
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioinspired intramolecular spirocyclopropanation of quinones as an interrupted photoredox process†‡
Alexander A.
Fadeev
 a,
Daniel
Bím
b,
Ivana
Císařová
c and
Martin
Kotora
*a
a,
Daniel
Bím
b,
Ivana
Císařová
c and
Martin
Kotora
*a
aDepartment of Organic Chemistry, Faculty of Science, Charles University, Hlavova 8, 128 00 Praha 2, Czech Republic. E-mail: kotora@natur.cuni.cz
bInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, 166 10 Praha 6, Czech Republic
cDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 8, 128 00 Praha 2, Czech Republic
First published on 20th August 2024
Abstract
Intramolecular photoreactions of quinones can be interrupted by proton transfer using small molecules, such as trimethylamine N-oxide. This interruption de-excites the reactive spirocyclopropyl intermediates, the structures of which were for the first time confirmed by isolating them in their neutral form. The mild conditions of this process allow the conversion of a broad spectrum of quinones possessing linear and branched substituents to spirocyclopropanes in a catalytic, diastereoselective, and atom-conserving manner. Density functional theory (DFT) calculations were performed to investigate the possible reaction pathways and the origin of stereoselectivity. The established spirocyclopropanation route might be used to perform unconventional transformations of the side chains of quinones and to provide clues for the co-occurrence of certain natural quinones, hydroquinones, and spirocyclopropanes.
Introduction
Quinones are a large class of natural compounds known for their redox and photoredox properties.1 This knowledge has served to create numerous synthetic quinones with diverse applications based on the underlying redox transformations.2 The ability of quinones to undergo intramolecular photoreduction through their side chains into hydroquinones is a powerful transformation of this kind, which enabled several biomimetic total syntheses of structurally diverse natural compounds.3,4 However, the mechanistic understanding of this process still lacks experimental and computational evidence. In the 1960s, photoreactions of quinones containing enough flexible side chains were suggested to proceed through spirocyclic intermediates, but attempts to detect or isolate such intermediates have not been fruitful yet (Scheme 1).5 At the same time, the co-occurrence of quinones and hydroquinones possessing three-carbon ring substituents (e.g. royleanones, allylroyleanones, and coleons) together with spirocoleons in plants6–8 raised the question of a biogenetic relationship between these natural product classes. Presumably, the spirocyclopropane moiety in spirocoleons is formed by heterolytic9 or homolytic10 pathways and could act as an intermediate responsible for the side chain variations in the natural quinones and hydroquinones. Although neither pathway has received an experimental validation so far, the above observations suggest that a yet unexplored way to convert an alkyl group of a (hydro)quinone into a spirocyclopropyl unit exists in nature. In this respect, quinones represent a starting point for the most atom-economical approach towards spirocyclic compounds and derivatives thereof.11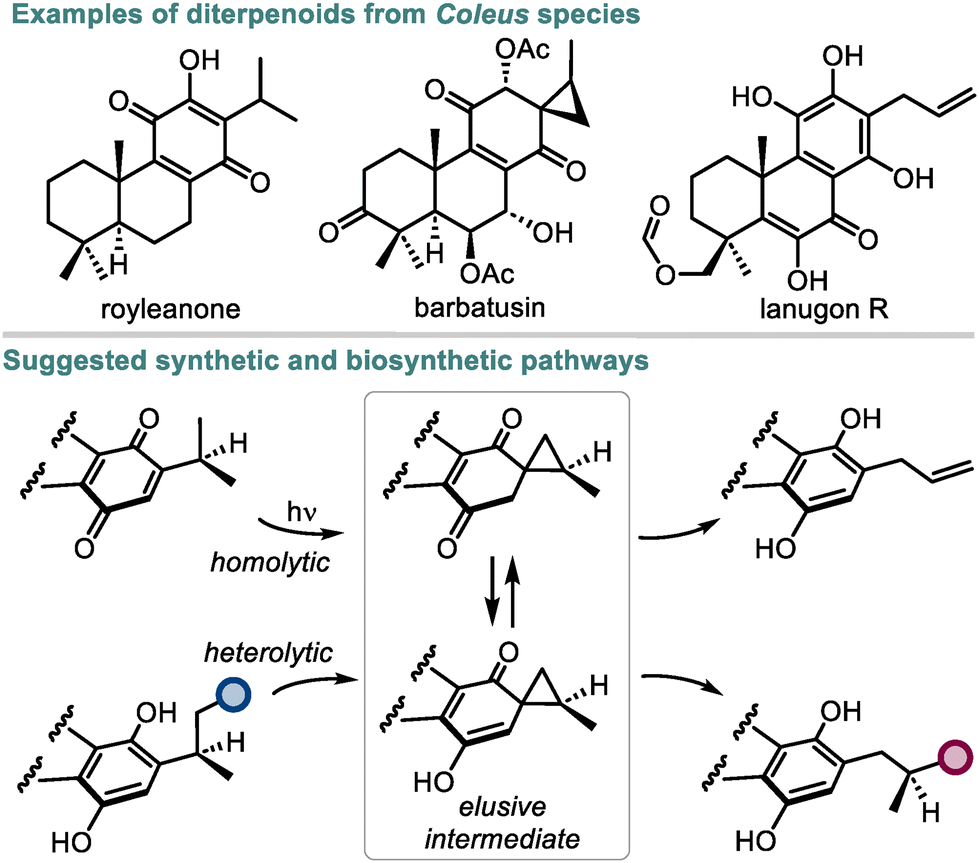 | ||
| Scheme 1 Natural diterpenoids from Coleus species and their suggested synthetic and biosynthetic pathways. | ||
We hypothesized that the previous attempts to establish such a transformation in solution using the energy of light5,10 failed due to a problematic de-excitation of the photochemically generated intermediates leading to their decomposition. Conversely, if the light energy is able to excite quinones under considerably more complex biological conditions,12 interruptions in the quinone photoredox reactivity by enzymes or small molecules are highly likely. Although only a few examples of interrupted photoredox processes have been reported in the literature,13 they exhibit vast potential for neutralizing reactive species or rerouting the established photoreactivity towards new products. Herein, we show that by interrupting the photoreactivity of quinones with natural and unnatural small molecules in catalytic amounts, the reaction outcome can be directed toward the spirocyclopropanation pathway avoiding aromatization. This transformation comprises a unique one-step atom-conserving approach for constructing spirocyclopropanes from quinones in a diastereoselective manner and under mild conditions, thus expanding the toolbox of current spirocyclopropanation strategies, which are mainly based on utilizing ylide or carbene precursors.14,15
Results and discussion
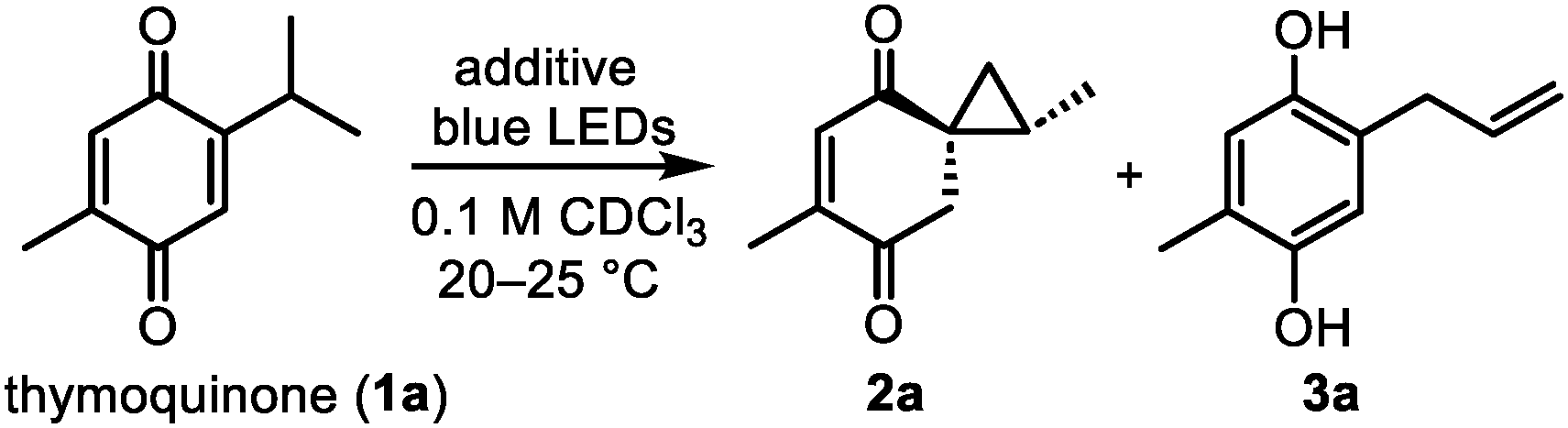
We started our investigation by elucidating the photochemistry of thymoquinone (1a) as one of the simplest quinones found in nature. Since 1877, thymoquinone has been known to undergo [2 + 2] photodimerization in the solid state,16 whereas unselective intramolecular photoredox reactions were later observed in solution.17 In order to suppress such processes in the favor of securing the desired spirocyclopropane intermediate 2a, we irradiated 1a with blue light in chloroform as a non-nucleophilic solvent in the presence of various additives that could presumably convert 2a from an excited state to its ground state (Table 1). To our surprise, several small molecules of different classes (onium salts, N-oxides, and amines) enabled the stereoselective formation of 2a in good yields, among which trimethylamine N-oxide (TMAO) gave the highest yield of 2a (84%). However, the low chemical stability of 2a did not allow us to isolate it in a higher than 51% yield. Allylhydroquinone 3a was the major reaction by-product, which predominated in the absence of additives (49% yield) and was also detected in decomposed samples of 2a. Interestingly, 3a was recently isolated together with thymoquinone from Nigella sativa.7|
|
|||
|---|---|---|---|
| Entry | Additivea | Yield of 2ab, % | Yield of 3ab, % |
a Reactions were performed on the 0.05 mmol scale using 10 mol% of additive unless noted otherwise (see the ESI‡ for the complete list of experiments).
b ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1H NMR yield.
c Isolated yield on the 0.5 mmol scale is given in parenthesis.
d 5 mol%.
e 15 mol%.
f No irradiation. 1H NMR yield.
c Isolated yield on the 0.5 mmol scale is given in parenthesis.
d 5 mol%.
e 15 mol%.
f No irradiation.
|
|||
| 1 | — | 0 | 49 |
| 2 | Berberine | 64 | 8 |
| 3 | Canadine | 44 | 27 |
| 4 | Caffeine | 0 | 46 |
| 5 | L-Phenylalanine | 0 | 41 |
| 6 | TBAB | 48 | 5 |
| 7 | Me3SOI | 64 | 19 |
| 8 | PPTS | 48 | 33 |
| 9 | Et3N | 82 | 12 |
| 10 | [bmim]Cl | 54 | 5 |
| 11 | NMO | 72 | 18 |
| 12 | TMAO | 84 (51) | 14 |
| 13 | TMAOd | 76 | 19 |
| 14 | TMAOe | 84 | 14 |
| 15 | TMAOf | 0 | 0 |
Next, we studied the scope of quinones that can undergo the spirocyclopropanation reaction with TMAO as an additive (Scheme 2). First, we established that spirocyclopropane 2a can be similarly prepared from thymoquinone's linear isomer (2-methyl-5-propyl-p-benzoquinone 1a′), albeit with slightly lower diastereoselectivity and yield (44%, 12:1 dr). The stability of spirocyclopropanes such as 2a is affected by the substitution character of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. Thus, less substituted compound 2b derived from 2-isopropyl-p-benzoquinone (1b) was not stable to isolation and could only be observed in solution (44% spectral yield). In contrast, spirocyclopropanes with sterically hindered CC bonds 2c and 2d were isolated in yields close to the spectral ones (42% and 47%, respectively). Products possessing annulated CC bonds 2e and 2f exhibited divergent behavior: benzoannulated 2e was isolated in 71% yield and with 7:1 dr, whereas pyridoannulated 2f was only stable in solution (80% spectral yield).
C bond. Thus, less substituted compound 2b derived from 2-isopropyl-p-benzoquinone (1b) was not stable to isolation and could only be observed in solution (44% spectral yield). In contrast, spirocyclopropanes with sterically hindered CC bonds 2c and 2d were isolated in yields close to the spectral ones (42% and 47%, respectively). Products possessing annulated CC bonds 2e and 2f exhibited divergent behavior: benzoannulated 2e was isolated in 71% yield and with 7:1 dr, whereas pyridoannulated 2f was only stable in solution (80% spectral yield).
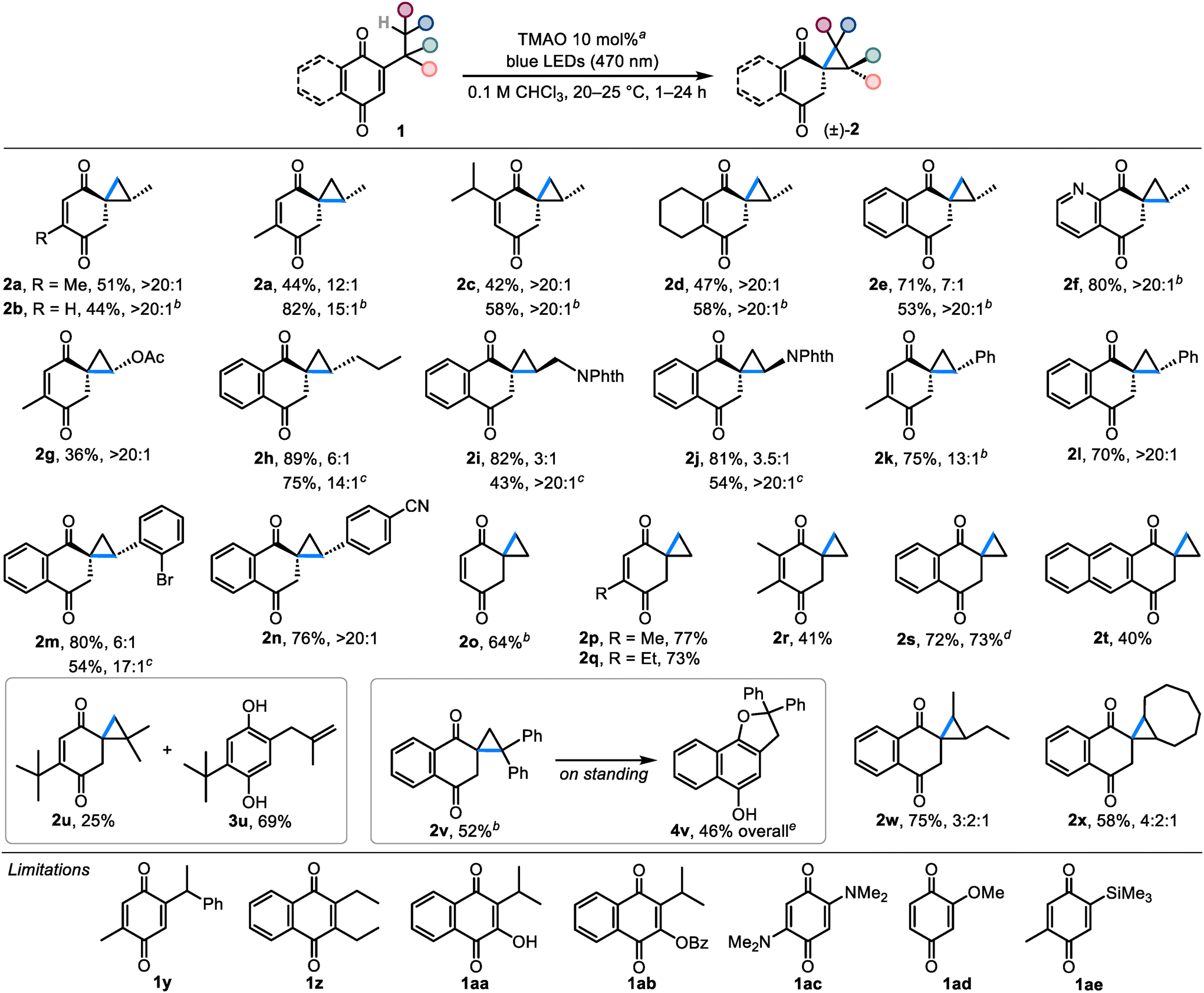 | ||
| Scheme 2 The scope of quinones participating in the spirocyclopropanation reaction (the newly formed bonds are highlighted in blue). aTypical reaction setup: quinone 1 (0.5 mmol) and TMAO (0.05 mmol) were irradiated in CHCl3 (5 mL) at 20–25 °C for 1–24 h; isolated yields and diastereomeric ratios of the products are given. bDetermined by 1H NMR analysis of the reaction mixture. cAfter subsequent separation of isomers. dIsolated yield on the 5 mmol scale. eIsolated as the O-acetate derivative. | ||
Then, we turned our attention to the substitution of the cyclopropane ring in 2. The results show that products decorated with various carbon- and heteroatom-substituted cyclopropane rings can be assembled from the corresponding quinones. For instance, acetoxy-substituted spirocyclopropane 2g was prepared in 36% yield and with >20:1 dr. Only minor by-products were observed, and the yield of 2g was largely affected by isolation. Stable benzoannulated products 2h–2j bearing alkyl and phthalimide groups were synthesized in 81–89% yields, although in lower diastereomeric ratios (3:1–6:1). Nonetheless, the diastereomeric purity of these compounds can be successfully increased by a subsequent chromatographic separation or crystallization. Aryl-substituted spirocyclopropane 2k was formed in 75% yield and with 13:1 dr, but underwent the Cloke–Wilson rearrangement18 into 4k upon work-up (30% yield). In contrast, benzoannulated analogs 2l–2n exhibited good stability and were obtained in 54–76% yields after separating the stereoisomers. The relative stereochemistry of compounds 2a–2n was determined on the basis of single-crystal X-ray diffraction analysis of 2j and 2l in conjunction with their characteristic peaks in 1H NMR spectra (see ESI Table S3‡). Interestingly, despite the fact that the anti-relationship between the carbonyl group connected to the cyclopropane ring and the adjacent exocyclic substituent was typical of most compounds (≥6:1 dr), phthalimides 2i and 2j preferred the syn-relationship (≥3:1 dr).
Ethylquinones can be used to prepare compounds 2 with an unsubstituted spirocyclopropyl moiety. Thus, the expectedly labile parent compound 2o can be generated from 2-ethyl-p-benzoquinone in 64% spectral yield (attempts to isolate it in pure form led to a complex product mixture). The stability of this scaffold is considerably improved by simply introducing a methyl or an ethyl group to the CC bond: bench-stable compounds 2p and 2q were synthesized in 77% and 73% isolated yields, respectively. On the other hand, the reaction leading to the doubly methylated compound 2r was sluggish and the product was obtained in only 41% yield. The synthesis of benzoannulated spirocyclopropane 2s gave similar results on 0.5 and 5 mmol scales (72–73% isolated yield), whereas naphthoannulated product 2t was obtained in a lower yield (40%) due to an incomplete conversion of the starting quinone even after extending the reaction time to 24 h.
Next, we studied the synthetic possibilities toward the products having geminal and vicinal substituents in the cyclopropane ring. Thus, 2,6-di-t-butyl-p-benzoquinone (1u) yielded isolable gem-dimethyl cyclopropane 2u (25%) together with an uninterrupted reaction product 3u (69%). Less stable gem-diphenyl cyclopropane 2v was observed in 52% spectral yield and similarly to 2k, it slowly underwent the Cloke–Wilson rearrangement into 4v on standing. At the same time, vicinally substituted spirocyclopropanes 1w and 1x were formed in 75% and 58% yields as diastereomeric mixtures and exhibited good stability to isolation and storage.
Several quinones were found to be unsuitable for the spirocyclopropanation reaction. For example, despite the fact that 2a could be prepared from both linear and branched quinones, only the linear quinone 1k gave product 2k, while the isomeric branched quinone 1y gave a complex product mixture. Disubstituted naphthoquinones, such as 1z, 1aa and 1ab, did not react under the standard reaction conditions. Electron-rich 2,5-bis(dimethylamino)-p-benzoquinone (1ac) reacted slowly forming the previously reported benzoxazoline,5b although the plausible spiroaziridine intermediate was not detected. 2-Methoxy-p-benzoquinone (1ad) was less reactive and only minor decomposition products were observed. Lastly, the trimethylsilyl group in 1ae also did not participate in the reaction, likely due to the distancing of the methyl groups caused by the large silicon atom.
After examining the scope of the spirocyclopropanation reaction, we aimed to shed light on its mechanistic details. The data presented so far indicate that spirocyclopropanes 2 do not readily undergo ring opening upon prolonged irradiation neither under the reaction conditions nor after isolation, therefore ground-state molecules 2 are not the reactive intermediates in the photoredox chemistry of quinones. Similarly, by-product 3a did not react under the standard reaction conditions, thus excluding the possibility of it forming 2a by the abnormal Claisen rearrangement.19 Hence, the formation of spirocyclopropanes 2 is a result of an interruption of the photoreactivity of quinones by small molecules. Conceivably, such interruption may commence with electron donor–acceptor (EDA) complexation or electron transfer between an electron-poor quinone and an electron-rich additive. However, the UV-Vis spectrum of the solution containing thymoquinone and TMAO did not show a bathochromic shift that could be attributed to an EDA complex (see ESI Fig. S1‡). Electron transfer between photoexcited quinones having β-H-atoms in the side chain and amines, such as triethylamine, was earlier ruled out.20 In any case, an interaction between a quinone and an additive is not crucial for the formation of the cyclopropane ring: among spirocyclopropanes 2, compounds 2f and 2o–2t were also formed when the reactions were conducted without any additives, albeit in lower yields (Fig. 1). These findings indicate that (a) the key action of an additive likely happens after the photoinduced formation of the cyclopropane ring, (b) in the absence of additives, reactive spirocyclopropyl intermediates generally undergo ring opening into allylhydroquinones 3 or give complex product mixtures instead of providing spirocyclopropanes 2, and (c) in specific cases, spirocyclopropanes 2 may be formed autocatalytically (e.g.2f) or when the cyclopropyl-allyl rearrangement is impossible (compounds 2o–2t).
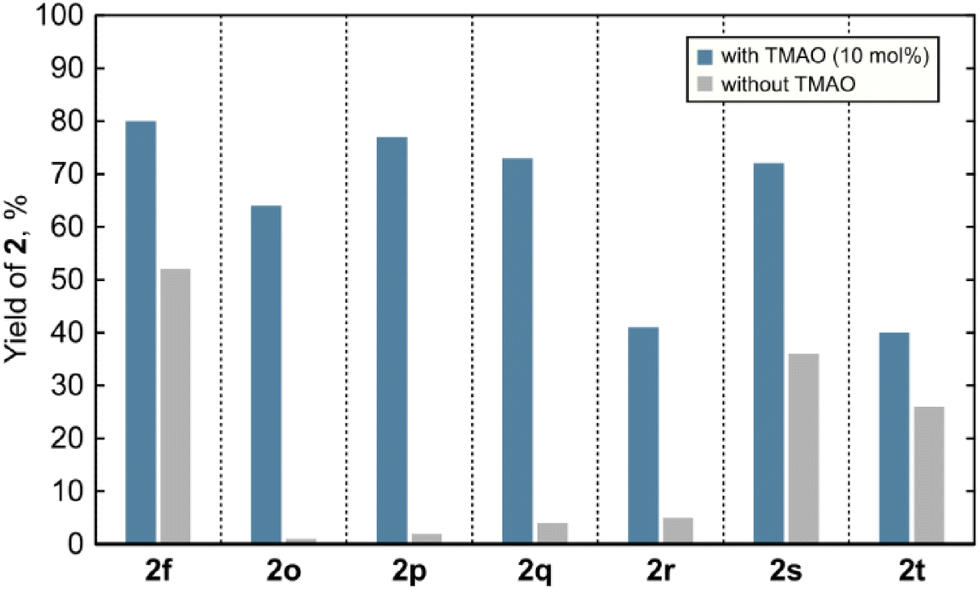 | ||
| Fig. 1 1H NMR yields of 2 in the reactions conducted with and without TMAO. | ||
The mechanism for the photoinduced formation of the reactive spirocyclopropyl intermediates from quinones was proposed in 19685b and has remained substantially unchanged to date. As illustrated in Scheme 3 for 1a, the reaction begins with n–π* photoexcitation of the quinone into delocalized biradical I, and the intramolecular 1,5-HAT process leads to biradical II. This intermediate was suggested to undergo neophyl-type ring closure5,21 into biradical III (pathway A). The subsequent conversion of biradical III into zwitterion IV is currently regarded as an electron transfer event.3
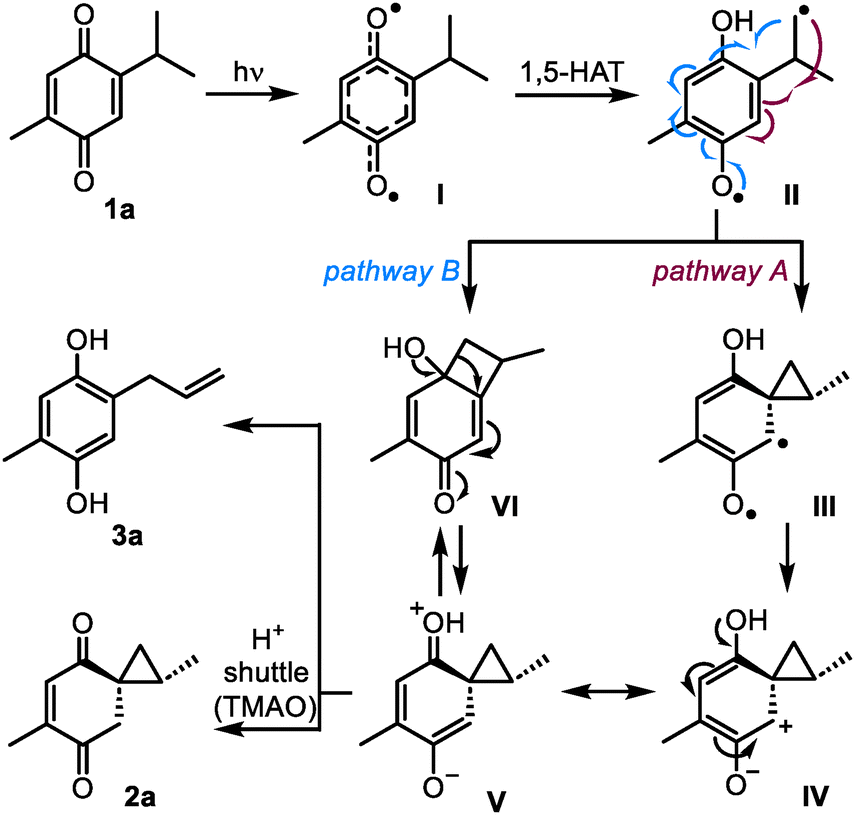 | ||
| Scheme 3 Plausible spirocyclopropanation pathways. | ||
The formation of zwitterionic intermediates in the process was confirmed using time-resolved UV-Vis spectroscopy by Görner,20 although the structure of these species and their ability to produce spirocyclopropanes 2 remained unclear. Nevertheless, the evidence collected so far points at zwitterionic species such as IV or its resonance form V as the main candidates to be intercepted by ionic additives, such as TMAO. In this respect, the most straightforward and sufficiently general interception pathway is the collapse of V into 2a by proton transfer. Indeed, if the proton shuttle is not available, the driving force of aromatization leads to ring-opening product 3a. An unconsidered alternative reaction pathway towards 2a and 3a could involve intramolecular recombination of biradical II into cyclobutanol VI, ring contraction of which would directly provide zwitterion V (pathway B).
In order to procure the missing mechanistic evidence, we first attempted to trap the reactive intermediates with stable radicals. The photoreactivity of 1a was expectedly shut down in the presence of galvinoxyl, whereas TEMPO surprisingly interrupted the reaction in the same way as TMAO: 2a was formed as the major product in 80% yield (Scheme 4a). The latter observation could be explained by the participation of the in situ generated oxammonium chloride of TEMPO.22 In the pursuit of cyclobutanol intermediates (such as VI, Scheme 3), we spotted the signs of a minor product possibly having a cyclobutane ring in the reaction mixture of quinone 1g, although in an amount not sufficient for characterization (see ESI Fig. S2‡). Luckily, when analogous naphthoquinone 1af was used as the starting material, the desired cyclobutanol 5 was observed as the major product regardless of the presence of TMAO (Scheme 4b). However, the attempted isolation of 5 only gave 26% of dihydrofuran 4af, which could be formed by the ring expansion directly or through the initial ring contraction into spirocyclopropane 2af (detected after the reaction). This exceptional behavior of quinone 1af forming 5 appears to be the result of the stabilization of 5 induced by the benzene ring and the acetate group. Interestingly, a similar transformation producing 4-membered oxetanol was noticed previously during the photolysis of 12-O-methyl royleanone.10 Lastly, to track the protons transferred in the spirocyclopropanation reaction, the conversion of deuterium-labelled thymoquinone d-1a into d-2a was monitored by 1H NMR (the use of CDCl3 as a solvent did not result in any deuterium incorporation in previous experiments). The results depicted in Scheme 4c show that the deuterium atoms from the labelled isopropyl group were mainly transferred to the methylene group in the 6-membered ring of d-2a (30% D, 60% of the theoretical amount). Additionally, the cyclopropane ring of d-2a was partially deuterated in the methine position (20% D), possibly caused by a reversible [1,2-H] shift in the isopropyl group during the process.23 However, this event appears unproductive, otherwise the 1-phenethyl group in 1y would participate in the reaction similarly to the 2-phenethyl group in 1k giving rise to 2k. Overall, the preservation of the deuterium label in d-2a is consistent with the proposed mechanism and indicates that TMAO only transfers the protons originating from the quinone itself.
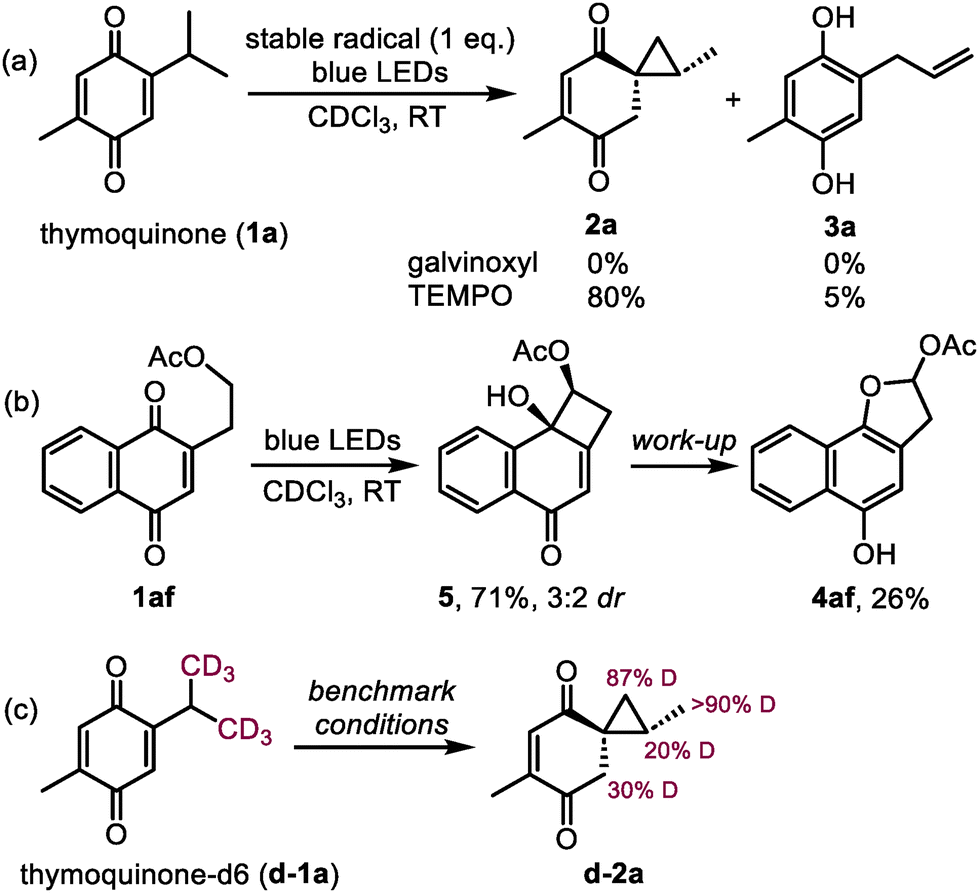 | ||
| Scheme 4 Mechanistic experiments. | ||
To understand the energy requirements of the reaction pathways outlined in Scheme 3, we performed density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations on 1a. First, we investigated the photoactivation of 1a and performed a relaxed geometry optimization scan along the 1,5-HAT coordinate. We observed that thermal 1,5-HAT is inaccessible due to a high activation energy exceeding ∼40 kcal mol−1 (see ESI Fig. S7‡). However, biradical II can be accessed through an excited-state process from 1a. Following the initial population of S1 or S2 excited states with the n–π* or π–π* character, the intersystem crossing to the lower-lying T1 state can drive the conversion of 1a to II. Subsequently, we assessed the energetics of biradical II reactivity, both with and without TMAO (Fig. 2). At the DFT level, biradical II is described with one unpaired electron at the methylene group and another electron delocalized over the semi-quinone moiety with the highest spin density at C4, C6, C8 and O12 atoms (cf.Fig. 2). The addition of TMAO resulted in H-bonding interaction but not proton transfer to TMAO, with a free energy gain of ∼7.7 kcal mol−1, minimally affecting the electronic structure of biradical II. The electronic distribution suggests potential reactivity toward closing the spirocyclopropane ring (pathway A in Scheme 3) or radical recombination, yielding two possible cyclobutanes (see ESI Fig. S8 and S9‡). Pathway A, involving spirocyclopropane ring closure into biradical III was found to have substantially lower activation free energy than radical recombination pathways regardless of the presence of TMAO. Considering stereochemistry, the activation free energy was lower for the anti-isomer by ∼1.8 kcal mol−1 (both with and without TMAO), leading to preferential anti-orientation of the spirocyclopropyl intermediate III. According to the Eyring equation (see ESI section 3.2.2‡), the difference in activation free energies favors anti-orientation of the spirocyclopropyl intermediate III in the ratio of ∼20:1 at 25 °C, in agreement with the experiment. Biradical III was found to be energetically higher than zwitterionic species IV/V, suggesting a barrier-less relaxation to IV/Vvia intramolecular electron transfer. The calculated electron density of the zwitterion IV/V suggests an electronic distribution that is an average between the resonance forms IV and V. Intermediate IV/V is stabilized by deprotonation by TMAO, which can further shuttle protons to the enol moiety and yield product 2a. In the absence of a proton shuttle, the push–pull character of zwitterionic intermediate IV/V promotes heterolytic ring opening,24 leading to aromatic product 3a.
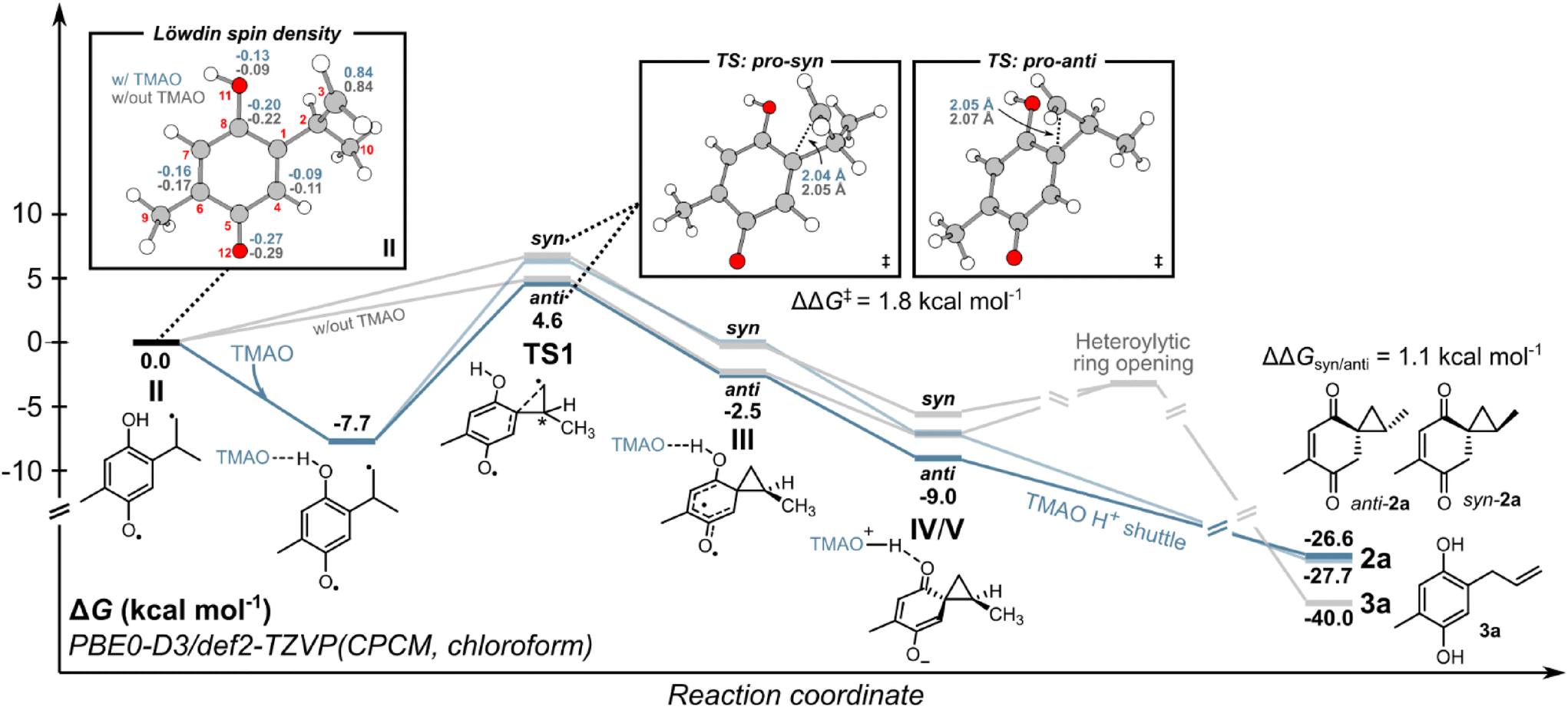 | ||
| Fig. 2 Computed energy profile for the formation of 2a and 3a from II. All computational details are provided in section 3 of the ESI.‡ | ||
Finally, to demonstrate how the side chains of quinones can be modified through spirocyclopropanes 2 as the reaction intermediates, we developed a series of sequential transformations. First, when 2a was generated from thymoquinone, the addition of methanol to the reaction mixture caused slow solvolysis that was accelerated thermally (60 °C) and provided hydroquinone 6 in 79% yield (Scheme 5). This hydroquinone was previously isolated from Nigella sativa and exhibited platelet aggregation activity.8 Second, the isopropyl group of thymoquinone was converted into the allyl group in 48% yield (compound 1ag) by treating the in situ formed 1a with phenyliodine(III) diacetate (PIDA) at room temperature. Benzoannulated spirocyclopropane 1s showed similar reactivity to 1a. Thus, a product of a formal C–H acetoxylation 1af was obtained from 2-ethylnaphthoquinone via2s in a three-step semi-one-pot sequence in 40% overall yield. Lastly, the dihydrofuran ring of 4s was constructed from the ethyl group of 2-ethylnaphthoquinone in 69% yield over two steps. Overall, the above transformations represent simple and efficient synthetic routes to compounds that are relatively difficult to prepare by other methods, thus opening numerous opportunities for using spirocyclopropanes 2 as acceptor-activated cyclopropanes.18,24 Furthermore, this reactivity may help to explain the co-occurrence and the biosynthetic origin of natural quinones and hydroquinones bearing alkyl, oxyalkyl and allyl groups.6–8 In particular, the fact that thymoquinone was earlier isolated together with hydroquinones 3a and 6 suggests that spirothymoquinone 2a could also be a natural compound, which remained elusive to isolate due to its chemical sensitivity. Similarly, the interrupted photoredox reactivity of quinones may play a role in the formation of other spirocyclopropanes in nature.6,25
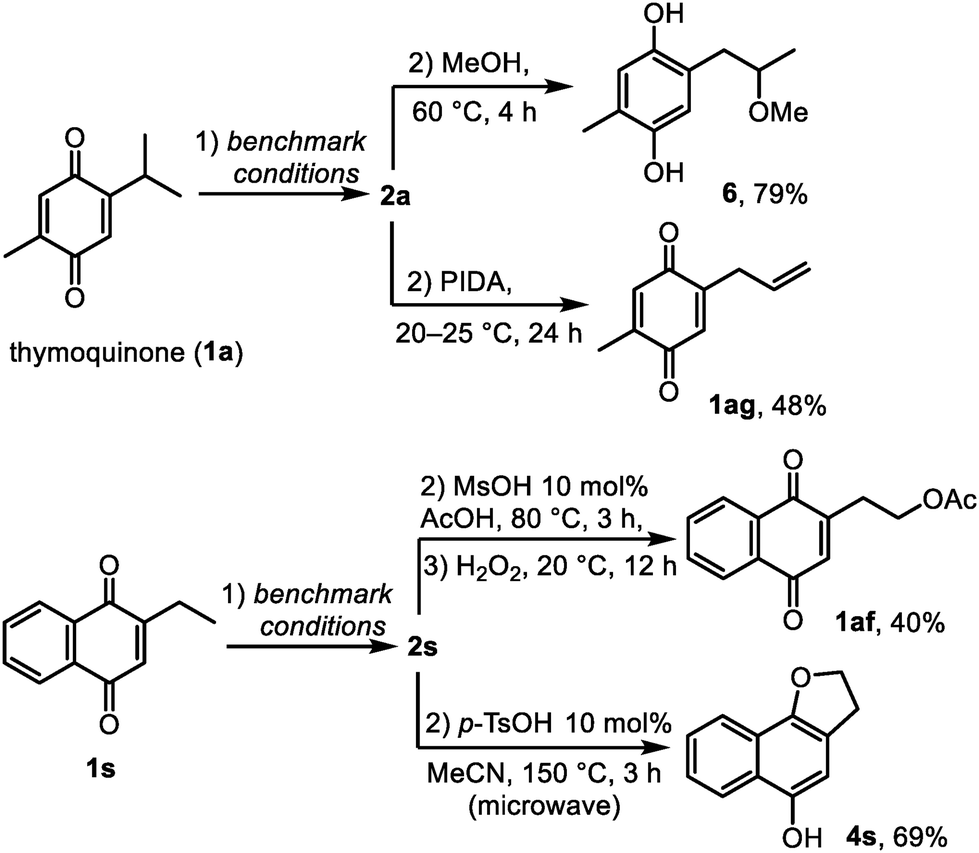 | ||
| Scheme 5 Transformations of quinones via spirocyclopropanes 2. | ||
Conclusions
In summary, the photoredox reactivity of quinones was successfully steered away from the aromatization route toward the spirocyclopropanation pathway by introducing a catalytic amount of TMAO to the reactions. The thus formed spirocyclopropanes were earlier suggested as the reaction intermediates, yet we showed that they are isolable compounds, the stability of which is strongly affected by the substitution character of the cyclopropane ring and of the enedione fragment. Still, the mild reaction conditions allow one to selectively convert a broad variety of benzo- and naphthoquinones having linear or branched side chains into the corresponding spirocyclopropanes that can be directly used in synthesis. Based on the experimental and computational data, TMAO enables the spirocyclopropanation pathway by incorporating a proton transfer step into the non-catalytic photoredox process. We believe that interruptions in the photoredox chemistry of quinones by such a universal mechanism may also occur in nature, and certain natural spirocyclopropanes could be biosynthesized accordingly and anticipated through synthesis.Author contributions
A. A. F. – synthetic investigation, D. B. – computational investigation, I. C. – single-crystal X-ray diffraction analysis, and M. K. – supervision and funding acquisition. The manuscript was composed with input and approval from all authors.Data availability
The data supporting this article can be found in the ESI.‡ Crystallographic data for compounds 2j, 2l and 2m have been deposited at the CCDC repository (https://www.ccdc.cam.ac.uk) under accession numbers 2352748, 2352747, and 2352749, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (Lead Agency Grant No. 21-39639L). The authors would like to thank Prof. Jiří Mosinger for helpful discussions.References
- (a) R. H. Thomson, Naturally Occurring Quinones IV, Springer, 1996 CrossRef; (b) N. El-Najjar, H. Gali-Muhtasib, R. A. Ketola, P. Vuorela, A. Urtti and H. Vuorela, The chemical and biological activities of quinones: overview and implications in analytical detection, Phytochem. Rev., 2011, 10, 353 CrossRef CAS; (c) S. N. Sunassee and M. T. Davies-Coleman, Cytotoxic and antioxidant marine prenylated quinones and hydroquinones, Nat. Prod. Rep., 2012, 29, 513 RSC; (d) M. Sarewicz and A. Osyczka, Electronic Connection Between the Quinone and Cytochrome c Redox Pools and Its Role in Regulation of Mitochondrial Electron Transport and Redox Signaling, Physiol. Rev., 2015, 95, 219 CrossRef; (e) J. R. Widhalm and D. Rhodes, Biosynthesis and molecular actions of specialized 1,4-naphthoquinone natural products produced by horticultural plants, Hortic. Res., 2016, 3, 16046 CrossRef.
- (a) W. Ma and Y.-T. Long, Quinone/hydroquinone-functionalized biointerfaces for biological applications from the macro- to nano-scale, Chem. Soc. Rev., 2014, 43, 30 RSC; (b) A. E. Wendlandt and S. S. Stahl, Quinone-Catalyzed Selective Oxidation of Organic Molecules, Angew. Chem., Int. Ed., 2015, 54, 14638 CrossRef PubMed; (c) Y. Wu, R. Zeng, J. Nan, D. Shu, Y. Qiu and S.-L. Chou, Quinone Electrode Materials for Rechargeable Lithium/Sodium Ion Batteries, Adv. Energy Mater., 2017, 7, 1700278 CrossRef; (d) G. G. Dias, A. King, F. de Moliner, M. Vendrell and E. N. da Silva Júnior, Quinone-based fluorophores for imaging biological processes, Chem. Soc. Rev., 2018, 47, 12 RSC; (e) C. Han, H. Li, R. Shi, T. Zhang, J. Tong, J. Li and B. Li, Organic quinones towards advanced electrochemical energy storage: recent advances and challenges, J. Mater. Chem. A, 2019, 7, 23378 RSC; (f) P. Natarajan and B. König, Excited-State 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ*) Initiated Organic Synthetic Transformations under Visible-Light Irradiation, Eur. J. Org. Chem., 2021, 2145 CrossRef CAS; (g) N. Monni, M. S. Angotzi, M. Oggianu, S. A. Sahadevan and M. L. Mercuri, Redox-active benzoquinones as challenging “non-innocent” linkers to construct 2D frameworks and nanostructures with tunable physical properties, J. Mater. Chem. C, 2022, 10, 1548 RSC.
- For a review, see: Y. Ando and K. Suzuki, Photoredox Reactions of Quinones, Chem. – Eur. J., 2018, 24, 15955 CrossRef CAS.
- (a) Y. Ando, A. Hanaki, R. Sasaki, K. Ohmori and K. Suzuki, Stereospecificity in Intramolecular Photoredox Reactions of Naphthoquinones: Enantioselective Total Synthesis of (−)-Spiroxin C, Angew. Chem., Int. Ed., 2017, 56, 11460 CrossRef CAS; (b) Y. Ando, T. Matsumoto and K. Suzuki, Intramolecular Photoredox Reaction of Naphthoquinone Derivatives, Synlett, 2017, 1040 CAS; (c) C. Thommen, M. Neuburger and K. Gademann, Collective Syntheses of Icetexane Natural Products Based on Biogenetic Hypotheses, Chem. – Eur. J., 2017, 23, 120 CrossRef; (d) W. He, Z. Zhang and D. Ma, A Scalable Total Synthesis of the Antitumor Agents Et-743 and Lurbinectedin, Angew. Chem., Int. Ed., 2019, 58, 3972 CrossRef; (e) Y. Ando, D. Tanaka, R. Sasaki, K. Ohmori and K. Suzuki, Stereochemical Dichotomy in Two Competing Cascade Processes: Total Syntheses of Both Enantiomers of Spiroxin A, Angew. Chem., Int. Ed., 2019, 58, 12507 CrossRef; (f) Y. Ando, D. Ogawa, K. Ohmori and K. Suzuki, Enantioselective Total Syntheses of Preussomerins: Control of Spiroacetal Stereogenicity by Photochemical Reaction of a Naphthoquinone through 1,6-Hydrogen Atom Transfer, Angew. Chem., Int. Ed., 2023, 62, e202213682 CrossRef PubMed; (g) M. Yokoya, M. Yamazaki-Nakai, K. Nakai, N. Sirimangkalakitti, S. Chamni, K. Suwanborirux and N. Saito, Transformation of Renieramycin M into Renieramycins T and S by Intramolecular Photoredox Reaction of 7-Methoxy-6-methyl-1,2,3,4-tetrahydroisoquinoline-5,8-dione Derivatives, J. Nat. Prod., 2023, 86, 222 CrossRef CAS PubMed.
- (a) C. M. Orlando Jr., H. Mark, A. K. Bose and M. S. Manhas, Photoreactions II. 2,2-dimethyl-5-hydroxycoumarans from t-butyl-p-benzoquinones, Tetrahedron Lett., 1966, 7, 3003 CrossRef; (b) C. M. Orlando Jr., H. Mark, A. K. Bose and M. S. Manhas, Photoreactions. V. Mechanism of the photorearrangement of alkyl-p-benzoquinones, J. Org. Chem., 1968, 33, 2512 CrossRef; (c) S. Farid, Photolysis of t-butyl-p-quinones: competing 1,4- and 1,5-dipolar cycloadditions of the photoproduct to nitriles and ketones, J. Chem. Soc. D, 1970, 303 RSC.
- (a) R. H. Alasbahi and M. F. Melzig, Plectranthus barbatus: A Review of Phytochemistry, Ethnobotanical Uses and Pharmacology – Part 1, Planta Med., 2010, 76, 653 CrossRef CAS; (b) R. J. Grayer, A. J. Paton, M. S. J. Simmonds and M.-J. R. Howes, Differences in diterpenoid diversity reveal new evidence for separating the genus Coleus from Plectranthus, Nat. Prod. Rep., 2021, 38, 1720 RSC; (c) M. Gáborová, K. Šmejkal and R. Kubínová, Abietane Diterpenes of the Genus Plectranthus sensu lato, Molecules, 2022, 27, 166 CrossRef.
- A. Benharref, R. Fdil, F. El Hanbali, A. Zeroual, M. Dakir and N. Mazoir, A New Monoterpene Isolated from Nigella sativa Essential Oil, Nat. Prod. Commun., 2017, 12, 881 CrossRef.
- S. Enomoto, R. Asano, Y. Iwahori, T. Narui, Y. Okada, A. N. B. Singab and T. Okuyama, Hematological Studies on Black Cumin Oil from the Seeds of Nigella sativa L., Biol. Pharm. Bull., 2001, 24, 307 CrossRef.
- P. Rüedi and C. H. Eugster, Struktur von Coleon E, einem neuen diterpenoiden Methylenchino aus der Coleus barbatus-Gruppe (Labiatae), Helv. Chim. Acta, 1972, 55, 1994 CrossRef.
- O. E. Edwards and P.-T. Ho, Photolysis of diterpenoid quinones, Can. J. Chem., 1978, 56, 733 CrossRef CAS.
- (a) F. Hu, Y.-B. Shen, L. Wang and S.-S. Li, Merging dearomatization with redox-neutral C(sp3)-H functionalization via hydride transfer/cyclization: recent advances and perspectives, Org. Chem. Front., 2022, 9, 5041 RSC; (b) Y. Chen, J. Xu and P. Gao, A route to carbon-sp3 bridging spiro-molecules: synthetic methods and optoelectronic applications, Org. Chem. Front., 2024, 11, 508 RSC; (c) S. Das, Annulations involving p-benzoquinones: stereoselective synthesis of fused, spiro and bridged molecules, New J. Chem., 2024, 48, 8243 RSC.
- Photochemical non-enzymatic pathways may be prevalent for quinones occurring on the skin surface: (a) H. Schildknecht, K. Holoubek, K. H. Weis and H. Krämer, Defensive Substances of the Arthropods, Their Isolation and Identification, Angew. Chem., Int. Ed. Engl., 1964, 3, 73 CrossRef; (b) Y. Saikawa, K. Hashimoto, M. Nakata, M. Yoshihara, K. Nagai, M. Ida and T. Komiya, The red sweat of the hippopotamus, Nature, 2004, 429, 363 CrossRef; (c) L. M. Bouthillette, V. Aniebok, D. A. Colosimo, D. Brumley and J. B. Macmillan, Nonenzymatic Reactions in Natural Product Formation, Chem. Rev., 2022, 122, 14815 CrossRef; (d) E. L. Bastos, F. H. Quina and M. S. Baptista, Endogenous Photosensitizers in Human Skin, Chem. Rev., 2023, 123, 9720 CrossRef CAS PubMed.
- (a) T. Duhamel, M. D. Martínez, I. K. Sideri and K. Muñiz, 1,3-Diamine Formation from an Interrupted Hofmann–Löffler Reaction: Iodine Catalyst Turnover through Ritter-Type Amination, ACS Catal., 2019, 9, 7741 CrossRef CAS; (b) M. H. Aukland, M. Šiaučiulis, A. West, G. J. P. Perry and D. J. Procter, Metal-free photoredox-catalysed formal C–H/C–H coupling of arenes enabled by interrupted Pummerer activation, Nat. Catal., 2020, 3, 163 CrossRef CAS; (c) M. V. Popescu, A. Mekereeya, J. V. Alegre-Requena, R. S. Paton and M. D. Smith, Visible-Light-Mediated Heterocycle Functionalization via Geometrically Interrupted [2 + 2] Cycloaddition, Angew. Chem., Int. Ed., 2020, 59, 23020 CrossRef CAS PubMed; (d) H.-M. Huang, P. Bellotti, P. M. Pflüger, J. L. Schwarz, B. Heidrich and F. Glorius, Three-Component, Interrupted Radical Heck/Allylic Substitution Cascade Involving Unactivated Alkyl Bromides, J. Am. Chem. Soc., 2020, 142, 10173 CrossRef CAS PubMed; (e) V. Trudel, C.-H. Tien, A. Trofimova and A. K. Yudin, Interrupted reactions in chemical synthesis, Nat. Rev. Chem., 2021, 5, 604 CrossRef PubMed; (f) Z. Liu, Y. Wei and M. Shi, Visible-light-mediated interrupted Cloke-Wilson rearrangement of cyclopropyl ketones to construct oxy-bridged macrocyclic framework, Tetrahedron Chem, 2022, 1, 100001 CrossRef.
- For reviews, see: (a) V. A. D'yakonov, O. A. Trapeznikova, A. De Meijere and U. M. Dzhemilev, Metal Complex Catalysis in the Synthesis of Spirocarbocycles, Chem. Rev., 2014, 114, 5775 CrossRef PubMed; (b) W. Wu, Z. Lin and H. Jiang, Recent advances in the synthesis of cyclopropanes, Org. Biomol. Chem., 2018, 16, 7315 RSC; (c) Q. Wang, H.-F. Liu, S.-Y. Ren, M.-X. He and Y.-M. Pan, Research Advances in Electrochemical Synthesis of Spirocyclic Skeleton Compounds, Synthesis, 2023, 2873 CAS; (d) J. Cao and S. P. Vincent, Spirocyclopropyl carbohydrates: Synthesis and applications, Tetrahedron, 2023, 140, 133465 CrossRef CAS; (e) J. Xie and G. Dong, Cyclopropylcarbinyl cation chemistry in synthetic method development and natural product synthesis: cyclopropane formation and skeletal rearrangement, Org. Chem. Front., 2023, 10, 2346 RSC.
- For representative examples, see: (a) U. Das, Y.-L. Tsai and W. Lin, An efficient organocatalytic enantioselective synthesis of spironitrocyclopropanes, Org. Biomol. Chem., 2013, 11, 44 RSC; (b) K. Gai, X. Fang, X. Li, J. Xu, X. Wu, A. Lin and H. Yao, Synthesis of spiro[2.5]octa-4,7-dien-6-one with consecutive quaternary centers via 1,6-conjugate addition induced dearomatization of para-quinone methides, Chem. Commun., 2015, 51, 15831 RSC; (c) P. Qian, B. Du, R. Song, X. Wu, H. Mei, J. Han and Y. Pan, N-Iodosuccinimide-Initiated Spirocyclopropanation of Styrenes with 1,3-Dicarbonyl Compound for the Synthesis of Spirocyclopropanes, J. Org. Chem., 2016, 81, 6546 CrossRef CAS; (d) E. M. D. Allouche and A. B. Charette, Non-stabilized diazoalkane synthesis via the oxidation of free hydrazones by iodosylbenzene and application in in situ MIRC cyclopropanation, Chem. Sci., 2019, 10, 3802 RSC; (e) J. Werth, K. Berger and C. Uyeda, Cobalt Catalyzed Reductive Spirocyclopropanation Reactions, Adv. Synth. Catal., 2020, 362, 348 CrossRef PubMed; (f) X. Li, R. J. Kutta, C. Jandl, A. Bauer, P. Nuernberger and T. Bach, Photochemically Induced Ring Opening of Spirocyclopropyl Oxindoles: Evidence for a Triplet 1,3-Diradical Intermediate and Deracemization by a Chiral Sensitizer, Angew. Chem., Int. Ed., 2020, 59, 21640 CrossRef; (g) Y.-H. Deng, W.-D. Chu, Y.-H. Shang, K.-Y. Yu, Z.-L. Jia and C.-A. Fan, P(NMe2)3-Mediated Umpolung Spirocyclopropanation Reaction of p-Quinone Methides: Diastereoselective Synthesis of Spirocyclopropane-Cyclohexadienones, Org. Lett., 2020, 22, 8376 CrossRef; (h) M. M. Lindner, M. W. Alachraf, B. Mitschke, P. Schulze, M. Leutzsch and B. List, Toward a Formyl-to-Phenyl Conversion: An Unexpected Photochemical Fulvene Rearrangement, Angew. Chem., Int. Ed., 2023, 62, e202303119 CrossRef; (i) K. E. Berger, R. J. Martinez, J. Zhou and C. Uyeda, Catalytic Asymmetric Cyclopropanations with Nonstabilized Carbenes, J. Am. Chem. Soc., 2023, 145, 9441 CrossRef CAS PubMed; (j) Y. Peng, Y. Zhang, M. Wu, X. Chen, X. Liu, Y. Liu and L. Rong, An Efficient Synthesis of Spirocyclopropanes by [2 + 1] Annulation of α,β-Unsaturated Carbonyl Compounds with Nitroacetates, ChemistrySelect, 2023, 8, e202301733 CrossRef CAS; (k) V. George and B. König, Photogenerated donor–donor diazo compounds enable facile access to spirocyclopropanes, Chem. Commun., 2023, 59, 11835 RSC; (l) M. Li, Y. Wei and M. Shi, Thermally-induced intramolecular [2 + 2] cycloaddition of allene-methylenecyclopropanes: expedient access to two separable spiropolycyclic heterocycles, Org. Chem. Front., 2023, 10, 440 RSC.
- C. Liebermann, Ueber Polythymochinon, Ber. Dtsch. Chem. Ges., 1877, 10, 2177 CrossRef.
- C. M. Orlando Jr., H. Mark, A. K. Bose and M. S. Manhas, Photoreactions: Rearrangement of Thymoquinone, Chem. Commun., 1966, 714 RSC.
- For a recent review, see: U. Nazeer, A. Mushtaq, A. F. Zahoor, F. Hafeez, I. Shahzadi and R. Akhtar, Cloke–Wilson rearrangement: a unique gateway to access five-membered heterocycles, RSC Adv., 2023, 13, 35695 RSC.
- Z. Wang, in Comprehensive Organic Name Reactions and Reagents, ed. Z. Wang, John Wiley & Sons, Inc., 2010, pp. 1–4 Search PubMed.
- H. Görner, Photoreactions of 2-methyl-5-isopropyl-1,4-benzoquinone, J. Photochem. Photobiol., A, 2004, 165, 215 CrossRef.
- For reviews on neophyl rearrangement, see: (a) A. Studer and M. Bossart, Radical aryl migration reactions, Tetrahedron, 2001, 57, 9649 CrossRef; (b) Z.-M. Chen, X.-M. Zhang and Y.-Q. Tu, Radical aryl migration reactions and synthetic applications, Chem. Soc. Rev., 2015, 44, 5220 RSC; (c) A. R. Allen, E. A. Noten and C. R. J. Stephenson, Aryl Transfer Strategies Mediated by Photoinduced Electron Transfer, Chem. Rev., 2022, 122, 2695 CrossRef CAS.
- For the formation of oxammonium chloride of TEMPO in halogenated solvents upon irradiation, see: J. Chateauneuf, J. Lusztyk and K. U. Ingold, Photoinduced electron transfer from dialkyl nitroxides to halogenated solvents, J. Org. Chem., 1990, 55, 1061 CrossRef CAS The proton transfer step could be then accomplished by the chloride anion, which is a known proton transfer agent (see: V. Pilepić, C. Jakobušić, D. Vikić-Topić and S. Uršić, Evidence for proton transfer from carbon to chloride ion in solution, Tetrahedron Lett., 2006, 47, 371 CrossRef ). Nevertheless, a distinct reaction mechanism involving TEMPO itself cannot be excluded.
- A similar radical [1,2-H] shift was observed in photoexcited fulvenes by List and co-workers (ref. 15h). Alternatively, if biradical II could undergo a conversion into the corresponding zwitterion in a similar manner to biradical III, the slight deuteration of the methine position of d-2a could be explained by the reversible 1,2-proton shift. As exemplified by the reactivity of 1k and 1y, the phenyl group could stabilize both radical and ionic intermediates resulting from the [1,2-H] shift and inhibit the cyclopropanation in the case of 1y, but not in the case of 1k. The cyclopropanation is favored in other cases owing to the stabilization induced by the bridging effect (ref. 21).
- For recent reviews, see: (a) T. F. Schneider, J. Kaschel and D. B. Werz, A New Golden Age for Donor–Acceptor Cyclopropanes, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef PubMed; (b) O. A. Ivanova and I. V. Trushkov, Donor-Acceptor Cyclopropanes in the Synthesis of Carbocycles, Chem. Rec., 2019, 19, 2189 CrossRef PubMed; (c) V. Pirenne, B. Muriel and J. Waser, Catalytic Enantioselective Ring-Opening Reactions of Cyclopropane, Chem. Rev., 2021, 121, 227 CrossRef PubMed; (d) Y. Cohen, A. Cohen and I. Marek, Creating Stereocenters within Acyclic Systems by C–C Bond Cleavage of Cyclopropanes, Chem. Rev., 2021, 121, 140 CrossRef PubMed; (e) E. Reyes, U. Uria, L. Prieto, L. Carrillo and J. L. Vicario, Organocatalytic activation of cyclopropanes in asymmetric synthesis, Tetrahedron Chem, 2023, 7, 100041 CrossRef.
- D. R. McMullin, B. D. Green, N. C. Prince, J. B. Tanney and J. D. Miller, Natural Products of Picea Endophytes from the Acadian Forest, J. Nat. Prod., 2017, 80, 1475 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Igor V. Trushkov on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2352748, 2352747 and 2352749. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01291g |
| This journal is © the Partner Organisations 2024 |