
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modern photo- and electrochemical approaches to aryl radical generation
Krzysztof
Grudzień
a,
Andrei
Zlobin
 b,
Jan
Zadworny
b,
Katarzyna
Rybicka-Jasińska†
*a and
Bartłomiej
Sadowski†
*b
b,
Jan
Zadworny
b,
Katarzyna
Rybicka-Jasińska†
*a and
Bartłomiej
Sadowski†
*b
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: katarzyna.rybicka-jasinska@icho.edu.pl
bCentre of New Technologies, University of Warsaw, S. Banacha 2c, 02-097 Warsaw, Poland. E-mail: b.sadowski@cent.uw.edu.pl
First published on 8th August 2024
Abstract
Aryl rings appear in a plethora of natural products and drugs. The vast majority of arylation processes still rely on the utilization of transition-metal complexes as catalysts. Another important strategy for introducing aryl rings into an organic skeleton operates through a radical pathway. The chemistry of aryl radicals has witnessed rapid development during the last few decades, especially as these species are crucial intermediates in many synthetically meaningful organic transformations, such as the Sandmeyer or Pschorr reactions. Predominately, they are prepared employing conventional redox reagents (oxidants or reductants) that are often used in large excess. In turn, recent developments in photocatalysis and synthetic electrochemistry allow for the assembly of Ar˙ species in a more sustainable and straightforward way; however, these advancements have not been timely and critically summarised to date. This review fills this obvious gap and sums up recent advances in processes involving Ar˙ by highlighting some challenges and identifying potential areas for improvement, which might inspire future research endeavors in this topic.
 Krzysztof Grudzień | Krzysztof Grudzień received his undergraduate training and M.Sc. degree in chemistry from the University of Warsaw (UW) (2012) under the guidance of Michał Barbasiewicz, then a Ph.D. degree (2018) under the supervision of Karol Grela at the Biological and the Chemical Research Centre, UW (development of NHC ligands for transition metal catalysis). He is currently a post-doctoral researcher in the Katarzyna-Rybicka Jasińska team at the Institute of Organic Chemistry, Polish Academy of Sciences, working in the field of photoelectrocatalysis. |
 Andrei Zlobin | Andrei Zlobin received his B.Sc. in Medicinal Chemistry from the University of Warsaw, Poland in 2023, performing undergraduate research in Prof. Zbigniew Czarnocki's group. The same year he joined the group of Bartlomiej Sadowski, Ph.D. at the Centre of New Technologies UW (CeNT UW). Andrei's research interests broadly focus on the chemistry of natural compounds and electrochemically mediated organic synthesis. |
 Jan Zadworny | Jan Zadworny studied medicinal chemistry at the College of Inter-Faculty Individual Studies in Mathematics and Natural Sciences at the University of Warsaw. His interests include organic chemistry, electrochemistry, and biochemistry. He is currently studying at the Pomeranian Medical University in Szczecin. |
 Katarzyna Rybicka-Jasińska | Katarzyna Rybicka-Jasińska graduated with an M.S. degree in chemistry from the University of Warsaw (UW) and obtained a Ph.D. in organic chemistry from the Institute of Organic Chemistry Polish Academy of Sciences (IOC PAS) in 2018 under the supervision of Prof. Dorota Gryko. After completing her post-doctoral work at the University of California, Riverside in the group of Prof. Valentine Vullev, she started her independent research as an assistant professor at the Institute of Organic Chemistry, Polish Academy of Sciences. She is currently working in the fields of photoelectrocatalysis and electrosynthesis and their application in modern organic synthesis. |
 Bartłomiej Sadowski | Bartłomiej Sadowski was educated in chemistry at the Warsaw University of Technology, Poland. He then obtained his Ph.D. from the Institute of Organic Chemistry of the Polish Academy of Sciences in 2019 under the supervision of Prof. Daniel T. Gryko. After completing his post-doctoral work at Georg-August-Universität Göttingen in the group of Prof. Lutz Ackermann, he began his independent career at the Centre of New Technologies, University of Warsaw. His research interests focus on the electrochemically enabled synthesis of open-shell intermediates as well as the chemistry of functional aromatic molecules, notably dipyrrolonaphthyridinediones. |
1. Introduction
Radicals are open-shell species that are crucial in the fields of synthetic organic chemistry,1 materials chemistry2 and biology.3 The very first example of the utilization of an aryl radical as an intermediate was reported in 1866 by Griess;4 however, the first real development in radical chemistry in general was initiated in 1900 with a description of the triphenylmethyl radical.5 Since then, many methods for the preparation and synthetic applications of these useful, but specific, intermediates have been developed.6The analysis of the radical stabilization energies7 (RSEs) of a variety of radicals along with bond dissociation energies8 (BDEs) of different C–H bonds (Fig. 1a) leads to the conclusion that the phenyl radical is an exceptionally unstable, thus extremely reactive, open-shell intermediate of transient character,9 with a remarkably short lifetime of about 150 ns regarding its disruptive hydrogen-atom transfer (HAT) process with the solvent (acetonitrile, MeCN).10 Therefore, after its initial formation, it may undergo many follow-up processes, such as hydrogen removal from solvent or other molecules present nearby, halogen transfer or addition to various bonds (Fig. 1b).6a,b However, in situ-formed aryl radicals were successfully applied as mediators in the formation of more stable radicals, finally allowing catalytic functionalisation of amides11 or aliphatic alcohols,12 thus underlining their utility in synthetic organic chemistry.
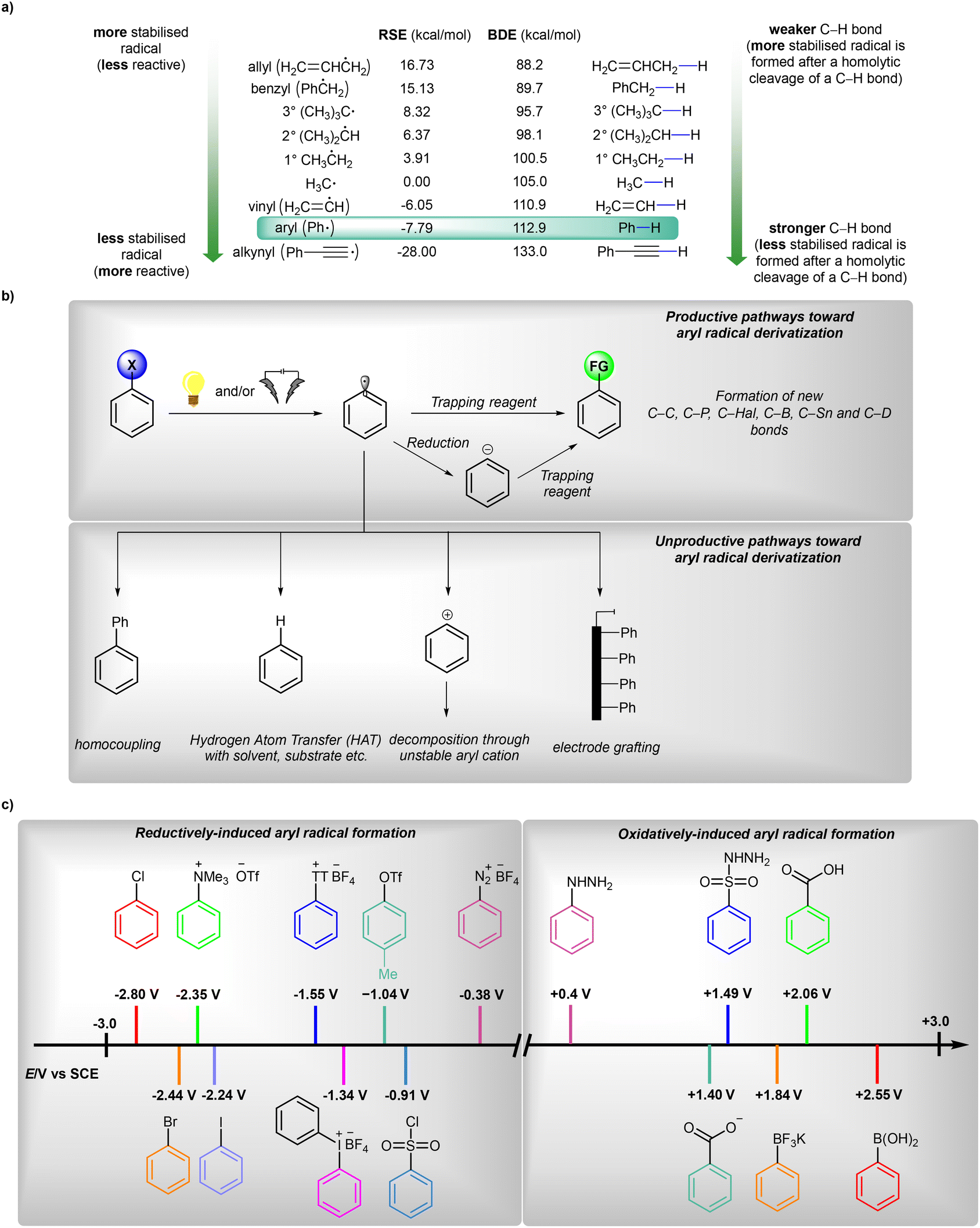 | ||
| Fig. 1 (a) General overview on generation and possible transformations of aryl radicals, (b) radical stabilization energies (RSE) and bond dissociation energies (BDE, given at 298 K) values for phenyl radical and its analogues and (c) comparison of oxidation18 and reduction19 potentials of typical aryl radical precursors. RSE values are given in reference to ˙CH3. | ||
In general, aryl radicals are produced via single-electron transfer (SET) with a variety of precursors (Fig. 1c).6,13 A homolytic cleavage event that leads to the formation of an aryl radical can be induced by conventional (chemical) reagents, visible light (photocatalysis) or electric current (synthetic electrochemistry). Classical methods6,13 for the generation of aryl radicals typically require the use of over-stoichiometric amounts of an oxidant or a reductant (expensive and/or toxic), as well as usually harsh reaction conditions (often elevated temperatures, etc.). While classical oxidative transformations are usually performed employing metal-based oxidants (Mn(III), Mn(VII), Fe(III), Mo(V) etc.), reductive transformations may be also initiated by a combination of an inorganic base and a small organic molecule.14 Along with the terminology proposed by Studer and Curran,15 these transformations can be classified as ‘electron-catalyzed reactions’. These seminal contributions set the stage for most developments within the modern reductive photochemistry.
During the last few decades, visible-light-photoinduced electron transfer methods (e.g. photoredox catalysis) and electrochemistry have enabled a tremendous array of new methods for the preparation of aryl radicals. In this way, these intermediates were successfully employed in C–C, C–B, and C–F bond-forming processes.6,13,16 Although some reviews6,13,17 have appeared recently that partially cover examples presented in our work, none of them evaluate all of the possible precursors of aryl radicals, with special emphasis given to modern photochemical/electrochemical approaches. This article aims to fill this obvious gap.
Herein, we review and analyse recent achievements in the generation of aryl radicals through the modern synthetic methods of photochemistry and synthetic electrochemistry, and their combination, photoelectrochemistry. To realize that intention, the article is divided into two main sections which are dedicated to reductively and oxidatively induced methodologies. Obviously, the terms ‘reductive’ and ‘oxidative’ do not refer to the entire process, but describe the nature of the first electron transfer event relative to a given precursor. Within the abovementioned sections, we showcase differences between synthetic photochemistry and electrochemistry for a given precursor. We believe that this structure of the review will provide a comprehensive overview of modern aryl radical chemistry and possibly stimulate new ideas about how to improve existing methods or even develop other ones, employing other plausible precursors.
2. Photochemistry versus electrochemistry versus photoelectrochemistry
2.1. Photochemistry
In recent years, photochemistry has become a powerful tool for the activation of small molecules through their direct excitation (direct photochemistry) or via energy or electron transfer from the photocatalyst.20 In the case of photon-coupled electron transfer (photoredox catalysis) in the general sense, the modus operandi relies on the fact that photoexcited redox catalysts (PC*) (such as metal complexes, organic dyes, or semiconductors) can simultaneously act as oxidants (reductive quenching catalytic cycle) and reductants (oxidative quenching catalytic cycle) (Scheme 1a). Therefore, photoredox catalysis makes it possible to continuously form small amounts of highly reactive species (radicals, radical ions, and charge transfer (CT) complexes) under mild conditions. The number of applications of photocatalysis grows every year because of the use of very mild conditions for highly selective and environmentally safe reactions. However, this methodology possesses some limitations: (1) challenges in scale-up due to limited light penetration; (2) a limited electrochemical potential window due to the energy limits resulting from the energy of visible photons; and (3) insufficient atom economy due to the addition of stoichiometric amounts of sacrificial reagents (oxidants and reductants). Nevertheless, it has indeed had a huge impact on aryl radical generation methods over the past decade.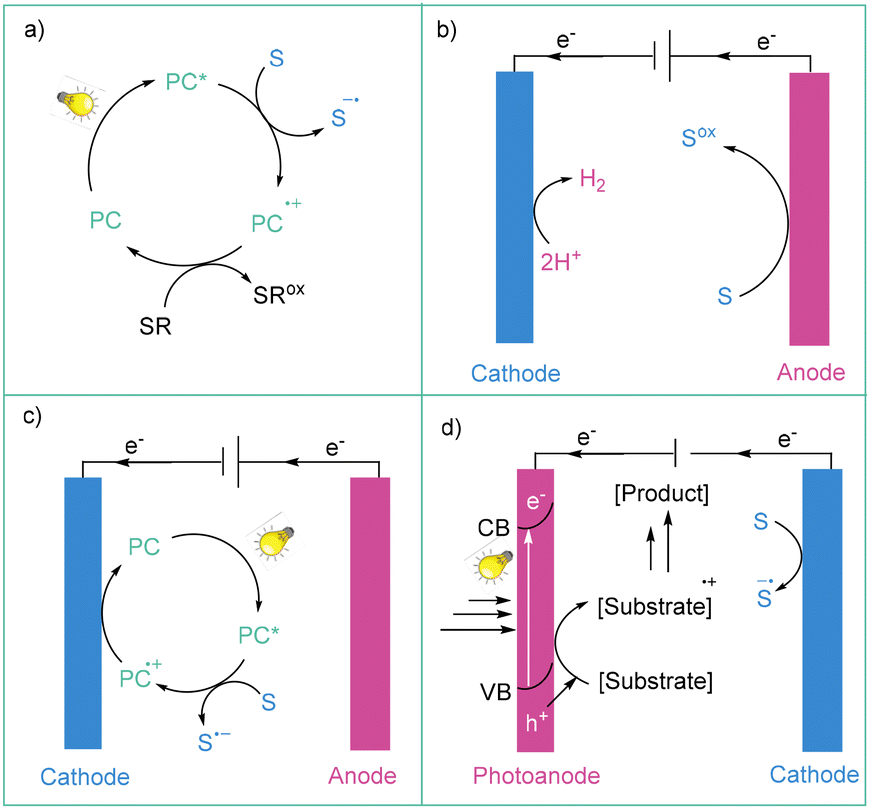 | ||
| Scheme 1 Comparison of (a) photoredox catalysis, (b) electrochemistry, (c) photoelectrocatalysis and (d) photoelectrochemical cells (PECs) for organic synthesis. PC – photocatalyst; S – substrate; SR – sacrificial reagent. | ||
2.2. Electrochemistry
Nowadays, more and more of the electrical energy produced by humans comes from renewable sources, such as wind or solar radiation. In this context, electrical energy has recently started to be considered as a green oxidant or a reductant in terms of synthetic methodology. Mainly due to the postulated green character of electricity, as well as the development of dedicated commercial equipment, the past decade has witnessed a renaissance in electrochemical organic synthesis.17b,21Electrochemistry is one of the most powerful tools for performing electron transfers available to chemists, as its biggest advantages are the ability to strictly control the applied current or potential, as well as the fact that the redox window is in theory only limited by the tolerance of a given solvent.17b,21a,22 More importantly, it constitutes a potent strategy for the generation of open-shell intermediates.21a,23 From the standpoint of redox processes applied toward the formation of aryl radicals, these species can be obtained via reduction at a cathode or oxidation at an anode. To date, most known electrochemical methods for the generation of aryl radicals are based on the electrochemical reduction of a given organic precursor (section 3); however, oxidation-based methodologies are also established in the literature (section 4).
The obvious difference between a given photochemical transformation and an electrochemical one is the presence of electrodes immersed in the solution between which a potential is applied. Additionally, a supporting electrolyte should be added to a cell to facilitate charge flow between electrodes, when the potential difference between them is too high. Importantly, the existence of electrodes in contact with the solution causes the electron transfer between an electrode and a given species in a solution to be interfacial in nature, which undoubtedly has a huge impact on the result/success of a designed electrochemical process. To give an example, for many electrochemical processes, the reaction rates are strongly dependent on the applied currents, indicating that a turnover-limiting electron transfer is operative.24 Therefore, the heterogeneous character of the electron transfer may affect the mechanistic aspects of the activation of a given precursor of aryl radicals.
Other major problems that chemists may encounter during an electrolysis toward aryl radicals are: (1) possible formation of phenyl anions (E°(Ph˙/Ph−) = 0.05 V vs. SCE)10a – particularly in the case of reductively induced methods – and (2) the possibility of electrografting.25 Still, many electrochemically based transformations toward aryl radicals were developed that produce these open-shell species in a selective and efficient manner (vide infra). Specifically, there are several experimental techniques that enable the efficient formation and transformation of aryl radicals. These include: (1) applying large excess (often >5 equivalents) of another reactant in order to immediately trap aryl radicals; (2) the presence of an electron mediator (a transfer of electrons occurs in a bulk solution, away from the electrode); (3) aryl radicals are trapped by a multiple bond within the same molecule in intramolecular radical cyclization reactions.
2.3. Photoelectrochemistry
Photoelectrochemistry is a constructive combination of both of the abovementioned methods (photochemistry and electrochemistry) (Scheme 1c). This merging seems to be the answer to the challenges of these two methods. From the point of view of aryl radical generation, two photoelectrochemical approaches can be involved: electrochemically-mediated photoredox catalysis (ePRC) and interfacial photoelectrochemistry (iPEC).26 Electrochemically-mediated photoredox catalysis (ePRC) can operate via two different strategies: photoexcitation of electrochemically generated ions where, for example, a photocatalyst can be firstly oxidised (or reduced) at the electrode and then get excited to the excited state, forming a super-oxidant (or a super-reductant); or by replacing sacrificial oxidants/reductants with the current (Scheme 1c).27 In the case of iPEC, the most commonly used systems are based on two electrodes (cathode and anode), and one of them is a photoelectrode, which is coated in a photoresponsive material (typically a semiconductor). A semiconducting photoelectrode is exposed to visible light and generates electron–hole pairs that are used to drive a redox reaction. In such a system, the applied potential followed by the irradiation promotes an electron (e−) from the valence band (VB) to the conductive band (CB), generating a hole (h+), which is used for oxidation, and the flow of electrons in the CB, which is used for the reduction (Scheme 1d). Photoelectrochemical cells are widely studied for solar energy conversion; however, their use in organic synthesis is relatively underexplored.26,27It should be emphasized that photoelectrochemical methods operate within low reduction or high oxidation potentials, concomitantly avoiding over-reduction to aryl anions or over-oxidation to aryl cations of aryl radicals. This is because an electron transfer event between PC*+/PC*− and substrate occurs in a bulk solution, that is, away from an electrode, therefore avoiding further reduction to the anion.
3. Reductively induced generation of aryl radicals
The formation of aryl radicals can generally be induced by a single-electron reduction of a given radical precursor. This can be realized by employing three different methodologies: photoinduced electron transfer (photoredox catalysis or through EDA complex formation and it subsequent reduction), organic electrochemistry28 and photoelectrochemistry.26,29 The reductively induced generation of aryl radicals is so far possible for aryl halides, aryl diazonium salts, sulfonyl chlorides, sulfonium salts, iodonium salts, aryl triflates and others (Scheme 2). The aforementioned precursors intrinsically differ in their ability to accept an electron, whereas charged species tend to be reduced much more easily than neutral molecules.30 The reduction of charged precursors depicted in Scheme 2, however, is characterised by lower atom economy due to the liberation of higher-weight molecules, such as NR3, ArI or R2S.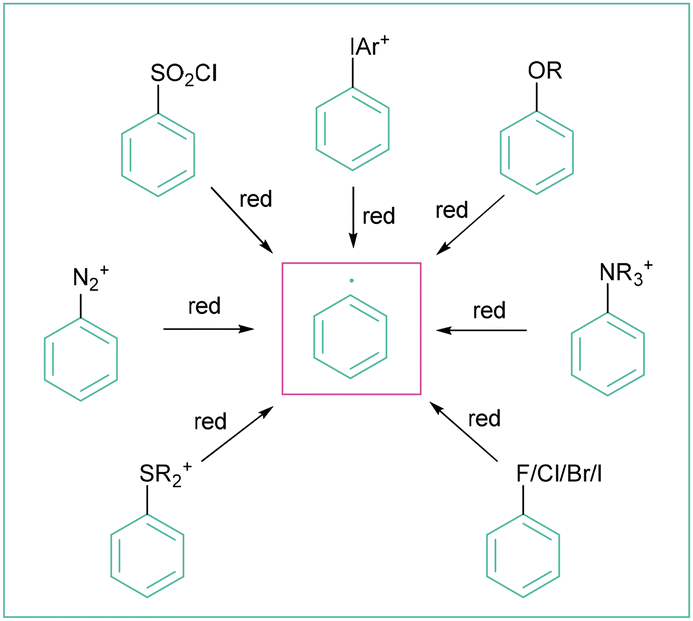 | ||
| Scheme 2 Reductive generation of aryl radicals from different precursors. | ||
3.1. Generation of aryl radicals from aryl halides
Aryl halides, although they are bench stable and readily available commercially and synthetically, have low reduction potentials (up to −2.8 V vs. SCE for electron-rich chlorides), which severely limits the application of photocatalytically generated aryl radicals. The most common applications of radicals generated photochemically from aryl halides include hydrodehalogenation, dehalogenative cyclization, intermolecular (hetero)arylation, phosphorylation, or borylation. Hereby, we present selected examples of pioneering and recent transformations of this type.Less active halides, including chlorides, often require nonstandard aryl radical generation strategies, which were recently summarised in an elegant review paper by Giedyk, Wenger, and Lu.33
As an example, aryl chlorides and fluorides were activated toward aryl radicals by employing indole thiolate organocatalyst, which upon excitation with 405 nm light, achieves strongly reducing properties.34 Using this strategy, dehalogenation, Birch reduction, phosphorylation and borylation were achieved featuring broad substrate scopes and moderate to good efficiency.
In 2014, one of these approaches to overcome the high potential barrier was based on a two-photon excitation process (sometimes also called ‘consecutive photoinduced electron transfer’, ConPET, see: Scheme 3)35 as seminally conceptualized by König.36 The strategy is based on a single photocatalyst (PC), in this case commercially available N,N′-bis(3-pentyl)perylene-3,4,9,10-bis(dicarboximide, PDI, 1), that is first excited by visible light (PC*) and then quenched by Et3N to give an anion radical (PC˙−), which is excited again, resulting in a highly reduced coloured anion radical (*PC˙−) which can transfer an electron to aryl iodides, bromides and some activated chlorides. The liberation of the halide anion results in an aryl radical that was subjected to a HAT from excess Et3N to give a hydrodehalogenation product or coupled with the pyrrole derivative when a radical trap was added in a very large excess (25–50 equiv.). A similar approach was later reported with the use of other photocatalysts (anthraquinone),37 9,10-dicyanonthracene (DCA),38 heteroleptic biphosphine/phenantroline Cu(I) complexes,39MesAcrBF4,40 and 2,4,5-tri(9H-carbazol-9-yl)-6-(ethyl(phenyl)amino)isophthalonitrile, 3CzEPAIPN19a.
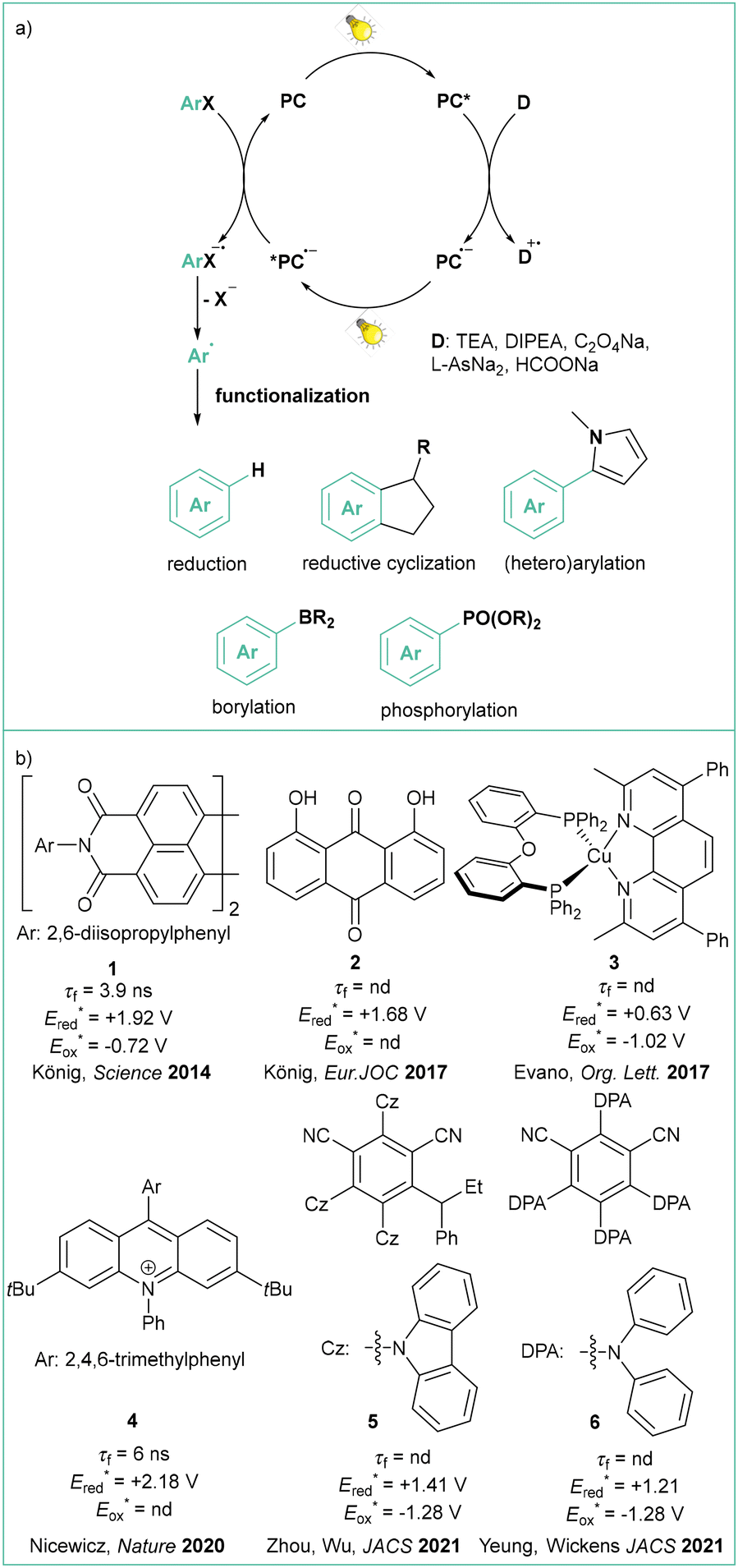 | ||
| Scheme 3 (a) General mechanism of ConPET in relation to the synthesis and functionalisation of aryl radicals from aryl halides and (b) selected photocatalysts studied in the ConPET reaction with aryl halides. Redox potentials are given vs. SCE. | ||
In the latter case, the authors successfully employed oxalate and ascorbate as inexpensive and environmentally friendly sacrificial electron donors. Gram-scale borylation of 4-chlorotoluene was easily achieved by employing more intensive light irradiation and a longer reaction time. Independently, Yeung and Wickens developed a very similar system using 1,3-dicyano-2,4,5,6-tetrakis(diphenylamino)-benzene (4DPAIPN, 6), 405 nm light irradiation, and sodium formate as an electron donor.41 Besides typical dehalogenation, borylation, and phosphorylation (photo-Arbuzov) of aryl chlorides (with redox potential as low as −3.4 V vs. SCE), the authors also carried out successful hydroarylation of unactivated olefins. Of note, the reaction was viable with tert-butylvinylcarbamate, which gives access to the arylethylamine pharmacophore scaffold. Shortly afterwards, Jui et al. published a similar methodology for iodides and activated chlorides employing phenothiazine as photocatalyst.42 Very recently, Gierschner, Wannemacher, and Kwon have studied the steric and photophysical parameters of popular cyanoarene catalysts including their related photodegradation processes (Scheme 4).43 Careful investigation led to the development of systems capable of complete hydrodehalogenation of 4-bromobenzonitrile using as low as 0.005 mol% catalyst loading of 4DPAIPN under visible light irradiation (455 nm). At 0.05 mol% loading, the catalyst enabled facile gram-scale dehalogenation without degassing the reaction medium, which underlines its abnormally high oxygen tolerance.
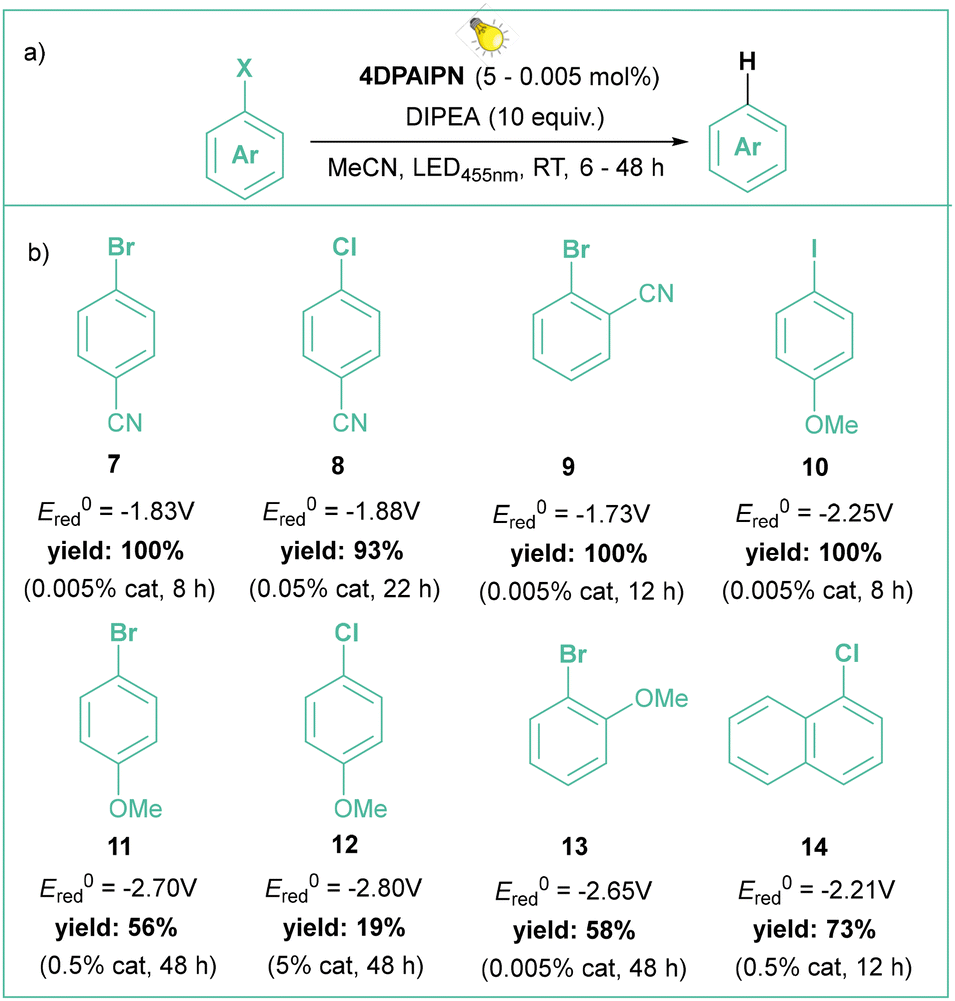 | ||
| Scheme 4 Hydrodehalogenation of aryl halides catalyzed by photoexcited 4DPAIPN: (a) model reaction and (b) representative products. | ||
Before the study of Yeung and Wickens was published, the ConPET mechanism was somewhat challenged by a similar examination of in situ-generated PC˙−. The authors noted very short excited lifetimes of DCA analogues that would make them rather poor photocatalysts.44
Nocera studied the excited state of the radical anion produced from naphthalenemonoimide NpMI, proposing that NpMI˙− is nonemissive and it rather derives from a Meisenheimer complex that is an active form of photocatalyst.45
In 2023, Scott's group managed to synthesize, isolate and fully characterize (including EPR and XRD) stable analogues of NpMI˙− and DCA˙− by reducing appropriate dyes with KC8 in the presence of a cryptand. Based on a series of simple experiments they proved their photostability and demonstrated that they are indeed active catalysts in visible light ConPET dehalogenation and phosphorylation of various aromatic chlorides, including electron-rich ones.46
In recent years, several groups found direct spectroscopic evidence for the thus far elusive pre-association of radical ions and substrate molecules that enables photoinduced electron transfer beyond the diffusion limit.47 These reports may finally end the discussion about whether excited radicals with picosecond lifetimes can be catalytically active in ConPET processes.
An interesting example of ConPET, reported by Jing, used dicyanoanthracene (DCA) that was incorporated within a metal-organic framework (MOF).48 The study proved that the ConPET was essential to form charge-transfer species and accelerate the conversion of interesting aryl halides (including relatively inactive species such as 1-chloro-4-methoxybenzene (Ered = −2.90 V vs. SCE) in moderate to good yields). Upon generation of the aryl radical using visible light, it was subsequently quenched with N-methylpyrrole and B2pin2 as radical traps. A similar incorporation of PDI into MOFs with further use in ConPET was performed by Duan and co-workers; however, it was limited to activated aryl chlorides that were employed in dehalogenation and heteroarylation reactions.49 Another way to overcome the high potential barrier of these challenging substrates is to use metal-free carbon dots (B-CDots) as photocatalyst.50 Namely, Yu and Huang achieved borylation of various aryl chlorides, iodides and bromides with very good to excellent yields. The authors proposed the aryl radical formation as a key step. Upon light irradiation B-CDots act as semiconductors – the electrons from the conductive band are able to reduce aryl halides toward radical anions followed by subsequent extrusion of a halide anion, eventually giving rise to aryl radicals that further react with a given boron source.
The application of photocatalyzed intramolecular aryl radical cyclizations toward the synthesis of polycyclic indolyl heterocycles was reported by Che and colleagues (Scheme 5a).51 Direct irradiation of model N-(2-chlorobenzoyl)indole with visible light (410 nm) in the presence of DIPEA (2.5 equiv.), which acts as a sacrificial electron donor, led to a yield of 76% for the Heck-type intramolecular reaction product. Variously substituted indoles were tested, and challenging reactions such as the use of aryl chlorides or the formation of quaternary carbon centres were indeed viable. This method has a high tolerance to functional groups, such as esters, amides, alcohols, or allyl moieties, and has been successfully applied in the synthesis of a number of complex natural product analogues (Scheme 5b). The use of UV light (365 nm) resulted in improved performance. According to the proposed mechanism (Scheme 5c), a long-lived excited state B generated from A undergoes a bimolecular reaction with an electron donor resulting in the formation of the anion radical C, which then undergoes intramolecular C–Cl bond activation to give the aryl radical D and Cl−. An intramolecular radical cyclization leads to the benzylic radical E, which then undergoes hydrogenation via a HAT or reduction/protonation to give the final product F. Abstraction of a bromide anion leading to aryl radicals that can further undergo intramolecular cyclization using diazaphospholene organocatalysts was recently reported independently by Cramer52 and Speed53 (Scheme 6a). The former approach relies on HBpin and DBU with SPO as a precatalyst, while the latter case uses PhSiH3, Et3N and Cs2CO3; in both cases visible light is required to regenerate the active DAP radical (DAP˙) (Scheme 6b). The reaction mechanism (Scheme 6c) is based on the phosphalene cycle in which DAP˙ abstracts the bromine atom from the substrate leading to aryl radical B and DAP-Br that undergoes 5-exo-trig cyclization to the radical C. DAP-Br is converted to DAP-Hvia a reaction with a hydrogen donor (either PSiH or HBpin). DAP-H acts as a HAT reagent that converts C to the final product D, regenerating DAP˙ at the same time.
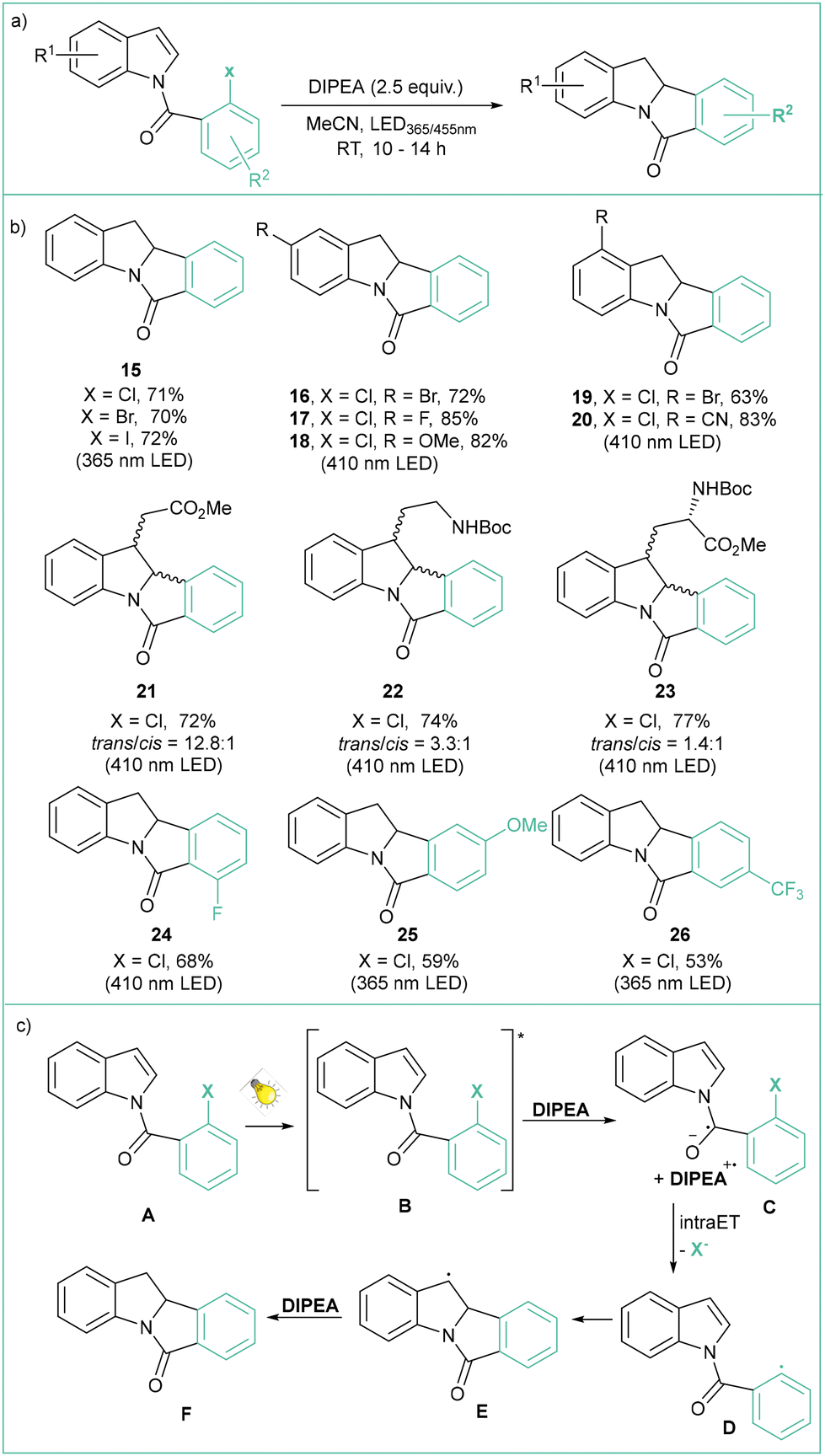 | ||
| Scheme 5 Photocatalyzed synthesis of polycyclic indolyl heterocycles: (a) model reaction, (b) representative products and (c) mechanistic proposal. | ||
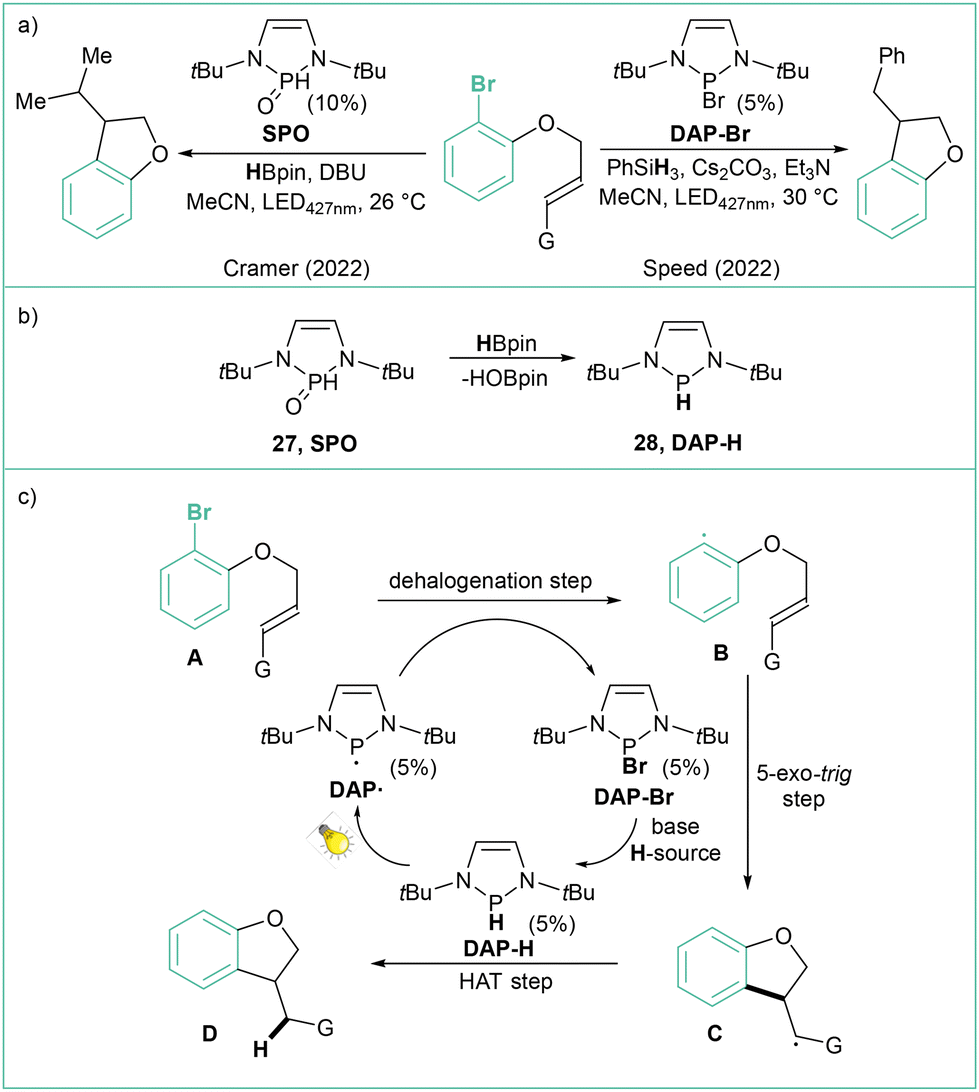 | ||
| Scheme 6 Approaches for intramolecular reductive cyclization using diazaphosholenes: (a) model reactions; (b) SPO precatalyst activation and (c) mechanistic proposal. | ||
Aryl radicals can also be generated via photoexcitation of an EDA complex and subjected to intermolecular transformations, as was recently presented by Xia (Scheme 7a).54 Namely, C–C cross-coupling of oxindoles and aryl halides was achieved under visible-light irradiation using CsOH as a base. The reaction of the aryl radical generated through a SET process from an excited EDA complex (A) leads to an anion radical B followed by loss of I− toward aryl radical C. Upon addition of C to D, the adduct E is formed which eventually gives the product G through a SRN1-type reaction55 (Scheme 7b).
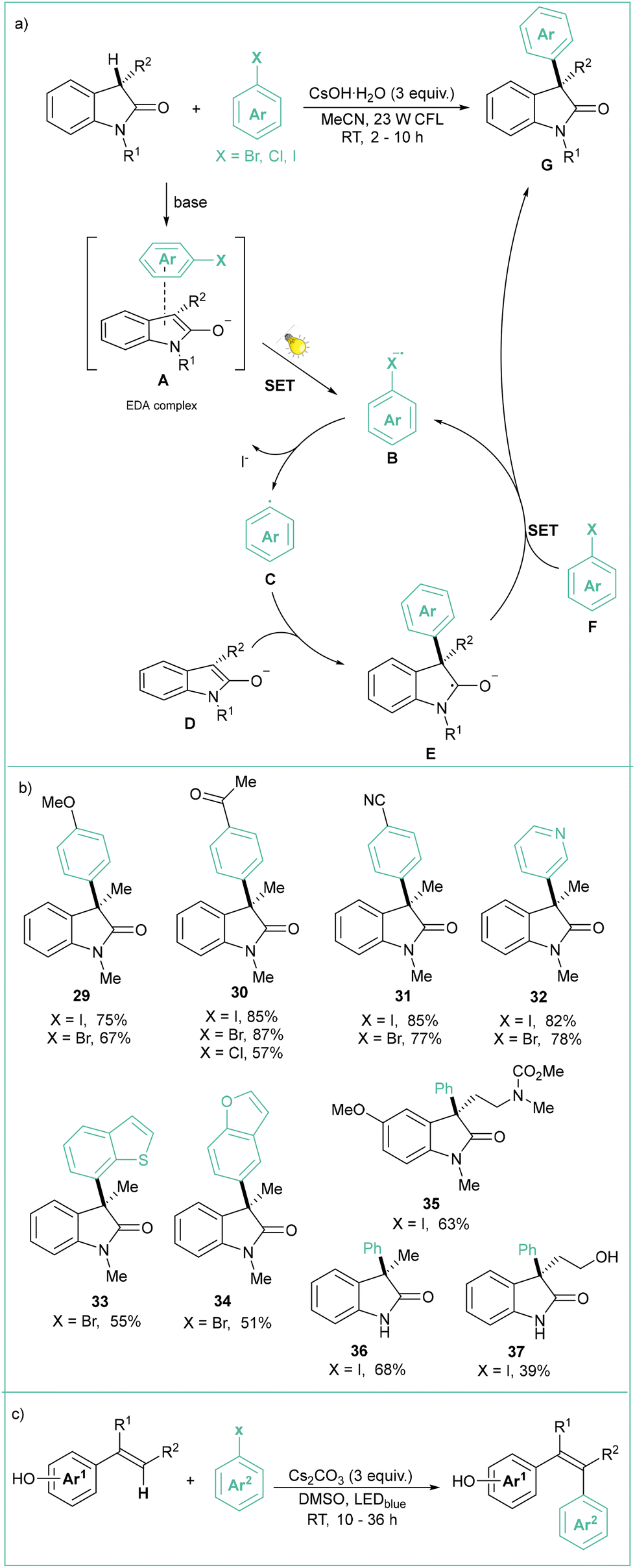 | ||
| Scheme 7 Photoarylation of oxoindoles: (a) model reaction with mechanistic proposal, (b) representative products and (c) photoarylation of vinylphenoles. | ||
Shortly thereafter, the authors extended the scope of this reaction by testing a wide range of vinylphenol derivatives (Scheme 7c).56 The protocol in general is very stereoselective (E/Z ratio 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and higher) and found utility in the late-stage functionalisation of complex natural products.
:1 and higher) and found utility in the late-stage functionalisation of complex natural products.
Recently, Larionov et al. developed a family of very potent SET photocatalysts based on simple, abundant and low molecular weight phenothiazines (Scheme 8).57 These catalysts can generate aryl radicals from a number of halides, including inactivated bromides and chlorides, which were further converted into various boronic esters and trifluoroborates, with a wide scope of functional groups tolerated and under visible light irradiation (Scheme 8b). The authors suggest that the reaction proceeds through a proton-coupled electron transfer (PCET) mechanism, where base-sensitive secondary ammonium catalysts PTH-1 and PTH-4 are more active than tertiary analogues PTH-2 and PTH-3 (Scheme 8a).
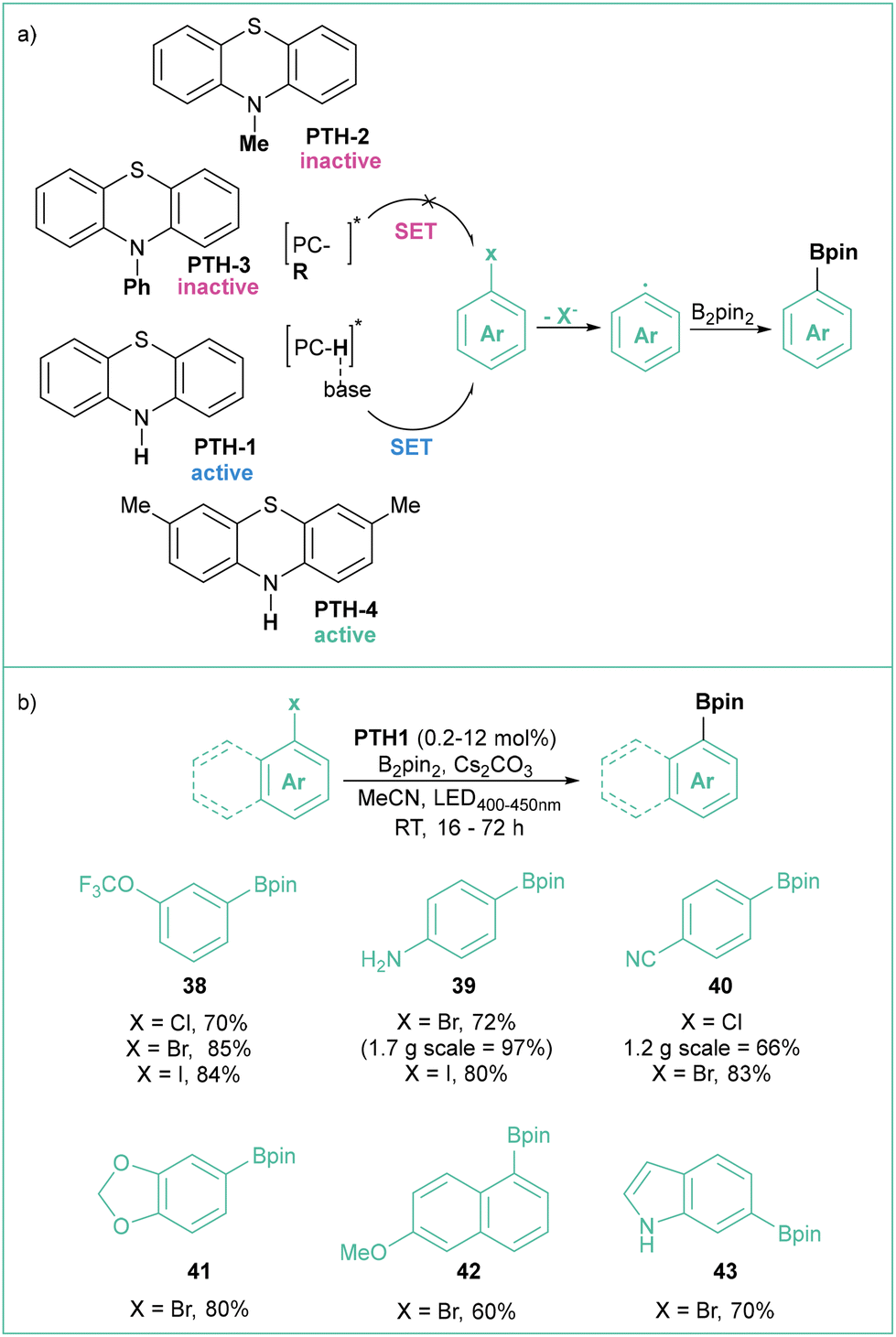 | ||
| Scheme 8 (a) Difference in the photocatalytic activity of phenothiazines in the generation of aryl radicals rationalized by the PCET mechanism. (b) Photoborylation of aryl halides using PTH1, model reaction, and representative products. | ||
Bakr and Reuping presented an interesting example of the generation of aryl radicals through photoexcitation of an intermediate in the photo-Ullmann reaction catalyzed by a Cu nanocluster [Cu61NC] (Scheme 9a).58 According to the proposed mechanism (Scheme 9b), upon deprotonation of carbazole, a Cu61-carbazolide complex [Cu61NC]-Cz forms, that is activated under visible light irradiation (λabs = 412 nm), in striking contrast to an earlier example of the photoUlmann reaction presented by Fu59 where higher-energy ultraviolet light was used for activation of the catalyst-nucleophile intermediate complex. A SET from the excited carbazolide-copper cluster [Cu61NC]-Cz* (via blue LED irradiation) to aryl halide generates the oxidised complex [Cu61NC]+-Cz, an aryl radical and a halide anion. This was rationalized by fluorescence and lifetime quenching experiments. Then, the radical interacts with the oxidised cluster giving rise to C(arene)–N(carbazolide) bond formation via a radical-radical bound or reductive elimination pathway, as well as regeneration of the original [Cu61NC]. The nucleophile scope includes carbazoles, indoles, and azaindole motifs. Moreover, electron-poor and electron-rich iodides, bromides and chlorides are well tolerated in this transformation (for selected examples, see Scheme 9c). The yields of the C–N coupling range from moderate to high, ranging from 48 to 87%, with 0.1 mol% catalyst loading, which translates to 6.1% clustered copper in the reaction medium.
 | ||
| Scheme 9 Photo-Ullmann reaction catalyzed by a copper(I) nanocluster: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
 | ||
| Scheme 10 (a) Commercial availability of different arene sources (according to SciFinder, accessed April 2, 2021) and reduction potentials of PhCl, PhBr and PhI as reported by Zhou, Wu and others;19a (b) general scheme for the direct electroreduction of aryl halides and (c) general scheme for the indirect electroreduction of aryl halides. | ||
Aryl radicals that are produced from ArXvia either a direct or indirect process may subsequently react with various trapping species such as B2pin2, O2, or H˙ donor. If the reaction system allows, the radicals can be reduced immediately to aryl anions (Scheme 10c) that are then trapped by relevant electrophiles such as CO2,63 an electrophile64 or even H+ donor.65
Direct electrolysis of aryl halides is rarely used, as such challenging conditions may be harmful to many functional groups. As an example, the introduction of the OH group is a particularly relevant method as phenols are important species that are present in many biologically relevant molecules.66 In the method developed by Ke et al.67 (Scheme 11), O2 from the air was the source of the hydroxyl function whilst a non-sacrificial anode was used. A wide range of phenols was assembled in an undivided cell employing water as a green solvent. The electrolyses occurred with good to excellent efficiencies (up to 95%) employing aryl chlorides, bromides or iodides.
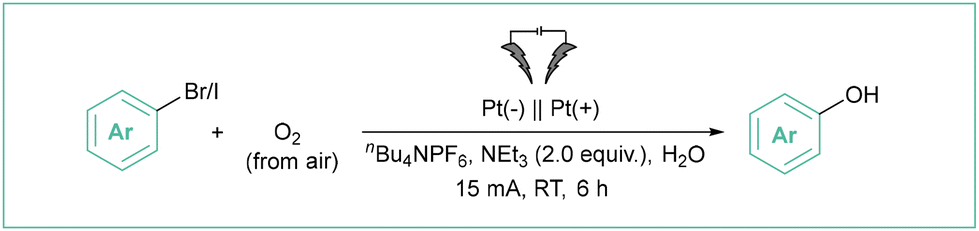 | ||
| Scheme 11 Hydroxylation of aryl halides via a direct electrolysis. | ||
In 1991, Savéant and colleagues studied the electroreduction of aryl halides in the presence of olefinic acceptors (styrene, butylvinylether or acrylonitrile).68 They found that the addition of some organic compounds (4,4′-bipyridine, naphthonitrile or methyl benzoate) significantly increases the efficiency of the hydroarylation reaction. Benzonitrile or m-tolunitrile were found to effectively mediate the electron transfer from a Pt-based cathode toward aryl halides in the preparation of 1-methylindans via an intramolecular cyclization of o-(3-butenyl)phenyl.69 In turn, methyl 4-tert-butyl benzoate appeared to be an appropriate mediator for the assembly of 2,3-dihydrobenzofuran-3-ylacetic acids starting from olefins (Scheme 12).70 Despite the rather narrow scope, the method constitutes an interesting example of employing aryl radicals in radical cascades, where the formed alkyl radical (via intramolecular addition of Ar˙ to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond) is reduced to a carbanion which then attacks an electrophilic CO2 molecule, giving rise to carboxylic acid derivative.
C double bond) is reduced to a carbanion which then attacks an electrophilic CO2 molecule, giving rise to carboxylic acid derivative.
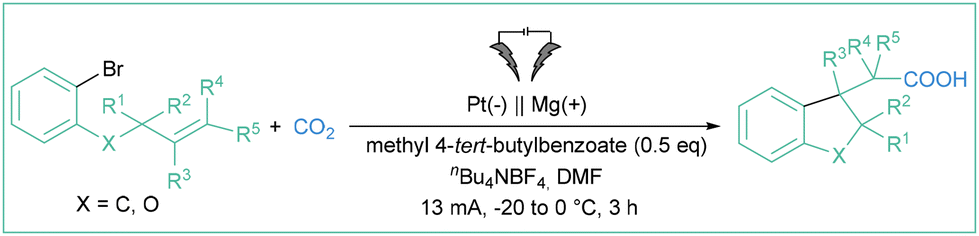 | ||
| Scheme 12 Synthesis of 2,3-dihydrobenzofuran-3-ylacetic acids described by Senboku et al. | ||
Direct arylation of pyrroles can be realized by indirect electroreduction of aryl halides bearing electron-withdrawing groups such as –COMe, –CO2Me or –CN.71 In this case, a perylene bisimide dye was used as an electron mediator.
Some other similar contributions underlined the importance of polycyclic aromatic hydrocarbons (PAHs, such as phenanthrene72 or fluorene derivatives73) as electron mediators in such cyclizations where the electroreduction of aryl halides constitutes an initial step. In 2022, Brown's group elegantly showcased the power of phenanthrene as an electron mediator in the assembly of cyclic derivatives (Scheme 13) by performing this transformation in flow.74 Here, aryl chlorides, bromides and iodides are compatible, along with substrates bearing easily reducible groups. Importantly, the developed conditions allow for the formation of rings of various sizes including 5- or 6-membered ones, or even spirocycles. The major difference between Brown's work and the aforementioned articles is the use of cost-effective electrodes and flow conditions, which should have positive impacts on the resource and energy economy of this process.
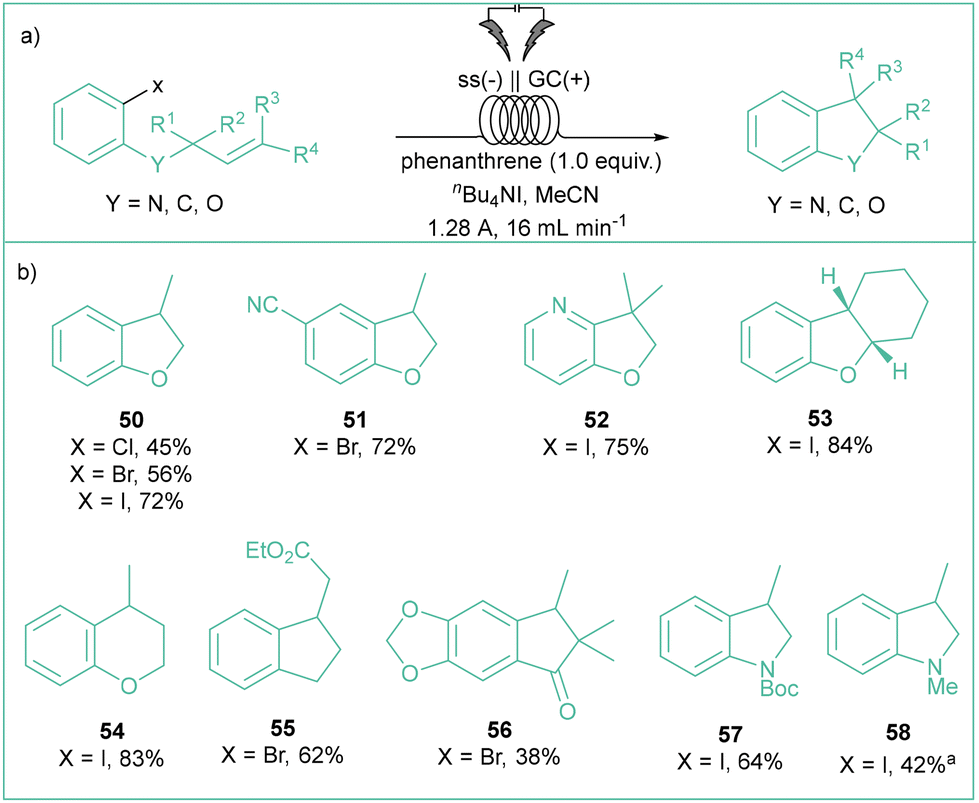 | ||
| Scheme 13 Synthesis of cyclic products xx in flow developed by Brown and colleagues in 2022: (a) model reaction and (b) representative products. aReaction carried out on 7.0 mmol scale in THF/MeCN (7:1). | ||
A new type of electron mediator, which is an analogue of cumulene (Ph4-5-CML, 59), was proposed by Milner and others to mediate the borylation of aryl halides in a potentiostatic regime (Scheme 14).75 The reaction tolerates a broad range of aryl halides (products 60–67) as well as different boron-based reagents (products 64–65). Despite the high reducing potentials needed for the ArX activation, all processes occur smoothly at relatively low cathodic potentials (Ucell = 2.5 V, approximately −1.9 V vs. Ag/AgCl) in comparison with those typically required for electrophotocatalytic or mediator-less electrochemical reactions. It should be emphasized here that even though the applied potential (−1.9 V) is way lower than the potential for aryl radical reduction (E°(Ph˙/Ph−) = 0.05 V vs. SCE),10a the formation of Ar− is avoided by means of mediated electrolysis – that is, the reduction occurs in bulk solution, not near the cathode surface. This constitutes a general advantage of mediated electrolysis over direct electrolysis that should be taken into account in the designing of new (photo)electrochemical synthetic methods based on aryl radical chemistry.
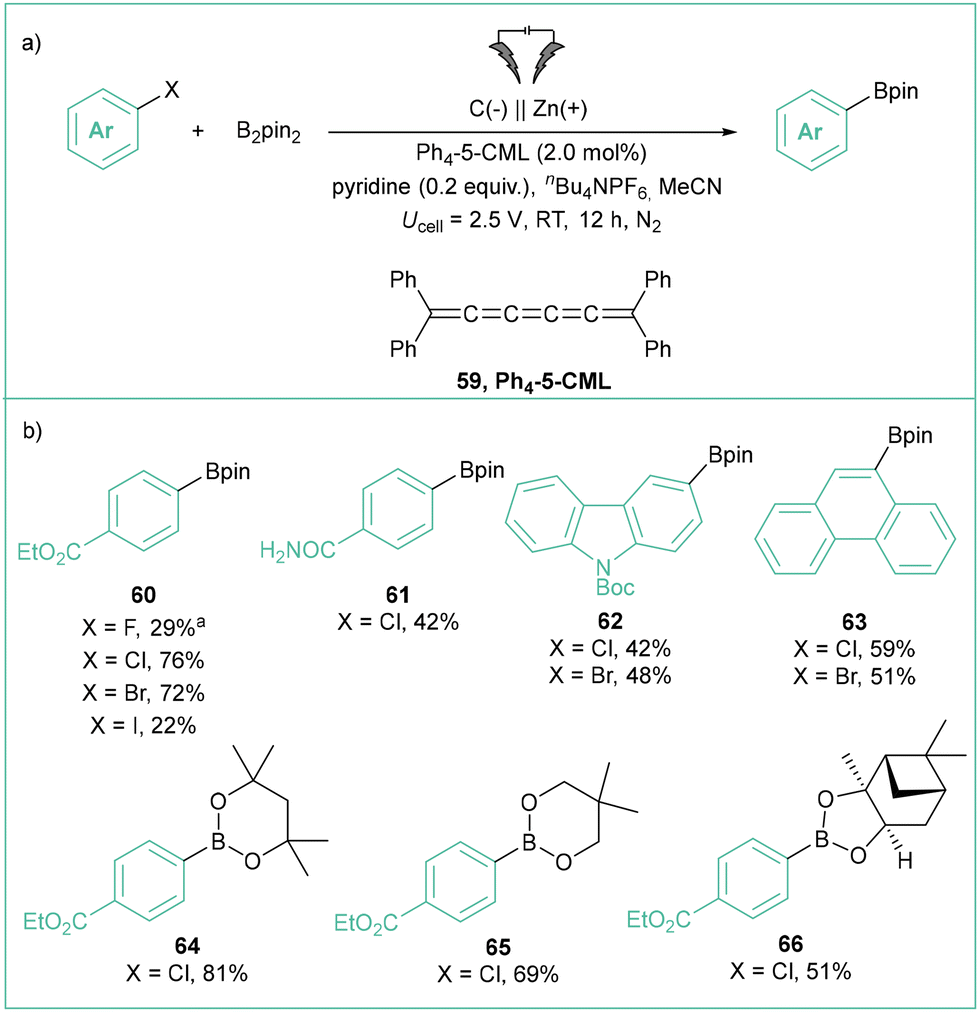 | ||
| Scheme 14 Employment of cumulenes as electron-mediators in borylation of aryl halides: (a) model reaction and (b) representative products. | ||
77) and is compatible with various aryl halides (Cl, Br).
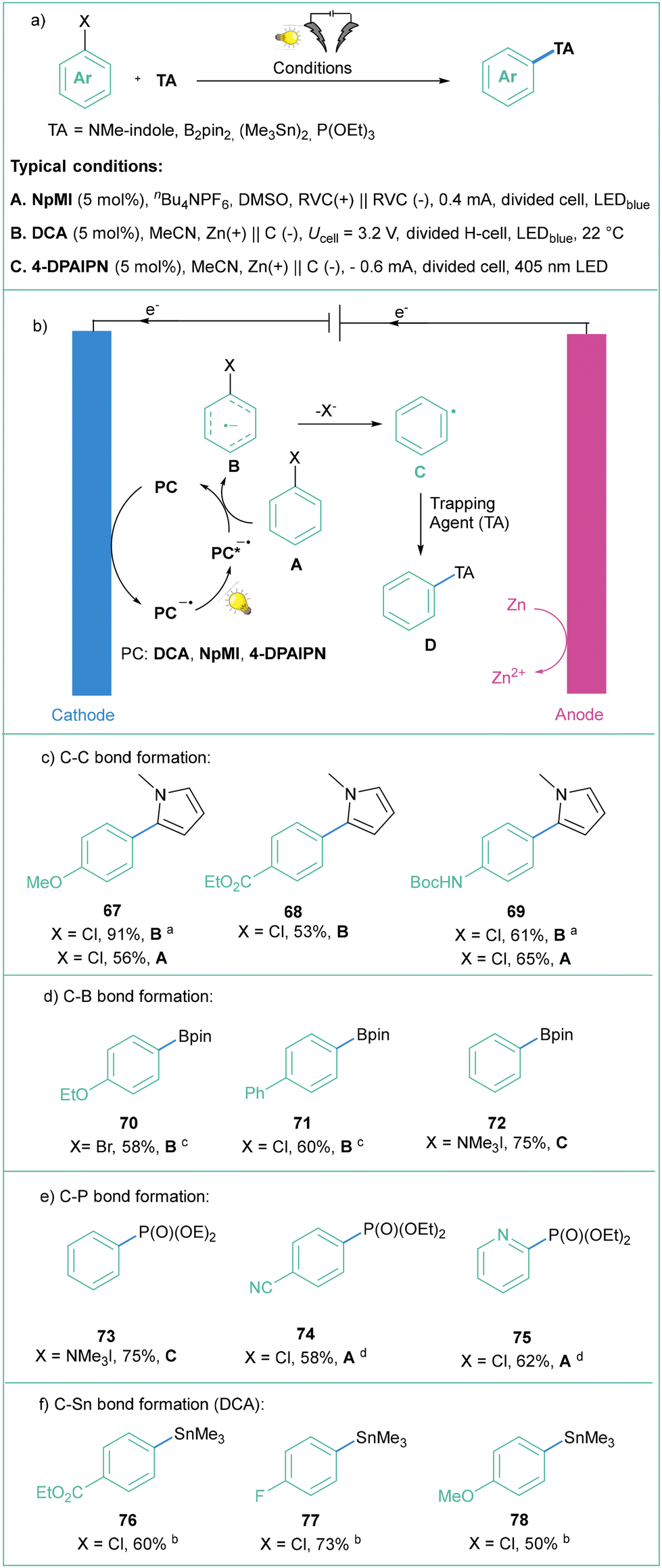 | ||
| Scheme 15 Photoelectrochemical generation of aryl radicals and subsequent C–C, C–B, C–P, and C–Sn bond-forming reactions. a10 mol% of DCA was used. b1HNMR is given. cPyridine (20 mol%) was used. dConstant current electrolysis at 0.8 mA was employed. | ||
On the other hand, Wu and co-workers reported an iPEC/photoredox system consisting of a Sb2(S,Se)3 photocathode and a PDI dye as a photoredox catalyst that was employed for very efficient and selective aryl functionalisations (Scheme 16).79 The proposed mechanism for this transformation assumes that after light irradiation (Vis-NIR), the photocathode gets excited and an electron-hole pair is generated (Scheme 16a). The photogenerated electrons from the conductive band reduce PDI to a radical anion (PDI˙−), which is then excited to form a highly reductive species (PDI*˙−). The photoexcited radical anion (PDI*˙−) is now able to reduce different aryl halides A and B, which subsequently undergo mesolytic cleavage to generate aryl radical C, which reacts with trapping reagents to yield the functionalised product D (Scheme 16b). Such an approach allows for the functionalisation of different (hetero)aryl halides in good to excellent yields, with many functional groups being tolerated (–CN, –CHO, –CO2Me, –COMe). On the other hand, the reaction is also efficient with different trapping agents such as pyrrole derivatives, P(OEt)3 and B2pin2 (Scheme 16c). The application of a dual system with a photocathode and a photoredox catalyst allows the production of high-value products in good yields. However, the same transformation can be performed in more simple way and without the use of the toxic antimony-covered photocathode (see Scheme 16). Moreover, the authors reported neither quantum efficiency measurements nor reusability studies for the Sb2(S,Se)3 photoelectrode, while a high-energy Xe lamp (500 W) was used as a light source. It is worth mentioning that the reaction still proceeds without electricity, albeit with lower efficiency (57% for compound 86).
 | ||
| Scheme 16 iPECs for the photoelectrochemical generation of aryl radicals: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
3.2. Generation of aryl radicals from diazonium salts
In comparison with aryl halides, aryl diazonium salts seem an even better source of aryl radicals due to their much lower redox potential and irreversible liberation of a nontoxic nitrogen molecule. Although they are certainly less stable than their respective halide analogues, they can be stored at low temperatures with good stability as long as a non-coordinating counterion, such as BF4, PF6 or OTs, is being used. They are also quite simple and cost-effective reagents to synthesize starting from abundant anilines and are often generated in situ.To give a recent example, Boixel, de Rouville, and Soulé developed a catalyst-free (or self-catalyzed) photoinduced arylation of N-substituted acridinium salts using various aryl diazonium tetrafluoroborates with high functional group tolerance (halide, ester, –CN, ketone and –NO2) (Scheme 17a).81 The synthesized acridinium salts were evaluated for their photoredox catalytic capabilities in the C–O bond fragmentation of ethers. The generation of the respective diol products was efficient, particularly when using the C9-(2-bromophenyl)-N-methyl dye, which was even able to outperform the commercial Fukuzumi catalyst for the reaction (Scheme 17b).
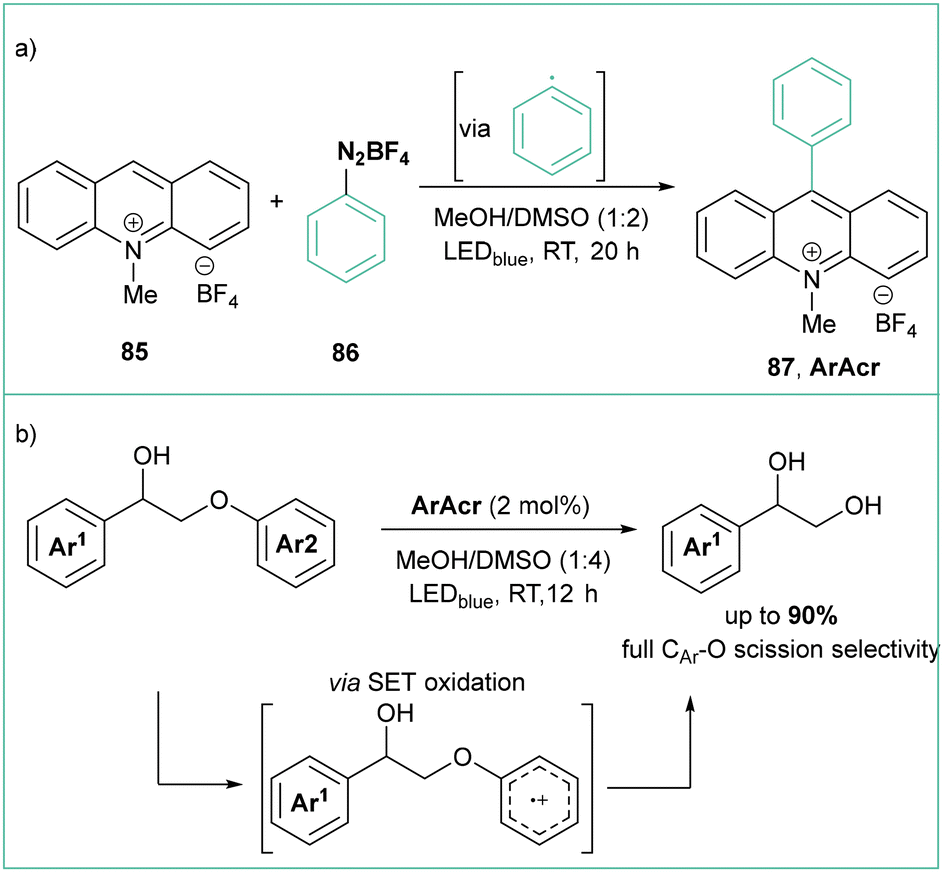 | ||
| Scheme 17 (a) Photoinduced arylation of N-methylacridinium salts to give new families of ArAcr photocatalysts. (b) Application of ArAcr in selective C–O bond fragmentation. | ||
The photoredox version of the classical Sandmeyer-type halogenation was realized by Tang and Luo (Scheme 18).82 They reported a facile conversion of anilines into acetates through the photoinduced generation of (hetero)aryl radicals using an excess of t-BuONO and simple diacetyl as a photosensitizer/trapping agent (Scheme 18a). More than 40 (hetero)aryl acetates were obtained this way with high functional group tolerance and moderate to good yields (Scheme 18b). Aryl bromides and iodides are also accessible when the appropriate trapping agent (CBr4 or NaI, respectively) is present in the reaction mixture, although the scope was rather narrow and bromination required pre-synthesized aryl diazonium tetrafluoroborates rather than in situ-generated intermediates.
 | ||
| Scheme 18 Photoredox approach for Sandmeyer reaction: (a) model reaction and (b) representative products. | ||
Another application of a reductive Sandmeyer-type reaction is the synthesis of sulfoxides from anilines under a photocatalytic regime.83 An, Wu and Zheng presented efficient sulfinylation of a variety of arenediazonium salts (formed in situ). The proposed mechanism is very similar to that described in the current chapter.
Wu reported the photocatalytic synthesis of E-stilbenes by coupling arenediazonium tetrafluoroborates with cinnamyl boronates (Scheme 19a).85 The reaction occurs employing only 2.0 mol% of a cost-effective organic dye (Eosin Y84), an EtOH/H2O mixture as a solvent and green (550 nm) light irradiation. More than 20 examples were presented, which mainly consisted of mono- and disubstituted stilbenes (Scheme 19b). Interestingly, changing the catalyst to a ruthenium complex (Ru(bpy)3Cl2) switched the selectivity to provide predominantly Z isomers, which might suggest a subsequent E/Z isomerization through an energy transfer process, since Ru(bpy)3Cl2 has a higher triplet state energy (Eosin Y ∼44 kcal mol−1;84,86 Ru(bpy)3Cl2 ∼49 kcal mol−1 (ref. 87)).
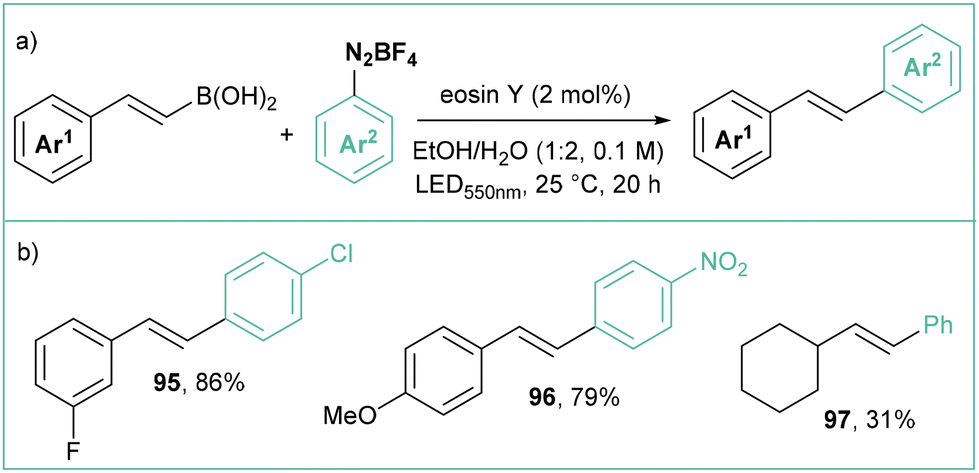 | ||
| Scheme 19 (E)-Selective photoredox synthesis of stilbenes from cinnamyl boronic acids using Eosin Y84 as a catalyst: (a) model reaction and (b) representative products. | ||
Decarboxylative, visible-light photoredox synthesis of trans-oxiranes from (E)-cinnamic acids using arenediazonium salts was developed recently by Singh (Scheme 20a).88 Eosin Y was also employed as a photocatalyst generating the aryl radical, while CuCl and TBHP served as the oxidant system. More than 20 examples of variously substituted diaryloxiranes, consisting mainly of mono and disubstituted products, were synthesized. The method tolerates many functionalities such as halides, nitro, cyano, ketone and ester groups. According to the proposed mechanism (Scheme 20c), the deprotonated cinnamate B reacts with the photogenerated aryl radical to give anion radical E. This intermediate forms a Cu(III) chelate F arising from a SET process with Cu(II). In the presence of TBHB and DBU, the chelate releases a Cu(I) species and a peroxide intermediate G, which liberates a t-butoxide anion and carbon dioxide, eventually leading to trans-oxirane product H.
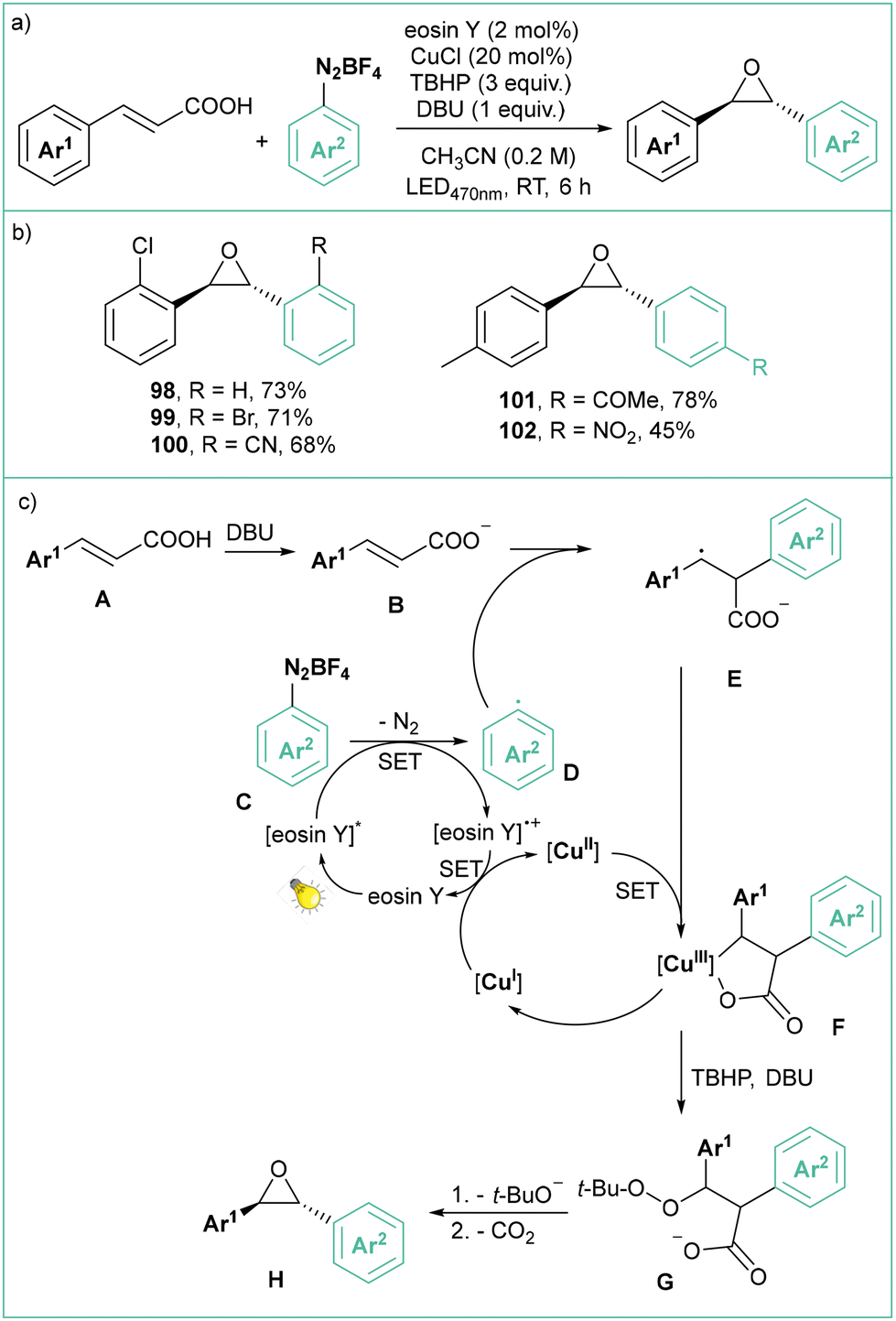 | ||
| Scheme 20 Decarboxylative photoorganometallic synthesis of trans-oxiranes using eosin Y and CuCl as co-catalysts: (a) model reaction and (b) representative products and (c) mechanistic proposal. | ||
Reactions based on sulfur(VI) fluoride exchange (SuFEx) are the basis of the second-generation click reactions developed by the Sharpless group and employed widely in biorthogonal processes including protein labelling.89 The photocatalytic procedure for the conversion of aryl diazonium tetrafluoroborates to the corresponding sulfonyl fluorides was developed by Troian-Gautier and Tambar (Scheme 21a).90 The optimised conditions employ Ru(bpy)3Cl2·6H2O as a catalyst, DABSO as a source of SO2 and NFSI as a fluorinating agent in MeCN. Using this procedure, the authors synthesized 10 electron-rich and electron-poor mono-, di-, and trisubstituted arylsulfonyl fluorides with yields ranging from 26 to 73% (Scheme 21b). According to the proposed mechanism (Scheme 21c), the photogenerated aryl radical B reacts with DABSO to give the mono-(sulfur dioxide) adduct C and sulfonyl radical D, which is fluorinated by NFSI to the final product. According to the authors, the Ru(III) species, resulting from the oxidative quenching of the photoexcited catalyst with the aryldiazonium salt, is reduced back to the ground state from a SET process with either C or DABSO.
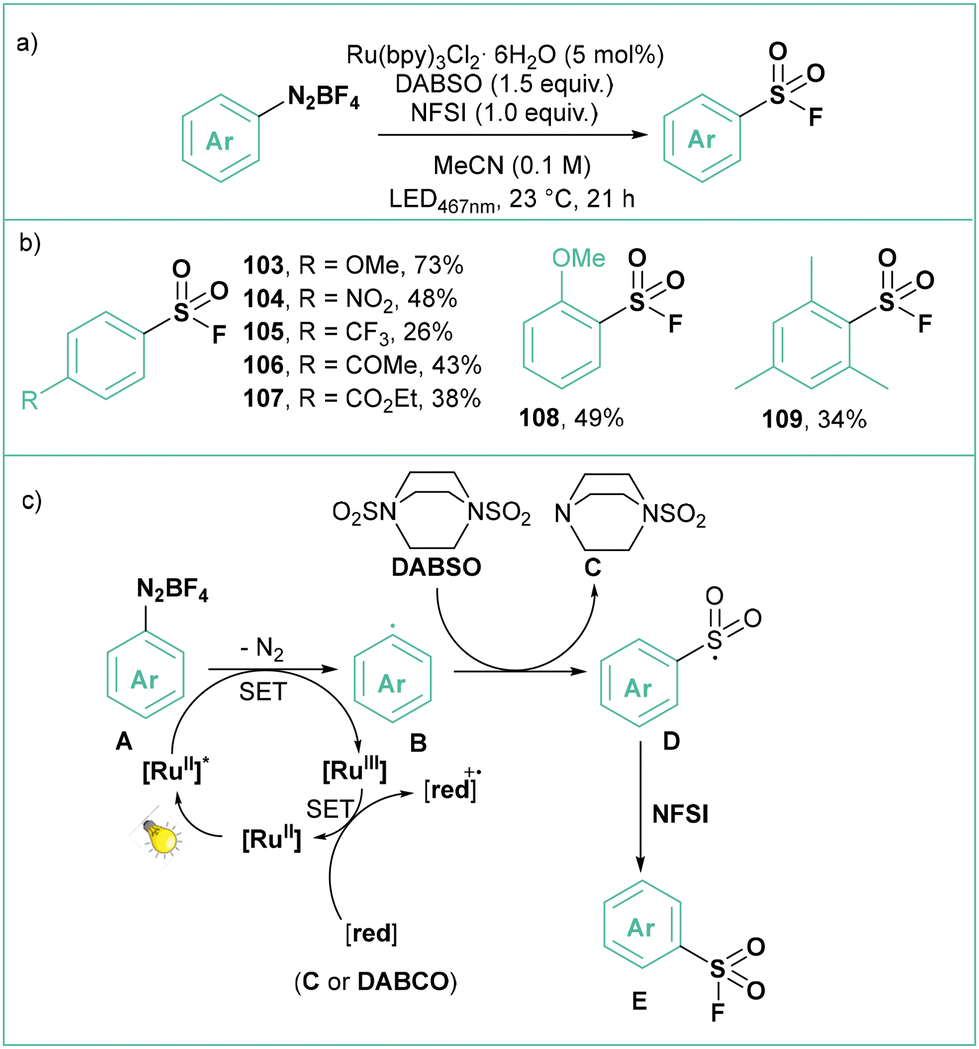 | ||
| Scheme 21 Photocatalytic synthesis of arylsulfonyl fluorides from aryldiazonium salts (a) model reaction, (b) representative products and (c) mechanistic proposal. | ||
It is worth mentioning that aryl radicals can be liberated from arylazosulfones; however, this transformation occurs through direct irradiation of the latter compounds.91–94
In 2018, Mo and co-workers demonstrated that the classic Sandmeyer reaction can also be performed under the influence of electric power (Scheme 22a) using non-sacrificial Pt electrodes.98 Namely, aryl diazonium salts are efficiently converted into their halogenated analogues using a variety of halogenation reagents such as NBS, NaBr, CH2I2, CBrCl3 or LiCl (Scheme 22b). In contrast to the classical conditions, there is no need to use metal-based species, such as copper(I) salts. Importantly, the process can be performed on a large scale (up to 15 mmol of a substrate) with efficiencies reaching 70–80% (Scheme 22b). Here, Pt anodes were successfully replaced with cost-effective graphite plates. This process can also be performed in a two-step, one-pot fashion via the initial formation of an aryldiazonium salt followed by electrolysis. From the mechanistic point of view, the reaction was described as a cross-coupling between two radicals: a halogen-based radical that was produced via oxidation at an anode and an aryl radical produced via reduction of ArN2+.
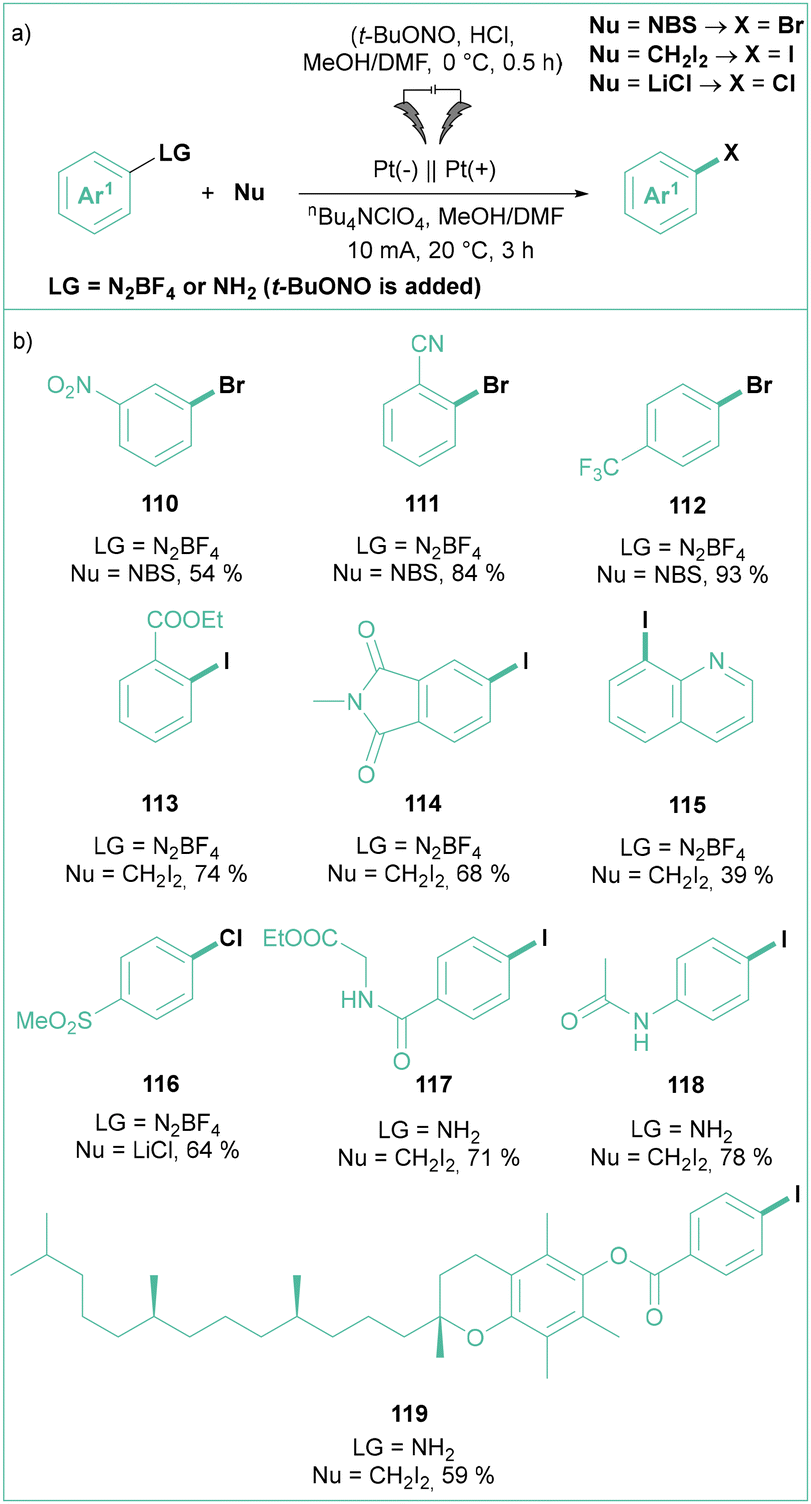 | ||
| Scheme 22 Electrochemically driven Sandmeyer reaction: (a) model reaction and (b) representative products. | ||
A variety of heteroarenes can react with the aryl radicals produced by the electroreduction of aryl diazonium salts. Specifically, Zhang et al. employed diazonium tetrafluoroborates for the electrochemical arylation of electron-deficient arenes (Scheme 23a).99 In this transformation, salts possessing electron-donating or withdrawing groups were reactive, although the latter featured reduced yields. A broad range of electron-deficient arenes, including quinoxalines, quinoxalinones, pyrazines, and other N-heteroarenes, were also tolerated.
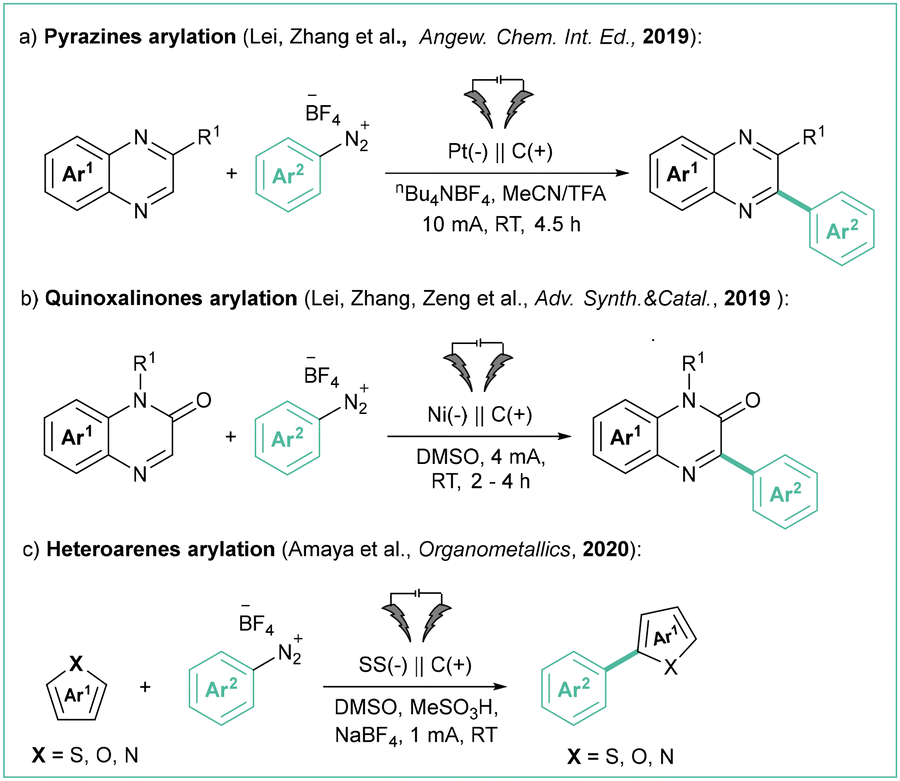 | ||
| Scheme 23 Electrochemical coupling of different heteroarenes with aryldiazonium salts: (a) pyrazines arylation, (b) quinoxaliones arylation and (c) heteroarenes. | ||
Along this line, Zeng et al. reported the arylation of quinoxalinones with aryldiazonium salts (Scheme 23b).100 The reaction shows very good scalability with high functional group tolerance, and proceeds without an electrolyte.
Similarly, Amaya et al. achieved the arylation of simple, electron-rich heteroarenes (Boc-protected pyrrole, thiophene, and furan) employing a stainless steel anode (Scheme 23c).101 Unfortunately, the scope is rather limited, and only moderate yields can be achieved, probably due to the simultaneous decomposition of easily oxidisable heteroarenes at the anode.
In 2023, Wang, Zhou and co-workers developed a straightforward method to synthesize polycyclic (hetero)aromatic compounds via [4 + 2] benzannulation (Scheme 24a).102 The reaction involved alkynes reacting with aryl diazonium salts, including 2-(hetero)aryl diazonium salts, or they can be generated in situ from the corresponding aniline. The reaction furnished the products in moderate to good yields, accepting differentiated substitution of both substrates but showed no selectivity for unsymmetrical heteroaryl diazonium salts (Scheme 24b). According to the proposed mechanism (Scheme 24c), the reaction starts with the reduction of the aryldiazonium salt A to an aryl radical B, which subsequently undergoes addition to an alkyne C giving rise to a vinyl radical D. The radical then undergoes cyclization followed by anodic oxidation, and bicarbonate-assisted deprotonation leads to products of type G. A similar transformation that led to a series of phenanthridines was developed by Sharma et al.19b Here, 2-isocyanobiphenyls were used as coupling partners for the in situ-formed aryldiazonium salts. The reaction occurs with good to excellent efficiencies at room temperature in an MeCN/HFIP (10:1) mixture employing a platinum cathode and an RVC anode.
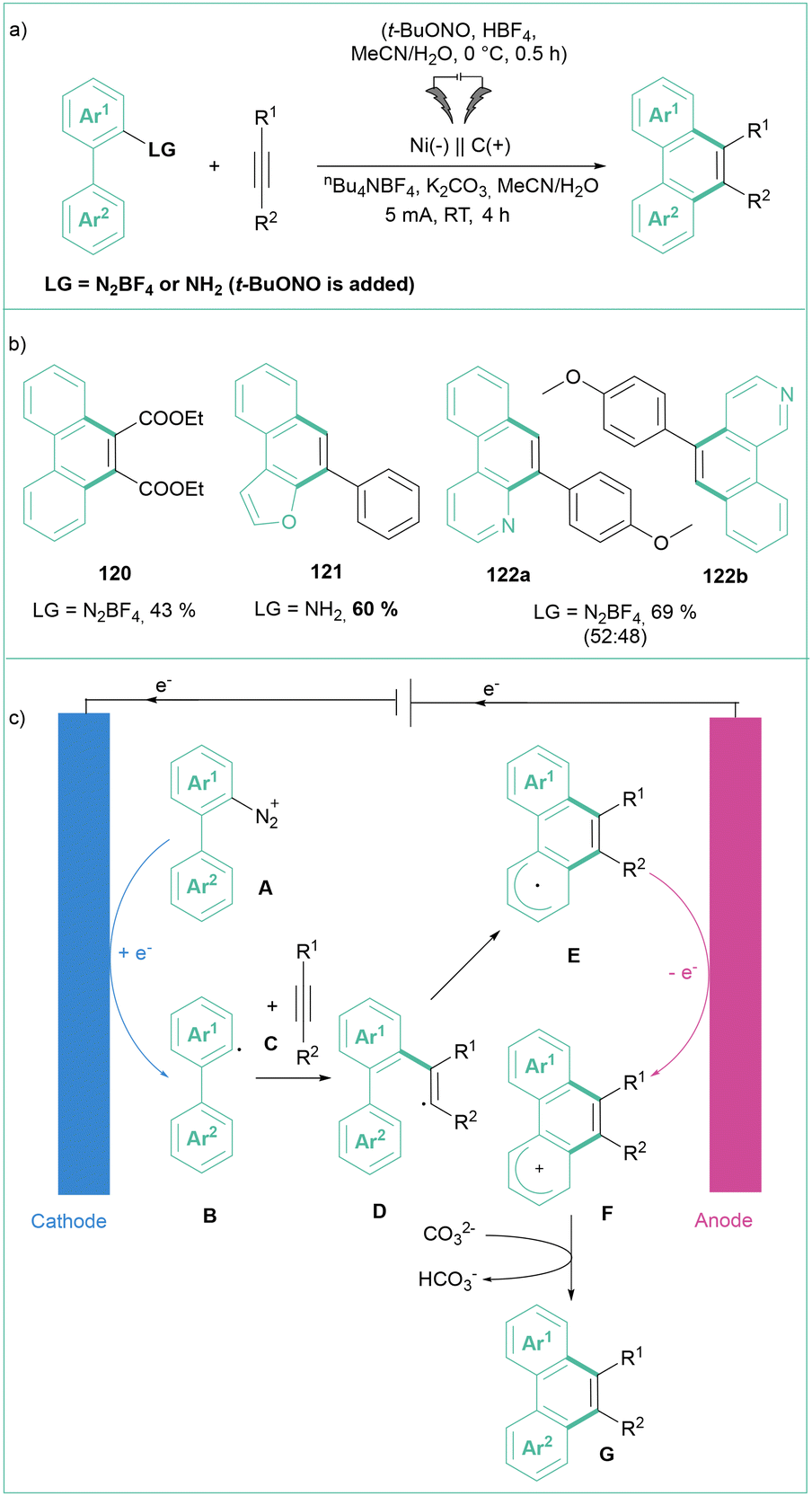 | ||
| Scheme 24 [4 + 2] Benzannulation of aryldiazonium salts and alkynes: (a) model reaction, (b) representative products and (c) mechanistic proposal. | ||
3.3. Generation of aryl radicals from sulfonium salts
Sulfonium salts, which are in an isoelectronic relationship with phosphines, have attracted considerable attention as versatile reagents in organic synthesis for many years,103 since they play an important role in various C–C bond-forming reactions that involve C–S bond cleavage.104Within this section, many methods that are based on thianthrenium chemistry are presented. Although extremely useful (vide infra), this kind of aryl radical precursor delivers high-weight by-product which may be re-isolated, however again with additional solvent waste. This should be also taken into account when planning the synthetic route involving aryl radical generation via reduction of thianthrenium salts.
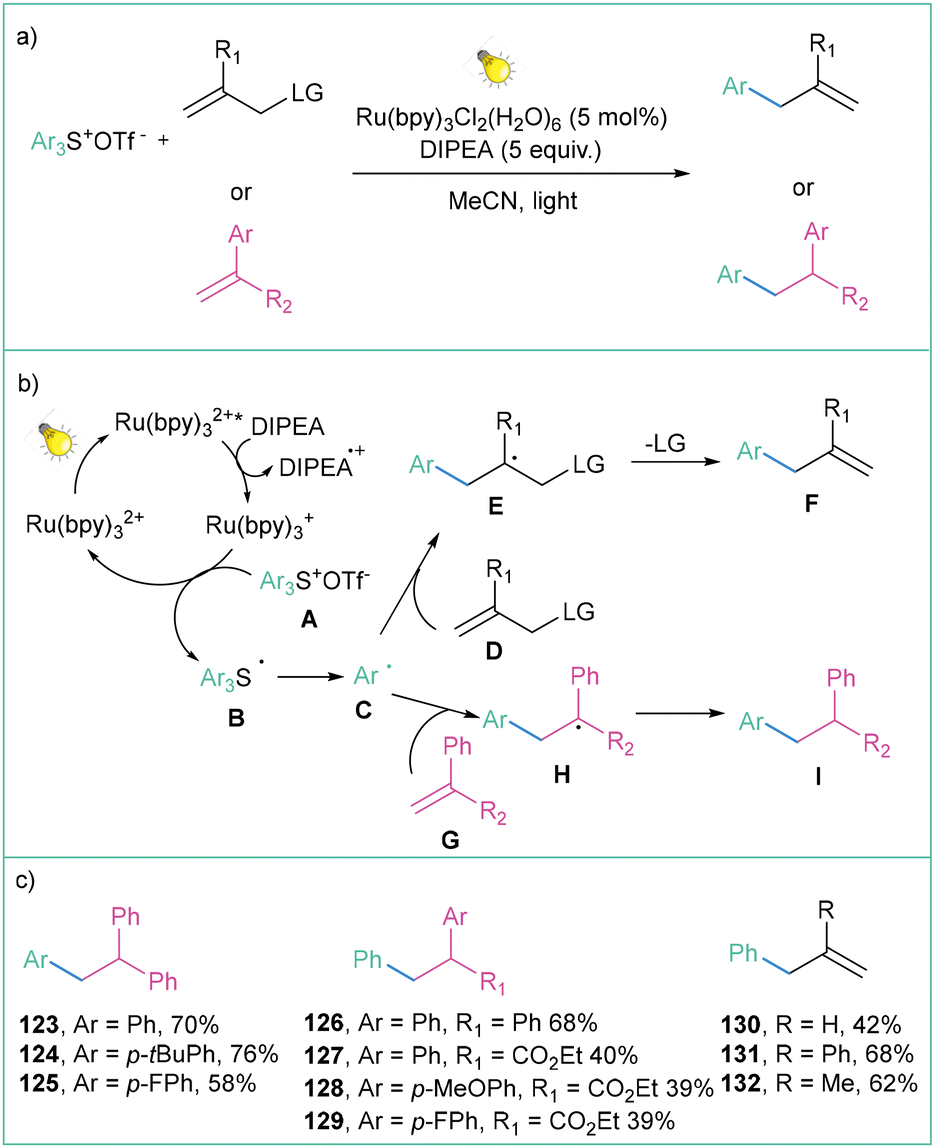 | ||
| Scheme 25 Triarylosulfonium salts as radical precursors: (a) model reaction, (b) mechanistic proposal and (c) representative examples. | ||
One of the most straightforward routes toward the synthesis of arylsulfonium salts is direct sulfenylation of C–H bonds with the use of thianthrene sulfoxide (143a). Such an approach was used for the preparation of arylsulfonium salts and their application as trifluomethylation agents as well as electrophilic partners in transition-metal-catalyzed C–C cross-couplings.107 Recently, Ritter with co-workers developed a highly para-selective C–H trifluoromethylation of arenes employing 136a (Scheme 26).108 In the next step, the generated salts were engaged in a dual catalytic system that involves a photochemical, Ru-catalyzed oxidation of Cu(I)–CF3 to Cu(II)–CF3 followed by copper-catalyzed C–C bond formation involving the Cu(III) intermediate.
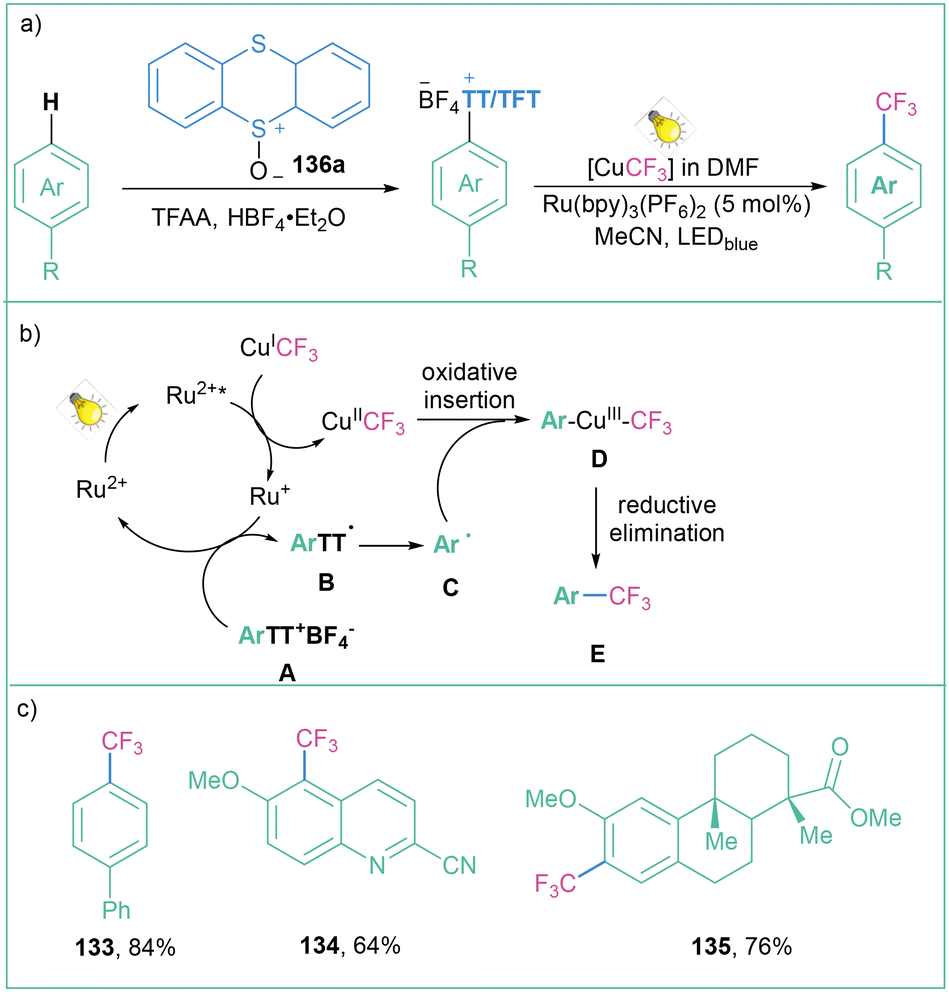 | ||
| Scheme 26 Aryl sulfonium salts for site-selective trifluoromethylation: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
The mechanistic proposal assumes that after light absorption, the ruthenium-based photocatalyst is excited to the excited state (Ru(bpy)32+*), which is capable of oxidising CuICF3 to CuIICF3. The RuI species subsequently reduces the tetrafluorothianthrene sulfonium salt A to radical B, which immediately decomposes to the aryl radical C. The generated radical C reacts with CuIICF3 during oxidative ligation to form complex D, after the subsequent formation of the reductive elimination product E (Scheme 26b). The reaction is generally high-yielding, and both electron-deficient and electron-donating groups are well tolerated (Scheme 26c).
The abovementioned findings set the stage for the general application of tetrafluorothianthrene sulfonium salts for the generation of aryl radicals under light irradiation in the presence of a photoredox catalyst. First, the synthesis of thianthrene sulfonium salts takes place, and then a photocatalyzed reduction to the aryl radical is performed, which subsequently can be engaged in further transformations (Scheme 27).
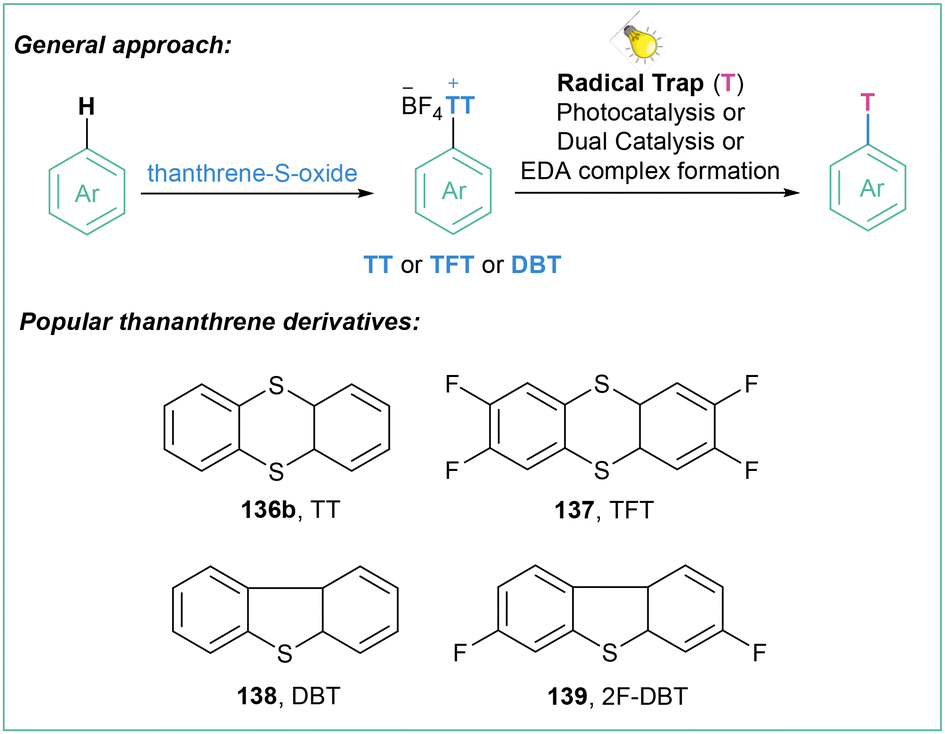 | ||
| Scheme 27 Generation of aryl radicals from tetrafluorothianthrene sulfonium salts. | ||
Such a methodology was already applied under different conditions (different photoredox catalyst and application of dual photoredox metal catalysis) in many transformations such as photocatalyzed trifluoromethylation108 (Scheme 28a), Cu-catalyzed fluorination19c (Scheme 28b), borylation109 (Scheme 28c), Sonogashira-type reaction110 (Scheme 28d), hydroarylation of vinyl acetamides111 (Scheme 28e), arylation of indoles112 (Scheme 28f), Meerwin-type bromoarylation113 (Scheme 28g), gem-difluoroallylation114 (Scheme 28h) and three-component synthesis of arenesulfonohydrazines115 (Scheme 28i). All of the transformations are high yielding and site selective. The same methodology can also be expanded to the application of aryl selenium salts as aryl radical precursors.116
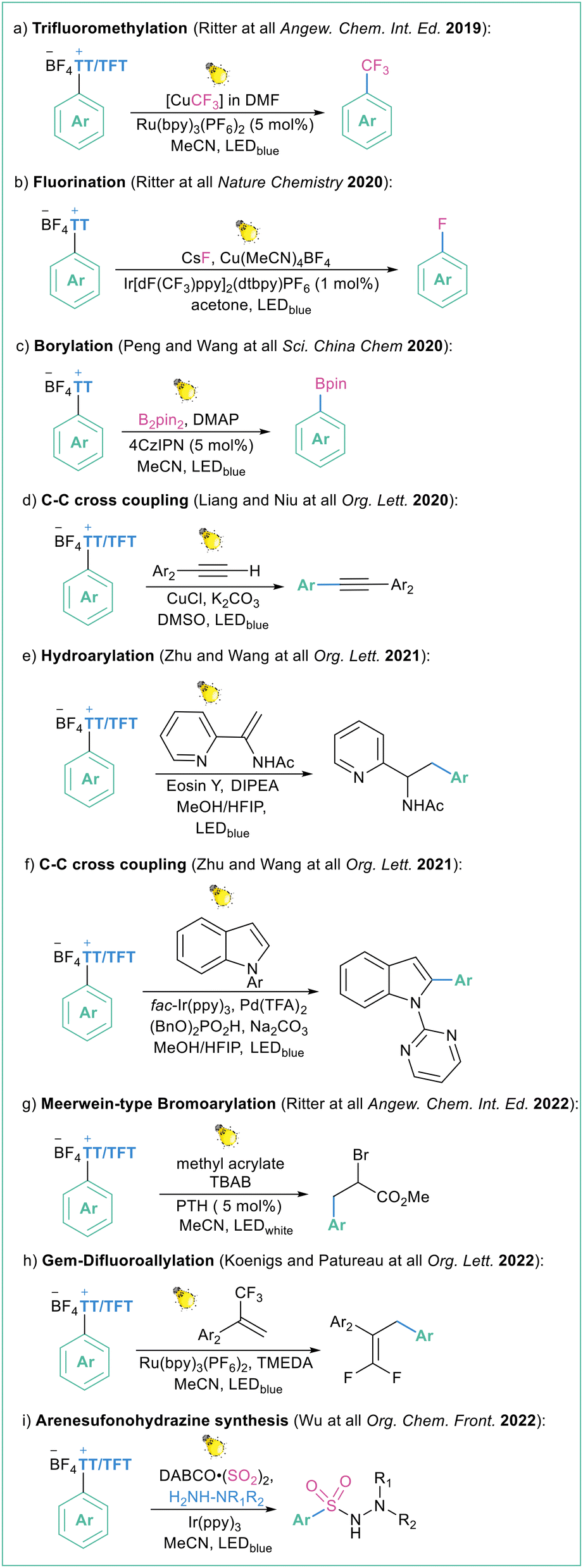 | ||
| Scheme 28 Scope of application of tetrafluorothianthrene sulfonium salts as arylating reagents. | ||
Importantly, all of the aforementioned transformations require prior salt synthesis and, in the next step, photoreduction of the sulfonium salt to finally assemble an aryl radical. Procter and co-workers described a successful modification of this approach, where the sulfonium salt is prepared in situ (Scheme 29).117 The authors demonstrated a one-pot strategy for the rapid synthesis of (hetero)biaryls from non-prefunctionalised partners via merging photoredox catalysis (with the use of an organic dye (10-phenylphenothiazine) as a photocatalyst) and the activation of the substrate via an interrupted Pummerer reaction (Scheme 29a). The mechanism for this transformation assumes in the first step Pummerer activation of arene A with DBT-S-oxide B in order to form sulfonium salt C, which after single-electron reduction (by the photocatalyst in the excited state) forms aryl radical D. Subsequently, reaction with heteroarene E forms radical F, which is oxidised by the photocatalyst radical cation (PTH˙+) to form G. Finally, rearomatization leads to the biaryl product H (Scheme 29b). Generally, this metal-free, one-pot reaction proceeds with moderate to good yields for different radical traps (N-Me-pyrroles, N-Boc-pyrroles, furans, and thiophens) and also for different arene substrates. Worth mentioning is the 10 mmol-scale synthesis for 148, performed with a yield similar to that performed for the smaller scale (0.2 mmol) (Scheme 29c).
 | ||
| Scheme 29 One-pot, catalyst-free photoinduced synthesis of biaryls: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
Another useful application of sulfonium salts is the generation of alkyl radicals via direct irradiation of an EDA complex, as demonstrated for the first time by Shi et al. (Scheme 30a).118 One year later, the same methodology was presented for the generation of aryl radicals.109b In this case, mechanistic studies suggest that the reaction begins with the formation of a straw-coloured EDA complex (B) between the DBT salt A and B2cat2 in DMF. Under light irradiation, the EDA complex (B) triggers a single-electron transfer (SET) from B2cat2 to DBT that forms the aryl radical D. After complexation with one molecule of DMF, the aryl radical D can interact with B2cat2 to form a radical intermediate E. B–B bond cleavage produces product F, which after the addition of pinacol forms the final Ar–Bpin product. After B–B cleavage, the salt radical H is also formed. The subsequent SET event between H and another molecule of DBT salt A regenerates the aryl radical D and forms a stable ionic salt C to complete the cycle (Scheme 30b).
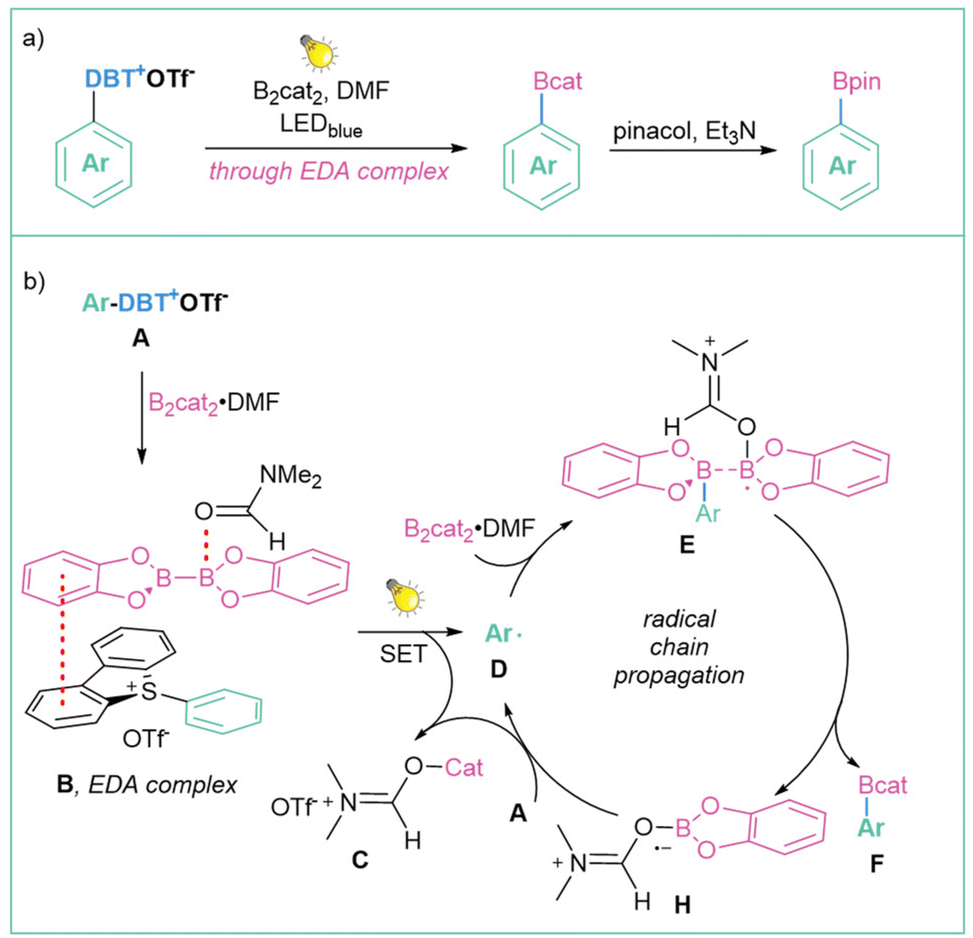 | ||
| Scheme 30 Photoinduced desulfurative borylation: (a) model reaction and (b) mechanistic proposal. | ||
This methodology of irradiating an EDA complex containing a TT-based salt was already applied in many successful transformations such as borylation109b (Scheme 31a), C–H-functionalisation119 (Scheme 31b), synthesis of aromatic disulphides120 (Scheme 31c), phosphorylation121 (Scheme 31d), trifluoromethylselenolation122 (Scheme 31e) and arylation of heteroarenes123 (Scheme 31f). Even though such a strategy is limited to substrates that are able to form EDA-type complexes in situ, it is extremely useful, economical, and environmentally friendly (metal and photocatalyst-free).
 | ||
| Scheme 31 The scope of transformations is enabled via the formation of EDA complex: (a) borylation, (b) C–H functionalization, (c) thioetherification, (d) phosphorylation, (e) trifluoromethylselenolation and (f) arylation of heteroarenes. | ||
3.4. Generation of aryl radicals from sulfonyl chlorides
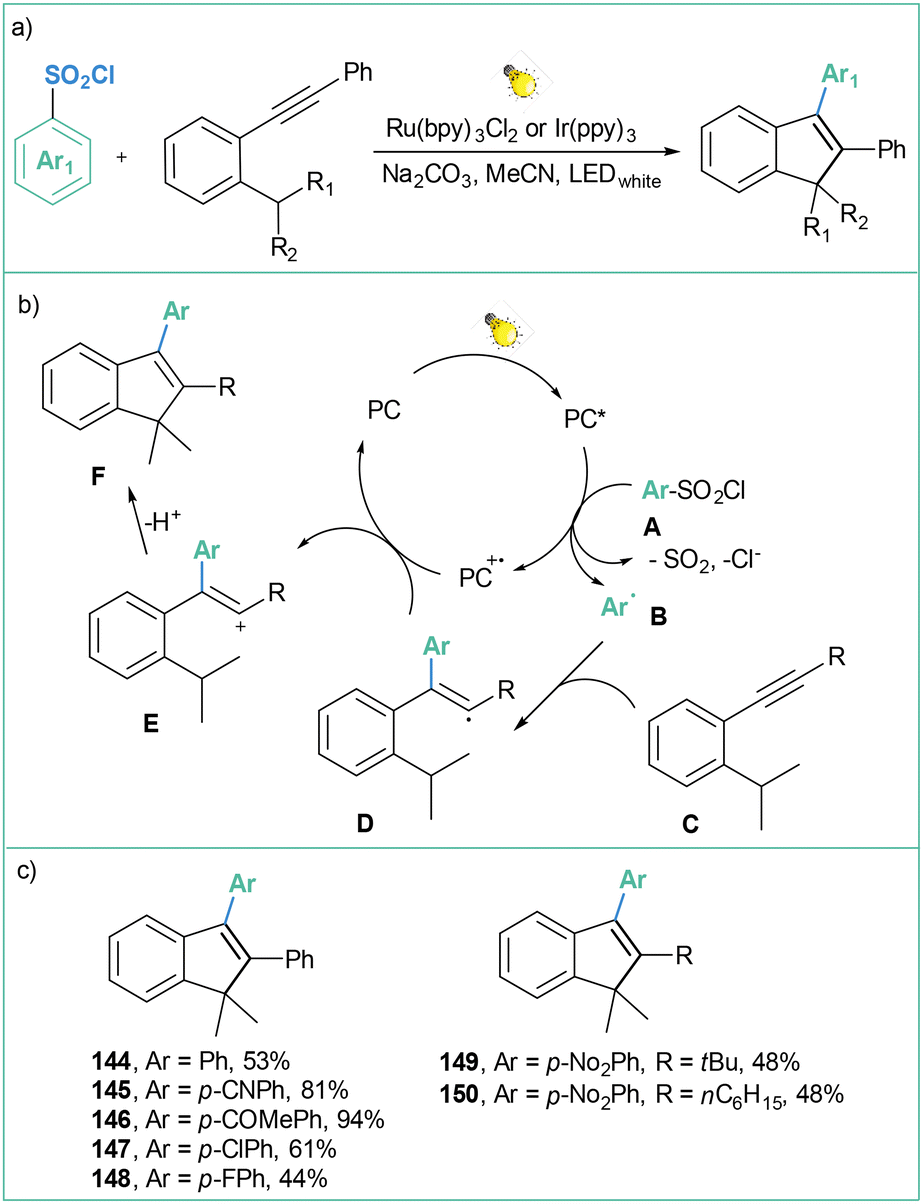 | ||
| Scheme 32 Photocatalyzed reaction of arylsulfonyl chlorides with alkynes: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
 | ||
| Scheme 33 Photocatalyzed production of aryl radicals from arylsulfonyl chlorides and their application in (a) carbocyclization, (b) tandem cyclization, (c) synthesis of phenanthridines, (d) synthesis of 2,3-diarylsubstituted indoles and (e) arylation of heteroarenes. | ||
3.5. Generation of aryl radicals from phenol derivatives (aryl triflates, phosphates)
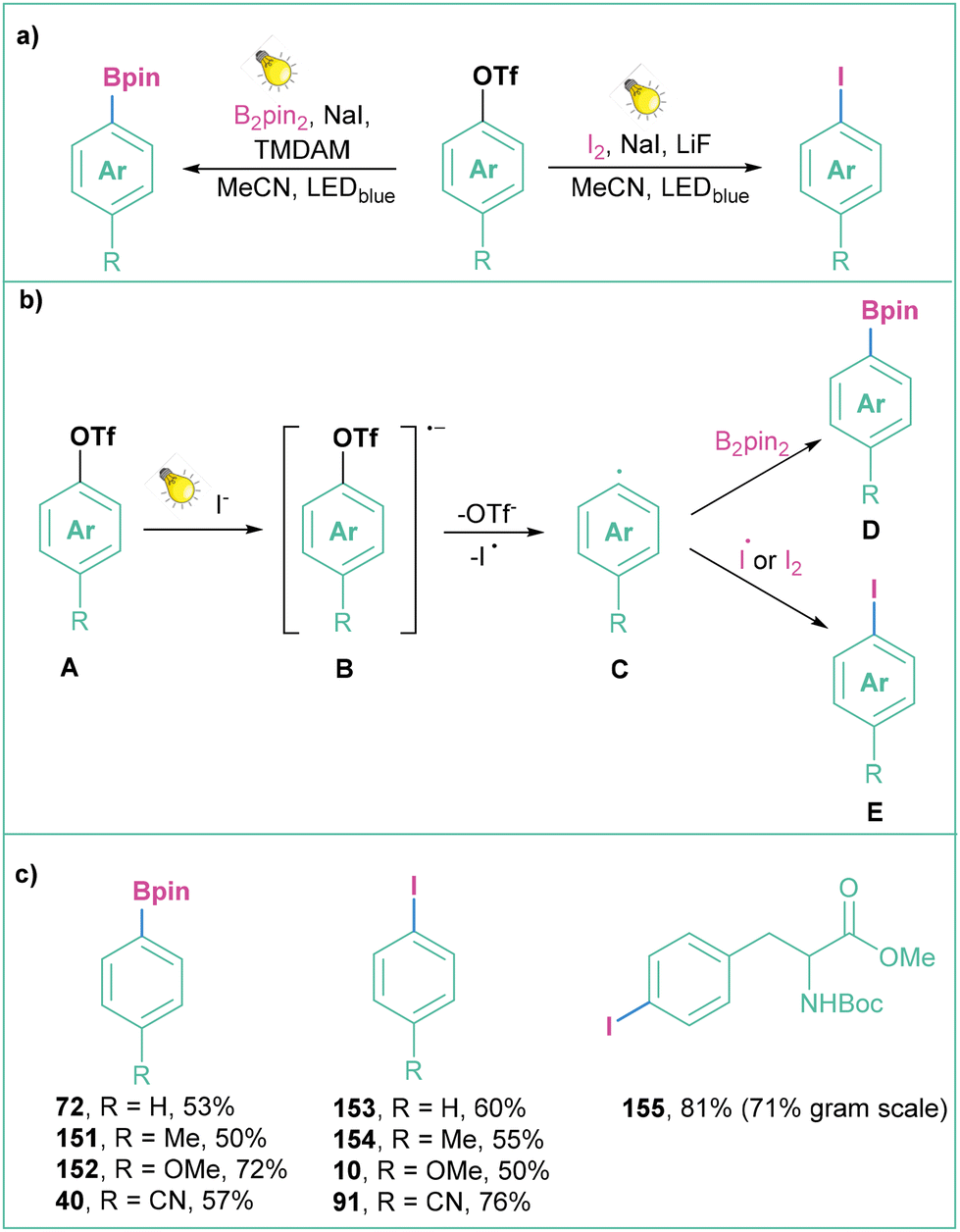 | ||
| Scheme 34 Generation of aryl radicals from aryl triflates: (a) model reaction, (b) mechanistic approach and (c) representative products. | ||
A similar mode of action was applied for the photoinduced Arbuzov-type reaction of aryl triflates with trialkyl phosphite, where NaI was replaced with TBAI (Scheme 35)132 and for the deuteration of aryls.133
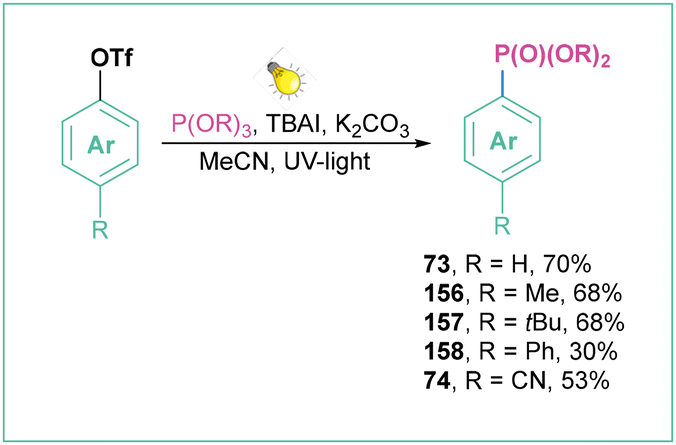 | ||
| Scheme 35 Photoinduced Arbuzov-type reaction of aryl triflates with trialkyl phosphite. | ||
Another way of activating aryl triflates toward the generation of aryl radicals was presented by Gevorgyan and co-workers (Scheme 36a).134 This methodology allows for efficient intramolecular C–H arylation of amides. The proposed mechanism depicted in Scheme 36b assumes the formation of a Pd-radical intermediate B, which after a 1,5-HAT produces radical C, which upon intramolecular cyclization and rearomatization forms product E. It is postulated that the formation of intermediate B can occur through different pathways: (a) a SET between the Pd catalyst in the excited state and aryl triflate (via standard reduction to a radical anion and then C–O cleavage to an aryl radical); (b) oxidative addition of the Pd catalyst into substrate A, forming a Pd complex and after subsequent light absorption, formation of intermediate B; (c) ligand exchange, formation of the PdI-catalyst, and subsequent homolysis to form an aryl radical; or (d) reductive elimination and generation of an aryl iodide that undergoes SET with the photoexcited Pd-catalyst, leading to B. Although the mechanism has been studied in detail, it was not possible to determine which path is the most reliable one. However, this transformation proved to be very efficient in forming different oxindoles (Scheme 36c) and isoindoline-1-ones.
 | ||
| Scheme 36 Visible-light-induced palladium-catalyzed generation of aryl radicals from aryl triflates: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
Aryl triflates are not the only phenol derivatives that can be activated to promote efficient aryl radical formation.135 Larionov and co-workers proved that it is possible to photoactivate other derivatives such as aryl diethyl phosphates (Ered ∼−3.0 V vs. SCE) (Scheme 37a), as well as aryl halides and quaternary aryl ammonium salts.57 Proton-coupled electron transfer (PCET) was utilized in order to activate the substrates with strongly negative reduction potentials (Scheme 37b). Generally, this approach possesses an enormously broad scope of application: EDG, EWG, heteroarenes, and natural compounds, among them amino acids and hormones (Scheme 37c).
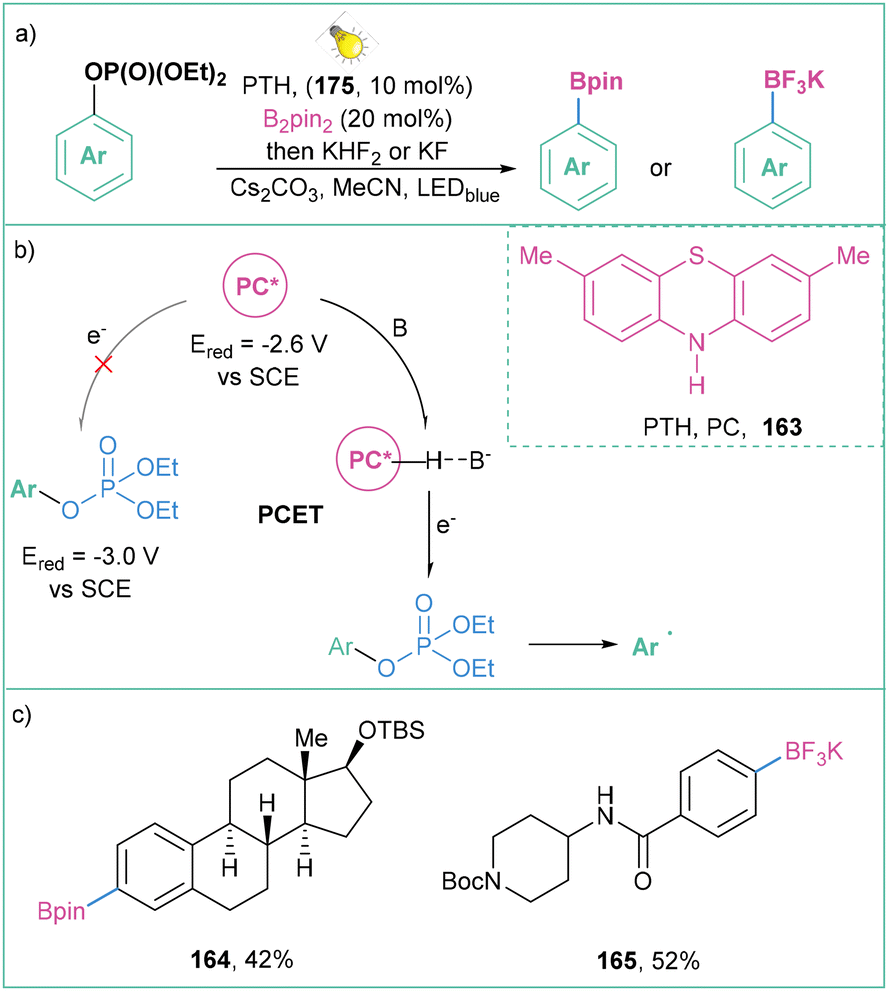 | ||
| Scheme 37 PCET as a mode of activation of phenol derivatives: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
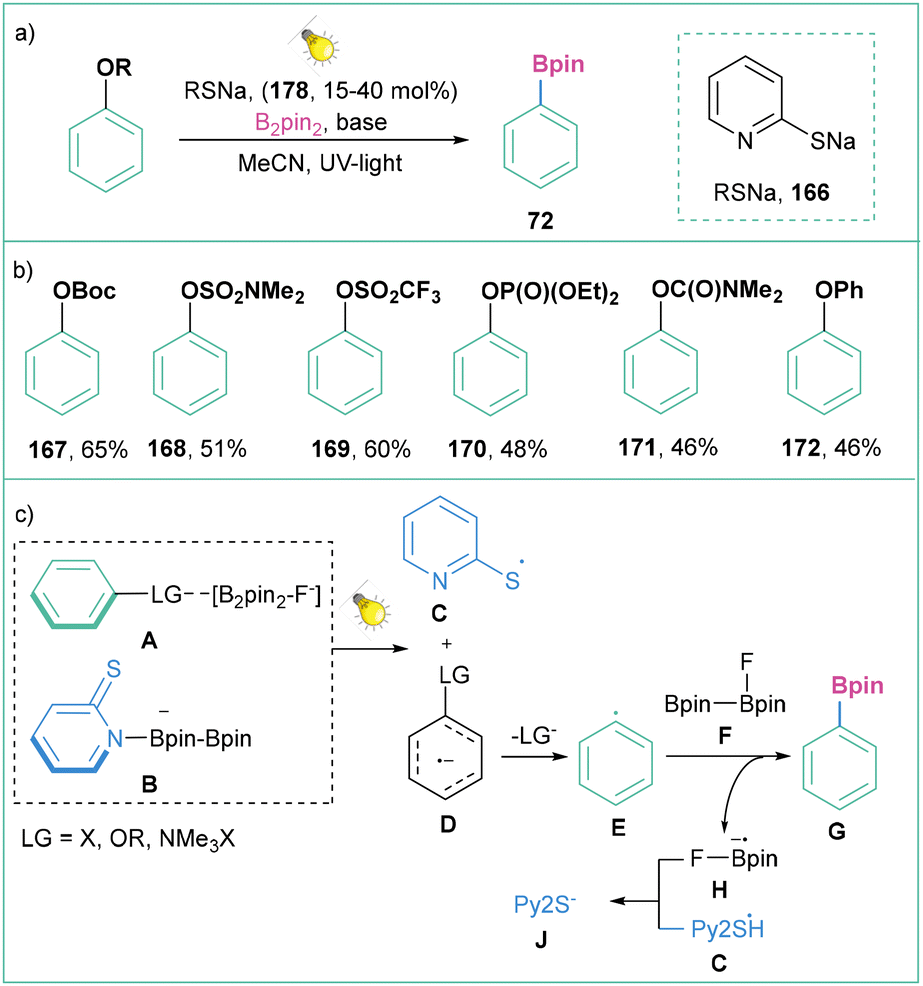
Another strategy for the activation of phenol derivatives that possess highly negative reduction potentials (OBoc, OSO2NMe2, OSO2CF3, OP(O)(OEt)2, OC(O)NMe2, OPh) is the application of thiolate as a catalyst (Scheme 38).136 Here, the reaction is initiated by a photoinduced electron transfer process between an activated boryl-anion A (LG = X, O or N) and the adduct 2-PySNa/B2pin2B to generate a thiyl radical C and radical anion D. The resulting radical anion D undergoes cleavage of the C–LG bond to form the aryl radical E, which then reacts with a diboron species [F, F-B2pin2] to give the borylation product G and the boryl radical anion H. Finally, the thiyl radical C is reduced by the boryl radical anion H to regenerate thiolate J with the closure of the cycle (Scheme 38c).
 | ||
| Scheme 38 PCET for the activation of phenol derivatives: (a) model reaction, (b) representative substrates and (c) mechanistic proposal. | ||
Generally, this strategy allows for the activation of not only a very broad range of inert C–O bonds within phenol derivatives (carbonate, sulfamate, phosphate, and carbamate) but also non-activated C–F bonds, C–N bonds (ammonium salts), and C–S bonds (sulfone, sulfoxide, and sulfide), with very negative reduction potentials, which are challenging for photoredox activation. Despite the high reducing power generated, this reaction shows broad functional group tolerance and yields borylated products in moderate to very good yields (Scheme 38b).
3.6. Generation of aryl radicals from quaternary arylammonium salts
The application of unprotected amines as building blocks in organic synthesis has been a central research topic for decades. However, the Caryl–N bond cleavage of amines represents a very challenging problem, because of the high bond dissociation energy. The preactivation of the amine into other functional groups is one way to solve this problem. Transformation of an amine to the corresponding quaternary ammonium salts is easily achievable synthetically and relatively inexpensive. They show good reactivity in various organic transformations, especially via C–N bond cleavage.137In 2019, Manthiram and colleagues investigated the electroreductive transformation of benzylammonium salts toward benzylcarboxylic acids.141 During the investigation, they also tested phenyltrimethylammonium triflate (173) (Scheme 39) as a possible aryl radical precursor. Indeed, benzoic acid (174) was isolated albeit in 15% yield, showcasing the synthetic potential of 180 at the same time. The important obstacle here is the high cell potential (Ucell = 4.5 V) that was required for the indicated efficiency.
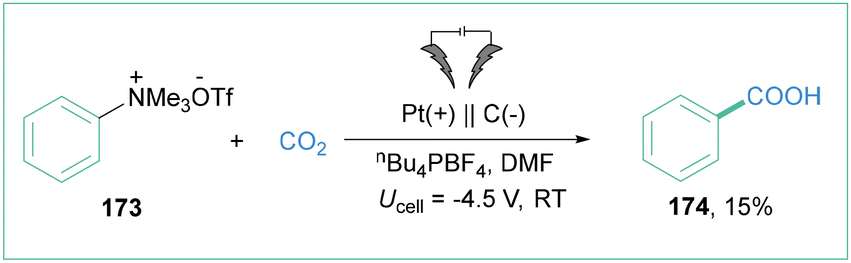 | ||
| Scheme 39 Electrolysis of phenyltrimethylammonium triflate toward benzoic acid formation. | ||
In 2021, Xu's group described the direct electrolysis of aryltrimethylammonium triflates toward synthetically meaningful organoboronates (Scheme 40a).142 Here, potentiostatic conditions were applied to produce products in moderate to good yields showing broad applicability (Scheme 40b). They proved that the presence of MeOH is essential for this transformation as the efficiency dropped significantly in the absence of this protic solvent. Mechanistically, the process starts with the formation of an aryl radical via direct electroreduction along with Me3N. Subsequently, Ar˙ reacts with B2pin2 in the presence of MeO−, giving rise to the expected product C and MeOBpin (Scheme 40c).
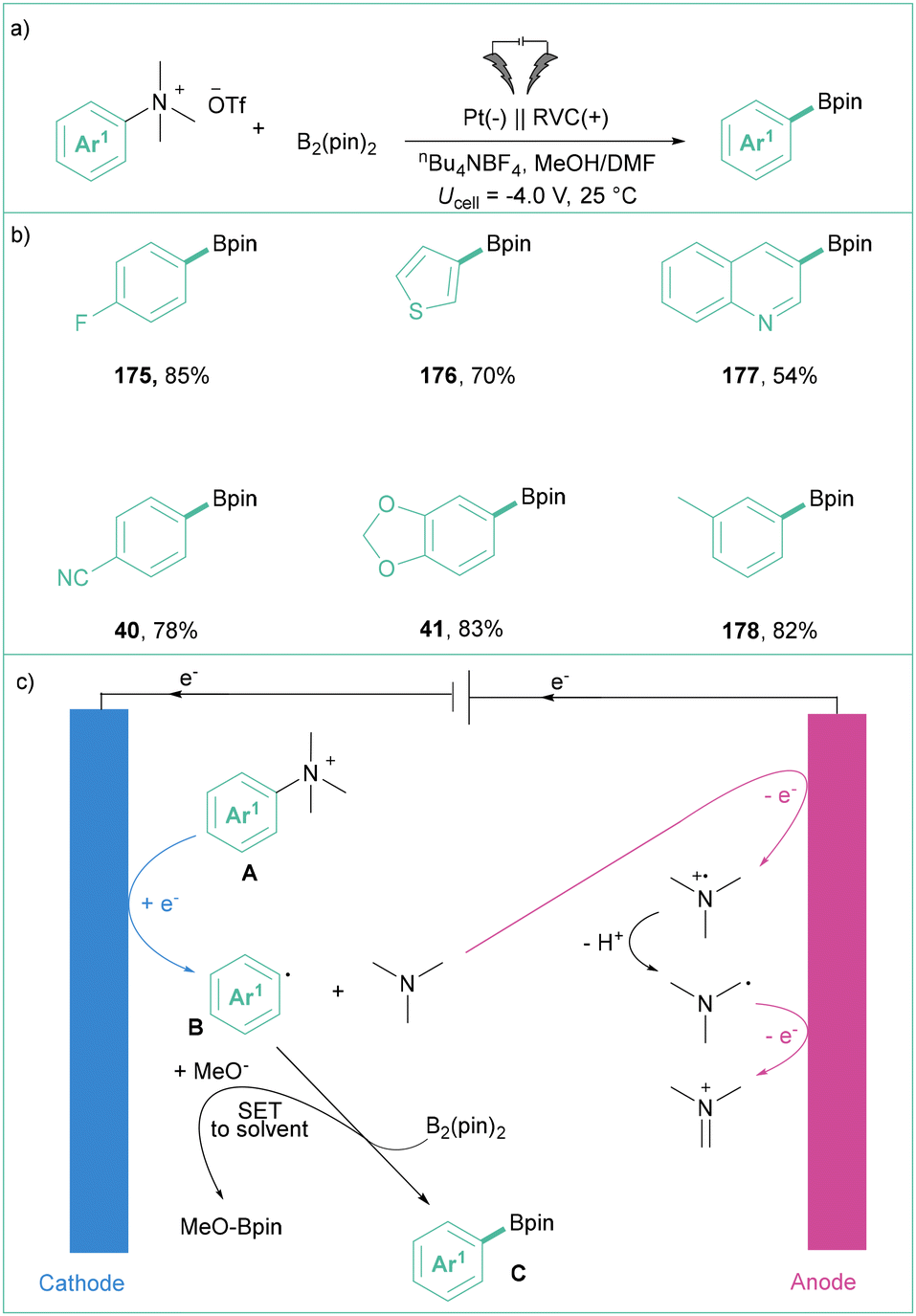 | ||
| Scheme 40 Electrolysis of phenyltrimethylammonium triflate toward benzoic acid: (a) model reaction, (b) representative products and (c) mechanistic proposal. | ||
The same group also proved that quaternary arylammonium salts can be transformed into a variety of benzonitriles (181) as well as benzylnitriles (182) by employing tosyl cyanide (TsCN) or azido allyl alcohol as cyanation or cyanomethylation reagents (Scheme 41a).143 The efficiency of cyanation was generally higher, in comparison with the cyanomethylation reaction. The method tolerates a broad range of substrates, including those bearing easily reducible functional groups (–CN, –CO2Alk, –NO2), S-, O- or N-based heterocyclic motifs, as well as natural product derivatives (Scheme 41b). Importantly, the authors found that the obtained nitriles of type 181–182 are stable during the electrolysis as they feature reversible electron-transfer characteristics (based on the CV studies). The proposed mechanism postulates the formation of an aryl radical Bvia a reductive process, which then reacts with electrophilic TsCN or 187, giving rise to N-centered radicals in both cases (C or D). Finally, E and I are formed with the release of Ts–Ts or acetone (both assembled from the respective radicals) (Scheme 41c).
 | ||
| Scheme 41 Electrochemical cyanation and cyanomethylation of aryltrimethylammonium salts: (a) model reaction, (b) representative products and (c) mechanistic proposal. | ||
The common feature of the electrochemical methods described above is the extremely high working potential (up to −5.0 V) that significantly hampers efficient energy utilization. Therefore, this sets the stage for more sustainable methods for the reduction of aryltrimethylammonium salts toward aryl radicals.
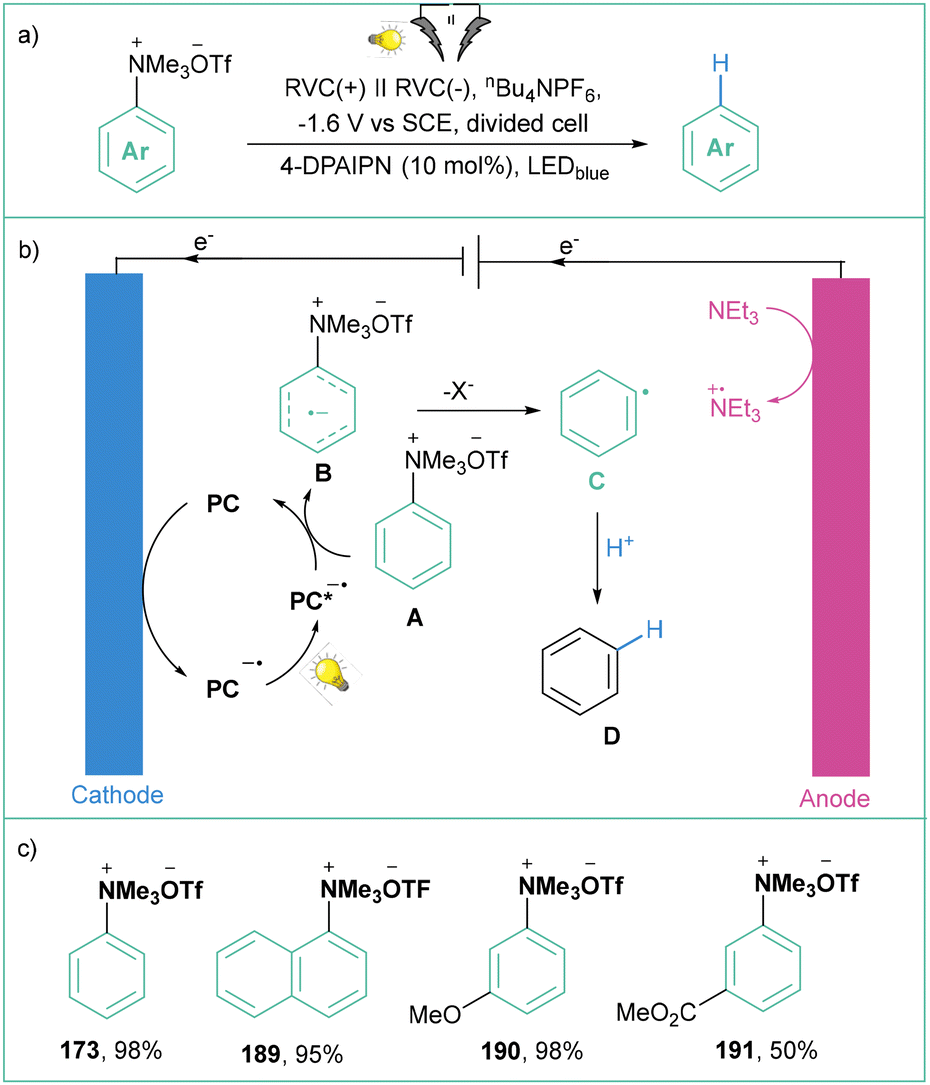 | ||
| Scheme 42 Photoelectrocatalytic activation of quaternary arylammonium salts: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
3.7. Generation of aryl radicals from diaryliodonium salts
Diaryliodonium salts, similarly to diazonium salts, can also be used for the generation of aryl radicals, although they generally feature lower reduction potentials (usually −0.4 to −0.8 V vs. SCE in MeCN).144 These compounds have recently received considerable attention as they are mild, nontoxic, selective, and compatible with many functional groups, and are low-cost reagents in organic synthesis. That's why diaryliodonium salts have become a good alternative to toxic, expensive, heavy-metal-based (Hg, Pb, Pd) catalysts in many organic transformations.145Li and co-workers developed a visible-light-promoted C2-selective arylation of quinoline and pyridine N-oxides using diaryliodonium tetrafluoroborates as arylation reagents (Scheme 43).154 Here, the application of 10 mol% of eosin Y as a photocatalyst and 5 W blue LEDs (450–480 nm) ensures satisfactory yields. BQ (1,4-benzoquinone) or K2S2O8 were used as stoichiometric co-oxidants.
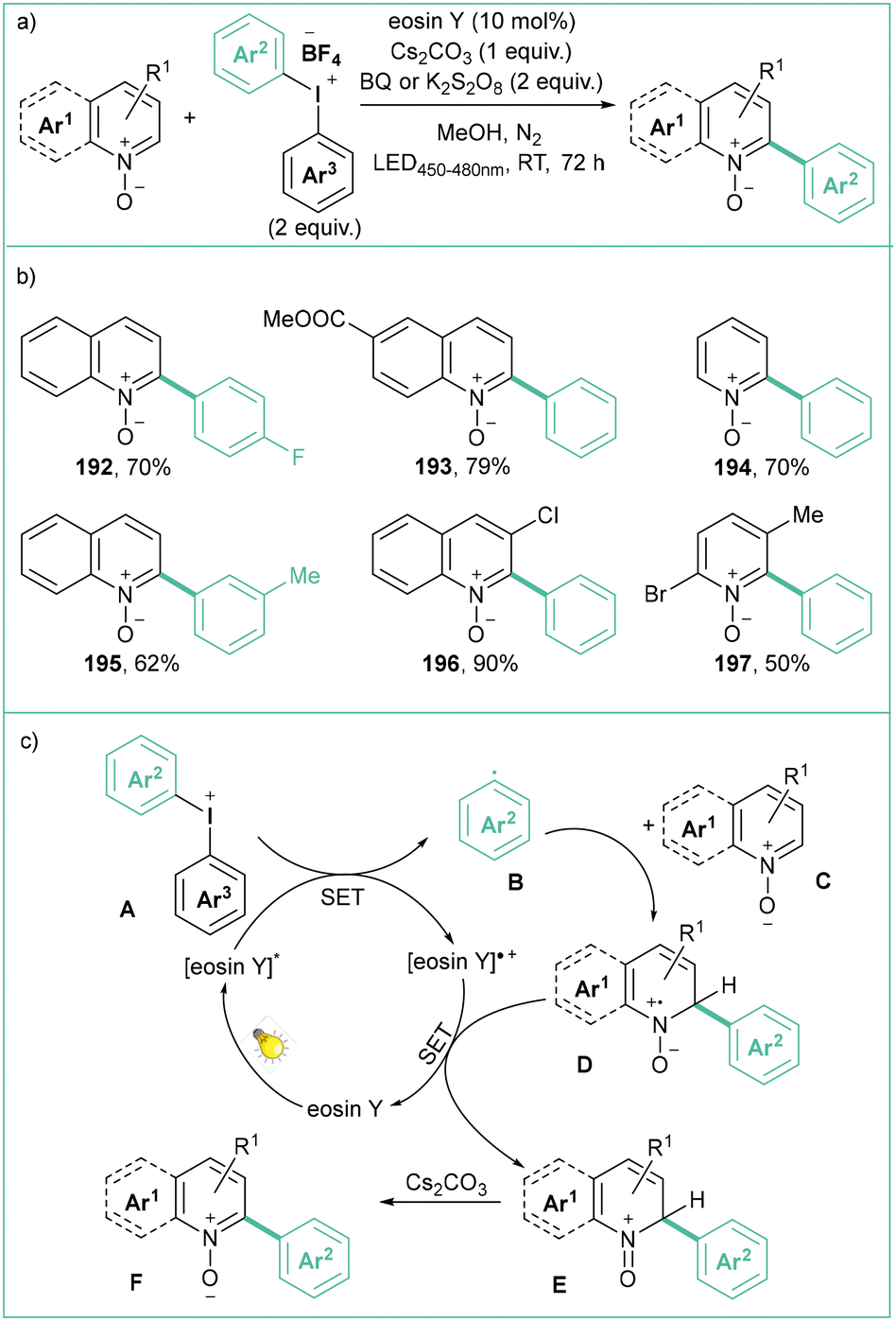 | ||
| Scheme 43 Arylation of quinoline and pyridine N-oxides with diaryliodonium tetrafluoroborates: (a) model reaction, (b) representative products and (c) mechanistic proposal. | ||
According to the mechanistic proposal, eosin Y undergoes excitation by visible light to its excited state and at the same time it reduces aryl iodonium salt A to aryl radical B. Then the radical B undergoes addition to pyridine N-oxide C (or quinoline) to form D, which after single-electron reduction forms E. Product F forms after rearomatization.
Similarly to the method mentioned above, the Ru(II)-based photocatalyst enables direct C3 arylation of quinoxalin-2-(1H)-ones employing easily available and stable diaryliodonium triflates (Scheme 44a).155 Specifically, the reaction proceeds in the presence of Ru(bpy)3Cl2·6H2O (5 mol%) under blue LED irradiation (467 nm) in MeCN. The scope is rather broad, and allyl/propargyl groups are well tolerated in this protocol (Scheme 44b). Importantly, if non-symmetrical diaryliodonium salts are used, the selective transfer of an electron-deficient and less sterically hindered part is preferred. In terms of mechanism, the reaction proceeds in a way analogous to that described earlier (see Scheme 43c).
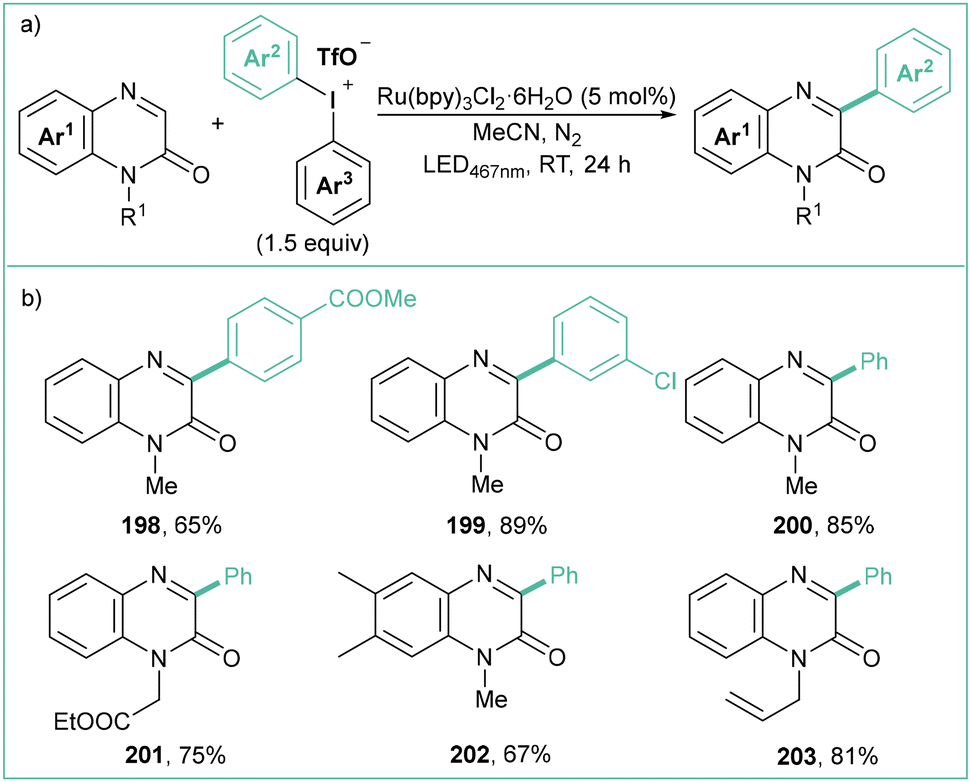 | ||
| Scheme 44 Arylation of quinoxalin-2(1H)-ones with diaryliodonium salts: (a) model reaction and (b) representative products. | ||
In turn, the iridium-based catalyst (Ir(ppy)2(dtbbpy)PF6) proved to be effective in the photocatalytic acylarylation of unactivated alkenes to cyclic ketones (Scheme 45a).156 The aldehyde substitution pattern has no significant impact on the reaction outcome and, at the same time, excellent trans-diastereoselectivity is observed. Initially, the Ir(III) catalyst is excited by visible light to the excited state Ir(III)*. Then, a SET process between Ir(III)* and A leads to an aryl radical B. Such an open-shell species B undergoes addition to the double bond within C, and the resultant radical D attacks the aldehyde functionality to form E. Oxidative quenching of Ir(IV) by this radical eventually gives rise to G (Scheme 45c).
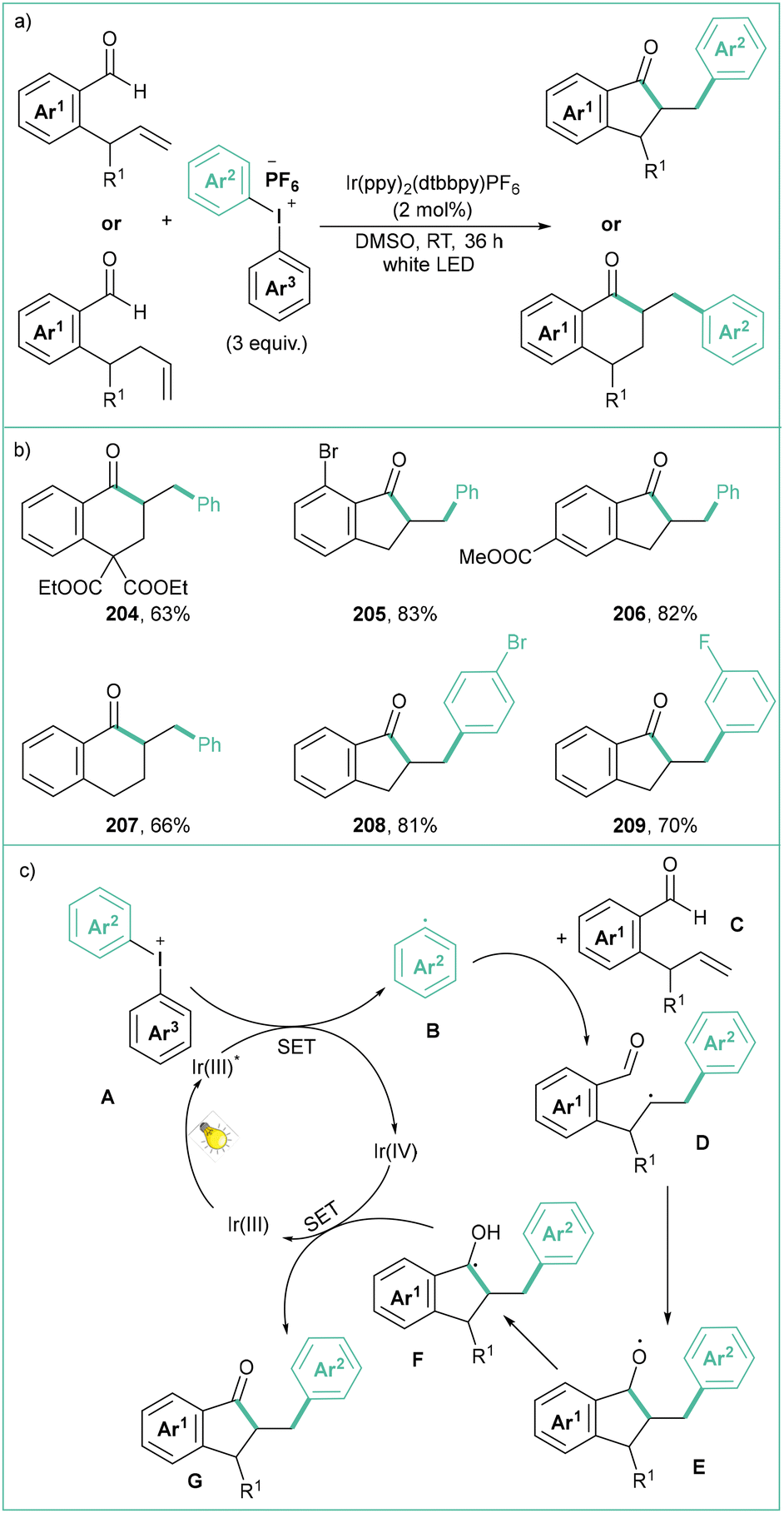 | ||
| Scheme 45 Acylarylation of unactivated alkenes with diaryliodonium salts: (a) model reaction, (b) representative products and (c) mechanistic proposal. | ||
N-Substituted 2-arylbenzoimidazoles show a similar reactivity pattern toward diphenyliodonium triflates affording arylated-benzimidazo[2,1-a]isoquinolin-6(5H)-ones (Scheme 46a).155 This cascade operates under mild conditions and offers a broad substrate scope with appreciable functional group tolerance (Scheme 46b). For non-symmetrical diphenyliodonium triflates, the transfer of an electron-deficient and sterically less hindered aryl ring prevails. The mechanism resembles the one described for the arylation of 2H-quinoxalines (Scheme 46c). The whole process is initiated by visible light photoexcitation of the Ru(II)-based photocatalyst in its excited form. Then, an aryl radical B is produced with the concomitant formation of the Ru(III) complex, which is reduced by a radical D formed via an addition of B to C. The final deprotonation step leads to G.
 | ||
| Scheme 46 Arylative cyclization of N-acryloyl-2-arylbenzoimidazoles with diaryliodonium salts: (a) model reaction, (b) representative products and (c) mechanistic proposal. | ||
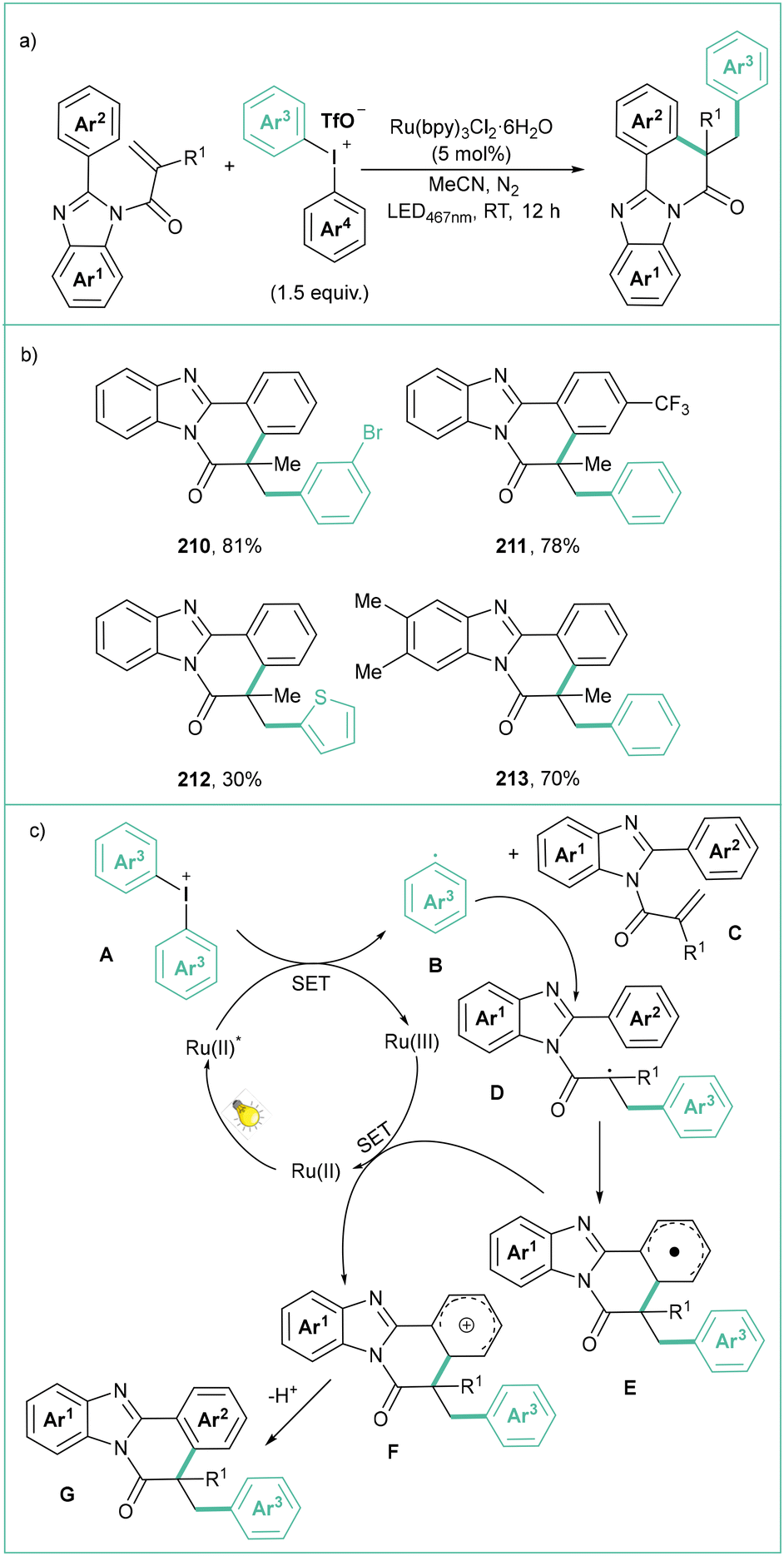
Liu and co-workers reported a three-component visible light photoredox-catalyzed synthesis of N-aminosulfonamides starting from diaryliodonium salts, hydrazines and different sulphur dioxide sources (Scheme 47).157 Here, diaryliodonium salts react with 4-aminomorpholine and sulphur dioxide (generated in situ from K2S2O5 and TFA) in the presence of 2 mol% PDI1 upon blue light irradiation (445 nm), giving rise to N-aminosulfonamides in a selective manner (Scheme 47a). The proposed mechanism (Scheme 47c) starts with a PDI1 photoexcitation and reductive quenching of PDI* with the hydrazine-sulphur dioxide complex B providing a radical adduct C and reduced catalyst PDI1−. Next, a SET between PDI1˙− and the diaryliodonium salt E gives an aryl radical F that then reacts with the sulphur-based radical D to finally give the expected product G.
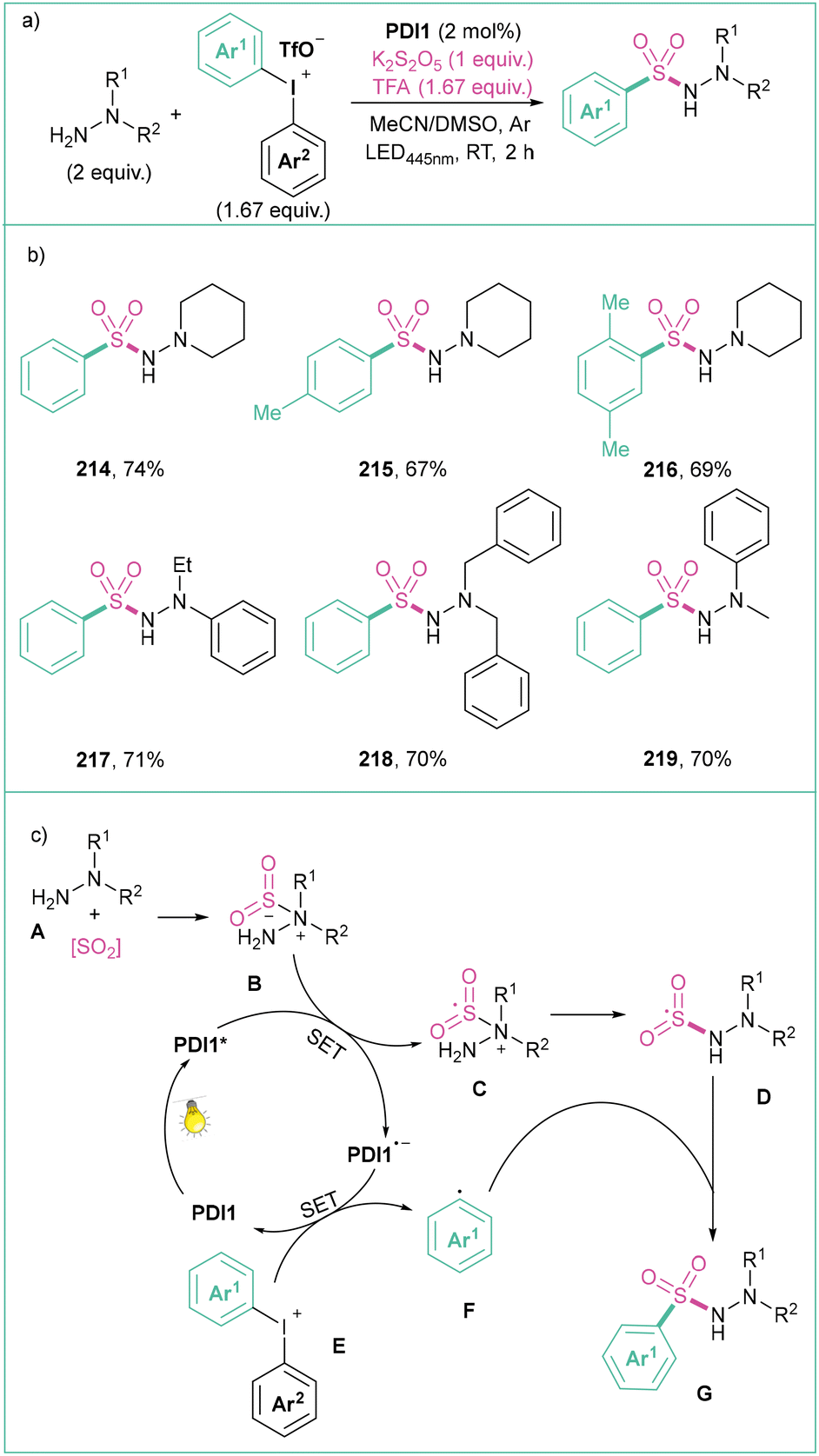 | ||
| Scheme 47 Synthesis of N-aminosulfonamides from diaryliodonium salts: (a) model reaction, (b) representative products and (c) mechanistic proposal. | ||
An efficient protocol for the synthesis of aryl sulfonyl fluorides was presented by Ma and colleagues (Scheme 48a).158 The reaction proceeds in MeCN under blue LED (440–445 nm) irradiation in the presence of a photosensitizer, namely camphorquinone (CQ), with diaryliodonium salts as a source of aryl radicals, alongside DABSO and KHF2 as the fluorine source. The process is compatible with a wide range of electron-donating, neutral, and electron-withdrawing functional groups that are tolerated in this transformation (Scheme 48b). The proposed reaction mechanism starts with the photoexcitation of CQ followed by a SET with salt A to give an aryl radical B. The radical B then interacts with DABSO (C), giving rise to a sulphur-based radical F. This species then reacts with the DABSO radical cation (E) which, under the influence of KHF2, produces G.
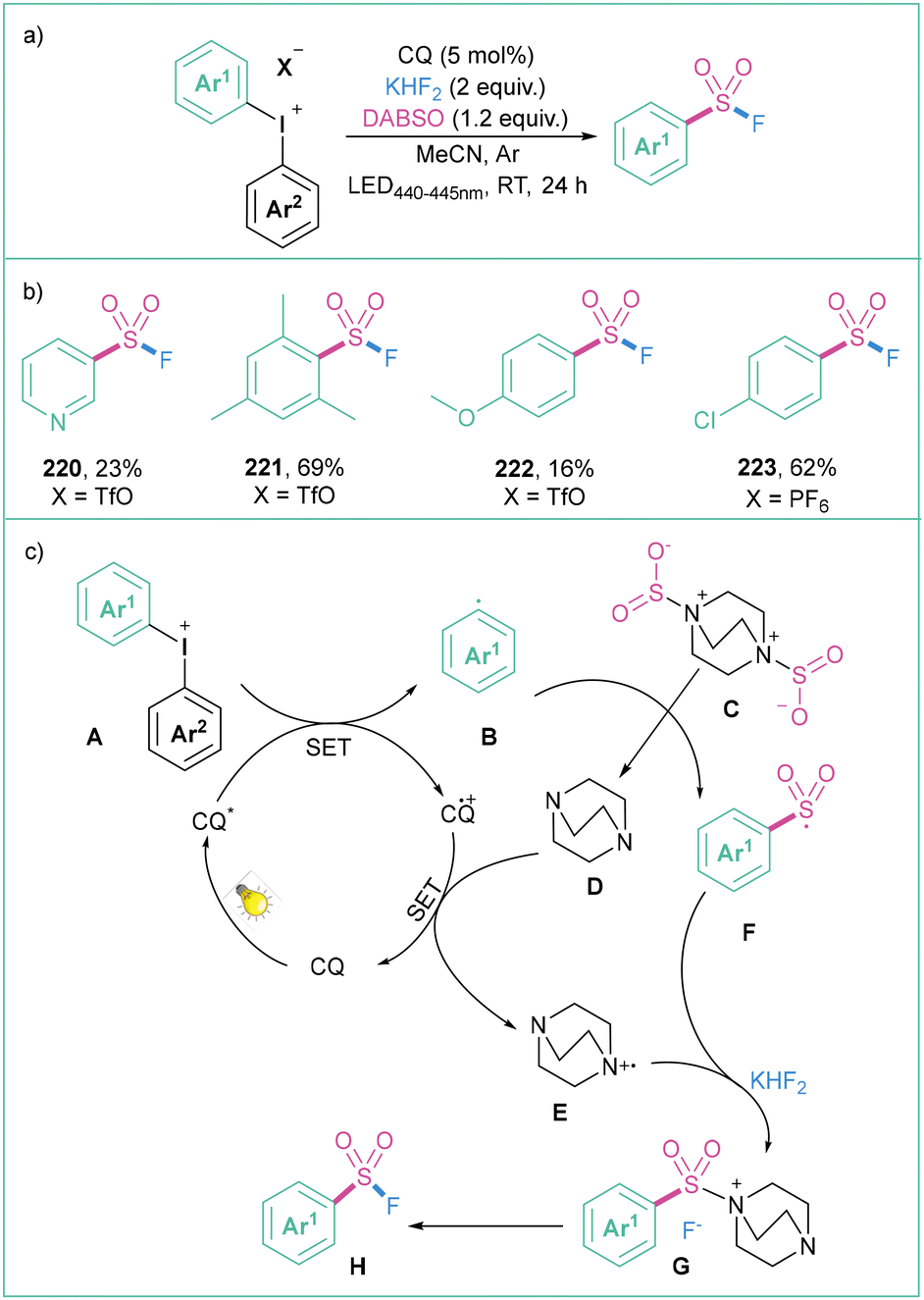 | ||
| Scheme 48 Synthesis of aryl sulfonyl fluorides from diaryliodonium salts: (a) model reaction, (b) representative products and (c) mechanistic proposal. | ||
4. Oxidatively induced generation of aryl radicals
Taking into account three SET methods: PET (photoinduced electron transfer) (e.g. photoredox catalysis and reactions accruing through formation of EDA complexes), organic electrochemistry, and photoelectrochemistry, the oxidative generation of aryl radicals is to date possible from aromatic boronic and carboxylic acids160 as well as aryl hydrazines (Scheme 49). Synthetic methods that use electric current for oxidative functionalisation reactions21b,23,161 have recently received considerable attention and aryl radicals can be generated in that way as well.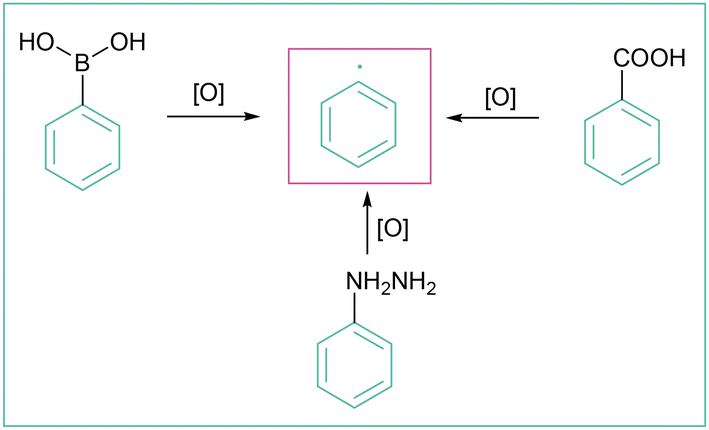 | ||
| Scheme 49 Oxidatively induced generation of aryl radicals from different precursors. | ||
4.1 Generation of aryl radicals from aromatic carboxylic acids and their derivatives
Benzoic acids seem to constitute the most promising precursors of aryl radicals, as they are easily accessible, bench-stable and structurally diverse reagents. However, until very recently, decarboxylative transformations were largely mediated by the use of high temperatures and stoichiometric amounts of heavy metals.6a,160 Among the biggest challenges associated with the oxidative decarboxylation of benzoic acids is the relative kinetic stability of the corresponding carboxyl radicals. Namely, PhCOO˙ was classified by Barton162 as a ‘nondecarboxylating acyloxy radical’, that is, the decarboxylation process should occur only at temperatures above 120–130 °C, which can eventually lead to the formation of unwanted side products. Nevertheless, these shortcomings were resolved partially by means of photoredox catalysis (vide infra), while there is still no method for the generation of Ar˙ in a purely electrochemical way. | ||
| Scheme 50 Decarboxylation of aryl carboxylic acids: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
The mechanistic scenario includes the initial oxidation of the deprotonated aromatic carboxylic acid A by the Ir-based catalyst in an excited state to form radical B. Then, in situ formation of an acyl hypobromite C occurs, which after reduction and concerted cleavage of the C–C and O–Br bonds produces CO2 and aryl radical E. Subsequent trapping of the aryl radical E by a heteroarene generates the rearomatization product F (Scheme 50b). The described protocol is mild, high yielding, and tolerates a broad range of substrates (Scheme 50c); however, the application of an expensive and toxic Ir photocatalyst remains its limitation.
This problem was partially overcome by Yoshimi and co-workers with the use of two active organic photocatalysts: benzophenone and dicyanonaphthalene (DCN) or dicyanoanthracene (DCA) (Scheme 51a).18d Photon-induced electron transfer between benzophenone (BP) and DCN (or DCA) enables the oxidation of BP to its radical cation (BP˙+), which can then oxidise carboxylic acid A to radical B, which after decarboxylation provides aryl radical C. The resultant aryl radical C reacts with electron-deficient olefins D to produce radical E, which after the reduction (enabled by the DCN-based radical anion formed by the catalytic cycle of BP) and protonation forms product G. When the aryl radical C is subjected to the reaction with B2pin2, borylated product H can be assembled employing this method (Scheme 51b). The methodology constitutes a facile synthetic protocol for alkylation and borylation of aromatic rings; however, yields remain moderate to good (Scheme 51c).
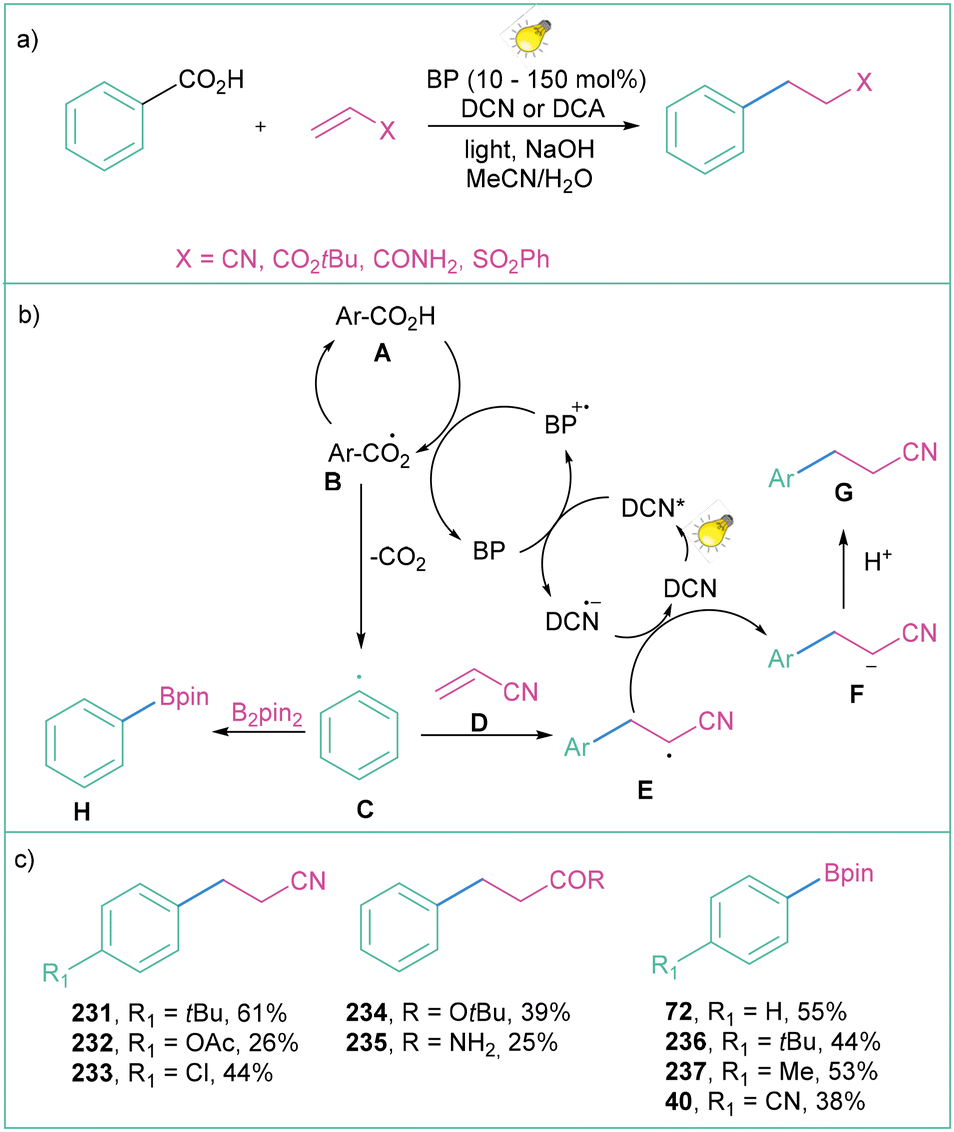 | ||
| Scheme 51 Photoinduced decarboxylative generation of aryl radicals from carboxylic acids: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
The search for a more efficient and sustainable photocatalyst for this transformation brought about an unexpected result: it is possible not to use a photocatalyst at all. The application of N-hydroxyphthalimide esters as aryl radical precursors enables catalyst-free conditions (Scheme 52a).165 The proposed strategy assumes the formation of an EDA complex between the photoexcited acceptor (B) and pyridine adduct (D), which subsequently undergoes an electron transfer, rapid decarboxylation and finally borylation to produce the product C (Scheme 52b). Although in this approach aromatic carboxylic acids need to be functionalised with a NHPI group, the method remains relatively cost-effective and does not require the addition of a photocatalyst. The reaction is operationally simple, scalable, and tolerates a broad range of substrates (Scheme 52c), but most importantly, it is transition-metal- and photocatalyst-free.
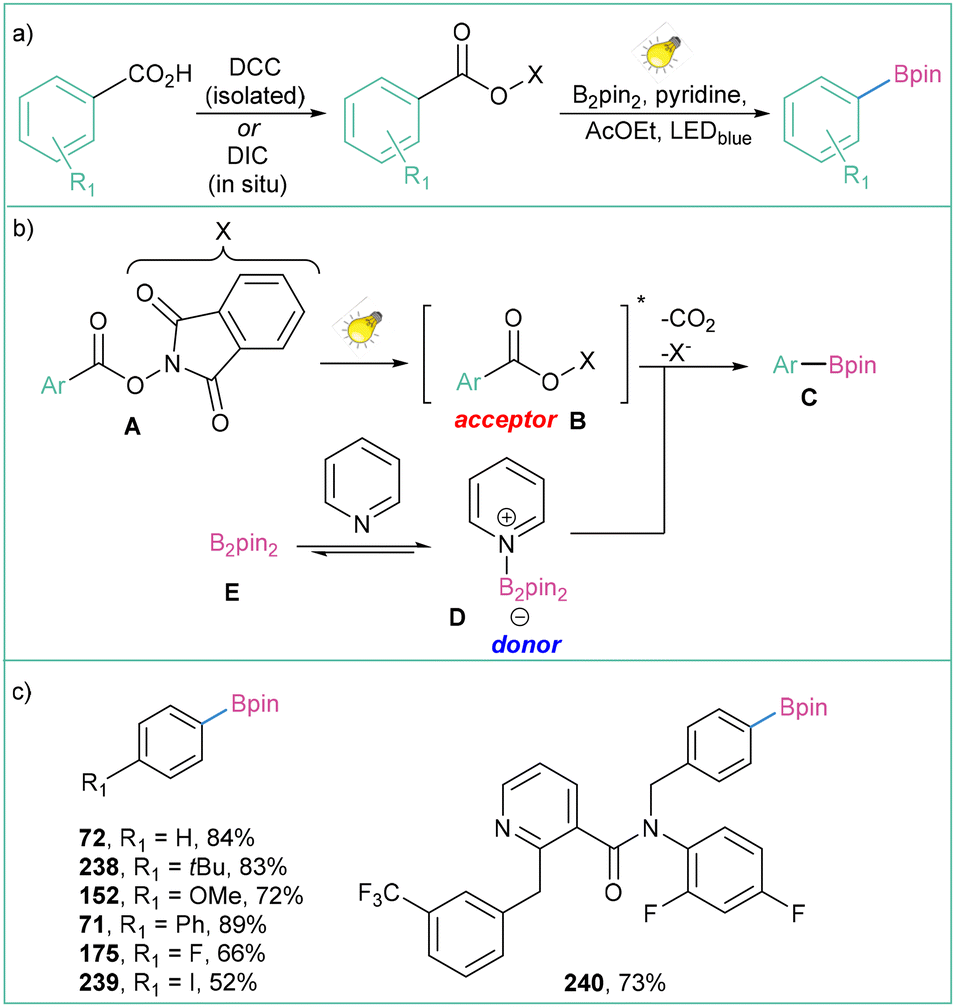 | ||
| Scheme 52 Decarboxylative borylation of aryl N-hydroxyphthalimide esters: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
Over the past decade, photoinduced intramolecular ligand to metal charge transfer (LMCT) has become a powerful strategy for the generation of oxygen-centred radicals through homolysis of oxygen–metal bonds,166 and its applications in the decarboxylation of alkyl carboxylates have been well documented.167 Ritter and co-workers proved that such an approach can also be applied for oxidative aryl radical generation. LMCT enables a mild and general decarboxylation of aryl carboxylic acids that works through high-valent arylcopper(III) intermediates. Subsequently, when generated in this way, aryl radicals can further react, giving rise to fluorinated16 (Scheme 53a) and hydroxylated168 products (Scheme 53b).
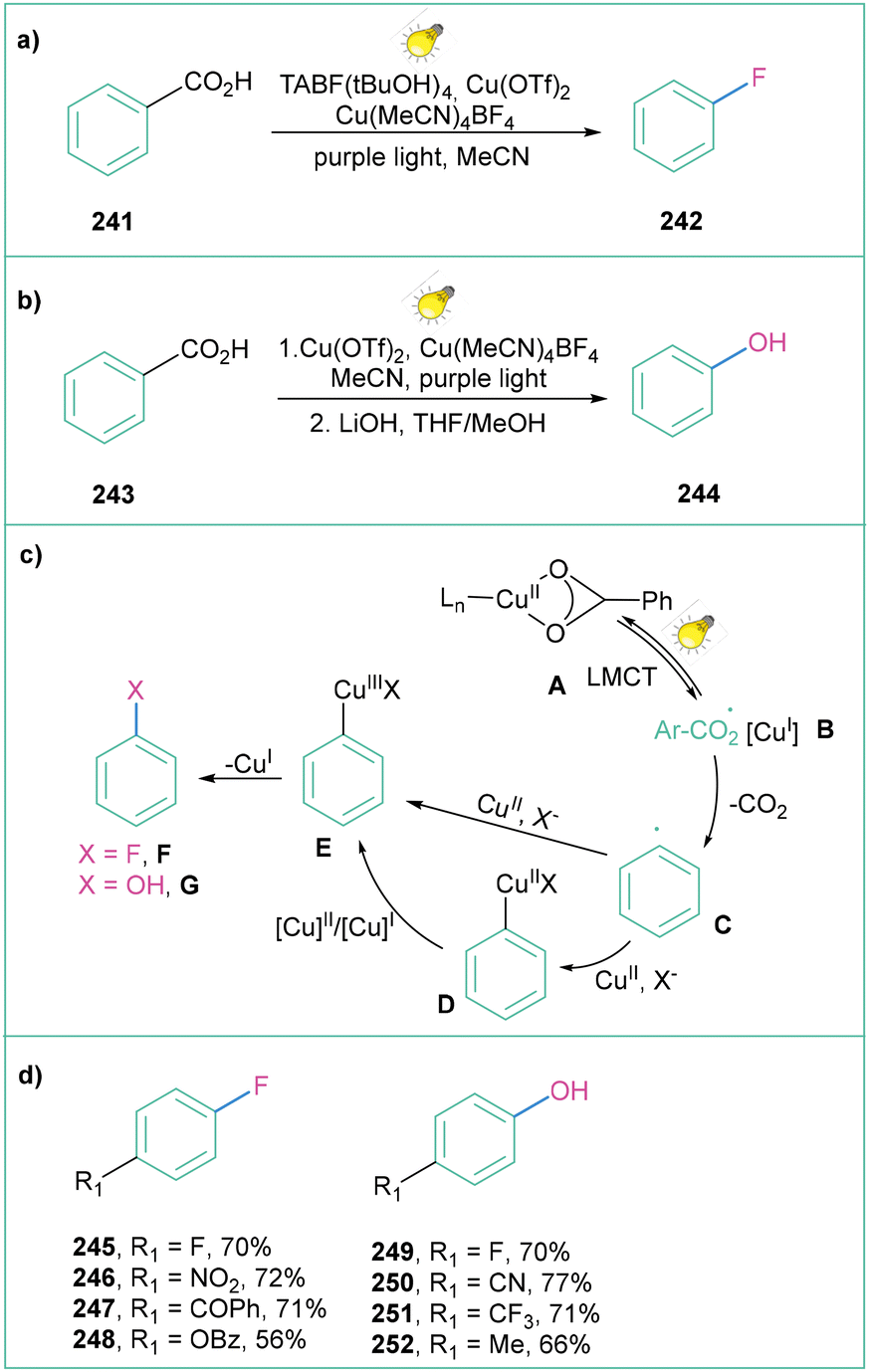 | ||
| Scheme 53 Decarboxylative fluorination and hydroxylation enabled by Cu-LMCT catalysis: (a) fluorination, (b) hydroxylation, (c) mechanistic proposal and (d) representative products. | ||
For both processes, the mechanistic proposal assumes that irradiation of Cu(II) carboxylate A results in carboxylate to CuII ligand to metal charge transfer (LMCT). Next, homolysis of the O–CuII bond results in the formation of aroyloxyl radical B, which after decarboxylation forms aryl radial C. The generated aryl radical C is trapped by a CuIIITC complex or by a CuITC complex with subsequent oxidation by CuII to afford arylcopper(III)TC. In both cases, reductive elimination of the C–O bond forms CuI and fluorinated product F or hydroxylated product G (depending on the reaction conditions) (Scheme 53c). Both protocols are operationally simple, Ir- and Ru-complex free, and moderate to high yielding (Scheme 53d).
MacMillan and co-workers presented a similar approach by developing a copper-catalyzed strategy for the decarboxylative halogenation (I, Br, Cl, F) of hetero(aryl) carboxylic acids (Scheme 54).169 Later, they showed that the reaction could be used for borylations via the merging of Cu-LMCT borylation/bromination cross-coupling (Scheme 54a).170 The proposed mechanism is similar to the one presented by the Ritter group (Scheme 53). The established methodology is general, mild, and moderate to high-yielding (see Scheme 54b); in addition, the synthesis of –Br, –Cl, –F and –I arenes and heteroarenes is compatible with a broad range of (hetero)aryl carboxylic acids (Scheme 54c).
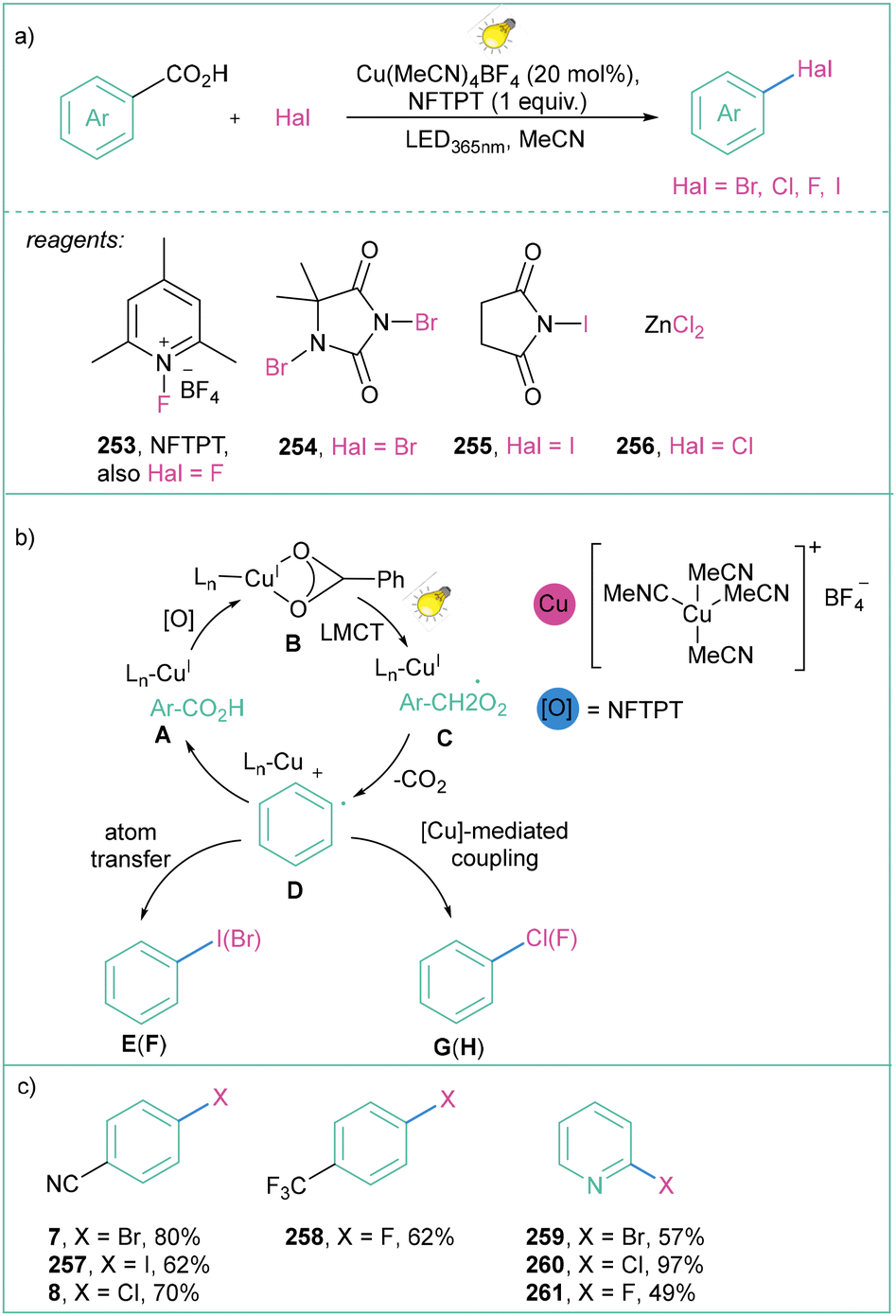 | ||
| Scheme 54 The general approach for the decarboxylative halogenation of aryl(heteroaryl) carboxylic acids: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
The MacMillan group demonstrated further achievements in the area of Cu-LMCT catalysis for the oxidative aryl radical generation and their subsequent application toward the synthesis of halogenated compounds and their cross-coupling. Namely, the authors established a methodology for the borylation of carboxylic acids.170 These boron-based derivatives were subsequently merged with aryl halides (obtained by Cu-LMCT halogenation169) in Pd-catalyzed cross-coupling170 (Scheme 55). Without a doubt, this approach facilitates the direct transformation of aryl carboxylic acid feedstocks into value-added products (pharmaceutically relevant compounds) under mild conditions.
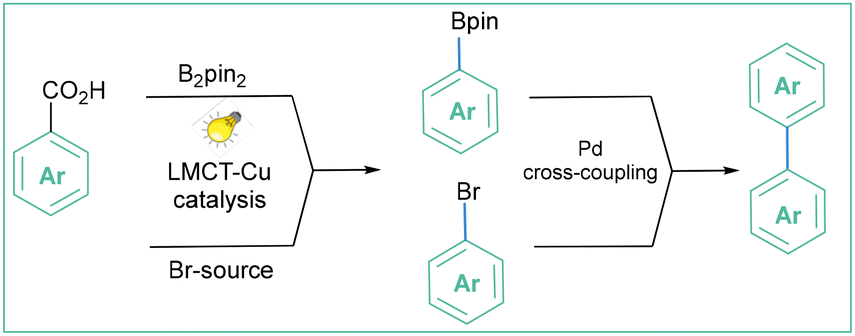 | ||
| Scheme 55 Cu-LMCT decarboxylative borylation/Pd-cross coupling sequence toward value-added compounds. | ||
4.2 Generation of aryl radicals from aromatic boronic acid derivatives
Arylboronic acid derivatives are highly important chemical entities, both in organic synthesis171 and medicinal chemistry.172 The radical chemistry of boron-based compounds has been intensively studied over the past years, resulting in broad synthetic applications thereof.173 Generally, organoboronic acids can serve as practical alkyl or aryl radical precursors via an oxidatively induced carbon–boron bond cleavage. Different catalytic systems based on metal catalysts, e.g. Mn(III), Ag(I), or Fe(II/III) have been demonstrated to be effective for such transformations;174 however, these methods often depend on superstoichiometric amounts of co-oxidants. | ||
| Scheme 56 Photoinduced Meerwin reaction between the aryl boronic acids and electron-deficient olefins: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
The generation of aryl radicals from aromatic boronic acids is also possible without the use of a photocatalyst. Zeng et al. have demonstrated that direct UV irradiation induced the generation of aryl radicals.135a Such an approach was applied toward the deuteration of aromatic compounds with D2O. Differently substituted arylboronic acids (both electron-withdrawing and electron-donating groups) were deuterated with very good yields (Scheme 57a). The mechanistic proposal assumes that the hydroxide ion coordinates to the empty p orbital of the boron and forms anion B. Subsequently, after promotion of the base, the hydroxyl hydrogens in the aryl boronic acid are exchanged for deuterium with D2O. Subsequently, two scenarios can occur: (a) upon UV irradiation, intermediate C is formed, which after rearrangement produces the deuterated product D (path a). On the other hand, intermediate B can undergo cleavage of the C–B bond to form the aryl radical E and radical B(OD)3, and then rapid electron transfer inside the cage between the aryl radical and the boric acid radical anion generates the aryl anion and the boric acid complex F. Finally, the phenyl anion captures deuterium from deuterated boric acid or D2O to form the deuterated product D (Scheme 57b).
 | ||
| Scheme 57 UV-light-induced deuteration of arylboronic acids with D2O: (a) model reaction with representative products and (b) mechanistic proposal. | ||
The application of arylboronic acid is not limited to only Meerwin-type reactions. Anandhan and co-workers displayed an efficient synthesis of 1,2-diketones and internal alkynes from terminal alkynes and arylboronic acids (Scheme 58a).135b
 | ||
| Scheme 58 Photoinduced synthesis of diketones and alkynes: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
The Cu-based complex plays a dual role in this transformation: it acts as a photoredox catalyst and metal catalyst. In the photoredox cycle, the Cu complex is excited and acts as a photooxidant to form aryl radical B′ from aryl boronic acid A′. At the same time, a phenylacetylene A forms phenylacetylide complex B in the presence of Cu(I)Ln. The formed complex B is excited under light irradiation and subsequently reacts with oxygen to form the Cu(II)Ln-phenylacetylide complex D and the oxygen radical anion (O2˙−). Next, the independently generated phenyl radical B′ attacks Cu(II)-phenylacetylide D to form α-stilbene radical Cu(II)-complex E. The radical complex E can take part in two different reaction pathways (paths A, B) depending on the substitution pattern. Path A dominates for substrates with electron-withdrawing groups (such as –NO2), where complex E undergoes rearrangement to the more thermodynamically stable radical F and, following reductive elimination, generates internal alkyne G as the product with regeneration of the [Cu(I)Ln] complex (Scheme 58b). On the other hand, path B dominates for substrates with electron-donating substituents (such as OMe), where complex E undergoes a direct reaction with the oxygen radical anion, followed by rearrangement and elimination toward the formation of diketone G. The photoinduced, Cu-catalyzed reaction of aryl boronic acids with terminal alkynes is efficient, moderate to high yielding and, most importantly, it is chemoselective (depending on the substitution pattern) (Scheme 58c).
Another interesting example of the application of arylboronic acids as precursors of aryl radicals is the synthesis of 3-cyanopyridines (285–288) and 3-cyanopyrroles (289–292) (Scheme 59a).135c The reaction also proceeds via the merging of photo and metal catalysis, with the use of a Pd complex. The cascade reaction proceeds under visible-light irradiation of the Pd catalyst, which subsequently undergoes metal-to-ligand charge transfer (MLCT), followed by redox transmetallation with arylboronic acids. Both types of products are formed in good yields for both electron-withdrawing and electron-donating substituents within the aromatic ring of aryl boronic acid (Scheme 59b).
 | ||
| Scheme 59 Synthesis of 3-cyanopyridines and 3-cyanopyrrole analogues from boronic acids: (a) model reaction and (b) representative products. | ||
In 2016, the first ever example of using arylboronic acid derivatives in order to electrochemically generate aryl radicals was published by Fuchigami and others.18a Namely, anodic oxidation of potassium aryltrifluoroborates in 1.0 M NaOAc/AcOH resulted in acetoxylated products (Scheme 60) in 39–85% yields. In the Fuchigami method, a large amount of electricity was needed to complete the electrolysis (up to 60 F mol−1), yet opening up room for new methods featuring more effective energy and resource utilization.
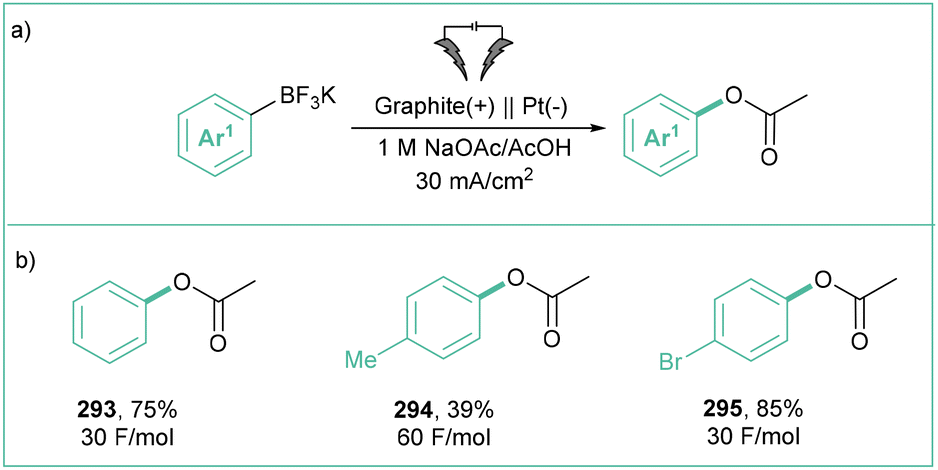 | ||
| Scheme 60 Electrochemical acetoxylation of trifluoroborates: (a) model reaction and (b) representative products. | ||
Later on, Lei's group developed a Mn(II)-mediated, electrochemically induced transformation toward functionalised benzo[4,5]imidazo[2,1-a]isoquinolin-6(5H)-one derivatives180 where alkyl boronic acids were principally used as radical precursors. In this work, the authors also tested phenylboronic acid as a radical precursor, and it appeared that a mixture of products was obtained (Scheme 61). In addition to the expected product 298, a side cyclization product 299 was obtained in 29% yield, where a reactive radical was derived from the interaction of an initially formed aryl radical and acetonitrile.
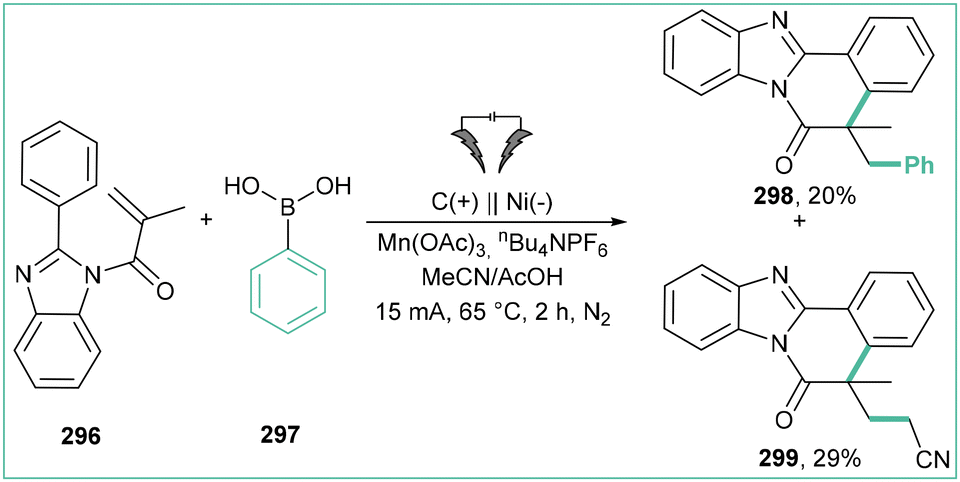 | ||
| Scheme 61 Mn(II)-catalyzed cascade reaction toward the formation of benzo[4,5]imidazo[2,1a]isoquinolin-6(5H)one derivatives. | ||
4.3 Generation of aryl radicals from aryl hydrazines
The generation of aryl radicals from aryl hydrazines is an attractive methodology because the substrates can be readily synthesized via a variety of well-established methods. These include a diazotisation/reduction sequence of anilines, palladium or copper-catalyzed cross-couplings of aryl halides with hydrazine, or aromatic nucleophilic substitution of aryl halides with hydrazine.181 Although synthetically useful, as described below, aryl hydrazines have been proved to be highly toxic/carcinogenic182 which may limit their synthetic usefulness.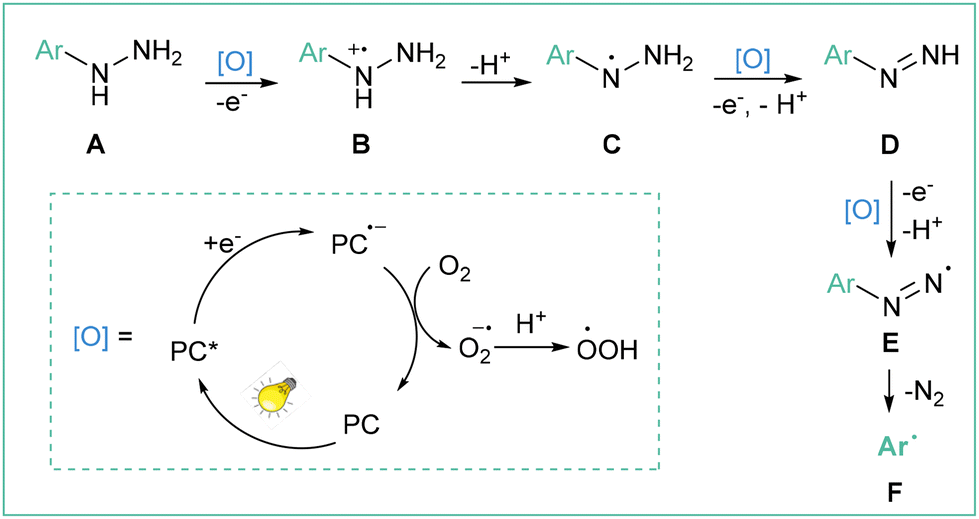 | ||
| Scheme 62 General mechanistic proposal for the generation of aryl radicals from aryl hydrazines. | ||
The construction of 6-substituted phenanthridines constitutes an early example of the use of aryl hydrazines as precursors of aryl radicals under light irradiation (Scheme 63a).135e The mechanistic proposal assumes the photocatalyzed formation of aryl radical B from aryl hydrazine A in a stepwise manner (−3e−, −3H+, –N2). Next, the aryl radical B reacts with 2-isocyanobiphenol C to form radical D, which after cyclization, oxidation, and deprotonation forms the product G (Scheme 63b). The photocyclization proceeds under mild conditions and is very efficient, since various functional groups are tolerant, and the reaction proceeds in moderate to good yields (Scheme 63c). Later, it was also demonstrated that the same transformation can also be catalyzed by the covalent organic framework (COF) as a photocatalyst.183 The photoinduced reaction of aryl hydrazines with α-substituted styrene derivatives in the presence of a photocatalyst and oxygen leads to hydroperoxyarylation of styrenes (Scheme 64a).184 The same process, but applied to unsubstituted styrene derivatives, gives rise to benzyl ketones (Scheme 64b).185
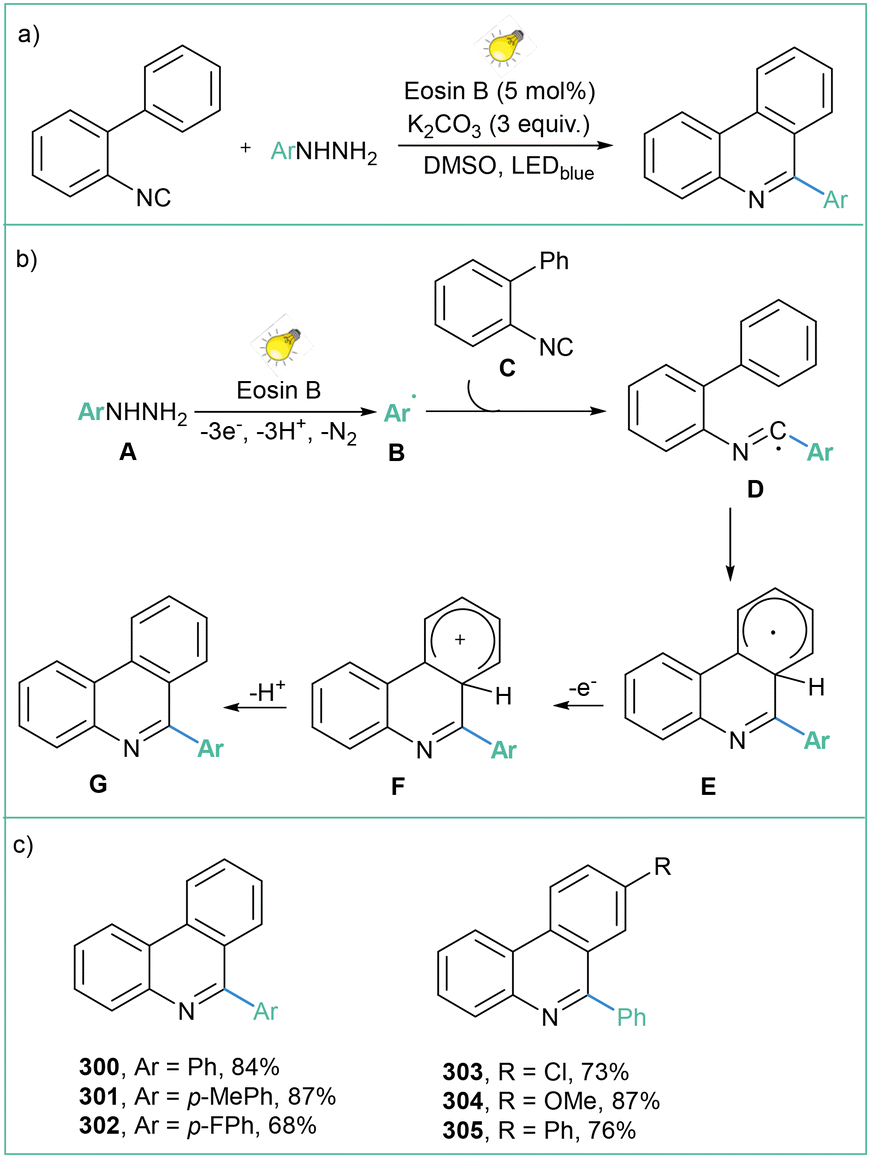 | ||
| Scheme 63 Synthesis of C6-substituted phenanthridines from aryl hydrazines: (a) model reaction, (b) mechanistic proposal and (c) representative products. | ||
 | ||
| Scheme 64 Photoinduced reaction of aryl hydrazines with styrene derivatives: (a) hydroperoxyarylation and (b) synthesis of benzyl ketones. | ||
Aryl hydrazines can also be used as radical precursors for transition metal-free borylation (Scheme 65a).186 Zhang and co-workers demonstrated a comparison of photocatalyzed and electrochemical approaches for the borylation of aryl hydrazines. Generally, the photoredox approach is more efficient than the electrochemical one; however, the electrochemical method is more scalable (Scheme 65b).
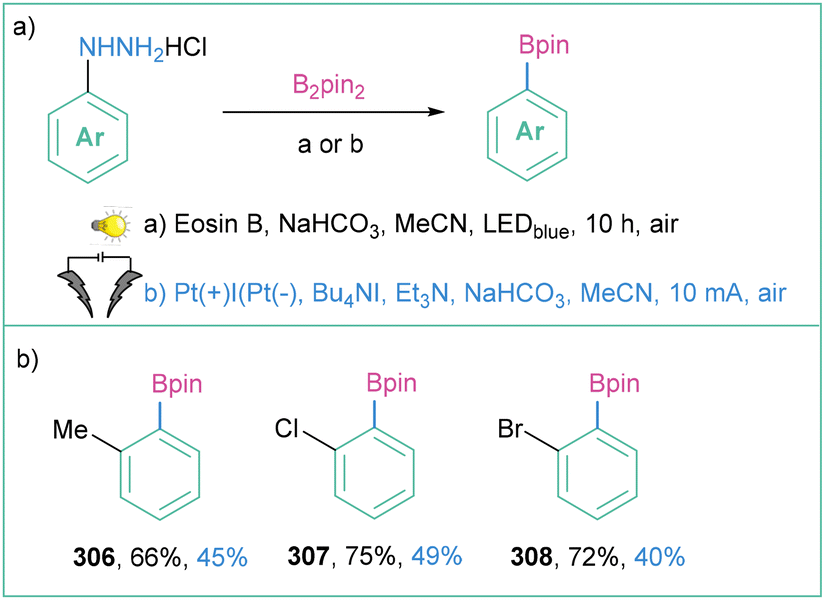 | ||
| Scheme 65 Photochemical and electrochemical borylation of arylhydrazines hydrochlorides: (a) model reactions and (b) representative products. | ||
4.3.2.1 Oxidation of aryl hydrazines. In 2018, Yu et al. developed a cobalt-catalyzed C–H/C–N oxidative coupling between arylhydrazines and N-arylacrylamides that gives oxindoles at room temperature (Scheme 66a).187 The method tolerates a wide range of electron-withdrawing and electron-donating groups within both substrates, although no regioselectivity was observed for unsymmetrically substituted N-acrylamides (Scheme 66b).
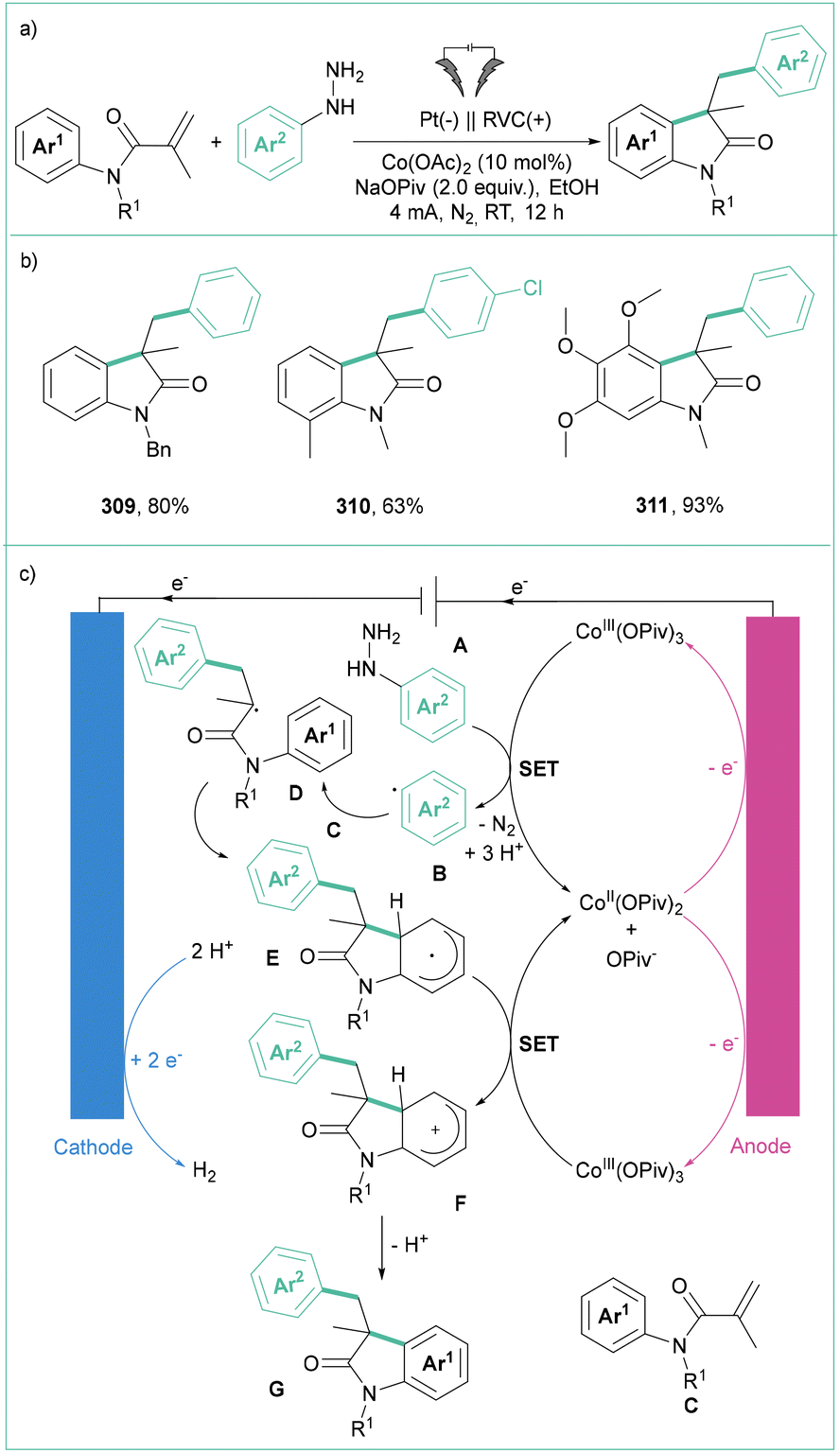 | ||
| Scheme 66 Synthesis of oxoindoles via cobalt-catalyzed electrochemical C–H/C–N oxidation: (a) model reaction, (b) representative products and (c) mechanistic proposal. | ||
According to the proposed mechanism, once aryl radical B is generated from arylhydrazine A by Co-mediated anodic oxidation, it attacks N-arylacrylamide C to gain radical intermediate D. Intramolecular cyclization within D leads to E, which is further oxidised by Co(III)-based species to cationic intermediate F. Deprotonation of F provides the final product G. The presence of the cobalt(II) catalyst was shown to be crucial.
In 2022, Zhou and Li et al. reported an arylation of quinoxalin(on)es with arylhydrazine hydrochlorides under galvanostatic conditions (Scheme 67a).188 The reaction demonstrates a broad functional group tolerance and provides the corresponding aryl products in moderate to excellent yields (Scheme 67b). This may be a result of self-coupling to Ar–Ar under oxidative conditions. The mechanism proposed by the authors closely reassembles the one shown in Scheme 66c, but this time no metal-based catalyst is necessary.
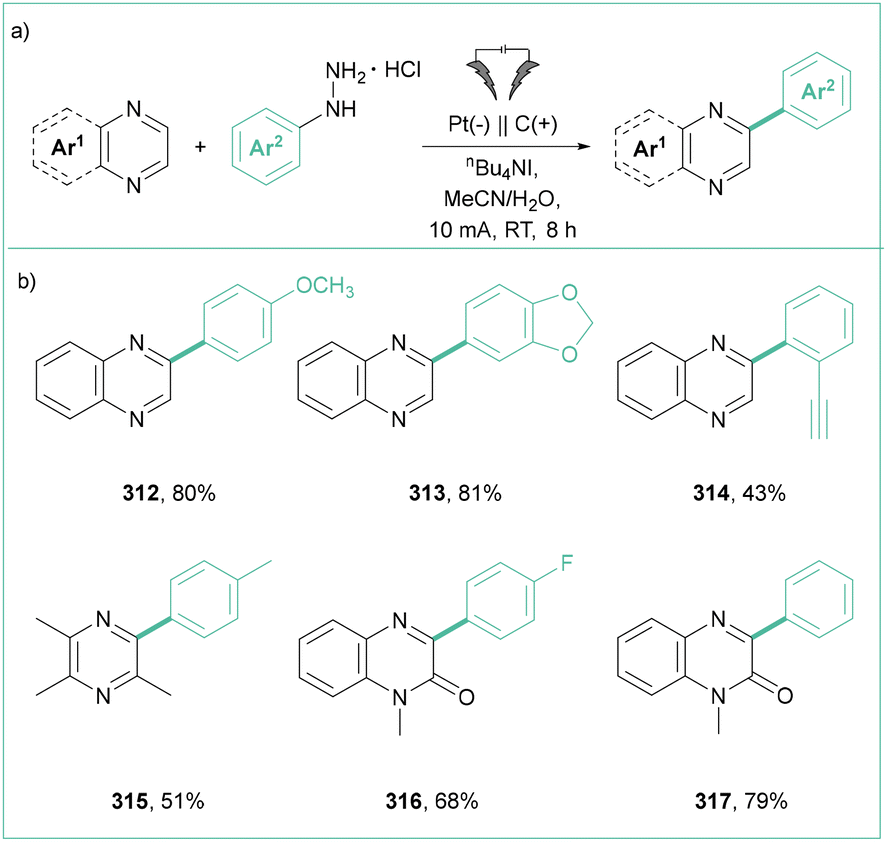 | ||
| Scheme 67 Oxidative C–H arylation of quinoxalin(on)es: (a) model reaction and (b) representative products. | ||
A similar reaction system proved viable in an electrochemical C–N borylation of arylhydrazines (Scheme 68a).186 Substrates containing electron-withdrawing or electron-donating groups in the ortho, meta and para positions are well tolerated (Scheme 68b). The authors found that the use of nBu4NI as an electrolyte is the key to ensure good efficiencies. As depicted in Scheme 68c, once the aryl radical B is formed, it then attacks adduct F (formed from B2pin2E and a bicarbonate ion) to give the desired product D and a radical anion G, which is then electrochemically oxidised toward the neutral species H.
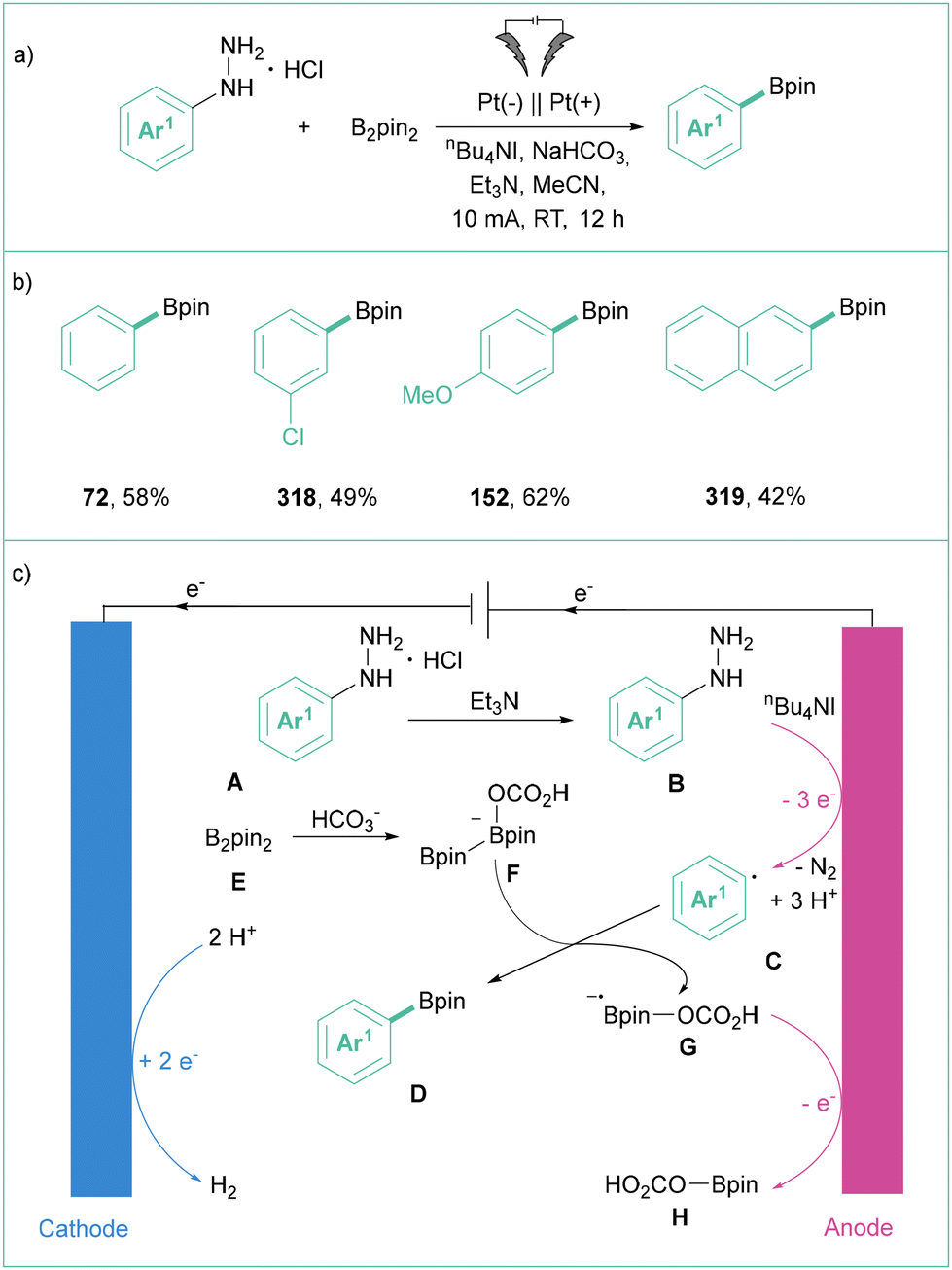 | ||
| Scheme 68 Electrochemical borylation of arylhydrazines: (a) model reaction, (b) representative products and (c) mechanistic proposal. | ||
4.3.2.2 Oxidation of arylsulfonyl hydrazides. Sulfonyl hydrazides are generally known to form sulfonyl radicals under electrochemical conditions thus leading to a variety of sulfonylation reactions.189 However, Lei and Wang et al. have proved that aromatic sulfonyl hydrazides possess the ability to readily form aryl radicals as well (Scheme 69a).190 Namely, electrolysis of arylsulfonyl hydrazine in the presence of isoquinoline N-oxide in a MeCN/HFIP mixture leads to C2-arylated quinolines with moderate to good efficiencies (Scheme 69b). While the procedure tolerates a wide range of variously substituted quinoline N-oxides, isoquinoline N-oxide is not reactive at all. The reaction is initiated by the anodic oxidation of the hydrazine moiety, releasing N2 and SO2 and giving rise to an aryl radical C. This open-shell intermediate then undergoes addition to arylsulfonyl hydrazine leading to intermediate E which, after the reduction/protonation/dehydration sequence, gives the final arylation product H (Scheme 69c). This process constitutes an example of linear paired electrolysis where two electrode processes (oxidation and reduction) are essential to form the final product H. The application of such an approach undoubtedly increases the energy and resource economy of a given process.
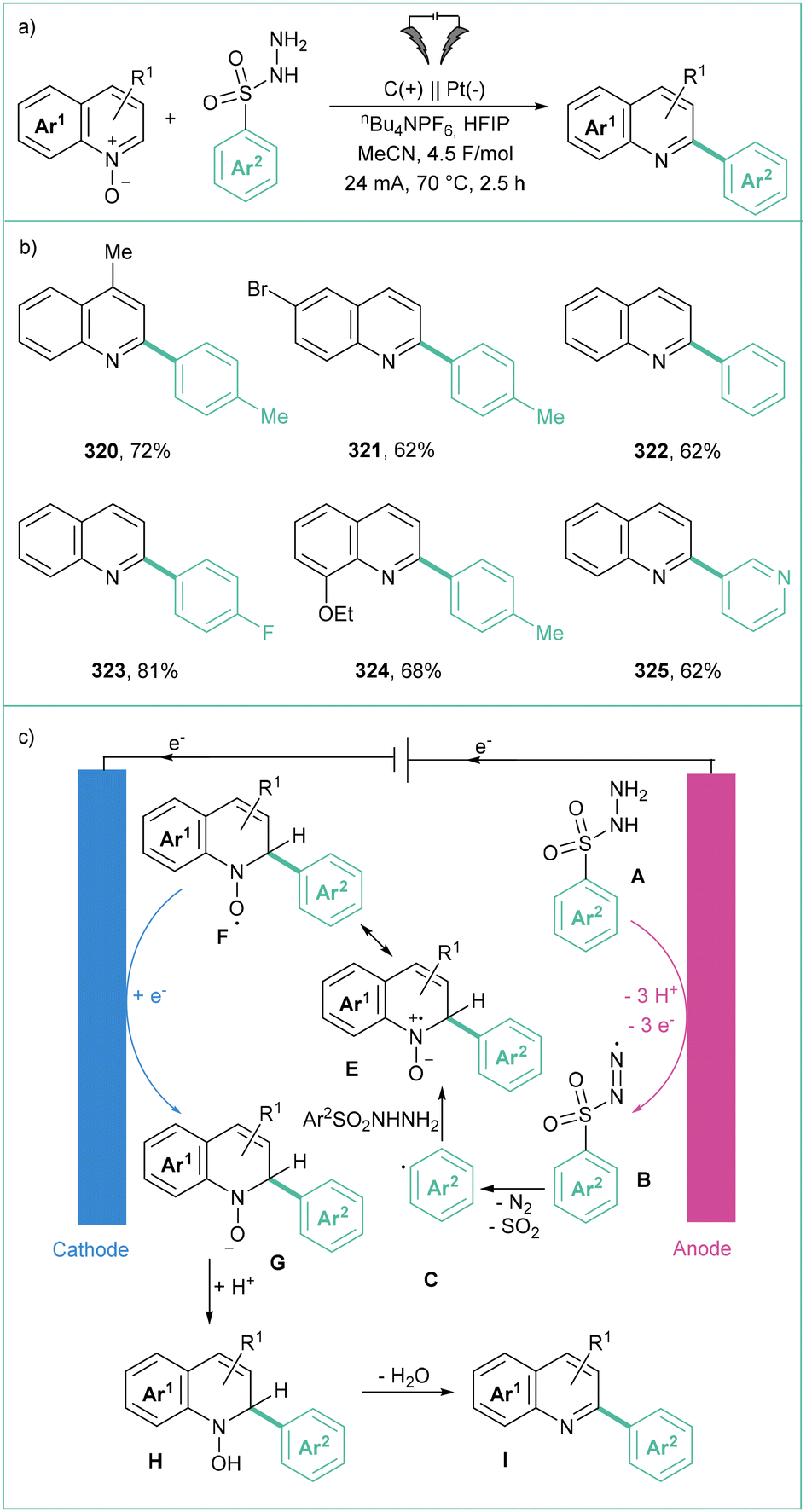 | ||
| Scheme 69 Proposed mechanism for the deoxygenative C2 arylation of quinoline N-oxides: (a) model reaction, (b) representative products and (c) mechanistic proposal. | ||
5. Summary and outlook
Recent numerous achievements in the field of aryl radical chemistry place demand for a clear and systematized survey that critically and timely sums them up in terms of a given synthetic approach and sustainability (energy and resource utilization). In this review article, we summarise recent achievements in the field of aryl radical generation by means of photoredox catalysis, electrochemistry and their constructive combination – photoelectrochemistry. Importantly, the number of methods that use light or electric power to induce chemical transformations leading to aryl radicals is growing rapidly. The overwhelming majority of them are light-driven reactions. Despite the many benefits of photoredox catalysis, this approach also has some issues that should be solved: challenges in scale-up, due to the limited light penetration; limited electrochemical potential window, due to the energy limits resulting from the energy of visible photons; and insufficient atom economy, due to the addition of stoichiometric amounts of sacrificial reagents.In some cases electrochemistry and light-assistance electrochemistry started to be an answer to these problems. However, at this time, there are still no examples in the literature of electrochemically driven organic reactions that employ arylboronic acids, phenol derivatives, diarylsulfonium salts, diaryliodonium salts, or benzoic acids as precursors. This fact can be partially explained by limited access to suitable electrode materials, potentiostats, or cells. In the case of photoredox catalysis, only a source of light is needed to start research on this topic. As mentioned in the Introduction, the presence of electrodes that are immersed in a solution sets the stage for possible side reactions such as reduction of Ar˙ to Ar− or electrografting. These side reactions may be prevented by using a divided cell, different electrode material, or an electron mediator.63,191 One may notice that for reductively induced aryl radical preparation, metal-based cathodes are crucial for a successful transformation. In contrast, oxidatively induced methods are based on carbon anodes, strongly increasing the possibility of electrografting occurring within the electrode surface. A possible solution for this particular challenge may be the application of electrodes with already modified surfaces, as in the work of Berben and others.192 Such modification may prevent direct arylation of the surface as well as modulate the electron-transfer event itself.
Most of electrochemical reactions presented herein (besides photoelectrochemical ones) are performed in an undivided cell, which significantly simplifies the formation of aryl radical using electricity.
Still, redox potential mismatch is a serious problem; however, recent developments in photoelectrochemistry26,29 have unlocked some challenging aryl radical precursors toward selective electrochemical transformations. Although some organic dyes have been already tested in photoelectrochemical transformations,26,29,79,193 we believe that new types of photoelectrocatalysts, either organic or inorganic, will be found to unlock unexplored redox windows as well as dramatically expand the synthetic possibilities offered by this field.
Needless to say, stereoselective transformations are pivotal in assembling molecules with high structural complexity. Despite recent developments in the assembling of molecules via “an aryl radical” pathway, no stereoselective methods have been developed so far. This is a massively underexplored research area, and we believe that this review will contribute to its further development.
Importantly, an overwhelming number of reactions described in this review were performed in solvents featuring a similar value of dipole moment (∼3.9 D) – MeCN or DMF (often as mixtures with protic solvents – MeOH, AcOH or H2O). This fact underlines the need for applying a polarized environment that will facilitate processes that occur via electron transfer and/or through charged intermediates.
Taking into account all of the above considerations, we certainly can say that electrochemistry has started to compete with photoredox catalysis in the area of aryl radical generation research; however, it is still at the very early stage of development in practical organic synthesis. We hope that this review will help to advance research on the design of new reaction systems that will allow the generation of aryl radicals and their use in further transformations in a more efficient, selective and controllable way.
Author contributions
Krzysztof Grudzień writing-original draft, writing-review & editing; Andrei Zlobin writing-original draft, writing-review & editing; Jan Zadworny writing-original draft; Katarzyna Rybicka-Jasińska conceptualization, funding acquisition, writing-original draft, writing-review & editing; Bartłomiej Sadowski conceptualization, funding acquisition, project administration, supervision, writing-original draft, writing-review & editing.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the Polish National Science Centre (Sonata 2021/43/D/ST4/02267 to B. S.) and Sonata (UMO-2020/39/D/ST4/01510 to K. R.-J.). B. S. is a recipient of a scholarship awarded by the Polish Ministry of Education and Science to outstanding young scientists. The authors would like to thank Joseph P. Milton (Institute of Organic Chemistry, Polish Academy of Sciences) for proofreading the manuscript.References
- (a) K. Matyjaszewski and J. Xia, Atom Transfer Radical Polymerization, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed; (b) K. J. Romero, M. S. Galliher, D. A. Pratt and C. R. J. Stephenson, Radicals in natural product synthesis, Chem. Soc. Rev., 2018, 47, 7851–7866 RSC; (c) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Recent Advances in Radical C–H Activation/Radical Cross-Coupling, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed.
- E. P. Tomlinson, M. E. Hay and B. W. Boudouris, Radical Polymers and Their Application to Organic Electronic Devices, Macromolecules, 2014, 47, 6145–6158 CrossRef CAS.
- J. Stubbe and D. G. Nocera, Radicals in Biology: Your Life Is in Their Hands, J. Am. Chem. Soc., 2021, 143, 13463–13472 CrossRef CAS PubMed.
- P. Griess, Ueber eine neue Klasse organischer Verbindungen, in denen Wasserstoff durch Stickstoff vertreten ist, Justus Liebigs Ann. Chem., 1866, 137, 39–91 CrossRef.
- M. Gomberg, An instance of trivalent carbon: triphenylmethyl, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- (a) D. I. Bugaenko, A. A. Volkov, A. V. Karchava and M. A. Yurovskaya, Generation of aryl radicals by redox processes. Recent progress in the arylation methodology, Russ. Chem. Rev., 2021, 90, 116–170 CrossRef CAS; (b) N. Kvasovs and V. Gevorgyan, Contemporary methods for generation of aryl radicals, Chem. Soc. Rev., 2021, 50, 2244–2259 RSC; (c) G. Pratsch and M. R. Heinrich, in Radicals in Synthesis III, ed. M. Heinrich and A. Gansäuer, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 33–59 Search PubMed.
- (a) H. Zipse, Top. Curr. Chem., 2006, 263, 163–189 CrossRef CAS; (b) M. D. Wodrich, W. C. McKee and P. V. R. Schleyer, On the Advantages of Hydrocarbon Radical Stabilization Energies Based on R−H Bond Dissociation Energies, J. Org. Chem., 2011, 76, 2439–2447 CrossRef CAS PubMed.
- Y.-R. Luo, Handbook of Bond Dissociation Energies, CRC Press, New York, 2003 Search PubMed.
- D. Griller and K. U. Ingold, Persistent carbon-centered radicals, Acc. Chem. Res., 1976, 9, 13–19 CrossRef CAS.
- (a) C. P. Andrieux and J. Pinson, The Standard Redox Potential of the Phenyl Radical/Anion Couple, J. Am. Chem. Soc., 2003, 125, 14801–14806 CrossRef CAS PubMed; (b) B. Branchi, C. Galli and P. Gentili, Reactivity of Aryl and Vinyl Radicals: Abstraction of Hydrogen Atom or Reaction with a Nucleophile, Eur. J. Org. Chem., 2002, 2844–2854 CrossRef CAS.
- Y. Matsuki, N. Ohnishi, Y. Kakeno, S. Takemoto, T. Ishii, K. Nagao and H. Ohmiya, Aryl radical-mediated N-heterocyclic carbene catalysis, Nat. Commun., 2021, 12, 3848 CrossRef CAS PubMed.
- A. Cai, W. Yan and W. Liu, Aryl Radical Activation of C–O Bonds: Copper-Catalyzed Deoxygenative Difluoromethylation of Alcohols, J. Am. Chem. Soc., 2021, 143, 9952–9960 CrossRef CAS PubMed.
- G. Yan, M. Yang and X. Wu, Synthetic applications of arylboronic acid via an aryl radical transfer pathway, Org. Biomol. Chem., 2013, 11, 7999–8008 RSC.
- (a) S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Potassium t-Butoxide Alone Can Promote the Biaryl Coupling of Electron-Deficient Nitrogen Heterocycles and Haloarenes, Org. Lett., 2008, 10, 4673–4676 CrossRef CAS PubMed; (b) E. Shirakawa, K.-I. Itoh, T. Higashino and T. Hayashi, tert-Butoxide-Mediated Arylation of Benzene with Aryl Halides in the Presence of a Catalytic 1,10-Phenanthroline Derivative, J. Am. Chem. Soc., 2010, 132, 15537–15539 CrossRef CAS PubMed; (c) C.-L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, B.-J. Li and Z.-J. Shi, An efficient organocatalytic method for constructing biaryls through aromatic C–H activation, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS PubMed; (d) J. A. Murphy, Discovery and Development of Organic Super-Electron-Donors, J. Org. Chem., 2014, 79, 3731–3746 CrossRef CAS PubMed; (e) G. Nocera and J. A. Murphy, Ground State Cross-Coupling of Haloarenes with Arenes Initiated by Organic Electron Donors, Formed in situ: An Overview, Synthesis, 2020, 327–336 CAS; (f) N. Bashir, O. Callaghan, J. A. Murphy, T. Ravishanker and S. J. Roome, On The Mechanism of the Tetrathiafulvalene-Mediated Radical-Polar Crossover Reactions, Tetrahedron Lett., 1997, 38, 6255–6258 CrossRef CAS; (g) N. Bashir and J. A. Murphy, The role of neighbouring group participation in TTF-mediated ‘radical-polar crossover’ reactions: trapping of aliphatic radicals by TTF+·, Chem. Commun., 2000, 627–628 RSC; (h) E. Cahard, F. Schoenebeck, J. Garnier, S. P. Y. Cutulic, S. Zhou and J. A. Murphy, Electron Transfer to Benzenes by Photoactivated Neutral Organic Electron Donor Molecules, Angew. Chem., Int. Ed., 2012, 51, 3673–3676 CrossRef CAS PubMed; (i) J. A. Murphy, T. A. Khan, S.-z. Zhou, D. W. Thomson and M. Mahesh, Highly Efficient Reduction of Unactivated Aryl and Alkyl Iodides by a Ground-State Neutral Organic Electron Donor, Angew. Chem., Int. Ed., 2005, 44, 1356–1360 CrossRef CAS PubMed; (j) S. S. Hanson, E. Doni, K. T. Traboulsee, G. Coulthard, J. A. Murphy and C. A. Dyker, Pushing the Limits of Neutral Organic Electron Donors: A Tetra(iminophosphorano)-Substituted Bispyridinylidene, Angew. Chem., Int. Ed., 2015, 54, 11236–11239 CrossRef CAS PubMed; (k) J. P. Barham, G. Coulthard, R. G. Kane, N. Delgado, M. P. John and J. A. Murphy, Double Deprotonation of Pyridinols Generates Potent Organic Electron–Donor Initiators for Haloarene–Arene Coupling, Angew. Chem., Int. Ed., 2016, 55, 4492–4496 CrossRef CAS PubMed.
- A. Studer and D. P. Curran, The electron is a catalyst, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed.
- P. Xu, P. López-Rojas and T. Ritter, Radical Decarboxylative Carbometalation of Benzoic Acids: A Solution to Aromatic Decarboxylative Fluorination, J. Am. Chem. Soc., 2021, 143, 5349–5354 CrossRef CAS PubMed.
- (a) X. Tian, Y. Liu, S. Yakubov, J. Schütte, S. Chiba and J. P. Barham, Photo- and electro-chemical strategies for the activations of strong chemical bonds, Chem. Soc. Rev., 2024, 53, 263–316 RSC; (b) N. E. S. Tay, D. Lehnherr and T. Rovis, Photons or Electrons? A Critical Comparison of Electrochemistry and Photoredox Catalysis for Organic Synthesis, Chem. Rev., 2022, 122, 2487–2649 CrossRef CAS PubMed; (c) R. H. Verschueren and W. M. De Borggraeve, Electrochemistry and Photoredox Catalysis: A Comparative Evaluation in Organic Synthesis, Molecules, 2019, 24, 2122 CrossRef PubMed.
- (a) J. Suzuki, M. Tanigawa, S. Inagi and T. Fuchigami, Electrochemical Oxidation of Organotrifluoroborate Compounds, ChemElectroChem, 2016, 3, 2078–2083 CrossRef CAS; (b) T. S. Calderwood, C. L. Johlman, J. L. Roberts, C. L. Wilkins and D. T. Sawyer, Oxidation of substituted hydrazines by superoxide ion: the initiation step for the autoxidation of 1,2-diphenylhydrazine, J. Am. Chem. Soc., 1984, 106, 4683–4687 CrossRef CAS; (c) W. J. Wei, Y. J. Zhong, Y. F. Feng, L. Gao, H. T. Tang, Y. M. Pan, X. L. Ma and Z. Y. Mo, Electrochemically Mediated Direct C(sp3)−H Sulfonylation of Xanthene Derivatives, Adv. Synth. Catal., 2022, 364, 726–731 CrossRef CAS; (d) S. Kubosaki, H. Takeuchi, Y. Iwata, Y. Tanaka, K. Osaka, M. Yamawaki, T. Morita and Y. Yoshimi, Visible- and UV-Light-Induced Decarboxylative Radical Reactions of Benzoic Acids Using Organic Photoredox Catalysts, J. Org. Chem., 2020, 85, 5362–5369 CrossRef CAS PubMed; (e) A. Arts, K. P. van den Berg, M. T. de Groot and J. van der Schaaf, Electrochemical oxidation of benzoic acid and its aromatic intermediates on boron doped diamond electrodes, Curr. Res. Green Sustainable Chem., 2021, 4, 100217 CrossRef CAS.
- (a) J. Xu, J. Cao, X. Wu, H. Wang, X. Yang, X. Tang, R. W. Toh, R. Zhou, E. K. L. Yeow and J. Wu, Unveiling Extreme Photoreduction Potentials of Donor–Acceptor Cyanoarenes to Access Aryl Radicals from Aryl Chlorides, J. Am. Chem. Soc., 2021, 143, 13266–13273 CrossRef CAS PubMed; (b) B. K. Malviya, K. Singh, P. K. Jaiswal, M. Karnatak, V. P. Verma, S. S. Badsara and S. Sharma, Catalyst-free synthesis of phenanthridines via electrochemical coupling of 2-isocyanobiphenyls and amines, New J. Chem., 2021, 45, 6367–6378 RSC; (c) J. Li, J. Chen, R. Sang, W.-S. Ham, M. B. Plutschack, F. Berger, S. Chabbra, A. Schnegg, C. Genicot and T. Ritter, Photoredox catalysis with aryl sulfonium salts enables site-selective late-stage fluorination, Nat. Chem., 2020, 12, 56–62 CrossRef CAS PubMed; (d) S.-P. Liu, Y.-H. He and Z. Guan, Photoredox-Catalyzed Radical–Radical Cross-Coupling of Sulfonyl Chlorides with Trifluoroborate Salts, J. Org. Chem., 2023, 88, 11161–11172 CrossRef CAS PubMed; (e) X. Kong, Y. Chen, X. Chen, C. Ma, M. Chen, W. Wang, Y.-Q. Xu, S.-F. Ni and Z.-Y. Cao, Organomediated electrochemical fluorosulfonylation of aryl triflates via selective C–O bond cleavage, Nat. Commun., 2023, 14, 6933 CrossRef CAS PubMed; (f) J. Annibaletto, C. Jacob and C. Theunissen, Ammonium Salts as Convenient Radical Precursors Using Iridium Photoredox Catalysis, Org. Lett., 2022, 24, 4170–4175 CrossRef CAS PubMed; (g) R. Shaw, N. Sihag, S. Jain, R. Sharma and M. R. Yadav, Photoinduced Alkyl/Aryl Radical Cascade for the Synthesis of Quaternary CF3-Containing Oxindoles and Indoline Alkaloids, J. Org. Chem., 2023, 88, 5652–5660 CrossRef CAS PubMed.
- (a) C. K. Prier, D. A. Rankic and D. W. C. Macmillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) J. W. Tucker and C. R. J. Stephenson, Shining Light on Photoredox Catalysis: Theory and Synthetic Applications, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed; (c) K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (d) M. Schmalzbauer, M. Marcon and B. König, Excited State Anions in Organic Transformations, Angew. Chem., Int. Ed., 2021, 60, 6270–6292 CrossRef CAS PubMed; (e) N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (f) B. König, Photocatalysis in Organic Synthesis – Past, Present, and Future, Eur. J. Org. Chem., 2017, 1979–1981 CrossRef.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (b) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Electrochemical Arylation Reaction, Chem. Rev., 2018, 118, 6706–6765 CrossRef CAS PubMed; (c) K. Kuciński, Electrifying synthesis of organosilicon compounds – from electrosynthesis to electrocatalysis, Inorg. Chem. Front., 2023, 10, 1382–1394 RSC; (d) J. E. Nutting, J. B. Gerken, A. G. Stamoulis, D. L. Bruns and S. S. Stahl, “How Should I Think about Voltage? What Is Overpotential?": Establishing an Organic Chemistry Intuition for Electrochemistry, J. Org. Chem., 2021, 86, 15875–15885 CrossRef CAS PubMed.
- L. M. Barton, L. Chen, D. G. Blackmond and P. S. Baran, Electrochemical borylation of carboxylic acids, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2109408118 CrossRef CAS PubMed.
- Y. Yuan, J. Yang and A. Lei, Recent advances in electrochemical oxidative cross-coupling with hydrogen evolution involving radicals, Chem. Soc. Rev., 2021, 50, 10058–10086 RSC.
- (a) I. Choi, A. M. Messinis, X. Hou and L. Ackermann, A Strategy for Site– and Chemoselective C−H Alkenylation through Osmaelectrooxidative Catalysis, Angew. Chem., Int. Ed., 2021, 60, 27005–27012 CrossRef CAS PubMed; (b) U. Dhawa, C. Tian, T. Wdowik, J. C. A. Oliveira, J. Hao and L. Ackermann, Enantioselective Pallada–Electrocatalyzed C−H Activation by Transient Directing Groups: Expedient Access to Helicenes, Angew. Chem., Int. Ed., 2020, 59, 13451–13457 CrossRef CAS PubMed; (c) P. Gandeepan, L. H. Finger, T. H. Meyer and L. Ackermann, 3d metallaelectrocatalysis for resource economical syntheses, Chem. Soc. Rev., 2020, 49, 4254–4272 RSC; (d) W. J. Kong, L. H. Finger, J. C. A. Oliveira and L. Ackermann, Rhodaelectrocatalysis for Annulative C−H Activation: Polycyclic Aromatic Hydrocarbons through Versatile Double Electrocatalysis, Angew. Chem., Int. Ed., 2019, 58, 6342–6346 CrossRef CAS PubMed; (e) W. J. Kong, Z. Shen, L. H. Finger and L. Ackermann, Electrochemical Access to Aza–Polycyclic Aromatic Hydrocarbons: Rhoda–Electrocatalyzed Domino Alkyne Annulations, Angew. Chem., Int. Ed., 2020, 59, 5551–5556 CrossRef CAS PubMed; (f) Y. Qiu, A. Scheremetjew and L. Ackermann, Electro-Oxidative C–C Alkenylation by Rhodium(III) Catalysis, J. Am. Chem. Soc., 2019, 141, 2731–2738 CrossRef CAS PubMed; (g) Y. Qiu, C. Tian, L. Massignan, T. Rogge and L. Ackermann, Electrooxidative Ruthenium-Catalyzed C−H/O−H Annulation by Weak O -Coordination, Angew. Chem., Int. Ed., 2018, 57, 5818–5822 CrossRef CAS PubMed; (h) Y. Wang, J. C. A. Oliveira, Z. Lin and L. Ackermann, Electrooxidative Rhodium–Catalyzed [5+2] Annulations via C−H/O−H Activations, Angew. Chem., Int. Ed., 2021, 60, 6419–6424 CrossRef CAS PubMed; (i) Y. Zhang, J. Struwe and L. Ackermann, Rhodium–Catalyzed Electrooxidative C−H Olefination of Benzamides, Angew. Chem., Int. Ed., 2020, 59, 15076–15080 CrossRef CAS PubMed.
- D. Bélanger and J. Pinson, Electrografting: a powerful method for surface modification, Chem. Soc. Rev., 2011, 40, 3995–4048 RSC.
- J. P. Barham and B. Konig, Synthetic Photoelectrochemistry, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed.
- H. Huang, K. A. Steiniger and T. H. Lambert, Electrophotocatalysis: Combining Light and Electricity to Catalyze Reactions, J. Am. Chem. Soc., 2022, 144, 12567–12583 CrossRef CAS PubMed.
- H. Xiang, J. He, W. Qian, M. Qiu, H. Xu, W. Duan, Y. Ouyang, Y. Wang and C. Zhu, Electroreductively Induced Radicals for Organic Synthesis, Molecules, 2023, 28, 857 CrossRef CAS PubMed.
- S. Wu, J. Kaur, T. A. Karl, X. Tian and J. P. Barham, Synthetic Molecular Photoelectrochemistry: New Frontiers in Synthetic Applications, Mechanistic Insights and Scalability, Angew. Chem., Int. Ed., 2022, 61, e202107811 CrossRef CAS PubMed.
- (a) H. G. Roth, N. A. Romero and D. A. Nicewicz, Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry, Synlett, 2016, 714–723 CAS; (b) L. Koefoed, K. H. Vase, J. H. Stenlid, T. Brinck, Y. Yoshimura, H. Lund, S. U. Pedersen and K. Daasbjerg, On the Kinetic and Thermodynamic Properties of Aryl Radicals Using Electrochemical and Theoretical Approaches, ChemElectroChem, 2017, 4, 3212–3221 CrossRef CAS.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Engaging unactivated alkyl, alkenyl and aryl iodides in visible-light-mediated free radical reactions, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- H. Kim and C. Lee, Visible-Light-Induced Photocatalytic Reductive Transformations of Organohalides, Angew. Chem., Int. Ed., 2012, 51, 12303–12306 CrossRef CAS PubMed.
- (a) M. Cybularczyk-Cecotka, J. Szczepanik and M. Giedyk, Photocatalytic strategies for the activation of organic chlorides, Nat. Catal., 2020, 3, 872–886 CrossRef CAS; (b) F. Glaser, C. Kerzig and O. S. Wenger, Multi–Photon Excitation in Photoredox Catalysis: Concepts, Applications, Methods, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS PubMed; (c) J. Lan, R. Chen, F. Duo, M. Hu and X. Lu, Visible-Light Photocatalytic Reduction of Aryl Halides as a Source of Aryl Radicals, Molecules, 2022, 27, 5364 CrossRef CAS PubMed.
- S. Wu, F. Schiel and P. Melchiorre, A General Light–Driven Organocatalytic Platform for the Activation of Inert Substrates, Angew. Chem., Int. Ed., 2023, 62, e202306364 CrossRef CAS PubMed.
- (a) M. Lepori, S. Schmid and J. P. Barham, Photoredox catalysis harvesting multiple photon or electrochemical energies, Beilstein J. Org. Chem., 2023, 19, 1055–1145 CrossRef CAS PubMed; (b) S. Caby, L. M. Bouchet, J. E. Argüello, R. A. Rossi and J. I. Bardagi, Excitation of Radical Anions of Naphthalene Diimides in Consecutive– and Electro–Photocatalysis, ChemCatChem, 2021, 13, 3001–3009 CrossRef CAS.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Reduction of aryl halides by consecutive visible light-induced electron transfer processes, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- J. I. Bardagi, I. Ghosh, M. Schmalzbauer, T. Ghosh and B. König, Anthraquinones as Photoredox Catalysts for the Reductive Activation of Aryl Halides, Eur. J. Org. Chem., 2018, 34–40 CrossRef CAS.
- M. Neumeier, D. Sampedro, M. Májek, V. A. De La Peña O’Shea, A. J. von Wangelin and R. Pérez-Ruiz, Dichromatic Photocatalytic Substitutions of Aryl Halides with a Small Organic Dye, Chem. – Eur. J., 2018, 24, 105–108 CrossRef CAS PubMed.
- B. Michelet, C. Deldaele, S. Kajouj, C. Moucheron and G. Evano, A General Copper Catalyst for Photoredox Transformations of Organic Halides, Org. Lett., 2017, 19, 3576–3579 CrossRef CAS PubMed.
- I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Discovery and characterization of an acridine radical photoreductant, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- A. F. Chmiel, O. P. Williams, C. P. Chernowsky, C. S. Yeung and Z. K. Wickens, Non-innocent Radical Ion Intermediates in Photoredox Catalysis: Parallel Reduction Modes Enable Coupling of Diverse Aryl Chlorides, J. Am. Chem. Soc., 2021, 143, 10882–10889 CrossRef CAS PubMed.
- A. J. Boyington, C. P. Seath, A. M. Zearfoss, Z. Xu and N. T. Jui, Catalytic Strategy for Regioselective Arylethylamine Synthesis, J. Am. Chem. Soc., 2019, 141, 4147–4153 CrossRef CAS PubMed.
- Y. Kwon, J. Lee, Y. Noh, D. Kim, Y. Lee, C. Yu, J. C. Roldao, S. Feng, J. Gierschner, R. Wannemacher and M. S. Kwon, Formation and degradation of strongly reducing cyanoarene-based radical anions towards efficient radical anion-mediated photoredox catalysis, Nat. Commun., 2023, 14, 92 CrossRef CAS PubMed.
- J. S. Beckwith, A. Aster and E. Vauthey, The excited-state dynamics of the radical anions of cyanoanthracenes, Phys. Chem. Chem. Phys., 2022, 24, 568–577 RSC.
- A. J. Rieth, M. I. Gonzalez, B. Kudisch, M. Nava and D. G. Nocera, How Radical Are “Radical” Photocatalysts? A Closed-Shell Meisenheimer Complex Is Identified as a Super-Reducing Photoreagent, J. Am. Chem. Soc., 2021, 143, 14352–14359 CrossRef CAS PubMed.
- S. J. Horsewill, G. Hierlmeier, Z. Farasat, J. P. Barham and D. J. Scott, Shining Fresh Light on Complex Photoredox Mechanisms through Isolation of Intermediate Radical Anions, ACS Catal., 2023, 13, 9392–9403 CrossRef CAS PubMed.
- (a) B. Pfund, D. Gejsnæs-Schaad, B. Lazarevski and O. S. Wenger, Picosecond reactions of excited radical ion super-reductants, Nat. Commun., 2024, 15, 4738 CrossRef CAS PubMed; (b) A. Kumar, P. Malevich, L. Mewes, S. Wu, J. P. Barham and J. Hauer, Transient absorption spectroscopy based on uncompressed hollow core fiber white light proves pre-association between a radical ion photocatalyst and substrate, J. Chem. Phys., 2023, 158, 144201 CrossRef CAS PubMed; (c) S. Wu, J. Žurauskas, M. Domański, P. S. Hitzfeld, V. Butera, D. J. Scott, J. Rehbein, A. Kumar, E. Thyrhaug, J. Hauer and J. P. Barham, Hole-mediated photoredox catalysis: tris(p-substituted)biarylaminium radical cations as tunable, precomplexing and potent photooxidants, Org. Chem. Front., 2021, 8, 1132–1142 RSC; (d) D. Y. Jeong, D. S. Lee, H. L. Lee, S. Nah, J. Y. Lee, E. J. Cho and Y. You, Evidence and Governing Factors of the Radical-Ion Photoredox Catalysis, ACS Catal., 2022, 12, 6047–6059 CrossRef CAS.
- L. Hou, X. Jing, H. Huang and C. Duan, Merging Charge Transfer into Metal–Organic Frameworks to Achieve High Reduction Potentials via Multiphoton Excitation, ACS Appl. Mater. Interfaces, 2022, 14, 15307–15316 CrossRef CAS PubMed.
- L. Zeng, T. Liu, C. He, D. Shi, F. Zhang and C. Duan, Organized Aggregation Makes Insoluble Perylene Diimide Efficient for the Reduction of Aryl Halides via Consecutive Visible Light-Induced Electron-Transfer Processes, J. Am. Chem. Soc., 2016, 138, 3958–3961 CrossRef CAS PubMed.
- T. He, H. Wei, Y. Zhou, L.-Y. Jiang, J. B. Baell, Y. Yu and F. Huang, Visible light-induced borylation and arylation of small organic molecules using carbon dots, Org. Chem. Front., 2023, 10, 2918–2926 RSC.
- D. Yu, W.-P. To, Y. Liu, L.-L. Wu, T. You, J. Ling and C.-M. Che, Direct photo-induced reductive Heck cyclization of indoles for the efficient preparation of polycyclic indolinyl compounds, Chem. Sci., 2021, 12, 14050–14058 RSC.
- J. Klett, Ł. Woźniak and N. Cramer, 1,3,2-Diazaphospholene-Catalyzed Reductive Cyclizations of Organohalides, Angew. Chem., Int. Ed., 2022, 61, e202202306 CrossRef CAS PubMed.
- R. D. Riley, B. S. N. Huchenski, K. L. Bamford and A. W. H. Speed, Diazaphospholene-Catalyzed Radical Reactions from Aryl Halides, Angew. Chem., Int. Ed., 2022, 61, e202204088 CrossRef CAS PubMed.
- K. Liang, N. Li, Y. Zhang, T. Li and C. Xia, Transition-metal-free α-arylation of oxindoles via visible-light-promoted electron transfer, Chem. Sci., 2019, 10, 3049–3053 RSC.
- M. P. Drapeau, I. Fabre, L. Grimaud, I. Ciofini, T. Ollevier and M. Taillefer, Transition-Metal-Free α-Arylation of Enolizable Aryl Ketones and Mechanistic Evidence for a Radical Process, Angew. Chem., Int. Ed., 2015, 54, 10587–10591 CrossRef PubMed.
- K. Liang, T. Li, N. Li, Y. Zhang, L. Shen, Z. Ma and C. Xia, Redox-neutral photochemical Heck-type arylation of vinylphenols activated by visible light, Chem. Sci., 2020, 11, 2130–2135 RSC.
- S. Jin, H. T. Dang, G. C. Haug, R. He, V. D. Nguyen, V. T. Nguyen, H. D. Arman, K. S. Schanze and O. V. Larionov, Visible Light-Induced Borylation of C–O, C–N, and C–X Bonds, J. Am. Chem. Soc., 2020, 142, 1603–1613 CrossRef CAS PubMed.
- A. Sagadevan, A. Ghosh, P. Maity, O. F. Mohammed, O. M. Bakr and M. Rueping, Visible-Light Copper Nanocluster Catalysis for the C–N Coupling of Aryl Chlorides at Room Temperature, J. Am. Chem. Soc., 2022, 144, 12052–12061 CrossRef CAS PubMed.
- D. T. Ziegler, J. Choi, J. M. Muñoz-Molina, A. C. Bissember, J. C. Peters and G. C. Fu, A Versatile Approach to Ullmann C–N Couplings at Room Temperature: New Families of Nucleophiles and Electrophiles for Photoinduced, Copper-Catalyzed Processes, J. Am. Chem. Soc., 2013, 135, 13107–13112 CrossRef CAS PubMed.
- (a) L. Pause, M. Robert and J.-M. Savéant, Can Single-Electron Transfer Break an Aromatic Carbon−Heteroatom Bond in One Step? A Novel Example of Transition between Stepwise and Concerted Mechanisms in the Reduction of Aromatic Iodides, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS; (b) C. Costentin, M. Robert and J.-M. Savéant, Fragmentation of Aryl Halide π Anion Radicals. Bending of the Cleaving Bond and Activation vs Driving Force Relationships, J. Am. Chem. Soc., 2004, 126, 16051–16057 CrossRef CAS PubMed; (c) J. S. Jaworski, P. Leszczyński and S. Filipek, Rates of the halide ion cleavage from halo-9,10-diphenylanthracene anion radicals in DMF, J. Electroanal. Chem., 1997, 440, 163–167 CrossRef CAS.
- (a) D. Hetemi, C. Combellas, F. Kanoufi and F. I. Podvorica, Direct vs Indirect Grafting of Alkyl and Aryl Halides, ChemPhysChem, 2021, 22, 1844–1849 CrossRef CAS PubMed; (b) J. Pinson and F. I. Podvorica, Surface modification of materials: Electrografting of organic films, Curr. Opin. Electrochem., 2020, 24, 44–48 CrossRef CAS.
- (a) C. P. Andrieux, J. Badoz-Lambling, C. Combellas, D. Lacombe, J. M. Saveant, A. Thiebault and D. Zann, Electron transfer catalyzed reactions. Electrochemical induction of the hydrogen atom transfer oxidation of alcohols and other substrates by aromatic halides, J. Am. Chem. Soc., 1987, 109, 1518–1525 CrossRef CAS; (b) C. Amatore, J. Badoz-Lambling, C. Bonnel-Huyghes, J. Pinson, J. M. Saveant and A. Thiebault, Hydrogen atom transfer oxidation of primary and secondary alcoholates into aldehydes and ketones by aromatic halides in liquid ammonia. A new electrochemically induceable reaction, J. Am. Chem. Soc., 1982, 104, 1979–1986 CrossRef CAS.
- Y. Wang, Z. Zhao, D. Pan, S. Wang, K. Jia, D. Ma, G. Yang, X. S. Xue and Y. Qiu, Metal–Free Electrochemical Carboxylation of Organic Halides in the Presence of Catalytic Amounts of an Organomediator, Angew. Chem., Int. Ed., 2022, 61, e202210201 CrossRef CAS PubMed.
- (a) J. Hong, Q. Liu, F. Li, G. Bai, G. Liu, M. Li, O. S. Nayal, X. Fu and F. Mo, Electrochemical Radical Borylation of Aryl Iodides, Chin. J. Chem., 2019, 37, 347–351 CrossRef CAS; (b) C. Laza, C. Pintaric, S. Olivero and E. Dunach, Electrochemical reduction of polyhalogenated aryl derivatives in the presence of pinacolborane: Electrosynthesis of functionalised arylboronic esters, Electrochim. Acta, 2005, 50, 4897–4901 CrossRef CAS; (c) C. Laza, E. Duñach, F. Serein-Spirau, J. J. E. Moreau and L. Vellutini, Novel synthesis of arylboronic acids by electroreduction of aromatic halides in the presence of trialkyl borates, New J. Chem., 2002, 26, 373–375 RSC.
- J. Ke, H. Wang, L. Zhou, C. Mou, J. Zhang, L. Pan and Y. R. Chi, Hydrodehalogenation of Aryl Halides through Direct Electrolysis, Chem. – Eur. J., 2019, 25, 6911–6914 CrossRef CAS PubMed.
- (a) D. M. Pereira, P. Valentão, J. A. Pereira and P. B. Andrade, Phenolics: From Chemistry to Biology, Molecules, 2009, 14, 2202–2211 CrossRef CAS; (b) A. E. Q. Sauceda, S. G. Sáyago-Ayerdi, J. F. Ayala-Zavala, A. Wall-Medrano, L. A. de la Rosa, G. A. González-Aguilar and E. Álvarez-Parrilla, in Fruit and Vegetable Phytochemicals, 2017, pp. 125–138 Search PubMed.
- L. Yang, Q. Zhuang, M. Wu, H. Long, C. Lin, M. Lin and F. Ke, Electrochemical-induced hydroxylation of aryl halides in the presence of Et3N in water, Org. Biomol. Chem., 2021, 19, 6417–6421 RSC.
- Z. Chami, M. Gareil, J. Pinson, J. M. Savéant and A. Thiebault, Aryl radicals from electrochemical reduction of aryl halides. Addition on olefins, J. Org. Chem., 1991, 56, 586–595 CrossRef CAS.
- M. D. Koppang, G. A. Ross, N. F. Woolsey and D. E. Bartak, Cyclization reactions of electrochemically generated o-(3-butenyl)phenyl anions and radicals to 1-methylindan, J. Am. Chem. Soc., 1986, 108, 1441–1447 CrossRef CAS.
- H. Senboku, J.-y. Michinishi and S. Hara, Facile Synthesis of 2,3-Dihydrobenzofuran-3-ylacetic Acids by Novel Electrochemical Sequential Aryl Radical Cyclization-Carboxylation of 2-Allyloxybromobenzenes Using Methyl 4-tert-Butylbenzoate as an Electron-Transfer Mediator, Synlett, 2011, 1567–1572 CrossRef CAS.
- G. Sun, S. Ren, X. Zhu, M. Huang and Y. Wan, Direct Arylation of Pyrroles via Indirect Electroreductive C–H Functionalization Using Perylene Bisimide as an Electron-Transfer Mediator, Org. Lett., 2016, 18, 544–547 CrossRef CAS PubMed.
- (a) N. Kurono, E. Honda, F. Komatsu, K. Orito and M. Tokuda, Regioselective synthesis of substituted 1-indanols, 2,3-dihydrobenzofurans and 2,3-dihydroindoles by electrochemical radical cyclization using an arene mediator, Tetrahedron, 2004, 60, 1791–1801 CrossRef CAS; (b) X.-Q. Zhou, P.-B. Chen, Q. Xia, T.-K. Xiong, X.-J. Li, Y.-M. Pan, M.-X. He and Y. Liang, Indirect electrochemical reductive cyclization of o-halophenylacrylamides mediated by phenanthrene, Org. Chem. Front., 2023, 10, 2039–2044 RSC.
- K. Mitsudo, Y. Nakagawa, J.-i. Mizukawa, H. Tanaka, R. Akaba, T. Okada and S. Suga, Electro-reductive cyclization of aryl halides promoted by fluorene derivatives, Electrochim. Acta, 2012, 82, 444–449 CrossRef CAS.
- A. A. Folgueiras-Amador, A. E. Teuten, M. Salam-Perez, J. E. Pearce, G. Denuault, D. Pletcher, P. J. Parsons, D. C. Harrowven and R. C. D. Brown, Cathodic Radical Cyclisation of Aryl Halides Using a Strongly-Reducing Catalytic Mediator in Flow, Angew. Chem., Int. Ed., 2022, 61, e202203694 CrossRef CAS PubMed.
- Y. Lai, A. Halder, J. Kim, T. J. Hicks and P. J. Milner, Electroreductive Radical Borylation of Unactivated (Hetero)Aryl Chlorides Without Light by Using Cumulene–Based Redox Mediators, Angew. Chem., Int. Ed., 2023, 62, e202310246 CrossRef CAS PubMed.
- N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, Potent Reductants via Electron-Primed Photoredox Catalysis: Unlocking Aryl Chlorides for Radical Coupling, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS PubMed.
- C. P. Chernowsky, A. F. Chmiel and Z. K. Wickens, Electrochemical Activation of Diverse Conventional Photoredox Catalysts Induces Potent Photoreductant Activity, Angew. Chem., Int. Ed., 2021, 60, 21418–21425 CrossRef CAS PubMed.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, Reductive Electrophotocatalysis: Merging Electricity and Light To Achieve Extreme Reduction Potentials, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed.
- Y.-J. Chen, T. Lei, H.-L. Hu, H.-L. Wu, S. Zhou, X.-B. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Tandem photoelectrochemical and photoredox catalysis for efficient and selective aryl halides functionalization by solar energy, Matter, 2021, 4, 2354–2366 CrossRef CAS.
- (a) F. Mo, D. Qiu, L. Zhang and J. Wang, Recent Development of Aryl Diazonium Chemistry for the Derivatization of Aromatic Compounds, Chem. Rev., 2021, 121, 5741–5829 CrossRef CAS PubMed; (b) E. L. S. De Souza and C. C. Oliveira, Selective Radical Transformations with Aryldiazonium Salts, Eur. J. Org. Chem., 2023, 26, e202300073 CrossRef CAS; (c) S. S. Babu, P. Muthuraja, P. Yadav and P. Gopinath, Aryldiazonium Salts in Photoredox Catalysis – Recent Trends, Adv. Synth. Catal., 2021, 363, 1782–1809 CrossRef CAS.
- Y.-X. Cao, G. Zhu, Y. Li, N. Le Breton, C. Gourlaouen, S. Choua, J. Boixel, H.-P. J. de Rouville and J.-F. Soulé, Photoinduced Arylation of Acridinium Salts: Tunable Photoredox Catalysts for C–O Bond Cleavage, J. Am. Chem. Soc., 2022, 144, 5902–5909 CrossRef CAS PubMed.
- Y.-M. Yang, W. Yan, H.-W. Hu, Y. Luo, Z.-Y. Tang and Z. Luo, Photoinduced Acetylation of Anilines under Aqueous and Catalyst-Free Conditions, J. Org. Chem., 2021, 86, 12344–12353 CrossRef CAS PubMed.
- G. Peng, H. Cheng, X. Cheng, Y. He, Y. An, J. Wu and D. Zheng, A reductive Sandmeyer-type reaction for the synthesis of sulfoxides from anilines under photocatalysis, Org. Chem. Front., 2023, 10, 3033–3038 RSC.
- D. Singla, V. Luxami and K. Paul, Eosin Y mediated photo-catalytic C–H functionalization: C–C and C–S bond formation, Org. Chem. Front., 2023, 10, 237–266 RSC.
- T. Meyer, J. X. Xu, J. Rabeah, A. Brückner and X. F. Wu, Photocatalytic Synthesis of Stilbenes via Cross–Coupling of Alkenyl Boronic Acids and Arenediazonium Tetrafluoroborate Salts, ChemPhotoChem, 2020, 4, 713–720 CrossRef CAS.
- D. P. Hari and B. König, Synthetic applications of eosin Y in photoredox catalysis, Chem. Commun., 2014, 50, 6688–6699 RSC.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Energy transfer catalysis mediated by visible light: principles, applications, directions, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- S. Chand, A. K. Sharma, A. K. Pandey and K. N. Singh, Visible-Light Photoredox-Catalyzed Synthesis of trans-Oxiranes via Decarboxylative Stereospecific Epoxidation of trans-Cinnamic Acids by Aryldiazonium Salts, Org. Lett., 2022, 24, 6423–6427 CrossRef CAS PubMed.
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- C. A. Vincent, A. Ripak, L. Troian-Gautier and U. K. Tambar, Photocatalytic conversion of aryl diazonium salts to sulfonyl fluorides, Tetrahedron, 2023, 139, 133364 CrossRef CAS PubMed.
- S. Crespi, S. Protti and M. Fagnoni, Wavelength Selective Generation of Aryl Radicals and Aryl Cations for Metal-Free Photoarylations, J. Org. Chem., 2016, 81, 9612–9619 CrossRef CAS PubMed.
- Y. Xu, X. Yang and H. Fang, Additive- and Photocatalyst-Free Borylation of Arylazo Sulfones under Visible Light, J. Org. Chem., 2018, 83, 12831–12837 CrossRef CAS PubMed.
- C. Lian, G. Yue, J. Mao, D. Liu, Y. Ding, Z. Liu, D. Qiu, X. Zhao, K. Lu, M. Fagnoni and S. Protti, Visible-Light-Driven Synthesis of Arylstannanes from Arylazo Sulfones, Org. Lett., 2019, 21, 5187–5191 CrossRef CAS PubMed.
- L. Di Terlizzi, S. Scaringi, C. Raviola, R. Pedrazzani, M. Bandini, M. Fagnoni and S. Protti, Visible Light-Driven, Gold(I)-Catalyzed Preparation of Symmetrical (Hetero)biaryls by Homocoupling of Arylazo Sulfones, J. Org. Chem., 2022, 87, 4863–4872 CrossRef CAS PubMed.
- (a) T. Menanteau, M. Dias, E. Levillain, A. J. Downard and T. Breton, Electrografting via Diazonium Chemistry: The Key Role of the Aryl Substituent in the Layer Growth Mechanism, J. Phys. Chem. C, 2016, 120, 4423–4429 CrossRef CAS; (b) A. Mattiuzzi, Q. Lenne, J. C. Padilha, L. Troian-Gautier, Y. R. Leroux, I. Jabin and C. Lagrost, Strategies for the Formation of Monolayers From Diazonium Salts: Unconventional Grafting Media, Unconventional Building Blocks, Front. Chem., 2020, 8, 559 CrossRef CAS PubMed.
- K. Kher, M. Dhaker and P. K. Baroliya, Recent advances in electrochemical functionalization using diazonium salts, Org. Biomol. Chem., 2023, 21, 7052–7061 RSC.
- J. D. Firth and I. J. S. Fairlamb, A Need for Caution in the Preparation and Application of Synthetically Versatile Aryl Diazonium Tetrafluoroborate Salts, Org. Lett., 2020, 22, 7057–7059 CrossRef CAS PubMed.
- Q. Liu, B. Sun, Z. Liu, Y. Kao, B.-W. Dong, S.-D. Jiang, F. Li, G. Liu, Y. Yang and F. Mo, A general electrochemical strategy for the Sandmeyer reaction, Chem. Sci., 2018, 9, 8731–8737 RSC.
- P. Wang, Z. Yang, Z. Wang, C. Xu, L. Huang, S. Wang, H. Zhang and A. Lei, Electrochemical Arylation of Electron–Deficient Arenes through Reductive Activation, Angew. Chem., 2019, 131, 15894–15898 CrossRef.
- Y. Y. Jiang, G. Y. Dou, L. S. Zhang, K. Xu, R. D. Little and C. C. Zeng, Electrochemical Cross–Coupling of C(sp2)−H with Aryldiazonium Salts via a Paired Electrolysis: an Alternative to Visible Light Photoredox–Based Approach, Adv. Synth. Catal., 2019, 361, 5170–5175 CrossRef CAS.
- D. Hata, M. Tobisu and T. Amaya, Electrolytic Cross-Coupling of Arenediazonium Salts and Heteroarenes, Bull. Chem. Soc. Jpn., 2018, 91, 1749–1751 CrossRef CAS.
- Y. Liu, P. Zhou, Y. Xu, Z. Yang and D. Wang, Electrochemically driven [4+2] benzannulation: synthesis of polycyclic (hetero)aromatic compounds, Chem. Commun., 2023, 59, 1681–1684 RSC.
- S. I. Kozhushkov and M. Alcarazo, Synthetic Applications of Sulfonium Salts, Eur. J. Inorg. Chem., 2020, 2020, 2486–2500 CrossRef CAS PubMed.
- (a) S. Donck, A. Baroudi, L. Fensterbank, J.-P. Goddard and C. Ollivier, Visible-Light Photocatalytic Reduction of Sulfonium Salts as a Source of Aryl Radicals, Adv. Synth. Catal., 2013, 355, 1477–1482 CrossRef CAS; (b) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Bond-Forming and -Breaking Reactions at Sulfur(IV): Sulfoxides, Sulfonium Salts, Sulfur Ylides, and Sulfinate Salts, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed; (c) Á. Péter, G. J. P. Perry and D. J. Procter, Radical C−C Bond Formation using Sulfonium Salts and Light, Adv. Synth. Catal., 2020, 362, 2135–2142 CrossRef.
- J. L. Dektar and N. P. Hacker, Photochemistry of triarylsulfonium salts, J. Am. Chem. Soc., 1990, 112, 6004–6015 CrossRef CAS.
- J. Gao, J. Feng and D. Du, Shining Light on C−S Bonds: Recent Advances in C−C Bond Formation Reactions via C−S Bond Cleavage under Photoredox Catalysis, Chem. – Asian J., 2020, 15, 3637–3659 CrossRef CAS PubMed.
- Z.-Y. Tian, Y.-T. Hu, H.-B. Teng and C.-P. Zhang, Application of arylsulfonium salts as arylation reagents, Tetrahedron Lett., 2018, 59, 299–309 CrossRef CAS.
- F. Ye, F. Berger, H. Jia, J. Ford, A. Wortman, J. Börgel, C. Genicot and T. Ritter, Aryl Sulfonium Salts for Site–Selective Late–Stage Trifluoromethylation, Angew. Chem., Int. Ed., 2019, 58, 14615–14619 CrossRef CAS PubMed.
- (a) J. Wu, Z. Wang, X.-Y. Chen, Y. Wu, D. Wang, Q. Peng and P. Wang, Para-selective borylation of monosubstituted benzenes using a transient mediator, Sci. China: Chem., 2020, 63, 336–340 CrossRef CAS; (b) B. Li, K. Wang, H. Yue, A. Drichel, J. Lin, Z. Su and M. Rueping, Catalyst-Free C(sp2)-H Borylation through Aryl Radical Generation from Thiophenium Salts via Electron Donor–Acceptor Complex Formation, Org. Lett., 2022, 24, 7434–7439 CrossRef CAS PubMed.
- L. Liang, H.-Y. Niu, R.-L. Li, Y.-F. Wang, J.-K. Yan, C.-G. Li and H.-M. Guo, Photoinduced Copper-Catalyzed Site-Selective C(sp2)–C(sp) Cross-Coupling via Aryl Sulfonium Salts, Org. Lett., 2020, 22, 6842–6846 CrossRef CAS PubMed.
- Y.-L. Zhang, G.-H. Wang, Y. Wu, C.-Y. Zhu and P. Wang, Construction of α-Amino Azines via Thianthrenation-Enabled Photocatalyzed Hydroarylation of Azine-Substituted Enamides with Arenes, Org. Lett., 2021, 23, 8522–8526 CrossRef CAS PubMed.
- X. Wang, X. Xun, H. Song, Y. Liu and Q. Wang, Palladium Metallaphotoredox-Catalyzed 2-Arylation of Indole Derivatives, Org. Lett., 2022, 24, 4580–4585 CrossRef CAS PubMed.
- Y. Cai and T. Ritter, Meerwein-type Bromoarylation with Arylthianthrenium Salts, Angew. Chem., Int. Ed., 2022, 61, e202209882 CrossRef CAS PubMed.
- Y. Zhao, C. Empel, W. Liang, R. M. Koenigs and F. W. Patureau, Gem-Difluoroallylation of Aryl Sulfonium Salts, Org. Lett., 2022, 24, 8753–8758 CrossRef CAS PubMed.
- Q. Li, J. Huang, Z. Cao, J. Zhang and J. Wu, Photoredox-catalyzed reaction of thianthrenium salts, sulfur dioxide and hydrazines, Org. Chem. Front., 2022, 9, 3781–3785 RSC.
- Q. Wang, X. Hao, K. Jin, R. Zhang, C. Duan and Y. Li, Visible-light-catalyzed C–H arylation of (hetero)arenes via arylselenonium salts, Org. Biomol. Chem., 2022, 20, 4427–4430 RSC.
- M. H. Aukland, M. Šiaučiulis, A. West, G. J. P. Perry and D. J. Procter, Metal-free photoredox-catalysed formal C–H/C–H coupling of arenes enabled by interrupted Pummerer activation, Nat. Catal., 2020, 3, 163–169 CrossRef CAS.
- C. Chen, Z.-J. Wang, H. Lu, Y. Zhao and Z. Shi, Generation of non-stabilized alkyl radicals from thianthrenium salts for C–B and C–C bond formation, Nat. Commun., 2021, 12, 4526 CrossRef CAS PubMed.
- A. Dewanji, L. van Dalsen, J. A. Rossi-Ashton, E. Gasson, G. E. M. Crisenza and D. J. Procter, A general arene C–H functionalization strategy via electron donor–acceptor complex photoactivation, Nat. Chem., 2023, 15, 43–52 CrossRef CAS PubMed.
- M. J. Cabrera-Afonso, A. Granados and G. A. Molander, Sustainable Thioetherification via Electron Donor–Acceptor Photoactivation Using Thianthrenium Salts, Angew. Chem., Int. Ed., 2022, 61, e202202706 CrossRef CAS PubMed.
- M. Zhang, B. Wang, Y. Cao, Y. Liu, Z. Wang and Q. Wang, Visible-Light-Driven Synthesis of Aryl Xanthates and Aryl Dithiocarbamates via an Electron Donor–Acceptor Complex, Org. Lett., 2022, 24, 8895–8900 CrossRef CAS PubMed.
- Z.-Y. Tian and C.-P. Zhang, Visible-light-initiated catalyst-free trifluoromethylselenolation of arylsulfonium salts with [Me4N][SeCF3], Org. Chem. Front., 2022, 9, 2220–2227 RSC.
- K. Sun, A. Shi, Y. Liu, X. Chen, P. Xiang, X. Wang, L. Qu and B. Yu, A general electron donor–acceptor complex for photoactivation of arenes via thianthrenation, Chem. Sci., 2022, 13, 5659–5666 RSC.
- R. Chaudhary and P. Natarajan, Visible Light Photoredox Activation of Sulfonyl Chlorides: Applications in Organic Synthesis, ChemistrySelect, 2017, 2, 6458–6479 CrossRef CAS.
- J.-D. Xia, G.-B. Deng, M.-B. Zhou, W. Liu, P. Xie and J.-H. Li, Reusable Visible Light Photoredox Catalysts; Catalyzed Benzylic C(sp3)–H Functionalization/Carbocyclization Reactions, Synlett, 2012, 2707–2713 CAS.
- G.-B. Deng, Z.-Q. Wang, J.-D. Xia, P.-C. Qian, R.-J. Song, M. Hu, L.-B. Gong and J.-H. Li, Tandem Cyclizations of 1,6-Enynes with Arylsulfonyl Chlorides by Using Visible-Light Photoredox Catalysis, Angew. Chem., Int. Ed., 2013, 52, 1535–1538 CrossRef CAS PubMed.
- L. Gu, C. Jin, J. Liu, H. Ding and B. Fan, Transition-metal-free, visible-light induced cyclization of arylsulfonyl chlorides with 2-isocyanobiphenyls to produce phenanthridines, Chem. Commun., 2014, 50, 4643–4645 RSC.
- L. Gu, C. Jin, W. Wang, Y. He, G. Yang and G. Li, Transition-metal-free, visible-light induced cyclization of arylsulfonyl chlorides with o-azidoarylalkynes: a regiospecific route to unsymmetrical 2,3-disubstituted indoles, Chem. Commun., 2017, 53, 4203–4206 RSC.
- P. Natarajan, A. Bala, S. K. Mehta and K. K. Bhasin, Visible-light photocatalyzed synthesis of 2-aryl N-methylpyrroles, furans and thiophenes utilizing arylsulfonyl chlorides as a coupling partner, Tetrahedron, 2016, 72, 2521–2526 CrossRef CAS.
- Z. Qiu and C.-J. Li, Transformations of Less-Activated Phenols and Phenol Derivatives via C–O Cleavage, Chem. Rev., 2020, 120, 10454–10515 CrossRef CAS PubMed.
- W. Liu, X. Yang, Y. Gao and C.-J. Li, Simple and Efficient Generation of Aryl Radicals from Aryl Triflates: Synthesis of Aryl Boronates and Aryl Iodides at Room Temperature, J. Am. Chem. Soc., 2017, 139, 8621–8627 CrossRef CAS PubMed.
- Q. Dou, L. Geng, B. Cheng, C.-J. Li and H. Zeng, Photoinduced transition-metal and external photosensitizer free cross-coupling of aryl triflates with trialkyl phosphites, Chem. Commun., 2021, 57, 8429–8432 RSC.
- B. Yan, Y. Zhou, J. Wu, M. Ran, H. Li and Q. Yao, Catalyst-free reductive hydrogenation or deuteration of aryl–heteroatom bonds induced by light, Org. Chem. Front., 2021, 8, 5244–5249 RSC.
- M. Ratushnyy, N. Kvasovs, S. Sarkar and V. Gevorgyan, Visible–Light–Induced Palladium–Catalyzed Generation of Aryl Radicals from Aryl Triflates, Angew. Chem., Int. Ed., 2020, 59, 10316–10320 CrossRef CAS PubMed.
- (a) Y. Lang, X. Peng, C.-J. Li and H. Zeng, Photoinduced catalyst-free deborylation–deuteration of arylboronic acids with D2O, Green Chem., 2020, 22, 6323–6327 RSC; (b) N. Neerathilingam, K. Prasanth and R. Anandhan, Substituent-controlled selective synthesis of 1,2-diketones and internal alkynes from terminal alkynes and arylboronic acids via α-stilbene radicals obtained from heteroleptic Cu(I) complexes under visible light, Green Chem., 2022, 24, 8685–8690 RSC; (c) A. Rakshit, P. Kumar, T. Alam, H. Dhara and B. K. Patel, Visible-Light-Accelerated Pd-Catalyzed Cascade Addition/Cyclization of Arylboronic Acids to γ- and β-Ketodinitriles for the Construction of 3-Cyanopyridines and 3-Cyanopyrrole Analogues, J. Org. Chem., 2020, 85, 12482–12504 CrossRef CAS PubMed; (d) Y. Kong, K. Wei and G. Yan, Radical coupling reactions of hydrazines via photochemical and electrochemical strategies, Org. Chem. Front., 2022, 9, 6114–6128 RSC; (e) T. Xiao, L. Li, G. Lin, Q. Wang, P. Zhang, Z.-W. Mao and L. Zhou, Synthesis of 6-substituted phenanthridines by metal-free, visible-light induced aerobic oxidative cyclization of 2-isocyanobiphenyls with hydrazines, Green Chem., 2014, 16, 2418–2421 RSC.
- S. Wang, H. Wang and B. König, Photo-induced thiolate catalytic activation of inert Caryl-hetero bonds for radical borylation, Chem, 2021, 7, 1653–1665 CAS.
- (a) Z.-X. Wang and B. Yang, Chemical transformations of quaternary ammonium salts via C–N bond cleavage, Org. Biomol. Chem., 2020, 18, 1057–1072 RSC; (b) X. Kong, Z.-Y. Cao, X. Chen, N.-Z. Wang and Y.-M. Cheng, Recent Advances in Electro- or Photochemical Driven Transformations via Cleavage of the C–N Bond of Quaternary Ammonium Salts, Synthesis, 2023, 2833–2842 CrossRef.
- A. M. Mfuh, J. D. Doyle, B. Chhetri, H. D. Arman and O. V. Larionov, Scalable, Metal- and Additive-Free, Photoinduced Borylation of Haloarenes and Quaternary Arylammonium Salts, J. Am. Chem. Soc., 2016, 138, 2985–2988 CrossRef CAS PubMed.
- (a) B. Emmert, Über die Elektrolyse quaternärer Pyridinium- und Chinoliniumsalze, Ber. Dtsch. Chem. Ges., 1909, 42, 1997–1999 CrossRef CAS; (b) B. Emmert, Über die Elektrolyse von Dialkyloxäthyl-phenyl-ammoniumjodiden und über einige Homologe des Cholins, Ber. Dtsch. Chem. Ges., 1912, 45, 430–433 CrossRef; (c) B. Emmert, Über die Elektrolyse von Trialkylphenyl-ammoniumjodiden, Ber. Dtsch. Chem. Ges., 1909, 42, 1507–1510 CrossRef CAS.
- (a) J. S. Mayell and A. J. Bard, The Electroreduction of Quaternary Ammonium Compounds, J. Am. Chem. Soc., 1963, 85, 421–425 CrossRef CAS; (b) S. D. Ross, M. Finkelstein and R. C. Petersen, The Electrochemical Degradation of Quaternary Ammonium Salts. II. The Mechanism of the Coupling Reaction, J. Am. Chem. Soc., 1960, 82, 1582–1585 CrossRef CAS; (c) S. D. Ross, M. Finkelstein and R. C. Petersen, Mechanism of the electroreduction of benzyltriethylammonium nitrate in dimethylformamide at aluminum and platinum cathodes, J. Am. Chem. Soc., 1970, 92, 6003–6006 CrossRef CAS.
- D.-T. Yang, M. Zhu, Z. J. Schiffer, K. Williams, X. Song, X. Liu and K. Manthiram, Direct Electrochemical Carboxylation of Benzylic C–N Bonds with Carbon Dioxide, ACS Catal., 2019, 9, 4699–4705 CrossRef CAS.
- X. Kong, L. Lin, Q. Chen and B. Xu, Radical generation from electroreduction of aryl and benzyl ammonium salts: synthesis of organoboronates, Org. Chem. Front., 2021, 8, 702–707 RSC.
- X. Kong, Y. Wang, Y. Chen, X. Chen, L. Lin and Z.-Y. Cao, Cyanation and cyanomethylation of trimethylammonium salts via electrochemical cleavage of C–N bonds, Org. Chem. Front., 2022, 9, 1288–1294 RSC.
- (a) P. P. Romańczyk and S. S. Kurek, Reliable reduction potentials of diaryliodonium cations and aryl radicals in acetonitrile from high-level ab initio computations, Electrochim. Acta, 2020, 351, 136404 CrossRef; (b) L. Koefoed, S. U. Pedersen and K. Daasbjerg, Grafting of Aryl Diazonium, Iodonium, and Sulfonium Salts in Unusual Patterns by Exploiting the Potential Gradient in Bipolar Electrochemistry, ChemElectroChem, 2016, 3, 495–501 CrossRef CAS.
- (a) E. A. Merritt and B. Olofsson, Diaryliodonium Salts: A Journey from Obscurity to Fame, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed; (b) X. Mi, C. Pi, W. Feng and X. Cui, Recent progress in the application of iodonium ylides in organic synthesis, Org. Chem. Front., 2022, 9, 6999–7015 RSC.
- (a) T. Koike, M. Akita and Y. Li, Radical Pentafluorosulfanylphenylation of Styrenes by Photoredox Catalysis, Synlett, 2016, 736–740 CrossRef; (b) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Merging Visible Light Photoredox and Gold Catalysis, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef CAS PubMed; (c) G. Fumagalli, S. Boyd and M. F. Greaney, Oxyarylation and Aminoarylation of Styrenes Using Photoredox Catalysis, Org. Lett., 2013, 15, 4398–4401 CrossRef CAS PubMed.
- M. Tobisu, T. Furukawa and N. Chatani, Visible Light-mediated Direct Arylation of Arenes and Heteroarenes Using Diaryliodonium Salts in the Presence and Absence of a Photocatalyst, Chem. Lett., 2013, 42, 1203–1205 CrossRef CAS.
- A. Baralle, L. Fensterbank, J. P. Goddard and C. Ollivier, Aryl Radical Formation by Copper(I) Photocatalyzed Reduction of Diaryliodonium Salts: NMR Evidence for a CuII/CuI Mechanism, Chem. – Eur. J., 2013, 19, 10809–10813 CrossRef CAS PubMed.
- S. R. Neufeldt and M. S. Sanford, Combining Transition Metal Catalysis with Radical Chemistry: Dramatic Acceleration of Palladium–Catalyzed C-H Arylation with Diaryliodonium Salts, Adv. Synth. Catal., 2012, 354, 3517–3522 CrossRef CAS PubMed.
- (a) D. Sun, K. Yin and R. Zhang, Visible-light-induced multicomponent cascade cycloaddition involving N-propargyl aromatic amines, diaryliodonium salts and sulfur dioxide: rapid access to 3-arylsulfonylquinolines, Chem. Commun., 2018, 54, 1335–1338 RSC; (b) M. Wang, Q. Fan and X. Jiang, Transition-Metal-Free Diarylannulated Sulfide and Selenide Construction via Radical/Anion-Mediated Sulfur–Iodine and Selenium–Iodine Exchange, Org. Lett., 2016, 18, 5756–5759 CrossRef CAS PubMed.
- Y. Li, M. Wang and X. Jiang, Controllable Sulfoxidation and Sulfenylation with Organic Thiosulfate Salts via Dual Electron- and Energy-Transfer Photocatalysis, ACS Catal., 2017, 7, 7587–7592 CrossRef CAS.
- X. Gong, J. Chen, J. Liu and J. Wu, Synthesis of thiophosphates through a three-component reaction by using sulfur dioxide as the sulfur source, Org. Chem. Front., 2017, 4, 2221–2225 RSC.
- F. Rammal, A. C. Gaumont and S. Lakhdar, Metal–Free Visible–Light–Mediated Aromatization of 1,2−Dihydronaphthalenes, Eur. J. Org. Chem., 2020, 1482–1485 CrossRef CAS.
- D. Li, C. Liang, Z. Jiang, J. Zhang, W.-T. Zhuo, F.-Y. Zou, W.-P. Wang, G.-L. Gao and J. Song, Visible-Light-Promoted C2 Selective Arylation of Quinoline and Pyridine N-Oxides with Diaryliodonium Tetrafluoroborate, J. Org. Chem., 2020, 85, 2733–2742 CrossRef CAS PubMed.
- R. K. Samanta, P. Meher and S. Murarka, Visible Light Photoredox-Catalyzed Direct C–H Arylation of Quinoxalin-2(1H)-ones with Diaryliodonium Salts, J. Org. Chem., 2022, 87, 10947–10957 CrossRef CAS PubMed.
- Y. Chen, C. Shu, F. Luo, X. Xiao and G. Zhu, Photocatalytic acylarylation of unactivated alkenes with diaryliodonium salts toward indanones and related compounds, Chem. Commun., 2018, 54, 5373–5376 RSC.
- N. W. Liu, S. Liang and G. Manolikakes, Visible–Light Photoredox–Catalyzed Aminosulfonylation of Diaryliodonium Salts with Sulfur Dioxide and Hydrazines, Adv. Synth. Catal., 2017, 359, 1308–1319 CrossRef CAS.
- Y. Ma, Q. Pan, C. Ou, Y. Cai, X. Ma and C. Liu, Aryl sulfonyl fluoride synthesis via organophotocatalytic fluorosulfonylation of diaryliodonium salts, Org. Biomol. Chem., 2023, 21, 7597–7601 RSC.
- (a) C. D. Christiansen, L. A. Sørensen and T. Lund, Modification of fluorine-doped tin oxide-electrodes by electrochemical reduction of di(4-nitrophenyl)iodonium tetrafluoroborate - And its application as a photo-anode in dye-sensitized solar cells, J. Electroanal. Chem., 2018, 809, 44–51 CrossRef CAS; (b) K. H. Vase, A. H. Holm, S. U. Pedersen and K. Daasbjerg, Immobilization of Aryl and Alkynyl Groups onto Glassy Carbon Surfaces by Electrochemical Reduction of Iodonium Salts, Langmuir, 2005, 21, 8085–8089 CrossRef CAS PubMed.
- X.-Q. Hu, Z.-K. Liu, Y.-X. Hou and Y. Gao, Single Electron Activation of Aryl Carboxylic Acids, iScience, 2020, 23, 101266 CrossRef CAS PubMed.
- (a) L. Ackermann, Metalla-electrocatalyzed C-H Activation by Earth-Abundant 3d Metals and Beyond, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed; (b) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Recent Advances in Oxidative R(1)-H/R(2)-H Cross-Coupling with Hydrogen Evolution via Photo-/Electrochemistry, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed; (c) J. L. Rockl, D. Pollok, R. Franke and S. R. Waldvogel, A Decade of Electrochemical Dehydrogenative C,C-Coupling of Aryls, Acc. Chem. Res., 2020, 53, 45–61 CrossRef PubMed.
- (a) D. H. R. Barton and M. Ramesh, Generation and fate of nondecarboxylating acyloxy radicals derived from the photolysis of acyl derivatives of N-hydroxy-2-thiopyridone, Tetrahedron Lett., 1990, 31, 949–952 CrossRef CAS; (b) D. H. R. Barton, B. Lacher and S. Z. Zard, The invention of radical reactions: Part XVI. Radical decarboxylative bromination and iodination of aromatic acids, Tetrahedron, 1987, 43, 4321–4328 CrossRef CAS.
- J. Chateauneuf, J. Lusztyk and K. U. Ingold, Spectroscopic and kinetic characteristics of aroyloxyl radicals. 2. Benzoyloxyl and ring-substituted aroyloxyl radicals, J. Am. Chem. Soc., 1988, 110, 2886–2893 CrossRef CAS.
- L. Candish, M. Freitag, T. Gensch and F. Glorius, Mild, visible light-mediated decarboxylation of aryl carboxylic acids to access aryl radicals, Chem. Sci., 2017, 8, 3618–3622 RSC.
- L. Candish, M. Teders and F. Glorius, Transition-Metal-Free, Visible-Light-Enabled Decarboxylative Borylation of Aryl N-Hydroxyphthalimide Esters, J. Am. Chem. Soc., 2017, 139, 7440–7443 CrossRef CAS PubMed.
- (a) Y. Abderrazak, A. Bhattacharyya and O. Reiser, Visible-Light-Induced Homolysis of Earth-Abundant Metal-Substrate Complexes: A Complementary Activation Strategy in Photoredox Catalysis, Angew. Chem., Int. Ed., 2021, 60, 21100–21115 CrossRef CAS PubMed; (b) P.-Z. Wang, J.-R. Chen and W.-J. Xiao, Emerging Trends in Copper-Promoted Radical-Involved C–O Bond Formations, J. Am. Chem. Soc., 2023, 145, 17527–17550 CrossRef CAS PubMed.
- F. Juliá, Ligand–to–Metal Charge Transfer (LMCT) Photochemistry at 3d–Metal Complexes: An Emerging Tool for Sustainable Organic Synthesis, ChemCatChem, 2022, 14, e202200916 CrossRef.
- W. Su, P. Xu and T. Ritter, Decarboxylative Hydroxylation of Benzoic Acids, Angew. Chem., Int. Ed., 2021, 60, 24012–24017 CrossRef CAS PubMed.
- T. Q. Chen, P. S. Pedersen, N. W. Dow, R. Fayad, C. E. Hauke, M. C. Rosko, E. O. Danilov, D. C. Blakemore, A.-M. Dechert-Schmitt, T. Knauber, F. N. Castellano and D. W. C. Macmillan, A Unified Approach to Decarboxylative Halogenation of (Hetero)aryl Carboxylic Acids, J. Am. Chem. Soc., 2022, 144, 8296–8305 CrossRef CAS PubMed.
- N. W. Dow, P. S. Pedersen, T. Q. Chen, D. C. Blakemore, A.-M. Dechert-Schmitt, T. Knauber and D. W. C. Macmillan, Decarboxylative Borylation and Cross-Coupling of (Hetero)aryl Acids Enabled by Copper Charge Transfer Catalysis, J. Am. Chem. Soc., 2022, 144, 6163–6172 CrossRef CAS PubMed.
- (a) A. Suzuki, Cross-Coupling Reactions Of Organoboranes: An Easy Way To Construct C-C Bonds (Nobel Lecture), Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed; (b) J. W. B. Fyfe and A. J. B. Watson, Recent Developments in Organoboron Chemistry: Old Dogs, New Tricks, Chem, 2017, 3, 31–55 CrossRef CAS.
- (a) P. C. Trippier and C. McGuigan, Boronic acids in medicinal chemistry: anticancer, antibacterial and antiviral applications, MedChemComm, 2010, 1, 183–198 RSC; (b) J. Plescia and N. Moitessier, Design and discovery of boronic acid drugs, Eur. J. Med. Chem., 2020, 195, 112270 CrossRef CAS PubMed.
- (a) S. Pillitteri, P. Ranjan, E. V. Van Der Eycken and U. K. Sharma, Uncovering the Potential of Boronic Acid and Derivatives as Radical Source in Photo(electro)chemical Reactions, Adv. Synth. Catal., 2022, 364, 1643–1665 CrossRef CAS; (b) V. Corcé, C. Ollivier and L. Fensterbank, Boron, silicon, nitrogen and sulfur-based contemporary precursors for the generation of alkyl radicals by single electron transfer and their synthetic utilization, Chem. Soc. Rev., 2022, 51, 1470–1510 RSC.
- (a) G. Yan, M. Yang and X. Wu, Synthetic applications of arylboronic acid via an aryl radical transfer pathway, Org. Biomol. Chem., 2013, 11, 7999 RSC; (b) X. Shang and Z.-Q. Liu, Advances in free-radical alkylation and arylation with organoboronic acids, Org. Biomol. Chem., 2022, 20, 4074–4080 RSC.
- Y. Iwata, Y. Tanaka, S. Kubosaki, T. Morita and Y. Yoshimi, A strategy for generating aryl radicals from arylborates through organic photoredox catalysis: photo-Meerwein type arylation of electron-deficient alkenes, Chem. Commun., 2018, 54, 1257–1260 RSC.
- M. Caldarelli, L. Laze, L. Piazza, G. Caputo, M. De Amici and G. Papeo, Use of acridinium-based photocatalyst in the Giese-type coupling of arylboronic acids with electron poor olefins, Tetrahedron Lett., 2022, 103, 153978 CrossRef CAS.
- J. R. Immel, M. Chilamari and S. Bloom, Combining flavin photocatalysis with parallel synthesis: a general platform to optimize peptides with non-proteinogenic amino acids, Chem. Sci., 2021, 12, 10083–10091 RSC.
- M. Chilamari, J. R. Immel and S. Bloom, General Access to C-Centered Radicals: Combining a Bioinspired Photocatalyst with Boronic Acids in Aqueous Media, ACS Catal., 2020, 10, 12727–12737 CrossRef CAS.
- M. Yamawaki, R. Hashimoto, Y. Kawabata, M. Ichihashi, Y. Nachi, R. Inari, C. Sakamoto, T. Morita and Y. Yoshimi, Visible–Light–Induced Decarboxylative and Deboronative Radical Addition to Alkenes in Two–Molecule Photoredox System Using Dibenzo[g,p]chrysene, Eur. J. Org. Chem., 2022, e202201225 CrossRef CAS.
- Y. Yuan, Y. Zheng, B. Xu, J. Liao, F. Bu, S. Wang, J.-G. Hu and A. Lei, Mn-Catalyzed Electrochemical Radical Cascade Cyclization toward the Synthesis of Benzo[4,5]imidazo[2,1-a]isoquinolin-6(5H)-one Derivatives, ACS Catal., 2020, 10, 6676–6681 CrossRef CAS.
- A. Hosseinian, R. Mohammadi, S. Ahmadi, A. Monfared and Z. Rahmani, Arylhydrazines: novel and versatile electrophilic partners in cross-coupling reactions, RSC Adv., 2018, 8, 33828–33844 RSC.
- J. H. Powell and P. M. Gannett, Mechanisms of Carcinogenicity of Aryl Hydrazines, Aryl Hydrazides, and Arenediazonium Ions, J. Environ. Pathol., Toxicol. Oncol., 2002, 21, 31 CrossRef.
- S. Liu, W. Pan, S. Wu, X. Bu, S. Xin, J. Yu, H. Xu and X. Yang, Visible-light-induced tandem radical addition–cyclization of 2-aryl phenyl isocyanides catalysed by recyclable covalent organic frameworks, Green Chem., 2019, 21, 2905–2910 RSC.
- Y.-H. Chen, M. Lee, Y.-Z. Lin and D. Leow, Remazol-Catalyzed Hydroperoxyarylation of Styrenes, Chem. – Asian J., 2015, 10, 1618–1621 CrossRef CAS PubMed.
- Y. Ding, W. Zhang, H. Li, Y. Meng, T. Zhang, Q.-Y. Chen and C. Zhu, Metal-free synthesis of ketones by visible-light induced aerobic oxidative radical addition of aryl hydrazines to alkenes, Green Chem., 2017, 19, 2941–2944 RSC.
- L. Du, L. Sun and H. Zhang, Photochemical and electrochemical C–N borylation of arylhydrazines, Chem. Commun., 2022, 58, 1716–1719 RSC.
- Y. Yu, P. Zheng, Y. Wu and X. Ye, Electrochemical cobalt-catalyzed C–H or N–H oxidation: a facile route to synthesis of substituted oxindoles, Org. Biomol. Chem., 2018, 16, 8917–8921 RSC.
- S. Song, X. Shi, Y. Zhu, Q. Ren, P. Zhou, J. Zhou and J. Li, Electrochemical Oxidative C–H Arylation of Quinoxalin(on)es with Arylhydrazine Hydrochlorides under Mild Conditions, J. Org. Chem., 2022, 87, 4764–4776 CrossRef CAS PubMed.
- W. Liu, L. Hao, J. Zhang and T. Zhu, Progress in the Electrochemical Reactions of Sulfonyl Compounds, ChemSusChem, 2022, 15, e202102557 CrossRef CAS PubMed.
- Y. Yuan, M. Jiang, T. Wang, Y. Xiong, J. Li, H. Guo and A. Lei, Synergy of anodic oxidation and cathodic reduction leads to electrochemical deoxygenative C2 arylation of quinoline N-oxides, Chem. Commun., 2019, 55, 11091–11094 RSC.
- (a) R. Francke and R. D. Little, Optimizing Electron Transfer Mediators Based on Arylimidazoles by Ring Fusion: Synthesis, Electrochemistry, and Computational Analysis of 2-Aryl-1-methylphenanthro[9,10-d]imidazoles, J. Am. Chem. Soc., 2014, 136, 427–435 CrossRef CAS PubMed; (b) A. J. J. Lennox, J. E. Nutting and S. S. Stahl, Selective electrochemical generation of benzylic radicals enabled by ferrocene-based electron-transfer mediators, Chem. Sci., 2018, 9, 356–361 RSC; (c) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Electrocatalysis as an enabling technology for organic synthesis, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC; (d) N. Salardón, G. Otárola, C. Mañas, E. Merino and B. Batanero, Electrochemically driven one-pot oxidative conversion of arylhydrazines into aromatic iodides, J. Environ. Chem. Eng., 2022, 10, 107486 CrossRef; (e) W. Shao, B. Lu, J. Cao, J. Zhang, H. Cao, F. Zhang and C. Zhang, The Use of Redox Mediators in Electrocatalysis and Electrosynthesis, Chem. – Asian J., 2023, 18, e202201093 CrossRef CAS PubMed; (f) B. R. Walker, S. Manabe, A. T. Brusoe and C. S. Sevov, Mediator-Enabled Electrocatalysis with Ligandless Copper for Anaerobic Chan–Lam Coupling Reactions, J. Am. Chem. Soc., 2021, 143, 6257–6265 CrossRef CAS PubMed.
- B. M. Bruce, R. Francke, R. D. Little and L. A. Berben, High turnover in electro-oxidation of alcohols and ethers with a glassy carbon-supported phenanthroimidazole mediator, Chem. Sci., 2017, 8, 6493–6498 RSC.
- J. Li and N. Wu, Semiconductor-based photocatalysts and photoelectrochemical cells for solar fuel generation: a review, Catal. Sci. Technol., 2015, 5, 1360–1384 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © the Partner Organisations 2024 |