
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Z-shaped polycyclic aromatic hydrocarbons with embedded five-membered rings and their application in organic thin-film transistors†‡
Sven M.
Elbert
a,
Evelin
Bolgert
a,
Owen T. A.
Paine
a,
Farhad
Ghalami
b,
Wen-Shan
Zhang
 a,
Ute
Zschieschang
c,
Frank
Rominger
a,
Dennis
Popp
a,
Hagen
Klauk
c,
Marcus
Elstner
b and
Michael
Mastalerz
*a
a,
Ute
Zschieschang
c,
Frank
Rominger
a,
Dennis
Popp
a,
Hagen
Klauk
c,
Marcus
Elstner
b and
Michael
Mastalerz
*a
aInstitute of Organic Chemistry, Ruprecht-Karls-Universtity Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany. E-mail: michael.mastalerz@oci.uni-heidelberg.de
bInstitute of Physical Chemistry and Theoretical Chemical Chemistry, Karlsruhe Institute of Technology, Kaiserstraße 12, D-76131, Karlsruhe, Germany
cMax Planck Institute for Solid State Research, Heisenbergstr. 1, 70569 Stuttgart, Germany
First published on 30th July 2024
Abstract
Z-shaped polycyclic hydrocarbons with embedded five-membered rings as well as their diaza-analogues have been synthesized. Soluble congeners were obtained by the introduction of mesityl substituents and studied by cyclo- as well as differential pulse voltammetry revealing an amphoteric redox behavior. Unsubstituted congeners showed close π-stacking in single crystal X-ray diffraction analyses and thus promising characteristics for the application as semiconducting materials in p-channel thin-film transistors (TFTs). The hole transport characteristics of the corresponding devices were investigated by GIWAXS.
Introduction
Polycyclic aromatic hydrocarbons with fused five-membered rings (also named cyclopenta-fused or short cp-PAHs) are a subclass of non-alternant PAHs with unique electronic and photophysical properties.1,2 cp-PAHs often represent cut-outs of fullerenes such as C60 or C70 and have a high electron affinity.3–8 Thus, cp-PAHs found application as semiconductors in electronic devices.9–11 For example, the dibenzo[a,m]rubicene (Fig. 1),12 a cut-out of C70 with two five-membered rings in the aromatic backbone was used as p-type organic semiconductor with a field effect mobility of 1.0 cm2 V−1 s−1 in thin-film transistors (TFTs).13–15 Another cp-PAH with two five-membered rings, a derivative of acenaphto[1,2-k]fluoranthene16 (Fig. 1), was used as a green emissive dye in organic light-emitting diodes (OLEDs).17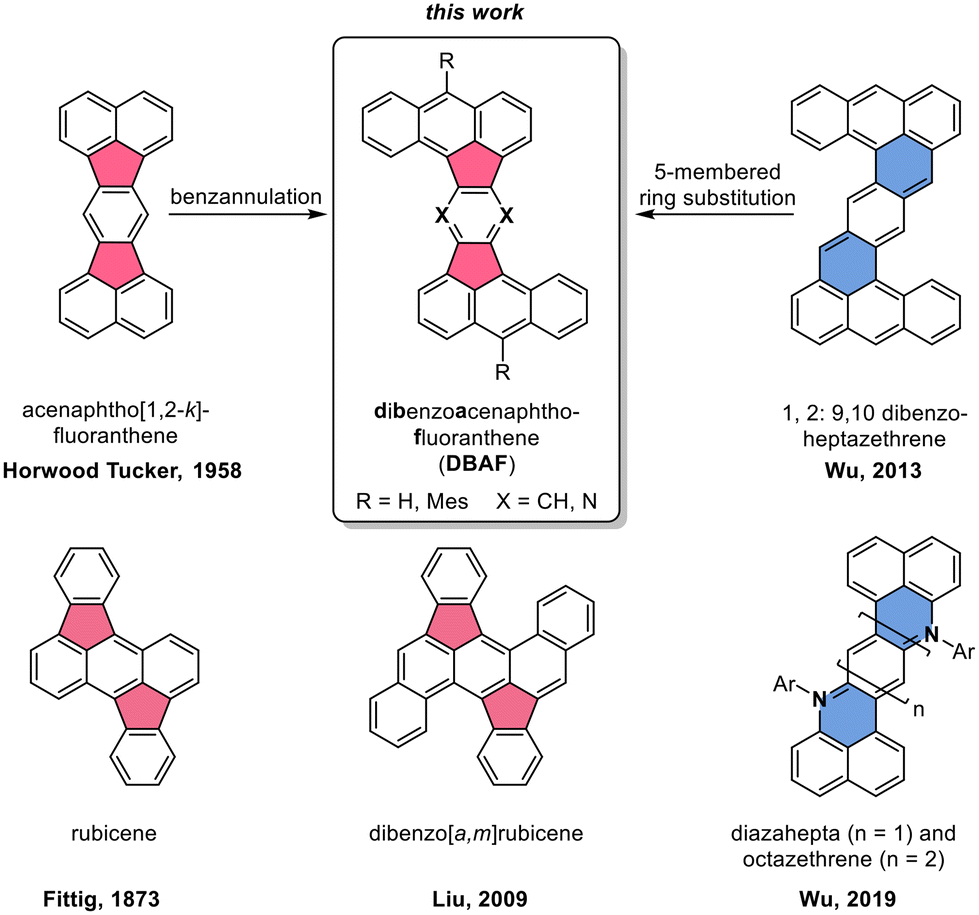 | ||
| Fig. 1 Top: Molecular structures of acenaphtho[1,2-k]fluoranthene16 (left) and 1,2:9,10-dibenzoheptazethrene20 (right) and the hypothetical structural relationship to the dibenzoacenaphthofluoranthenes (DBAFs; middle). Bottom: Structure of rubicene (left),22,23 dibenzo[a,m]rubicene12 and diaza zethrenes.27 | ||
Besides cp-PAHs, zethrenes are another class of Z-shaped PAHs with exceptional properties.18,19 They have biradical character with indices up to γ0 = 0.58. The γ0-values were tuned by benzannulation as shown in the cases of the 1,2:9,10-, and the 5,6:13,14-dibenzoheptazethrenes (short: DBHZ; Fig. 1).20 Furthermore, for organic field effect transistors (OFETs) based on single crystals of a DBHZ, a hole mobility up to 0.15 cm2 V−1 s−1 was obtained.21
Rubicenes14,22–25 and some of their extended congeners are rare examples combining both, a molecular Z-shape with embedded five-membered rings which is created by a phenyl–anthracenyl–phenyl scaffold (Fig. 1).26 We envisioned to extend the portfolio of PAHs (or cp-PAHs) in this respect by a series of compounds which can be understood either as benzannulated acenaphtofluoranthenes or as 1,2:9,10-dibenzoheptazethrenes with two six-membered rings formally substituted by five-membered ones (shown in blue and red in Fig. 1). Therefore, these compounds are named dibenzoacenaphthofluoranthenes, abbreviated as DBAFs. The DBAFs have an “inverted” anthracenyl-phenyl-anthracenyl scaffold in comparison to rubicenes (Fig. 1).26
By simple modifications of the molecular precursors, nitrogen substituted analogues (the DBAF-N2s) are accessible to investigate the influence of nitrogen substitution on the compounds' properties which was for example realized for diazahepta and octazethrenes27–29 (Fig. 1) and is generally rare in the field of cp-PAHs.30,31
Results and discussion
The Z-shaped five-membered ring containing PAHs were obtained by Suzuki–Miyaura cross-coupling reactions followed by palladium-catalysed arylations under C–H activation starting from dichlorodibromobenzene 132 or -pyrazine 233 (Scheme 1). The cross-coupling of 1 or 2 with 9-anthracenylboronic acid 3 gave the corresponding products 5 and 6 in 80% and 56% yield respectively. The mesityl-substituted congeners 7 and 8 were obtained from 1 and 2 with 10-mesityl-anthracenyl boronic pinacol ester 4 in 34% and 68% yield. Compounds 5–8 were fully characterized by NMR as well as IR spectroscopy, mass spectrometry and their constitution was unambiguously proven by single crystal X-ray diffraction (SCXRD; for details see ESI‡).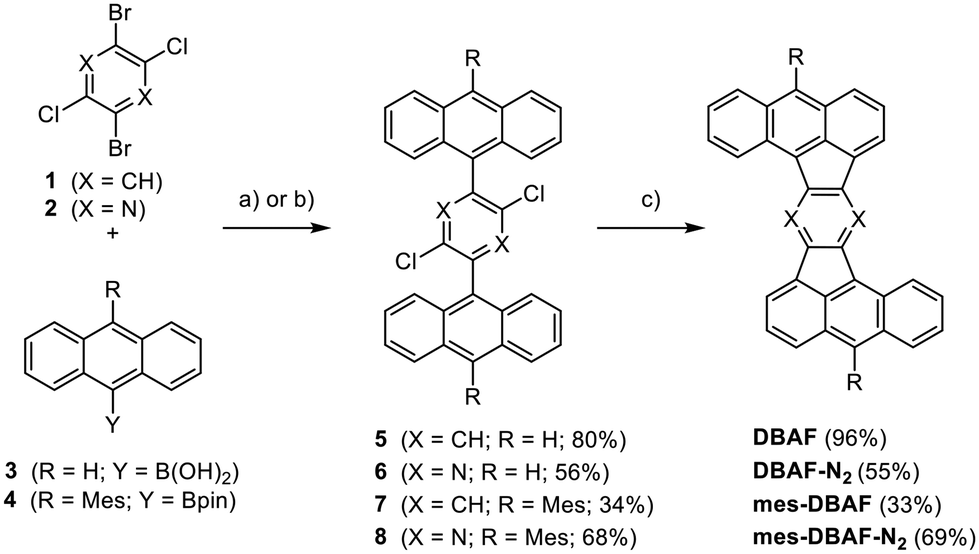 | ||
| Scheme 1 Synthesis of DBAF, DBAF-N2, mes-DBAF and mes-DBAF-N2 from dibromodichlorobenzene 1 and dibromodichloropyrazine 2. (a) For 5 and 7: Pd(OAc)2 (2.5–3 mol%), SPhos (5–6 mol%), K2CO3 aq. (1 M), THF, 85 °C 16 h; for 6 and 8: (b) Pd(dppf)Cl2 (10 mol%), K2CO3 (1 M), THF, 85 °C, 16 h, (c) PdCl2(PCy3)2, DBU, DMAc, 200 °C, 16 h. | ||
Subjecting the four dichlorinated precursors 5–8 to typical conditions for arylations under C–H-activation (PdCl2(PCy3)2, DBU, DMAc, 200 °C, 48 h)34–41 gave after sublimation unsubstituted DBAF in 96% and DBAF-N2 in 55% yield as well as the mesityl substituted mes-DBAF in 33% and mes-DBAF-N2 in 69% after column chromatography (Scheme 1). The parent DBAF and DBAF-N2 are poorly soluble in common organic solvents at room temperature and NMR spectroscopy had to be performed in ortho-dichlorobenzene-d4 at elevated temperatures (323 K; see ESI‡). The successful pentannulations have been proven by 1D- and 2D-NMR spectroscopy as well as mass spectrometry (see ESI‡) and by SCXRD analyses (see discussion below).
The optical properties of the DBAF series were investigated by UV/vis- as well as fluorescence spectroscopy in oDCB (DBAF and DBAF-N2) or dichloromethane (mes-DBAF and mes-DBAF-N2) (Fig. 2 and Table 1). DBAF and DBAF-N2 were only soluble in oDCB after heating and ultrasonication. DBAF shows an intensive absorption maximum at λabs = 349 nm and additional low energy absorption maxima at λabs = 461, 491, 518 and 550 nm. A red emission can be detected by bare eye, which corresponds to a maximum at λem = 572 nm and a shoulder at λem ∼ 620 nm with a Stokes shift of ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 699 cm−1 and a photoluminescence quantum yield of φ = 21%.
= 699 cm−1 and a photoluminescence quantum yield of φ = 21%.
 | ||
| Fig. 2 UV/vis absorption (solid lines) and fluorescence (dashed lines) spectra of DBAF (red) and mes-DBAF (orange) (top) and DBAF-N2 (blue) and mes-DBAF-N2 (violet) (bottom), measured in o-DCB (DBAF and DBAF-N2) or CH2Cl2 (mes-DBAF) and mes-DBAF-N2 at room temperature. | ||
| Cmpd | λ max , [nm] | λ onset [nm] | E gap,opt [eV] | λ em [nm] |
Stokes
[cm−1] |
PLQY [%] | E IP,CV [eV] | E EA,CV [eV] | E gap,CV [eV] | E HOMO,DFT [eV] | E LUMO,DFT [eV] | E Diff,DFT [eV] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Most-red-shifted absorption maximum. b DBAF and DBAF-N2 were measured in o-DCB at room temperature; mes-DBAF and mes-DBAF-N2 were measured in CH2Cl2 at room temperature. c Estimated from the corresponding λonset by Egap,opt = 1240/λonset. d Cyclic voltammograms (CV) of mes-DBAF and mes-DBAF-N2 in dichloromethane (c = 1 mM) using [Bu4N][PF6] (0.1 M) as electrolyte. CV scan speed 100 mV s−1. EIP,CV = −(EOx1/2 + 4.8 eV); EEA,CV = −(Ered1/2 + 4.8 eV). e Calculated at the u-B3LYP/6-31G(d,p) level of theory. f Not determined due to solubility issues. | ||||||||||||
| DBAF | 550 | 581 | 2.1 | 572 | 699 | 21 | —f | —f | —f | −5.0 | −2.4 | 2.6 |
| DBAF-N2 | 555 | 582 | 2.1 | 577 | 687 | 5 | —f | —f | —f | −5.2 | −2.6 | 2.6 |
| mes-DBAF | 561 | 589 | 2.1 | 588 | 819 | 20 | −5.3 | −3.0 | 2.3 | −4.9 | −2.4 | 2.6 |
| mes-DBAF-N2 | 561 | 592 | 2.1 | 593 | 961 | 29 | −5.6 | −3.2 | 2.4 | −5.1 | −2.5 | 2.6 |
mes-DBAF shows similar spectroscopic properties as its non-mesitylated analogue with a high intensity absorption band at λabs = 350 nm (DBAF: 349 nm) and again poorly resolved maxima at λabs = 460, 493, 526 and 561 nm and a slightly red-shifted emission with λem = 588 nm (DBAF: 572 nm) leading to a larger Stokes shift of = 819 cm−1 (φ = 20%, Table 1).
DBAF-N2 shows two maxima at λabs = 379 and 399 nm and a less resolved pattern of overlapping adsorption maxima with a most red-shifted maximum at λabs = 519 nm with a shoulder at λabs = 555 nm. The corresponding emission maximum is found at λem = 577 nm with a shoulder at λem = 621 nm and a Stokes shift of = 687 cm−1 with φ = 5% (Table 1). Similarly, as for mes-DBAF and DBAF the spectral differences of mes-DBAF-N2 and DBAF-N2 are negligible (Fig. 2 and Table 1), indicating that the mesityl groups change the solubility without significantly influencing the optoelectronic properties of the molecular scaffolds.
As already mentioned, DBAF and DBAF-N2 are poorly soluble in organic solvents so that decent cyclovoltammograms could not be recorded. Therefore, only the mesitylated derivatives mes-DBAF and mes-DBAF-N2 have been investigated by cyclic as well as differential pulse voltammetry revealing an amphoteric redox behaviour in both cases (Fig. 3). Two reversible oxidations can be found for mes-DBAF at E1Ox = 0.52 V and E2Ox = 0.83 V as determined by DPV (CV: EOx,11/2 = 0.56 V und EOx,21/2 = 0.86 V) and one reversible reduction was detected at Ered = −1.76 V (CV: Ered1/2 = −1.80 V). For mes-DBAF-N2 the corresponding oxidation waves appear at approx. 0.2–0.3 V higher potentials with E1Ox = 0.76 V and E2Ox = 1.11 V (CV: EOx,11/2 = 0.76 V und EOx,21/2 = 1.13 V). The reduction is shifted by 0.14 V to higher potentials with Ered = −1.62 V (CV: Ered1/2 = 1.63 V) indicating the electrochemical stabilization of PAHs by nitrogen substitution.42–44 This is further displayed in the differences of the corresponding ionization potentials (mes-DBAF: IP = −5.32 eV; mes-DBAF-N2: IP = −5.56 eV) as well as electron affinities (mes-DBAF: EA = −3.04 eV; mes-DBAF-N2: EA = −3.18 eV) derived from this data. By UV/vis spectrsocopy it was demonstrated that the mesityl substituents, which are oriented orthogonal to the π backbone, do not significantly influence these electronically. Therefore, it is assumed that the redox behaviour of DBAF and DBAF-N2 are comparable to mes-DBAF and mes-DBAF-N2. By DFT calculations (u-B3LYP/6-31G(d,p); Fig. 4 and Table 1) a 0.19 eV lower HOMO energy of mes-DBAF-N2 (EHOMO = −5.12 eV) in comparison to mes-DBAF (EHOMO = −4.93 eV) is found as well as a 0.14 eV lower LUMO energy (mes-DBAF-N2: ELUMO = −2.52 eV; mes-DBAF: ELUMO = −2.38 eV), which corroborates with the trend found by CV.
 | ||
| Fig. 3 Cyclic voltammograms (CV) and differential pulse voltammograms (DPV) of mes-DBAF (top) and mes-DBAF-N2 (bottom) in dichloromethane (c = 1 mM) using [Bu4N][PF6] (0.1 M) as electrolyte. CV scan speed 100 mV s−1; DPV: step size of 0.005 V, a modulation amplitude of 0.025 V a modulation time of 0.05 s and an interval time of 0.5 s. | ||
 | ||
| Fig. 4 DFT-calculated frontier molecular orbitals on the u-B3LYP/6-31G(d,p) level of theory (LUMOs top; HOMOs bottom) of (from left to right) DBAF, DBAF-N2, mes-DBAF and mes-DBAF-N2 with the calculated energy levels. | ||
Again, the mesityl substituents seem to only have a small influence to the energy levels with the HOMOs being lowered by only 0.09–0.1 eV and the LUMOs by only 0.04–0.06 eV for DBAF and DBAF-N2 in comparison to their mesitylated congeners (Fig. 4 and Table 1) and the distribution of frontier molecular orbital coefficients are nearly the same for all four compounds (Fig. 4).
By thermal crystallization from ortho-dichlorobenzene as well as by sublimation isomorphic single crystals of DBAF suitable for SCXRD were obtained. DBAF crystallizes in the monoclinic space group P21/n with Z = 2. The crystalline packing is dominated by slipped π-stacked molecular columns (Fig. 5b and c) with a spacing of dπ–π = 3.33 Å propagating along the crystallographic a-axis. Edge-to-face π stacking with dC–H⋯π = 2.70 Å and an angle of the π-systems of θ = 51.8° (Fig. 5c) leads to a herringbone like motif (Fig. 5d). Suitable crystals of DBAF-N2 have also been obtained by sublimation. DBAF-N2 also crystallized in the P21/n space group and again slipped π-stacked columns can be found, here with a slightly larger distance of dπ–π = 3.40 Å but with a larger overlap of two adjacent π systems (compare Fig. 5b and f). Similar edge-to-face π stacking is found but with a larger angle of θ = 73.1° and a larger distance between adjacent molecules (dC–H⋯π = 2.87 Å), leading to a herringbone type packing as well (Fig. 5g and h). Single-crystals of mes-DBAF were obtained by slow evaporation of a chloroform solution and crystallized in the monoclinic space group P21/n with Z = 2. This time with one enclathrated disordered molecule of chloroform per mes-DBAF (Fig. 5). Due to the almost perpendicular oriented (87.6°) mesityl groups, no direct π stacking of the DBAF central backbone is found (Fig. 5i). Instead, the crystalline packing is dominated by C–H⋯π-interactions of the para-methyl-group of a mesityl substituent with the central benzene ring of the DBAF backbone (dC–H⋯π = 2.34 Å) leading to a loose herringbone-like arrangement with chloroform molecules in its voids (Fig. 5j–l). Crystals of mes-DBAF-N2 were obtained by vapor phase diffusion of n-hexane into a saturated chloroform solution. mes-DBAF-N2 also crystallized in the monoclinic space group P21/n with Z = 2. In contrast to mes-DBAF, mes-DBAF-N2 shows linear columns of face-to-face π stacks along the crystallographic a-axis (Fig. 5n and p). Within these stacks two symmetrically independent molecules with unparallel π-planes and a shortest distance of dπ–π = 3.22 Å are found in a rare twisted offset45–47 with an angle of ∼32° between the central molecular axes to maximize orbital overlaps by avoiding the steric repulsion of the mesityl groups. The columns themselves interact with each other by dispersion interactions between the methyl groups of the mesityl substituents.
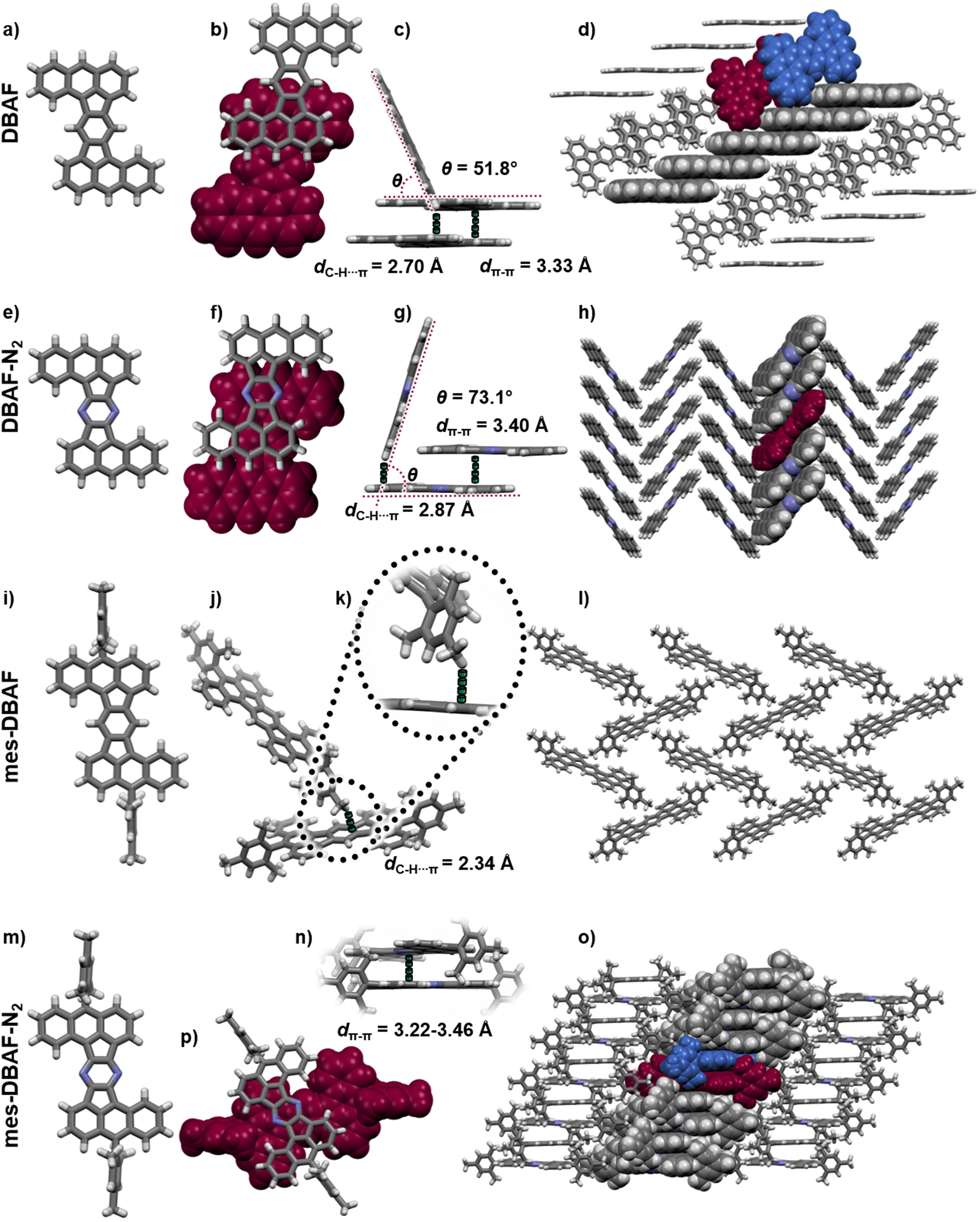 | ||
| Fig. 5 Single crystal X-ray structures of DBAF (a–d), DBAF-N2 (e–h), mes-DBAF (i–l) and mes-DBAF-N2. a, e, i and m: molecular structures. b, f and p: top-view on face-to-face π-stacked dimers. c, g and j: side-view on edge-to-face interactions. d, h, l and o: molecular packings. k: zoom-in on the C–H⋯π-interaction of mes-DBAF. n: side-view on the face-to-face π-stacked dimer of mes-DBAF-N2. | ||
Charge transfer integrals (t) between the molecules as well as intrinsic charge carrier mobilities (hole and electron) were calculated via fragment based non-adiabatic molecular dynamic simulations (for a detailed description of the method see ESI‡).48,49 Only hole transport properties will be discussed (calculated electron transport properties can be found in the ESI‡), due to the fact, that the experimentally determined electron affinities of EA = −3.04 to −3.18 eV exclude a potential use in n-type semiconducting devices due to a mismatch with the work functions of commonly used electrode materials such as gold.50 The hole transfer integral of DBAF along the axis of the face-to-face stacking was calculated using the non-self-consistent density functional tight binding (DFTB) method with special parameter set51 to be t‖,h = 20 ± 14 meV (Fig. 5d). In the direction of the edge-to-face stacking only small transfer integrals were found (t⊥,h = 1 ± 3 meV). The corresponding theoretical hole mobility of μh = 0.31 cm2 V−1 s−1 calculated by the fewest switches surface hopping algorithm with implicit relaxation (FSSH-IR), where the reorganization energy is calculated by DFT with B3LYP functional and 6-31G(d,p) level of theory to be λh, B3LYP = 153 meV indicates the potential use of DBAF in such devices. In the case of DBAF-N2 (Fig. 5h), a higher electronic coupling with transfer integrals of t‖,h = 70 ± 24 meV was found. The corresponding hole mobilities of μh = 14.41 cm2 V−1 s−1 are based on a reorganization energy of λh, B3LYP = 167 meV. The mesitylated congeners mes-DBAF and mes-DBAF-N2 showed either no or only negligible theoretical mobilities and have thus been excluded for device fabrication (see ESI‡ for further details).
Organic p-channel TFTs of DBAF gave hole mobilities of 1 × 10−4 to 1 × 10−3 cm2 V−1 s−1 independent of the type of the substrate (silicon or flexible polyethylene naphthalate (PEN)) and the substrate temperature during the vacuum deposition of the semiconductor (60 or 80 °C) when using a pentadecafluoro-octadecylphosphonic acid self-assembled monolayer (F-SAM) as part of the gate dielectric.52
These mobilities significantly increased using an n-tetradecylphosphonic acid SAM (H-SAM) instead. In this case, mobilities of 0.08 up to 0.4 cm2 V−1 s−1 have been obtained, again independent of the type of substrate and the substrate temperature during the semiconductor deposition. The best TFT performance was obtained on a PEN substrate using a substrate temperature of 80 °C during the semiconductor deposition (for details see ESI‡), with a mobility of μh,exp = 0.4 cm2 V−1 s−1 (Fig. 6), which is in good agreement to the theoretical mobility discussed above (μh,theo = 0.31 cm2 V−1 s−1). Although DBAF-N2 showed even higher theoretical hole mobilities (μh,theo = 14.41 cm2 V−1 s−1, see discussion above), the mobilities obtained experimentally are much smaller, only 4 × 10−5 to 5 × 10−4 cm2 V−1 s−1, independent of the type of substrate, the type of SAM, and the substrate temperature during the semiconductor deposition. The calculated values represent the intrinsic mobility in the perfect crystalline structures of DBAF and DBAF-N2, where no grain boundaries or other defects are present. However, in thin-films, such defects exist, and the thin-film structures may deviate from the ideal single-crystal form. Consequently, the calculated mobilities provide a useful estimation of how closely the deposited thin-films approximate a perfect single crystal. This can be inferred from the similarity of their charge-transport properties. The significant difference between the experimental and theoretical mobilities suggests that the deposited DBAF-N2 structure deviates considerably from the perfect crystal structure.
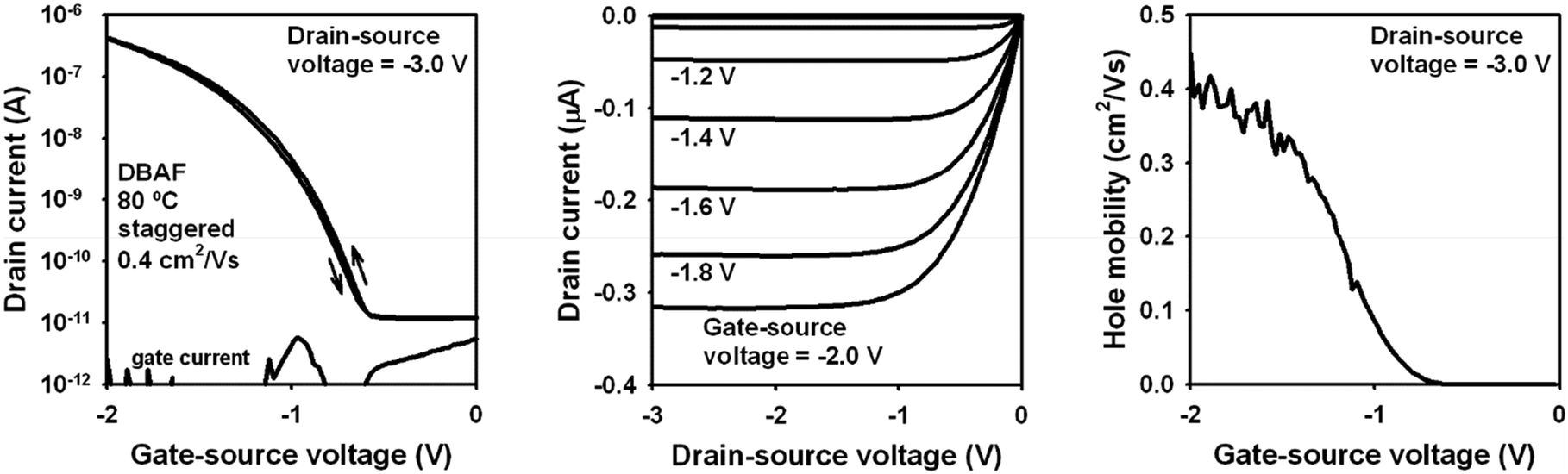 | ||
| Fig. 6 Electrical characteristics of a DBAF TFT fabricated on a flexible PEN substrate in the inverted staggered (bottom-gate, top-contact) device architecture using an n-tetradecylphosphonic acid SAM as part of the gate dielectric, with the substrate held at a temperature of 80 °C during the semiconductor deposition. The effective charge-carrier mobility is 0.4 cm2 V−1 s−1. | ||
To better understand the measured TFT characteristics (schematic device structure shown in Fig. 7a), grazing-incidence wide-angle X-ray scattering (GIWAXS) was employed. The critical angles (αc) of DBAF and DBAF-N2 are estimated at 0.17° and 0.18°, respectively.53 Therefore, the incident angle (αi) of the incoming X-ray was set at 0.3° (αi > αc) in order to probe microstructures of the deeper-lying organic/SAM/inorganic interface, rather than those from the topmost surface. Though the AFM and SEM investigations reveal the polycrystallinity of the thin-films with non-specific orientations, the small grain size (∼0.1–0.5 μm) makes the in-plane information difficult or impossible to be detected by GIWAXS. Instead, we obtain the strong out-of-plane signals and are thus able to analyze the interface configurations. Two sets of out-of-plane signals have been observed. As exemplified in Fig. 7c and d, the lower one at ∼21 Å stems from the self-assembled monolayer that is part of the gate dielectric, while the upper one at ∼10 Å belongs to the organic semiconductor thin-films. Specifically, for DBAF, the reflex at 9.99 Å is assigned to the (001) plane (10.5 Å calculated from the signal crystal data) and for DBAF-N2, the reflex at 9.75 Å to the (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) plane (9.77 Å calculated from the signal crystal data). Both molecules show an edge-on orientation and a layered structure. DBAF stays more tilted (43°–44°) than DBAF-N2 (77°–78°) to the substrate, which could slightly lower the HOMO level. Within one layer, both DBAF and DBAF-N2 molecules stack in lines. The small π–π distance (DBAF 3.3 Å and DBAF-N2 3.4 Å) of two adjacent molecules and a reasonable overlap of their electron clouds (∼1/3 for DBAF and ∼2/3 for DBAF-N2) favor the horizontal charge-carrier hopping along the stack-propagating direction (Fig. 7b). This agrees with the values of the calculated transfer integrals (for DBAFt‖,h = 20 ± 14 meV and for DBAF-N2t‖,h = 70 ± 24 meV). The main difference is found in the vertical perspective (parallel to the substrate surface normal) between the adjacent layers. DBAF shows an edge-to-face π–π-arrangement (t⊥,h = 1 ± 3 meV), whereas DBAF-N2 has an edge-to-edge arrangement, giving rise to a negligible overlap of the π-electron cloud of molecules stemming from different layers (Fig. 7i). Thus, the vertical charge-carrier transport in the DBAF-N2 thin-films is inefficient, leading to a larger contact resistance and thereby to a smaller effective charge-carrier mobility54 compared to the DBAF TFTs. Another explanation for the substantial difference in measured charge-carrier mobilities is that unlike DBAF, DBAF-N2 does not form a completely closed film on the substrate surface (as indicated by the AFM images in the ESI in Fig. S74‡), which is expected to greatly impede the lateral charge transport.
) plane (9.77 Å calculated from the signal crystal data). Both molecules show an edge-on orientation and a layered structure. DBAF stays more tilted (43°–44°) than DBAF-N2 (77°–78°) to the substrate, which could slightly lower the HOMO level. Within one layer, both DBAF and DBAF-N2 molecules stack in lines. The small π–π distance (DBAF 3.3 Å and DBAF-N2 3.4 Å) of two adjacent molecules and a reasonable overlap of their electron clouds (∼1/3 for DBAF and ∼2/3 for DBAF-N2) favor the horizontal charge-carrier hopping along the stack-propagating direction (Fig. 7b). This agrees with the values of the calculated transfer integrals (for DBAFt‖,h = 20 ± 14 meV and for DBAF-N2t‖,h = 70 ± 24 meV). The main difference is found in the vertical perspective (parallel to the substrate surface normal) between the adjacent layers. DBAF shows an edge-to-face π–π-arrangement (t⊥,h = 1 ± 3 meV), whereas DBAF-N2 has an edge-to-edge arrangement, giving rise to a negligible overlap of the π-electron cloud of molecules stemming from different layers (Fig. 7i). Thus, the vertical charge-carrier transport in the DBAF-N2 thin-films is inefficient, leading to a larger contact resistance and thereby to a smaller effective charge-carrier mobility54 compared to the DBAF TFTs. Another explanation for the substantial difference in measured charge-carrier mobilities is that unlike DBAF, DBAF-N2 does not form a completely closed film on the substrate surface (as indicated by the AFM images in the ESI in Fig. S74‡), which is expected to greatly impede the lateral charge transport.
 | ||
| Fig. 7 Molecular orientation of DBAF and DBAF-N2 at the organic/SAM/inorganic interface of the OTFT devices. (a) The OTFT device used in this work has a bottom-gate-top-contact structure. (b) Illustration of charge transport taking place in a working device of DBAF. (c) and (d) are typical GIWAXS data for the DBAF- and DBAF-N2-based devices, respectively. (e) Side view and (g) top view of the orientation of DBAF molecules. (f) Side view and (h and i) top views of the orientation of DBAF-N2 molecules. (h) shows the first layer (L1) at the SAM/inorganic substrate (bright green). (g) and (i) show the first two layers, in which molecules colored in grey come from the first layer (L1) and molecules in various blue colors are from the second layer (L2). | ||
Conclusions
The PAHs presented in this study have a Z-shape conformation and can be tuned in terms of their solubility by peripheral substituents without influencing the electronic properties of the core structure as proven by spectroscopic methods and quantum chemical calculations. This made the investigation in solution as e.g. by electrochemical methods possible and revealed suitable characteristics for the application in p-type thin-film transistors. Corresponding devices of DBAF showed experimental mobilities up to μh,exp = 0.4 cm2 V−1 s−1 which is in agreement to quantum chemical calculations based on single crystal X-ray structure data. A substitution of two carbon atoms in the aromatic backbone with nitrogen was possible by simple modification of the molecular precursors. While an expected stabilization of the frontier molecular orbitals was achieved and higher theoretical hole mobilities were obtained, proving the beneficial intrinsic properties of DBAF-N2, the corresponding devices suffered from a disadvantageous orientation of the crystalline films on the devices' surface resulting in three orders of magnitude smaller experimental mobilities. This study shows on the one hand the potential of this class of PAHs for the application as semiconducting materials in organic electronics and on the other hand the importance of the interplay of suitable intrinsic molecular properties and the controllable device morphology upon structural changes on a molecular level.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors are grateful for financial support to this project by Deutsche Forschungsgemeinschaft (DFG) in the framework of the Collaborative Research Center SFB1249 (TP-A04, -B02, and -C01). Support by the state of Baden-Württemberg through bwHPC and the DFG through grant no. INST 40/575-1 FUGG (JUSTUS 2 cluster) is acknowledged. Jan Lennart Peters is acknowledged for assistance with synthesis. We thank the Kivala group (Heidelberg) for access to their fluorimeter for PLQY determination.References
- R. Zahradník, Electronic Structure and Properties of Non-Alternant Hydrocarbons, Angew. Chem., Int. Ed. Engl., 1965, 4, 1039–1050 CrossRef.
- K. N. Plunkett, What About the Five-Membered Ring? Cyclopenta-fused Polycyclic Aromatic Hydrocarbons as a Building Block for Functional Materials, Synlett, 2013, 24, 898–902 CrossRef CAS.
- R. G. Lawton and W. E. Barth, Synthesis of corannulene, J. Am. Chem. Soc., 1971, 93, 1730–1745 CrossRef CAS.
- A. M. Butterfield, B. Gilomen and J. S. Siegel, Kilogram-Scale Production of Corannulene, Org. Process Res. Dev., 2012, 16, 664–676 CrossRef CAS.
- X. Li, F. Kang and M. Inagaki, Buckybowls: Corannulene and Its Derivatives, Small, 2016, 12, 3206–3223 CrossRef CAS PubMed.
- J. D. Wood, J. L. Jellison, A. D. Finke, L. Wang and K. N. Plunkett, Electron Acceptors Based on Functionalizable Cyclopenta[hi]aceanthrylenes and Dicyclopenta[de,mn]tetracenes, J. Am. Chem. Soc., 2012, 134, 15783–15789 CrossRef CAS PubMed.
- G. C. Kulkarni, J. L. Morales-Cruz, W. A. Hussain, I. J. Garvey and K. N. Plunkett, Electron Acceptors Based on Cyclopentannulated Tetracenes, Synlett, 2018, 29, 2572–2576 CrossRef CAS.
- E. Misselwitz, J. Spengler, F. Rominger and M. Kivala, Indenoannulated Tridecacyclene: An All-Carbon Seven-Stage Redox-Amphoter, Chem. – Eur. J., 2024, 30, e202400696 CrossRef CAS PubMed.
- S. M. Elbert, A. Haidisch, T. Kirschbaum, F. Rominger, U. Zschieschang, H. Klauk and M. Mastalerz, 2,7,11,16-Tetra-tert-Butyl Tetraindenopyrene Revisited by an “Inverse” Synthetic Approach, Chem. – Eur. J., 2020, 26, 10585–10590 CrossRef CAS PubMed.
- H. Tanoguchi, T. Yuki, S. Yokokura, T. Yanase, M. Jin, H. Ito, T. Nagahama and T. Shimada, Single Crystal Growth of Cyclopenta-Fused Polycyclic Aromatic Hydrocarbon by the Naphthalene Flux Method: 2D Ambipolar Charge Transport Properties and NIR Absorption, ACS Appl. Electron. Mater., 2023, 5, 6266–6274 CrossRef CAS.
- A. Naibi Lakshminarayana, J. Chang, J. Luo, B. Zheng, K.-W. Huang and C. Chi, Bisindeno-annulated pentacenes with exceptionally high photo-stability and ordered molecular packing: simple synthesis by a regio-selective Scholl reaction, Chem. Commun., 2015, 51, 3604–3607 RSC.
- H.-H. Hseuh, M.-Y. Hsu, T.-L. Wu and R.-S. Liu, Efficient Synthesis and Characterization of Dibenzo[a,m]rubicenes and Tetrabenzo[a,f,r,m]rubicenes, J. Org. Chem., 2009, 74, 8448–8451 CrossRef CAS PubMed.
- X. Gu, X. Xu, H. Li, Z. Liu and Q. Miao, Synthesis, Molecular Packing, and Thin Film Transistors of Dibenzo[a,m]rubicenes, J. Am. Chem. Soc., 2015, 137, 16203–16208 CrossRef CAS PubMed.
- L. Ma, Y. Han, Q. Shi and H. Huang, The design, synthesis and application of rubicene based polycyclic aromatic hydrocarbons (PAHs), J. Mater. Chem. C, 2023, 11, 16429–16438 RSC.
- H. Sakurai, T. Daiko and T. Hirao, A Synthesis of Sumanene, a Fullerene Fragment, Science, 2003, 301, 1878–1878 CrossRef CAS PubMed.
- S. H. Tucker, Synthesis of fluoranthenes. Part XIII. Synthesis of benzo[1:2-a, 3:4-a′]- and of benzo[1:2-a, 4:5-a′]-diacenaphthylene, J. Chem. Soc., 1958, 1462–1465 RSC.
- L. Han, K. Ye, C. Li, Y. Zhang, H. Zhang and Y. Wang, Thermally Stable and Highly Luminescent Green Emissive Fluorophores with Acenaphtho[1,2-k]fluoranthene Cores and Aromatic Amine Groups, ChemPlusChem, 2017, 82, 315–322 CrossRef CAS PubMed.
- Y.-C. Hsieh, H.-Y. Fang, Y.-T. Chen, R. Yang, C.-I. Yang, P.-T. Chou, M.-Y. Kuo and Y.-T. Wu, Zethrene and Dibenzozethrene: Masked Biradical Molecules?, Angew. Chem., Int. Ed., 2015, 54, 3069–3073 CrossRef CAS PubMed.
- L. Shan, Z. Liang, X. Xu, Q. Tang and Q. Miao, Revisiting zethrene: synthesis, reactivity and semiconductor properties, Chem. Sci., 2013, 4, 3294–3297 RSC.
- Z. Sun, S. Lee, K. H. Park, X. Zhu, W. Zhang, B. Zheng, P. Hu, Z. Zeng, S. Das, Y. Li, C. Chi, R.-W. Li, K.-W. Huang, J. Ding, D. Kim and J. Wu, Dibenzoheptazethrene Isomers with Different Biradical Characters: An Exercise of Clar's Aromatic Sextet Rule in Singlet Biradicaloids, J. Am. Chem. Soc., 2013, 135, 18229–18236 CrossRef CAS PubMed.
- C. Zong, X. Zhu, Z. Xu, L. Zhang, J. Xu, J. Guo, Q. Xiang, Z. Zeng, W. Hu, J. Wu, R. Li and Z. Sun, Isomeric Dibenzoheptazethrenes for Air-Stable Organic Field-Effect Transistors, Angew. Chem., Int. Ed., 2021, 60, 16230–16236 CrossRef CAS PubMed.
- R. Fittig and E. Ostermayer, Ueber das Phenanthren, einen neuen Kohlenwasserstoff im Steinkohlentheer, Liebigs Ann. Chem., 1873, 166, 361–382 CrossRef.
- A. Schmitz and I. Ueber, hochsiedende Kohlenwasserstoffe des Steinkohlentheers, Liebigs Ann. Chem., 1878, 193, 115–128 CrossRef.
- V. Sachweh and H. Langhals, Synthese von Reinst-Rubicen und Rubicen-Derivaten, Chem. Ber., 1990, 123, 1981–1987 CrossRef CAS.
- M. Kawamura, E. Tsurumaki and S. Toyota, Facile Synthesis of Rubicenes by Scholl Reaction, Synthesis, 2018, 134–138 CAS.
- S. Toyota, S. Ban, M. Hara, M. Kawamura, H. Ikeda and E. Tsurumaki, Synthesis and Properties of Rubicene-Based Aromatic π-Conjugated Compounds as Five-Membered Ring Embedded Planar Nanographenes, Chem. – Eur. J., 2023, 29, e202301346 CrossRef CAS PubMed.
- C. H. E. Chow, Y. Han, H. Phan and J. Wu, Nitrogen-doped Heptazethrene and Octazethrene Diradicaloids, Chem. Commun., 2019, 55, 9100–9103 RSC.
- K. Fujimoto, S. Takimoto, S. Masuda, T. Inuzuka, K. Sanada, M. Sakamoto and M. Takahashi, 5,11-Diazadibenzo[hi,qr]tetracene: Synthesis, Properties, and Reactivity toward Nucleophilic Reagents, Chem. – Eur. J., 2021, 27, 8951–8955 CrossRef CAS PubMed.
- K. Tajima, K. Matsuo, H. Yamada, N. Fukui and H. Shinokubo, Diazazethrene bisimide: a strongly electron-accepting π-system synthesized via the incorporation of both imide substituents and imine-type nitrogen atoms into zethrene, Chem. Sci., 2023, 14, 635–642 RSC.
- Y. S. Park, D. J. Dibble, J. Kim, R. C. Lopez, E. Vargas and A. A. Gorodetsky, Synthesis of Nitrogen-Containing Rubicene and Tetrabenzopentacene Derivatives, Angew. Chem., Int. Ed., 2016, 55, 3352–3355 CrossRef CAS PubMed.
- Y. Morimoto, Y. H. Koo, K. Otsubo, H. Kitakado, S. Seki, A. Osuka and T. Tanaka, Dibenzodiazapyracylenes: Doubly N-Doped Cyclopenta-Fused Polycyclic Molecules That Exhibit High Carrier Mobility, Angew. Chem., Int. Ed., 2022, 61, e202200341 CrossRef CAS PubMed.
- H. W. H. Lai, Y. C. Teo and Y. Xia, Functionalized Rigid Ladder Polymers from Catalytic Arene-Norbornene Annulation Polymerization, ACS Macro Lett., 2017, 6, 1357–1361 CrossRef CAS PubMed.
- P. Meti, E.-S. Lee, J.-W. Yang and Y.-D. Gong, Regioselective synthesis of dipyrrolopyrazine (DPP) derivatives via metal free and metal catalyzed amination and investigation of their optical and thermal properties, RSC Adv., 2017, 7, 18120–18131 RSC.
- B. D. Steinberg, E. A. Jackson, A. S. Filatov, A. Wakamiya, M. A. Petrukhina and L. T. Scott, Aromatic π-Systems More Curved Than C60. The Complete Family of All Indenocorannulenes Synthesized by Iterative Microwave-Assisted Intramolecular Arylations, J. Am. Chem. Soc., 2009, 131, 10537–10545 CrossRef CAS PubMed.
- M.-K. Chen, H.-J. Hsin, T.-C. Wu, B.-Y. Kang, Y.-W. Lee, M.-Y. Kuo and Y.-T. Wu, Highly Curved Bowl-Shaped Fragments of Fullerenes: Synthesis, Structural Analysis, and Physical Properties, Chem. – Eur. J., 2014, 20, 598–608 CrossRef CAS PubMed.
- T. Kubo, S. Miyazaki, T. Kodama, M. Aoba, Y. Hirao and H. Kurata, A facile synthesis of trinaphtho[3.3.3]propellane and its π-extension and the formation of a two-dimensional honeycomb molecular assembly, Chem. Commun., 2015, 51, 3801–3803 RSC.
- Y.-C. Hsieh, T.-C. Wu, J.-Y. Li, Y.-T. Chen, M.-Y. Kuo, P.-T. Chou and Y.-T. Wu, Dinaphthozethrene and Diindenozethrene: Synthesis, Structural Analysis, and Properties, Org. Lett., 2016, 18, 1868–1871 CrossRef CAS PubMed.
- X. Tian, L. M. Roch, K. K. Baldridge and J. S. Siegel, Diindenocorannulenes: Curved Aromatics Blending Bowl-in-Bowl Assembly and Nanocarbon Material Properties, Eur. J. Org. Chem., 2017, 2801–2805 CrossRef CAS.
- Y. Wang, O. Allemann, T. S. Balaban, N. Vanthuyne, A. Linden, K. K. Baldridge and J. S. Siegel, Chiral Atropisomeric Indenocorannulene Bowls: Critique of the Cahn–Ingold–Prelog Conception of Molecular Chirality, Angew. Chem., Int. Ed., 2018, 57, 6470–6474 CrossRef CAS PubMed.
- S. Seifert, D. Schmidt, K. Shoyama and F. Würthner, Base-Selective Five- versus Six-Membered Ring Annulation in Palladium-Catalyzed C–C Coupling Cascade Reactions: New Access to Electron-Poor Polycyclic Aromatic Dicarboximides, Angew. Chem., Int. Ed., 2017, 56, 7595–7600 CrossRef CAS PubMed.
- X.-Y. Wang, X. Yao and K. Müllen, Polycyclic aromatic hydrocarbons in the graphene era, Sci. China: Chem., 2019, 62, 1099–1144 CrossRef CAS.
- U. H. F. Bunz and J. Freudenberg, N-Heteroacenes and N-Heteroarenes as N-Nanocarbon Segments, Acc. Chem. Res., 2019, 52, 1575–1587 CrossRef CAS PubMed.
- T. Kirschbaum, F. Rominger and M. Mastalerz, A Chiral Polycyclic Aromatic Hydrocarbon Monkey Saddle, Angew. Chem., Int. Ed., 2020, 59, 270–274 CrossRef CAS PubMed.
- T. Kirschbaum, F. Rominger and M. Mastalerz, An Isosteric Triaza Analogue of a Polycyclic Aromatic Hydrocarbon Monkey Saddle, Chem. – Eur. J., 2020, 26, 14560–14564 CrossRef CAS PubMed.
- X. Roy, J. H. Chong, B. O. Patrick and M. J. MacLachlan, Molecular Scaffolding of Prussian Blue Analogues Using a Phenanthroline-Extended Triptycene Ligand, Cryst. Growth Des., 2011, 11, 4551–4558 CrossRef CAS.
- L. Ueberricke, J. Schwarz, F. Ghalami, M. Matthiesen, F. Rominger, S. M. Elbert, J. Zaumseil, M. Elstner and M. Mastalerz, Triptycene End-Capped Benzothienobenzothiophene and Naphthothienobenzothiophene, Chem. – Eur. J., 2020, 26, 12596–12605 CrossRef CAS PubMed.
- L. Ueberricke, I. Ciubotaru, F. Ghalami, F. Mildner, F. Rominger, M. Elstner and M. Mastalerz, Di- and Tetracyano-Substituted Pyrene-Fused Pyrazaacenes: Aggregation in the Solid State, Chem. – Eur. J., 2020, 26, 11634–11642 CrossRef CAS PubMed.
- S. Roosta, F. Ghalami, M. Elstner and W. Xie, Efficient Surface Hopping Approach for Modeling Charge Transport in Organic Semiconductors, J. Chem. Theory Comput., 2022, 18, 1264–1274 CrossRef CAS PubMed.
- W. Xie, D. Holub, T. Kubař and M. Elstner, Performance of Mixed Quantum-Classical Approaches on Modeling the Crossover from Hopping to Bandlike Charge Transport in Organic Semiconductors, J. Chem. Theory Comput., 2020, 16, 2071–2084 CrossRef CAS PubMed.
- H. Klauk, Organic thin-film transistors, Chem. Soc. Rev., 2010, 39, 2643–2666 RSC.
- A. Heck, J. J. Kranz, T. Kubař and M. Elstner, Multi-Scale Approach to Non-Adiabatic Charge Transport in High-Mobility Organic Semiconductors, J. Chem. Theory Comput., 2015, 11, 5068–5082 CrossRef CAS PubMed.
- U. Kraft, U. Zschieschang, F. Ante, D. Kälblein, C. Kamella, K. Amsharov, M. Jansen, K. Kern, E. Weber and H. Klauk, Fluoroalkylphosphonic acid self-assembled monolayer gate dielectrics for threshold-voltage control in low-voltage organic thin-film transistors, J. Mater. Chem., 2010, 20, 6416–6418 RSC.
- M. Birkholz, in Thin Film Analysis by X-Ray Scattering, WILEY-VCH Verlag GmbH & Co. KGaA, 2006 Search PubMed.
- B. Peng, K. Cao, A. H. Y. Lau, M. Chen, Y. Lu and P. K. L. Chan, Crystallized Monolayer Semiconductor for Ohmic Contact Resistance, High Intrinsic Gain, and High Current Density, Adv. Mater., 2020, 32, 2002281 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Dr. Frank Würthner on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2353527–2353534. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01252f |
| This journal is © the Partner Organisations 2024 |