
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyridine-based tricarboxamides: complementary monomers for supramolecular copolymerization with C3-symmetric oligophenylenetricarboxamides†
L.
López-Gandul
 ,
L.
Sánchez
* and
F.
García
*
,
L.
Sánchez
* and
F.
García
*
Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: lusamar@ucm.es; fatgar02@ucm.es
First published on 8th July 2024
Abstract
The supramolecular copolymerization of triangular-shaped oligophenylenetricarboxamides 1 with the electronically and geometrically complementary pyridine tricarboxamides 2 is investigated. Moreover, amplification of asymmetry between both tricarboxamides is also studied both experimentally and by making use of mathematical models. In the modified seargents and soldiers (mSaS) experiments between achiral tricarboxamide a-1 and chiral Py-based (S)-2 or (R)-2, the existence of amplification of asymmetry is proved. In these experiments, the point chirality embedded in the side chains of (S)-2 or (R)-2 yields homochiral copolymers upon mixing with achiral a-1. The formation of the heteropolymers poly-(S)-1-co-(R)-2 or poly-(R)-1-co-(S)-2 is also studied by mixing the monomeric species with opposite point chirality [(S)-1 + (R)-2 or (R)-1 + (S)-2] in different ratios but keeping the total concentration constant. These experiments reveal a weak amplification of asymmetry that could be indicative of an inefficient supramolecular copolymerization of both chiral species. However, the application of the copolymerization model shows that these geometrically and electronically complementary species form copolymers with a block microstructure, in which large blocks of homopolymers poly-1 and poly-2 coexist with a very low number of heterocontacts. This arrangement of the blocks of opposite helicity finally results in the experimentally observed weak amplification of asymmetry.
Introduction
Self-assembly, the spontaneous and reversible organization of molecular units into ordered structures by non-covalent interactions, occurs spontaneously in Nature, giving rise to well-organized, functional structures.1 Unlike self-assembly, which involves only one chemical species, co-assembly requires the non-covalent interaction of two or more species. Cell membranes and virus capsids exemplify the level of sophistication and functionality that natural co-assembled structures can reach.2–4 For researchers, understanding and harnessing this process enables the development of advanced materials and technologies with a wide range of applications, from medicine to electronics and sustainable energy solutions.Supramolecular polymers (SPs), in which the monomeric units are individual molecules held together by non-covalent interactions, have arisen as a very useful benchmark for investigating self-assembly processes.5 These non-covalent interactions provide the systems with dynamism, in contrast to their traditional covalent congeners. Therefore, SPs can respond to environmental changes—concentration, temperature or solvent nature—that favour the formation/disruption of intermolecular interactions and show enhanced and advanced functionalities in comparison to the isolated monomers.6 A vast majority of the examples reported for SPs are based on the self-assembly of a single monomeric species that, following an isodesmic or a cooperative supramolecular polymerization mechanism,5 yields highly organized, functional structures.6 Importantly, in the last few years, a number of examples describing the formation of supramolecular block copolymers (SBCPs), that is, macromolecules formed by the non-covalent interaction of two or more monomeric species, have been reported.7 These SBCPs exhibit a higher level of complexity in comparison to SPs due to the increased number of components. The formation of supramolecular copolymers requires a high level of control over the supramolecular interactions between the different monomers and the environment. To obtain supramolecular copolymers, the different monomers should meet the following requirements: (i) structural similarity; (ii) low exchange dynamics and (iii) thermodynamically favoured heterorecognition.7 A key issue on the further development of SBCPs is unravelling the microstructure of such co-assembled structures. If the monomers do not copolymerize, narcissistic self-sorting takes place, yielding individual homopolymer chains formed exclusively by a single monomer. However, if copolymerization takes place, the resulting SBCPs can be blocky, periodic, statistical, or alternating.7
A key point in the development and study of SPs relies on the use of mathematical models reported for elucidating the supramolecular polymerization mechanism governing the process and the derivation of the corresponding thermodynamic parameters.8 These mathematical models are also used for unveiling the microstructure of SBCPs under the condition that both the homo- and heteropolymerization processes are thermodynamically controlled.8,9 Using the co-assembly mathematical model reported by ten Eikelder and coworkers,9 in which the thermodynamic parameters associated with the homopolymerization process of the pristine components are required, the microstructure and a number of thermodynamically controlled SBCPs can be elucidated.10–15 It is worth mentioning that elegant examples of kinetically controlled SBCPs, in which the corresponding thermodynamic parameters associated with heteropolymerization cannot be derived, have been reported to afford alternating16–18 or blocky heteropolymers.19
An active line of research within the field of SPs is the achievement of chiral supramolecular polymers.20 These systems hold applicability as new functional materials, showing exciting properties such as the emission of circularly polarized light21–24 or the generation of a spin-filtering effect due to chirality-induced spin selectivity properties.25–27 At the same time, chiral SPs are useful platforms to investigate relevant processes such as the transfer and amplification of asymmetry, closely related to the origin of natural homochirality.28–30 Amplification of asymmetry is a process in which the enantiomeric excess (ee) of a macromolecule is higher than the ee of the monomer counterparts. It was firstly reported by Green and coworkers for polyisocyanates, where minute amounts of chiral monomers bias the helicity of the whole polymer.31,32 Soon after, amplification of asymmetry was also reported for SPs formed by benzenetricarboxamides in the seminal reports by Meijer and coworkers.33 Formally, the amplification of asymmetry in supramolecular polymers involves the formation of SBCPs, in which an achiral and an enantioenriched monomeric species, in the case of sergeants and soldiers (SaS) experiments, or two enantiomers in different ratios, for majority rules (MRs) experiments, are mixed up together.33,34,37 In fact, the above-mentioned mathematical model reported by ten Eikelder and coworkers includes these experiments as a relevant part in the study of SBCPs.9
Our research group has actively worked on the supramolecular polymerization, transfer, and amplification of asymmetry of oligo(phenyleneethynylene)tricarboxamides (compounds 1 in Fig. 1a). These tricarboxamides self-assemble into columnar, helical aggregates through the operation of a triple array of intermolecular hydrogen bonds between amide groups and the π-stacking of the central aromatic skeletons.35,38 Very recently, we have also reported the synthesis of C2v-symmetric tricarboxamide (S)-2 (Fig. 1a), in which a benzene ring is replaced by an electron-withdrawing pyridine moiety in comparison to 1. The Py-based tricarboxamide (S)-2, upon self-assembly, gives rise to helical supramolecular polymers, poly-(S)-2, as P-helices in methylcyclohexane (MCH) as a solvent, following a cooperative mechanism. Moreover, the electronic complementarity and similar geometry allowed the successful copolymerization of 1 and (S)-2 as periodical type P-helical heteropolymers.36
 | ||
| Fig. 1 (a) Chemical structures of tricarboxamides 1 and 2; (b) and (c) schematic illustration of the mSaS (b) and mMRs co-assembly experiments (c) performed by using mixtures of 1 and 2. In the former, a molar fraction of 0.3 for the chiral enantiomer (S)-2 is enough to achieve a homochiral copolymer, whilst in the latter, the co-assembly yields block SBCPs with large domains of opposite helicity of both enantiomers. | ||
Herein, we further investigate the co-assembly of chiral monomeric species under thermodynamic control, exhibiting geometric and electronic complementarity. In particular, we report the co-assssembly of pyridine-based tricarboxamides 2 and tricarboxamides 1 to form the corresponding heteropolymers. Through a combination of spectroscopy and atomic force microscopy, we study the phenomena of amplification of asymmetry through the SaS and MRs experiments and establish relationships with their microstructure by application of the co-assembly model to assess the microstructure of the copolymers. The studies carried out with these tricarboxamides contribute to increase the comprehension of the requirements that self-assembling monomeric units should fulfil to generate homochiral supramolecular copolymers.
Results and discussion
Synthesis of Py-based tricarboxamide (R)-2 and supramolecular homo-polymerization
The synthesis of both tricarboxamides 1 and Py-based tricarboxamide (S)-2 was reported previously.35,36 By following an analogous synthetic procedure, pyridine tricarboxamide (R)-2 has been obtained, and all new compounds have been fully characterized using the standard techniques, as detailed in the ESI.†The self-assembly studies performed previously by our group with tricarboxamides 1 in MCH solution demonstrated the cooperative formation of helical aggregates with enantioenriched P- or M-type helices for chiral congeners, (S)-1 or (R)-1, respectively.35C2v-Symmetric tricarboxamide (S)-2 self-assembles following a cooperative mechanism into enantioenriched P-helical aggregates, as reported previously.36
To elucidate the self-assembling features of (R)-2 and the non-covalent forces involved in its supramolecular homopolymerization, we firstly recorded FTIR spectra in different solvents. A first indication of the supramolecular polymerization by the operation of a triple array of H-bonding interactions between the amide functional groups was obtained from the FTIR spectrum of (R)-2 in a bad solvent like MCH at a total concentration cT = 1 mM. This spectrum displays values for the N–H and the amide I stretching bands centred at 3267 and 1633 cm−1, respectively, indicating the formation of intermolecular H-bonds (Fig. S1†).36,39–42 On the other hand, the FTIR spectrum of (R)-2 in chloroform, a good solvent that favours the disassembly of the aggregated species, displays the N–H and the amide I stretching band centred at 3452 and 1658 cm−1, values ascribable to the molecularly dissolved state (Fig. S1†).36,39–42 The variable temperature (VT) UV-Vis spectrum of (R)-2 in MCH demonstrates that the self-assembly of this tricarboxamide is reinforced by the π-stacking of the aromatic cores. Thus, and in good analogy with its (S)-enantiomer,36 the absorption pattern of (R)-2 in MCH solution at cT = 10 μM and at 20 °C displays a maximum centred at 301 nm that splits into two bands centred at 316 and 298 nm on heating the sample up to 90 °C, evidencing the formation of H-type aggregates (Fig. 2a). Plotting the variation of the absorbance of the band centred at 315 nm versus temperature yields non-sigmoidal curves characteristic of cooperative mechanisms (Fig. 2c).36 The global fitting of the data allows the calculation of the thermodynamic parameters associated with the supramolecular polymerization of (R)-2 by applying the one-component equilibrium (EQ) model reported by ten Eikelder and coworkers43 (Fig. 2c and Table 1). The supramolecular polymerization of (R)-2 efficiently yields M-type helical aggregates, as demonstrated by the corresponding CD spectrum of (R)-2 in MCH solution, with a clear −/+ bisignated Cotton effect. This spectrum shows maxima at λ = 304 and 277 nm and a zero-crossing point at λ = 294 nm, a mirror image of its enantiomer (S)-2 (Fig. 2b).
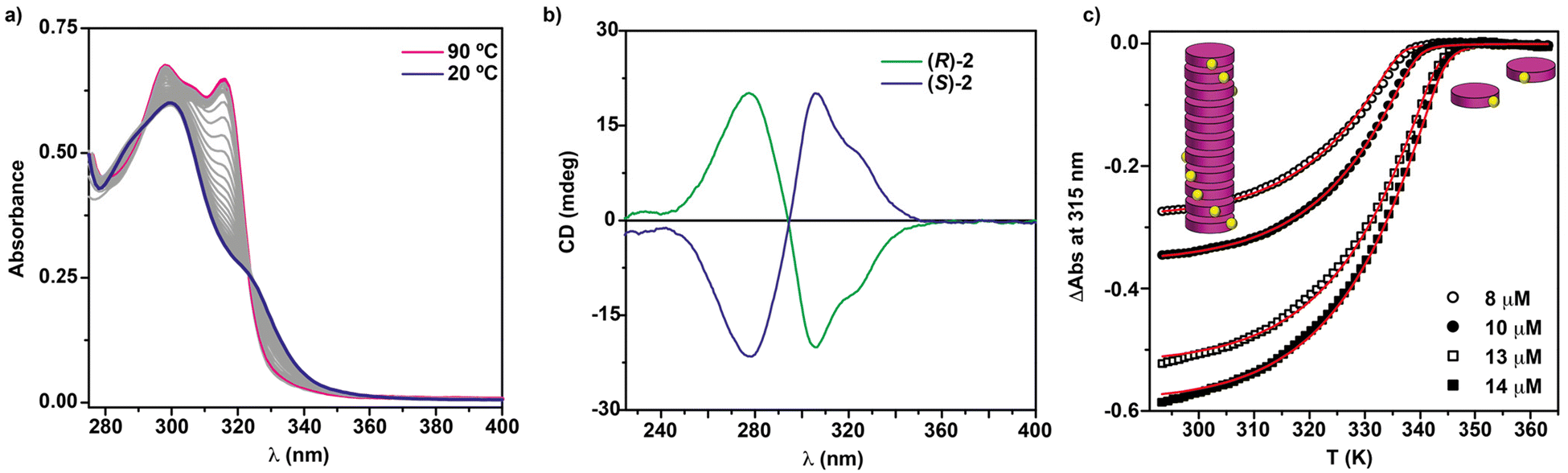 | ||
| Fig. 2 (a) UV-vis spectra of (R)-2 at different temperatures (MCH, cT = 5 μM); (b) CD spectra of (S)-2 and (R)-2 (20 °C, MCH, cT = 10 μM); (c) changes in the band centred at λ = 315 nm upon cooling 90 °C solutions of (R)-2 (MCH, cooling rate: 1 K min−1). The red lines in (c) correspond to the fitting to the one component EQ model.43 | ||
Modified SaS experiments
In our previous contribution, it was established that a-1 and (S)-2 efficiently co-assemble, giving rise to poly-(a-1)0.5-co-((S)-2)0.5, and the thermodynamic parameters associated with the co-assembly process were extracted by using the copolymerization model.9 To further investigate the co-assembly of these two geometrically and electronically complementary monomeric species, we have performed a modified SaS (mSaS) experiment, in which these two tricarboxyamides, with a similar but not identical chemical structure, are mixed together at different ratios but keeping the total concentration (cT = 10 μM) constant (Fig. 3a–c). Thus, upon increasing additions of chiral sergeant (S)-2 to a solution of the achiral soldier a-1, keeping the total concentration constant (10 μM),‡ a nonlinear increase of the dichroic signal is observed and the maximum dichroic response is reached upon addition of ∼30% of the chiral sergeant (Fig. 3b and c). It is worth mentioning that, unlike the CD response that increases upon the addition of the chiral sergeant, the corresponding UV-Vis spectra experience minute changes in the absorption maxima (Fig. S2†). This experiment corroborates an efficient co-assembly between both species and the amplification of asymmetry exerted by the pyridine tricarboxamide (S)-2 to a-1, yielding enantioenriched P-helical copolymers. | ||
| Fig. 3 mSaS experiments between a-1 and (S)-2. (a) CD spectra of tricarboxamides 1 (blue, solid line), (S)-2 (purple, dashed line) and mixtures of both (grey lines) at different ratios; (b) plot of the variation of the CD response at λ = 304 nm versus the molar fraction (X) of chiral (S)-2. Experimental conditions: MCH as solvent, 20 °C, cT = 10 μM; the red line in (b) corresponds to a sigmoidal fit of the plotted data to guide the eye. (c) Speciation plot. The MMP parameter for this plot is MMP = 0.05 kJ mol−1. | ||
To further rationalize the co-assembly of a-1 and (S)-2, we have used the co-assembly model and the previously derived thermodynamic parameters for pristine poly-(a-1) and poly-(S)-2 (Table 1).36 Thus, a speciation plot of the monomers and the M- and P-helices was simulated for different values of the energy penalty when a chiral monomer is introduced in a stack of its unpreferred helicity (mismatch penalty, MMP). The best fit with the experimental data was found for an energy penalty value of 0.05 kJ mol−1 (see the ESI† for details and Fig. 3c). This value is one order of magnitude lower than that reported for benzene tricarboxamides (BTAs), with an energy penalty of ∼0.5 kJ mol−1.9 This low value can explain the relatively less efficient amplification of asymmetry found for (S)-2 and a-1 in comparison to BTAs. In this copolymer, the energy penalty for the incorporation of a monomer within the opposite helicity polymer is so small that more monomers will be incorporated into the wrong helix and, therefore, ending in a decrease of the amplification of asymmetry phenomenon compared to BTAs. The efficient intercalation of a-1 and (S)-2 along the stacks, giving rise to blocks with alternating fragments of 3 units of monomeric species, allows an efficient amplification of asymmetry, as reported previously for poly-(a-1)0.5-co-((S)-2)0.5.36 To further corroborate the validity of the applied co-assembly model, we have also utilized different values of the MMP (0.5, 0.075 and 0.025). The co-assembly model reveals that higher values of the MMP would result in a more efficient amplification of asymmetry (for more details, see part 5 in the ESI†).
A similar mSaS experiment has been carried out by utilizing the Py-based tricarboxamide (R)-2 as a chiral sergeant. In very good agreement with the above-mentioned experiment, the CD spectra of the mixtures of achiral a-1 with chiral (R)-2 in MCH at cT = 10 μM and at different ratios show a non-linear increase of the dichroic response due to the amplification of asymmetry, concomitant with the co-assembly of both species. Once again, the homochiral mixture yielding an M-type helical aggregate is achieved at a molar fraction of (R)-2 of ∼0.3 (Fig. S3†).
Majority rules and modified majority rules
Once we proved the ability of Py-based tricarboxamides 2 to amplify asymmetry in achiral tricarboxamide a-1, we studied the phenomenon of amplification of asymmetry between chiral tricarboxamides (S)-2 and (R)-2 through a MRs experiment. Upon mixing solutions of (S)-2 and (R)-2 at different ratios in MCH (cT = 10 μM), CD spectra showing the Cotton effect with maxima centred at λ = 304 and 277 nm and a zero-crossing point at λ = 295 nm are observed. Plotting the variation of the CD response as a function of the ee displays a straight line, indicating no amplification of asymmetry between (S)-2 and (R)-2 (Fig. S4†). The application of the two-component EQ model reported by ten Eikelder and coworkers43 and the thermodynamic parameters collected in Table 1 for (S)-2 and (R)-2 results in a high value for the MMP (6.9 kJ mol−1). This MMP value is similar to that previously reported for C2v-symmetric tricarboxamides endowed with only one stereogenic centre per monomeric unit, which do not experience amplification of asymmetry.38 Taking into account these results, two different scenarios could be considered: (i) the low ability of these enantiomers to co-assemble, giving rise to a narcissistic polymerization in which P- and M-helices are formed exclusively by (S)-2 and (R)-2 monomers, respectively; and (ii) the copolymer formed has different sections of opposite helicity within the same stack (Fig. S4c†). Both situations would result in a CD response that increases linearly upon increasing the ratio of one of the enantiomers in the mixture. The high value of the derived MMP could account for option (i), in which a narcissistic self-sorting takes place, which seems to be the most plausible situation for the mixture of both enantiomers. The high energy penalty that the system should overcome when an enantiomer stacks in a helical aggregate of its unpreferred handedness provokes each enantiomer to be incorporated in a polymer helical stack formed by its own congeners (Fig. S4†).To complement the co-assembly studies, and based on the positive results on the amplification of asymmetry between (S)-2 and a-1, we investigated if chiral Py-based tricarboxamides 2 could also dictate the asymmetry of mixtures of these Py-based tricarboxamides and chiral tricarboxamides 1 by performing modified MRs (mMRs) experiments. Before preparing the mixtures of these two tricarboxamides, we noticed that, despite the high similarity of the dichroic pattern of both systems, with maxima at λ ∼ 275 and 300 nm and a zero-crossing point at λ ∼ 295 nm, the intensity of the CD spectra of 2 is lower, almost a threefold decrease in comparison to those recorded for chiral tricarboxamides 1 under the same experimental conditions (Fig. 4a and S5a†).
 | ||
Fig. 4 Co-assembly of (R)-1 and (S)-2; (a) CD spectra of pristine poly-(R)-1 (blue line), poly-(S)-2 (purple line) and mixtures of both at different ratios (the blue and purple arrows indicate the changes in the CD spectra upon the addition of increasing amounts of (R)-1 or (S)-2, respectively); (b) plot of the variation of the CD response versus the ee (the red lines correspond to sigmoidal fits to guide the eye). Experimental conditions for the mMRs experiment: MCH as solvent, 20 °C; cT = 10 μM; (c) UV/Vis cooling curves (1 K min−1) of MCH solutions of poly-(R)-1 (blue line) and poly-(S)-2 (purple line) at cT = 5 μM and a MCH solution of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of (R)-1 and (S)-2 (red line) at cT = 10 μM. The dashed line represents the arithmetic sum of the experimental curves recorded for poly-(R)-1 and 5 μM poly-(S)-2 at cT = 5 μM in MCH; (d) cooling curves calculated using the supramolecular copolymerization model for poly-(R)-1, poly-(S)-2 and poly-(R)-1-co-(S)-2 in MCH. :1 mixture of (R)-1 and (S)-2 (red line) at cT = 10 μM. The dashed line represents the arithmetic sum of the experimental curves recorded for poly-(R)-1 and 5 μM poly-(S)-2 at cT = 5 μM in MCH; (d) cooling curves calculated using the supramolecular copolymerization model for poly-(R)-1, poly-(S)-2 and poly-(R)-1-co-(S)-2 in MCH. | ||
Considering this difference, we performed mMRs experiments by mixing equimolar MCH solutions of (R)-1 and (S)-2 at cT = 10 μM and recorded their CD spectra (Fig. 4a). The dissimilar dichroic intensity results in the absence of the zero response in CD for zero ee. Plotting the variation of the dichroic response versus ee shows a non-linear increase that could be indicative of a slight amplification of asymmetry (Fig. 4b). This non-linear trend is more noticeable for negative ee values, wherein the maximum dichroic response is obtained for an ee value of −0.6. This ee value corresponds to a 1:3 ratio of (R)-1:(S)-2. Similar findings are observed by performing these mMRs experiments between (S)-1 and (R)-2 (Fig. S5†).§
The low ability of Py-based tricarboxamides 2 to provoke an amplification of asymmetry in C3-symmetric tricarboxamides 1 and vice versa raises reasonable doubts about the actual co-assembly of these two species to form the corresponding heteropolymers poly-(R)-1-co-(S)-2 or poly-(R)-2-co-(S)-1. To shed light on the formation of these copolymers, we have prepared a 1:1 mixture of (R)-1 and (S)-2 at cT = 10 μM in MCH and recorded a cooling curve at a cooling rate of 1 K min−1. This cooling curve presents a non-sigmoidal shape and a single temperature of elongation, Te, of 64 °C (Fig. 4c and Table 2). Furthermore, the cooling curves of each (R)-1 and (S)-2 at cT = 5 μM in MCH were also recorded by using the same cooling rate. These two curves also present a non-sigmoidal shape and Te values of 60 and 64 °C for poly-(R)-1 and poly-(S)-2, respectively (Fig. 4c and Table 2). Importantly, the cooling curve of the mixture of (R)-1 and (S)-2 is different from the arithmetic sum of the cooling curves of poly-(R)-1 and poly-(S)-2 (dashed line in Fig. 4c), which indicates the co-assembly of both components to form the heteropolymer poly-(R)-1-co-(S)-2.9,36
| T e/°C | poly-(R)-1 | poly-(S)-2 | poly-(R)-1-co-(S)-2 |
|---|---|---|---|
| Experimental | 60 | 64 | 64 |
| Calculated | 60.9 | 62.8 | 63.7 |
To gain insight into the microstructure of poly-(R)-1-co-(S)-2, we have applied the supramolecular copolymerization model by using the thermodynamic parameters derived for pristine poly-(R)-1 and poly-(S)-2 (Table 1) and considering intermediate values for the elongation enthalpy, ΔHe, and entropy, ΔS. The best fit for the copolymerization model has been obtained by assigning values for ΔHe and ΔS of −73.5 kJ mol−1 and −140 J K−1mol−1, respectively. These values are in good analogy with those previously reported for the heteropolymer poly-a-1-co-(S)-2.36 Using these thermodynamic parameters, the cooling curves of the homo- and heteropolymers were simulated. These simulated cooling curves display Te values of 60.9, 62.8 and 63.7 °C for homopolymers poly-(R)-1 and poly-(S)-2 and heteropolymer poly-(R)-1-co-(S)-2, respectively, in very good accordance with the experimental values (Fig. 4d and Table 2).
The supramolecular copolymerization model not only corroborates the formation of the heteropolymers poly-(R)-1-co-(S)-2 but also provides insights about the blocky microstructure of this copolymer. Thus, the length of the blocks in homopolymers poly-(R)-1 and poly-(S)-2 gradually increases upon decreasing the temperature until reaching a length of ∼8 units per block (Fig. S6a†). In contrast, the block length for the copolymer experiences a negligible change upon decreasing the temperature, showing only two units per block (Fig. S6a†). Additionally, the bond fraction of the homopolymers is four-fold in comparison to that observed for the corresponding heteropolymer poly-(R)-1-co-(S)-2. Therefore, the small number of heterocontacts can also justify its blocky nature and, therefore, the lack of amplification of asymmetry found for this heteropolymer (Fig. S6b†). As expected, the copolymerization model also reflects that the length of the copolymer increases upon decreasing the temperature, although this increase is lower than that predicted for the previously reported heteropolymer poly-a-1-co-(S)-2 (Fig. S6c†).36 In short, the predicted microstructure of the heteropolymer poly-(R)-1-co-(S)-2 justifies the limited amplification of asymmetry observed experimentally since large blocks of opposite helicity coexist in the same columnar aggregate (Fig. 1c).
Finally, we have used Atomic Force Microscopy (AFM) to visualize the morphology of new homo- and heteropolymers. Spin-coating a 10 μM solution of (R)-2 in MCH onto the HOPG surface reveals the formation of long and thin, rope-like fibres with heights of around 4 nm. In addition, these fibres intertwine to yield thicker fibres with heights of around 25 nm (Fig. 5a–c). These dimensions are in very good agreement with those previously reported for the enantiomer (S)-2.22 Analogously, the AFM images of a spin-coated MCH solution of the copolymer poly-(S)-1-co-(R)-2 onto HOPG, obtained upon mixing equimolecular amounts of pristine homopolymers, show the formation of long and thin fibrillar, rope-like supramolecular aggregates with heights of ∼3 nm (Fig. 5d and e). In addition, intertwined, thicker rope-like aggregates are also visible in the AFM images with heights of ∼7 nm that, taking into account the molecular dimensions of these tricarborxamides,25 would correspond to individual fibres and bundles of fibrillar aggregates (Fig. 5d and e).
 | ||
| Fig. 5 AFM images of poly-(R)-2 (a and b) and copolymer poly-(R)-2-co-(S)-1 (d and e). (a) and (d) Height images. (b) and (e) Phase images. Height profiles along the green lines in (a) and (d) of the fibers formed by poly-(R)-2 (c) and poly-(R)-2-co-(S)-1 (f). Experimental conditions: cT = 10 μM, HOPG as surface, MCH as solvent. | ||
Conclusions
In this contribution, we describe the synthesis and a complete study of the supramolecular polymerization of the triangular-shaped, Py-based tricarboxamide (R)-2 in MCH solution. The thermodynamic parameters associated with the self-assembly of (R)-2 are very similar to those previously reported for its S-enantiomer, (S)-2, although with mirror image CD spectra, indicating the formation of supramolecular polymers of opposite handedness. A detailed investigation of the co-assembly of these electron-withdrawing tricarboxamides with the electron-donating tricarboxamides 1 is presented. Firstly, we have developed mSaS experiments that prove the efficient co-assembly of chiral (S)-2 or (R)-2 with their achiral congener a-1. The application of the co-assembly mathematical model reveals a low MMP factor (0.05 kJ mol−1) for these experiments on the amplification of asymmetry. MRs experiments between (S)-2 and (R)-2 proved the lack of amplification of asymmetry between them, justified by the large value of the MMP (−6.9 kJ mol−1). Finally, we have investigated the co-assembly of the chiral congeners of 1 and 2 to form the corresponding heteropolymers poly-(S)-1-co-(R)-2 and poly-(R)-1-co-(S)-2. Interestingly, the mMRs experiments, performed by mixing (S)-1 and (R)-2 or (R)-1 and (S)-2 in different ratios but keeping the total concentration constant, reveal a weak amplification of asymmetry that could be diagnostic of a lack of efficient co-assembly. However, the application of the co-assembly model shows that these geometrically and electronically complementary species form copolymers with a block microstructure with large blocks of homopolymers poly-1 and poly-2 and a small number of hetero-contacts. This microstructure prevents an efficient transfer of the asymmetry between these two chiral species. The results presented herein contribute to shed light on the thermodynamically controlled co-assembly of chiral monomeric species with geometric and electronic complementarity, yielding heteropolymers with a controlled microstructure.Data availability
The data that support the findings of this study have been included in the main text and ESI† and are available from the corresponding author upon reasonable request.Author contributions
L. López-Gandul (first author): investigation, data curation, formal analysis, visualization and revising the original draft; Dr L. Sánchez (corresponding author): investigation, writing – review & editing the original draft, and funding acquisition; Dr F. García (corresponding author): conceptualization, supervision, validation, writing – review & editing and funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the MCIN/AEI of Spain (CNS2022-136058, PID2020-113512GB-I00 and TED2021-130285B-I00) and Comunidad de Madrid (P27/21-008) is acknowledged.References
- D. Philp and J. F. Stoddart, Self-Assembly in Natural and Unnatural Systems, Angew. Chem., Int. Ed. Engl., 1996, 35, 1154 CrossRef.
- P. Makam and E. Gazit, Minimalistic peptide supramolecular co-assembly: expanding the conformational space for nanotechnology, Chem. Soc. Rev., 2018, 47, 3406 RSC.
- I. R. Sasselli and Z. Syrgiannis, Small Molecules Organic Co-Assemblies as Functional Nanomaterials, Eur. J. Org. Chem., 2020, 5305 CrossRef CAS.
- P. Xing and Y. Zhao, Controlling Supramolecular Chirality in Multicomponent Self-Assembled Systems, Acc. Chem. Res., 2018, 51, 2324 CrossRef CAS.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Supramolecular Polymers, Chem. Rev., 2001, 101, 4071 CrossRef CAS.
- T. Aida, E. W. Meijer and S. I. Stupp, Functional Supramolecular Polymers, Science, 2012, 335, 813 CrossRef CAS PubMed.
- B. Adelizzi, N. J. Van Zee, L. N. J. de Windt, A. R. A. Palmans and E. W. Meijer, Future of Supramolecular Copolymers Unveiled by Reflecting on Covalent Copolymerization, J. Am. Chem. Soc., 2019, 141, 6110 CrossRef CAS.
- H. M. M. ten Eikelder and A. J. Markvoort, Mass-Balance Models for Scrutinizing Supramolecular (Co)polymerizations in Thermodynamic Equilibrium, Acc. Chem. Res., 2019, 52, 3465 CrossRef CAS PubMed.
- H. M. M. ten Eikelder, B. Adelizzi, A. R. A. Palmans and A. J. Markvoort, Equilibrium Model for Supramolecular Copolymerizations, J. Phys. Chem. B, 2019, 123, 6627 CrossRef CAS PubMed.
- B. Adelizzi, A. Aloi, A. J. Markvoort, H. M. M. Ten Eikelder, I. K. Voets, A. R. A. Palmans and E. W. Meijer, Supramolecular Block Copolymers under Thermodynamic Control, J. Am. Chem. Soc., 2018, 140, 7168 CrossRef CAS PubMed.
- L. N. J. de Windt, C. Kulkarni, H. M. M. ten Eikelder, A. J. Markvoort, E. W. Meijer and A. R. A. Palmans, Detailed Approach to Investigate Thermodynamically Controlled Supramolecular Copolymerizations, Macromolecules, 2019, 52, 7430 CrossRef CAS PubMed.
- L. N. J. de Windt, Z. Fernández, M. Fernández-Míguez, F. Freire and A. R. A. Palmans, Elucidating the Supramolecular Copolymerization of N- and C-Centered Benzene-1,3,5-Tricarboxamides: The Role of Parallel and Antiparallel Packing of Amide Groups in the Copolymer Microstructure, Chem. – Eur. J., 2022, 28, e202103691 CrossRef CAS.
- B. Adelizzi, P. Chidchob, N. Tanaka, B. A. G. Lamers, S. C. J. Meskers, S. Ogi, A. R. A. Palmans, S. Yamaguchi and E. W. Meijer, Long-Lived Charge-Transfer State from B–N Frustrated Lewis Pairs Enchained in Supramolecular Copolymers, J. Am. Chem. Soc., 2020, 142, 16681 CrossRef CAS PubMed.
- H. Su, S. A. H. Jansen, T. Schnitzer, E. Weyandt, A. T. Rösch, J. Liu, G. Vantomme and E. W. Meijer, Unraveling the Complexity of Supramolecular Copolymerization Dictated by Triazine–Benzene Interactions, J. Am. Chem. Soc., 2021, 143, 17128 CrossRef CAS PubMed.
- R. Liao, F. Wang, Y. Guo, Y. Han and F. Wang, Chirality-Controlled Supramolecular Donor–Acceptor Copolymerization with Distinct Energy Transfer Efficiency, J. Am. Chem. Soc., 2022, 144, 9775 CrossRef CAS.
- D. Görl, X. Zhang, V. Stepanenko and F. Würthner, Supramolecular block copolymers by kinetically controlled co-self-assembly of planar and core-twisted perylene bisimides, Nat. Commun., 2015, 6, 7009 CrossRef PubMed.
- A. Sarkar, R. Sasmal, C. Empereur-mot, D. Bochicchio, S. V. K. Kompella, K. Sharma, S. Dhiman, B. Sundaram, S. S. Agasti, G. M. Pavan and S. J. George, Self-Sorted, Random, and Block Supramolecular Copolymers via Sequence Controlled, Multicomponent Self-Assembly, J. Am. Chem. Soc., 2020, 142, 7606 CrossRef CAS PubMed.
- A. Sarkar, T. Behera, R. Sasmal, R. Capelli, C. Empereur-mot, J. Mahato, S. S. Agasti, G. M. Pavan, A. Chowdhury and S. J. George, Cooperative Supramolecular Block Copolymerization for the Synthesis of Functional Axial Organic Heterostructures, J. Am. Chem. Soc., 2020, 142, 11528 CrossRef CAS PubMed.
- W. Zhang, W. Jin, T. Fukushima, A. Saeki, S. Seki and T. Aida, Supramolecular Linear Heterojunction Composed of Graphite-Like Semiconducting Nanotubular Segments, Science, 2011, 334, 340 CrossRef CAS PubMed.
- F. García, R. Gómez and L. Sánchez, Chiral supramolecular polymers, Chem. Soc. Rev., 2023, 52, 7524 RSC.
- Y. Wang, Y. Jiang, X. Zhu and M. Liu, Significantly Boosted and Inversed Circularly Polarized Luminescence from Photogenerated Radical Anions in Dipeptide Naphthalenediimide Assemblies, J. Phys. Chem. Lett., 2019, 10, 5861 CrossRef CAS PubMed.
- S. Lee, K. Y. Kim, S. H. Jung, J. H. Lee, M. Yamada, R. Sethy, T. Kawai and J. H. Jung, Finely Controlled Circularly Polarized Luminescence of a Mechano-Responsive Supramolecular Polymer, Angew. Chem., Int. Ed., 2019, 58, 18878 CrossRef CAS PubMed.
- Y. Wang, K. Wan, F. Pan, X. Zhu, Y. Jiang, H. Wang, Y. Chen, X. Shi and M. Liu, Bamboo-like π-Nanotubes with Tunable Helicity and Circularly Polarized Luminescence, Angew. Chem., Int. Ed., 2021, 60, 16615 CrossRef CAS.
- L. López-Gandul, C. Naranjo, C. Sánchez, R. Rodríguez, R. Gómez, J. Crassous and L. Sánchez, Stereomutation and chiroptical bias in the kinetically controlled supramolecular polymerization of cyano-luminogens, Chem. Sci., 2022, 13, 11577 RSC.
- J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Recent Progress on Circularly Polarized Luminescent Materials for Organic Optoelectronic Devices, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef.
- C. Kulkarni, A. K. Mondal, T. K. Das, G. Grinbom, F. Tassinari, M. F. J. Mabesoone, E. W. Meijer and R. Naaman, Highly Efficient and Tunable Filtering of Electrons’ Spin by Supramolecular Chirality of Nanofiber-Based Materials, Adv. Mater., 2020, 32, 1904965 CrossRef CAS PubMed.
- R. Rodríguez, C. Naranjo, A. Kumar, P. Matozzo, T. K. Das, Q. Zhu, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, Mutual Monomer Orientation To Bias the Supramolecular Polymerization of [6]Helicenes and the Resulting Circularly Polarized Light and Spin Filtering Properties, J. Am. Chem. Soc., 2022, 144, 7709 CrossRef.
- A. R. A. Palmans and E. W. Meijer, Amplification of Chirality in Dynamic Supramolecular Aggregates, Angew. Chem., Int. Ed., 2007, 46, 8948 CrossRef CAS.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Supramolecular Helical Systems: Helical Assemblies of Small Molecules, Foldamers, and Polymers with Chiral Amplification and Their Functions, Chem. Rev., 2016, 116, 13752 CrossRef CAS.
- Y. Dorca, E. E. Greciano, J. S. Valera, R. Gómez and L. Sánchez, Hierarchy of Asymmetry in Chiral Supramolecular Polymers: Toward Functional, Helical Supramolecular Structures, Chem. – Eur. J., 2019, 25, 5848 CrossRef CAS PubMed.
- M. M. Green, M. P. Reidy, R. D. Johnson, G. Darling, D. J. O'Leary and G. Willson, Macromolecular stereochemistry: the out-of-proportion influence of optically active comonomers on the conformational characteristics of polyisocyanates. The sergeants and soldiers experiment, J. Am. Chem. Soc., 1989, 111, 6452 CrossRef.
- H. Gu, Y. Nakamura, T. Sato, A. Teramoto, M. M. Green, S. K. Jha, C. Andreola and M. P. Reidy, Optical Rotation of Random Copolyisocyanates of Chiral and Achiral Monomers: Sergeant and Soldier Copolymers, Macromolecules, 1998, 31, 6362 CrossRef CAS.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Sergeants-and-Soldiers Principle in Chiral Columnar Stacks of Disc-Shaped Molecules with C3 Symmetry, Angew. Chem., Int. Ed. Engl., 1997, 36, 2648 CrossRef CAS.
- M. M. J. Smulders, P. J. M. Stals, T. Mes, T. F. E. Paffen, A. P. H. J. Schenning, A. R. A. Palmans and E. W. Meijer, Probing the Limits of the Majority-Rules Principle in a Dynamic Supramolecular Polymer, J. Am. Chem. Soc., 2010, 132, 620 CrossRef CAS.
- F. García, P. M. Viruela, E. Matesanz, E. Ortí and L. Sánchez, Cooperative Supramolecular Polymerization and Amplification of Chirality in C3-Symmetrical OPE-Based Trisamides, Chem. – Eur. J., 2011, 17, 7755 CrossRef PubMed.
- L. López-Gandul, A. Morón-Blanco, F. García and L. Sánchez, Supramolecular Block Copolymers from Tricarboxamides. Biasing Co-assembly by the Incorporation of Pyridine Rings, Angew. Chem., Int. Ed., 2023, 62, e202308749 CrossRef.
- M. M. J. Smulders, I. A. W. Filot, J. M. A. Leenders, P. van der Schoot, A. R. A. Palmans, A. P. H. J. Schenning and E. W. Meijer, Tuning the Extent of Chiral Amplification by Temperature in a Dynamic Supramolecular Polymer, J. Am. Chem. Soc., 2010, 132, 611 CrossRef CAS.
- E. E. Greciano, J. Calbo, J. Buendía, J. Cerdá, J. Aragó, E. Ortí and L. Sánchez, Decoding the Consequences of Increasing the Size of Self-Assembling Tricarboxamides on Chiral Amplification, J. Am. Chem. Soc., 2019, 141, 7463 CrossRef CAS.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, Mechanism of Self-Assembly Process and Seeded Supramolecular Polymerization of Perylene Bisimide Organogelator, J. Am. Chem. Soc., 2015, 137, 3300 CrossRef CAS PubMed.
- E. E. Greciano and L. Sánchez, Seeded Supramolecular Polymerization in a Three-Domain Self-Assembly of an N-Annulated Perylenetetracarboxamide, Chem. – Eur. J., 2016, 22, 13724 CrossRef CAS PubMed.
- E. E. Greciano, S. Alsina, G. Ghosh, G. Fernández and L. Sánchez, Alkyl Bridge Length to Bias the Kinetics and Stability of Consecutive Supramolecular Polymerizations, Small Methods, 2020, 4, 1900715 CrossRef CAS.
- J. Matern, Z. Fernández, N. Bäumer and G. Fernández, Expanding the Scope of Metastable Species in Hydrogen Bonding-Directed Supramolecular Polymerization, Angew. Chem., Int. Ed., 2022, 61, e202203783 CrossRef CAS PubMed.
- H. M. M. ten Eikelder, A. J. Markvoort, T. F. A. de Greef and P. A. J. Hilbers, An Equilibrium Model for Chiral Amplification in Supramolecular Polymers, J. Phys. Chem. B, 2012, 116, 5291 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedures, synthetic details, spectroscopic characterization, NMR spectra of all new products and other experimental details. See DOI: https://doi.org/10.1039/d4qo01065e |
| ‡ To efficiently co-assemble both tricarboxamides, after preparation, the solutions were heated up to 90 °C and then allowed to reach room temperature. |
| § A positive ee value corresponds to an excess of the (R)-enantiomer. |
| This journal is © the Partner Organisations 2024 |