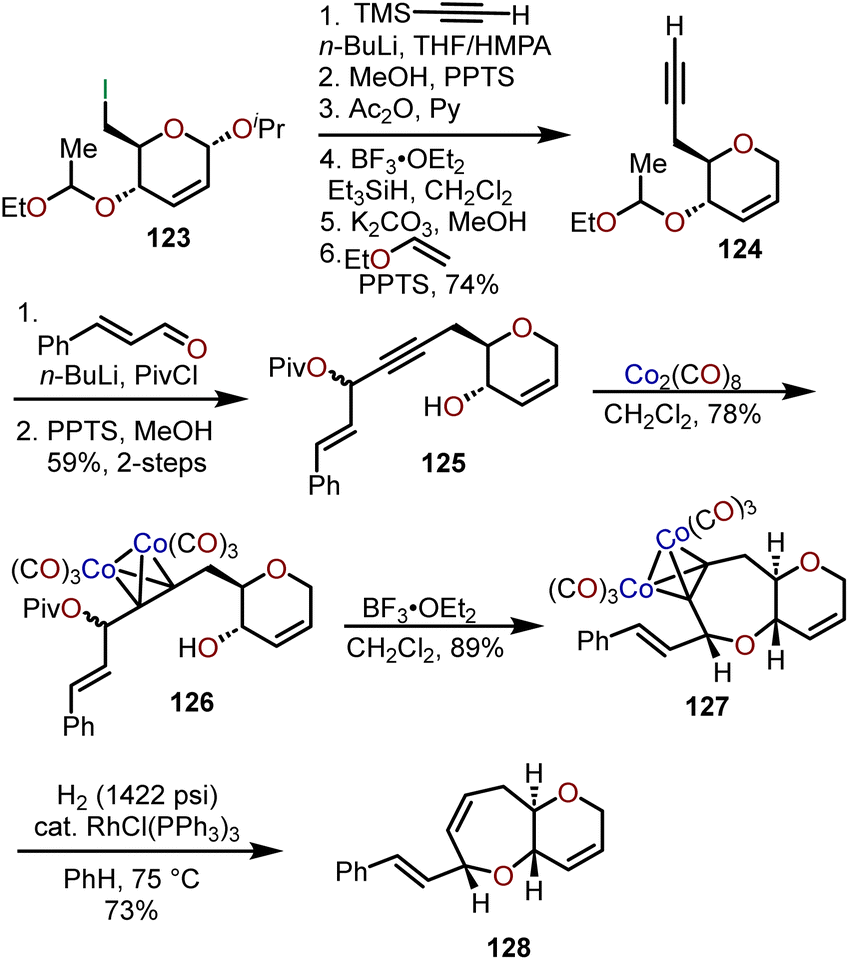 | ||
| Scheme 21 Access to syn–trans fused 6/7-bicyclic ethers through Lewis acid-catalyzed generation of propargyl cations from dicobalt intermediates.57 | ||
When the method was applied to systems (e.g., 129), which lacked additional stabilization through conjugation with an aryl system as in 126, adjustments in temperature, concentration and time were needed to decrease the formation of the more stable open chain isomer 131 and shift the equilibrium towards formation of polycyclic ether 130; which contains the ABC ring skeleton of ciguatoxin (Scheme 22).57,62 Of note, the use of TfOH resulted in primarily open chain isomer 131 and only a 10% yield of 130. Given the utility of 130 as a skeletal fragment of ciguatoxin, Isobe and coworkers developed alternative methods for decomplexation of dicobalt hexacarbonyl acetylenes that were more mild than previously employed with high pressure hydrogenation conditions (Scheme 22).63 The use of an excess of n-Bu3SnH as a reductant and heating to 65 °C affords the resultant oxepine (e.g., 132) with the cobalt complex functioning as the radical initiator for decomplexation. Alternatively, the use of Et3SiH enabled a hydrosilative decomplexation to afford vinylsilane-containing oxepines with high regio- and stereoselectivities resulting in the silyl group oriented away from the more sterically encumbered substituent (e.g., 133). Variations of this approach have been advanced using dicobalt hexacarbonyl acetylenes to construct polyoxygenated oxepanes through the recyclization of sugar acetylenes,64,65 the opening of dihydropyrans and recyclization,60 and in the synthesis of the natural product ciguatoxin 1B.61,62,66,67
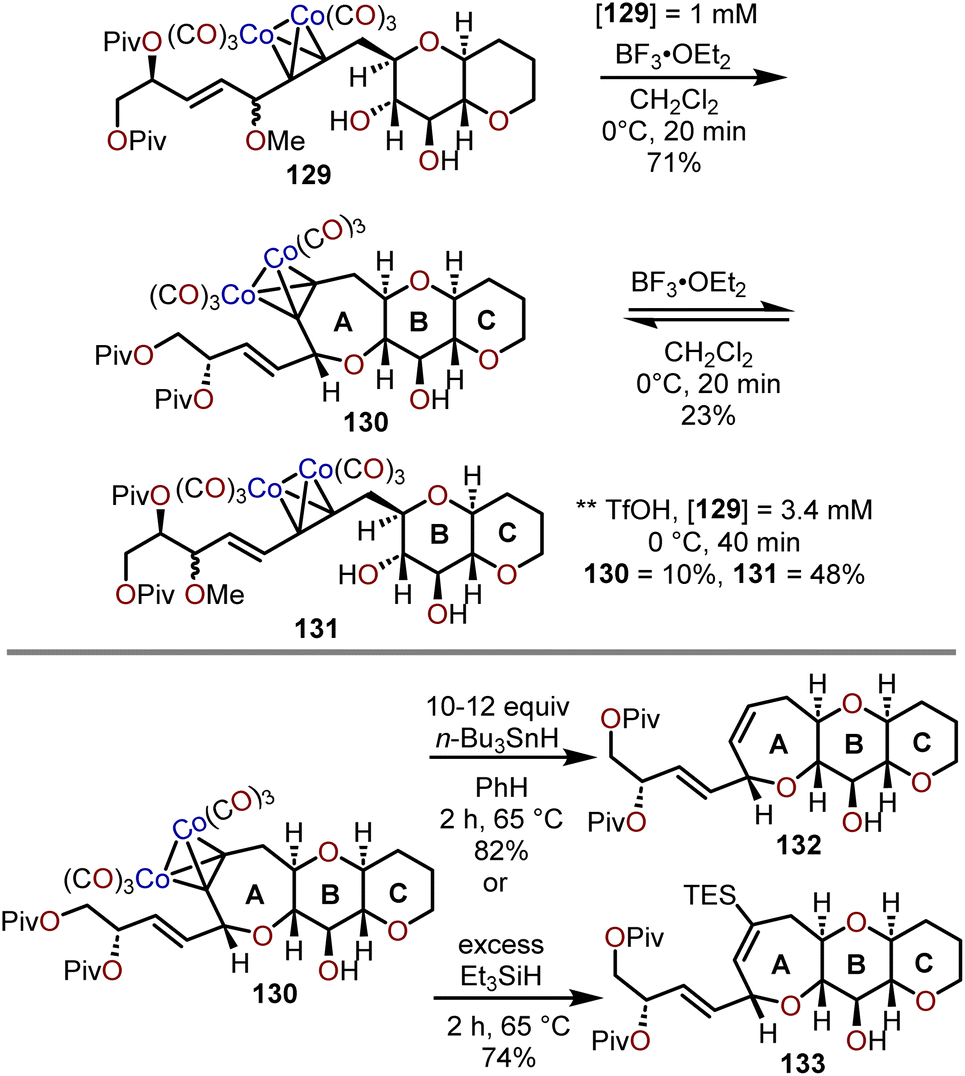 | ||
| Scheme 22 Access to syn–trans,trans-fused 7/6/6-tricyclic ethers systems via Lewis acid-catalyzed cyclization of dicobalt hexacarbonyl acetylenes and reductive methods for decomplexation.57,63 | ||
Bouché and coworkers used enantiopure nitrones (e.g., 134) that are readily prepared from erythrose sugars to access chiral 1,2-oxazines through a [3 + 3]-cyclization with lithiated TMSE-allene (135) and advanced the resulting 1,2-oxazines to poly(hydroxy)aminooxepanes through a Lewis acid-mediated rearrangement and reduction sequence (Scheme 23).68 The key 1,2-oxazine intermediate 136 was formed with excellent diastereoselectivity (dr = 96:4) from the addition of 135 to enantiopure nitrone 134. The high degree of diastereoselectivity observed for this transformation is attributable to Re-side attack of the nitrone by the lithiated TMSE-allene and is rationalized by a Felkin–Ahn model with coordination of the lithium cation to the nitrone oxygen further assisting to enhance selectivity.68 Addition of TMSOTf to 1,2-oxazine 136 consequently forms ketone 137 in moderate yield via a Lewis acid-mediated rearrangement that proceeds through an intramolecular aldol reaction of an enol ether with an activated acetal in a Prins-type cyclization. Subsequent reduction of ketone 137 with sodium borohydride (NaBH4) and desilylation with TBAF afforded triol 138. Subsequent cleavage of the benzyl group in 138 under hydrogenolysis conditions followed by filtration through an acidic DOWEX® resin, and elution with aqueous ammonia afforded poly(hydroxy)aminooxepane 139 in a 90% yield. The route offers a modular approach to highly oxygenated aminooxepanes allowing for additional azide, alkynyl, and aryl-containing derivatives to be prepared.68
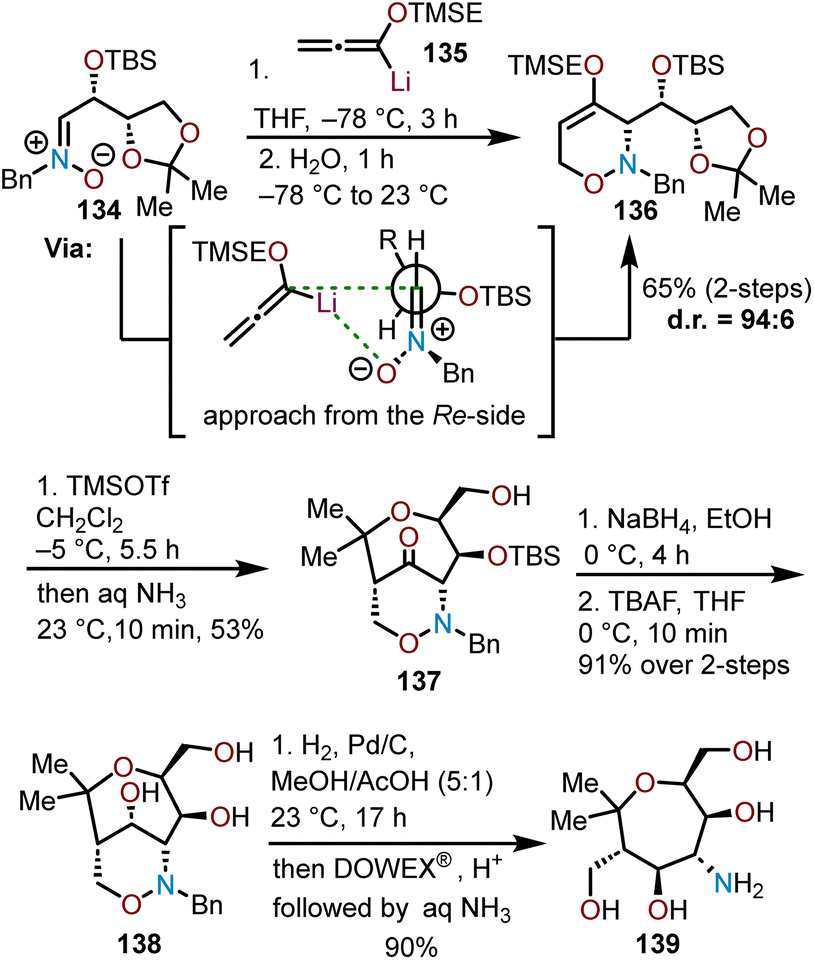 | ||
| Scheme 23 Diastereoselective Lewis-acid rearrangement to afford enantiopure poly(hydroxy)aminooxepanes.68 | ||
3. Synthesis of oxepanes from non-sugar-based starting materials
The inherent oxygenation and ability to relay stereochemical information provided by sugar-based starting materials has substantial advantages in the synthesis of polyoxygenated oxepanes. In the same vein, the use of non-sugar-based feedstocks can offer modularity and orthogonal avenues for diversification of the oxepane scaffold leading to polyoxygenated oxepanes that are not easily accessed from sugar-based feedstocks. The approaches used in de novo syntheses of oxepanes are often built upon strategies developed for sugar-based approaches and include cyclization by RCM, Lewis acid-mediated cyclizations, and ring-expansions through skeletal rearrangements.3.1 Ring closing metathesis approaches
Access to polyoxygenated oxepanes through oxidation of seven-membered oxepines, which are easily accessible from RCM methods using functionalized dienes as starting materials prepared via olefination and O-allylation. The position of the olefin within the oxepine product can be easily varied by choice of starting materials.An example of an RCM strategy utilizing readily available, non-sugar based starting materials by Yu, Blagg and coworkers is shown in Scheme 24.69 Through vinylation of 4-pentene-1-ol (140) with benzyloxy allene 141 in presence of Pd(OAc)2 as a catalyst, RCM precursor 142 was accessed in 95% yield.29 RCM using 2 mol% Grubbs II catalyst provides oxepine 143 in 71% yield and advancement through Upjohn oxidation to the cis-diol and trapping with 1,1-carbonyl diimidazole (CDI) affords the carbonate-containing oxepane 144 in 68% yield over two steps. Unfortunately, removal of the benzyl protecting group results in 145 being isolated as a 3:2 mixture of anomers (Scheme 24). This strategy has also been used by others to prepare small libraries of oxepanes by changing the substitution pattern on the diene and allene.29,70
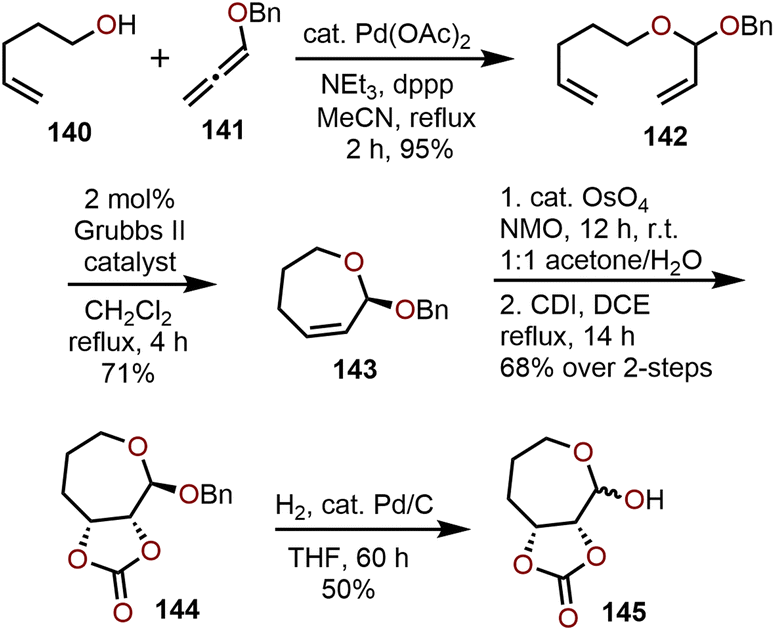 | ||
| Scheme 24 Synthesis of oxepanes via RCM using Grubbs II catalyst by Yu, Blagg, and coworkers.69 | ||
Using a RCM strategy, Das and coworkers have been able to develop a synthesis of enantiopure oxepanes as carbohydrate mimics (Scheme 25).71 Beginning with achiral β-hydroxyester 146, a biocatalytic reduction with a ketoreductase from Klebsiella pneumoniae allows for a dynamic kinetic reductive resolution to afford essentially enantiopure 147. The alcohol in 147 is protected as its O-allyl ether using allyl trichloroimidate 148 to obtain 149 in 80% yield, which was followed by an RCM reaction using 10 mol% of the Grubbs first-generation catalyst to produce an oxepine 150 in a 65% yield. Sharpless asymmetric dihydroxylation of the olefin affords bis-hydroxyoxepane 151 in a 72% yield, which was then treated with DIBAL-H to reduce the ester group to the primary alcohol affording polyhydroxylated oxepane 152 in a 78% yield. A unique feature of this synthetic strategy is, by utilizing biocatalytically derived β-hydroxy esters, synthesis of enantiopure polyhydroxylated oxepanes is possible with a good overall yield.
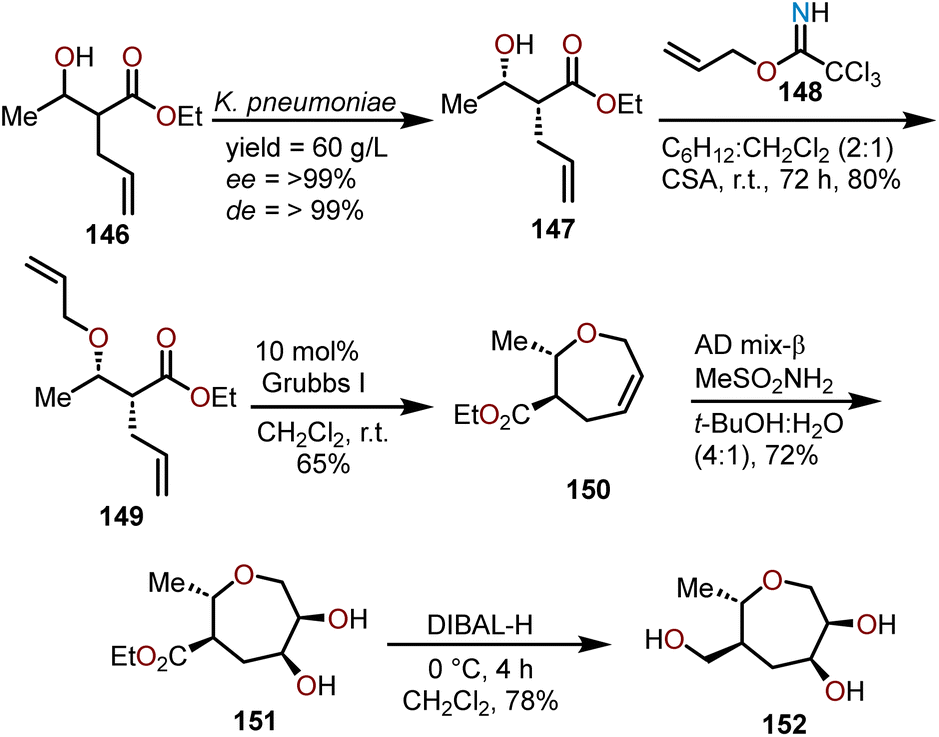 | ||
| Scheme 25 Synthesis of oxepanes using Grubbs I catalyst by Das and coworkers.71 | ||
3.2 Lewis acid-mediated cyclizations of bis-epoxides
The synthesis of various polycyclic ethers through tandem oxacyclizations have been reported. Naturally, exo-oxacyclizations predominant over endo-oxacyclizations, which have been used to form polycyclic ethers. McDonald and coworkers expanded on this concept for the synthesis of polyhydroxylated oxepanes via a Lewis acid-mediated tandem endo-selective oxacyclization of 1,5-diepoxides.72The starting materials for the cyclization reactions were prepared from commercially available geranyl acetone and it was derivatized via enantioselective Shi epoxidation conditions to obtain the 1,5-diketone system. After screening several Lewis acid-mediated cyclizations, BF3·OEt2 in DCM at −40 °C was identified as the best condition for the majority of the substrates evaluated.72,73 The 1,5-diepoxyketone 153 was able to be cyclized using B(C6H5)3 in DCM solvent, which provided a 40% yield of 154, whereas using the general conditions using BF3·OEt2 work-up with acetic anhydride was required to isolate corresponding acetate 155 (Scheme 26, I).73 With diepoxide acetate esters 156 and 158, the yields were improved (60% and 46%) using the original conditions (i.e., BF3·OEt2 in DCM at −40 °C). McDonald and coworkers further demonstrated the stereospecificity of the oxacyclization from diastereomeric epoxide diacetate 158 (Scheme 26, III), which produced 159 with a slightly lower yield (46%).
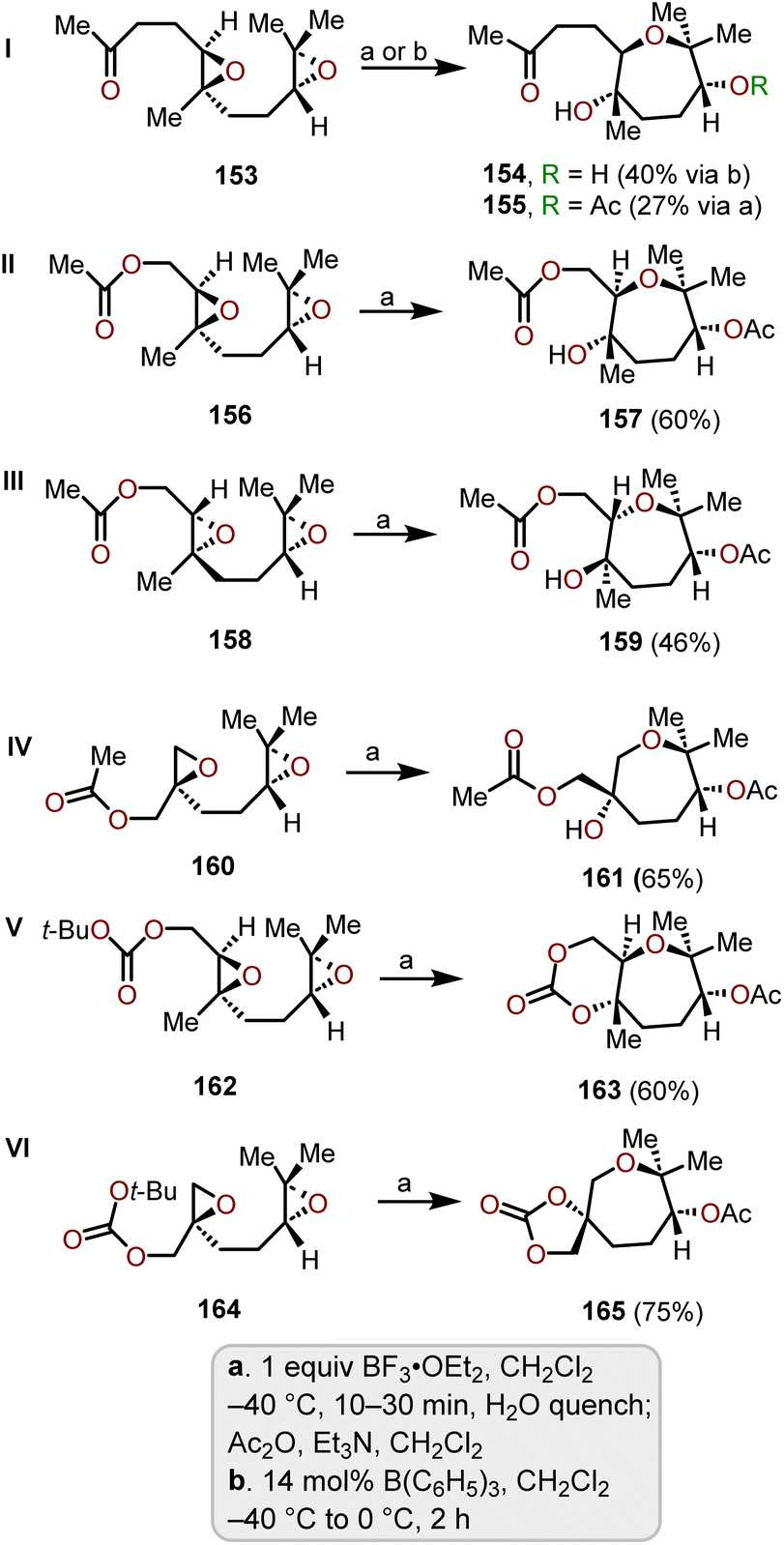 | ||
| Scheme 26 Lewis acid-mediated cyclization of 1,5-diepoxides to synthesize oxepanes.72,73 | ||
Diepoxide acetate 160 bearing a different substitution pattern underwent cyclization to obtain 161 with a slightly better 65% yield (Scheme 26, IV). Altering the terminal functional group from acetate to tert-butyl carbonate provided fused- and spiro-bicyclic carbonate oxepane systems 163 and 165 with higher yields (60% and 75%) than the acetate bearing starting material (Scheme 26, V and VI). The mechanism of these bis-epoxide oxacyclizations involve Lewis acid activation of the terminal epoxide followed by an intramolecular nucleophilic addition from the internal epoxide oxygen to obtain a bicyclic intermediate. This intermediate is then attacked by a carbonyl group on the side chain to form the seven-membered oxacycles. Nucleophilic attack of the internal epoxide oxygen is primarily endo in the highlighted examples, which is governed by the carbonyl group of the side chain. Using the above strategy affords oxepanes and bis-oxepanes with high selectivity via tandem endo-selective and trans-stereoselective cyclization processes, which makes this transformation a powerful tool for synthetic chemists. Furthermore, this approach is viable from an abundant variety of commercially available starting materials.
3.3 Ring expansion via skeletal rearrangements
One of the less intuitive techniques to synthesize polyhydroxylated oxepanes is via skeletal rearrangement of functionalized tetrahydropyrans. Such strategies are very useful for the synthesis of natural products containing polycyclic ethers like brevetoxin B or isolaurepinnacin.74,75 In addition to these natural product syntheses, such strategies can also be utilized for the elaboration of functionalized oxepanes. Hori and coworkers have demonstrated that a Zn(OAc)2 mediated rearrangement-ring expansion strategy of functionalized tetrahydropyrans to obtain oxepane 173 and 174. Six membered cyclic ethers 167 and 168 were prepared from geraniol derivatives 166a and 166bvia a three-step procedure involving epoxidation, exo-cyclization then silyl deprotection and subsequent acetate protection with AcCl-ZnCl2 (Scheme 27). Intermediates (167 and 168) were further reacted with monochlate chloride (McCl) or methanesulfonyl chloride (MsCl) to obtain the desired precursor for the rearrangement (e.g., 169–172).76 Treatment of 169 with Zn(OAc)2 under reflux in AcOH:H2O solvent for 30 min produced 2,3-trans oxepane 173a and 173c with 89% combined yield. Maintaining the same reaction for a longer period of time (3.5 h) at 80 °C produced a mixture of products 173a (68%) and 173b (19%). When the same transformation was carried out without the presence of Zn(OAc)2, oxepanes 173a and 173c were produced in a 72% combined yield. The corresponding mesylate 170 produced 173a and 173b in 53% and 42% yield, respectively, when refluxed in AcOH:H2O solvent in presence of Zn(OAc)2.
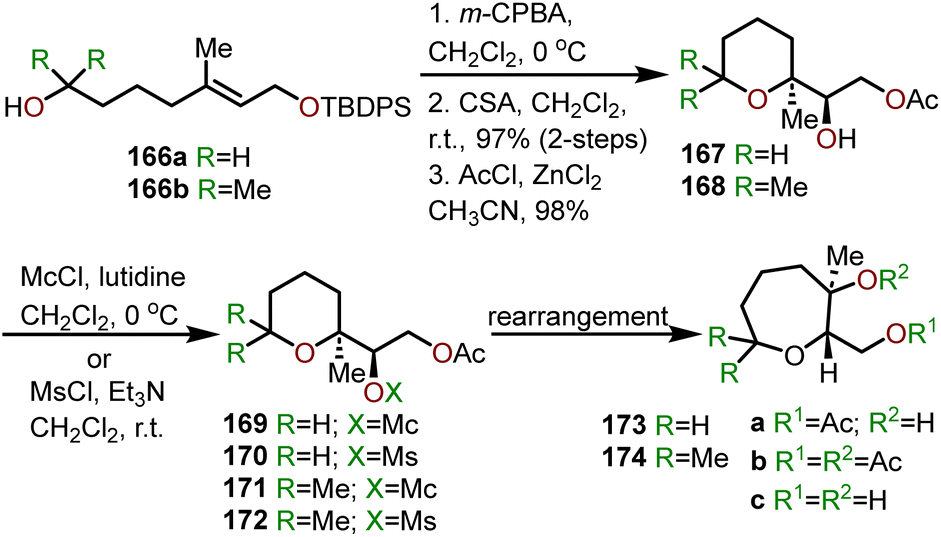 | ||
| Scheme 27 Oxepanes via skeletal rearrangement of functionalized tetrahydropyrans.75,76 | ||
The rearrangement of monochlate 171 produced oxepane 174a exclusively in a 92% yield when refluxed for 24 h in AcOH:H2O in the presence of Zn(OAc)2, whereas 174c is exclusively produced when refluxed in a dioxane:H2O mixture for 4 days. Using Sc(OTf)3 under dioxane:H2O reflux conditions, monochlate 171 produced an 82% combined yield of 174a and 174c after 6 h. Mesylate 172, when refluxed with Zn(OAc)2 in AcOH:H2O produced 174a with 90% overall yield in 2 h. The above stereoselective rearrangement is proposed to occur via a concerted mechanism to afford a single diastereomer as shown in Scheme 28. It was concluded that the ring expansion proceeds for monochlates under milder conditions, which were more effective than the corresponding mesylates.75,76 The rearrangement reactions using monochlate with Zn(OAc)2 in AcOH:H2O have been successfully applied to the synthesis of hemibrevetoxin B by Morimoto and coworkers.77 Mujica and coworkers have demonstrated a similar strategy for the synthesis of enantiopure oxepanes (Scheme 29).78 Using (R)-glyceraldehyde acetonide as a chiral pool starting material, cyclic ether 175, can be accessed via a Diels–Alder reaction with the requisite diene. This cycloadduct was then transformed into a linear ether via a three-step reaction sequence. The cyclic ether 175 was elaborated via ozonolysis followed by NaBH4 reduction of the resulting aldehyde and methylation of the acid using diazomethane to afford 176. Silyl protection of 176 with tert-butyldiphenylsilyl chloride (TBDPSCl), followed by reduction of the methyl ester and deoxygenation of the resulting primary alcohol was accomplished via tosylation and treatment with LiAlH4. Acetonide deprotection and a final epoxide formation afforded enantiopure oxepane precursor 177.78 When 177 was treated with four equivalents of LDA in THF at −65 °C, the oxepane 178 was obtained as a 1:1 mixture of epimers at C4 (oxepane numbering) with 96% yield. Despite the mixture of epimers, the sulfone of 178 can be interconverted into a prochiral ketone after silyl protection of the free hydroxyl, treatment with LDA, then subsequent oxidation with MoOPh.78 A key benefit of such transformation allows the established stereocenters to dictate nucleophilic additions into the oxepane C4.
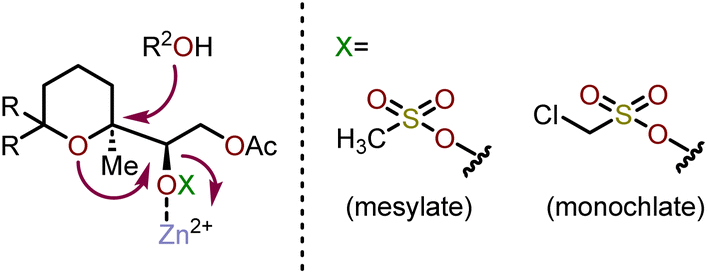 | ||
| Scheme 28 Concerted mechanism for synthesis of oxepanes via skeletal rearrangement.76 | ||
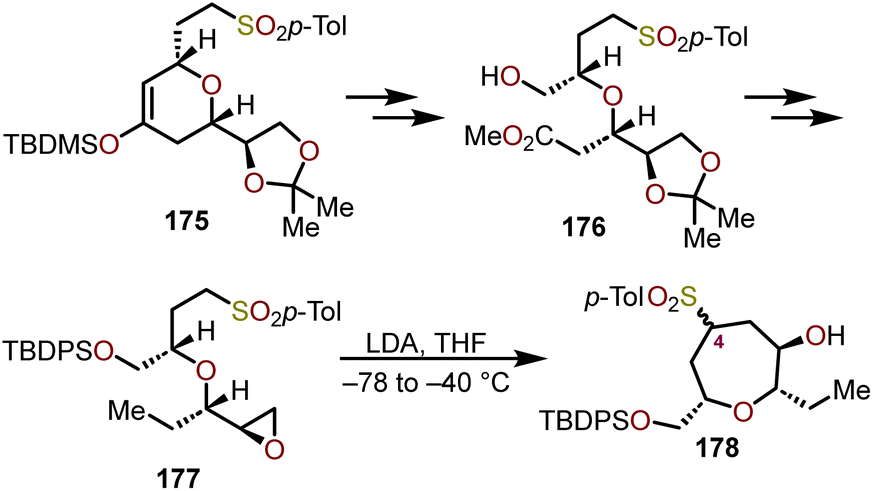 | ||
| Scheme 29 Synthesis of enantiopure oxepanes via epoxide ring opening.78 | ||
In the above methodology, it is important to note that formation of the carbanion and attack on the electrophilic epoxide are two major transformations which give rise to oxepane system. Although both endo and exo attack of the nucleophile on the epoxide can occur, the formation of products from exo attack were not observed. This article has demonstrated a strategy for making functionalized oxepanes with a high degree of stereocontrol which can be useful for the synthesis of a variety oxepane-containing natural products.
Armbruster and coworkers have demonstrated a strategy to synthesize meso persubstituted oxepanes using cyclooctatetraene via a modified skeletal rearrangement and ring contraction.79 Synthesis of polyhydroxylated oxepane 181 or 182 started via cyclooctene 179 which containing two adjacent dimethyl ethers within a bis-epoxide system (Scheme 30). Intermediate 179 is obtained from a cyclooctatetraene, which was treated with trifluoroperacetic acid to yield a bis-1,5-epoxide system followed by a selective dihydroxylation using OsO4/NMO.80,81 For installation of O-functionality via allylic substitution in the above diepoxide system, a monofunctional nucleophile such as water was selected. The nucleophilic attack of water and ensuing epoxide-opening sequence affords bicycle 180, thus providing the starting material to access fully functionalized oxepanes. Subjection of 180 to ozonolysis conditions followed by a reductive work-up afforded 181 with the stereochemistry shown in Scheme 30. Acetylation allowed isolation of polyoxygenated oxepane 182 with isolated yields in the range of 50–60%. Above is an excellent strategy to form polyhydroxylated oxepanes with a high degree of stereocontrol, with no observed epimerization in the intermediate steps. Modifying the nucleophile from water to primary amines allows access to N-substituted azepanes via a similar reaction sequence.79
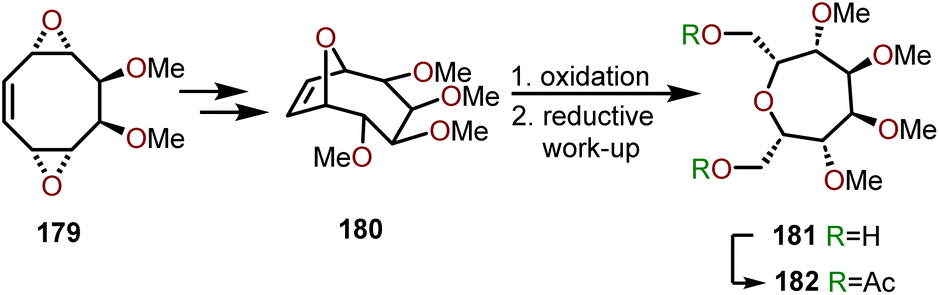 | ||
| Scheme 30 Synthesis of oxepanes via functionalized cyclooctatetraene.79–81 | ||
4. Synthesis of polyhydroxylated thiepanes
Unnatural sugar analogs such as thiepanes have been synthesized in similar fashion to their carbohydrate counterparts. Shih and coworkers developed a procedure to readily prepare 3,4,6-trihydroxythiepanes from D-(–)-quinic acid (Scheme 31).82 After sequential dihydroxylation and oxidative cleavage of 183 with RuCl3 and NaIO4, and reduction with NaBH4 gave diol 184 in an 72% yield. After mesylation of 184 to afford bis-mesylate 185, incorporation of the sulfur with Na2S·9H2O provides thiepane 186 in a 94% yield. Deprotection of the benzyl group was carried out with DDQ, followed by acetal deprotection with 2 N HCl for 3.5 h to afford the desired 3,4,6-trihydroxythiepane 189 in an 83% yield. Longer reaction times led to the formation of the ring-contracted thiopyranyl isomers 187 and 188. Specifically, with the reaction stirring for 3 days, the relative ratio of 187 to 188 to 189 was reported as 5.8:0:1. Alternatively, Shih and coworkers investigated the use of acetal protected thiepane 190, which upon treatment with 2 N HCl for 3.5 h, exclusively provided polyhydroxylated thiepane 191 in a 54% yield. Decomposition of thiepane 191 was observed with longer reaction times affording thiopyrans. Nonetheless, literature is scarce in the development of synthetic methods that afford polyhydroxylated thiepanes and should be considered as an area of research in the future.
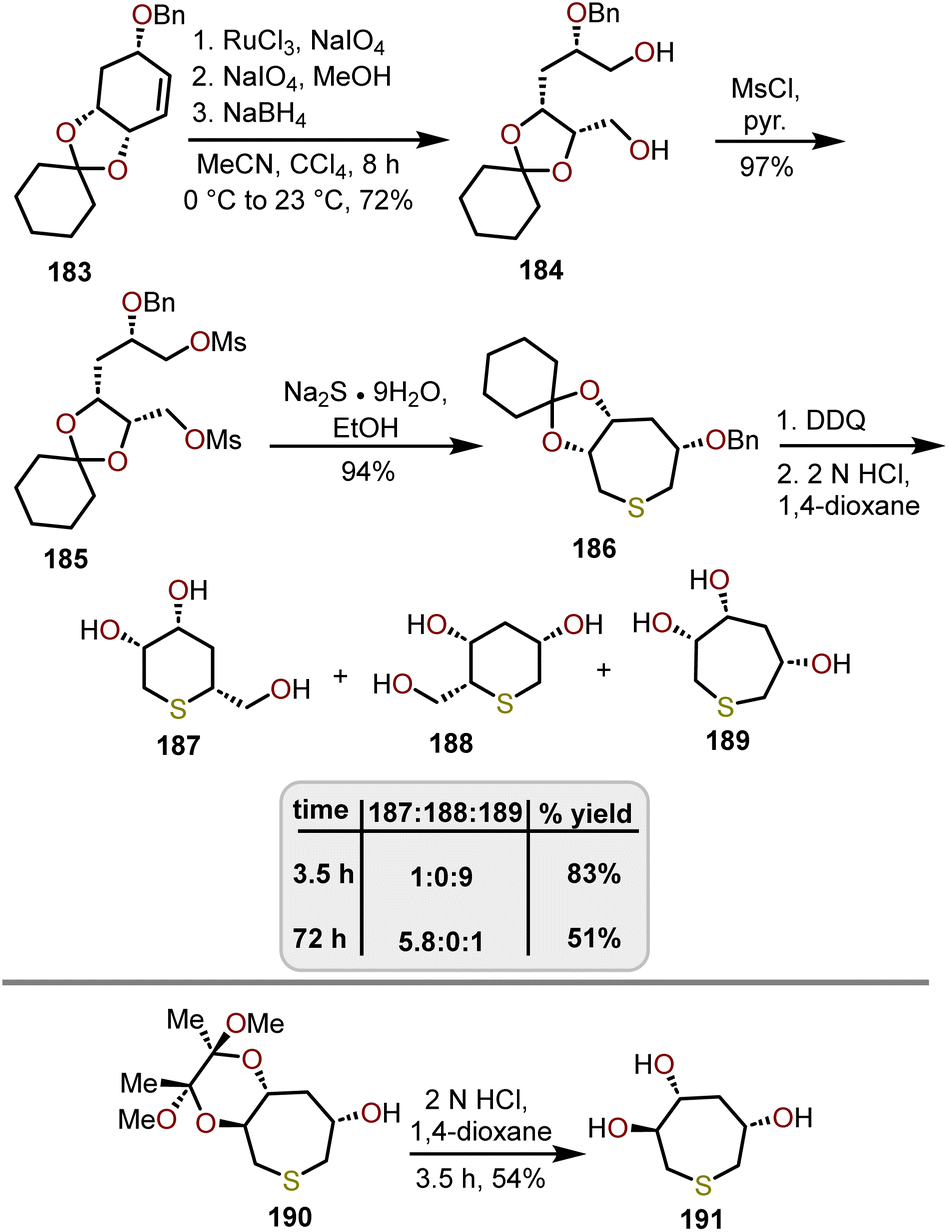 | ||
| Scheme 31 Synthetic route to thiepane via a ring-expansion strategy.82 | ||
5. Applications to natural product total synthesis
A key application of the highlighted synthetic methods for accessing polyoxygenated oxepanes is found in target-oriented synthesis. These seven-membered oxacycles are found within a variety of advanced scaffolds in natural products, whose assembly is a challenging task to synthetic chemists. In addition to the inherent structural complexity of oxepane-containing target molecules, some express notable bioactivities which makes them relevant for pharmaceuticals. Consequently, the discovery of these natural products has served as motivation for the development of novel synthetic methodologies to form oxepanes in various complex systems, which showcases the robustness of these transformations. This section will focus on the key synthetic efforts utilized to access natural products that possess polyoxygenated oxepane motifs.5.1. Total synthesis of hemibrevetoxin B
A large family of natural products bearing the oxepane moiety are the brevetoxins. These highly bioactive compounds possess a ladder of polycyclic ethers and have been noted for their potent neurotoxicity. Amongst these biologically relevant molecules are the hemibrevetoxins, which are speculated to be biosynthetic metabolites to the brevetoxins. In 1989, Prasad and Shimizu elucidated the structure of hemibrevetoxin B, a trans-fused tetracyclic scaffold with two tetrahydropyrans adjacent to two oxepanes.83 Hemibrevetoxin B contains ten stereogenic centers embedded within the 6/6/7/7-polycyclic array. Moreover, hemibrevetoxin B (1) was shown to exhibit cytotoxicity towards mouse neuroblastoma cells with an IC50 = 5 μM.83 As a result of its bioactivity and its structural complexity, hemibrevetoxin B became a target molecule which was initially targeted and synthesized by the groups of Nicolaou, Yamamoto, Nakata, and Mori.50,77,84,85The first total synthesis of hemibrevetoxin B was accomplished in 1992 by Nicolaou and coworkers whom used a linear, sugar-based approach starting from pyranose 192, which is derived from readily available D-mannose (Scheme 32).84 This allowed for the sequential construction of the 6/6 ring system after several transformations to afford key intermediate 193 and setting the stage for oxepane formation.
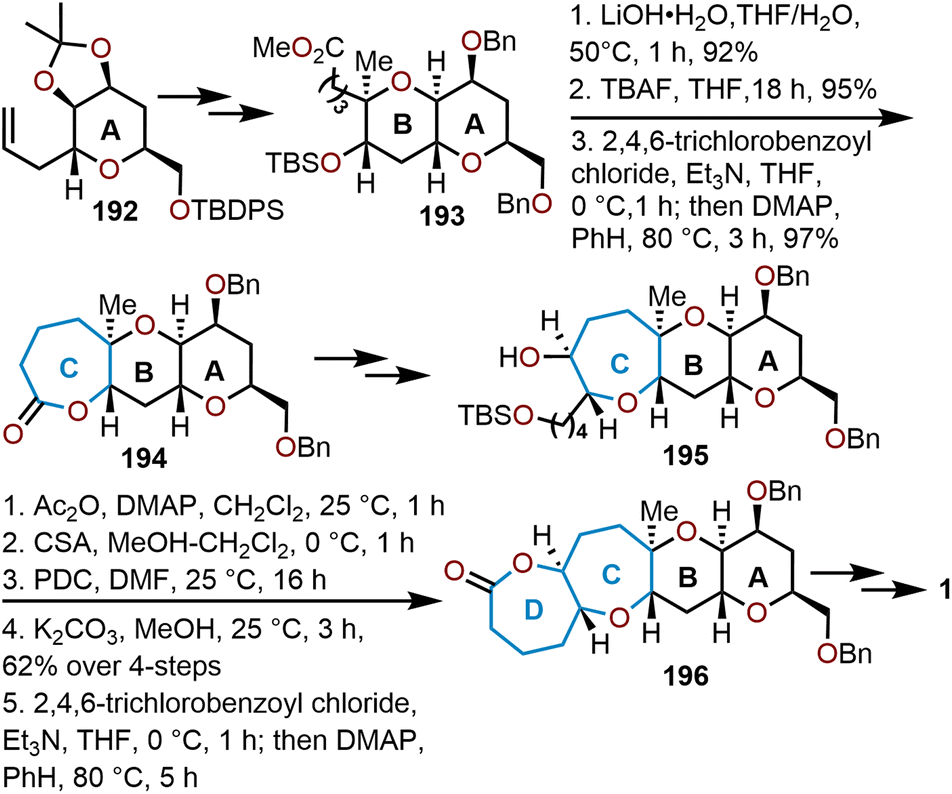 | ||
| Scheme 32 Nicolaou and coworkers’ strategy to access hemibrevetoxin B.84 | ||
Sequential saponification, desilylation, then Yamaguchi lactonization gives the seven-membered lactone 194 from 193 in an 85% yield over the three-step sequence (Scheme 32). Elaboration of the tricyclic intermediate led Nicolaou and coworkers to the TBS-ether 195. The free hydroxyl of 195 was capped with acetic anhydride and the TBS silyl-ether was cleaved with camphorsulfonic acid (CSA), whereupon oxidation of resulting primary alcohol with pyridinium dichromate (PDC) affords a carboxylic acid. Lastly, potassium carbonate was used to cleave the acetate protecting group and set the stage for the macrolactonization under Yamaguchi's standard protocol to give dioxepane 196. The synthesis of hemibrevetoxin B (1) was completed in twenty additional steps from 196. The work from Nicolaou's group demonstrates that dioxepane systems can be successfully installed through Yamaguchi macrolactonizations, allowing for an iterative elaboration of the polycyclic system.
Another linear, sugar-based approach for the total synthesis of hemibrevetoxin B was accomplished by Yamamoto and coworkers in 1995.85 Their route for the 6/6 ring system was closely inspired by Nicolaou's group, but the construction of the 7/7 rings were executed in a different manner. From D-mannose, known pyranose 197 was derived, whereupon several transformations provided key intermediate 198. Aldehyde 198 underwent a stereoselective Lewis acid-mediated intramolecular condensation with the tethered allylstannane to rapidly access oxepane 199 in a 94% yield (Scheme 33).85 The second oxepane ring 201 was also constructed via an intramolecular Lewis-acid mediated cyclization of 200, which was elaborated over 14 additional steps to eventually afford hemibrevetoxin B. Kadota and Yamamoto reported an improved and stereocontrolled route to 1 in 1998 that constructed the A and B ring oxepanes in a similar manner.86
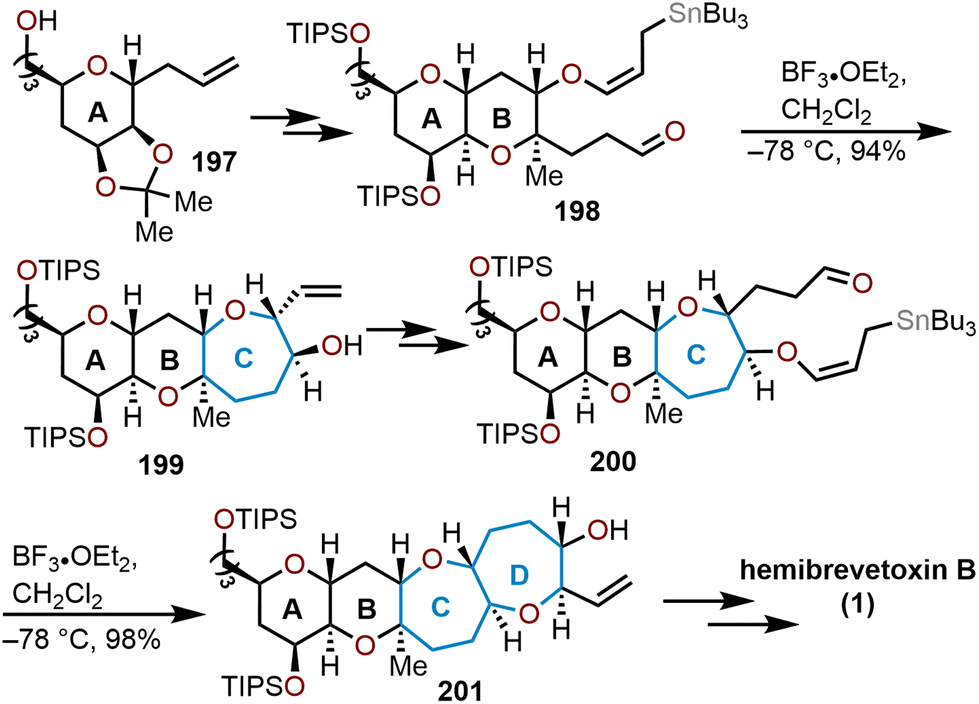 | ||
| Scheme 33 Yamamoto and coworkers’ strategy to access hemibrevetoxin B.85,86 | ||
In their total synthesis of hemibrevetoxin B reported in 1996, Nakata and coworkers relied on a double ring expansion of the pyranyl moieties to construct the bis-oxepane (Scheme 34).77 The bicyclic ether 202 was treated with chloromethanesulfonyl chloride in the presence of 2,6-lutidine to give a bis-chloromethanesulfonate that was then treated with zinc(II) acetate in acetic acid to perform the double rearrangement-ring expansion, thus yielding bicyclic septanoside 203 as shown in Scheme 34.77 A unique feature of this transformation was the retention of stereochemistry, which results from the proposed concerted mechanism shown in Scheme 28.
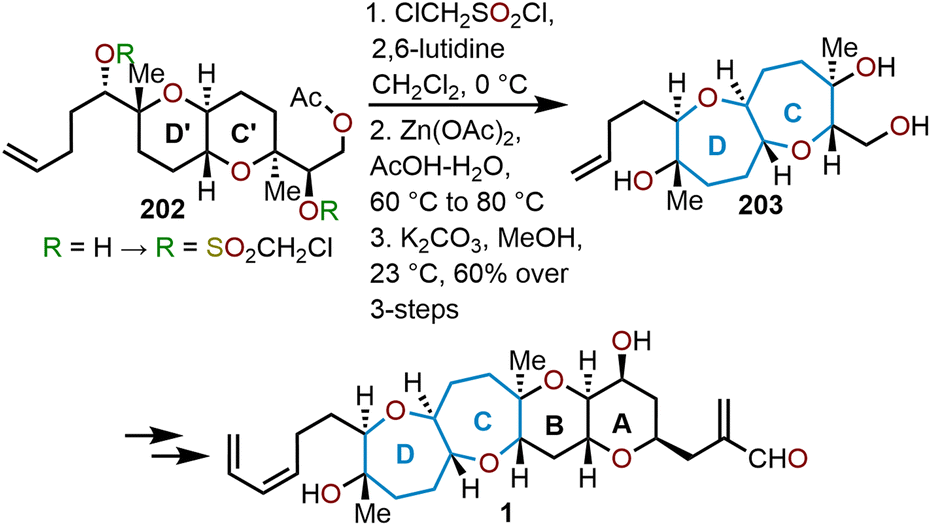 | ||
| Scheme 34 Nakata and coworkers’ strategy to access hemibrevetoxin B.77 | ||
Mori and coworkers demonstrate the high utility of oxiranyl anions in their 1997 formal synthesis of hemibrevetoxin B (Scheme 35).50 Their innovative approach offers a stereocontrolled synthesis of 1 by leveraging their developed protocols with sulfonyl-stabilized oxiranyl anions (Scheme 19). For example, readily prepared epoxysulfone 205 is deprotonated with n-butyllithium at cold temperatures to form the oxiranyl anion, which reacts with aldehyde 204 in a stereoselective fashion (3:1 mixture) to afford the desired alcohol 206 in 63% yield. Lewis acid-mediated cyclization with boron trifluoride etherate (BF3·OEt2) led to the formation of a tricyclic hydroxy ketone, whereupon addition of SmI2 removes the hydroxyl group to produce ketone 207.
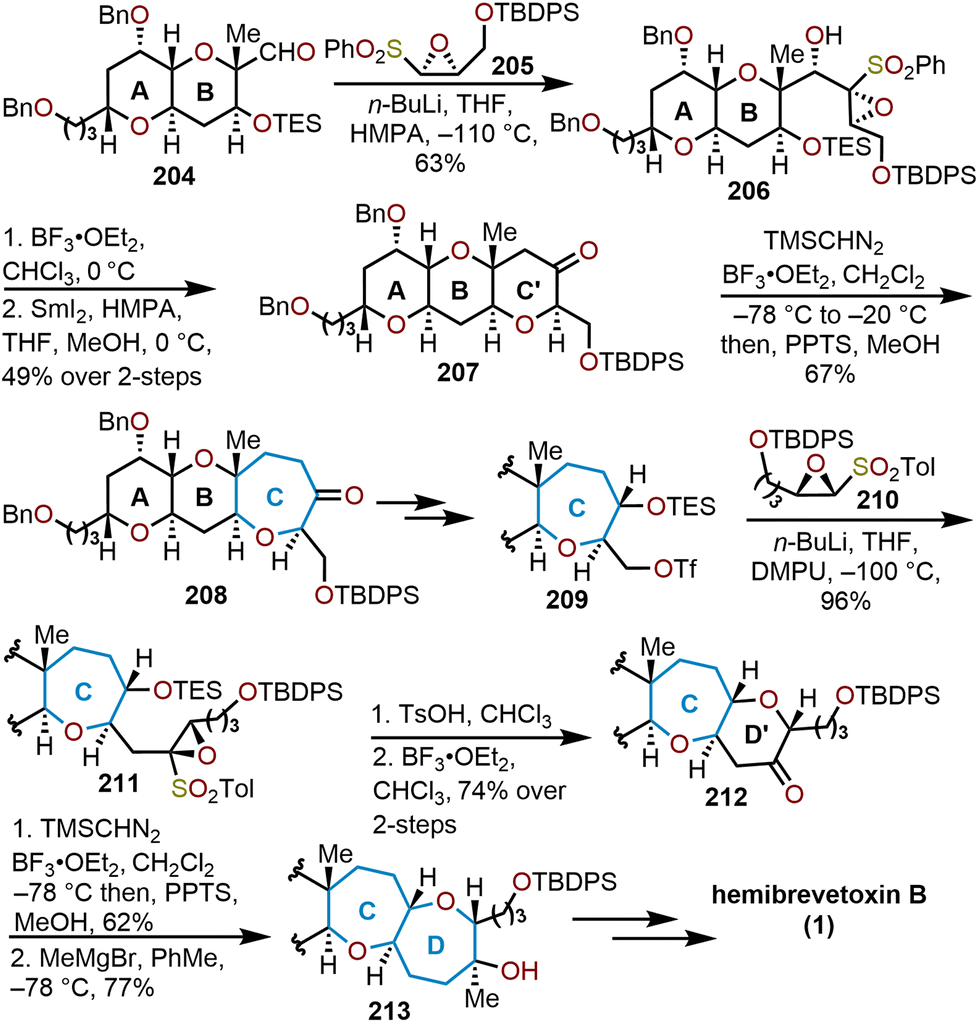 | ||
| Scheme 35 Mori and coworkers’ strategy to access hemibrevetoxin B.50 | ||
Oxepane formation was then achieved through a homologation event with TMSCHN2 in the presence of BF3·OEt2 (see Scheme 18 above) to give the 6/6/7-tricyclic ketone 208 in 67% yield. After several transformations Mori and coworkers performed this sequence again with triflate 209. Coupling of the triflate and the epoxy sulfone 210 proceeded smoothly to afford the tethered tricycle 211 in great yield. Treatment of 211 with p-TsOH and BF3·OEt2 formed the tetracyclic ketone 212 in 74% over two steps. Subsequent homologation of 212 with TMSCHN2 gave the 6/6/7/7-tetracyclic system, which upon treatment with methylmagnesium bromide afforded alcohol 213. After the complete formation of the tetracyclic scaffold, protecting group manipulation allowed access to a common intermediate employed by Yamamoto and coworkers85 enabling a formal synthesis of hemibrevetoxin B in nine steps from 213. The strategy used by Mori and coworkers displays the unique approach to form six-membered ethers via addition of oxiranyl anions to electrophiles, followed by a Lewis acid-mediated cyclization. This synthetic strategy was nicely used to prepare for the homologation to arrive at the seven-membered rings.
Two additional formal syntheses of hemibrevetoxin B were completed by the Rainier87 and Nelson88 groups in 2001, and in 2003 the Holton89 group completed a shorter, convergent total synthesis of 1 in 39 steps with a 4% overall yield. Rainier and coworkers' formal synthesis87 used an annulation reaction that proceeded through a mixed acetal to construct the C ring oxepane and a RCM reaction using Grubbs I catalyst to construct the D ring oxepane, which allowed advancement to a common intermediate employed by Mori and coworkers.50 Nelson and coworkers' formal synthesis did not tackle the construction of oxepanes, but used a desymmetrization of a centrosymmetric diepoxide through an enantioselective epoxide hydrolysis to access an epoxy acetal that contained both A and B ring tetrahydropyrans and was used by Nakata and coworkers.77 In Holton and coworkers' convergent synthesis of 189 the C ring oxepane was formed through a biomimetic epoxy alcohol cyclization72,73 (see Scheme 26 above) using N-(phenylseleno)phthalimide as the electrophile in HFIP solvent, and the D ring oxepane was constructed through a RCM reaction using Grubbs II catalyst.
5.2 Total synthesis of brevetoxin B
The brevetoxins are a widely known family of polycyclic ethers that are potent neurotoxins found in the marine organism Gymnodinium breve Davis.90 Brevetoxin B (Fig. 2, 214) contains 11 trans-fused oxacycles with 6,7, and 8-membered rings with 23 stereocenters, thereby presents a challenging target for total synthesis and attracted the interest of several synthetic groups. Nicolaou and coworkers were the first to access 214 in 1995 from a sugar-based convergent synthetic route (Scheme 36).91–93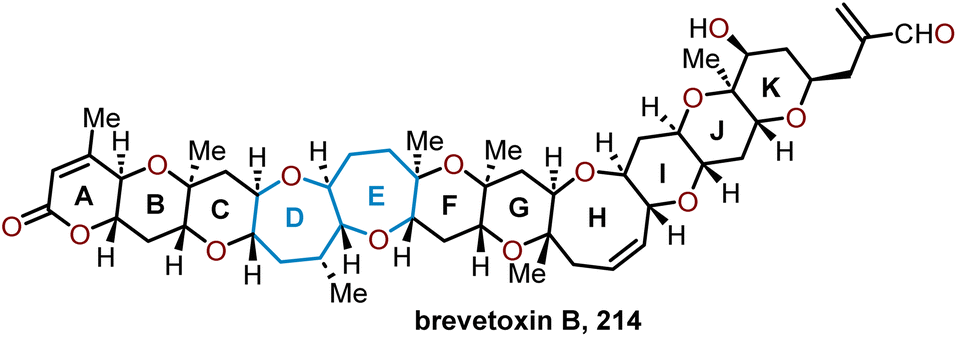 | ||
| Fig. 2 Structure of brevetoxin B. | ||
 | ||
| Scheme 36 Nicolaou and coworkers’ route to brevetoxin B.91–93 | ||
Their approach involved the assembly of the ABCDEFG and IJK ring systems, whereupon after convergence, the H ring would be stitched together via a hydroxydithioketal cyclization. The route towards 214 began with 2-deoxy-D-ribose as the starting material to construct the FG ring system in hydroxy acid 215 enabling macrolactonization under Yamaguchi conditions to give the seven-membered lactone 216 in 90% yield and stitch on the E ring (Scheme 36).91 This intermediate was advanced to form a second hydroxy acid 217 to perform another Yamaguchi macrolactonization to furnish bis-oxepane 218 in 60% yield, now containing the D ring. Elaboration of 218 to oxepine 219 and hydroboration-oxidation to hydroxy oxepane 220, which contains an alkyl tether with the requisite atoms for C-ring construction to further advance the left flank and eventually arrive at brevetoxin B.
The total synthesis of brevetoxin B was also accomplished by Nakata and coworkers in 2004.94 A bidirectional strategy consisting of a double radical-induced reductive cyclization method was successfully employed to construct the CDE ring system as depicted in Scheme 37. Starting with commercially available tri-O-acetyl-D-glucal, the α,β-unsaturated ester 221 was produced after several steps. Oxidation of the alcohol with TEMPO and PIDA forms the aldehyde whereupon treatment with SmI2 in methanol promotes their developed radical cyclization (Scheme 14) to furnish the 5/7/6 tricyclic lactone 222 with an 83% over the two steps.
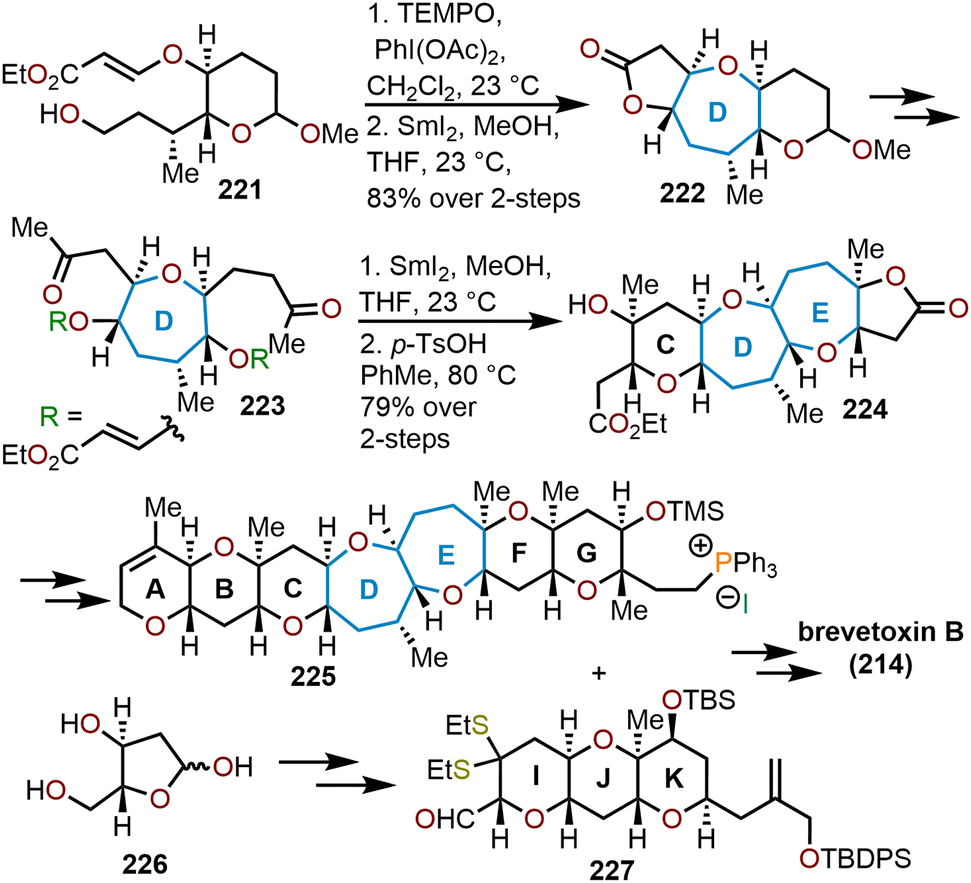 | ||
| Scheme 37 Nakata and coworkers’ route to brevetoxin B.94 | ||
Cleavage of the lactone and acetal led Nakata and coworkers to eventually arrive at the bis(methyl ketone) 223 on both flanks of the D ring. The bidirectional radical cyclization was able to furnish the CDE ring system 224 in a stereoselective fashion after treatment with p-TsOH. From the CDE lactone, the outer rings were constructed to afford the ABCDEFG ring system in 225, which was coupled to the IJK ring system 227 available from 2-deoxy-D-ribose (226), eventually arriving at brevetoxin B.
5.3 Total synthesis of brevetoxin A
The most potent sodium channel activator of the brevetoxins is brevetoxin A, whose scaffold is composed of polycyclic ether ladder with one 5-membered lactone, four pyranyl units, one oxepane, three 8-membered oxacycles, and one 9-membered oxacycle (Fig. 3). The structural complexity of brevetoxin A is further compounded from the slow conformational changes within its skeletal to cause a 90° twist at one of the rings.95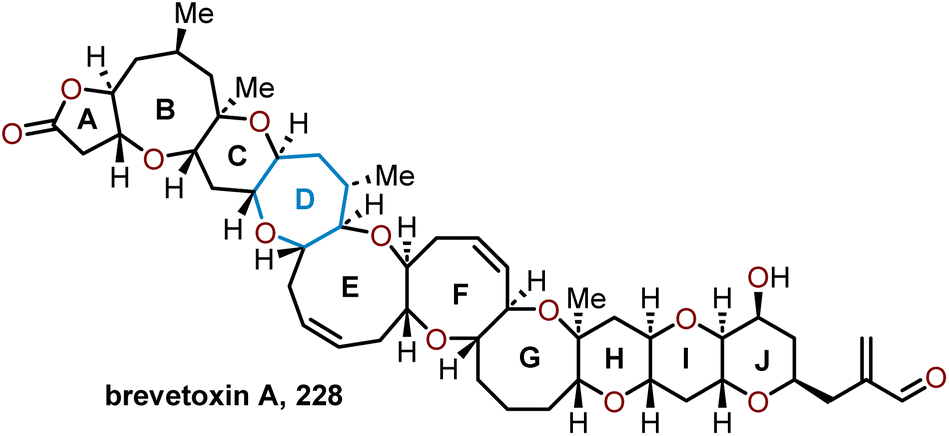 | ||
| Fig. 3 Structure of brevetoxin A. | ||
The first total synthesis of brevetoxin A (228) was accomplished by Nicolaou and coworkers in 1998.95 A sugar-based, convergent synthetic route was employed to stitch together the BCDE and the GHIJ ring systems. In the construction of the BCDE ring system, D-glucose was appointed as the starting material to obtain diacid 229 over 18 steps (Scheme 38). The B and D rings were formed in a single step via a double Yamaguchi macrolactonization to give bis-lactone 230 in 82% yield. Desilylation, followed by hydrogenation of the exo-methylene in 230 afforded the key intermediate 231. A bidirectional approach was taken by Nicolaou and coworkers to afford bis(vinylstannane) 232 from 231 in 65% yield in a one-pot sequence. Tin-lithium exchange of the bis(vinylstannane) 232 with n-butyllithium followed by transmetallation with the copper acetylide of 1-pentyne provided a mixed cuprate, which was treated with benzyloxyethyl triflate to afford an intermediate bis(vinylether) that was subjected to hydroboration with thexylborane and basic hydrogen peroxide work-up to give diol 233 in 52% yield over three sequential steps. From the 8/6/7 tricycle, Nicolaou and coworkers were able to utilize the functionality on both flanks to complete the total synthesis of brevetoxin A.
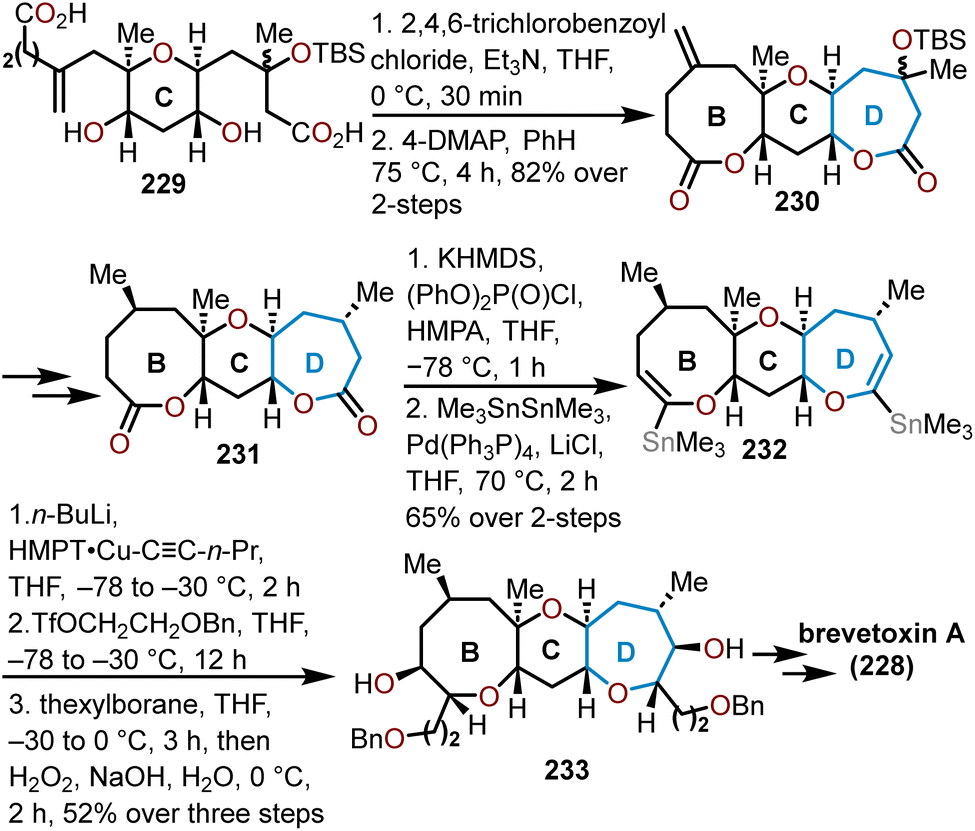 | ||
| Scheme 38 Nicolaou and coworkers’ route to brevetoxin A.95 | ||
The second group to accomplish the total synthesis of brevetoxin A was Crimmins and coworkers in 2009 (Scheme 39).96 From their efforts, a stereoselective synthesis of the BCDE fragment was realized via a glycolyl thioimide auxiliary 234 (Scheme 39A).97 After several synthetic transformations the BCE ring system was afforded and set the stage for an intramolecular cyclization event of the keto alcohol 235 using pyridinium p-toluenesulfonate (PPTS) to construct the D-ring. The cyclization event resulted in a 35% yield of oxepane 236 or 70% based on recovered starting material (brsm). Given bottlenecks in this original route, Crimmins and coworkers developed a second-generation route to access brevetoxin A from glycocyl imide 237 (Scheme 39B).98 The formation of the oxepane in ring D was improved upon by employing camphorsulfonic acid (CSA), which greatly increased the overall efficiency by cleaving the TBS-silyl ether in 238 to promote direct cyclization and afford 239 in 76% yield. Reduction of the mixed methyl ketals was carried out with BF3·Et2O and dimethylphenylsilane (see Scheme 20 above) to complete the construction of the BCDE fragment 240 in a 78% yield.
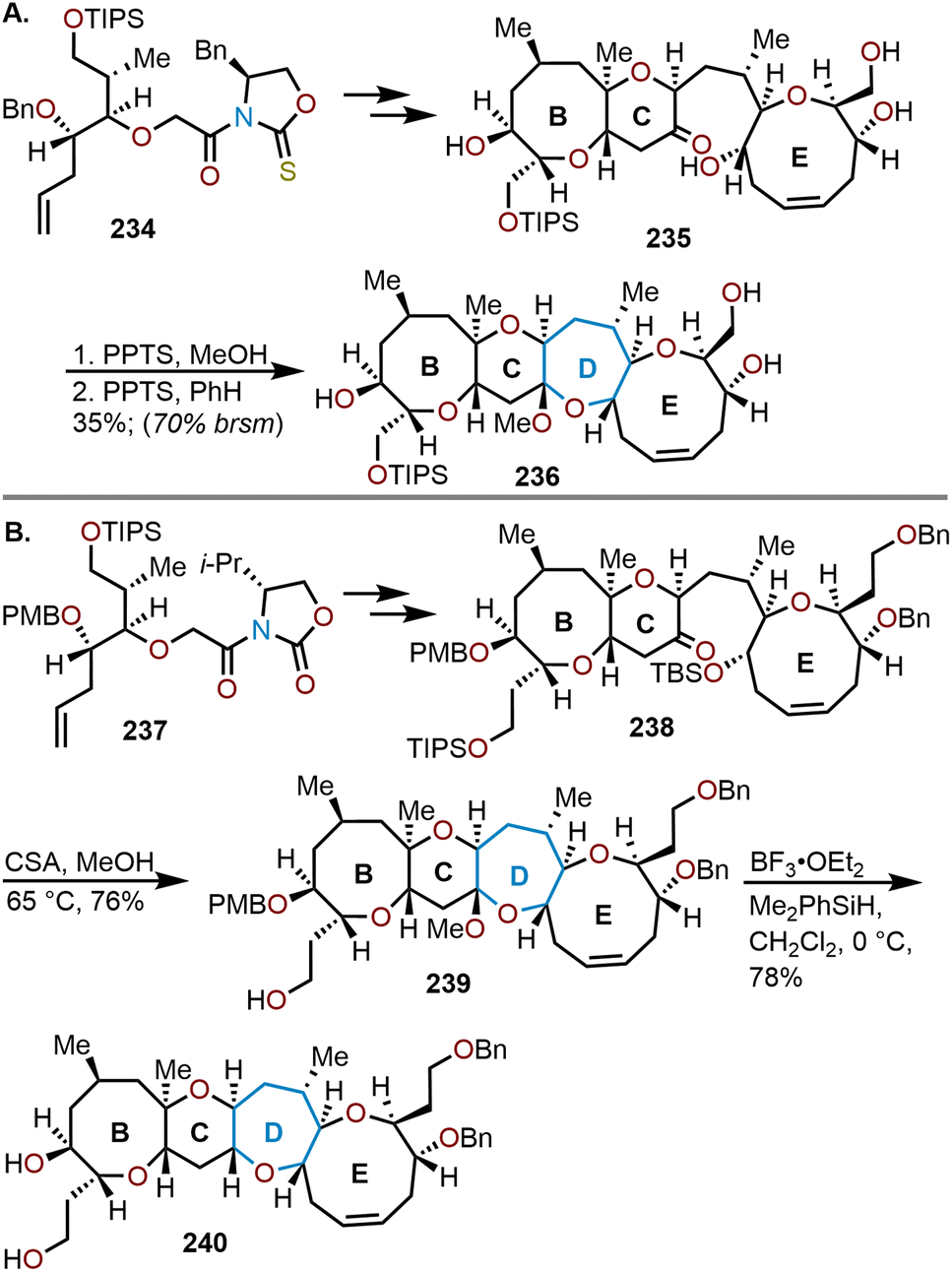 | ||
| Scheme 39 Crimmins and coworkers’ synthesis of brevetoxin A.97,98 | ||
5.4 Total synthesis of brevenal
Brevenal (Fig. 4, 241) was also isolated from the same species of dinoflagellate as the brevetoxins thus shares the same structural and stereochemical complexity inherent to this class of molecules. This pentacyclic polyether consists of two pyranyl units and three oxepane units with 13 stereogenic centers. An important feature of this natural product was observed from its ability to improve tracheal mucus velocity in picomolar concentrations, which could be harnessed as a therapeutic for treating a variety of lung diseases.99 The potential of medicinal applications made brevenal a target molecule for several groups.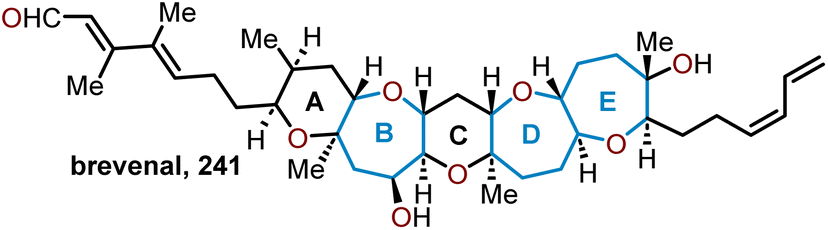 | ||
| Fig. 4 Structure of brevenal. | ||
Sasaki and coworkers were the first group to complete the total synthesis of brevenal in 2006 (Scheme 40).100 A convergent synthesis strategy was employed to couple the AB and DE fragments (244 and 248) through a Suzuki cross-coupling to afford 249 (Scheme 40C). The AB fragment was derived from a non-sugar starting material, which was eventually converted to α,β-unsaturated pyranyl ester 242 (Scheme 40A). Deprotection of the benzyl ester via hydrogenolysis followed by Yamaguchi macrolactonization afforded the AB ring system 243 in an 88% yield over the two steps. The lactone was then converted to enol phosphate 244 in preparation for the Suzuki cross-coupling. The DE ring system was derived starting from Nicolaou and coworkers known oxepane 245.
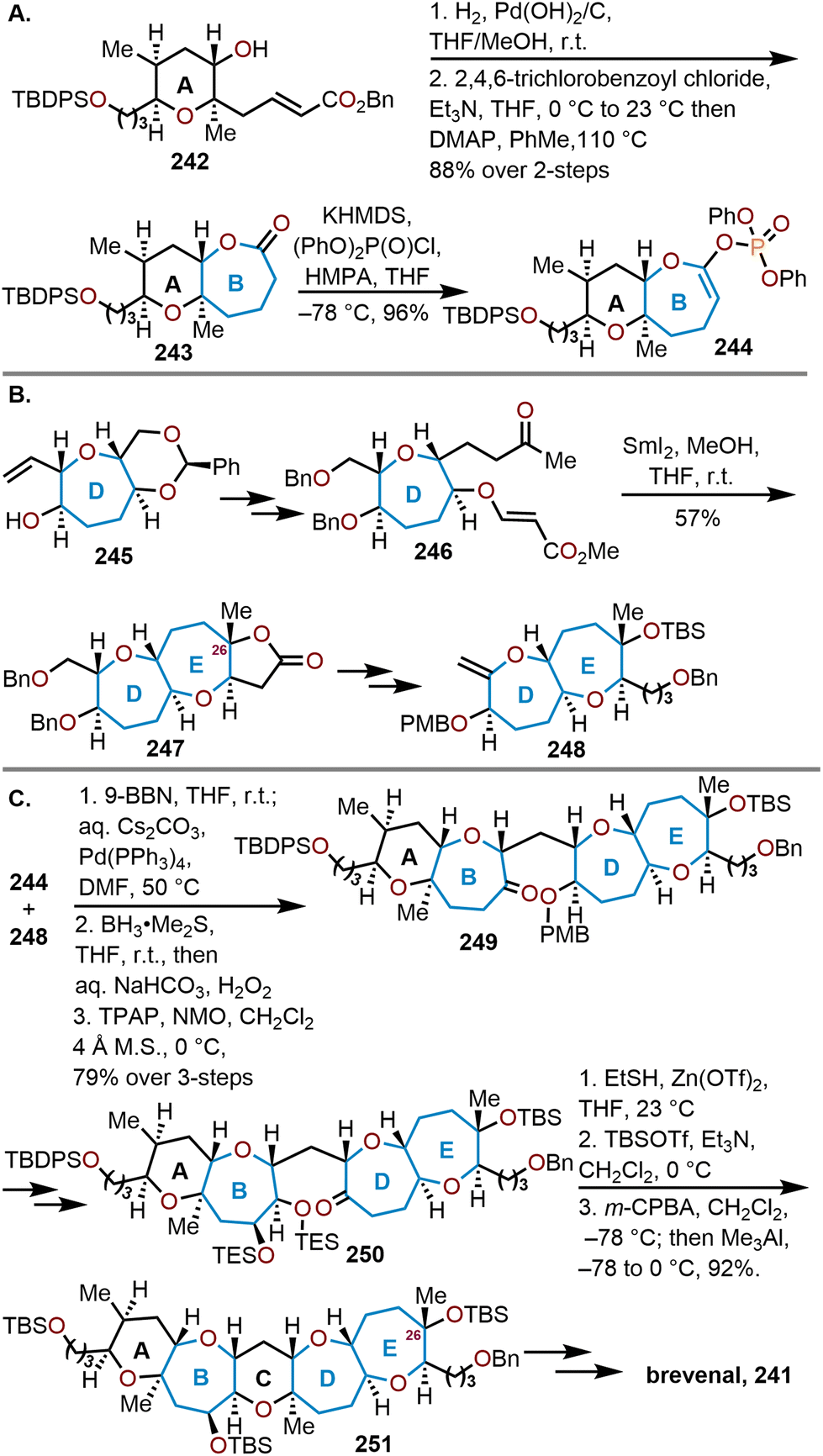 | ||
| Scheme 40 Sasaki and coworkers’ convergent strategy to access brevenal.100 | ||
After several transformations, α,β-unsaturated ester 246 was prepared (Scheme 40B). The E ring was formed via a reductive cyclization of 246 with SmI2 (see Scheme 14 above) to afford dioxepane lactone 247 in 57% yield. Advancement to dioxepane 248 was accomplished over 12 steps, which enabled the hydroboration of the exocyclic enol ether in 248 with 9-BBN to provide a suitable coupling partner for Suzuki coupling with enol phosphate 244, which was accomplished in the presence of Pd(PPh3)4 and Cs2CO3 to give a single stereoisomer (Scheme 31C). A hydroboration–oxidation of the resulting endocyclic enol ether furnishes an alcohol, which is afterwards oxidized to form the ABDE ring system 249 in an 79% yield over the three steps. Six additional steps from 249 were required to arrive at the bis-triethylsilyl ether 250 to carry out the final cyclization of ring C. The cyclization event proceeded with zinc triflate [Zn(OTf)2] and ethanethiol to form a trapped thioketal. With the thioketal, a one-pot oxidation–methylation reaction with m-CPBA and trimethylaluminum (AlMe3) was sequentially carried out to produce the completed pentacyclic scaffold 251. Brevenal would be afforded in 18 additional synthetic steps after several more peripheral transformations of the sidechains and included the structural reassignment of the C26 stereocenter.
Another total synthesis of brevenal was completed in 2009 by Kadota and coworkers as shown in Scheme 41.101 A linear route to brevenal was envisioned from the central C ring. Tetrahydropyran 252 was readily prepared from known procedures and advanced to a hydroxy acid via a hydroboration–oxidation, stepwise oxidation, and silyl deprotection in preparation for a Yamaguchi macrolactonization to afford the seven-membered lactone 253. After 21 additional synthetic transformations, Kadota and coworkers arrived at allylstannane 254. In the presence of magnesium bromide, an intramolecular allylation closes the D ring to give the 6/7/6/7-tetracyclic diene 255 in a stereoselective manner. Furthermore, this prepares for the formation of the E ring via a Grubbs cross-metathesis to form oxepine 256. From intermediate 256, brevenal was afforded in 12 additional steps.
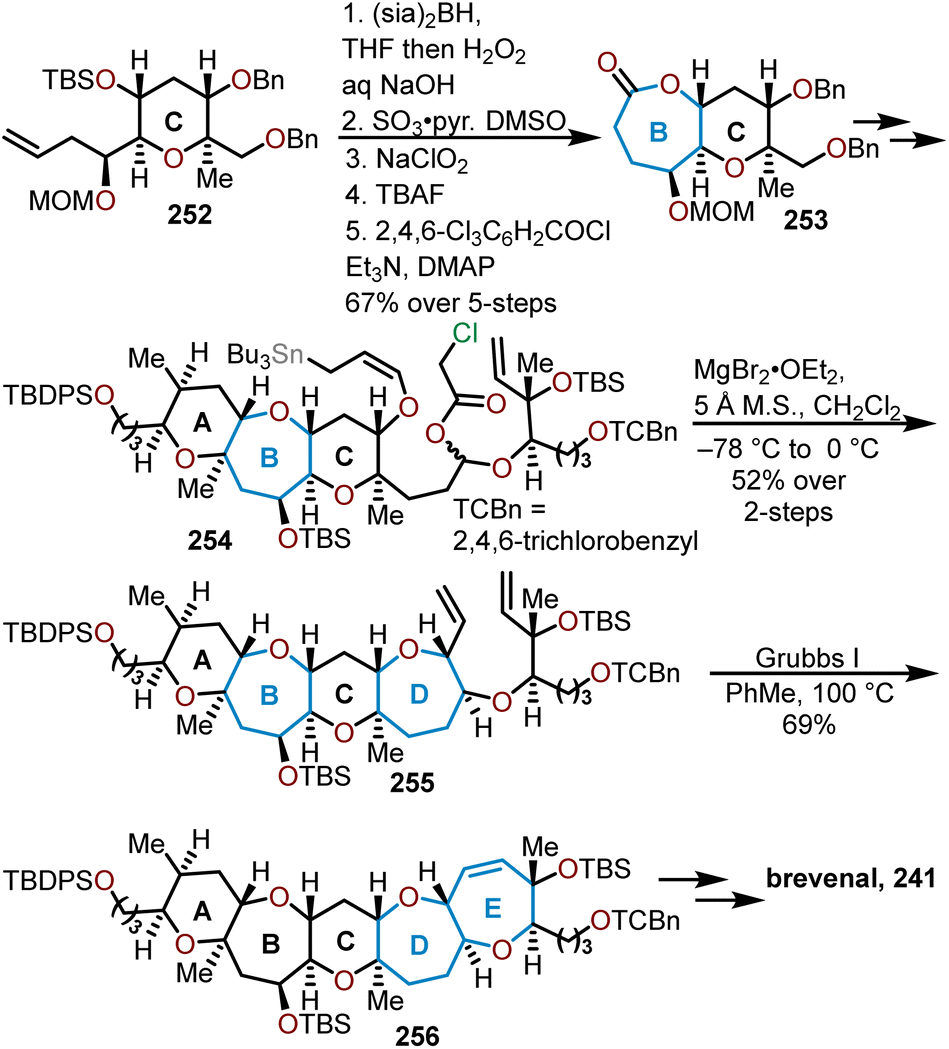 | ||
| Scheme 41 Kadota and coworkers’ linear approach to brevenal.101 | ||
The Rainier group was also completed the total synthesis of brevenal (241) in 2011.102 A convergent route was taken to couple the AB and E ring systems, where they would fuse the central C and D rings to complete the pentacyclic scaffold as shown in Scheme 42. A unique, unprecedented cyclization strategy using olefinic-esters served as inspiration to construct brevenal.103 Starting with simple building blocks, acetal 257 was derived in seven linear steps utilizing previous chemistry developed in their efforts towards hemibrevetoxin B.104 A two-step acid-mediated cyclization to a mixed acetal, followed by elimination of methanol afforded oxepine 258 in an 82% yield over the two steps. This two-step approach was more effective than a previously investigated one-step approach using PPTS and pyridine due to the sensitivity of 257 to PPTS at 130 °C.105,106 Epoxidation of the olefin with DMDO, and subsequent allylation with allyl Grignard furnished allyl oxepane 259 in an 87% yield and 10:1 mixture of diastereomers. This intermediate was further advanced through a Ley oxidation of the C15 hydroxyl group, Rubottom oxidation to install the C14 hydroxyl group as a 6:1 mixture of the desired stereoisomer, a stereoselective DIBAL-H reduction to reinstate the C15 hydroxyl group, silyl protection of the diol with TESCl, dihydroxylation of the olefin, lead tetraacetate diol cleavage and oxidation afforded key acid 259 in 56% yield over the sequence. After constructing the AB core, Rainier and coworkers began to forge the DE ring system. The olefinic-ester 260 is available from L-glyceraldehyde acetonide in four linear steps and served as the starting point for the E ring. Rainier and coworkers optimized the olefinic-ester cyclization to form oxepine 261 in 66% yield, using an in situ-generated reduced titanium ethylidene reagent from titanium tetrachloride, zinc, lead(II) chloride, and 1,1-dibromoethane. After eight synthetic transformations, hydroxyl-containing oxepane 262 was afforded to be coupled to the AB ring system in 259via a Shiina esterification. After successful coupling, ketone 263 was subjected to the same olefinic-ester cyclization conditions for preparing oxepine 261 to give ABDE ring system 264 in 65% yield. Epoxidation, in situ reductive opening with DIBAL-H, and Ley oxidation gives ketone 265 in 65% yield over the two steps. The central C ring was fused together via a Lewis acid-mediated cyclization with concomitant cleavage of the triethylsilyl (TES) ethers in 265. Addition of ethanethiol forms a trapped thioketal allowing methylation to be accomplished from the procedure developed by Kadota and coworkers.85 After completion of the pentacyclic system, 10 additional synthetic steps were required for side chain incorporation, which allowed Rainier and coworkers to complete the shortest synthesis of brevenal to-date in 38 linear steps and 0.99% overall yield.
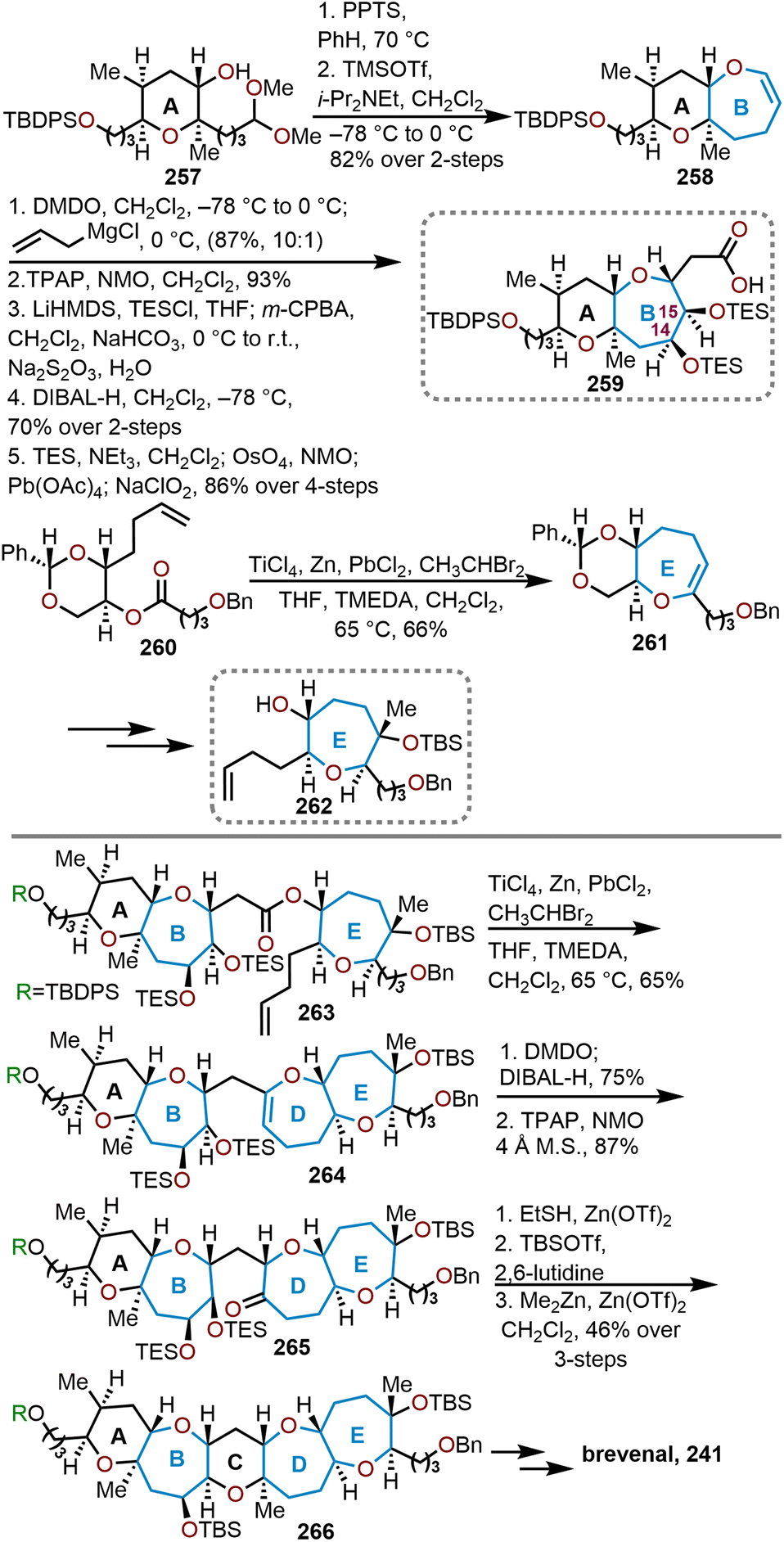 | ||
| Scheme 42 Rainier and coworkers’ strategy to access brevenal.102 | ||
5.5 Total synthesis of ciguatoxin 1B (CTX1B)
Ciguatoxins are a family of polycyclic ethers, which have been known to be highly potent neurotoxins found in marine dinoflagellate Gambierdiscus toxicus. The prominent ciguatoxin is ciguatoxin 1B (CTX1B, 267a, Fig. 5) and its structure was elucidated in 1989 by Yasumoto and coworkers.107 CTX1B was found to possess a trans-fused polycyclic ring system consisting of 13 oxacycles of various ring sizes (five to nine) and 33 stereogenic centers. Due to its unprecedented ladder-like ring system and its scarcity in nature, CTX1B became a target molecule for many groups. The first group to complete a total synthesis of CTX1B (267a) was that of Inoue, Hirama and coworkers in 2006.108 | ||
| Fig. 5 Relevant ciguatoxin target molecules. | ||
Inoue, Hirama and coworkers were able to synthesize CTX1B via a convergent synthesis using sugar-based starting materials. Their efforts are highlighted throughout several publications that entail the routes taken to synthesize each fragment.108–118 The seven-membered oxacycles (rings A, D, E, G, and K) were afforded by various chemical routes as shown in Schemes 43 and 44. For example, in the construction of the AB fragment, Hirama and coworkers started from the sugar-derived tri-O-benzyl-D-glucal 25 and in three steps via bromination with NBS, basic epoxide formation and opening with allylmagnesium bromide, and O-allylation with allyl bromide provided a reliable route to diene 268 (Scheme 43). This sequence set the stage for RCM with Grubbs’ first-generation catalyst to form the A ring 269 in 84% yield.110,114 Oxepine 269 is then advanced in 13 synthetic steps to diene 270 containing the ABC ring system, which was then poised for a second RCM for D-ring formation. Of note, to afford a high yield of RCM product, the free hydroxy groups and Grubbs’ second-generation catalyst were required.
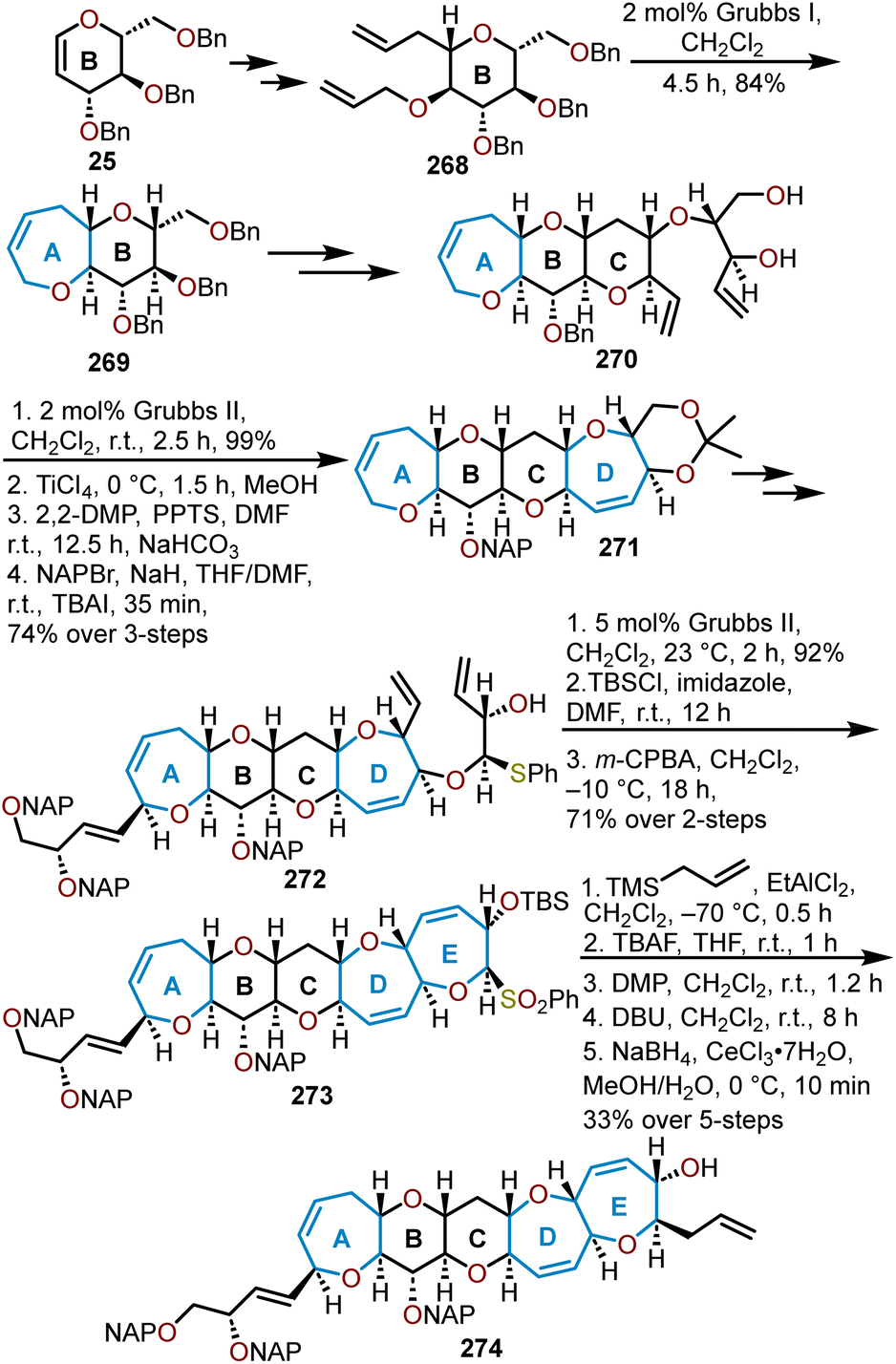 | ||
| Scheme 43 Inoue, Hirama, and coworkers’ synthesis of the A, D, and E oxepanes in CTX1B.110,114,115 | ||
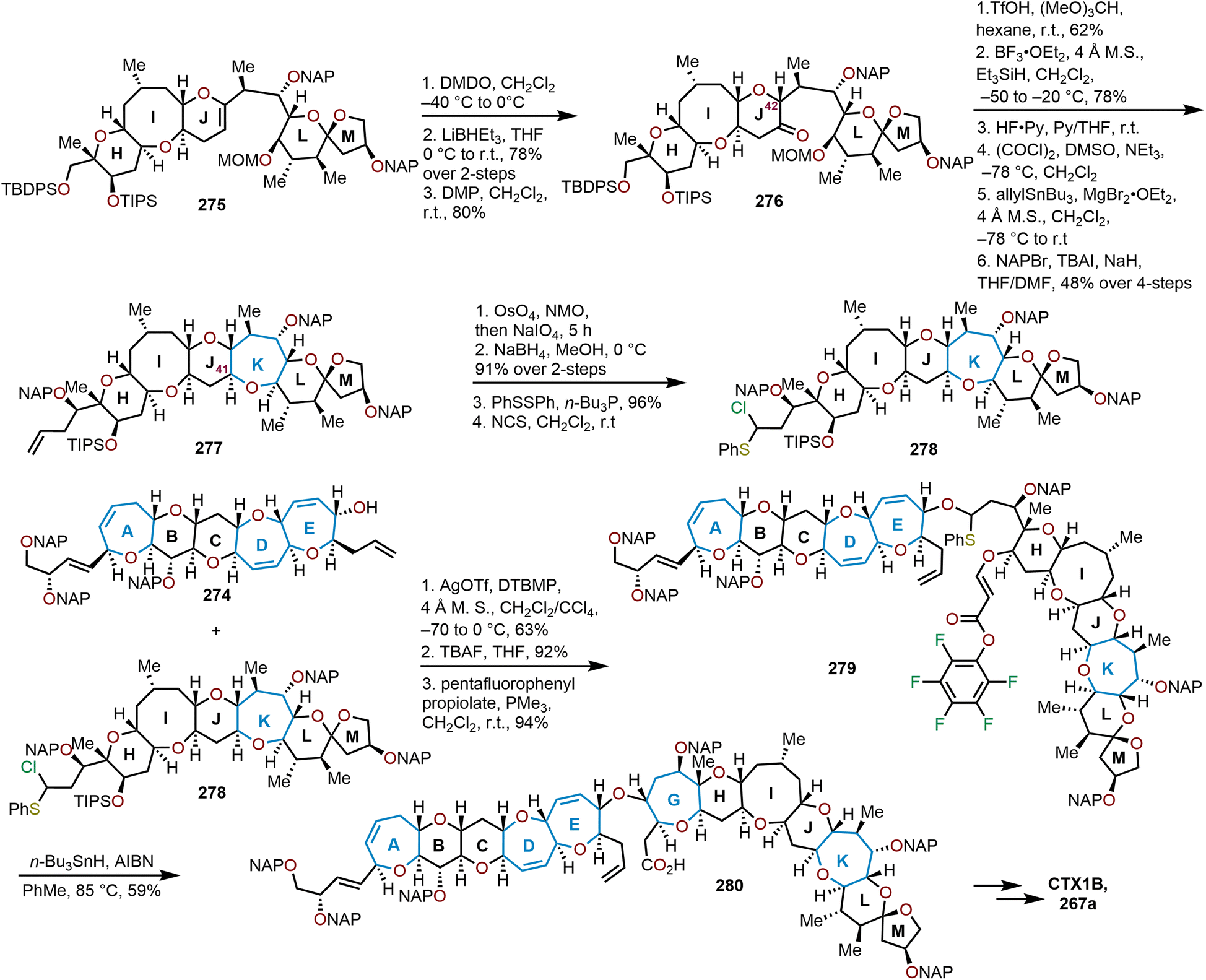 | ||
| Scheme 44 Inoue, Hirama, and coworkers’ synthesis of the K and G oxepanes of CTX1B (267a).108,116–118 | ||
Subsequent debenzylation, acetal protection with 2,2-dimethoxypropane (2,2-DMP), and reprotection of the secondary alcohol a 2-napthylmethyl group (NAP) afforded acetal 271 in 74% yield.110 Acetal 271 served as the starting point for side-chain elaboration and E ring construction. Side chain installation was accomplished via olefin migration within the A ring with Wilkinson's catalyst and DBU, enol ether oxidation with lead tetraacetate, and a stereoselective nickel-catalyzed coupling of the resulting allyl acetate with an alkenylborate variant of the TBS-protected sidechain. An additional 10 synthetic steps were required to access diene 272, which was able to undergo RCM with Grubbs’ second-generation catalyst to construct the E-ring of CTX1B in 92% yield. Protection of the secondary alcohol with TBSCl and m-CPBA oxidation of the sulfide afforded sulfone 273. Allylation of sulfone 273 allyltrimethylsilane and EtAlCl2, TBS removal, Dess–Martin periodinane oxidation, epimerization with DBU, and stereoselective reduction under Luche conditions afforded key ABCDE coupling fragment 274 in 33% yield over the five steps.110 After construction of the left wing of CTX1B, Inoue, Hirama, and coworkers turned their attention towards the right wing fragment (rings HIJKLM) as shown in Scheme 44.
The strategy used to make the right wing fragment of 267a was to couple the HI and LM ring systems via a Yamaguchi protocol followed by J ring construction with a low-valent titanium reagent to afford 275, and finally fusion of the seven-membered K ring allowed completion of the ring-wing.116,117 Fusion of the K ring was initiated by DMDO oxidation of the enol ether in 275 followed by a stereoselective, reductive-opening of the epoxide with LiBHEt3 to afford the desired C42 stereochemistry, which subsequent oxidation of the hydroxyl group with Dess–Martin periodinane afforded ketone 276. In the presence of triflic acid (TfOH) and trimethyl orthoformate, the K ring was formed via acetalization to a seven-membered methoxy acetal, which was followed by a reductive etherification to set the C41 stereocenter. Sidechain elaboration was then initiated with deprotection of the primary alcohol, followed by Swern oxidation, allylation with allyltributylstannane, and protection of the resulting secondary alcohol with a 2-napthylmethyl group to afford 277. Further elaboration of 277 to requisite α-chlorosulfide 278 was accomplished via dihydroxylation, NaIO4 cleavage to an aldehyde, reduction with NaBH4, thioether formation, and chlorination with N-chlorosuccinimide (NCS). The pinnacle of the first total synthesis of CTX1B 267a was observed in the coupling of the ABCDE 274 (from Scheme 43) and HIJKLM 278 fragments.108 The coupling was accomplished in a 63% yield in the presence of an excess of silver(I) triflate and di-tert-butylmethylpyridine (DTBMP). Removal of the TIPS ether with TBAF and treatment with pentafluorophenyl propiolate and trimethylphosphine provided pentafluorophenyl acrylate 279 for G ring construction via a stereoselective, 7-exo radical cyclization that proceeded in 59% yield to give 280.
Ciguatoxin 1B was afforded after subsequent elaboration of the carboxylic acid to a terminal olefin to allow for a Grubbs cross-metathesis to form the final F ring and removal of the NAP protecting groups provided 267a in six additional steps from 280. The highlights of the oxepane construction within Inoue, Hirama, and coworkers’ ciguatoxin 1B synthesis were A, D, and E ring construction via RCM, K ring creation via methyl acetal formation and reductive etherification, and G ring construction via a 7-exo radical cyclization mediated by tributyltin hydride and AIBN.
A few years later in 2009, Isobe and coworkers became the second group to accomplish the total synthesis of CTX1B (267a).67 The Isobe group sought to employ their recently developed methods for preparing cyclic ethers via acetylene-dicobalt hexacarbonyl complexes, which has shown to give syn, trans polycyclic systems in a stereoselective manner (see Schemes 21 and 22 above).47,55–64,66,119,120 Their unique cyclization strategy was used to construct the BCDE66,121 and HIJKLM120 fragments, and to forge the central F and G rings (Schemes 45–47). The construction of the BCDE ring system (Scheme 45) began with the addition of dicobalt octacarbonyl and boron trifluoride etherate known propargylic alcohol 281, which was available in 31 synthetic steps from methyl-α-D-glucoside.66,121 Cyclization of 281 to form the D ring resulted in Nicholas adduct 282, which underwent hydrosilylation with triethylsilane in propargyl alcohol/dichloroethane (DCE) solvent to afford vinylsilane 283 in 75% yield over two steps. Sidechain elaboration and oxidation of the oxepine over 14 additional synthetic steps led to propargylic alcohol 284, wherein the iterative cyclization–hydrosilylation sequence via the dicobalt hexacarbonyl adduct forms the E ring in 285 in 71% yield over three steps. Jones oxidation of the primary alcohol, iodolactonization, protection of the methyl ketone to afford the dithioketal, DIBAL-H reduction of the lactone, and subsequent treatment with DBU led to the formation of epoxysilane 286 in 52% over five steps. The requisite BCDE coupling fragment 287 was completed from an additional nine synthetic steps from 286 and was used for the coupling to the HIJKLM fragment. Previously Isobe and coworkers optimized routes to access vinyl sulfone 288 in 22 steps from methyl-a-D-glucoside and acetylene 289 in 20 steps from tri-O-acetyl-D-glucal which served as starting points for HIJKLM ring construction as illustrated in Scheme 46.120,122 A heteroconjugate addition123 of the lithium acetylide of 289 to vinylsulfone 288 and TBS deprotection provided alkyne 290.
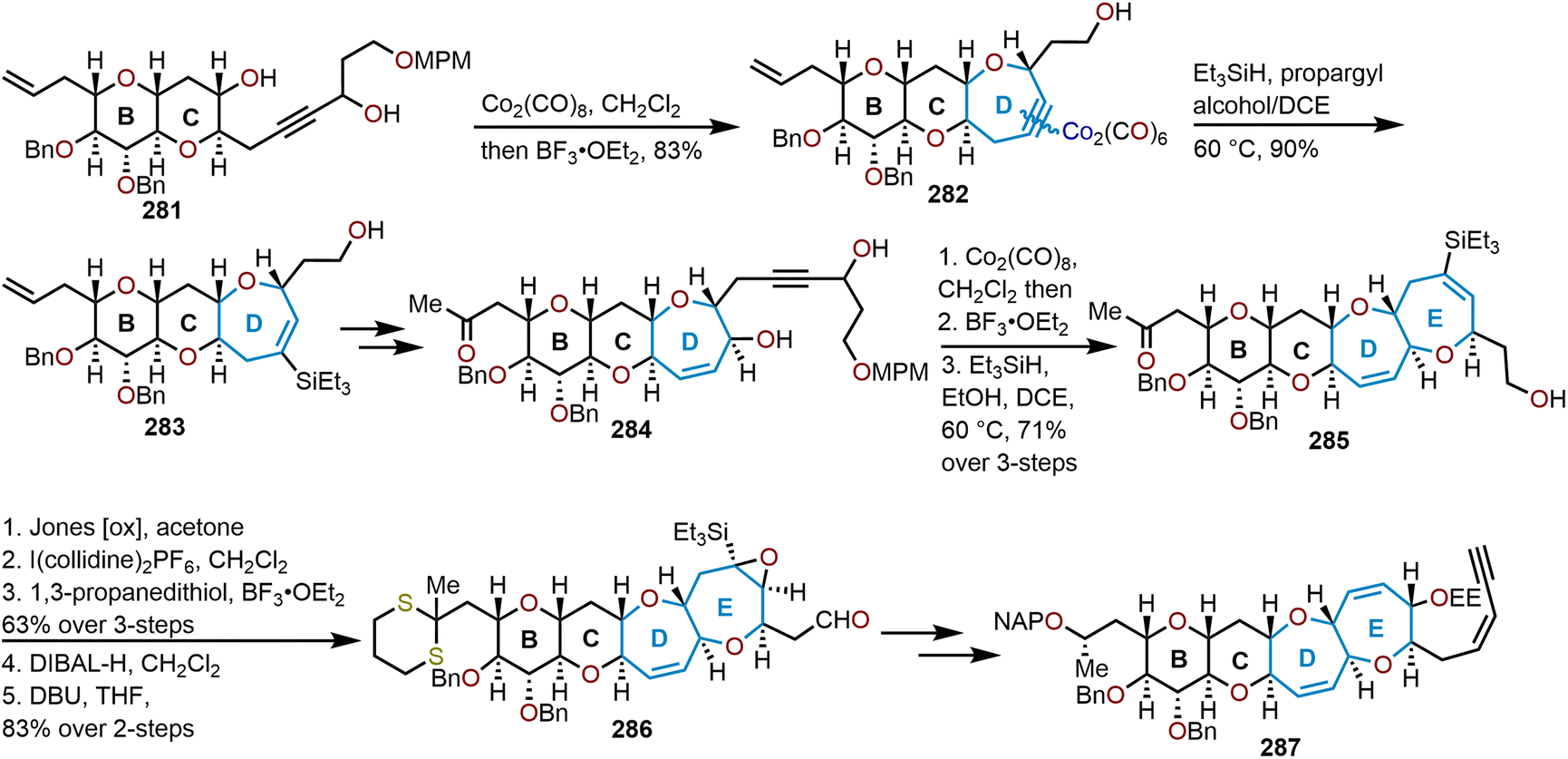 | ||
| Scheme 45 Isobe and coworkers’ route to access the BCDE fragment.66 | ||
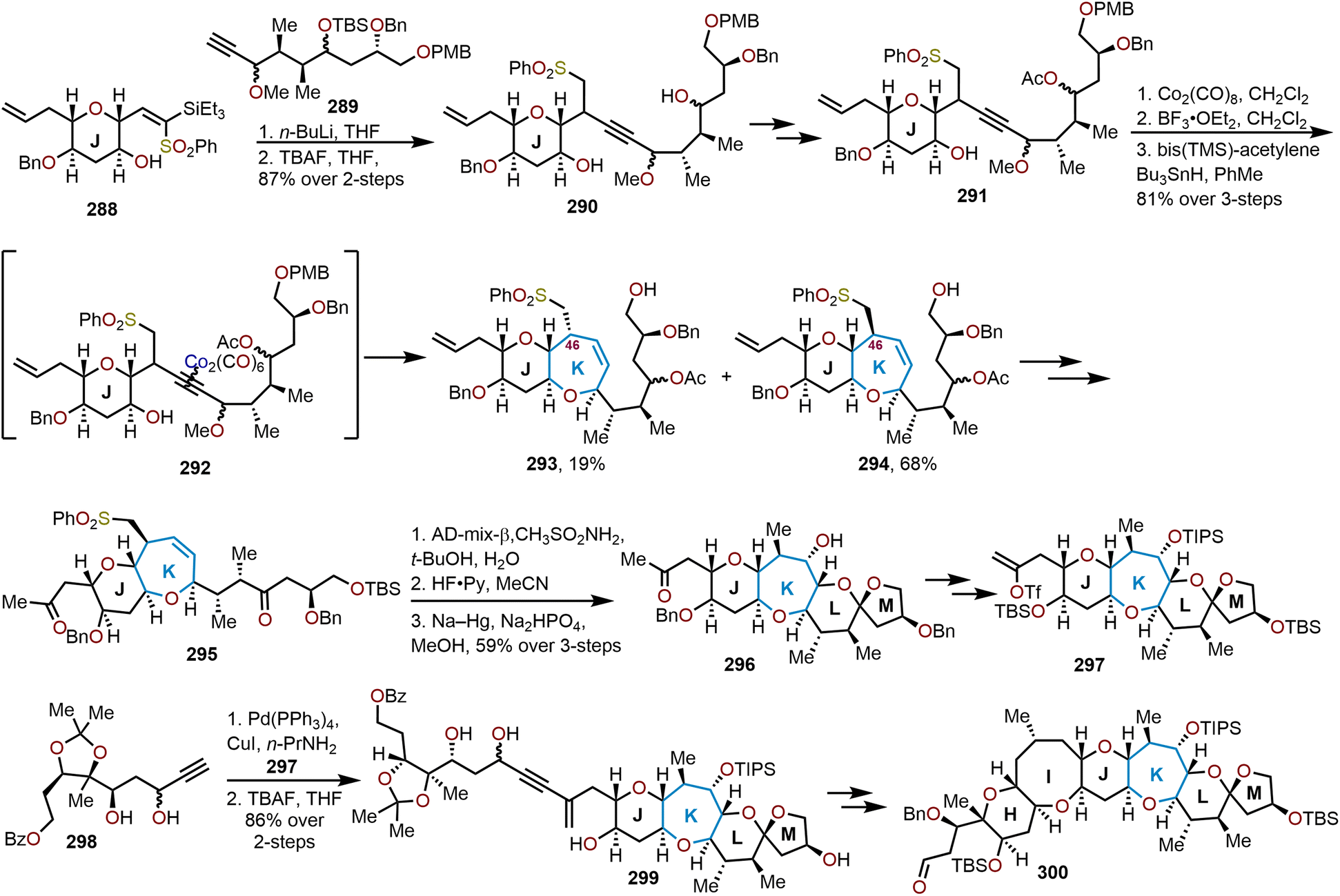 | ||
| Scheme 46 Isobe and coworkers’ route to access the HIJKLM fragment.67,120,122 | ||
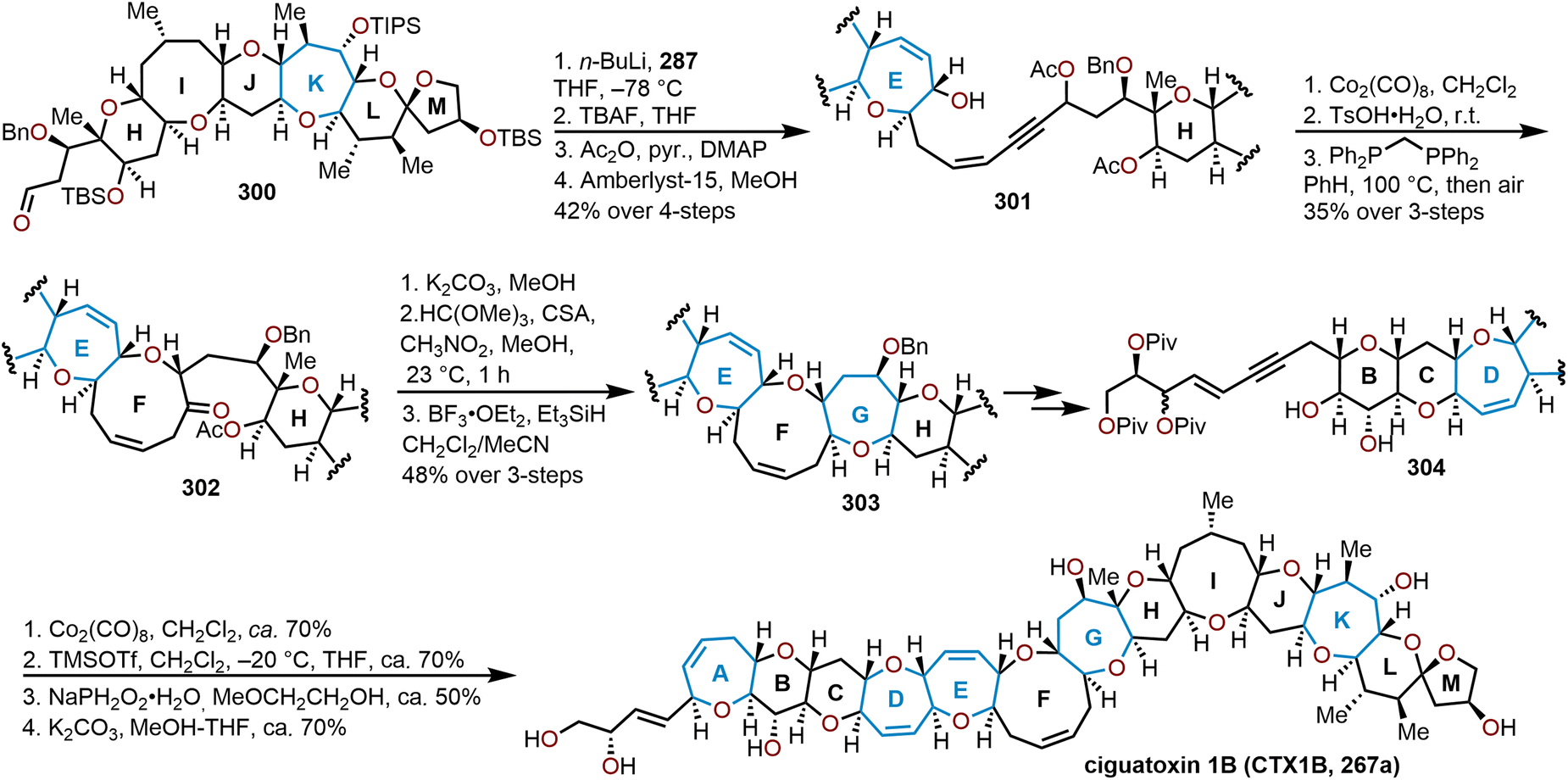 | ||
| Scheme 47 Isobe and coworkers’ convergent route to complete CTX1B.67 | ||
After further protecting group manipulation to afford acetate 291 and using their iterative cyclization–hydrosilylation sequence via the dicobalt hexacarbonyl adduct 292 the K ring was installed. Epimerization during the process at C46 led to the formation of diastereomers, 293 and 294 in 19% and 68% yields, respectively. Fortunately, the major product 294 was needed to move forward in the sequence. Following protecting group manipulations, IBX oxidation of the secondary alcohol, and Wacker oxidation afforded dione 295. Dihydroxylation of the oxepine under Sharpless conditions followed by removal of the TBS ether promoted spiroketalization to furnish the L and M rings and subsequent desulfurization provided 296 in 59% over the three steps.
Lastly, silyl ether protection of the free hydroxyl with TIPSCl, debenzylation/reprotection with TBSCl, and vinyl triflate formation provided 297, which was poised for a Sonogashira coupling with known alkyne 298 available from tri-O-acetyl-D-glucal.120,124 From the coupled alkyne 299, the dicobalt hexacarbonyl mediated cyclization–hydrosilylation strategy fused together the I ring, enabled H ring construction, and allowed further advancement to aldehyde 300 in 15 steps from 299 in order to set up the coupling with the BCDE ring system 287.
The coupling of BCDE and HIJKLM fragments was achieved via 1,2-addition of the lithium acetylide of 287 to aldehyde 300, which was followed by global deprotection of the TBS ethers, global acetate installation, and removal of the ethoxyethyl acetal (EE) to afford enyne 301 (Scheme 47).67 Treatment of 301 with dicobalt octacarbonyl, p-TsOH, and oxidative decomplexation using bis(diphenylphosphino)methane (dppm) with exposure of air produced ketone 302 now containing the F ring. After acetate deprotection of 302, acetalization using trimethyl orthoformate and camphorsulfonic acid (CSA) followed by a reductive etherification resulted in construction of the G-ring oxepane to afford 303. Functionalization of the left flank on the B ring in 7 synthetic steps provides enyne 304 in prelude to the final cyclization event using dicobalt octacarbonyl. Formation of the propargyl cation required TMSOTf, but THF was also needed to scavenge excess Lewis acid in order to promote the cyclization to form the A ring oxepine. Lastly, reductive decomplexation of the cobalt complex and global acetate deprotection affords ciguatoxin 1B (CTX1B; 267a).
The highlights of the oxepane construction within Isobe and coworkers’ ciguatoxin 1B synthesis were the use of an iterative set of cyclizations of acetylene-dicobalt hexacarbonyl complexes followed by a hydrosilylation or decomplexation to form and elaborate the A, D, E, and K rings as well as an acetalization and reductive etherification sequence to form the G ring oxepane.
5.6 Total synthesis of ciguatoxin 3C (CTX3C)
The structure of ciguatoxin 3C (CTX3C; 267b), a simplified congener of CTX1B, was elucidated in 1975 by Yasumoto and coworkers.125 The scaffold of 267b consists of 13 embedded oxacycles of 5, 6, 7, 8, and 9-membered rings that are trans-fused, and contains 31 stereocenters (Fig. 5). Inoue, Hirama and coworkers were able to synthesize CTX3C (267b) in 2001 through a convergent route (Scheme 48).113,115,118,126,127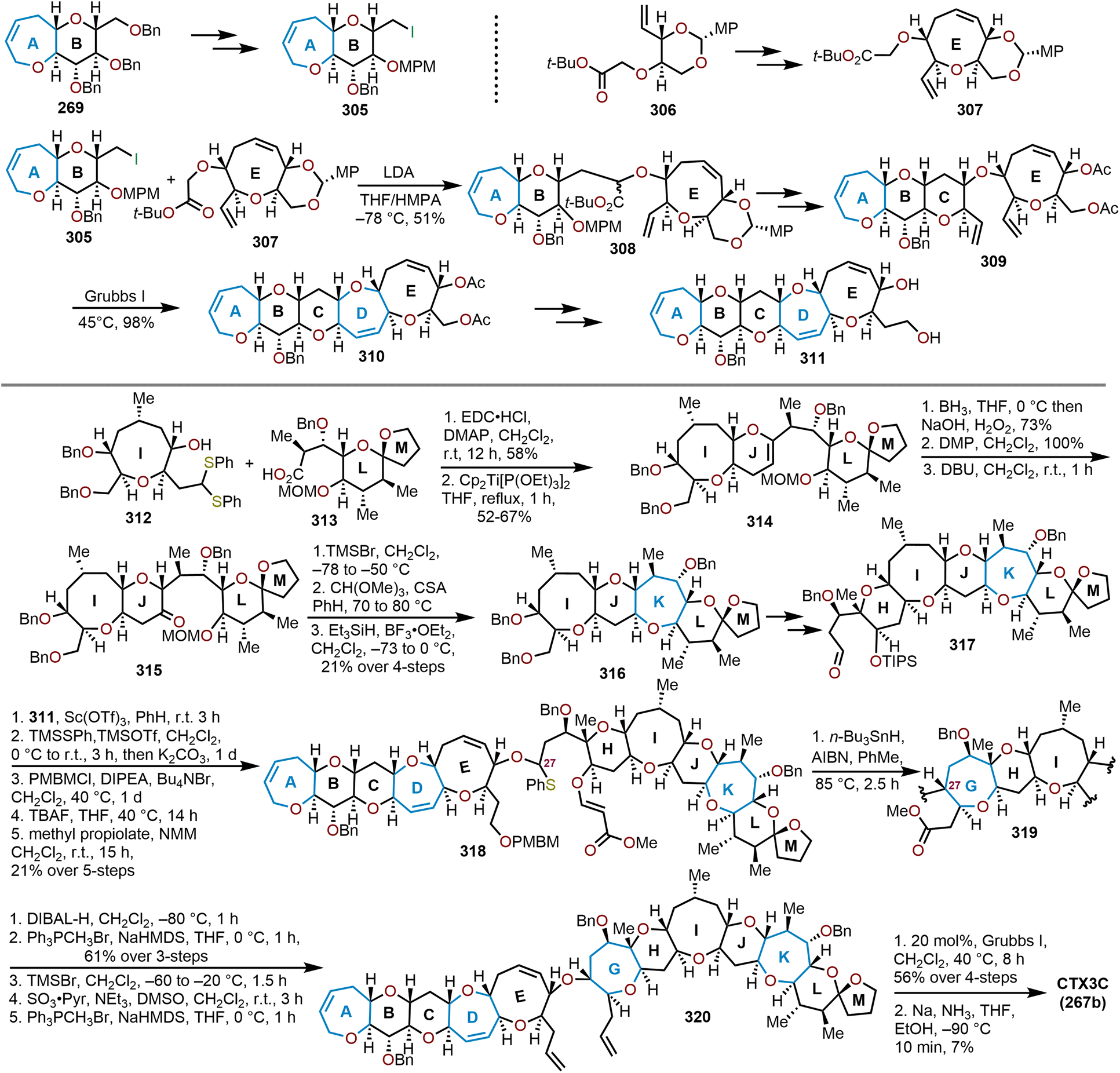 | ||
| Scheme 48 Hirama and coworkers’ efforts to synthesize CTX3C.113,115,118,126,127 | ||
The left wing fragment consisting of the ABCDE ring system126,128311 and the right wing fragment consisting of the HIJKLM ring system126,129,130317 were individually prepared, then coupled together. The remaining F and G were fused lastly to complete the scaffold. The left wing fragment was constructed by implementing an RCM strategy using a Grubbs catalyst. The A ring in 269 was constructed using the RCM strategy described in Scheme 43 above from CTX1B, and then advanced to iodide 305.115 Methoxybenzylidene (MP) acetal 306 was advanced to construct the E ring and afford 307, which upon treatment of LDA generated the lithium enolate that was coupled to iodide 305 to eventually arrive at 308. Following C ring construction and advancement over 6 synthetic steps to diene 309, cyclization of the D ring was possible via another intramolecular Grubbs cross-metathesis to furnish the ABDE ring system 310.128 Five additional synthetic steps were required to afford diol 311. To construct the right wing portion of CTX3C, dithioacetal 312 containing the I ring and carboxylic acid 313 containing the L and M rings were condensed to allow for J ring construction using the Takeda low-valent titanium reagent to afford 314.131 Oxidation of the J ring enol ether and epimerization afforded ketone 315. Acetalization and reductive etherification allowed construction of the K ring oxepane to afford IJKLM fragment 316, which after 16 synthetic steps was advanced to aldehyde 317.126,130 The key coupling of 311 and 317 was accomplished via Sc(OTf)3-catalyzed O,O-acetalization and conversion to a O,S-acetal using phenylthiotrimethylsilane and TMSOTf. Protection of the primary hydroxyl as a p-methoxybenzyloxymethyl (PMBM) ether, deprotection of the TIPS group, and installation of the methyl acrylate afforded 318 in 21% over the five-step sequence. Formation of the G ring oxepane was accomplished by treatment with tributyltin hydride and AIBN to promote a stereo- and chemo-selective radical cyclization of the C27 radical with the α,β-unsaturated ester to afford 319. Further advancement to diene 320 enabled the formation of the final F ring oxepine through a RCM with Grubbs’ first-generation catalyst, and global debenzylation using Birch reduction conditions provided the first total synthesis of CTX3C (267b). Inoue, Hirama, and coworkers later reported an improved protecting-group strategy to enable a shorter total synthesis of CTX3C.115–118,127
The highlights of the oxepane construction within Inoue, Hirama, and coworkers’ ciguatoxin 3C syntheses were the use of a RCM strategy to prepare the A and D rings, an acetalization/reductive etherification to construct the K ring, and a stereo- and chemoselective radical cyclization to construct the G ring.
5.7 Total synthesis of gymnocin A
The isolation of gymnocin A from the dinoflagellate Karenia mikimotoi, was reported in 2002 by Satake and coworkers.132 The architecture of gymnocin A contains a ladder of 14 contiguous cyclic ethers embedded with 31 stereogenic centers (Fig. 6, 321). While gymnocin A is a potent toxin found in the red tide algae, the natural product exhibits an EC50 value of 1.26 μM against P388 leukemia cells.133 As a result of its structural complexity and its inherent bioactivities, gymnocin A became a target molecule for many groups. The first total synthesis of gymnocin A was completed in 2003 by Sasaki and coworkers.133 A convergent route was taken to stitch together ABCD (Scheme 49),134 and the GHI and KLMN fragments (Scheme 50)135via a Suzuki cross-coupling (Scheme 51).133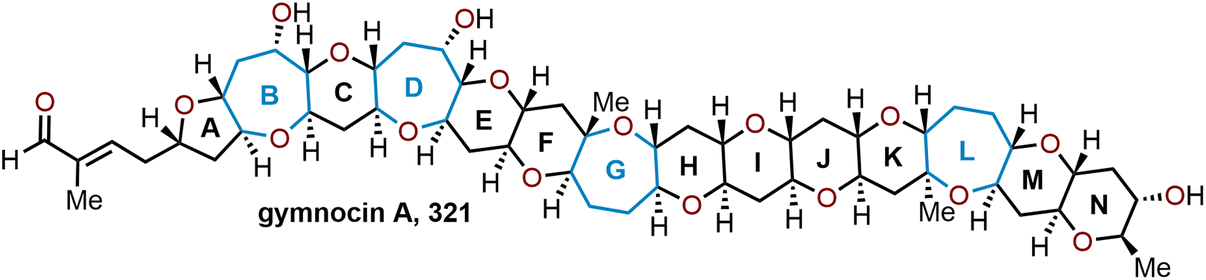 | ||
| Fig. 6 Structure of gymnocin A. | ||
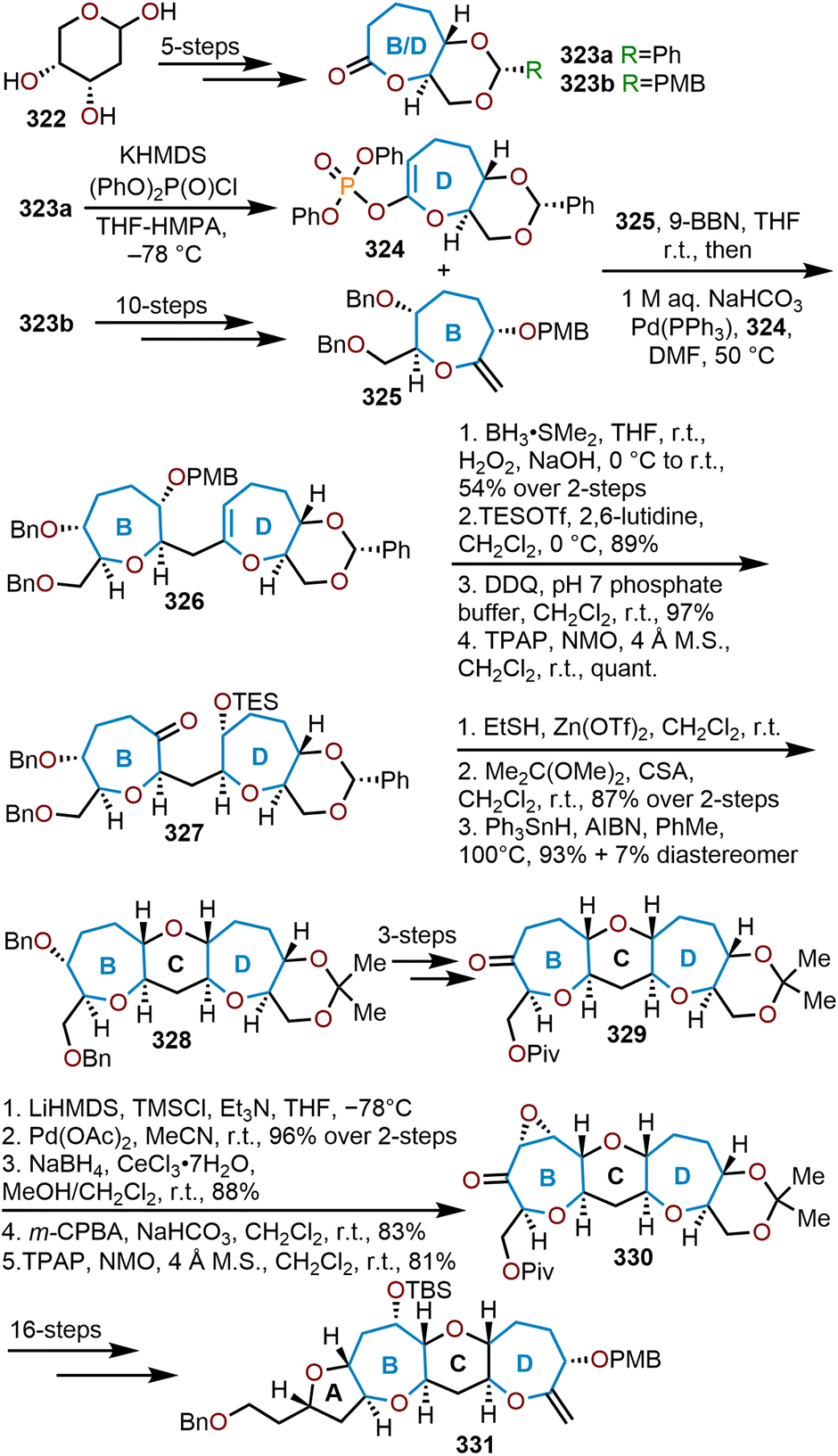 | ||
| Scheme 49 Sasaki and coworkers’ synthesis of the ABCD fragment for gymnocin A.133,134 | ||
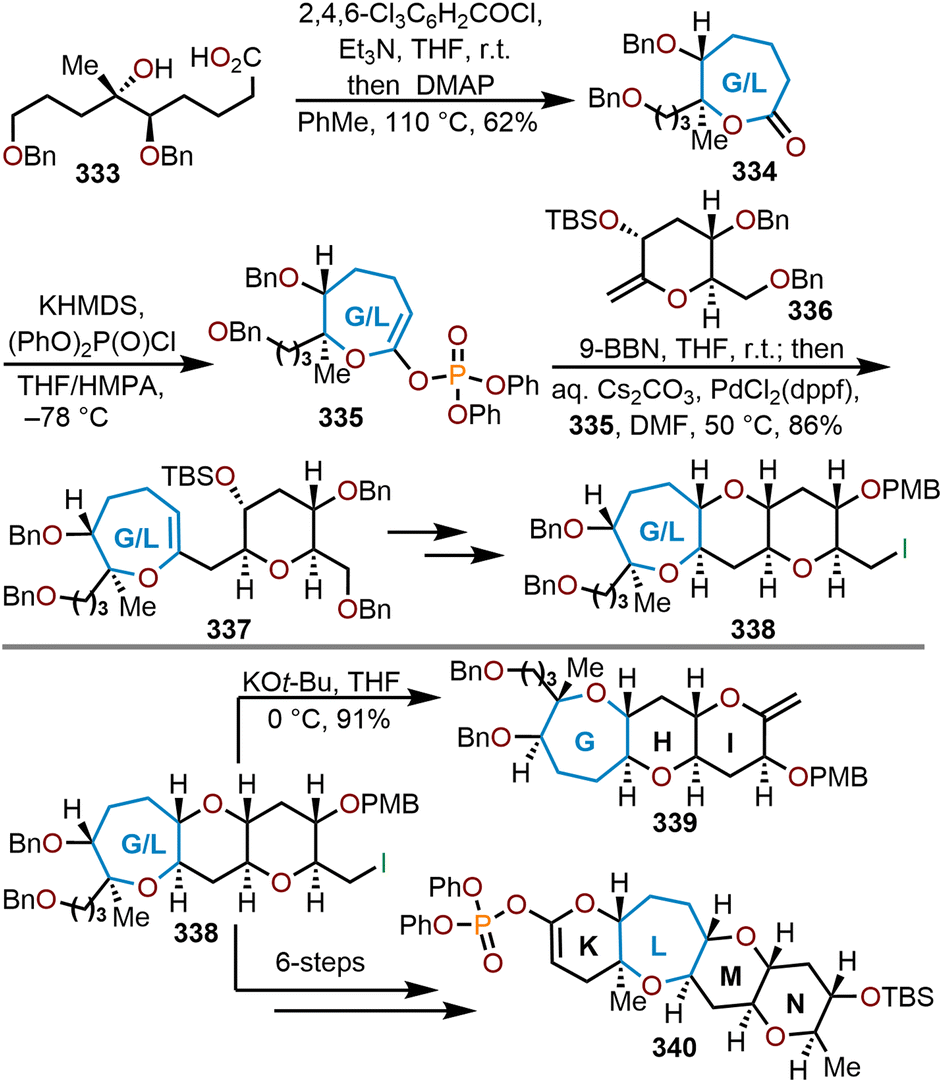 | ||
| Scheme 50 Sasaki and coworkers’ synthesis of the GHI and KLMN fragments for gymnocin A.135 | ||
 | ||
| Scheme 51 Sasaki and coworkers’ union of all fragments to complete the total synthesis of gymnocin A.135 | ||
Initial efforts were carried out to construct the ABCD fragment (Scheme 49).134 Their route began with 2-deoxy-D-ribose (322) which was elaborated to 323a or 323b over five steps, with ring closure accomplished via a Yamaguchi lactonization. From 323a, access to vinyl phosphonate ester 324 in one step and from 323b access to oxepane 325 in 10 synthetic steps was possible. Hydroboration of the olefin in 325 and subsequent Suzuki coupling with vinyl phosphonate ester 324 joined the two B and D oxepane rings to allow for D ring oxygenation and oxidation of the B ring to afford 327 in preparation for C ring construction; which was accomplished via a thioketalization followed by desulfurization to afford 328. Advancement of 328 to ketone 329 allowed for oxygenation of the B ring oxepane via a Saegusa–Ito oxidation to an enone, followed by a Luche reduction. Epoxidation of the corresponding allylic alcohol with m-CPBA and Ley oxidation provided epoxy ketone 330, which upon further advancement in 16-synthetic steps provided the ABCD ring system and key olefin 331 for coupling to the GHIJKLMN fragment. Concurrent to their efforts to the ABCD ring system, Sasaki and coworkers constructed the GHI and KLMN fragments (Scheme 50), which were advanced to the GHIJKLMN fragment (Scheme 51).135
Oxepane formation was achieved by Yamaguchi lactonization of hydroxy acid 333 to seven-membered lactone 334 in 62% yield, which was used to provide the G and L oxepane rings in Gymnocin A. Phosphonation of lactone 334 gave vinyl phosphonate 335, which set the stage for an in situ Suzuki cross-coupling with olefin 336 to afford 337 in 86% yield. After 9 or 11 (recycling an undesired diastereomer) synthetic steps from 337, iodide 338 was obtained, which served as a versatile intermediate. Given the pseudo-symmetry of the GHI and KLMN fragments, Sasaki and coworkers elaborated 338 to form the GHI fragment (339) by elimination of the iodide and the KLMN fragment (340) after 6 synthetic steps as illustrated in Scheme 50. They used their optimized in situ Suzuki cross-coupling method to stitch the GHI (339) and KLMN (340) fragments together to arrive at polycycle 341 (Scheme 51). The F and J rings were subsequently installed over 9 synthetic steps to give vinyl triflate 342. The corresponding vinyl phosphonate failed to couple to the ABCD fragment (331), thus 342 was used to accomplish the Suzuki coupling with 331 to afford the desired product 343 in 81% yield. From 343, the E ring installation, protecting group manipulation as well as installation of the aldehyde sidechain required 18 additional synthetic steps to achieve the first total synthesis of gymnocin A.
Oxepane construction and elaboration in Sasaki and coworkers’ gymnocin A synthesis was facilitated by construction of seven-membered lactones (e.g., 323a, 323b, and 334) via a Yamaguchi lactonization of hydroxy ketones. The lactone was then able to be elaborated to a vinyl triflate, vinyl phosphonate, or exocyclic olefin to enable construction of the polycyclic ether core through a key B-alkyl Suzuki–Miyaura coupling strategy. Lactones 323a and 323b were diverged to provide both the B and D oxepane rings, and due to the pseudo symmetry of the GHI and KLMN fragments, lactone 334 was able to provide the G and L oxepane rings. Further oxygenation of the intermediate oxepines was accomplished using hydroboration conditions and thioketalization followed by desulfurization fused the oxepanes to the C, H and M tetrahydropyran rings. Sasaki and coworkers further detailed this developed chemistry in a full account published in 2005.136
Mori and coworkers were also able to complete the total synthesis of gymnocin A in 2015 (Scheme 52),137 by implementing their developed oxiranyl anion approach to polycyclic ethers (see Schemes 18 and 19).
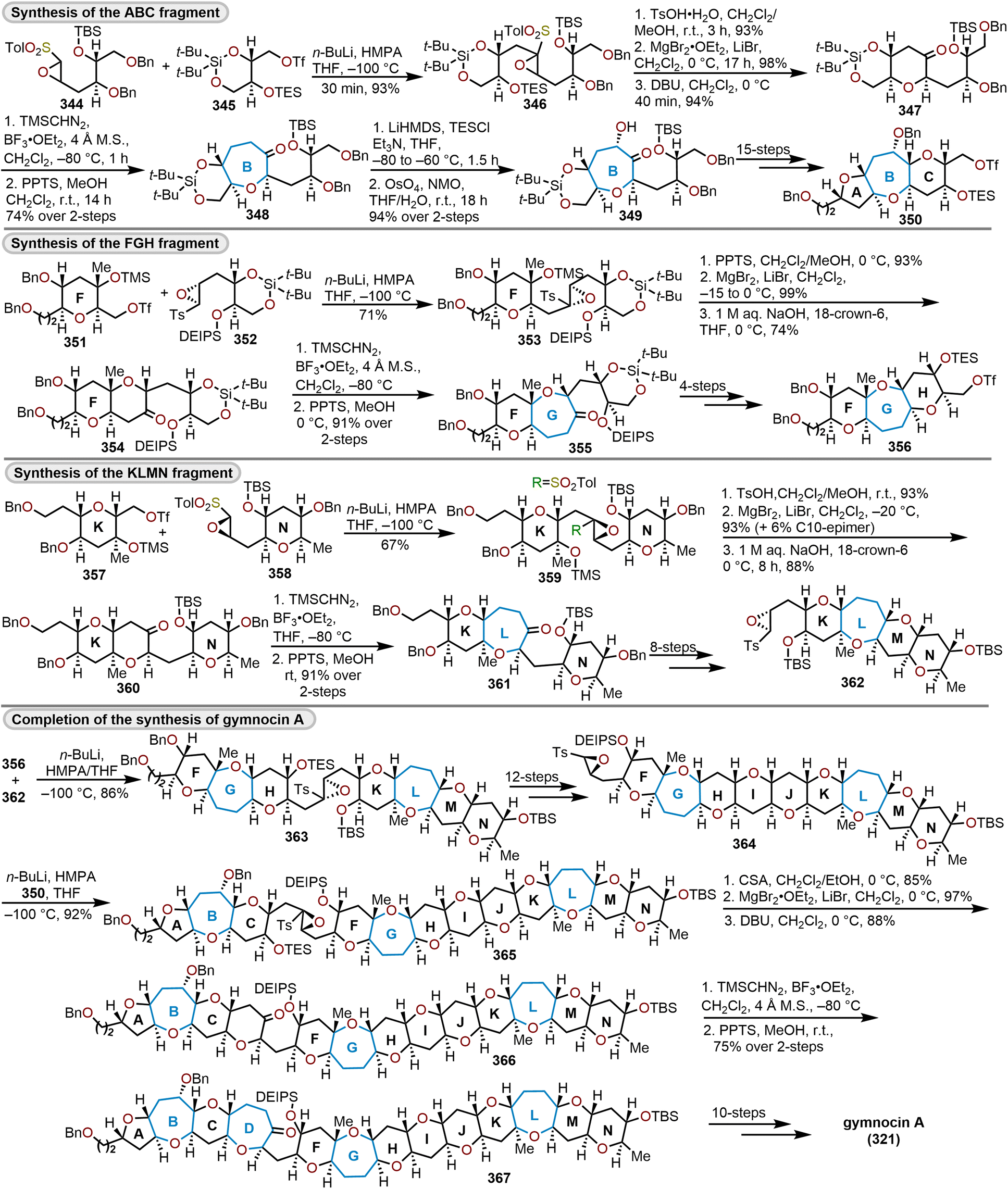 | ||
| Scheme 52 Mori and coworkers’ total synthesis of gymnocin A.137–139 | ||
Beginning with epoxy sulfone 344 available in 6 synthetic steps from 2-deoxy-D-ribose, the oxiranyl anion was generated using n-butyllithium with HMPA as an additive in THF solvent, then added to cyclic triflate 345 to form epoxide 346. The use of the cyclic-protected triflate 345 was important since the acyclic benzyl-protected triflate was unstable. Removal of the triethylsilyl protecting group, treatment with magnesium bromide diethyl etherate furnished an intermediate α-bromoketone, that upon treatment with DBU provided cyclic ketone 347. Then using their developed methodology for Lewis acid-catalyzed ring expansions of cyclic ketones using TMSCHN2 (Schemes 18 and 19),35,48,49,51347 was homologated to afford oxepane 348 containing the B ring.
Oxygenation of 348 was accomplished through silyl enol ether formation and dihydroxylation under Upjohn conditions to afford hydroxylated oxepane 349 as single diastereomer. Further advancement of 349 to key ABC coupling fragment 350 was accomplished after 15 additional synthetic steps.138 The FGH ring fragment 356 was constructed in a similar manner from triflate 351 and epoxy sulfone 352, which required a diethylisopropylsilyl (DEIPS) for selective deprotection of the trimethylsilyl group in the subsequent step.137 The utility of the oxiranyl anion strategy also enabled the construction of the KLMN fragment 362 from triflate 357 and epoxy sulfone 358.139 Mori and coworkers’ Lewis acid-catalyzed ring expansions of cyclic ketones using TMSCHN2 also enabled the construction of the G and L oxepanes embedded within these fragments. Further demonstrating the iterative nature of this strategy, the FGH 356 and KLMN 362 fragments were joined using the oxiranyl anion chemistry to provide 363 that was advanced in 12 synthetic steps to epoxy sulfone 364 containing the FGHIJKLMN rings to be joined to the ABC ring fragment 350 using the same chemistry, which provided epoxide 365. Silyl deprotection of 365, α-bromoketone formation, and treatment with DBU gave ketone 366 which was ring expanded using TMSCHN2 to afford 367 containing the D ring oxepane. An additional 10 synthetic steps were required from 367 for E ring formation, protecting group manipulations, and side chain installation to access gymnocin A (321).137
5.8 Total synthesis of gymnocin B
The second largest polycyclic ether of marine origin was isolated by Yasumoto and coworkers in 2005 from the cells of dinoflagellate Karenia mikimotoi and was found to exhibit cytotoxicity against mouse lymphoide P388 cells at 1.7μg mL−1.140 Gymnocin B (368) contains a ladder-like scaffold consisting of 15 oxacycles, 5 of which are oxepanes, along with 33 stereogenic centers (Fig. 7). The first total synthesis of gymnocin B was accomplished by Sittihan and Jamison in 2019, who utilized a biomimetic two-phase synthetic approach.141 A unique biosynthesis of these marine ladder polyethers was proposed by Nakanishi in 1985, which involved an epoxide-opening cascade.142 This plausible biosynthesis inspired Sittihan and Jamison to develop bromonium-mediated, Lewis acid-catalyzed, water promoted,143 and base-mediated epoxide-opening cascades to construct 10 out of 15 oxacycles in gymnocin B.141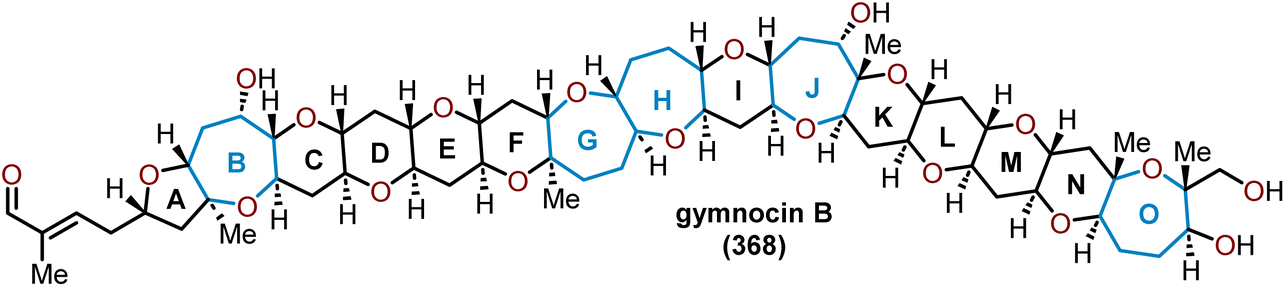 | ||
| Fig. 7 Structure of gymnocin B. | ||
Four different strategies were used to construct the B, G, H, J, and O ring oxepanes in gymnocin B (Scheme 53). The first oxepane targeted in their synthetic efforts was the B ring oxepane, which was prepared through a bromonium-initiated 7-endo-5-exo epoxide cascade from hydroxy epoxide 369, available in 16 linear synthetic steps from 2-deoxy-D-ribose (Scheme 53).141 Using N-bromosuccinimide as the initiator in hexafluoroisopropanol, the AB rings were forged with complete regio- and diastereoselectivity to give the 5/7/6/6 tetracyclic ABCD system 370 in 68% yield. Advancement of 370 in 7 additional synthetic steps provided 371 in 24-steps from 2-deoxy-D-ribose.
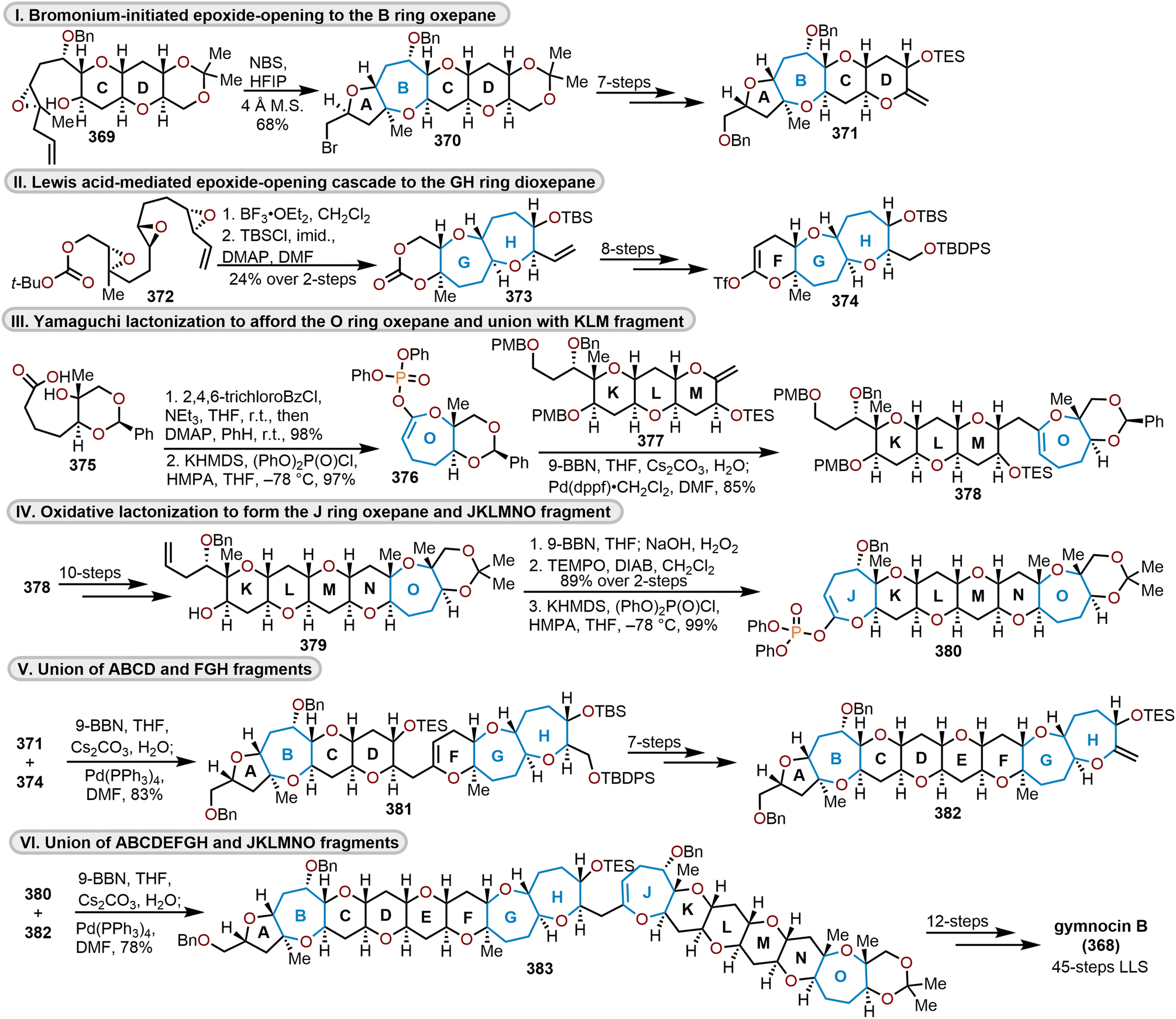 | ||
| Scheme 53 Jamison and coworkers’ various strategies to access oxepane motifs in gymnocin B.141 | ||
In another sequence the GH dioxepane system was forged via an epoxide-opening cascade of triepoxide 372, which was available from geraniol in 8 linear synthetic steps. A Lewis-acid mediated cyclization using boron trifluoride etherate assembled the GH dioxepane system and TBSCl was then added to protect the free hydroxyl to give 373 in 24% over two steps. Advancement of 373 to enol triflate 374 required for attachment of the FGH ring system to the ABCD ring system required 8 synthetic steps.
Assembly of the O ring oxepane was achieved via a Yamaguchi lactonization of hydroxy acid 375,144 available from 2-deoxy-D-ribose in 5 synthetic steps,145 which was followed by vinyl phosphonate formation to provide known oxepine 376.146 Adopting the B-alkyl Suzuki–Miyaura coupling strategy advanced by Sasaki and coworkers for their gymnocin A synthesis, Sittihan and Jamison were able to couple 376 to olefin 377 to adjoin the KLM and O ring fragments to provide polycycle 378.
Advancement of 378 to alcohol 379 to allow for construction of the J ring oxepane required 10 additional synthetic steps. A regioselective hydroboration/oxidation of the olefin in 379 was employed followed by an oxidative lactonization with the nitroxyl oxidant TEMPO to afford the corresponding lactone in 89% yield over the two steps, which was then converted to vinyl phosphonate 380 in 99% yield.
Olefin 371 containing the ABCD ring fragment and enol triflate 374 containing the FGH ring fragment were also united by a B-alkyl Suzuki–Miyaura coupling to afford 381 that was further advanced in 7 synthetic steps to olefin 382. Vinyl phosphonate 380 with the JKLMNO ring system was then joined to olefin 382 possessing the ABCDEFGH ring system in yet another a B-alkyl Suzuki–Miyaura coupling to afford polycycle 383 containing all five oxepane rings. The construction of the I ring, protecting group manipulations, and sidechain installation required 12 additional synthetic steps to complete the first total synthesis of gymnocin B in 45-steps from the longest linear sequence (LLS).
Highlights of the oxepane construction in Sittihan and Jamison's gymnocin B synthesis included B ring formation through an NBS-mediated epoxide cyclization cascade, a Lewis acid-mediated epoxide cyclization cascade to afford the GH dioxepane ring system, a Yamaguchi lactonization to prepare the O ring oxepane, and a TEMPO-mediated oxidative lactonization provided access to the J ring oxepane.
5.9 Total synthesis of gambierol
Gambierol (Fig. 8, 3) is another potent toxin responsible for ciguatera poisoning. This trans-fused octacyclic ether was isolated in 1993 by Yasumoto and coworkers, whose characterization determined the presence of two seven-membered oxacycles, 18 stereogenic centers, and a triene sidechain containing a (Z,Z)-diene.147,148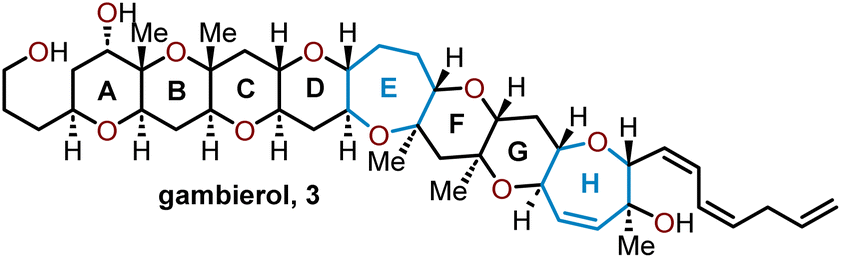 | ||
| Fig. 8 Structure of gambierol. | ||
The first total synthesis of 3 was completed by Sasaki and coworkers in 2002.149 A convergent route was taken to couple the ABC and DEFGH fragments via a B-alkyl Suzuki–Miyaura cross-coupling strategy (Scheme 54). Two strategies were used to construct the oxepine (ring H) and oxepane (ring E), a SmI2-mediated reductive cyclization (see Scheme 14)32,34 and a Yamaguchi lactonization, respectively. Sasaki and coworkers began constructing the DEFGH fragment with known ester150384.149 After 6 synthetic transformations, aldehyde 385 was afforded, which allowed for a SmI2 reductive cyclization to give lactone 386, containing the H ring oxepane, in 70% yield (Scheme 54).149 Lactone 386 was advanced to 387 now containing the FGH ring system, which is poised for E ring formation. Respective deprotection of the trimethylsilyl (TMS) ether and benzyl ester reveals a hydroxy acid, which underwent a Yamaguchi lactonization. The corresponding lactone was converted to ketene acetal phosphate 388 in 92% yield over the four steps in preparation for the coupling of the ABC (389) and EFGH (388) fragments via a B-alkyl Suzuki–Miyaura cross-coupling to afford polycycle 390. Following the coupling, Sasaki and coworkers successfully stitched the D ring via Lewis acid-mediated cyclization, installed the triene sidechain, and completed protecting group manipulations in 22 additional synthetic steps to complete the first total synthesis of gambierol (3).149
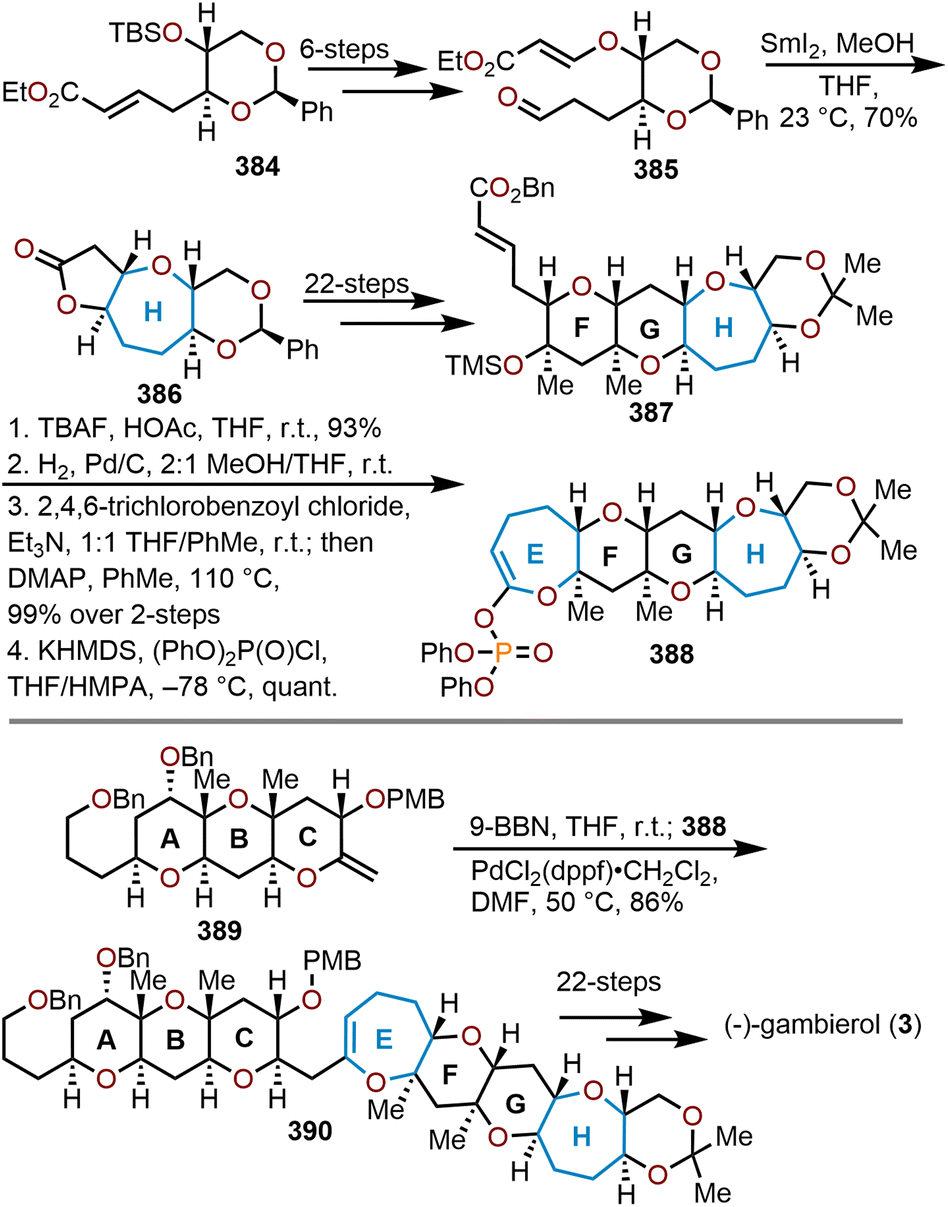 | ||
| Scheme 54 Sasaki and coworkers’ strategy to access gambierol.149 | ||
A few years later in 2005, Rainier and coworkers published the second total synthesis of gambierol (3).151 An iterative C-glycoside/enol ether-olefin ring closing metathesis strategy was employed to construct the subunits of gambierol and to stitch together the octacyclic core (Scheme 55). Aldehyde 392, prepared in 18-steps from D-glucal derivative 391,152 was subjected to HCl in MeOH to afford both cyclic and acyclic acetals, which upon treatment with pyridinium p-toluenesulfonate, pyridine and heat105 resulted in the formation of oxepine 393 in 37% yield over the two steps.
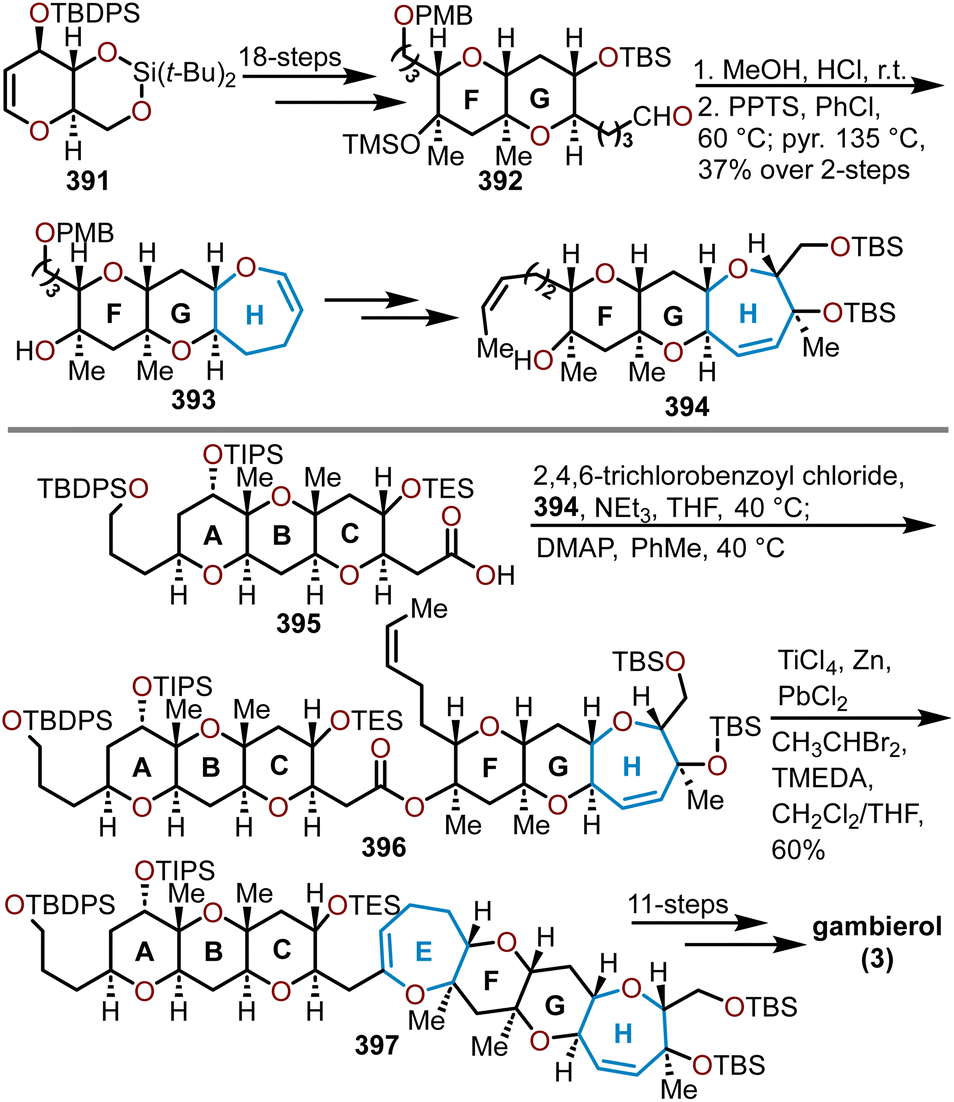 | ||
| Scheme 55 Rainier and coworkers’ strategy to access gambierol.151 | ||
After swapping protecting groups and oxidizing the ring H oxepine, Rainier and coworkers accessed tricycle 394. Yamaguchi coupling of the free hydroxyl group in 394 to the acid-containing ABC fragment 395153 afforded the necessary precursor 396 to attempt their enol ether-olefin ring-closing metathesis strategy to construct the E ring oxepane. Initially, the Takai–Utimoto titanium methylidene protocol154 proved to be insufficient for the cyclization of the E ring, as a key finding was that olefin degradation was prevailing over the cyclization. Rainier and coworkers overcame this obstacle by substituting the traditional 1,1-dibromomethane with 1,1-dibromoethane to generate the titanium alkylidene, which was able to afford the desired oxepine 397 in 60% yield from ester 396. The total synthesis of gambierol was completed in 11 additional steps from 397 following a reductive cyclization to forge the D ring, addition of the triene sidechain, and protecting group manipulations. The Rainier and coworkers’ synthesis of gambierol was completed in 44-steps (LLS) from D-glucal and in 1.2% yield.
Mori and coworkers also accomplished the total synthesis of gambierol (3) in 2009 as shown in Scheme 56.155 Similar to their strategy in the total synthesis of hemibrevetoxin B (Scheme 35), the strategy relied on the use of oxiranyl anions to carry out sulfonyl-assisted 6-endo cyclizations followed by a Lewis acid-promoted homologation with TMSCHN2 to afford the oxepanes. The addition of the oxiranyl anion of 399 to triflate 398, containing the ABCD ring system,156 yielded epoxy sulfone 400. The 6-endo cyclization was promoted by addition of BF3·OEt2 to give the pyranyl ring in 401, whereupon addition of TMSCHN2 in the presence of BF3·OEt2 furnished the E ring oxepane to yield 402. Installation of the F and G rings and advancement to triflate 403 required 19 synthetic steps from 402 and was set-up to use their oxiranyl addition/cyclization/expansion strategy. Addition of the anion of epoxy sulfone 404 to 403 afforded hydroxy epoxide 405, whereupon treatment with BF3·OEt2 furnished ketone 406. Again, a homologation with TMSCHN2 in the presence of BF3·OEt2 was used to access the H ring oxepane to afford 407 in 81% yield over two steps. From there, Mori and coworkers were able to complete the total synthesis of gambierol (3) in 11 additional steps following the addition of the triene sidechain and protecting group manipulations.
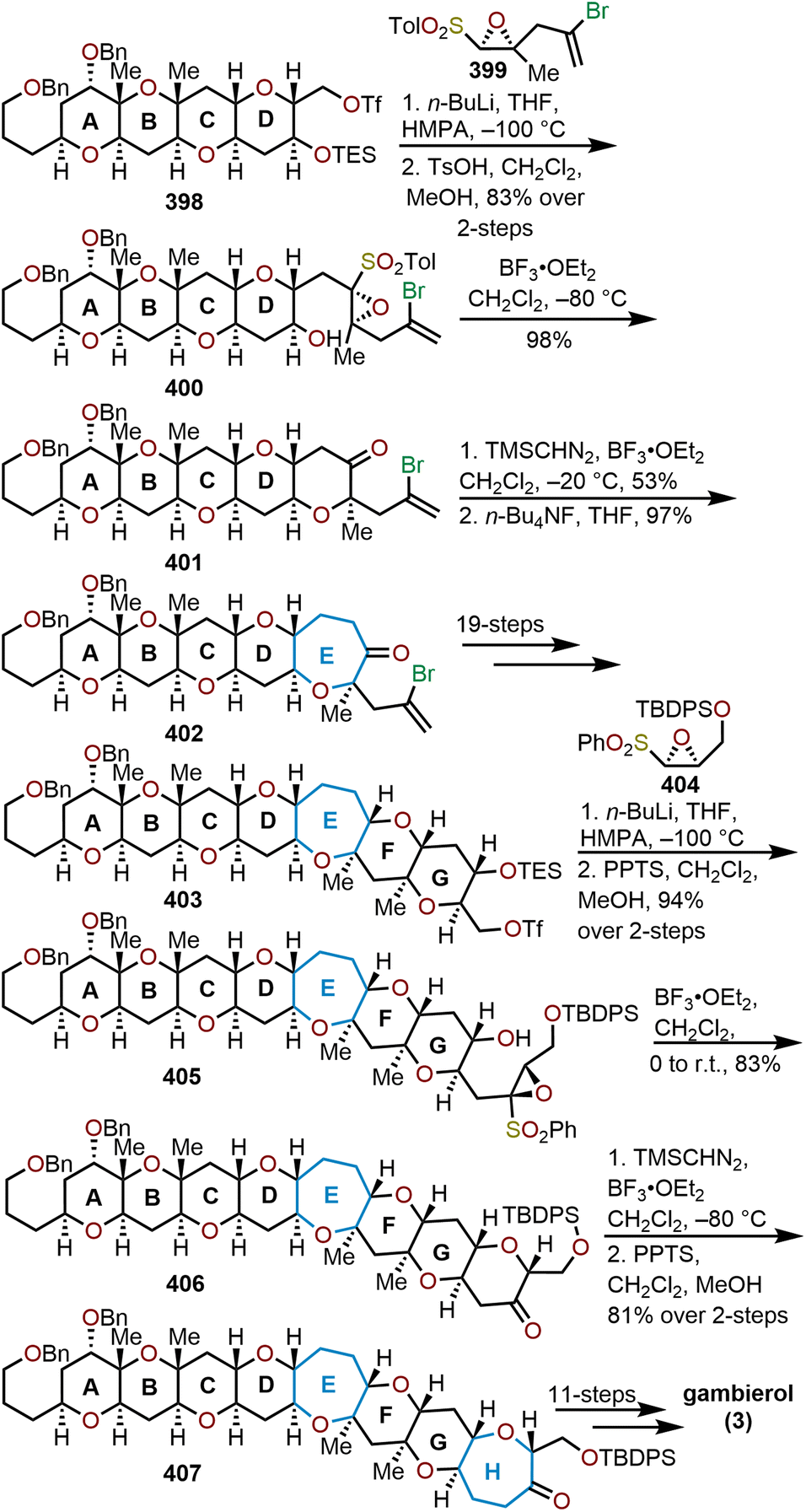 | ||
| Scheme 56 Mori and coworkers’ strategy to access gambierol.155 | ||
5.10 Total synthesis of gambieric acid A
In 1992, Nagai, Yasumoto and coworkers isolated a toxin from the dinoflagellate Gambierdiscus toxicus, whose structure consists of ten cyclic ethers of varying sizes (five, six, seven, and nine-membered) with 27 stereogenic centers (Fig. 9).157 Interestingly, (+)-gambieric acid A (408a) displays highly potent antifungal activities, thus making the gambieric acids sought after target molecules. Access to the A–E subunit of the gambieric acids was reported by Roberts and Rainier in 2007158 and more recently access to the A–D subunit was described in 2015 by Clark and coworkers,159 but thus far, a single total synthesis of gambieric acid A was reported by Fuwa, Sasaki and coworkers in 2012. Furthermore, during studies of model subunit systems Fuwa, Sasaki, and coworkers reassigned the absolute stereochemistry of the polycyclic ether region in 2008,160,161 which helped facilitate their efforts to complete 408a.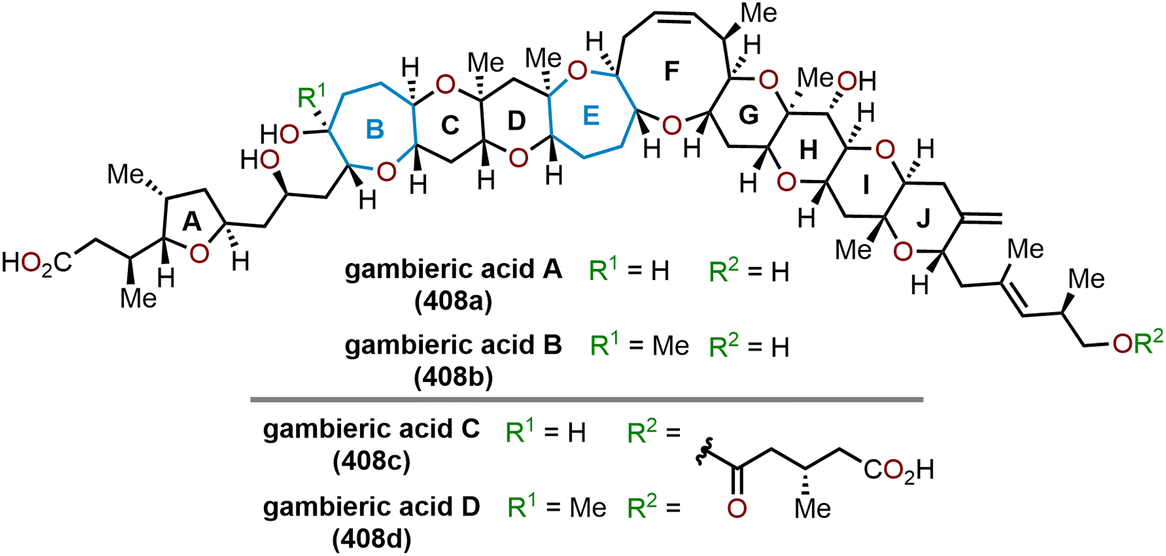 | ||
| Fig. 9 Structure of gambieric acids A–D. | ||
Their route began with known alcohol 409, which was advanced to diol 410 in three steps (Scheme 57).162 An oxoammonium salt-mediated oxidative lactonization procedure was carried out using TEMPO and PIDA to form the seven-membered lactone 411 in 86% yield. A vinyl phosphonate was prepared from the lactone to carry out a palladium-catalyzed methoxycarbonylation to afford the α,β-unsaturated ester 412. Reduction of the ester with DIBAL-H revealed a primary alcohol which was capped with a TBS protecting group, then hydroboration of the oxepine olefin with thexylborane and hydrogen peroxide afforded the desired alcohol 413 in a 57% yield. After several transformations, Fuwa, Sasaki, and coworkers were able to prepare olefin 414, which contained the B ring oxepane and A ring sidechain. A B-alkyl Suzuki–Miyaura coupling with 415, followed by a RCM using the Grubb's 2nd generation catalyst yielded 416 with the tethered the D ring that was advanced in 17 synthetic steps to the ABCD ring fragment 417.
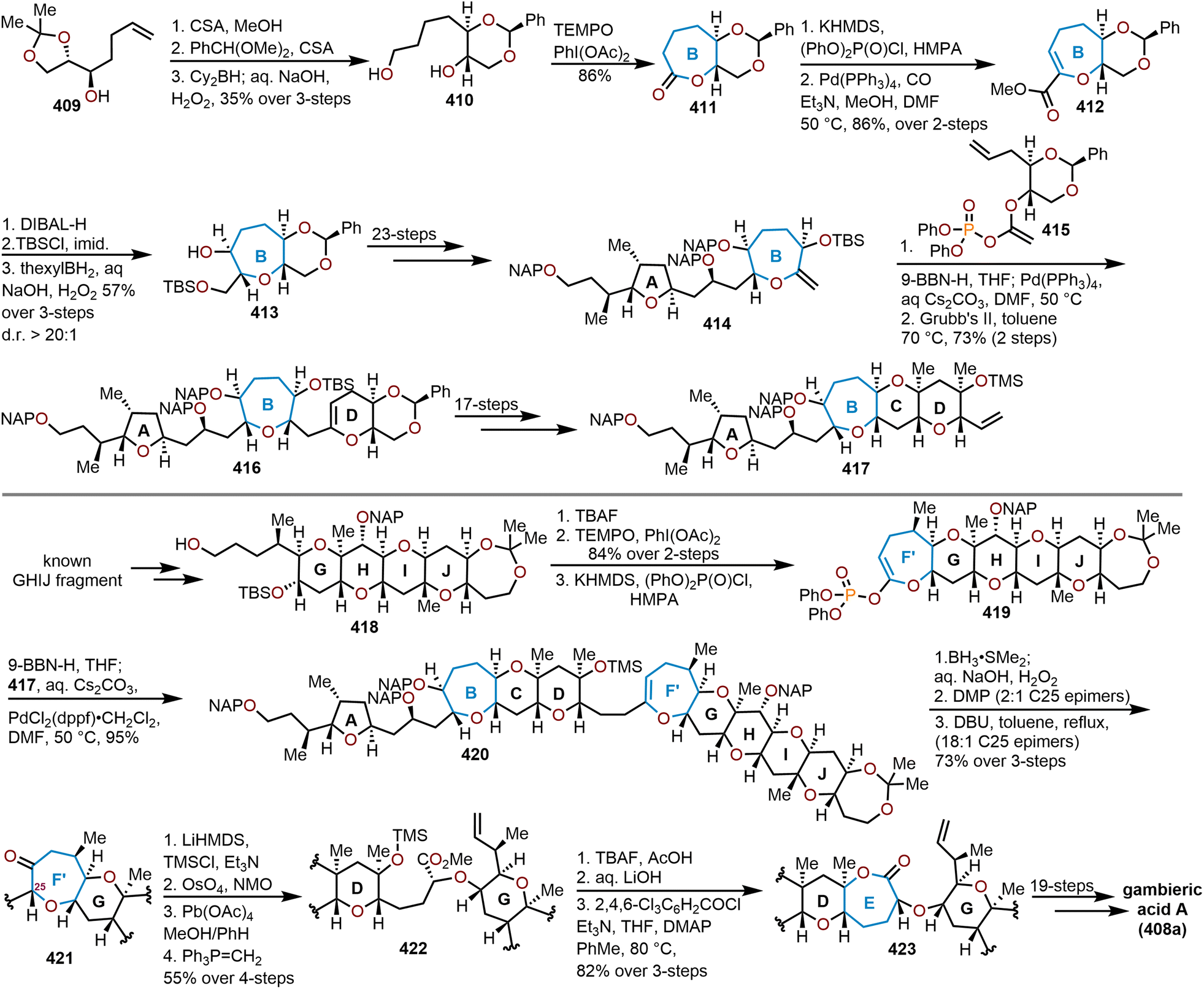 | ||
| Scheme 57 Fuwa, Sasaki, and coworkers’ route to gambieric acid A.162 | ||
Using their previously prepared GHIJ ring fragment,163,164 they were able to access polycycle 418. A three-step sequence of TBS deprotection, an oxoammonium salt-mediated oxidative lactonization with TEMPO and PIDA, and vinyl phosphonate formation provided oxepine 419 containing the F′ ring. Then another B-alkyl Suzuki–Miyaura coupling of 419 with 417 afforded 420 in 95% yield. The F′ ring oxepine was then subjected to a hydroboration–oxidation, oxidation of the resulting alcohol with the Dess Martin periodinane, and epimerization of the C25 center with DBU to arrive at the seven-membered ketone 421. Silyl enol ether formation, α-hydroxylation of the ketone, and Pb(OAc)4-mediated oxidative cleavage of the α-hydroxy ketone severed the F′ seven-membered ring. The intermediate aldehyde was then methylenated to produce 422 in a 55% yield over the 4-steps. Deprotection of D ring TMS-protected alcohol was carried out with TBAF, saponification of the methyl ester, and subsequent lactonization of the intermediate hydroxy acid under Yamaguchi conditions to yield lactone 423, containing the E ring oxepane, in 82% yield over the 3-steps. Following F-ring construction, the sidechain installation on the J ring, oxidation of the A ring sidechain, and protecting group manipulations, the first total synthesis and structural confirmation of gambieric acid A (408a) was completed by Fuwa, Sasaki, and coworkers.162 The oxepane rings within 408a were constructed by two methods. The first exploited an in situ generated oxoammonium salt prepared by using TEMPO and PIDA to mediate an oxidative lactonization providing versatile seven-membered lactones, which allowed the construction and elaboration of the B ring oxepane as well as set-up a key oxidative cleavage of an F′ oxepane to allow for E ring construction and a RCM strategy to construct the 9-membered F ring. The commonly employed Yamaguchi lactonization, as seen in other syntheses above, was the second method used for E ring oxepane construction.
5.11 Total synthesis of ent-nakorone
In the late 1990s, a family of highly condensed oxepane-cycloalkane terpenoids were isolated from Red Sea sponges and were shown to exhibit cytotoxic bioactivities. McDonald and coworkers successfully synthesized the tricyclic septanoside natural product, ent-nakorone (ent-2) inspired by a biomimetic approach through tandem oxa- and carbacyclizations (Scheme 58).165 The biomimetic synthesis began with farnesol to arrive at diepoxide 424 in 4-steps through lithiation of 1-farensyl p-tolyl sulfone and alkylation with 1-bromo-4-trimethylsilyl-2-butyne, a regio- and diastereoselective epoxidation using the Shi catalyst, and reductive desulfonylation. The oxepane ring was constructed from the enyne diepoxide 424 using TMSOTf in the presence of 2,6-di-tert-butyl-4-methylpyridine (DTBMP) to promote cyclization with the propargylsilane nucleophile to yield tricyclic allene 425. The allene was cleaved via ozonolysis to give tricyclic ketone 426, a versatile intermediate that was used by McDonald and coworkers to form ent-abudinol B an additional septanose natural product (see Scheme 59). The total synthesis of 2 was completed after removal of the trimethylsilyl ether in 426 by addition of TBAF.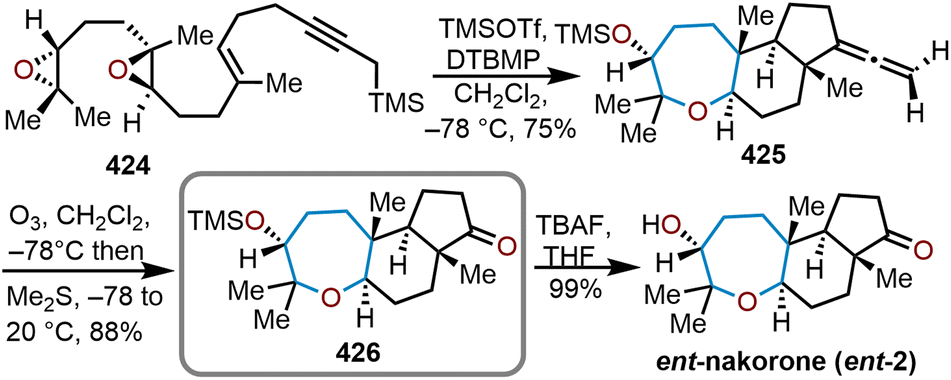 | ||
| Scheme 58 McDonald and coworkers’ strategy toward ent-nakorone.165 | ||
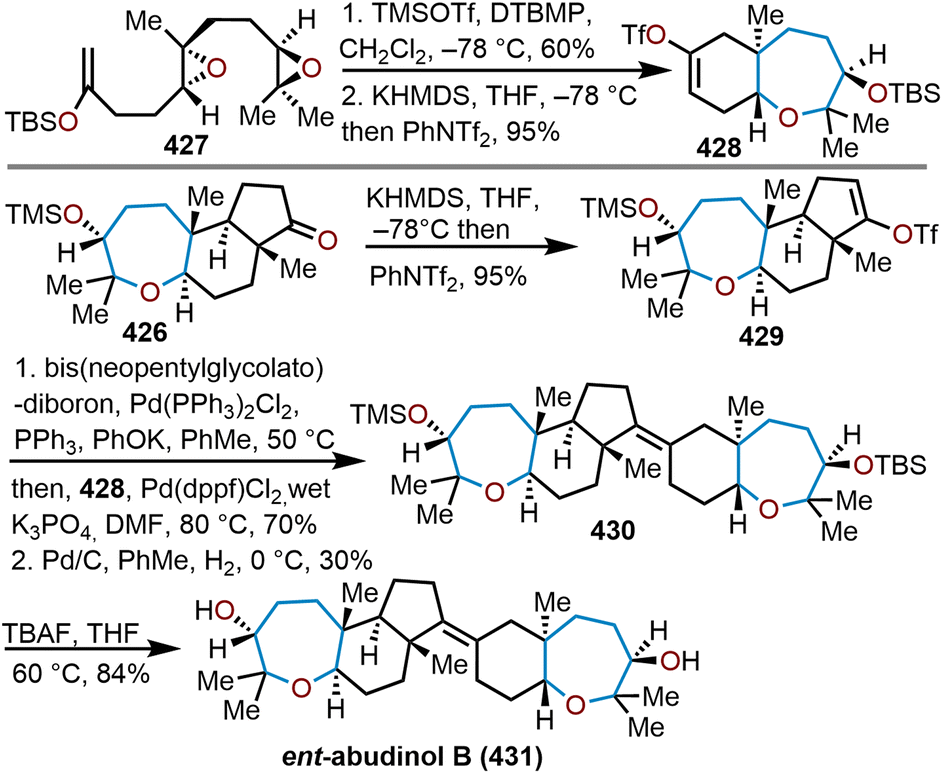 | ||
| Scheme 59 McDonald and coworkers’ strategy toward ent-Abudinol B.165 | ||
5.12 Total synthesis of ent-abudinol B
In addition to the total synthesis of ent-nakorone (ent-2), McDonald and coworkers were able to use 426 to afford ent-abudinol B (431) via a palladium-catalyzed cross-coupling (Scheme 59).165 Enolsilane diepoxide 427, accessible in four steps from geranylacetone, was subjected to a TMSOTf-mediated cyclization and triflation to afford oxepane-containing vinyl triflate 428. Tricyclic ketone 426 was also advanced to vinyl triflate 429 in a 95% yield. Miyaura borylation of vinyl triflate 429 was employed to enable a Suzuki coupling with 428, and a subsequent hydrogenation was successfully employed to derive the tetrasubstituted olefin 430. The synthesis of 431 was completed following desilylation of the TMS and TBS ethers of 430 with excess TBAF at reflux.5.13 Total synthesis of heliannuol C
Heliannuol C is a sesquiterpenoid isolated from the cultivar sunflower Helianthus annus that contains a hydroxylated oxepane core structure.166 Shishido and coworkers were the first to accomplish a total synthesis of (−)-heliannuol C (438) in 2003, in 16 linear steps from diol 432 (Scheme 60).167 Their synthesis relied on an enzymatic desymmetrization of 432 to afford hydroxy acetate 434. From there, 8 steps were required to access diene 435 that was used to construct oxepine 436via a RCM using Grubbs’ second generation catalyst. Dioxirane-mediated epoxidation of the olefin and reductive opening with LiAlH4 afforded hydroxylated oxepane 437 and four additional steps afforded (−)-heliannuol C (438).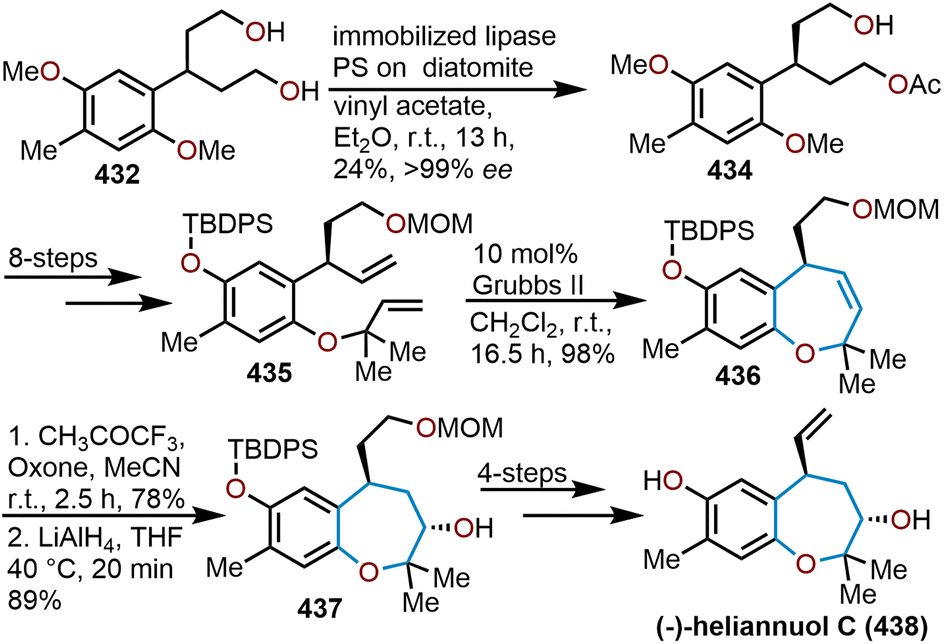 | ||
| Scheme 60 Shishido and coworkers’ synthetic strategy to (−)-heliannuol C.167 | ||
Vyvyan and coworkers were also successful in completing a 6-step total synthesis of (±)-heliannuol C in 2005, which relied on a regioselective aromatic Clasien rearrangement and a biomimetic 7-endo phenol epoxide cyclization (Scheme 61).168
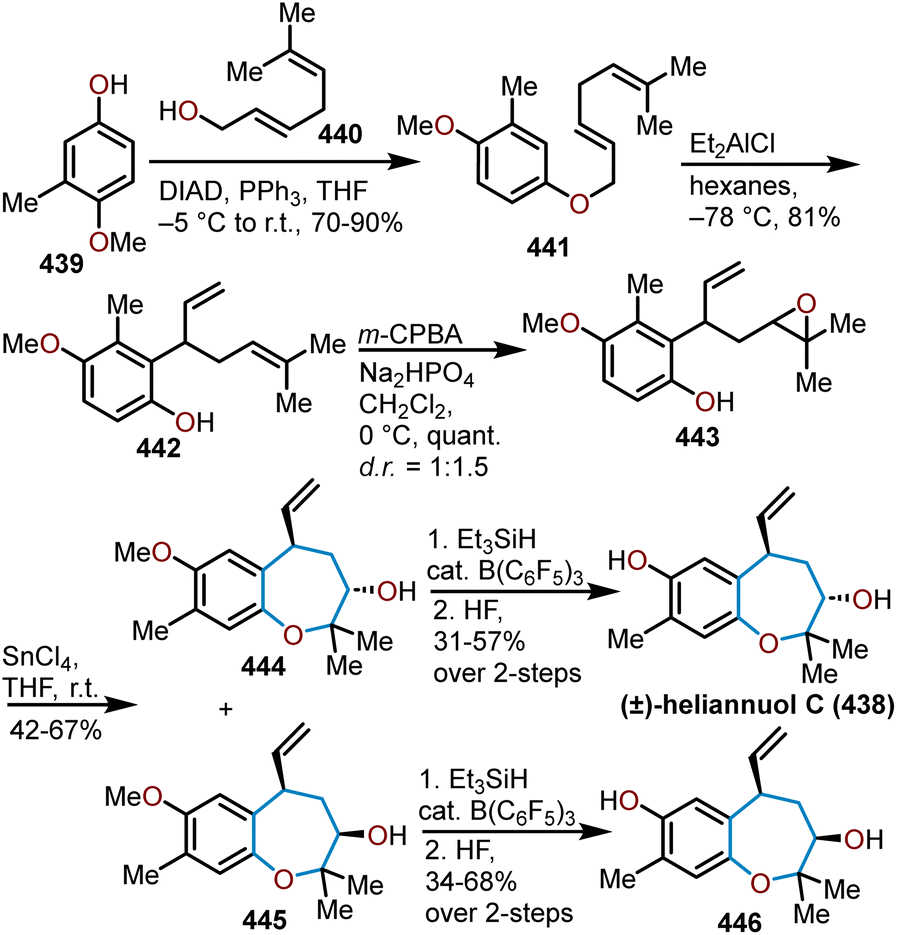 | ||
| Scheme 61 Vyvyan and coworkers’ total synthesis of (±)-heliannuol C.168 | ||
Mitsunobu etherification of phenol 439 with 440 provided diene 441 that when treated with Et2AlCl at low temperatures underwent a facile Claisen rearrangement to afford 442. Epoxidation of the more substituted olefin with m-CPBA afforded an inseparable 1:1.5 diasteromeric mixture of epoxides 443. Treatment with SnCl4 mediated a regioselective 7-endo-cyclization to afford a separable mixture of benzoxepanes (444 and 445). Demethylation of 444 afforded (±)-heliannuol C (438) and epimer 445 afforded epi-heliannuol C (446).
Venkateswaran and coworkers developed a formal synthesis of (±)-heliannuol C (438) in 2006 that used a Bargellini condensation, a Claisen rearrangement, and a Dieckmann cyclization of a diester to construct the benzoxepane in 438 (Scheme 62).169 Starting with 6-hydroxycoumarin 447, methylation and reduction afforded diol 448 that then underwent a selective Bargellini condensation with chloroform and acetone, and the resulting acid was then converted to methyl ester 449 using diazomethane. The allylic alcohol in 449 was then subjected to a Claisen orthoester rearrangement to afford diester 450. Treatment of the diester with LDA promoted a Dieckmann cyclization to afford β-ketoester 451. A Krapcho decarboxylation and sodium borohydride reduction of the ketone provided the same separable mixture of benzoxepanes (444 and 445) that Vyvyan and coworkers obtained (Scheme 61), thus formally provided access to 438. They also reported a slightly modified formal route to (±)-heliannuol C (438) in 2007, which also relied on a Dieckmann cyclization of 450 to afford 451 and the benzoxepane core of (438).170
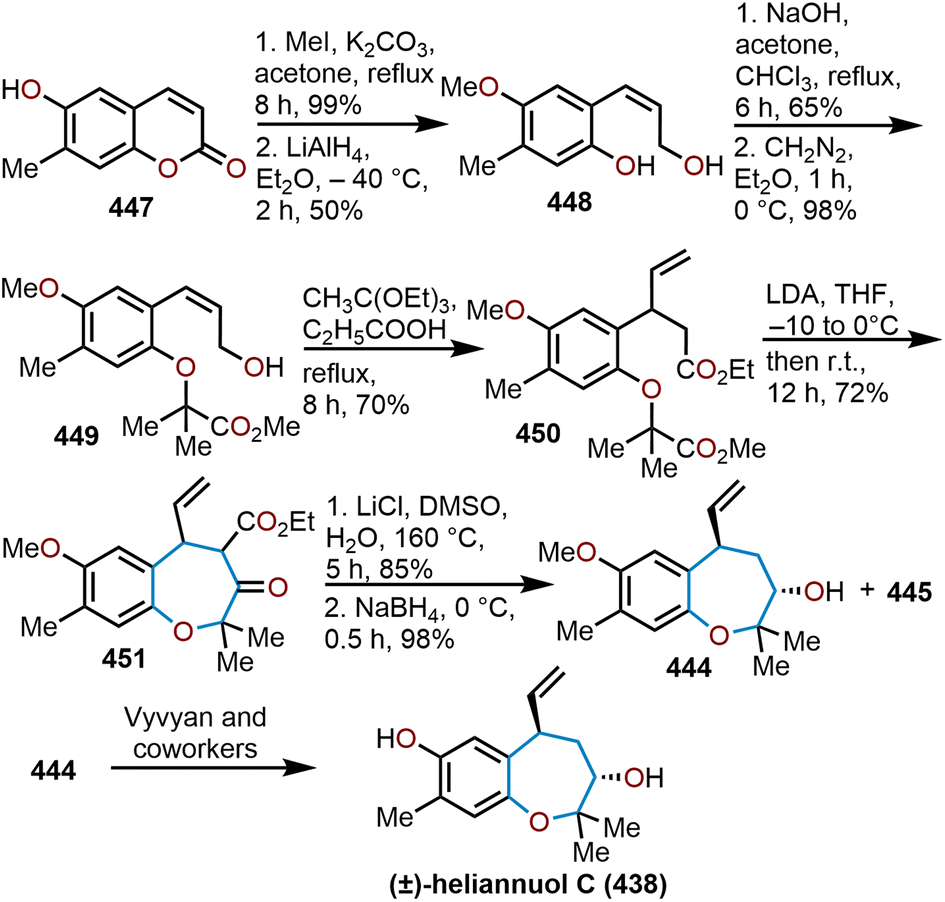 | ||
| Scheme 62 Venkateswaran and coworkers’ formal synthesis of (±)-heliannuol C.169 | ||
Roy and coworkers also completed a formal synthesis of (±)-heliannuol C 438 in 2017 that similarly constructed the benzoxepane via a Dieckmann cyclization of the diester 452 to give 453 (Scheme 63).171 After 4 additional synthetic steps, benzoxepane 454, was afforded. Epoxidation using m-CPBA afforded a (70:30) diastereomeric mixture of epoxide 455. After concomitant reduction of the ethyl ester and epoxide opening with LiAlH4, the dehydration of the primary alcohol via elimination of a p-nitrophenyl selenate provided 444 the same benzoxepane that Vyvyan and coworkers obtained (Scheme 61), thus formally provided access to (±)-heliannuol C (438).
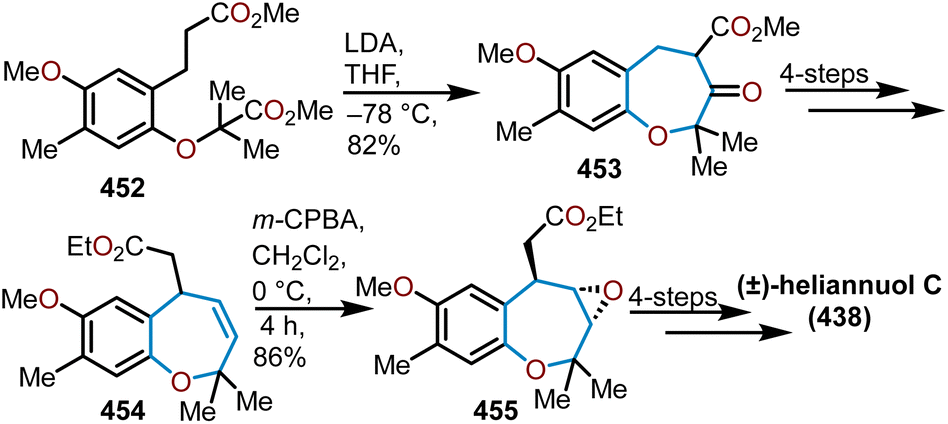 | ||
| Scheme 63 Roy and coworkers’ synthetic strategy to access (±)-heliannuol C.171 | ||
5.14 Total synthesis of zoapatanol
A family of diterpenoid oxepane natural products were isolated in 1979 from the Mexican-native plant Montanoa tomentosa, as this plant has been historically used in folk medicine as a form of contraceptive.172 Amongst these natural products zoapatanol 3 received the most attention owing to its potential as an antifertility agent, with several syntheses having been completed since its isolation.173 The different aspects of the oxepane construction and oxygenation will be highlighted here. In 1980, Chen and Rowand as well as Nicolaou and coworkers were the first two groups to complete the total synthesis of (±)-zoapatanol (4).174,175The approach taken by Nicolaou and coworkers revolved around a key epoxide-ring opening to construct the oxepane moiety (Scheme 64).174 This key cyclization was carried out with the dihydroxy epoxide 457 which was derived from commercially available epoxide 456. Exposure of 457 to dimsyl potassium in DMSO promoted a regioselective, intramolecular epoxide-opening to afford the desired oxepane 458 in 75% yield. The total synthesis of zoapatanol was completed in five additional steps following oxidative cleavage of the diol, a Horner–Wadsworth Emmons olefination, ester reduction, and protecting group removal. In contrast to Nicolaou and coworkers’ approach which used basic conditions to form the oxepane ring, Chen and Rowand used an acid-catalyzed cyclization of dihydroxy epoxide 460, available in 9 synthetic steps from myrcene (459), to afford diacetate 461. Deprotection of the THP alcohol and oxidation with Jones reagent afforded the acid that was treated with an excess of organolithium 462 to install the side chain and afford (±)-zoapatanol (4).175
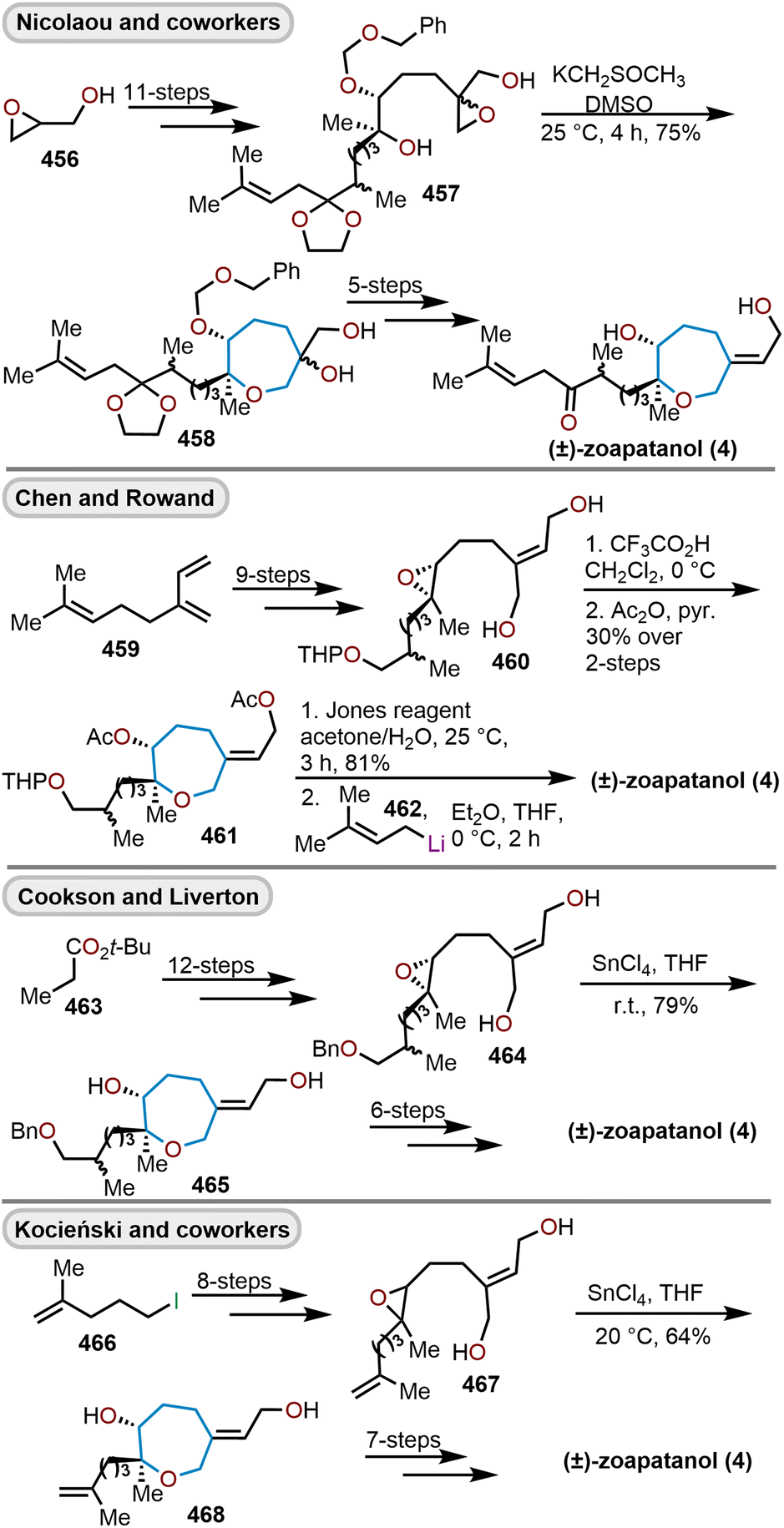 | ||
| Scheme 64 Routes to (±)-zoapatanol using epoxide openings to construct the oxepane ring.174–178 | ||
Cookson and Liverton also accessed 4 using a SnCl4 acid-catalyzed cyclization of dihydroxy epoxide 464, available in 12 synthetic steps from tert-butyl ester 463, to afford oxepane 465 in 79% yield, which required 6 additional steps to access (±)-zoapatanol (4).176 Beginning with iodide 466 Kocieński and coworkers accessed dihydroxy epoxide 467 in 8 synthetic steps, which also underwent a SnCl4 acid-catalyzed cyclization to afford 468 in 64% yield, which allowed them to prepare (±)-zoapatanol (4) after 7 additional synthetic steps.177,178 All four of these routes to 4 relied on an epoxide opening to construct the central oxepane ring.
Other approaches to the oxepane of 4 include a Baeyer–Villiger expansion of cyclohexanones, cyclization of alkyl triflates, RCM of dienes, and a Horner–Wadsworth–Emmons olefination (Scheme 65).179–183 Kane and Doyle employed the Wieland–Miescher ketone 469 to access substituted cyclohexanone 470 in 17 synthetic steps, which was then subjected to Baeyer–Villiger ring expansion conditions to access seven-membered lactone 471 in 70% yield (Scheme 65).179,180 From lactone 471, another 22-synthetic steps were required to access (±)-zoapatanol (4). In 1994, Trost and coworkers reported the first asymmetric synthesis of (+)-zoapatanol (4) from methallyl alcohol 472 (Scheme 65).181 A Sharpless epoxidation was used to introduce chirality along the way and provided access to diol 473, which upon conversion to the triflate cyclized to afford oxepane 474 with the correct absolute and relative stereochemistry. Eight additional steps were required to access (+)-zoapatanol (4).
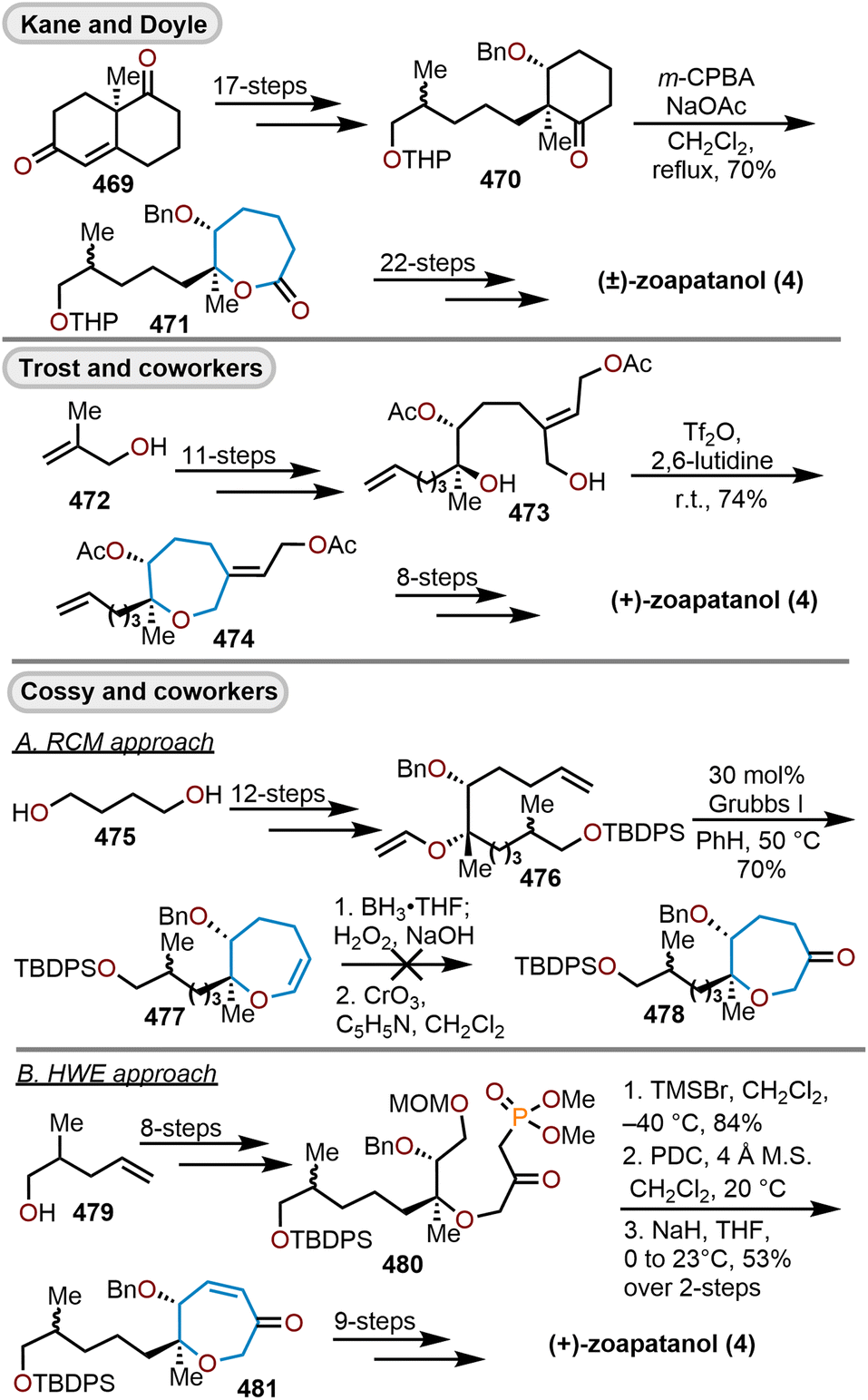 | ||
| Scheme 65 Various synthetic strategies investigated to access (+)-zoapatanol.179–183 | ||
Cossy and coworkers also completed an enantioselective total synthesis of 4 (Scheme 65).182,183 Starting with readily available 1,4-butanediol (475), Cossy and coworkers were able to afford diene 476 after an enantioselective Sharpless dihydroxylation was employed to set the desired stereocenters. A RCM was successful using the Grubbs first-generation catalyst to produce the oxepine 477 from 476 in 70% yield. Unfortunately, this initial route ended up being unsuccessful as oxepine 477 was unreactive under hydroboration and oxidation conditions, thus preventing access to ketone 478 that was needed to advance to 4. As a result, an enantioselective Sharpless dihydroxylation route was combined with a Horner–Wadsworth–Emmons (HWE) olefination strategy to circumvent these difficulties. Starting with 2-methylpent-4-en-1-ol (479), alkyl phosphonate 480 could be accessed in 8 synthetic steps. The pre-installation of the ketone in 480 avoided the difficulties with oxidation in the previous approach, while setting up the HWE olefination. Deprotection of the methoxymethyl (MOM) ether with TMSBr revealed the primary alcohol in 84%. Subsequent oxidation with pyridinium chromate (PDC) afforded the aldehyde, whereupon exposure to base allowed for an intramolecular HWE olefination producing oxepinone 481 in 53% yield over the two steps. Nine additional synthetic steps were required to access (±)-zoapatanol (4).
In 2012, Hou and coworkers were able to employ an RCM strategy to access the oxepane core and complete a formal asymmetric synthesis of (–)-zoapatanol (4) (Scheme 66A).184 Beginning with L-maleic acid (482) Hou and coworkers were able to access diene 483 in 10 steps to allow for an RCM to construct the oxepane ring 484 using the Grubbs second-generation catalyst. Hou and coworkers’ formal access to (–)-zoapatanol (4)184 requires 14 synthetic steps from 484 with 6 additional steps to intercept a common intermediate used in Cossy and coworkers’ synthesis.182 Likewise, Yadav and coworkers (Scheme 66B) were able to access 486 from geraniol (485) in 11 steps to construct the central oxepane using a Grubbs first-generation catalyst to access diacetate 487 in 70%, further advancement of 487 towards (+)-zoapatanol (4) was described, but efforts to install the sidechain have not yet been reported.185
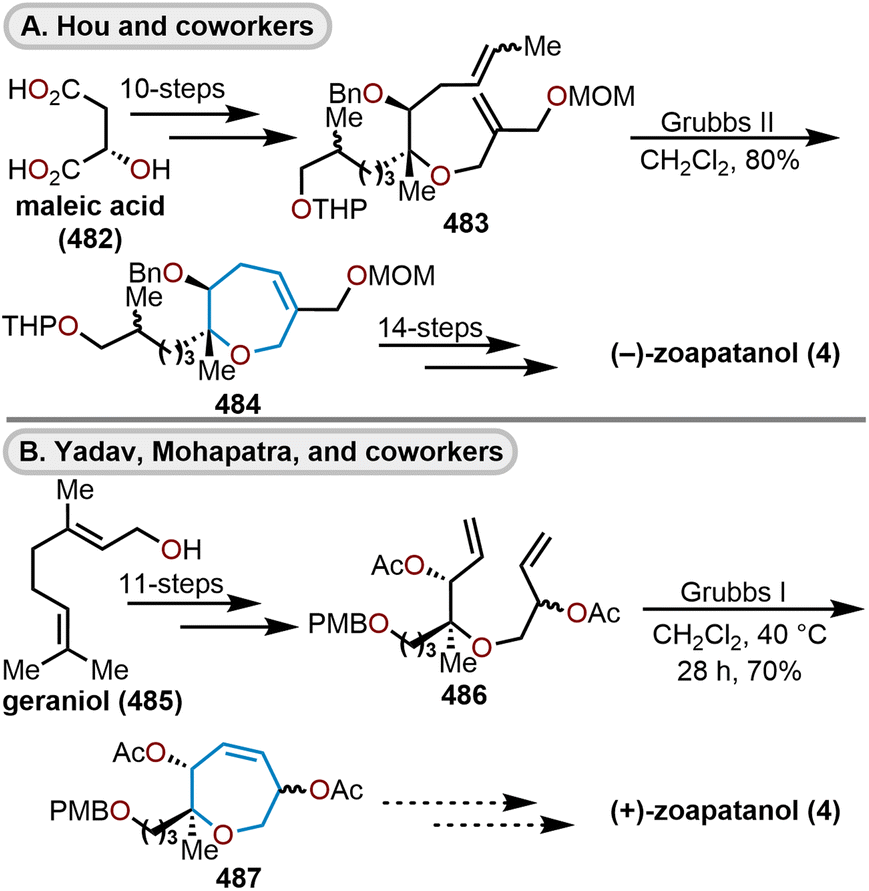 | ||
| Scheme 66 RCM strategies to access zoapatanol.184,185 | ||
In summary, methods for construction of the central oxepane in 4 include: acid and base-catalyzed cyclizations of dihydroxy epoxides (Scheme 64), Baeyer–Villiger ring expansions of cyclohexanones (Scheme 65), cyclization by displacement of alkyl triflates (Scheme 65), an intramolecular HWE-olefination (Scheme 65), and RCM methods (Schemes 65 and 66).
5.15 Total synthesis of (–)-acetylaranotin
Epidithiodiketopiperazines (ETPs) are a unique family of fungal metabolite natural products that exhibit various bioactivities and those containing a dihydrooxepine motif have posed a challenge for synthetic chemists. Over 40 years after its isolation, Reisman and coworkers accomplished the first enantioselective total synthesis of the dihydrooxepine ETP, (–)-acetylaranotin (492) in 2011.186 To construct the peripheral dihydrooxepine, Reisman and coworkers envisioned a transition metal-catalyzed heterocycloisomerization (Scheme 67). Initial studies were taken with aldehyde 488 available from ethyl glycinate in 9 steps; however, the dihydrooxepine 489 was never obtained with only recovery of epimerized starting material or complete decomposition being observed. As an alternative, chlorohydrin 490 was evaluated. The metal vinylidene-mediated 7-endo cycloisomerization was achieved with catalytic [Rh(cod)-Cl]2 and tris(4-fluorophenyl)phosphine in N,N-dimethylformamide (DMF) solvent at 85 °C to give the chlorotetrahydrooxepine 491 in 88% yield. Elimination of the chloride to give the dihydrooxepine core was achieved in the presence of lithium chloride and lithium carbonate at 100 °C in DMF solvent as part of seven additional synthetic steps needed to access 492 from 491.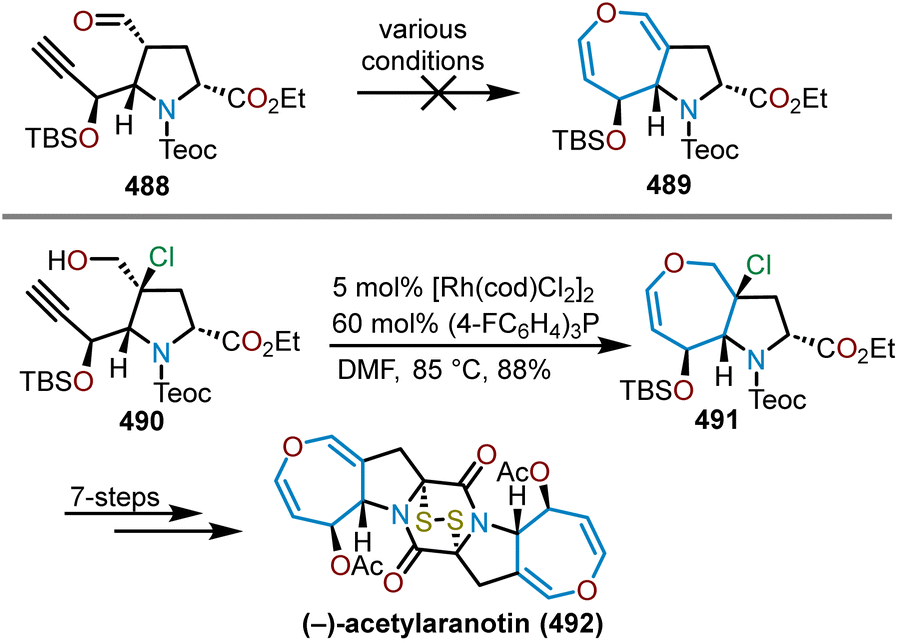 | ||
| Scheme 67 Reisman and coworkers’ heterocycloisomerization strategy to synthesize tetrahydrooxepine 491 enroute to (–)-acetylaranotin (492).186 | ||
5.16 Enantioselective total syntheses of (+)-isolaurepinnacin and (+)-neoisoprelaurefucin
(+)-Isolaurepinnacin (496) and (+)-neoisoprelaurefucin (503) are two halogenated marine natural products isolated from red seaweeds of the genus Laurencia that contain central seven-membered cyclic ethers. The first total synthesis of a member of this family of natural products was completed in 1993 by Overman and coworkers who enantioselectively prepared (+)-isolaurepinnacin (496) in 12 synthetic from cis-2-penten-1-ol (493) (Scheme 68).187,188 The key oxepane forming step in the synthesis was accomplished via a Prins-type cyclization of a β-haloacetal which was generated in situ from 494 by treatment with BCl3 at low temperature. Subsequent desilylation with TBAF afforded alcohol 495 that was advanced to the target molecule in 5 additional steps. In 2001, Suzuki and coworkers reported a formal synthesis of (+)-isolaurepinnacin (496) by intercepting one of Overman and coworkers’ late-stage intermediates (Scheme 68).189 While Suzuki and coworkers formal synthesis was much longer, their oxepane ring construction was accomplished by cyclization of hydroxy epoxide 498, available in 14 steps from chiral diol 497. Using their developed (Bu3Sn)2O/Zn(OTf)2 system,190,191 they were able to promote the cyclization of 498 to oxepane 499. From 499 another 20 steps were required to formally access (+)-isolaurepinnacin (496) including the steps of Overman's route.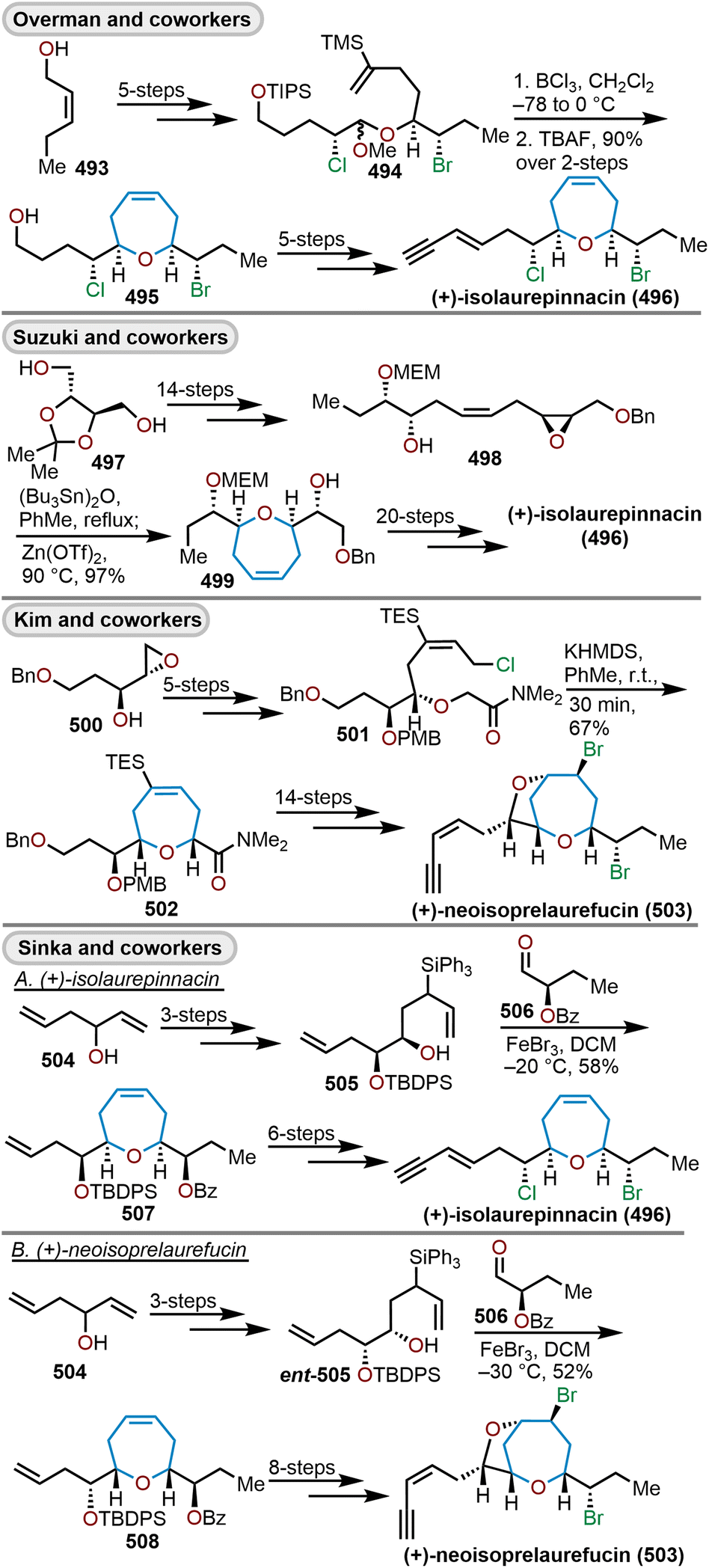 | ||
| Scheme 68 Synthetic routes to the oxepane cores in the syntheses of (+)-isolaurepinnacin and (+)-neoisoprelaurefucin.187–189,192,193 | ||
In 2003, Kim and coworkers reported the first total synthesis as well as confirmed the absolute configuration of (+)-neoisoprelaurefucin (503).192 Beginning with chiral epoxide 500, they were able to access amide 501, which upon treatment with KHMDS resulted in an intramolecular alkylation by displacement of the allylic chloride to afford the triethylsilyloxepine 502, which was advanced to (+)-neoisoprelaurefucin (503) in 14 steps. In 2022, Sinka and coworkers developed the shortest known enantioselective total syntheses of (+)-isolaurepinnacin (496) and (+)-neoisoprelaurefucin (503) to date.193 The cis-oxepane ring was constructed using a Prins–Peterson cyclization of chiral silyl alcohol 505 with chiral aldehyde 506 promoted by iron(III) bromide to afford 507 in a 58% yield, which was advanced to 496 in only 6 additional steps (Scheme 68). Beginning with the opposite enantiomer of silyl alcohol 505 the same Prins–Peterson cyclization with aldehyde 506 affords 508 that can be advanced to 503 in 8 additional steps (Scheme 68).
5.17 Synthesis of unnatural septanose analog of 7-oxostaurosporine
From a medicinal chemistry perspective, septanosides can be formed as unnatural analogs to biologically relevant natural products. In 2022, Rhee and coworkers showcased the successful synthesis of an unnatural septanoside analog of 7-oxostaurosporine (517) via sequential metal catalysis (Scheme 69).194 A palladium-catalyzed coupling of indolocarbazole 509 and alkoxyallene 510 was able to afford an acyclic allylic acetal 512 in near quantitative yield and in a 9:1 dr when ligand (R,R)-511 was used. Ring-closing metathesis (RCM) with the second-generation Hoveyda–Grubbs catalyst (HGII) was employed to form the seven-membered oxacycle, 513 which set the stage for a successful olefin migration facilitated by ruthenium hydride catalyst 514. Using palladium acetate (Pd(OAc)2) under an oxygen atmosphere allowed for an oxidative cyclization to install the second N-glycosidic bond to give compound 516. Lastly, osmium tetraoxide (OsO4) mediated syn-hydroxylation affords the desired unnatural septanose analog of 7-oxostaurosporine 517.
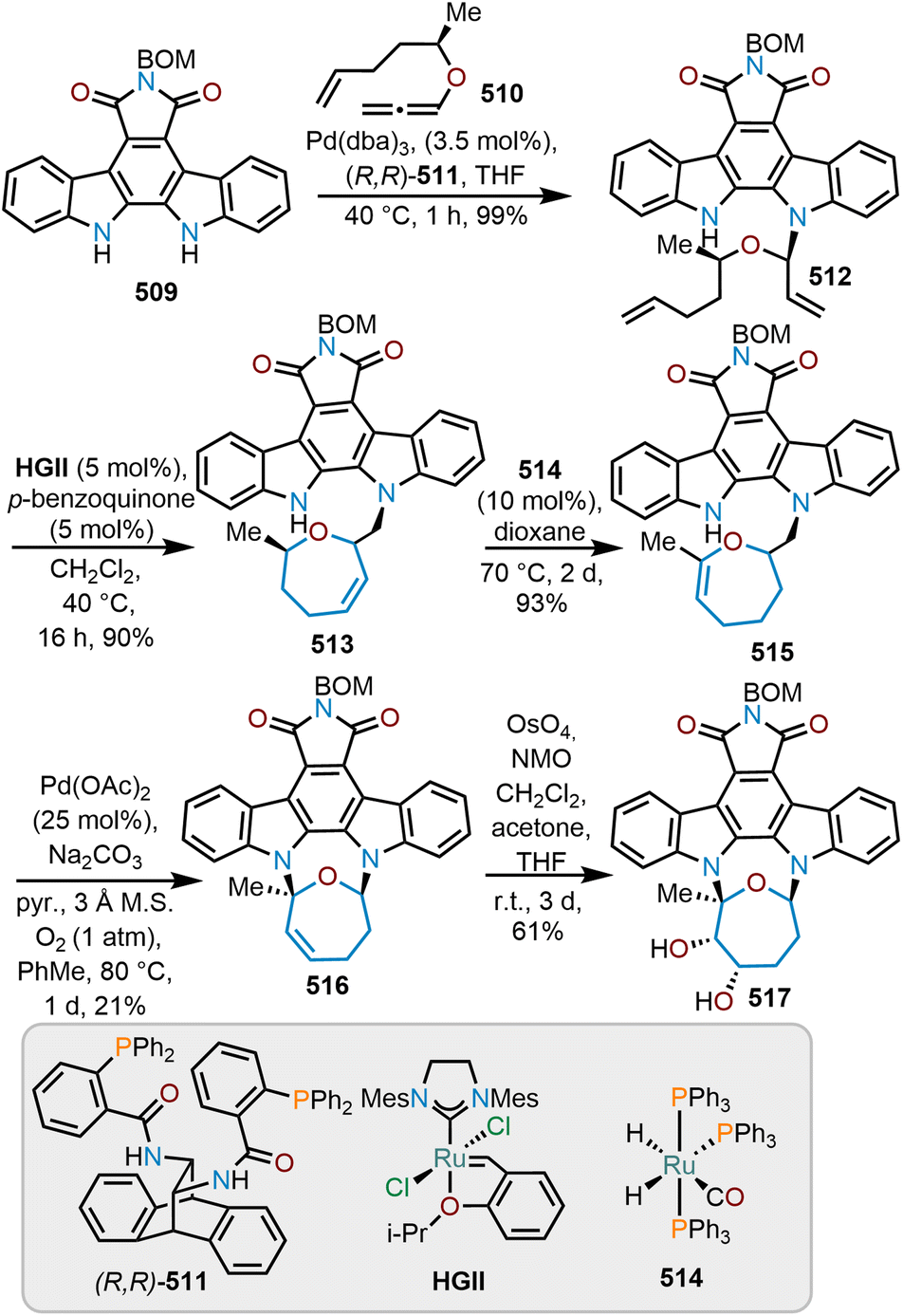 | ||
| Scheme 69 Oxidative cyclization strategy to access unnatural analog to 7-oxostaurosporine.194 | ||
5.18 Synthesis of artemisinin and its derivatives
Malaria remains as a prominent disease in many underdeveloped countries. Many therapeutics have been developed to combat malaria, but adverse side-effects have remained a pressing issue. As an alternative, artemisinin combination treatments (ACTs) have recently been the recommended therapeutic to combat malaria, and have been showcased in the World Health Organization's (WHO) list of “essential medicines”.195 Surprisingly, the antimalarial activity of artemisinin (Fig. 1, 5) is not stereospecific, thus making this natural product and its derivatives highly sought after potential therapeutic agents.196 These sesquiterpene endoperoxides possess an unprecedented scaffold and have been infamously challenging target molecules, as one must take necessary precautions when constructing the peroxide on a large scale. Since its isolation in 1979, artemisinin (5) and its derivatives became target molecules for many groups.From the 1980s through early 2000s many groups accomplished total syntheses of artemisinin.197–207 The first group to synthesize artemisinin was Schmid and Hofheinz at Hoffman-LaRoche in 1983 (Scheme 70).197 Starting with the commercially available terpene, (–)-isopulegol (518), Schmid and coworkers were able to arrive at the enol ether 519. From there, a key photooxygenation step of 519 through an ene reaction with singlet oxygen in the presence of methylene blue at cold temperatures afforded the proposed peroxide acetal intermediate 520, which upon acid hydrolysis forms the lactone, endoperoxide and oxepane in a single step to arrive at (+)-artemisinin (5) in a 30% yield. This installation of the endo peroxide coinciding with oxepane formation using singlet oxygen is common throughout the early synthetic routes to 5.204
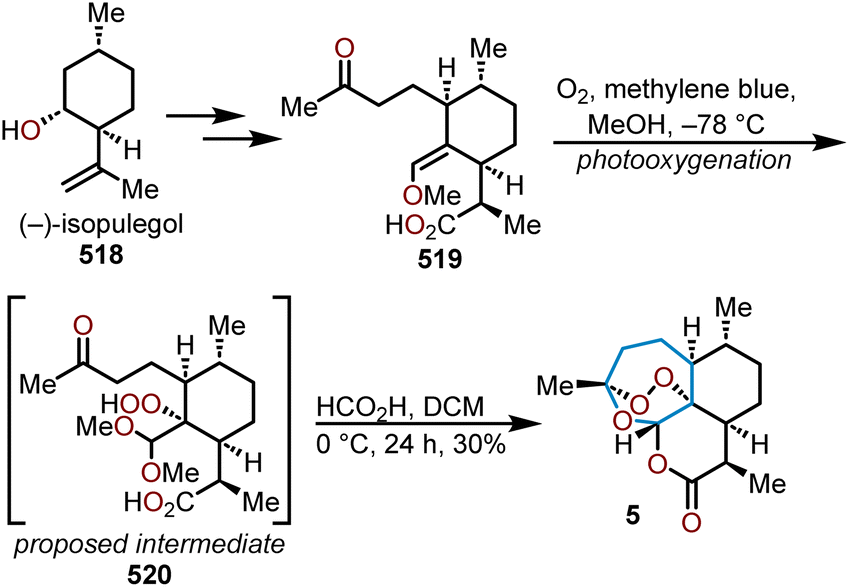 | ||
| Scheme 70 First reported total synthesis of (+)-artemisinin from (–)-isopulegol by Schmid and Hofheinz in 1983.197 | ||
A notable concise and enantioselective total synthesis of (+)-artemisinin (5) from inexpensive starting materials was achieved by Cook and coworkers in 2012 (Scheme 71).208 Their efforts began with commercially available cyclohexenone (521), which was elaborated via a key [4 + 2] annulation to the orthoester-containing methyl ketone 522 in 7 steps with reactions run on as high as 47 g scale. A unique aspect of the Cook synthesis is that by using ammonium molybdate a controlled decomposition of hydrogen peroxide into singlet oxygen allowed for the oxidation of the enol olefin in 522, which upon treatment with acid completed the final oxidative rearrangement to afford (+)-artemisinin 5 on gram scale.
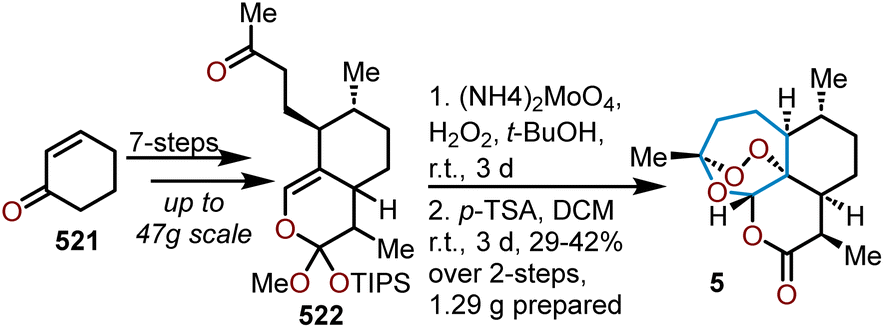 | ||
| Scheme 71 Cook and coworkers’ route to (+)-artemisinin.208 | ||
While the direct total syntheses of 5 are notable feats and have advanced new chemical methods, current routes are not scalable enough (e.g., high costs of terpene-based starting materials, elaborate synthetic routes, low-yielding reactions, and safety) to be able to meet the high demand of artemisinin which is currently accessed through plant-based extractions. To provide an alternative pathway to 5, Sanofi and others have pursued the development of alternative semi-syntheses from artemisinic acid (523); the proposed biosynthetic precursor to artemisinin (5). Biosynthetic access to artemisinic acid is available from the fermentation of sugar using genetically engineered yeast, which has been exploited to prepare over 60 metric tons of 5 in 2014 through a three-step semi-synthetic route developed by Sanofi that involves diastereoselective hydrogenation, acyl substitution, and photooxygenation (Scheme 72).209 Starting with artemisinic acid 523, the diastereoselective reduction was carried out with an optimized catalyst/ligand set {RuCl2[(R)-DTBM-Segphos](DMF)n} to obtain 524 and 525 with a respective dr of 95:5. Addition of ethyl chloroformate to the diastereomeric mixture gives the mixed anhydrides 526 and 527 in a quantitative yield. Next, a mercury vapor lamp is used to initiate a regioselective Schenck ene reaction, whereupon subsequent Hock cleavage and concomitant oxidative cyclization of proposed intermediate 528 afforded (+)-artemisinin (5) isolated in 55% yield over the three-steps (average batch isolation of 370 kg).
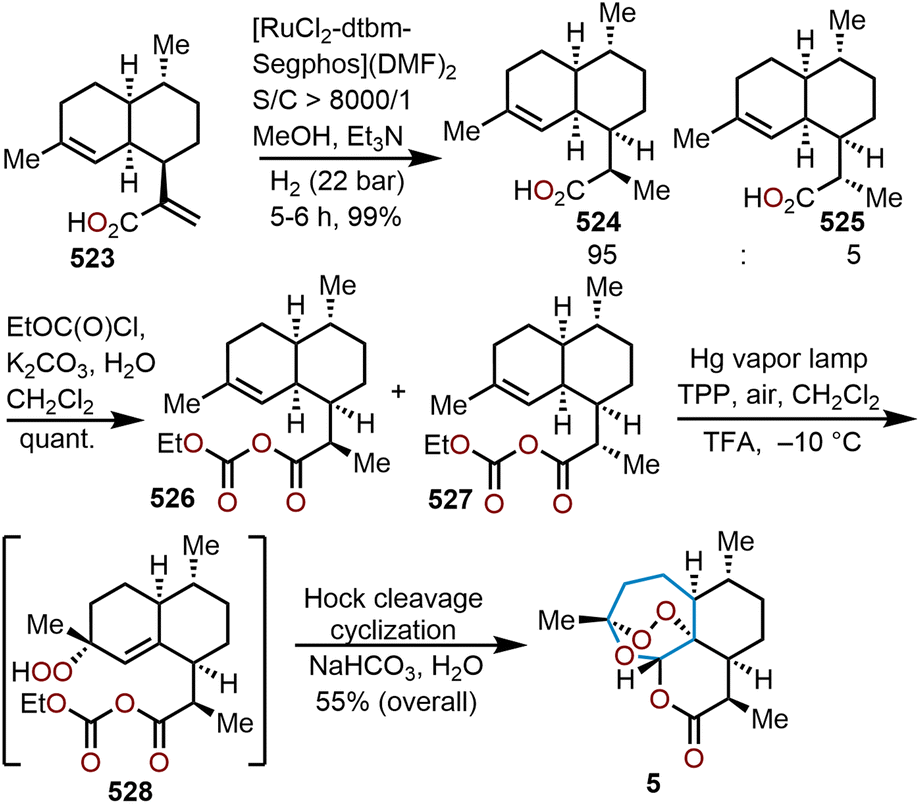 | ||
| Scheme 72 Optimized semi-synthesis of artemisinin from artemisinic acid.209 | ||
6. Summary and outlook
Recent advancements toward the preparation of oxepanes have been extensively covered. The synthetic strategies to access this moiety can arise from cyclic or acyclic start materials. Commonly, feedstock pyranoses are used in de novo syntheses to prepare oxepanes from cyclic precursors. Benefits to using sugar-based reagents is that they are often an affordable source of cyclic reagents with nearly all stereocenters established, which can be quickly elaborated. However, there are demerits for employing carbohydrate starting materials: often solubility and selectivity issues arise, which requires the extensive use of protecting groups to alleviate these difficulties. Alternatively, oxepanes have also been successfully prepared from acyclic, yet readily available starting materials.The use of acyclic, non-sugar-based starting materials have been strategically used in literature to circumvent the problems established during sugar-based syntheses. Typically, solubility and selectivity issues can be avoided by starting with carefully chosen reagents; however, this comes at the expense of having to set the desired stereocenters. Nonetheless, literature has shown over the years how both methods can be viable to carry out a total synthesis of biologically relevant natural products.
The formation of cyclopropanated glycals from the readily available sugar-based starting materials, allows rapid access to oxepanes. Furnishing the pyranose with a ketone opens the opportunity to carry out a homologation, thus affording the seven-membered oxacycles. This tactic was commonly employed by Mori and coworkers for their endeavors in natural product total synthesis of hemibrevetoxin B50 and gambierol.155 A unique approach is also offered from functionalization of glucals, whereupon a Nicholas–Ferrier rearrangement can execute the ring expansion of the pyranyl system. Isobe and coworkers showcased the utility of this transformation from their efforts in the total synthesis of the ciguatoxins.67
Various cyclization strategies have been developed to afford the seven-membered oxacycle. Amongst the plethora of tactics, Lewis acid-mediated cyclizations are the most widely applicable method. Epoxide-opening cascades provide an excellent example of this transformation, which has been notably used by Jamison and coworkers for their construction of gymnocin B.141 Radical cyclization strategies have also been advanced to afford oxepanes from the assistance of samarium iodide. This reductive radical cyclization was used extensively by Fuwa, Sasaki and coworkers as advertised from their works in the total synthesis of brevenal100 and gambierol.149 Ring closing metathesis is another common cyclization strategy for the construction of oxepanes from tethered dienes. Employing the RCM with Grubbs I or Grubbs II has been heavily pioneered in total synthesis, which was the prevailing method to preparing the seven-membered oxacycles by Inoue, Hirama and coworkers in efforts to the ciguatoxins.108
The current progression of advancements in the synthesis of oxepanes reveals that new methodologies and opportunities will continue to arise from endeavors in the total synthesis of biologically relevant natural products. While there have been extensive breakthroughs in oxepane synthesis as highlighted in this review, photocatalytic,210 biocatalytic, and biomimetic strategies211 that use cascade reactions and incorporate aspects of green chemistry are primed areas for advancement over the next decade to expedite access to complex oxepane natural products.
Author contributions
All authors conceptualized, discussed the concept of this article, and contributed to the scientific writing of the original manuscript. All authors have read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported through funds from the Old Dominion University Department of Chemistry and Biochemistry. S. M. W. is grateful for support from a Dominion Scholar fellowship provided by the Old Dominion University College of Sciences.References
- J. C. Valentine and F. E. McDonald, Biomimetic synthesis of trans,syn,trans-fused polycyclic ethers, Synlett, 2006, 1816–1828 CAS.
- G. A. Molander, Diverse Methods for Medium Ring Synthesis, Acc. Chem. Res., 1998, 31, 603–609 CrossRef CAS.
- T. Yasumoto and M. Murata, Marine Toxins, Chem. Rev., 1993, 93, 1897–1909 CrossRef CAS.
- A. S. Kleinke, D. Webb and T. F. Jamison, Recent progress in the synthesis of oxepanes and medium ring ethers, Tetrahedron, 2012, 68, 6999–7018 CrossRef CAS.
- J. Saha and M. W. Peczuh, Synthesis and properties of septanose carbohydrates, Adv. Carbohydr. Chem. Biochem., 2011, 66, 121–186 CrossRef CAS PubMed.
- S. Castro, M. Duff, N. L. Snyder, M. Morton, C. V. Kumar and M. W. Peczuh, Recognition of septanose carbohydrates by concanavalin A, Org. Biomol. Chem., 2005, 3, 3869–3872 RSC.
- J. O. Hoberg and J. J. Bozell, Cyclopropanation and ring-expansion of unsaturated sugars, Tetrahedron Lett., 1995, 36, 6831–6834 CrossRef CAS.
- J. O. Hoberg, Formation of Seven-Membered Oxacycles through Ring Expansion of Cyclopropanated Carbohydrates, J. Org. Chem., 1997, 62, 6615–6618 CrossRef CAS.
- R. Batchelor and J. O. Hoberg, Diastereoselective formation of seven-membered oxacycles by ring-expansion of cyclopropanated galactal, Tetrahedron Lett., 2003, 44, 9043–9045 CrossRef CAS.
- R. Batchelor, J. E. Harvey, P. T. Northcote, P. Teesdale-Spittle and J. O. Hoberg, Heptanosides from galactose-derived oxepenes via stereoselective addition reactions, J. Org. Chem., 2009, 74, 7627–7632 CrossRef CAS PubMed.
- R. Batchelor, J. E. Harvey, P. Teesdale-Spittle and J. O. Hoberg, Mechanistic studies of rearrangements during the ring expansions of cyclopropanated carbohydrates, Tetrahedron Lett., 2009, 50, 7283–7285 CrossRef CAS.
- D. Sabatino and M. J. Damha, Oxepane nucleic acids: Synthesis, characterization, and properties of oligonucleotides bearing a seven-membered carbohydrate ring, J. Am. Chem. Soc., 2007, 129, 8259–8270 CrossRef CAS PubMed.
- N. V. Ganesh and N. Jayaraman, Synthesis of Septanosides through an Oxyglycal Route, J. Org. Chem., 2007, 72, 5500–5504 CrossRef CAS PubMed.
- S. Dey and N. Jayaraman, Branching out at C, -2 of septanosides. Synthesis of 2-deoxy-2- C -alkyl/aryl septanosides from a bromo-oxepine, Beilstein J. Org. Chem., 2012, 8, 522–527 CrossRef CAS PubMed.
- A. M. Gómez, F. Lobo, C. Uriel and J. C. López, Recent Developments in the Ferrier Rearrangement: Recent Developments in the Ferrier Rearrangement, Eur. J. Org. Chem., 2013, 7221–7262 CrossRef.
- B. J. Teobald, The Nicholas reaction: the use of dicobalt hexacarbonyl-stabilised propargylic cations in synthesis, Tetrahedron, 2002, 58, 4133–4170 CrossRef CAS.
- A. M. Gómez, F. Lobo, D. Pérez De Las Vacas, S. Valverde and J. C. López, Formation and reactivity of new Nicholas–Ferrier pyranosidic cations: novel access to oxepanes via a 1,6-hydride shift/cyclization sequence, Chem. Commun., 2010, 46, 6159 RSC.
- C. Pavlik, A. Onorato, S. Castro, M. Morton, M. Peczuh and M. B. Smith, An unexpectedly facile cyclization of polyhydric alcohols, Org. Lett., 2009, 11, 3722–3725 CrossRef CAS PubMed.
- T. Satoh, T. Imai, S. Umeda, K. Tsuda, H. Hashimoto and T. Kakuchi, Regio- and stereoselective cyclizations of dianhydro sugar alcohols catalyzed by a chiral (salen)CoIII complex, Carbohydr. Res., 2005, 340, 2677–2681 CrossRef CAS PubMed.
- M. H. Wu, K. B. Hansen and E. N. Jacobsen, Regio- and enantioselective cyclization of epoxy alcohols catalyzed by a [CoIII(salen)] complex, Angew. Chem., Int. Ed., 1999, 38, 2012–2014 CrossRef CAS PubMed.
- J. E. Baldwin, Rules for Ring Closure, J. Chem. Soc., Chem. Commun., 1976, 734–736 RSC.
- R. Vannam and M. W. Peczuh, How to Homologate Your Sugar: Synthetic Approaches to Septanosyl Containing Carbohydrates, Eur. J. Org. Chem., 2016, 1800–1812 CrossRef CAS.
- R. Vannam and M. W. Peczuh, Synthesis of C-septanosides from pyranoses via vinyl addition and electrophilic cyclization, Org. Lett., 2013, 15, 4122–4125 CrossRef CAS PubMed.
- C. P. Sager, B. Fiege, P. Zihlmann, R. Vannam, S. Rabbani, R. P. Jakob, R. C. Preston, A. Zalewski, T. Maier, M. W. Peczuh and B. Ernst, The price of flexibility – a case study on septanoses as pyranose mimetics, Chem. Sci., 2018, 9, 646–654 RSC.
- M. W. Peczuh and N. L. Snyder, Carbohydrate-based oxepines: ring expanded glycals for the synthesis of septanose saccharides, Tetrahedron Lett., 2003, 44, 4057–4061 CrossRef CAS.
- S. D. Markad, S. Xia, N. L. Snyder, B. Surana, M. D. Morton, C. M. Hadad and M. W. Peczuh, Stereoselectivity in the epoxidation of carbohydrate-based oxepines, J. Org. Chem., 2008, 73, 6341–6354 CrossRef CAS PubMed.
- R. Vannam and M. W. Peczuh, A practical and scalable synthesis of carbohydrate based oxepines, Org. Biomol. Chem., 2016, 14, 3989–3996 RSC.
- J. C. Y. Wong, P. Lacombe and C. F. Sturino, A ring closing metathesis-osmylation approach to oxygenated oxepanes as carbohydrate surrogates, Tetrahedron Lett., 1999, 40, 8751–8754 CrossRef CAS.
- H. Ovaa, M. A. Leeuwenburgh, H. S. Overkleeft, G. A. Van Der Marel and J. H. Van Boom, An expeditious route to the synthesis of highly functionalized chiral oxepines from monosaccharides, Tetrahedron Lett., 1998, 39, 3025–3028 CrossRef CAS.
- D. J. Edmonds, D. Johnston and D. J. Procter, Samarium(II)-Iodide-Mediated Cyclizations in Natural Product Synthesis, Chem. Rev., 2004, 104, 3371–3404 CrossRef CAS PubMed.
- T. Nakata, SmI2-induced reductive cyclizations for the synthesis of cyclic ethers and applications in natural product synthesis, Chem. Soc. Rev., 2010, 39, 1955–1972 RSC.
- N. Hori, H. Matsukura and T. Nakata, Efficient Synthesis of trans-Fused Polycyclic Ethers Including Tetrahydropyrans and Oxepanes Based on SmI2-Induced Reductive Cyclization, Org. Lett., 1999, 1, 1099–1101 CrossRef CAS.
- N. Hori, H. Matsukura, G. Matsuo and T. Nakata, An efficient strategy for the iterative synthesis of a trans-fused polytetrahydropyran ring system via SmI2-induced reductive intramolecular cyclization, Tetrahedron Lett., 1999, 40, 2811–2814 CrossRef CAS.
- G. Matsuo, N. Hori and T. Nakata, Stereoselective syntheses of trans-fused 6,6- and 6,7-membered ether ring systems having an angular methyl group based on SmI2-induced reductive intramolecular cyclization, Tetrahedron Lett., 1999, 40, 8859–8862 CrossRef CAS.
- Y. Mori, K. Yaegashi and H. Furukawa, A New Strategy for the Reiterative Synthesis of trans-Fused Tetrahydropyrans Via Alkylation of Oxiranyl Anion and 6-endo Cyclization, J. Am. Chem. Soc., 1996, 118, 8158–8159 CrossRef CAS.
- K. C. Nicolaou, C. K. Hwang, B. E. Marrón, S. A. DeFrees, E. A. Couladouros, Y. Abe, P. J. Carroll and J. P. Snyder, Bridging of Macrodithionolactones to Bicyclic Systems. Synthesis and Modeling of Oxapolycyclic Frameworks, J. Am. Chem. Soc., 1990, 112, 3040–3054 CrossRef CAS.
- M. Sasaki, M. Inoue, T. Noguchi, A. Takeichi and K. Tachibana, Convergent and stereoselective method for synthesis of O-linked oxepane ring system by intramolecular radical cyclization, Tetrahedron Lett., 1998, 39, 2783–2786 CrossRef CAS.
- M. Sasaki, T. Noguchi and K. Tachibana, Synthesis of the FGH ring fragment of ciguatoxin, Tetrahedron Lett., 1999, 40, 1337–1340 CrossRef CAS.
- M. Sasaki, T. Noguchi and K. Tachibana, Intramolecular Radical Cyclization Ring-Closing Metathesis Approach to Fused Polycyclic Ethers. Convergent Synthesis and Conformational Analysis of the (E)FGH Ring System of Ciguatoxin, J. Org. Chem., 2002, 67, 3301–3310 CrossRef CAS PubMed.
- K. C. Nicolaou, C. K. Hwang, M. E. Duggan, K. B. Reddy, B. E. Marron and D. G. McGarry, Bridging of macrocycles to bicycles. New synthetic technology for the construction of cis- and trans-fused oxopolycyclic systems, J. Am. Chem. Soc., 1986, 108, 6800–6802 CrossRef CAS.
- K. C. Nicolaou, M. E. Duggan, C.-K. Hwang and P. K. Somers, Activation of 6-endo over 5-exo epoxide openings. Ring-selective formation of tetrahydropyran systems and stereocontrolled synthesis of the ABC ring framework of brevetoxin B, J. Chem. Soc., Chem. Commun., 1985, 1359–1362 RSC.
- K. C. Nicolaou, M. E. Duggan and C. K. Hwang, New synthetic technology for the construction of oxocenes, J. Am. Chem. Soc., 1986, 108, 2468–2469 CrossRef CAS PubMed.
- K. C. Nicolaou, C. K. Hwang, S. DeFrees and N. A. Stylianides, Novel chemistry of dithiatopazine, J. Am. Chem. Soc., 1988, 110, 4868–4869 CrossRef CAS.
- K. C. Nicolaou, C. K. Hwang and D. A. Nugiel, Synthetic studies on the dioxepane region of brevetoxin B. New synthetic technology for the construction of oxepanes and synthesis of a model for the CDEF ring skeleton of brevetoxin B, J. Am. Chem. Soc., 1989, 111, 4136–4137 CrossRef CAS.
- K. C. Nicolaou, C. K. Hwang, M. E. Duggan and P. J. Carroll, Dithiatopazine. The first stable 1,2-dithietane, J. Am. Chem. Soc., 1987, 109, 3801–3802 CrossRef CAS.
- N. Hashimoto, T. Aovamo and T. Shioiri, New methods and reagents in organic synthesis. 10. trimethylsilydiazomethane (TMSCHN2). A new, stable, and safe reagent for the homologation of ketones, Tetrahedron Lett., 1980, 21, 4619–4622 CrossRef CAS.
- T. Kawai, M. Isobe and S. Peters, Factors Affecting Reaction of 1,6-Anhydrohexos-2-ulose Derivatives, Aust. J. Chem., 1995, 48, 115 CrossRef CAS.
- K. Maruoka, A. B. Concepcion and H. Yamamoto, Organoaluminum-Promoted Homologation of Ketones with Diazoalkanes, J. Org. Chem., 1994, 59, 4725–4726 CrossRef CAS.
- K. Maruoka, A. B. Concepcion and H. Yamamoto, Selective Homologation of Ketones and Aldehydes with Diazoalkanes Promoted by Organoaluminum Reagents, Synthesis, 1994, 1283–1290 CrossRef CAS.
- Y. Mori, K. Yaegashi and H. Furukawa, Oxiranyl Anions in Organic Synthesis: Application to the Synthesis of Hemibrevetoxin B, J. Am. Chem. Soc., 1997, 119, 4557–4558 CrossRef CAS.
- Y. Mori, K. Yaegashi and H. Furukawa, Stereoselective synthesis of the 6,7,6- and 6,7,7-ring systems of polycyclic ethers by 6-endo cyclization and ring expansion, Tetrahedron, 1997, 53, 12917–12932 CrossRef CAS.
- K. C. Nicolaou, C.-K. Hwang and D. A. Nugiel, A Photolytic Entry into Oxepane Systems, Angew. Chem., Int. Ed. Engl., 1988, 27, 1362–1364 CrossRef.
- M. B. Sassaman, K. D. Kotian, G. K. S. Prakash and G. A. Olah, General ether synthesis under mild acid-free conditions. Trimethylsilyl iodide catalyzed reductive coupling of carbonyl compounds with trialkylsilanes to symmetrical ethers and reductive condensation with alkoxysilanes to unsymmetrical ethers, J. Org. Chem., 1987, 52, 4314–4319 CrossRef CAS.
- M. B. Sassaman, G. K. Surya Prakash, G. A. Olah, P. Donald and K. B. Loker, Ionic Hydrogenation with Organosilanes under Acid-Free Conditions. Synthesis of Ethers, Alkoxysilanes, Thioethers, and Cyclic Ethers via rganosilyl Iodide and Triflate Catalyzed Reductions of Carbonyl Compounds and Their Derivatives, Tetrahedron, 1988, 44, 3771–3780 CrossRef CAS.
- S. Takai, N. Sawada and M. Isobe, Convergent Synthesis of the E‘FGH Ring Fragment of Ciguatoxin 1B via an Acetylene Cobalt Complex Strategy, J. Org. Chem., 2003, 68, 3225–3231 CrossRef CAS PubMed.
- S. Takai and M. Isobe, Convergent Synthesis of the E‘FGH‘ Ring Fragment of Ciguatoxin 1B via an Acetylene Cobalt Complex Strategy, Org. Lett., 2002, 4, 1183–1186 CrossRef CAS PubMed.
- M. Isobe, S. Hosokawa and K. Kira, syn-trans Fused Bicyclic Ether Formation via Acetylene-biscobalthexacarbonyl Complex, Chem. Lett., 1996, 25, 473–474 CrossRef.
- M. Isobe, C. Yenjai and S. Tanaka, Medium Size Ether Ring Formation of C-Alkynylated Sugars via Dicobalthexacarbonyl Complexes, Synlett, 1994, 916–918 CrossRef CAS.
- S. Tanaka, T. Tsukiyama and M. Isobe, Epimerization of C-1 alkynyl group on pyranose ring through dicobalt hexacarbonyl complexes, Tetrahedron Lett., 1993, 34, 5757–5760 CrossRef CAS.
- S. Tanaka and M. Isobe, Opening of dihydropyran and recyclizing to dehydrooxepane through C-1 alkynyl cobalt complex—A new method toward marine polyether toxins, Tetrahedron Lett., 1994, 35, 7801–7804 CrossRef CAS.
- S. Hosokawa and M. Isobe, Synthesis of two Diastereoisomers of A/B Fragment of Ciguatoxin, Synlett, 1995, 1179–1180 CrossRef CAS.
- S. Hosokawa and M. Isobe, Partial Synthesis of Ciguatoxin, an A/B/C Fragment, Synlett, 1996, 351–352 CrossRef CAS.
- S. Hosokawa and M. Isobe, Reductive decomplexation of biscobalthexacarbonyl acetylenes into olefins, Tetrahedron Lett., 1998, 39, 2609–2612 CrossRef CAS.
- C. Yenjai and M. Isobe, One-step recyclization of sugar acetylenes to form medium ether rings via dicobalthexacarbonyl complexes, Tetrahedron, 1998, 54, 2509–2520 CrossRef CAS.
- J. Palazón and V. S. Martín, A stereoselective synthesis of medium-sized cyclic ethers by the intramolecular cyclization of linear hydroxyalkyl-propargylic alcohols assisted by Co2(CO)8, Tetrahedron Lett., 1995, 36, 3549–3552 CrossRef.
- K. Kira and M. Isobe, Synthesis of the BCDE rings of ciguatoxin 1B via an acetylene biscobalthexacarbonyl–vinylsilane strategy, Tetrahedron Lett., 2001, 42, 2821–2824 CrossRef CAS.
- A. Hamajima and M. Isobe, Total Synthesis of Ciguatoxin, Angew. Chem., Int. Ed., 2009, 48, 2941–2945 CrossRef CAS PubMed.
- L. Bouché, M. Kandziora and H.-U. Reissig, Synthesis of new enantiopure poly(hydroxy)aminooxepanes as building blocks for multivalent carbohydrate mimetics, Beilstein J. Org. Chem., 2014, 10, 213–223 CrossRef PubMed.
- X. M. Yu, H. Han and B. S. J. Blagg, Synthesis of mono- and dihydroxylated furanoses, pyranoses, and an oxepanose for the preparation of natural product analogue libraries, J. Org. Chem., 2005, 70, 5599–5605 CrossRef CAS PubMed.
- F. P. J. T. Rutjes, T. Martijn Kooistra, H. Hiemstra and H. E. Schoemaker, A novel transition metal-catalyzed route to functionalized dihydropyrans and tetrahydrooxepines, Synlett, 1998, 192–194 CrossRef CAS.
- D. Das, J. Halder, R. Bhuniya and S. Nanda, Stereoselective Synthesis of Enantiopure Oxetanes, a Carbohydrate Mimic, an ε-Lactone, and Cyclitols from Biocatalytically Derived β-Hydroxy Esters as Chiral Precursors: Stereoselective Synthesis of Enantiopure Oxetanes, Eur. J. Org. Chem., 2014, 5229–5246 CrossRef CAS.
- F. E. McDonald, X. Wang, B. Do and K. I. Hardcastle, Synthesis of oxepanes and trans-fused bisoxepanes via biomimetic, endo-regioselective tandem oxacyclizations of polyepoxides, Org. Lett., 2000, 2, 2917–2919 CrossRef CAS PubMed.
- F. E. McDonald, F. Bravo, X. Wang, X. Wei, M. Toganoh, J. R. Rodríguez, B. Do, W. A. Neiwert and K. I. Hardcastle, Endo-oxacyclizations of polyepoxides: Biomimetic synthesis of fused polycyclic ethers, J. Org. Chem., 2002, 67, 2515–2523 CrossRef CAS PubMed.
- M. T. Mujica, M. M. Afonso, A. Galindo and J. A. Palenzuela, A versatile approach to cyclic ethers. Synthesis of disubstituted oxepanes and oxocanes, Tetrahedron Lett., 1994, 35, 3401–3404 CrossRef CAS.
- N. Hori, K. Nagasawa, T. Shimizu and T. Nakata, Efficient synthesis of 2,3-trans-tetrahydropyrans and oxepanes: Rearrangement-ring expansion of cyclic ethers having a chloromethanesulfonate, Tetrahedron Lett., 1999, 40, 2145–2148 CrossRef CAS.
- T. Nakata, S. Nomura and H. Matsukura, Stereoselective Synthesis of Six- and Seven-Membered Ether Rings Based on the Ring Expansion, Tetrahedron Lett., 1996, 37, 213–216 CrossRef CAS.
- M. Morimoto, H. Matsukura and T. Nakata, Total synthesis of hemibrevetoxin B, Tetrahedron Lett., 1996, 37, 6365–6368 CrossRef CAS.
- M. T. Mujica, M. M. Afonso, A. Galindo and J. A. Palenzuela, Enantioselective Synthesis of α,β,α‘-Trisubstituted Cyclic Ethers, J. Org. Chem., 1998, 63, 9728–9738 CrossRef CAS.
- J. Armbruster, F. Stelzer, P. Landenberger, C. Wieber, D. Hunkler, M. Keller and H. Prinzbach, From cyclooctatetraene to chiral polyfunctionalized C8 building blocks—meso-persubstituted oxepanes and azepanes, Tetrahedron Lett., 2000, 41, 5483–5487 CrossRef CAS.
- R. W. Murray, M. Singh and N. P. Rath, The reaction of cyclooctatetraene with dimethyldioxirane, Tetrahedron, 1999, 55, 4539–4558 CrossRef CAS.
- E. Alvarez, M. T. Díaz, R. Pérez, J. L. Ravelo, A. Regueiro, J. A. Vera, D. Zurita and J. D. Martín, Simple Designs for the Construction of Complex Trans-Fused Polyether Toxin Frameworks. A Linear Strategy Based on Entropically Favored Oxirane Ring Enlargement in Epoxycycloalkenes Followed by Carbon-Carbon or Carbon-Oxygen Bond-Forming Cyclizations, J. Org. Chem., 1994, 59, 2848–2876 CrossRef CAS.
- T. Shih and Y. Fang, Expeditious Synthesis of New 3,4,6−Trihydroxythiepanes from d–(–)–Quinic Acid, Synth. Commun., 2007, 37, 3337–3349 CrossRef CAS.
- A. V. K. Prasad and Y. Shimizu, The structure of hemibrevetoxin-B: a new type of toxin in the Gulf of Mexico red tide organism, J. Am. Chem. Soc., 1989, 111, 6476–6477 CrossRef CAS.
- K. C. Nicolaou, K. R. Reddy, G. Skokotas, F. Sato and X. Y. Xiao, Total synthesis of hemibrevetoxin B, J. Am. Chem. Soc., 1992, 114, 7935–7936 CrossRef CAS.
- I. Kadota, P. Jung-Youl, N. Koumura, G. Pollaud, Y. Matsukawa and Y. Yamamoto, Total synthesis of Hemibrevetoxin B, Tetrahedron Lett., 1995, 36, 5777–5780 CrossRef CAS.
- I. Kadota and Y. Yamamoto, Stereocontrolled Total Synthesis of Hemibrevetoxin B, J. Org. Chem., 1998, 63, 6597–6606 CrossRef CAS.
- J. D. Rainier, S. P. Allwein and J. M. Cox, C-Glycosides to Fused Polycyclic Ethers. A Formal Synthesis of (±)-Hemibrevetoxin B, J. Org. Chem., 2001, 66, 1380–1386 CrossRef CAS PubMed.
- J. M. Holland, M. Lewis and A. Nelson, First Desymmetrization of a Centrosymmetric Molecule in Natural Product Synthesis: Preparation of a Key Fragment in the Synthesis of Hemibrevetoxin B, Angew. Chem., Int. Ed., 2001, 40, 3927–4105 CrossRef.
- A. Zakarian, A. Batch and R. A. Holton, A Convergent Total Synthesis of Hemibrevetoxin B, J. Am. Chem. Soc., 2003, 125, 7822–7824 CrossRef CAS PubMed.
- Y.-Y. Lin, M. Risk, S. M. Ray, D. Van Engen, J. Clardy, J. Golik, J. C. James and K. Nakanishi, Isolation and structure of brevetoxin B from the ‘red tide’ dinoflagellate Ptychodiscus brevis (Gymnodinium breve), J. Am. Chem. Soc., 1981, 103, 6773–6775 CrossRef CAS.
- K. C. Nicolaou, F. P. J. T. Rutjes, E. A. Theodorakis, J. Tiebes, M. Sato and E. Untersteller, Total Synthesis of Brevetoxin B. 3. Final Strategy and Completion, J. Am. Chem. Soc., 1995, 117, 10252–10263 CrossRef CAS.
- K. C. Nicolaou, E. A. Theodorakis, F. P. J. T. Rutjes, J. Tiebes, M. Sato, E. Untersteller and X.-Y. Xiao, Total Synthesis of Brevetoxin B. 1. CDEFG Framework, J. Am. Chem. Soc., 1995, 117, 1171–1172 CrossRef CAS.
- K. C. Nicolaou, F. P. J. T. Rutjes, E. A. Theodorakis, J. Tiebes, M. Sato and E. Untersteller, Total Synthesis of Brevetoxin B. 2. Completion, J. Am. Chem. Soc., 1995, 117, 1173–1174 CrossRef CAS.
- G. Matsuo, K. Kawamura, N. Hori, H. Matsukura and T. Nakata, Total Synthesis of Brevetoxin-B, J. Am. Chem. Soc., 2004, 126, 14374–14376 CrossRef CAS PubMed.
- K. C. Nicolaou, Z. Yang, G. Shi, J. L. Gunzner and K. A. Agrios, Total synthesis of brevetoxin A, Nature, 1998, 392, 264–269 CrossRef CAS PubMed.
- M. T. Crimmins, J. L. Zuccarello, J. M. Ellis, P. J. McDougall, P. A. Haile, J. D. Parrish and K. A. Emmitte, Total Synthesis of Brevetoxin A, Org. Lett., 2009, 11, 489–492 CrossRef CAS PubMed.
- M. T. Crimmins, P. J. McDougall and K. A. Emmitte, A Convergent Coupling Strategy for the Formation of Polycyclic Ethers: Stereoselective Synthesis of the BCDE Fragment of Brevetoxin A, Org. Lett., 2005, 7, 4033–4036 CrossRef CAS PubMed.
- M. T. Crimmins, P. J. McDougall and J. M. Ellis, Improved Synthesis of the ABCDE Fragment of Brevetoxin A, Org. Lett., 2006, 8, 4079–4082 CrossRef CAS PubMed.
- W. M. Abraham, A. J. Bourdelais, J. R. Sabater, A. Ahmed, T. A. Lee, I. Serebriakov and D. G. Baden, Airway Responses to Aerosolized Brevetoxins in an Animal Model of Asthma, Am. J. Respir. Crit. Care Med., 2005, 171, 26–34 CrossRef PubMed.
- H. Fuwa, M. Ebine, A. J. Bourdelais, D. G. Baden and M. Sasaki, Total Synthesis, Structure Revision, and Absolute Configuration of (−)-Brevenal, J. Am. Chem. Soc., 2006, 128, 16989–16999 CrossRef CAS PubMed.
- H. Takamura, S. Kikuchi, Y. Nakamura, Y. Yamagami, T. Kishi, I. Kadota and Y. Yamamoto, Total Synthesis of Brevenal, Org. Lett., 2009, 11, 2531–2534 CrossRef CAS PubMed.
- Y. Zhang, J. Rohanna, J. Zhou, K. Iyer and J. D. Rainier, Total Synthesis of Brevenal, J. Am. Chem. Soc., 2011, 133, 3208–3216 CrossRef CAS PubMed.
- K. Iyer and J. D. Rainier, Olefinic Ester and Diene Ring-Closing Metathesis Using a Reduced Titanium Alkylidene, J. Am. Chem. Soc., 2007, 129, 12604–12605 CrossRef CAS PubMed.
- J. D. Rainier, S. P. Allwein and J. M. Cox, A Highly Efficient Synthesis of the Hemibrevetoxin B Ring System, Org. Lett., 2000, 2, 231–234 CrossRef CAS PubMed.
- J. D. Rainier and S. P. Allwein, An Iterative Approach to Fused Ether Ring Systems, J. Org. Chem., 1998, 63, 5310–5311 CrossRef CAS.
- J. D. Rainier and S. P. Allwein, A highly efficient iterative approach to fused ether ring systems, Tetrahedron Lett., 1998, 39, 9601–9604 CrossRef CAS.
- M. Murata, A. M. Legrand, Y. Ishibashi and T. Yasumoto, Structures of ciguatoxin and its congener, J. Am. Chem. Soc., 1989, 111, 8929–8931 CrossRef CAS.
- M. Inoue, K. Miyazaki, Y. Ishihara, A. Tatami, Y. Ohnuma, Y. Kawada, K. Komano, S. Yamashita, N. Lee and M. Hirama, Total Synthesis of Ciguatoxin and 51-HydroxyCTX3C, J. Am. Chem. Soc., 2006, 128, 9352–9354 CrossRef CAS PubMed.
- H. Oguri, S. Hishiyama, T. Oishi and M. Hirama, Enantio-Controlled Synthesis of the AB Ring Moiety of Ciguatoxin, Synlett, 1995, 1252–1254 CrossRef CAS.
- S. Kobayashi, B. H. Alizadeh, S. Sasaki, H. Oguri and M. Hirama, Synthesis of the Fully Functionalized ABCDE Ring Moiety of Ciguatoxin, Org. Lett., 2004, 6, 751–754 CrossRef CAS PubMed.
- T. Oishi, S. Tanaka, Y. Ogasawara, K. Maeda, H. Oguri and M. Hirama, Highly Stereocontrolled Synthesis of the ABCD Ring Fragment of Ciguatoxin CTX3C, Synlett, 2001, 0952–0954 CrossRef CAS.
- K. Maeda, T. Oishi, H. Oguri and M. Hirama, Convergent synthesis of the ABCDE ring framework of ciguatoxin, Chem. Commun., 1999, 1063–1064 RSC.
- M. Maruyama, M. Inoue, T. Oishi, H. Oguri, Y. Ogasawara, Y. Shindo and M. Hirama, Convergent synthesis of the ABCDE ring system of ciguatoxin CTX3C, Tetrahedron, 2002, 58, 1835–1851 CrossRef CAS.
- S. Kobayashi, Y. Takahashi, K. Komano, B. H. Alizadeh, Y. Kawada, T. Oishi, S. Tanaka, Y. Ogasawara, S. Sasaki and M. Hirama, Stereocontrolled synthesis of the ABCDE ring moiety of ciguatoxin CTX3C, Tetrahedron, 2004, 60, 8375–8396 CrossRef CAS.
- M. Inoue and M. Hirama, Total Synthesis of Ciguatoxin CTX3C, a Causative Toxin of Ciguatera Seafood Poisoning, Synlett, 2004, 577–595 CrossRef CAS.
- A. Tatami, M. Inoue, H. Uehara and M. Hirama, A concise route to the right wing of ciguatoxin, Tetrahedron Lett., 2003, 44, 5229–5233 CrossRef CAS.
- M. Inoue, S. Yamashita, A. Tatami, K. Miyazaki and M. Hirama, A New Stereoselective Synthesis of Ciguatoxin Right Wing Fragments, J. Org. Chem., 2004, 69, 2797–2804 CrossRef CAS PubMed.
- M. Inoue and M. Hirama, Evolution of a Practical Total Synthesis of Ciguatoxin CTX3C, Acc. Chem. Res., 2004, 37, 961–968 CrossRef CAS PubMed.
- R. Saeeng and M. Isobe, Partial synthesis of ciguatoxin (5R)-ABC segment, Tetrahedron Lett., 1999, 40, 1911–1914 CrossRef CAS.
- A. Hamajima and M. Isobe, Convergent Synthesis of the Right-Hand Segment of Ciguatoxin, Org. Lett., 2006, 8, 1205–1208 CrossRef CAS PubMed.
- K. Kira, A. Hamajima and M. Isobe, Synthesis of the BCD-ring of ciguatoxin 1B using an acetylene cobalt complex and vinylsilane strategy, Tetrahedron, 2002, 58, 1875–1888 CrossRef CAS.
- T. Baba, G. Huang and M. Isobe, Synthesis of the JKLM-ring fragment of ciguatoxin, Tetrahedron, 2003, 59, 6851–6872 CrossRef CAS.
- T.-Z. Lui, B. Kirschbaum and M. Isobe, Heteroconjugate Addition Strategy for the Synthesis of the (HIJ)K-Ring Fragment of Ciguatoxin, Synlett, 2000, 0587–0590 Search PubMed.
- St. Baba, S. Takai, N. Sawada and M. Isobe, Stereoselective Synthesis of the Fully Functionalized HIJ-ring Framework of Ciguatoxin, Synlett, 2004, 603–608 Search PubMed.
- M. Satake, M. Murata and T. Yasumoto, The structure of CTX3C, a ciguatoxin congener isolated from cultured Gambierdiscus toxicus, Tetrahedron Lett., 1993, 34, 1975–1978 CrossRef CAS.
- M. Hirama, T. Oishi, H. Uehara, M. Inoue, M. Maruyama, H. Oguri and M. Satake, Total Synthesis of Ciguatoxin CTX3C, Science, 2001, 294, 1904–1907 CrossRef CAS PubMed.
- M. Inoue, H. Uehara, M. Maruyama and M. Hirama, Practical Total Synthesis of Ciguatoxin CTX3C by Improved Protective Group Strategy, Org. Lett., 2002, 4, 4551–4554 CrossRef CAS PubMed.
- M. Hirama, M. Maruyama, K. Maeda, T. Oishi and H. Oguri, Convergent Strategy for Synthesizing Polycyclic Ether Marine Toxins: Synthesis of the ABCDE Ring Fragment of Ciguatoxin CTX3C, Heterocycles, 2001, 54, 93 CrossRef PubMed.
- T. Oishi, Y. Nagumo, M. Shoji, J.-Y. L. Brazidec, H. Uehara and M. Hirama, Convergent synthesis of the IJKLM ring fragment of ciguatoxin CTX3C, Chem. Commun., 1999, 2035–2036 RSC.
- T. Oishi, H. Uehara, Y. Nagumo, M. Shoji, J.-Y. Le Brazidec, M. Kosaka and M. Hirama, Practical entry into the HIJKLM ring segment of ciguatoxin CTX3C, Chem. Commun., 2001, 381–382 RSC.
- A. Rahim, T. Fujiwara and T. Takeda, Titanocene(II)-Promoted Olefination of ω,ω-Bis(phenylthio)alkyl Alkanoates. A New Method for the Preparation of ω-Hydroxy Ketones, Tetrahedron, 2000, 56, 763–770 CrossRef.
- M. Satake, M. Shoji, Y. Oshima, H. Naoki, T. Fujita and T. Yasumoto, Gymnocin-A, a cytotoxic polyether from the notorious red tide dinoflagellate, Gymnodinium mikimotoi, Tetrahedron Lett., 2002, 43, 5829–5832 CrossRef CAS.
- C. Tsukano and M. Sasaki, Total Synthesis of Gymnocin-A, J. Am. Chem. Soc., 2003, 125, 14294–14295 CrossRef CAS PubMed.
- M. Sasaki, C. Tsukano and K. Tachibana, Synthetic entry to the ABCD ring fragment of gymnocin-A, a cytotoxic marine polyether, Tetrahedron Lett., 2003, 44, 4351–4354 CrossRef CAS.
- M. Sasaki, C. Tsukano and K. Tachibana, Studies toward the Total Synthesis of Gymnocin A, a Cytotoxic Polyether: A Highly Convergent Entry to the F–N Ring Fragment, Org. Lett., 2002, 4, 1747–1750 CrossRef CAS PubMed.
- C. Tsukano, M. Ebine and M. Sasaki, Convergent Total Synthesis of Gymnocin-A and Evaluation of Synthetic Analogues, J. Am. Chem. Soc., 2005, 127, 4326–4335 CrossRef CAS PubMed.
- T. Sakai, S. Matsushita, S. Arakawa, K. Mori, M. Tanimoto, A. Tokumasu, T. Yoshida and Y. Mori, Total Synthesis of Gymnocin-A, J. Am. Chem. Soc., 2015, 137, 14513–14516 CrossRef CAS PubMed.
- T. Sakai, S. Matsushita, S. Arakawa, A. Kawai and Y. Mori, Synthetic study of gymnocin-A: synthesis of the ABC ring fragment, Tetrahedron Lett., 2014, 55, 6557–6560 CrossRef CAS.
- T. Sakai, H. Asano, K. Furukawa, R. Oshima and Y. Mori, Synthesis of the KLMN Fragment of Gymnocin-A Using Oxiranyl Anion Convergent Methodology, Org. Lett., 2014, 16, 2268–2271 CrossRef CAS PubMed.
- M. Satake, Y. Tanaka, Y. Ishikura, Y. Oshima, H. Naoki and T. Yasumoto, Gymnocin-B with the largest contiguous polyether rings from the red tide dinoflagellate, Karenia (formerly Gymnodinium) mikimotoi, Tetrahedron Lett., 2005, 46, 3537–3540 CrossRef CAS.
- S. Sittihan and T. F. Jamison, Total Synthesis of the Marine Ladder Polyether Gymnocin B, J. Am. Chem. Soc., 2019, 141, 11239–11244 CrossRef CAS PubMed.
- K. Nakanishi, The chemistry of brevetoxins: A review, Toxicon, 1985, 23, 473–479 CrossRef CAS PubMed.
- I. Vilotijevic and T. F. Jamison, Epoxide-Opening Cascades Promoted by Water, Science, 2007, 317, 1189–1192 CrossRef CAS PubMed.
- I. Kadota, C. Kadowaki, C.-H. Park, H. Takamura, K. Sato, P. W. H. Chan, S. Thorand and Y. Yamamoto, Syntheses of the AB and EFGH ring segments of gambierol, Tetrahedron, 2002, 58, 1799–1816 CrossRef CAS.
- K. C. Nicolaou, C. A. Veale, C.-K. Hwang, J. Hutchinson, C. V. C. Prasad and W. W. Ogilvie, Novel Strategies for the Construction of Complex Polycyclic Ether Frameworks. Stereocontrolled Synthesis of the FGHIJ Ring System of Brevetoxin A, Angew. Chem., Int. Ed. Engl., 1991, 30, 299–303 CrossRef.
- T. Kuranaga, M. Satake, D. G. Baden, J. L. C. Wright and K. Tachibana, Synthesis of the BC/DE ring model of brevisin for confirmation of the structure around the acyclic junction, Tetrahedron Lett., 2010, 51, 4673–4676 CrossRef CAS.
- M. Satake, M. Murata and T. Yasumoto, Gambierol: a new toxic polyether compound isolated from the marine dinoflagellate Gambierdiscus toxicus, J. Am. Chem. Soc., 1993, 115, 361–362 CrossRef CAS.
- A. Morohashi, M. Satake and T. Yasumoto, The absolute configuration of gambierol, a toxic marine polyether from the dinoflagellate, Gambierdiscus toxicus, Tetrahedron Lett., 1999, 40, 97–100 CrossRef CAS.
- H. Fuwa, N. Kainuma, K. Tachibana and M. Sasaki, Total Synthesis of (−)-Gambierol, J. Am. Chem. Soc., 2002, 124, 14983–14992 CrossRef CAS PubMed.
- K. C. Nicolaou, P. A. Wallace, S. Shi, M. A. Ouellette, M. E. Bunnage, J. L. Gunzner, K. A. Agrios, G. Shi, P. Gärtner and Z. Yang, Total Synthesis of Brevetoxin A: Part 2: Second Generation Strategy and Construction of EFGH Model System, Chem. – Eur. J., 1999, 5, 618–627 CrossRef CAS.
- H. W. B. Johnson, U. Majumder and J. D. Rainier, The Total Synthesis of Gambierol, J. Am. Chem. Soc., 2005, 127, 848–849 CrossRef CAS PubMed.
- U. Majumder, J. M. Cox and J. D. Rainier, Synthesis of an F−H Gambierol Subunit Using a C -Glycoside-Centered Strategy, Org. Lett., 2003, 5, 913–916 CrossRef CAS PubMed.
- J. M. Cox and J. D. Rainier, C-Glycosides to Fused Polycyclic Ethers. An Efficient Entry into the A−D Ring System of Gambierol, Org. Lett., 2001, 3, 2919–2922 CrossRef CAS PubMed.
- K. Takai, T. Kakiuchi, Y. Kataoka and K. Utimoto, A Novel Catalytic Effect of Lead on the Reduction of a Zinc Carbenoid with Zinc Metal Leading to a Geminal Dizinc Compound. Acceleration of the Wittig-Type Olefination with the RCHX2-TiCl4-Zn Systems by Addition of Lead, J. Org. Chem., 1994, 59, 2668–2670 CrossRef CAS.
- H. Furuta, Y. Hasegawa and Y. Mori, Total Synthesis of Gambierol, Org. Lett., 2009, 11, 4382–4385 CrossRef CAS PubMed.
- H. Furuta, M. Hase, R. Noyori and Y. Mori, Synthesis of the ABCD Ring of Gambierol, Org. Lett., 2005, 7, 4061–4064 CrossRef CAS PubMed.
- H. Nagai, M. Murata, K. Torigoe, M. Satake and T. Yasumoto, Gambieric acids, new potent antifungal substances with unprecedented polyether structures from a marine dinoflagellate Gambierdiscus toxicus, J. Org. Chem., 1992, 57, 5448–5453 CrossRef CAS.
- S. W. Roberts and J. D. Rainier, Synthesis of an A−E Gambieric Acid Subunit with Use of a C -Glycoside Centered Strategy, Org. Lett., 2007, 9, 2227–2230 CrossRef CAS PubMed.
- J. S. Clark, F. Romiti, B. Sieng, L. C. Paterson, A. Stewart, S. Chaudhury and L. H. Thomas, Synthesis of the A–D Ring System of the Gambieric Acids, Org. Lett., 2015, 17, 4694–4697 CrossRef CAS PubMed.
- H. Fuwa, T. Goto and M. Sasaki, Stereocontrolled Synthesis of the A/B-Ring Fragment of Gambieric Acid B: Reassignment of the Absolute Configuration of the Polycyclic Ether Region, Org. Lett., 2008, 10, 2211–2214 CrossRef CAS PubMed.
- H. Fuwa, K. Ishigai, T. Goto, A. Suzuki and M. Sasaki, Synthetic Studies on Gambieric Acids, Potent Antifungal Polycyclic Ether Natural Products: Reassignment of the Absolute Configuration of the Nonacyclic Polyether Core by NMR Analysis of Model Compounds, J. Org. Chem., 2009, 74, 4024–4040 CrossRef CAS PubMed.
- H. Fuwa, K. Ishigai, K. Hashizume and M. Sasaki, Total Synthesis and Complete Stereostructure of Gambieric Acid A, J. Am. Chem. Soc., 2012, 134, 11984–11987 CrossRef CAS PubMed.
- K. Tsubone, K. Hashizume, H. Fuwa and M. Sasaki, Studies toward the total synthesis of gambieric acids, potent antifungal polycyclic ethers: convergent synthesis of a fully elaborated GHIJ-ring fragment, Tetrahedron, 2011, 67, 6600–6615 CrossRef CAS.
- K. Tsubone, K. Hashizume, H. Fuwa and M. Sasaki, Studies toward the total synthesis of gambieric acids: convergent synthesis of the GHIJ-ring fragment having a side chain, Tetrahedron Lett., 2011, 52, 548–551 CrossRef CAS.
- R. Tong, J. C. Valentine, F. E. McDonald, R. Cao, X. Fang and K. I. Hardcastle, Total Syntheses of Durgamone, Nakorone, and Abudinol B via Biomimetic Oxa- and Carbacyclizations, J. Am. Chem. Soc., 2007, 129, 1050–1051 CrossRef CAS PubMed.
- F. A. Macias, J. M. G. Molinillo, R. M. Varela, A. Torres and F. R. Fronczek, Structural Elucidation and Chemistry of a Novel Family of Bioactive Sesquiterpenes: Heliannuols, J. Org. Chem., 1994, 59, 8261–8266 CrossRef CAS.
- T. Kamei, M. Shindo and K. Shishido, First enantioselective total synthesis of (−)-heliannuol C, Tetrahedron Lett., 2003, 44, 8505–8507 CrossRef CAS.
- J. R. Vyvyan, J. M. Oaksmith, B. W. Parks and E. M. Peterson, Total synthesis of (±)-heliannuol C and E via aromatic Claisen rearrangement, Tetrahedron Lett., 2005, 46, 2457–2460 CrossRef CAS.
- B. Biswas, P. K. Sen and R. V. Venkateswaran, The synthesis of heliannuol C, an allelochemical from Helianthus annuus, Tetrahedron Lett., 2006, 47, 4019–4021 CrossRef CAS.
- B. Biswas, P. K. Sen and R. V. Venkateswaran, Bargellini condensation of coumarins. Expeditious route to o-carboxyvinylphenoxyisobutyric acids and application to the synthesis of sesquiterpenes helianane, heliannuol A and heliannuol C, Tetrahedron, 2007, 63, 12026–12036 CrossRef CAS.
- B. Biswas, P. K. Sen and A. Roy, Synthesis of (±)-heliannuol C, Synth. Commun., 2017, 47, 1692–1701 CrossRef CAS.
- S. D. Levine, R. E. Adams, R. Chen, M. L. Cotter, A. F. Hirsch, V. V. Kane, R. M. Kanojia, C. Shaw and M. P. Wachter, Zoapatanol and montanol, novel oxepane diterpenoids, from the Mexican plant zoapatle (Montanoa tomentosa), J. Am. Chem. Soc., 1979, 101, 3404–3405 CrossRef CAS.
- J. Cossy, V. Bellosta and C. Taillier, in Strategies and Tactics in Organic Synthesis, Elsevier, 2008, vol. 7, pp. 59–98 Search PubMed.
- K. C. Nicolaou, D. A. Claremon and W. E. Barnette, Total synthesis of (±)-zoapatanol, J. Am. Chem. Soc., 1980, 102, 6611–6612 CrossRef CAS.
- R. Chen and D. A. Rowand, Total synthesis of (±)-zoapatanol, J. Am. Chem. Soc., 1980, 102, 6609–6611 CrossRef CAS.
- R. C. Cookson and N. J. Liverton, A total synthesis of zoapatanol, J. Chem. Soc., Perkin Trans. 1., 1985, 1589–1595 RSC.
- P. Kocieński, C. Love, R. Whitby and D. A. Roberts, A synthesis of zoapatanol, Tetrahedron Lett., 1988, 29, 2867–2870 CrossRef.
- P. J. Kocieński, C. J. Love, R. J. Whitby, G. Costello and D. A. Roberts, A Total Synthesis of (±)-Zoapatanol and Demethyl-ORF13811, Tetrahedron, 1989, 45, 3839–3848 CrossRef.
- V. V. Kane and L. Doyle, Total synthesis of (±) zoapatanol: a stereospecific synthesis of a key intermediate, Tetrahedron Lett., 1981, 22, 3027–3030 CrossRef CAS.
- V. V. Kane and D. L. Doyle, Total synthesis of (±) zoapatanol, Tetrahedron Lett., 1981, 22, 3031–3034 CrossRef CAS.
- B. M. Trost, P. D. Greenspan, H. Geissler, J. H. Kim and N. Greeves, A Total Synthesis of (+)–2′S,3′R –Zoapatanol, Angew. Chem., Int. Ed. Engl., 1994, 33, 2182–2184 CrossRef.
- C. Taillier, V. Bellosta and J. Cossy, Total Synthesis of Natural (+)-(2′S,3′R)-Zoapatanol, Org. Lett., 2004, 6, 2149–2151 CrossRef CAS PubMed.
- C. Taillier, B. Gille, V. Bellosta and J. Cossy, Synthetic Approaches and Total Synthesis of Natural Zoapatanol, J. Org. Chem., 2005, 70, 2097–2108 CrossRef CAS PubMed.
- H.-Y. Cheng, Y.-S. Lin, C.-S. Sun, T.-W. Shih, H.-H. G. Tsai and D.-R. Hou, Ring-closing metathesis and palladium-catalyzed formate reduction to 3-methyleneoxepanes. Formal synthesis of (−)-zoapatanol, Tetrahedron, 2012, 68, 747–753 CrossRef CAS.
- J. S. Yadav, U. Dash, N. Guguloth and D. K. Mohapatra, Synthesis of the Major Oxepane Segment of Zoapatanol, Helv. Chim. Acta, 2013, 96, 663–674 CrossRef CAS.
- J. A. Codelli, A. L. A. Puchlopek and S. E. Reisman, Enantioselective Total Synthesis of (−)-Acetylaranotin, a Dihydrooxepine Epidithiodiketopiperazine, J. Am. Chem. Soc., 2012, 134, 1930–1933 CrossRef CAS PubMed.
- D. Berger, L. E. Overman and P. A. Renhowe, Enantioselective total synthesis of (+)-isolaurepinnacin, J. Am. Chem. Soc., 1993, 115, 9305–9306 CrossRef CAS.
- D. Berger, L. E. Overman and P. A. Renhowe, Total Synthesis of (+)-Isolaurepinnacin. Use of Acetal-Alkene Cyclizations To Prepare Highly Functionalized Seven-Membered Cyclic Ethers, J. Am. Chem. Soc., 1997, 119, 2446–2452 CrossRef CAS.
- T. Suzuki, R. Matsumura, K. Oku, K. Taguchi, H. Hagiwara, T. Hoshi and M. Ando, Formal synthesis of (+)-isolaurepinnacin, Tetrahedron Lett., 2001, 42, 65–67 CrossRef CAS.
- R. Matsumura, T. Suzuki, K. Sato, T. Inotsume, H. Hagiwara, T. Hoshi, V. P. Kamat and M. Ando, Stereospecific synthesis of α,ω-cis- and α,ω-trans-disubstituted oxepanes, Tetrahedron Lett., 2000, 41, 7697–7700 CrossRef CAS.
- R. Matsumura, T. Suzuki, K. Sato, K. Oku, H. Hagiwara, T. Hoshi, M. Ando and V. P. Kamat, Cyclization of hydroxy epoxides promoted by (Bu3Sn)2O/Lewis acid: efficient synthesis of oxepanes, Tetrahedron Lett., 2000, 41, 7701–7704 CrossRef CAS.
- H. Lee, H. Kim, S. Baek, S. Kim and D. Kim, Total synthesis and determination of the absolute configuration of (+)-neoisoprelaurefucin, Tetrahedron Lett., 2003, 44, 6609–6612 CrossRef CAS.
- V. Sinka, D. A. Cruz, V. S. Martín and J. I. Padrón, Shortest Enantioselective Total Syntheses of (+)-Isolaurepinnacin and (+)-Neoisoprelaurefucin, Org. Lett., 2022, 24, 5271–5275 CrossRef CAS PubMed.
- K. Seo, S. H. Jang and Y. H. Rhee, Sequential Metal Catalysis towards 7−Oxostaurosporine and Its Non–Natural Septanose Analogue, Angew. Chem., Int. Ed., 2022, 61, e202112524 CrossRef CAS PubMed.
- Y. Li, Qinghaosu (artemisinin): Chemistry and pharmacology, Acta Pharmacol. Sin., 2012, 33, 1141–1146 CrossRef CAS PubMed.
- J. Krieger, T. Smeilus, M. Kaiser, E. Seo, T. Efferth and A. Giannis, Total Synthesis and Biological Investigation of (−)–Artemisinin: The Antimalarial Activity of Artemisinin Is not Stereospecific, Angew. Chem., Int. Ed., 2018, 57, 8293–8296 CrossRef CAS PubMed.
- G. Schmid and W. Hofheinz, Total synthesis of qinghaosu, J. Am. Chem. Soc., 1983, 105, 624–625 CrossRef CAS.
- X. Xing-Xiang, Z. Jie, H. Da-Zhong and Z. Wei-Shan, Total synthesis of arteannuin and deoxyarteannuin, Tetrahedron, 1986, 42, 819–828 CrossRef.
- M. A. Avery, C. Jennings-White and W. K. M. Chong, The Total synthesis of (+)-artemisinin and (+)-9-desmethyltemesinin, Tetrahedron Lett., 1987, 28, 4629–4632 CrossRef CAS.
- M. A. Avery, W. K. M. Chong and C. Jennings-White, Stereoselective total synthesis of (+)-artemisinin, the antimalarial constituent of Artemisia annua L, J. Am. Chem. Soc., 1992, 114, 974–979 CrossRef CAS.
- L. Hsing-Jang, Y. Wen-Lung and Y. C. Sew, A total synthesis of the antimarial natural product (+)-qinghaosu, Tetrahedron Lett., 1993, 34, 4435–4438 CrossRef.
- W.-S. Zhou and X.-X. Xu, Total Synthesis of the Antimalarial Sesquiterpene Peroxide Qinghaosu and Yingzhaosu A, Acc. Chem. Res., 1994, 27, 211–216 CrossRef CAS.
- S. P. Cook, Artemisinin: A Case Study in the Evolution of Synthetic Strategy, Synlett, 2014, 751–759 CrossRef CAS.
- V. Vil’, I. Yaremenko, A. Ilovaisky and A. Terent'ev, Synthetic Strategies for Peroxide Ring Construction in Artemisinin, Molecules, 2017, 22, 117 CrossRef PubMed.
- J. S. Yadav, R. S. Babu and G. Sabitha, Stereoselective total synthesis of (+)-artemisinin, Tetrahedron Lett., 2003, 44, 387–389 CrossRef CAS.
- J. S. Yadav, B. Thirupathaiah and P. Srihari, A concise stereoselective total synthesis of (+)-artemisinin, Tetrahedron, 2010, 66, 2005–2009 CrossRef CAS.
- G. Li, M. Lou and X. Qi, A brief overview of classical natural product drug synthesis and bioactivity, Org. Chem. Front., 2022, 9, 517–571 RSC.
- C. Zhu and S. P. Cook, A Concise Synthesis of (+)-Artemisinin, J. Am. Chem. Soc., 2012, 134, 13577–13579 CrossRef CAS PubMed.
- J. Turconi, F. Griolet, R. Guevel, G. Oddon, R. Villa, A. Geatti, M. Hvala, K. Rossen, R. Göller and A. Burgard, Semisynthetic Artemisinin, the Chemical Path to Industrial Production, Org. Process Res. Dev., 2014, 18, 417–422 CrossRef CAS.
- H. Wu, S.-H. He, H.-T. Qin and F. Liu, Photoredox-catalyzed, oxygen-directed unactivated δ-C(sp3)–H functionalization toward oxepanes, Org. Chem. Front., 2023, 10, 3870–3874 RSC.
- M. Kourgiantaki, G. G. Bagkavou, C. I. Stathakis and A. L. Zografos, Selective preparative ‘oxidase phase’ in sesquiterpenoids: the radical approach, Org. Chem. Front., 2023, 10, 2095–2114 RSC.
Footnote |
| † A. R. P. and S. M. W. contributed equally. |
| This journal is © the Partner Organisations 2024 |