
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA pillar[5]arene-based three-component supramolecular copolymer for the fluorescence detection of spermine†
Martina
Mazzaferro
 ab,
Daniele
Crisafulli
a,
Francesca
Mancuso
a,
Marco
Milone
a,
Fausto
Puntoriero
a,
Anna
Irto
a,
Salvatore
Patanè
c,
Valentina
Greco
d,
Alessandro
Giuffrida
d,
Ilenia
Pisagatti
*a,
Anna
Notti
a,
Melchiorre F.
Parisi
a and
Giuseppe
Gattuso
*a
ab,
Daniele
Crisafulli
a,
Francesca
Mancuso
a,
Marco
Milone
a,
Fausto
Puntoriero
a,
Anna
Irto
a,
Salvatore
Patanè
c,
Valentina
Greco
d,
Alessandro
Giuffrida
d,
Ilenia
Pisagatti
*a,
Anna
Notti
a,
Melchiorre F.
Parisi
a and
Giuseppe
Gattuso
*a
aDipartimento di Scienze Chimiche, Biologiche, Farmaceutiche ed Ambientali, Università degli Studi di Messina, Viale F. Stagno d'Alcontres, 31, 98166 Messina, Italy. E-mail: ipisagatti@unime.it; ggattuso@unime.it
bDipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, via Elce di Sotto, 8, 06123 Perugia, Italy
cDipartimento di Scienze Matematiche e Informatiche, Scienze Fisiche e Scienze della Terra, Università degli Studi di Messina, Viale F. Stagno d'Alcontres 31, 98166 Messina, Italy
dDipartimento di Scienze Chimiche, Università di Catania, Viale A. Doria 6, 95125 Catania, Italy
First published on 18th September 2024
Abstract
The supramolecular polymerization of a bis-pillar[5]arene dicarboxylic acid monomer (H) in the presence of a mixture of complementary bis-guests 1,12-dodecanediyl-bis-1,1′-1H-imidazole (G1) and bis-N,N’-(6-(1H-imidazole)decyl)-perylene bisimide (G2), produces an AA/BB-type supramolecular copolymer H/G1/G2 that retains the properties of the parent bi-component systems, that is, H/G1 solubility and H/G2 photoresponsiveness. The supramolecular copolymer showed stimuli-responsiveness, reacting to the presence of the cancer marker, spermine (S), by disassemblying and releasing G2. Once released, the perylene bisimide monomer (quenched in the copolymer by host-to-guest electron transfer), showed a remarkable increase of emission intensity. ESI-MS data are fully consistent with the formation of the H/G1/G2 copolymer, and AFM investigations on films cast from H/G1/G2 and H/G1/G2 + S solutions demonstrated that the supramolecular copolymer sensing abilities are retained also in the solid state.
Introduction
Supramolecular polymers1–3 are ordered superstructures (or, as defined by J.-M. Lehn, ‘novel expressions of complex matter’4) composed of monomeric units held together by non-covalent interactions rather than covalent bonds, as it happens in classical polymeric materials. They are generally prepared by the supramolecular polymerization5–7 of small molecules that spontaneously self-assemble to produce large aggregates, that ultimately display polymeric properties.The main advantage of these novel materials over covalent polymers is that they can be produced from a wide variety of molecular monomers, which can be chosen depending on the properties the final product must possess. In this light, both the monomer and the type of interaction that will hold the supermolecules together can in principle be customized, providing at the same time function, as brought by the molecular features of the monomer(s) of choice, as well as robustness and conditional stability, deriving from the dynamic and reversible nature of the intermolecular – i.e., intermonomer – forces involved. In addition, the combination of the molecular and supramolecular properties of these materials often provides stimuli-responsiveness, the most desirable of their unique features.
Supramolecular polymers are long past the stage of scientific curiosity and have become a technological reality, with applications that span from materials science8,9 to the biomedical field.10–12 Research currently focus on the ability of these materials to perform specific functions in a targeted fashion. In particular, considerable effort has been devoted to the use of supramolecular polymers in the area of sensing and biosensing. Again, owing to their adaptive nature, they represent a significant advancement over conventional sensing systems based either on covalent (single-use) polymers or single-molecule reporters. In fact, supramolecular polymer sensors, in addition to the required sensitivity, selectivity and ease of read out, provide a reversibility that makes them reusable and capable of real-time monitoring of physical parameters or chemical agents. Among many, an elegant example is the one reported by Schenning and co-workers,13 an inkjet-printable sensing system based on a hydrogen-bonded supramolecular cholesteric liquid crystal network, able to return a fast response to changes in temperature and humidity by changing its reflection colour. Colour changes were determined by a change in the helix pitch as a result of water adsorption.
Supramolecular polymers can be used also for the detection of small organic molecules, as it was shown in the case of thin films formed from π-electron rich anthracene- or pyrene-containing tetracarboxylic acids.14 Exposure to vapours of electron-deficient nitroaromatic compounds resulted in a macroscopic quenching of fluorescence, thus allowing for the detection of explosives at the level of parts per trillion (ppt).15
Supramolecular polymers have been also used to detect intrinsic molecular properties such as chirality. Haino and co-workers16 reported that a porphyrin-based, hydrogen-bonded supramolecular polymer could reveal the enantiomeric excess of (+)- and (–)-α-pinene mixtures. The polymer was composed of achiral monomers that assembled in racemic (+)- and (–)-helical aggregates which reacted to the addition of nonracemic molecule with an intense response in their electronic circular dichroism spectra.
These highly representative examples show that, with the choice of suitable monomers, it is possible to program the properties of a material that has to execute a given function. When this is not directly achievable, though, it is possible to resort to blending or formulation of composite material that conjugate different properties arising from different components. It may be done by mixing supramolecular and classical covalent components (as it has been recently shown, for instance, in mechanochromic materials that display self-diagnostic properties17,18), or by the careful balancing of different monomers to obtain supramolecular copolymers.19–21 These are multicomponent, non-covalently bound systems – with varying degree of internal organization22 – comprising supramolecular domains with different molecular properties, that may even act in a synergistic manner.
Nanoarchitectures with 1D (linear) topology can be prepared by the supramolecular polymerization of heteroditopic AB-type, or pairs of complementary homoditopic AA/BB-type monomers. Among the wide range of monomeric units that have been employed,23 those relying on host (A) and guest (B) mutual recognition are the most fascinating,24 as a careful selection of the two counterparts may bring to the final material specific properties such as robustness, luminescence, pH responsiveness, molecular recognition etc.25,26 Owing to their ease of chemical modification, crown ethers,27,28 cyclodextrins,29 calix[n]arenes,30,31 pillar[n]arenes32–34 and other macrocyclic receptors have been employed in the construction of supramolecular polymers.
In the course of our studies in this field, we have actively investigated stimuli-responsive supramolecular polymers that self-assemble as a result of host–guest interactions between calix[5]arene-35–38 or pillar[5]arene-containing39 monomers and complementary diaminoalkanes or alkyl(di)ammonium ions. Herein, taking advantage of the affinity between pillar[5]arenes and alkylimidazole moieties,40,41 we describe a supramolecular copolymer composed of a novel bis-pillar[5]arene dicarboxylic acid H and, as complementary partners, 1,12-dodecanediyl-bis-1,1′-1H-imidazole G1 and bis-N,N’-(6-(1H-imidazole)decyl)-perylene bisimide G2 (Scheme 1). We will show how the H/G1/G2 copolymer, in retaining polymers H/G1 adaptivity and solubility and H/G2 photophysical properties, can act as an efficient OFF/ON luminescent sensing agent for the cancer marker, spermine (S),42 both in solution and in solid films.
 | ||
| Scheme 1 Chemical structures and cartoon representation of compounds H, G1, G2 and S. | ||
Results and discussion
Synthesis of the molecular components H, G1 and G2
Bis-pillararene H was synthesized in two steps starting from nonamethyl-pillar[5]arene431 by following a procedure recently optimized by us for the syntheses of similar calix[5]arene36 and pillar[5]arene39 monomers (Scheme S1, see the ESI†). Pillararene 1 was reacted with dimethyl meso-2,8-dibromononandioate442 and K2CO3 in refluxing acetonitrile to give bis-pillararene diester 3 in 46% yield. Diester 3 was then hydrolysed to H using LiOH in THF/H2O (30%). While G1 is a known compound,45G2 (Scheme S2, ESI†) was prepared from 10-(1H-imidazole)-1-N-phthalimidodecane 4,46 which was first converted into 10-(1H-imidazole)-1-decanamine 5 by treatment with hydrazine (66%), and then reacted with perylene-3,4,9,10-tetracarboxylic dianhydride 6 in refluxing DMF (80%).H/G1 Supramolecular polymer
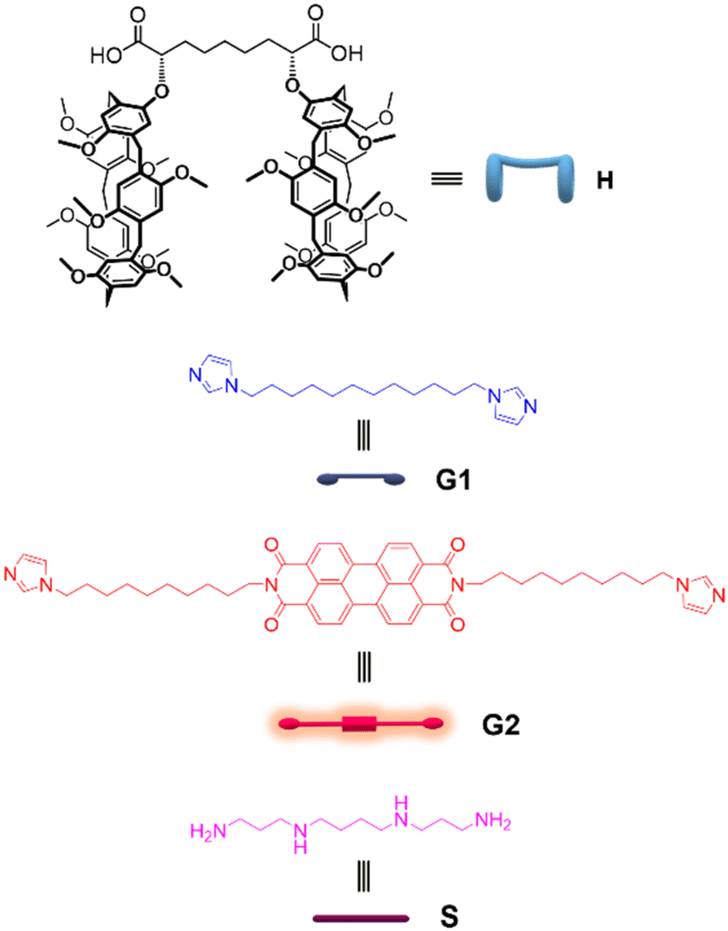
The ability of homoditopic H to act as a monomer for the self-assembly of supramolecular polymers was tested in the presence of G1 as the complementary homoditopic partner.Upon addition of 1 equiv. of monomer G1 to H, the 1H NMR resonances of the two species (Fig. 1, spectra a and c) undergo dramatic changes, confirming that the pillararene/alkylimidazole recognition is taking place. Notably, the aromatic resonances of the imidazole moieties and the aryl units present in the individual spectra of G1 and H disappear, leaving room to a broad resonance (spectrum b); at the same time, two new resonances appear at high fields (δ = −0.28 and 0.62 ppm, respectively). A 2D TOCSY spectrum (Fig. S2, ESI†) allowed for the assignment of the resonances of the alkyl chains of both G1 and H, showing that the high-field peaks belong to the imidazole guest placed within the cavity of a pillararene moiety experiencing – along with the adjacent methylene groups – the shielding effect of the aromatic rings of the host.
 | ||
| Fig. 1 1H NMR spectra (500 MHz, CDCl3, 25 °C) of: (a) [H] = 10 mM; (b) [H] = [G1] = 10 mM; (c) [G1] = 10 mM. *Asterisks indicate residual solvent peaks. | ||
Diffusion ordered spectroscopy (DOSY) NMR experiments shed light on the self-assembly of the H/G1 supramolecular polymer (Fig. S7–S10, ESI†). In keeping with previous studies,35–39 the self-diffusion coefficients (D) of the species present in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 samples of H and G1 were found to be concentration dependent. The D values were seen to decrease consistently upon increasing the concentration of H and G1 from 1.0 to 16.6 mM (D = 3.04 ± 0.08 to 1.45 ± 0.08 × 10–10 m2 s−1), respectively. Remarkably, over the entire concentration range investigated, both H and G1 diffuse with the same coefficient, indicating the formation of H/G1 aggregates (Table 1).
:1 samples of H and G1 were found to be concentration dependent. The D values were seen to decrease consistently upon increasing the concentration of H and G1 from 1.0 to 16.6 mM (D = 3.04 ± 0.08 to 1.45 ± 0.08 × 10–10 m2 s−1), respectively. Remarkably, over the entire concentration range investigated, both H and G1 diffuse with the same coefficient, indicating the formation of H/G1 aggregates (Table 1).
| [H] mM | [G1] mM | [3] mM | D (× 10−10 m2 s−1) | M (amu) |
|---|---|---|---|---|
| 1 | 1 | — | 3.04 ± 0.08 | 5350 |
| 5 | 5 | — | 2.43 ± 0.09 | 8250 |
| 10 | 10 | — | 1.84 ± 0.09 | 24100 |
| 16.6 | 16.6 | — | 1.45 ± 0.08 | 49300 |
| 16.6 | — | — | 3.87 ± 0.08 | 2600 |
| — | — | 16.6 | 4.50 ± 0.09 | 1684 |
The average molecular mass of the aggregates can be estimated from eqn (1)47 that, assuming the formation of random-coil, spherical-shaped supramolecular polymers, inversely correlates the self-diffusion coefficient D with the molecular mass M:
| Daggregate/Dmonomer = (Mmonomer/Maggregate)1/3 | (1) |
Given the natural tendency of carboxylic acids to form hydrogen-bonded dimers (especially in aprotic solvents such as CDCl3), the self-diffusion coefficients of H and the diester precursor 3 were measured and compared (Fig. S11, ESI†). The latter was found to diffuse with a D = 4.50 ± 0.09 × 10−10 m2 s−1, indicating that H (D = 3.87 ± 0.08 × 10−10 m2 s−1) is in equilibrium with its dimeric form. Simple calculations (using the D value of 3 as the reference value for a monomeric bis-pillar[5]arene) show that at 1 mM the H/G1 aggregates have an average M = 5350 amu whereas at 16.6 mM the average M increases to 49300 amu, roughly corresponding to an increase from ca. 3 to 25 H/G1 monomeric units.
H/G2 Supramolecular system
Perylene bisimide-containing monomer G2 was found to be sparingly soluble in CHCl3, preventing further studies on the H/G2 polymer in the millimolar range to allow for NMR characterization. However, the interaction between H and G2 could conveniently be investigated by UV-vis and fluorescence spectroscopy (Fig. 2). To this end, a G2 solution (7.83 × 10−6 M in CHCl3/CF3CH2OH (TFE) 97:3 v/v, vide infra) was titrated with a solution of bis-pillar[5]arene H. Progressive additions of H caused a corresponding hyperchromic effect on the band at λ = 292 nm belonging to the pillar[5]arene macrocycle, without affecting the absorption spectrum of G2 (characterized by the typical perylene bisimide absorptions in the 420–540 nm spectral range), thus indicating that the ground-state of the perylene bisimide moiety is not affected.
 | ||
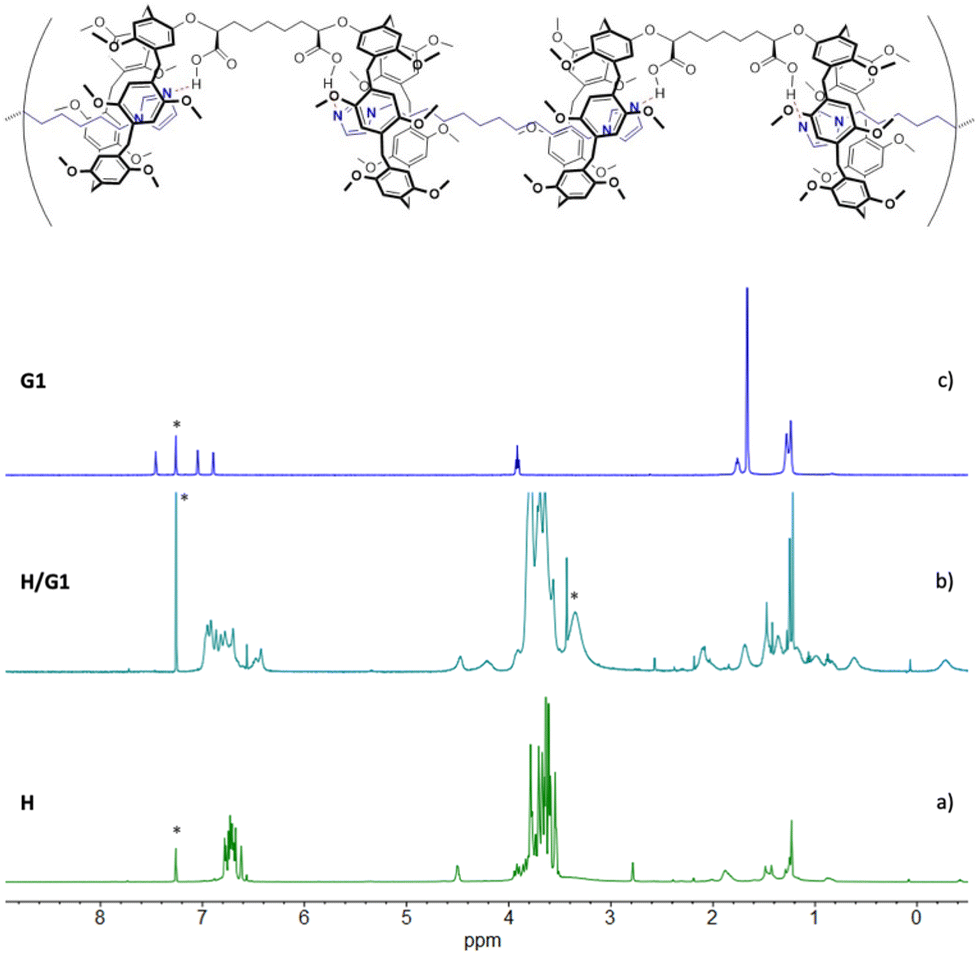
| Fig. 2 Emission and absorption (inset) titrations in CHCl3/TFE 97:3 v/v of [G2] = 7.83 × 10−6 M with [H] = 2.46 × 10−4 M solution (titrant concentration from 7.78 × 10−7 to 1.54 × 10−5 M). The excitation wavelength was set at λex = 456 nm. | ||
The emission spectrum of G2 (λex = 456 nm) features the three typical bands of perylene bisimide moieties at λ = 537, 578 and 627 nm, respectively. Upon addition of H, the fluorescence intensity of G2 was progressively quenched as the titration proceeded, probably as result of a host-to-guest photoinduced electron transfer (PET), favoured by the tendency of the perylene bisimide moiety to behave as a good electron acceptor (owing to its relatively low reducing potential).48–50
To evaluate in detail the effect of complexation on the photophysical properties of perylene bisimide, pump–probe transient absorption experiments were carried out by exciting at 400 nm (100 fs) a solution of H/G2. The transient absorption spectra (TAS) are shown in Fig. 3.
 | ||
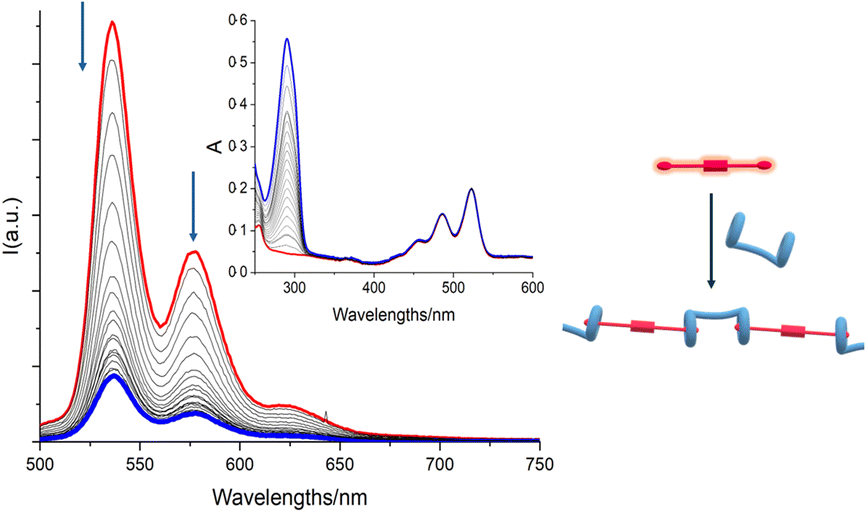
| Fig. 3 TAS matrix (bottom) and selected TASs (top) with kinetic fitting @ 530 nm in the inset (top) of [H/G2] = 3.83 × 10−5 M in C2H2Cl4/TFE 97:3 v/v solution. Chloroform was replaced with tetrachloroethane to avoid the formation of HCl under high-energy laser light excitation. The excitation wavelength was set at λex = 400 nm (pulse 100 fs, 100 mJ). | ||
The initial spectrum showed the characteristic bands of a perylene bisimide. Bleaching could be observed in the region between 420 and 540 nm and a stimulated emission contribution at lower energy, while above 600 nm an intense absorption typical of perylene bisimides could be distinguished.49,51
The spectrum of the optically populated species evolved in about 9 ps, albeit with little spectral variation, to a new species characterized by a weak absorption around 620 nm. The evolution of TAS remained almost constant in the first 90 ps, to leave the field to a spectrum, weak in intensity, attributable to a small fraction of the sample, that evolved with a lifetime on the order of 900 ps.
To rationalize these results, it should be kept in mind that pillararenes may undergo an oxidation process at about 0.75 V vs. Fc (ferrocenium/ferrocene) while perylene bisimides undergo a reduction process at potentials less negative than −1 V vs. Fc.49,51
The excited state energy for G2, evaluated from the intersection of the normalized absorption and emission spectra, resides at approximately 2.4 eV. Using the simplified Rehm–Weller equation52 it is possible to estimate the driving force for the photoinduced electron transfer process from the pillararene to the fluorophore which, despite all the used approximations, turns out to be strongly negative of about 0.7 eV.
In order to gain additional evidence, TAS experiments were carried out by recording the spectral changes in the near infrared and, as shown in Fig. S17, ESI,† within a few ps the formation of a peak at about 950 nm assignable (by comparison with literature data51) to the formation of the reduced G2 by the pillararene is observed and this species recombines to the fundamental state within about 100 ps, while an absorption tail is recovered on a longer time scale (about 1 ns).
Taken together, these results suggest that the excited state of G2 in the complexed form is quenched by electron transfer from the pillararene with a time constant of 8.6 ps and the charge-separated state so formed recombines with a time constant of about 85 ps.
The signal that evolves in a longer time scale can be attributed to the non-complexed portion of G2 or to a remote electron transfer effect from a pillararene cavity that is not directly complexed, but still in close proximity to the fluorophore. This species evolves with a time constant comparable to that of the residual luminescent excited state, which is found to be about 1 ns.
With the aim of testing the H/G2 system for a potential use as the sensing component in a biomedical device, a 3:1 H/G2 solution in CHCl3/TFE 97:3 v/v (excess H was added to make sure that G2 was conveniently quenched) was treated with increasing amounts of spermine (S). While the UV-vis absorption spectra showed no change whatsoever (Fig. 4), emission spectra showed a progressive increase of emission intensity (Fig. 4), indicating that G2 was being released from the pillararene cavities and it was regaining its emission properties, confirming that S may efficiently act as competitor for the bis-pillar[5]arene H, and therefore the H/G2 system may successfully act as a sensing agent for spermine detection.
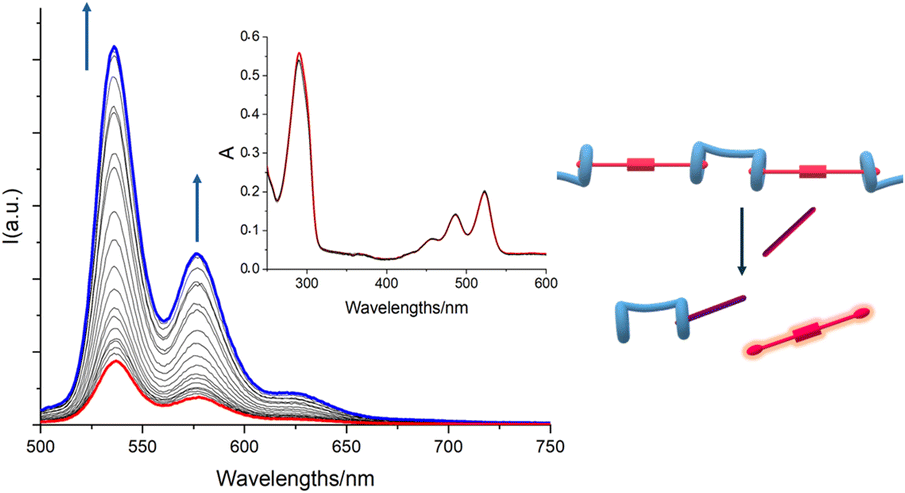 | ||
| Fig. 4 Absorption and emission (inset) titrations in CHCl3/TFE 97:3 v/v of a solution containing [G2] = 7.34 × 10−4 M and [H] = 2.19 × 10−3 M with a [S] = 2.3 × 10−4 M solution (titrant concentration from 7.73 × 10−7 to 1.57 × 10−5 M). The excitation wavelength was set at λex = 456 nm. | ||
H/G1/G2 Supramolecular copolymer
The construction of a functioning device requires the incorporation of the sparingly soluble H/G2 sensing system into an adaptive and easy-to-handle backbone. It was therefore envisaged that formulation of a three component H/G1/G2 copolymer could provide a material retaining both the H/G1 stability and H/G2 sensing properties. The copolymer was prepared by solid–liquid extraction of G2 with a solution of preformed H/G1 oligomers, containing a slight excess of H over G1 (1:0.9 host-to-guest ratio). Therefore, a [H] = 10 mM, [G1] = 9 mM in a CDCl3/TFE (97:3 v/v) solution was stirred for 2 h at room temperature in the presence of an excess of solid G2, and then filtered through a 0.45 μm PTFE membrane.
1H NMR spectra in Fig. 5 show that, after the extraction (spectrum c), the resonances of free G2 (spectrum d) underwent significant changes. The multiplet belonging to the perylene aromatic core (δ = 8.58–8.71 ppm) broadened, whereas the imidazole ring hydrogen atoms shifted up-field, merging with the broad resonance assigned to the pillararene aromatic hydrogen atoms. In addition, comparison between spectra (b) and (c) shows the appearance of a second set of resonances in the high field region consistent with the methylene groups of G2 included inside the aromatic cavities of the pillararene macrocycles (as demonstrated by a 2D TOCSY spectrum where the methylene chains of the three monomers are clearly identified, see Fig. S3 and S4, ESI†). Integration of the relevant peaks at δ = 4.43 ppm (for H), δ = –0.11 ppm (for G1) and δ = –0.28 ppm (for G2) in spectrum 5c confirms the 1:0.9:0.1 composition of the copolymer.
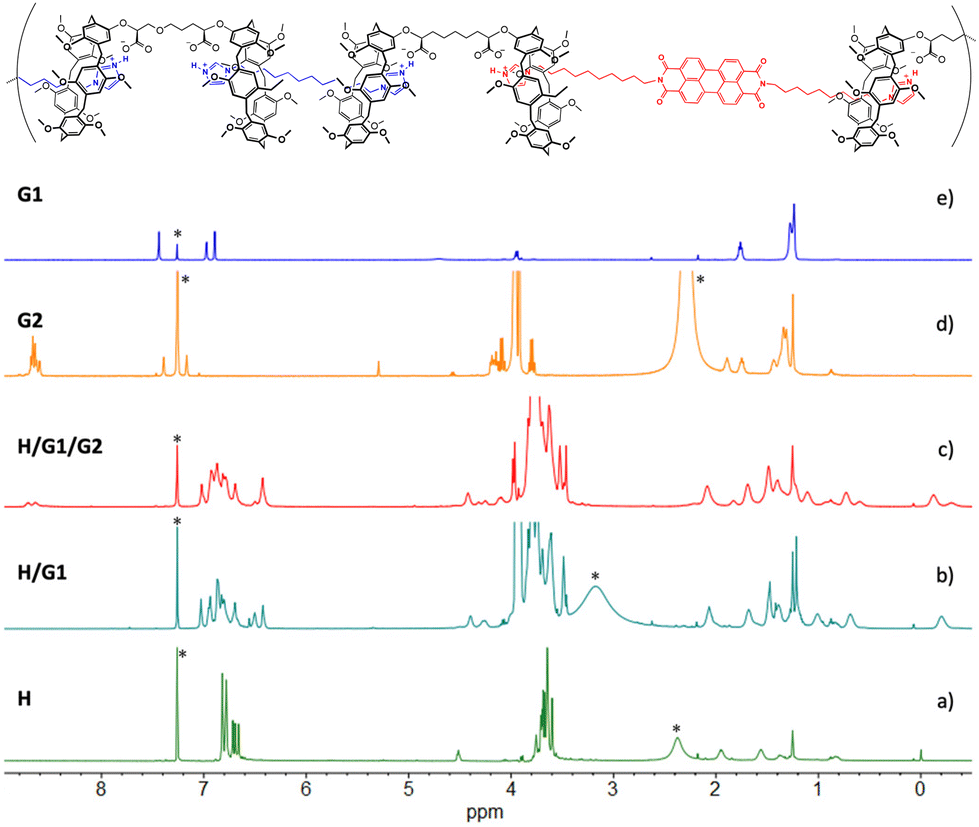 | ||
| Fig. 5
1H NMR spectra (500 MHz, 25 °C, CDCl3/TFE, 97:3, v/v) of: (a) [H] = 10 mM; (b) [H] = [G1] = 10 mM; (c) [H] = 10 mM, [G1] = 9 mM, [G2] = 1 mM; (d) [G2] = 0.6 mM. *Asterisks refer to residual solvent peaks. | ||
A series of DOSY NMR spectra provided conclusive evidence on the H/G1/G2 copolymer formation, rather than the coexistence of a mixture of H/G1 and H/G2 aggregates (Fig. 6). Careful inspection of Fig. 6c reveals that all the components of the copolymer diffuse with the same coefficient, indicating that they belong to the same aggregate and that they are not diffusing independently. Moreover, the diffusion coefficient of the H/G1/G2 copolymer (D = 1.47 ± 0.06 × 10−10 m2 s−1, at [H] = 10 mM, [G1] = 9 mM and [G2] = 1 mM in CDCl3/TFE, 97:3, v/v, Table 2, see also Fig. S12–14, ESI†) was found to be smaller than the one measured at the same concentration for the H/G1 polymer (1.83 ± 0.05 × 10−10 m2 s−1), showing that the addition of TFE (that improves G2 solubility) led to an increase of the average molecular mass from M = 24500 to 47300 amu. In fact, in line with previous studies,39,53 protic solvents promote host-to-guest proton transfer, with the result that electrostatic attraction between the charged moieties (COO−⋯HN+) increase the affinity of the pillararene cavity for the guest monomers.‡ Diacid H in this solvent mixture was found to diffuse with a D very close to that of the diester 3, which in turn was found to diffuse with same coefficient as in neat CDCl3.
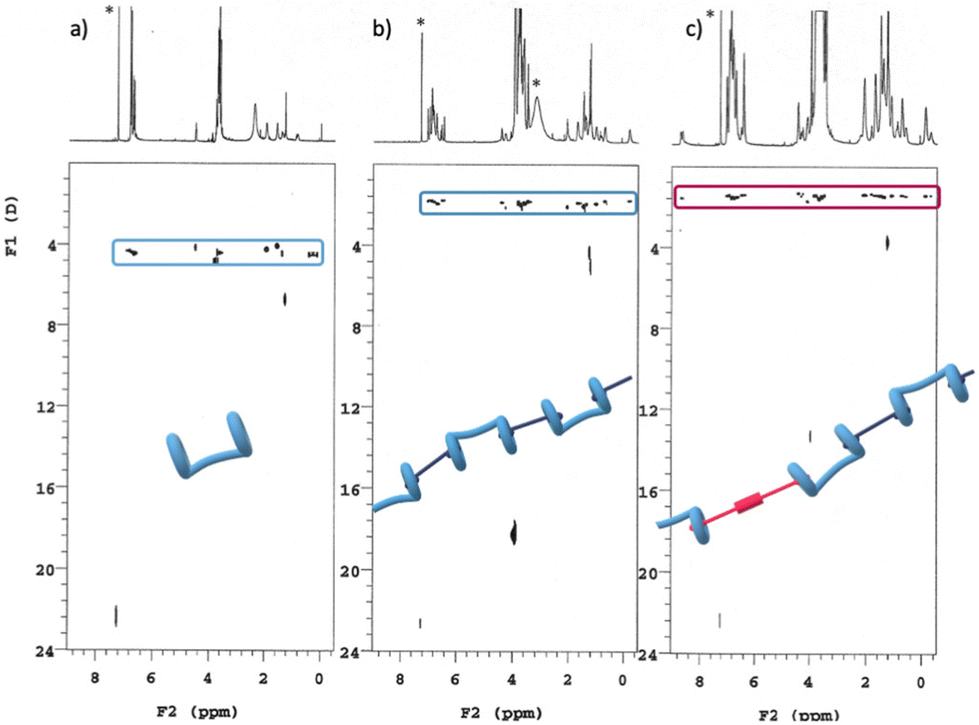 | ||
| Fig. 6 DOSY plots (500 MHz, 25 °C, CDCl3/TFE, 97:3, v/v) of (a): [H] = 10 mM; (b) [H] = [G1] = 10 mM; (c) [H] = 10 Mm, [G1] = 9 mM, [G2] = 1 mM. *Asterisks refer to residual solvent peaks. | ||
:3, v/v
| [H] mM | [G1] mM | [G2] mM | [3] mM | D (× 10−10 m2 s−1) | M (amu) |
|---|---|---|---|---|---|
| 10 | 10 | — | — | 1.83 ± 0.05 | 24500 |
| 5 | 4.5 | 0.5 | — | 1.91 ± 0.09 | 21600 |
| 10 | 9 | 1 | — | 1.47 ± 0.06 | 47300 |
| 10 | — | — | — | 4.41 ± 0.05 | 1780 |
| — | — | — | 10 | 4.50 ± 0.09 | 1684 |
The effect of spermine on the supramolecular copolymer H/G1/G2 was initially investigated by 1H NMR (Fig. 7). Addition of up to 2 eq. of S led to dramatic changes (spectrum b): the resonances for both included G1 and G2 methylene groups disappeared, and the peaks for the free imidazole ring(s) reappeared in the aromatic region of the spectrum. Likewise, no free spermine resonances were seen in spectrum b, indicating that it was recognized and complexed by the pillar[5]arene cavities of H.§
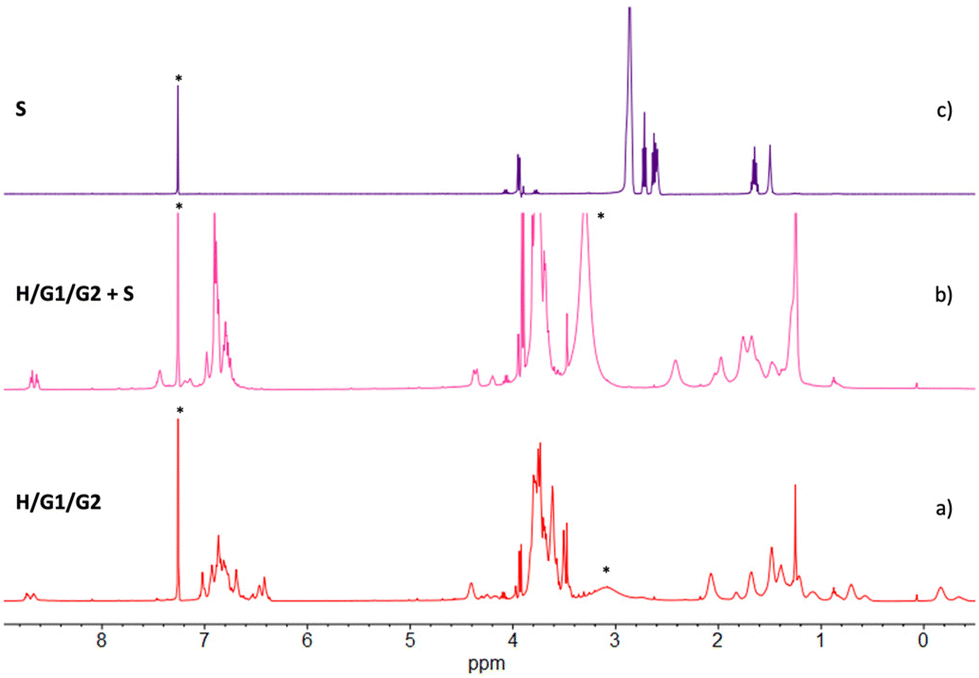 | ||
| Fig. 7
1H NMR spectra (500 MHz, 25 °C, CDCl3/TFE, 97:3, v/v) of (a) [H] = 5 mM, [G1] = 4.5 mM, [G2] = 0.5 mM; (b) [H] = 5 mM, [G1] = 4.5 mM, [G2] = 0.5 mM, [S] = 10 mM; (c) [S] = 10 mM. *Asterisks refer to residual solvent peaks. | ||
DOSY experiments confirmed the conclusions drawn from 1H NMR data (Fig. 8): addition of spermine results in the disassembly of the supramolecular copolymer. In fact, all the single species present in solution diffuse with a different self-diffusion coefficient, as expected for a mixture of compounds not interacting with each other and definitively not belonging to the same supramolecular species. In addition, the appearance in the TOCSY spectrum of the same mixture of spectra 7b and 8b (Fig. S5, ESI†) of seven cross-peaks (belonging to the central alkyl chain of a non-symmetrically complexed bis-pillararene) demonstrated that a 1:1 H/S is formed upon copolymer disaggregation.
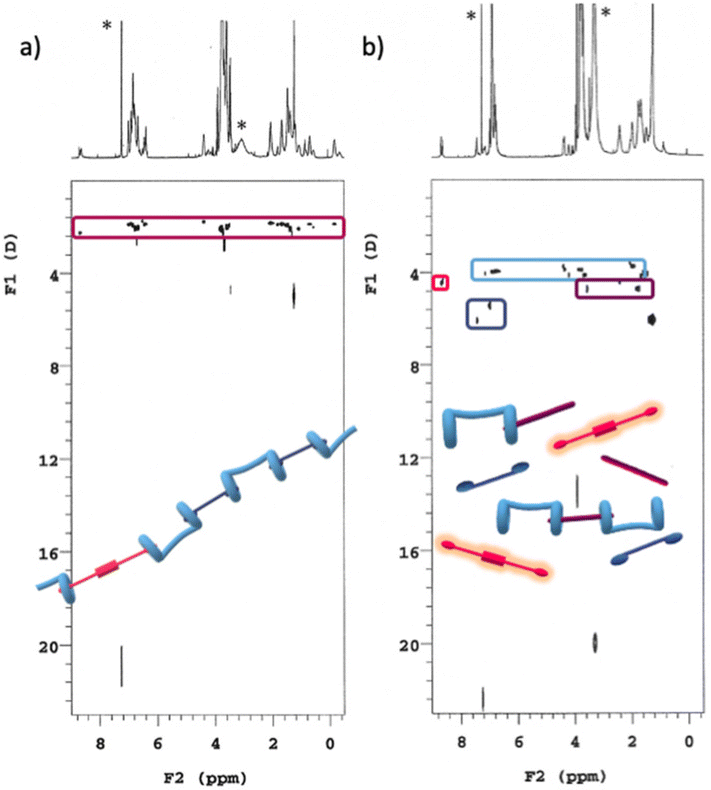 | ||
| Fig. 8 DOSY plots (500 MHz, 25 °C, CDCl3/TFE, 97:3, v/v) of (a) [H] = 5 mM, [G1] = 4.5 mM, [G2] = 0.5 mM; (b) [H] = 5 mM, [G1] = 4.5 mM, [G2] = 0.5 mM, [S] = 10 mM. *Asterisks refer to residual solvent peaks. | ||
The conclusive evidence on the ability of the H/G1/G2 copolymer to respond to and to reveal spermine S was provided by UV-vis and fluorescence data (Fig. 9). Again, while absorption spectra do not change significantly upon addition of S, emission spectra sharply increase in intensity when a 9:1 S/G2 molar ratio (e.g. 1:1 S/G1) is reached, suggesting that the displacement of G2 begins only after G1 is fully released. This observation also showed that upon release from the copolymer backbone, G2 fluorescence is switched on, confirming that the optical properties of the H/G2 sensing system are fully retained even when incorporated in the copolymer.
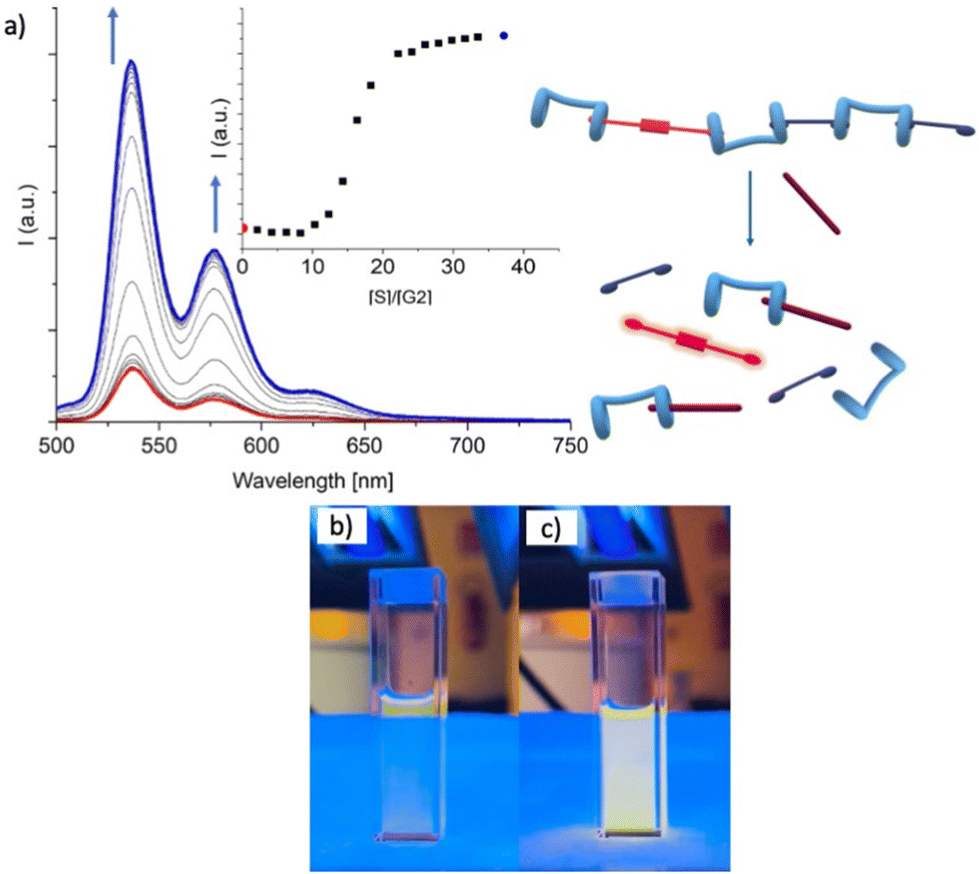 | ||
| Fig. 9 (a) Emission titration in CHCl3/TFE 97:3 v/v of a solution containing [G1] = 1.01 × 10−5 M, [G2] = 1.13 × 10−6 M and [H] = 1.13 × 10−5 M with a [S] = 1.54 × 10−3 M solution (titrant concentration from 6.0 × 10−6 to 1.0 × 10−4 M). The excitation wavelength was set at λex = 456 nm. Inset: emission intensity as a function of [S]/[G2] ratio. Pictures (b) and (c) show the cuvette at the beginning and at the end of the titration. | ||
Such a progressive displacement was explained by a series of 1H NMR titrations that, combined with the emission titrations, allowed us to quantify the strength, in terms of formation constants, of the different diads (AB) and triads (ABA, BAB) contained in the copolymer backbone (i.e., H/G1, H/G2, G1/H/G1, G2/H/G2, H/G1/H and H/G2/H). Data treatment provided the formation constants reported in Table 3 (for full details see Tables S1 and S2, ESI†).
:3 v/v or 1H NMR titrations in CDCl3/TFE 97:3 v/v (see Tables S1, S2, Fig. S15, S16 and S18–S20, ESI, for full details†)
| Complex | logβ |
logK |
K (M−1) |
|---|---|---|---|
| a Determined by 1H NMR. b Determined by emission data. c These constants refer to HX + H → H2X or HX + X → HX2 equilibria (X = G1, G2 or S). | |||
| H/G1a | 6.03 ± 0.01 | 6.03 ± 0.01 | 1.07 ± 0.05 × 106 |
| H/G2b | 6.42 ± 0.01 | 6.42 ± 0.01 | 2.69 ± 0.06 × 106 |
| H/G1/Ha | 8.51 ± 0.07 | 2.48 ± 0.08 | 3.02 ± 0.56 × 102c |
| G1/H/G1a | 11.10 ± 0.02 | 5.07 ± 0.03 | 1.17 ± 0.08 × 105c |
| H/G2/Hb | 12.66 ± 0.02 | 6.24 ± 0.03 | 1.74 ± 0.12 × 106c |
| G2/H/G2b | 10.80 ± 0.02 | 4.40 ± 0.03 | 2.51 ± 0.17 × 104c |
| H/Sa | 6.39 ± 0.01 | 6.39 ± 0.01 | 2.45 ± 0.06 × 106 |
| S/H/Sa | 10.08 ± 0.01 | 3.69 ± 0.02 | 4.90 ± 0.20 × 103c |
K for the H/G1 and H/G2 complexes are very close, indicating that the 1:1 interaction between the pillararene cavity and the alkylimidazole guest moiety – after host-to-guest proton transfer – is not significantly influenced by the remaining parts of the G1 and G2 guest monomers.¶ Little difference is also seen for the H-centered G1/H/G1 and G2/H/G2 triads (1.17 ± 0.08 × 105 and 2.51 ± 0.17 × 104, respectively), showing that the complexation of two alkylimidazole by a single ditopic H monomer is marginally influenced by the recognition events taking place in the two pillararene cavities. On the other hand, a remarkable difference is seen for the ABA (guest-centered) triads. H/G1/H is formed from H/G1 with a K = 3.02 ± 0.56 × 102 M−1, whereas H/G2/H is formed from H/G2 with a K = 1.74 ± 0.12 × 106 M−1, revealing that the ‘weak’ link where the attack of the spermine analyte takes place is the H/G1/H unit, probably because G1 is too short to avoid steric repulsion between the pillararene moieties of two different H units encircling the two ends of a G1 monomer (H/G1/H has a much lower percentage of formation than H/G2/H, see also the distribution diagrams in Fig. S19 and S20, ESI†). This evidence also explains the reason why G2 is released only after all G1 molecules have been replaced by S. Furthermore, even though S binds to H with a K = 2.45 ± 0.06 × 106, comparable to the one of the H/G1 and H/G2 complexes, smoothly displaces both G1 and G2, being primary amines by far more basic than imidazole.54–57 Remarkably, the chemical shifts observed during the titrations and the calculated ones are in excellent agreement, (Fig. S18, ESI†).
Mass spectrometry experiments allowed to further elucidate the interactions between spermine and the supramolecular copolymer. Analysis of the ESI-MS spectra corroborated data previously acquired by DOSY NMR (Fig. 10). In particular, the observation of peaks at m/z 1961.1 and 984.9, corresponding to the [H + G1 + H]+ ion and the [2H + 2G1 + H2O + 4H]4+ multicharged ion, as well as m/z 2461.3 and 1234.9, corresponding to the [H + G2 + H]+ and [2H + 2G2 + H2O + 4H]4+ ions, respectively, confirmed the formation of 1:1 and 2:2 complexes within the two distinct polymer systems (see Fig. S21–S24 for details†). The same species were observed in the spectrum of the H/G1/G2 copolymer.||
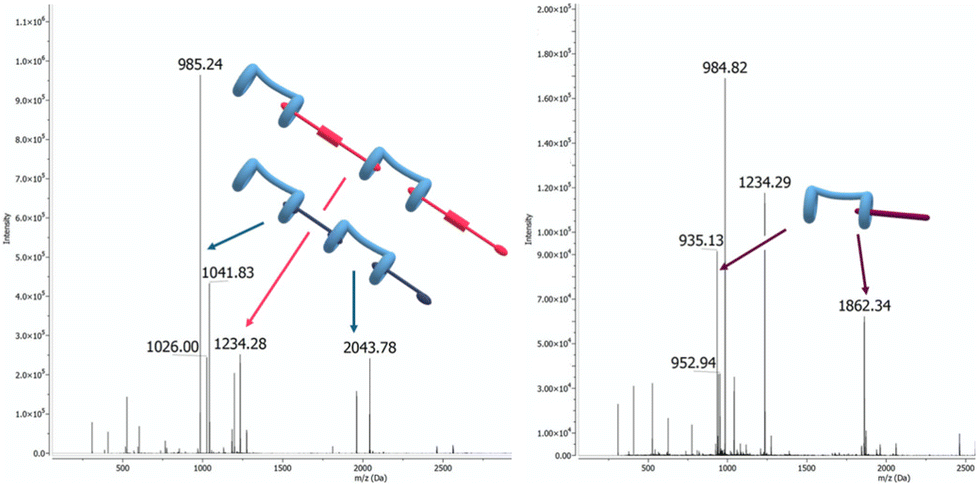 | ||
| Fig. 10 ESI-MS(+) spectra of left: H/G1/G2 ([H] = 10 μM, [G1] = 9 μM, [G2] = 1 μM), and right: H/G1/G2 treated with S ([S] = 10 μM). | ||
Furthermore, the interaction between H and spermine (S) was investigated by ESI-MS,58,59 and the formation of 1:1 H/S and larger complexes was confirmed by the observation of peaks at m/z 1862.4 and 935.4, corresponding to the [H + S + H]2+ and [2H + 2S + H2O + 4H]4+ ions, respectively.|| The displacement of G2 was demonstrated by treating the H/G1/G2 supramolecular copolymer with S. Subsequent analysis, under identical experimental conditions, clearly showed H/S complex formation.
Atomic Force Microscopy (AFM) and Raman spectroscopy shed further light on the ability of the H/G1/G2 copolymer to reveal spermine even in the solid state.60,61 As a preliminary screening, Raman spectra were collected for the three components and the copolymer (Fig. 11), revealing that both G2 and H/G1/G2 copolymer display between 1300 and 1600 cm−1 the typical Raman bands of perylene bisimide moieties (corresponding to C–H bending and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching, respectively),62,63 thus confirming that, also in the film, G2 is still part of the copolymer. Emission spectra recorded on G2 (which forms small clusters upon evaporation, Fig. 12a), and H/G1/G2 films (Fig. 12b) showed that the perylene bisimide fluorophore is quenched in the copolymer film but it provides an intense emission when cast as the free species.
C stretching, respectively),62,63 thus confirming that, also in the film, G2 is still part of the copolymer. Emission spectra recorded on G2 (which forms small clusters upon evaporation, Fig. 12a), and H/G1/G2 films (Fig. 12b) showed that the perylene bisimide fluorophore is quenched in the copolymer film but it provides an intense emission when cast as the free species.
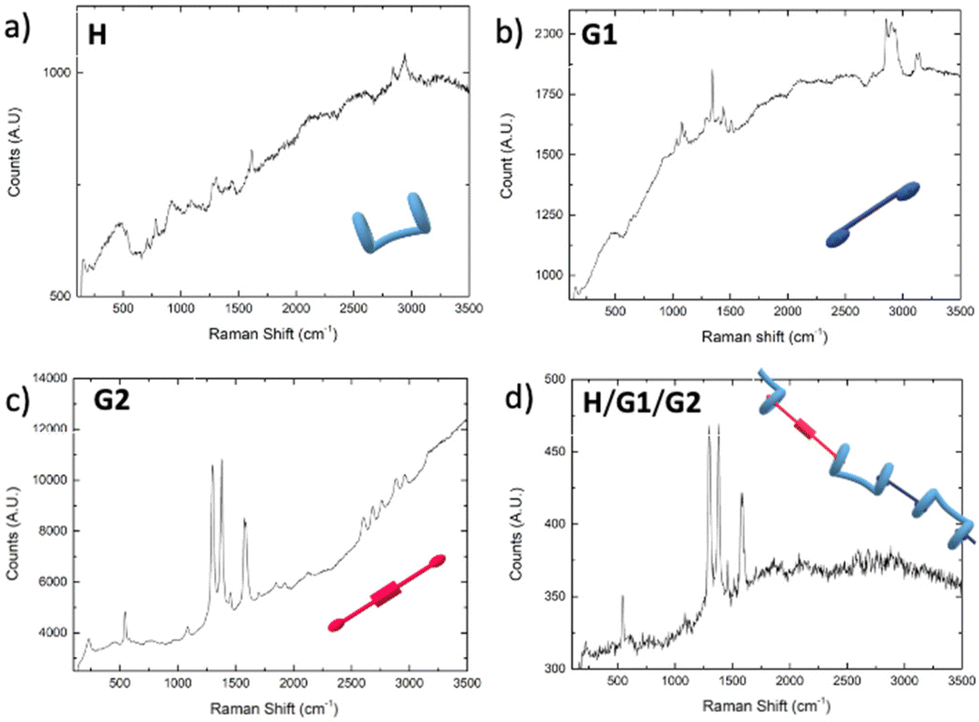 | ||
| Fig. 11 Raman spectra collected from solids deposited on silica surface from solutions (CHCl3/TFE, 97:3, v/v): (a) [H] = 3 mM; (b) [G1] = 2.7 mM; (c) [G2] = 0.3 mM; (d) [H] = 3 mM, [G1] = 2.7 mM, [G2] = 0.3 mM. | ||
 | ||
| Fig. 12 (a) Photoluminescence observed upon irradiation at λex = 472 nm of a cluster of G2. (b) AFM topography image of the grid obtained upon spin-coating of a [H] = 3 mM, [G1] = 2.7 mM, [G2] = 0.3 mM solution (CHCl3/TFE, 97:3, v/v) onto a silica surface. | ||
Finally, deposition of a solution containing the copolymer H/G1/G2 containing 2 eq. of spermine S produced a grid-like film in which G2 fluorescence has been restored (Fig. 13), demonstrating the viability of the supramolecular copolymer for the incorporation into sensing devices.
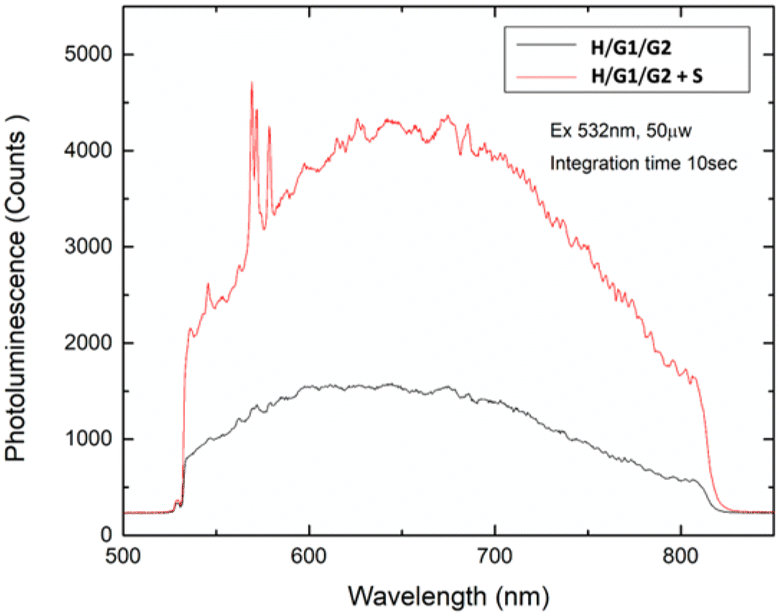 | ||
| Fig. 13 Emission spectra recorded on solid films obtained from [H] = 3 mM, [G1] = 2.7 mM, [G2] = 0.3 mM (black line), and [H] = 3 mM, [G1] = 2.7 mM, [G2] = 0.3 mM, [S] = 6 mM (red line) solutions. | ||
Conclusions
Data presented in this paper show that the supramolecular copolymer herein self-assembled from three monomers – a bis-pillar[5]arene dicarboxylic acid (H), a dodecanediyl-bis-imidazole (G1) and a bis-N,N’-imidazoledecyl-perylene bisimide (G2) retained both the solubility and adaptability of the H/G1 polymer and the OFF/ON fluorescence detection ability of the H/G2 system. This material was shown, by a variety of techniques, to be able to efficiently detect the cancer marker, spermine (S) in solution, as a result of a progressive displacement/disassembly process that restores the fluorescence of the perylene bis-imide signalling unit.The results presented demonstrate a key concept: formulation of supramolecular copolymers opens up avenues for the facile development of complex materials that may conjugate properties from different components, avoiding tiresome syntheses in favour of an easy approach of readily-made monomers ‘pick-and-mix.’
Further investigations will be aimed at the development of a portable sensing device for the direct detection of spermine in biological fluids, and to the assessment of the supramolecular copolymer potential as a carrier in drug-delivery.
Author contributions
Conceptualization: G. G., I. P.; data curation: A. I., F. P.; formal analysis: A. I., M. Mi., A. G., F. P., S. P.; funding acquisition: G. G., M. F. P., A. N.; investigation: M. Ma., D. C., F. M., M. Mi., V. G., S. P.; project administration: G. G., I. P., F. P.; resources: M. Ma., D. C.; supervision: I. P., M. F. P., A. N., F. P., A. G.; validation: I. P., F. P.; visualization: D. C., M. Ma.; writing – original draft: G. G., I. P., F. P.; writing – review & editing: G. G., M. F. P.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially funded by the European Union (NextGeneration EU), through the MUR-PNRR project SAMOTHRACE (SiciliAn MicronanOTecH Research And Innovation CEnter, ECS00000022).References
- U. S. Schubert, G. R. Newkome and A. Winter, Supramolecular Polymers and Assemblies – From Synthesis to Properties and Application, Wiley-VCH, Weinheim, 2021 Search PubMed.
- A. J. Savyasachi, O. Kotova, S. Shanmugaraju, S. J. Bradberry, G. M. O'Máille and T. Gunnlaugsson, Supramolecular Chemistry: A Toolkit for Soft Functional Materials and Organic Particles, Chem, 2017, 3, 764–811 CAS.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Supramolecular Polymers: Historical Development, Preparation, Characterization, and Functions, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed.
- J.-M. Lehn, Perspectives in Chemistry—Aspects of Adaptive Chemistry and Materials, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Supramolecular Polymerization, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- P. K. Hakshim, J. Bergueiro, E. W. Meijer and T. Aida, Supramolecular Polymerization: A Conceptual Expansion for Innovative Materials, Prog. Polym. Sci., 2020, 105, 101250 CrossRef.
- J. Matern, Y. Dorca, L. Sańchez and G. Fernańdez, Revising complex supramolecular polymerization under kinetic and thermodynamic control, Angew. Chem., Int. Ed., 2019, 58, 16730–16740 CrossRef CAS.
- F. Xu and B. L. Feringa, Photoresponsive Supramolecular Polymers: From Light-Controlled Small Molecules to Smart Materials, Adv. Mater., 2023, 35, 2204413 CrossRef CAS PubMed.
- A. D. O'Donnell, S. Salimi, L. R. Hart, T. S. Babra, B. W. Greenland and W. Hayes, Applications of supramolecular polymer networks, React. Funct. Polym., 2022, 172, 105209 CrossRef.
- D. Wang, G. Tong, R. Dong, Y. Zhou, J. Shen and X. Zhu, Self-assembly of supramolecularly engineered polymers and their biomedical applications, Chem. Commun., 2014, 50, 11994–12017 RSC.
- R. Dong, Y. Zhou, X. Huang, X. Zhu, Y. Lu and J. Shen, Functional Supramolecular Polymers for Biomedical Applications, Adv. Mater., 2015, 27, 498–526 CrossRef CAS PubMed.
- X. Li, M. Shen, J. Yang, L. Liu and Y.-W. Yang, Pillararene-Based Stimuli-Responsive Supramolecular Delivery Systems for Cancer Therapy, Adv. Mater., 2024, 36, 2313317 CrossRef CAS.
- N. Herzer, H. Guneysu, D. J. D. Davies, D. Yildirim, A. R. Vaccaro, D. J. Broer, C. W. M. Bastiaansen and A. P. H. J. Schenning, Printable Optical Sensors Based on H-Bonded Supramolecular Cholesteric Liquid Crystal Networks, J. Am. Chem. Soc., 2012, 134, 7608–7611 CrossRef CAS.
- B. Gole, S. Shanmugaraju, A. K. Bar and P. S. Mukherjee, Supramolecular polymer for explosives sensing: role of H-bonding in enhancement of sensitivity in the solid state, Chem. Commun., 2011, 47, 10046–10048 RSC.
- B. Gole, W. Song, M. Lackinger and P. S. Mukherjee, Explosives Sensing by Using Electron-Rich Supramolecular Polymers: Role of Intermolecular Hydrogen Bonding in Significant Enhancement of Sensitivity, Chem. – Eur. J., 2014, 20, 13662–13680 CrossRef CAS PubMed.
- T. Hirao, S. Kishino and T. Haino, Supramolecular chiral sensing by supramolecular helical polymers, Chem. Commun., 2023, 59, 2421–2424 RSC.
- Y. Sagara, M. Karman, A. Seki, M. Pannipara, N. Tamaoki and C. Weder, Rotaxane-Based Mechanophores Enable Polymers with Mechanically Switchable White Photoluminescence, ACS Cent. Sci., 2019, 5, 874–881 CrossRef CAS.
- A. E. Früh, F. Artoni, R. Brighenti and E. Dalcanale, Strain Field Self-Diagnostic Poly(dimethylsiloxane) Elastomers, Chem. Mater., 2017, 29, 7450–7457 CrossRef.
- L. Albertazzi, N. van der Veeken, M. B. Baker, A. R. A. Palmans and E. W. Meijer, Supramolecular copolymers with stimuli- responsive sequence control, Chem. Commun., 2015, 51, 16166–16168 RSC.
- H. Frisch, E.-C. Fritz, F. Stricker, L. Schmüser, D. Spitzer, T. Weidner, B. J. Ravoo and P. Besenius, Kinetically Controlled Sequential Growth of Surface-Grafted Chiral Supramolecular Copolymers, Angew. Chem., Int. Ed., 2016, 55, 7242–7246 CrossRef CAS.
- H. Li, Y. Yang, F. Xu, Z. Duan, R. Li, H. Wen and W. Tian, Sequence-controlled supramolecular copolymer constructed by self-sorting assembly of multiple noncovalent interactions, Org. Chem. Front., 2021, 8, 1117–1124 RSC.
- A. Sarkar, R. Sasmal, C. Empereur-mot, D. Bochicchio, S. V. K. Kompella, K. Sharma, S. Dhiman, B. Sundaram, S. S. Agasti, G. M. Pavan and S. J. George, Self-Sorted, Random, and Block Supramolecular Copolymers via Sequence Controlled, Multicomponent Self-Assembly, J. Am. Chem. Soc., 2020, 142, 7606–7617 CrossRef CAS PubMed.
- N. Roy, V. Schädler and J.-M. Lehn, Supramolecular Polymers: Inherently Dynamic Materials, Acc. Chem. Res., 2024, 57, 349–361 CrossRef CAS.
- S. Y. Dong, B. Zheng, F. Wang and F. Huang, Supramolecular Polymers Constructed from Macrocycle-Based Host−Guest Molecular Recognition Motifs, Acc. Chem. Res., 2014, 47, 1982–1994 CrossRef CAS PubMed.
- T. Haino, Designer supramolecular polymers with specific molecular recognitions, Polym. J., 2019, 51, 303–318 CrossRef CAS.
- A. Harada, Supramolecular Polymers (Host-Guest Interactions), in Encyclopedia of Polymeric Nanomaterials, ed. S. Kobayashi and K. Müllen, Springer, Berlin, Heidelberg, 2013, DOI:10.1007/978-3-642-36199-9_54-1.
- B. Zheng, F. Wang, S. Dong and F. Huang, Supramolecular polymers constructed by crown ether-based molecular recognition, Chem. Soc. Rev., 2012, 41, 1621–1636 RSC.
- Z. Duan, F. Xu, X. Huang, Y. Qian, H. Li and W. Tian, Crown Ether-Based Supramolecular Polymers: From Synthesis to Self-Assembly, Macromol. Rapid Commun., 2022, 43, 2100775 CrossRef CAS.
- A. Harada, Y. Takashima and H. Yamaguchi, Cyclodextrin-based supramolecular polymers, Chem. Soc. Rev., 2009, 38, 875–882 RSC.
- U. Schubert, A. Winter and G. Newkome, Supramolecular Polymers, Based on the Host–Guest Chemistry of Calixarenes, in Supramolecular Polymers and Assemblies, ed. U. Schubert, A. Winter and G. Newkome, Wiley VCH, 1st edn, 2021 Search PubMed.
- D.-S. Guo and Y. Liu, Calixarene-based supramolecular polymerization in solution, Chem. Soc. Rev., 2012, 41, 5907–5921 RSC.
- T. Ogoshi, T. A. Yamagishi and Y. Nakamoto, Pillar-Shaped Macrocyclic Hosts Pillar[n]arenes: New Key Players for Supra- molecular Chemistry, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed.
- H. Li, Y. Yang, F. Xu, T. Liang, H. Wen and W. Tian, Pillararene-based supramolecular polymers, Chem. Commun., 2019, 55, 271–285 RSC.
- T. Kakuta, T. Yamagishi and T. Ogoshi, Stimuli-Responsive Supramolecular Assemblies Constructed from Pillar[n]arenes, Acc. Chem. Res., 2018, 51, 1656–1666 CrossRef CAS.
- N. Manganaro, I. Pisagatti, A. Notti, M. F. Parisi and G. Gattuso, Self-sorting assembly of a calixarene/crown ether polypseudorotaxane gated by ion-pairing, New J. Chem., 2019, 43, 7936–7940 RSC.
- N. Manganaro, I. Pisagatti, A. Notti, A. Pappalardo, S. Patanè, N. Micali, V. Villari, M. F. Parisi and G. Gattuso, Ring/Chain Morphology Control in Overall-Neutral, Internally Ion-Paired Supramolecular Polymers, Chem. – Eur. J., 2018, 24, 1097–1103 CrossRef CAS PubMed.
- C. Capici, Y. Cohen, A. D'Urso, G. Gattuso, A. Notti, A. Pappalardo, S. Pappalardo, M. F. Parisi, R. Purrello, S. Slovak and V. Villari, Anion-Assisted Supramolecular Polymerization: From Achiral AB-Type Monomers to Chiral Assemblies, Angew. Chem., Int. Ed., 2011, 50, 11956–11961 CrossRef CAS PubMed.
- S. Pappalardo, V. Villari, S. Slovak, Y. Cohen, G. Gattuso, A. Notti, A. Pappalardo, I. Pisagatti and M. F. Parisi, Counterion-Dependent Proton-Driven Self-Assembly of Linear Supramolecular Oligomers Based on Amino-Calix[5]arene Building Blocks, Chem. – Eur. J., 2007, 13, 8164–8173 CrossRef CAS PubMed.
- A. Notti, I. Pisagatti, F. Nastasi, S. Patanè, M. F. Parisi and G. Gattuso, Stimuli-Responsive Internally Ion-Paired Supramolecular Polymer Based on a Bis-pillar[5]arene Dicarboxylic Acid Monomer, J. Org. Chem., 2021, 86, 1676–1684 CrossRef CAS PubMed.
- C. Li, S. C. J. Li, K. Han, M. Xu, B. Hu, Y. Yu and X. Jia, Novel neutral guest recognition and interpenetrated complex formation from pillar[5]arenes, Chem. Commun., 2011, 47, 11294–11296 RSC.
- B. Xia, B. Zheng, C. Han, S. Dong, M. Zhang, B. Hu, Y. Yu and F. Huang, A novel pH-responsive supramolecular polymer constructed by pillar[5]arene-based host–guest interactions, Polym. Chem., 2013, 4, 2019–2024 RSC.
- J. Chen, H. Ni, Z. Meng, J. Wang, X. Huang, Y. Dong, C. Sun, Y. Zhang, L. Cui, J. Li, X. Jia, Q. Meng and C. Li, Supramolecular trap for catching polyamines in cells as an anti-tumor strategy, Nat. Commun., 2019, 10, 3546 CrossRef.
- T. Ogoshi, K. Demachi, K. Kitajima and T. Yamagishi, Monofunctionalized pillar[5]arenes: synthesis and supramolecular structure, Chem. Commun., 2011, 47, 7164–7166 RSC.
- C. T. Nieto, M. M. Salgado, S. H. Domínguez, D. Díez and N. M. Garrido, Rapid access with diversity to enantiopure flexible PNA monomers following asymmetric orthogonal strategies, Tetrahedron: Asymmetry, 2014, 25, 1046–1060 CrossRef CAS.
- J. Xu, Y. Chen, L. Wu, C. Tung and Q. Yang, Dynamic Covalent Bond Based on Reversible Photo [4 + 4] Cycloaddition of Anthracene for Construction of Double-Dynamic Polymers, Org. Lett., 2013, 15, 6148–6151 CrossRef CAS.
- J. B. Press, W. B. Wright, Jr., P. S. Chan, J. W. Marsico, M. F. Haug, J. Tauber and A. S. Tomcufcik, Thromboxane synthetase inhibitors and antihypertensive agents. 2. N-[(1H-imidazol-1-yl)alkyl]-1H-isoindole-1,3(2H)-diones and N-[(1H-1,2,4-triazol-1-yl)alkyl]-1H-isoindole-1,3(2H)-diones as unique antihypertensive agents, J. Med. Chem., 1986, 29, 816–819 CrossRef CAS.
- A. R. Waldeck, P. W. Kuchel, A. J. Lennon and B. E. Chapman, Prog. NMR diffusion measurements to characterise membrane transport and solute binding, Nucl. Magn. Reson. Spectrosc., 1997, 30, 39–68 CrossRef CAS.
- B. A. Jones, M. J. Ahrens, M.-H. Yoon, A. Facchetti, T. J. Marks and M. R. Wasielewski, High–Mobility Air–Stable n–Type Semiconductors with Processing Versatility: Dicyanoperylene–3,4:9,10−bis(dicarboximides), Angew. Chem., Int. Ed., 2004, 43, 6363–6366 CrossRef CAS.
- N. Pearce, K. E. A. Reynolds, S. Kayal, X. Z. Sun, E. S. Davies, F. Malagreca, C. J. Schürmann, S. Ito, A. Yamano, S. P. Argent, M. W. George and N. R. Champness, Selective photoinduced charge separation in perylenediimide-pillar[5]arene rotaxanes, Nat. Commun., 2022, 13, 415 CrossRef CAS.
- I. Pisagatti, D. Crisafulli, A. Pappalardo, G. Trusso Sfrazzetto, A. Notti, F. Nastasi, M. F. Parisi, N. Micali, G. Gattuso and V. Villari, Photoinduced Electron Transfer in Host-Guest Interactions of a Viologen Derivative with a Didansyl-Pillar[5]arene, Mater. Today Chem., 2022, 24, 100841 CrossRef CAS.
- F. M. Toma, F. Puntoriero, T. V. Pho, M. La Rosa, Y.-S. Jun, B. J. T. D. Villers, J. Pavlovich, G. D. Stucky, S. Campagna and F. Wudl, Polyimide Dendrimers Containing Multiple Electron Donor–Acceptor Units and Their Unique Photophysical Properties, Angew. Chem., Int. Ed., 2015, 54, 6775–6779 CrossRef CAS.
- D. Rehm and A. Weller, Kinetics of Fluorescence Quenching by Electron and H-Atom Transfer, Ber. Bunsen-Ges., 1969, 73, 834–839 CrossRef CAS.
- G. Gattuso, D. Crisafulli, M. Milone, F. Mancuso, I. Pisagatti, A. Notti and M. F. Parisi, Proton transfer mediated recognition of amines by ionizable macrocyclic receptors, Chem. Commun., 2022, 58, 10743–10756 RSC.
- R. L. Benoit, D. Boulet, L. Séguin and M. Fréchette, Protonation of purines and related compounds in dimethylsulfoxide and water, Can. J. Chem., 1985, 63, 1228–1232 CrossRef CAS.
- B. N. Palmer and H. K. J. Powell, Polyamine Complexes with Seven-membered Chelate Rings: Complex Formation of 3-Azaheptane-1,7-diamine, 4-Azaoctane-1,8-diamine (Spermidine), and 4,9-Diazadodecane-1,12-diamine (Spermine) with Copper(II) and Hydrogen Ions in Aqueous Solution, J. Chem. Soc., Dalton Trans., 1974, 2089–2092 RSC.
- B. Barszcz, M. Gabryszewski, J. Kulig and B. Lenatcik, Potentiometric Studies on Complexes of Silver(I) in Solutions. Part 2. Correlation between the Stability of the Silver(I)-Azole Complexes and the Ligand Basicity, J. Chem. Soc., Dalton Trans., 1986, 2025–2028 RSC.
- M. J. Power, D. T. J. Morris, I. J. Vitorica-Yrezabal and D. A. Leigh, Compact Rotaxane Superbases, J. Am. Chem. Soc., 2023, 145, 8593–8599 CrossRef CAS PubMed.
- Z.-Y. Li, Y. Zhang, C.-W. Zhang, L.-J. Chen, C. Wang, H. Tan, Y. Yu, X. Li and H.-B. Yang, Cross-Linked Supramolecular Polymer Gels Constructed from Discrete Multi-pillar[5]arene Metallacycles and Their Multiple Stimuli-Responsive Behavior, J. Am. Chem. Soc., 2014, 136, 8577–8589 CrossRef CAS PubMed.
- J. Liu, X. Jiang, X. Huang, L. Zou and Q. Wang, Photo-responsive supramolecular polymer based on a CB[5] analogue, Colloid Polym. Sci., 2016, 294, 1243–1249 CrossRef CAS.
- R. Dobrawa, M. Lysetska, P. Ballester, M. Grüne and F. Würthner, Fluorescent Supramolecular Polymers: Metal Directed Self-Assembly of Perylene Bisimide Building Blocks, Macromolecules, 2005, 38, 1315–1325 CrossRef CAS.
- Y. Yamauchi, K. Yamada, N. N. Horimoto and Y. Ishida, Supramolecular self-assembly of an ABA-Triblock bottlebrush polymer: Atomic-force microscopy visualization of discrete oligomers, Polymer, 2017, 120, 68–72 CrossRef CAS.
- A. Blacha-Grzechnik, A. Drewniak, K. Z. Walczak, M. Szindler and P. Ledwon, Efficient generation of singlet oxygen by perylene diimide photosensitizers T covalently bound to conjugate polymers, J. Photochem. Photobiol., A, 2020, 388, 112161 CrossRef CAS.
- S. Winters, C. Backes, N. C. Berner, R. Mishra, K. C. Dumbgen, M. Hegner, A. Hirsch and G. S. Duesberg, On-surface derivatisation of aromatic molecules on graphene: the importance of packing density, Chem. Commun., 2015, 51, 16778–16781 RSC.
- C. Capici, G. Gattuso, A. Notti, M. Parisi, S. Pappalardo, G. Brancatelli and S. Geremia, Selective Amine Recognition Driven by Host−Guest Proton Transfer and Salt Bridge Formation, J. Org. Chem., 2012, 77, 9668–9675 CrossRef CAS PubMed.
- G. Brancatelli, G. Gattuso, S. Geremia, A. Notti, S. Pappalardo, M. F. Parisi and I. Pisagatti, Probing the Inner Space of Salt-Bridged Calix[5]arene Capsules, Org. Lett., 2014, 16, 2354–2357 CrossRef CAS PubMed.
- G. Brancatelli, G. Gattuso, S. Geremia, N. Manganaro, A. Notti, S. Pappalardo, M. F. Parisi and I. Pisagatti, α,ω-Alkanediyldiammonium dications sealed within calix[5]arene capsules with a hydrophobic bayonet-mount fastening, CrystEngComm, 2015, 17, 7915–7921 RSC.
- G. Brancatelli, G. Gattuso, S. Geremia, N. Manganaro, A. Notti, S. Pappalardo, M. F. Parisi and I. Pisagatti, Encapsulation of biogenic polyamines by carboxylcalix[5]arenes: when solid-state design beats recognition in solution, CrystEngComm, 2016, 18, 5012–5016 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedures, synthetic procedure, additional NMR, PM6, TASs, ESI-MS figures, distribution diagrams, additional tables. See DOI: https://doi.org/10.1039/d4qo01470g |
| ‡ Previous studies39 on the self-assembly of supramolecular polymers from analogous bis-pillar[5]arene dicarboxylic acids and α,ω-diaminoalkanes showed that, upon recognition, proton transfer from the host to the guest64 takes place if a protic solvent (e.g., trifluoroethanol (TFE), 3–10% v/v) is added to the chloroform solution containing both monomers, whereas no proton transfer can be observed in neat chloroform. In the present case, addition of trifluoroethanol to a H/G1 solution in CDCl3 result in the COOH group of the host protonating the basic imidazole nitrogen atom of the guest, as confirmed by the tell-tale upfield shift (from δ = 4.47 to 4.40 ppm) of the resonance belonging to the CH group adjacent to the carboxyl moiety (Fig S1, ESI,† shows the optimized geometry of the repeating unit of the H/G1 aggregate). This topic has been extensively reviewed recently.53 |
| § Linear (poly)amines are efficiently recognized by carboxyl-bearing receptors. Host-to-guest proton transfer has been demonstrated as a powerful tool for the formation of internally ion-paired complexes.53,65–67 |
| ¶ The association constant of decamethyl-pillar[5]arene and 1-decylimidazole was reported to be 2.3 ± 0.2 × 102 M−1, whereas between decamethyl-pillar[5]arene and 1-decylimidazolium trifluoroacetate was 1.0 ± 0.3 × 104 M−1 in chloroform.41 Proton-transfer mediated recognition explain the higher stability (>106 M−1) of the H/G1 and H/G2 complexes. |
| || Given the tendency of these polymeric systems to aggregate, the formation of larger multicharged ions (e.g., 2H + 2G1 + 2H]2+) cannot be ruled out. |
| This journal is © the Partner Organisations 2024 |