
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC–H acylation as an enabling tool to tag phenolic drugs†
Carlota
Girón-Elola
 and
Arkaitz
Correa
*
and
Arkaitz
Correa
*
Department of Organic Chemistry I, Joxe Mari Korta R&D Center, University of the Basque Country, (UPV/EHU), Avda. Tolosa 72, 20018 Donostia-San Sebastián, Spain. E-mail: arkaitz.correa@ehu.eus
First published on 28th October 2024
Abstract
The site-selective functionalization of value-added compounds while implementing atom-economical C–H coupling partners represents an unmet challenge of utmost importance within organic synthesis. Herein, we report a Pd-catalyzed directed C–H acylation of a collection of relevant phenol-containing compounds with ethanol and other alcohols and aldehydes. This tagging technique is distinguished by its water compatibility and predictable regioselectivity and features the use of ethanol as renewable feedstock for the modification of intricate phenols, including estrogens and other top-selling pharmaceuticals. Mechanistic studies support the intermediacy of a challenging 6-membered dimeric palladacycle that undergoes the addition of nucleophilic acyl radical species.
Introduction
Phenols are privileged scaffolds of widespread presence in a vast number of natural products, agrochemicals, pharmaceuticals, dyes, fragrance compounds and other value-added products (Fig. 1).1 As a result, the development of efficient synthetic tools aimed at increasing the molecular complexity of phenol-containing compounds poses a challenging task of capital importance within organic chemistry.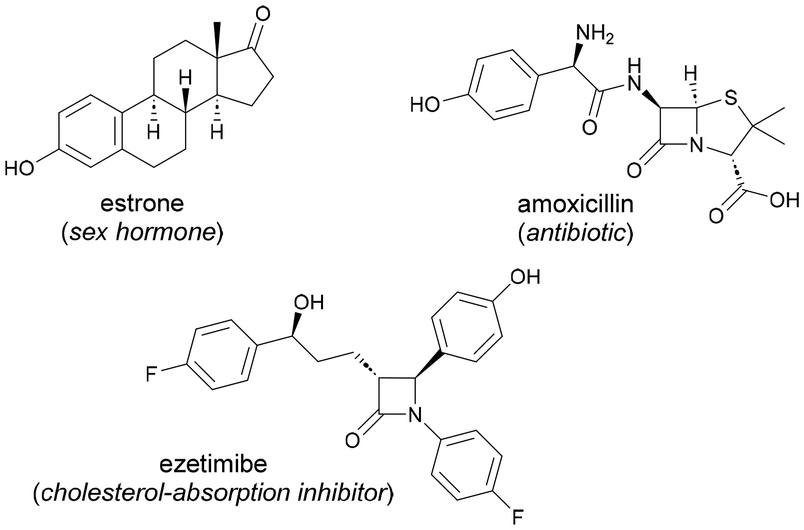 | ||
| Fig. 1 Phenol-containing bioactive molecules. | ||
Friedel–Crafts acylation2 is one of the most useful methods to functionalize phenols through a classical electrophilic aromatic substitution mechanism and is included in all the undergraduate-level organic chemistry textbooks (Scheme 1a). Likewise, Fries rearrangement offers an alternative route to ortho-hydroxyaryl ketones from the corresponding O-acylated phenols.3 Despite their extensive use even in industrial environments,4 these well-known transformations suffer from severe drawbacks such as the lack of regioselectivity, use of highly reactive acyl chlorides and toxic chemical waste derived from the use of stoichiometric amounts of halide-containing Lewis acids. Accordingly, the introduction of novel yet sustainable processes for the practical acetylation of phenols featuring other acyl surrogates represents a prime goal of paramount synthetic significance. Indeed, acetyl groups have been demonstrated to act as valuable linkers for further chemical ligation5 and hence they stand out as versatile handles within organic synthesis.
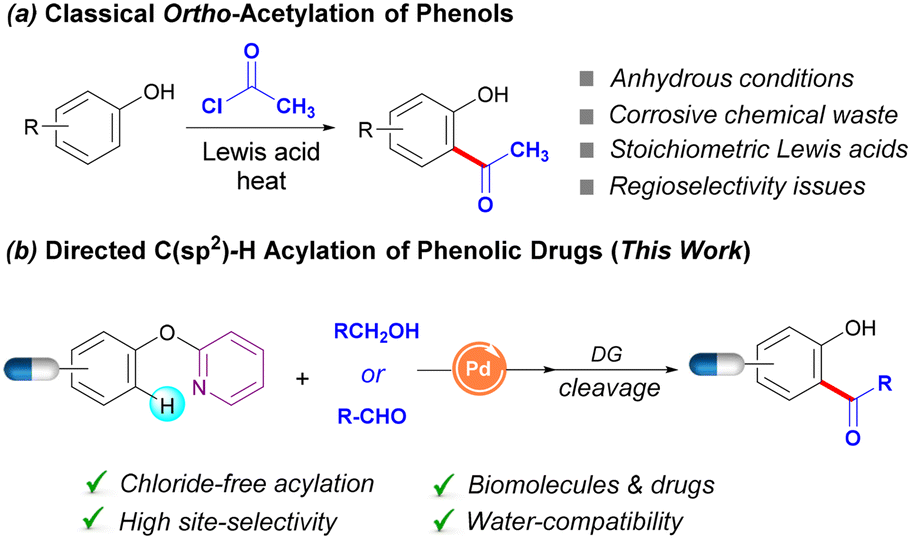 | ||
| Scheme 1 Acylation of phenols. | ||
C–H functionalization has changed the landscape of retrosynthetic analysis, thereby enabling the direct conversion of otherwise unreactive hydrocarbon backbones into functionalized compounds in a straightforward fashion.6 In this respect, the last few years have witnessed the surge of a plethora of metal-catalyzed C(sp2)–H functionalizations of phenols,7 which outcompete classical methods in terms of sustainability and site-selectivity. Most of the methods within the existing portfolio leverage the electron-rich nature of the phenol framework to further add electrophilic counterparts at the aromatic ring in a predictable manner. Conversely, the appendance of nucleophilic coupling partners requires the use of distinct tactics such as chelation assistance,8 which facilitates their addition toward electrophilic organometallic intermediates formed upon a directed C–H activation event.
In this context, while acyl radicals exhibit an amphiphilic character,9 they are predominantly nucleophilic in nature and hence they can undergo different fundamental reactions than those of the corresponding acylium species involved in classical Friedel–Crafts acylation. Therefore, the directed dehydrogenative radical C(sp2)–H acylation of strategically modified phenols represents an attractive alternative for accessing biologically relevant hydroxyaryl ketones. As part of our interest in metal-catalyzed radical C–H functionalization,10 we have recently unlocked the synthetic potential of simple and abundant ethanol as a C2-chemical feedstock11 to perform the late-stage acetylation of a library of tyrosine-containing peptides.12 Despite its importance within the realm of bioconjugation, the method was not explored for other value-added phenol-containing biomolecules or pharmaceuticals and the proposed mechanism was merely speculative based on indirect evidence.
Owing to their presence within female sex hormones, estrogens represent a key family of phenol-containing biomolecules including estrone and estradiol. Unfortunately, the chemical diversification of estrogens has been overlooked and general methods toward their late-stage modification are still elusive. In this communication, we explore the full synthetic potential of a Pd-catalyzed C(sp2)–H acylation manifold featuring EtOH and other alcohols as well as aldehydes to tag a collection of estrogens and other phenolic drugs. Notably, the required directing group (DG) can be easily installed and cleaved, which illustrates the utility of the method to prepare ortho-acylated phenols. Importantly, the full characterization of an acetate-bridged cyclopalladated dimer and its catalytic and stoichiometric experiments supported its intermediacy within a Pd(II)–Pd(IV) catalytic cycle.
Results and discussion
Driven by the successful use of 2-pyridyl ether as an efficient directing group (DG) in the modification of phenol compounds upon metal-catalyzed C–H functionalization,10c,12,13 we first selected estrone derivative 1a as the model substrate to test the feasibility of an acetylation reaction featuring the advantageous use of EtOH as a sustainable surrogate of corrosive acetyl chloride. After careful evaluation of the reaction conditions,14 we obtained the exclusive formation of mono-acetylated product 2a in 58% yield in the presence of 10 mol% of Pd(OAc)2 and an excess of an aqueous solution of cheap tert-butyl hydroperoxide (TBHP) at 120 °C (Table 1, entry 1). Control experiments underpinned the crucial role of both the catalyst and the oxidant, as the process was entirely inhibited in their absence (entries 2 and 3). The performance of the reaction at lower temperature or under air resulted in much lower yields (entries 4 and 5).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||
|---|---|---|
| Entry | Change from standard conditions |
2ab (%) |
| a Reaction conditions: 1a (0.15 mmol), EtOH (3.75 mmol, 0.2 mL), Pd(OAc)2 (10 mol%), and tBuOOH (0.90 mmol, 70 wt% in H2O) in PhCF3 (1 mL) at 120 °C for 16 h under Ar. b Yield of the isolated product after column chromatography. c Reaction performed with 1.44 mmol (500 mg) of 1a. d NMR yield obtained using the product as the internal standard. | ||
| 1 | None | 58 (46)c |
| 2 | Without Pd(OAc)2 | 0 |
| 3 | Without tBuOOH | 0 |
| 4 | Under air | 31 |
| 5 | T = 80 °C | 22 |
| 6 | With EtOH (15 equiv., 120 μL) | 27 |
| 7 | With EtOH (5 equiv., 40 μL) | Traces |
| 8 | Pd(OPiv)2 instead of Pd(OAc)2 | 43 |
| 9 | Pd(TFA)2 instead of Pd(OAc)2 | Traces |
| 10 | PdCl2 instead of Pd(OAc)2 | 26 |
| 11 | PhMe instead of PhCF3 as the solvent | 45 |
| 12 | 1,2-DCE instead of PhCF3 as the solvent | 41d |
| 13 | DCP instead of tBuOOH as the oxidant | 0 |
| 14 | DTBP instead of tBuOOH as the oxidant | 0 |
| 15 | t BuOOH (5.0 equiv.) | 36 |
| 16 | t BuOOH (5.0–6.0 M in decane) as the oxidant | 48 |
|
||
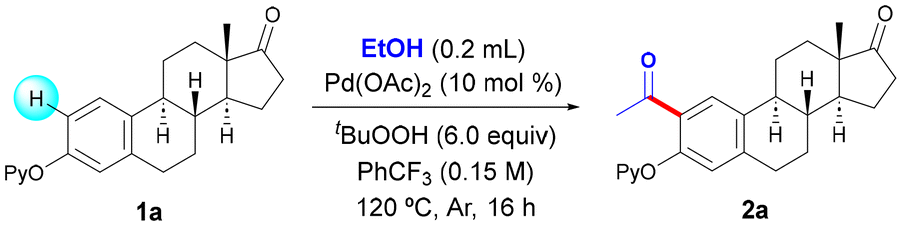
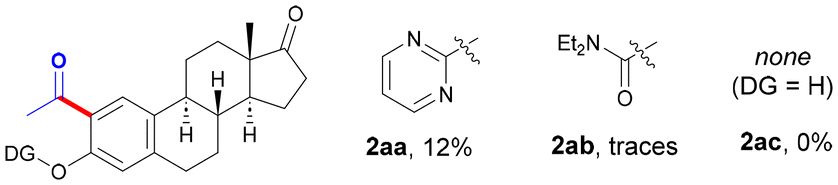
The optimal amount of EtOH was found to be 25 equiv., and the use of a lower (entries 6 and 7) or a higher amount of EtOH (up to 50 equiv.) resulted in comparatively lower yields. While other related Pd sources could be utilized, Pd(OAc)2 showed a superior catalytic activity (entries 8–10). Likewise, the nature of the solvent played a crucial role and trifluorotoluene outperformed other solvents such as toluene (entry 11) or 1,2-dichloroethane (entry 12), among others.14 Regarding the oxidant, while the use of dicumyl peroxide (entry 13) or di-tert-butyl peroxide (entry 14) resulted in no reaction, the use of a solution of tert-butyl hydroperoxide was key within the reaction outcome. Importantly, the inexpensive aqueous solution of TBHP afforded a higher yield than the use of the parent reagent in decane (entry 16), which represents an added bonus from a sustainable perspective. Accordingly, the drawback derived from using up to 6.0 equiv. of an oxidant (entry 1 vs. entry 15) is balanced by its low price and water-compatible character. In order to evaluate the role of the DG, control experiments were performed with other estrone derivatives. As expected, the absence of a DG resulted in the entire inhibition of the process; however, other related DGs such as pyrimidine (2aa) or a carbamate (2ab), previously used in the metal-catalyzed functionalization of phenol-based compounds,15 exhibited an inferior reactivity to that of the pyridyl motif. Notably, the reaction could be scaled up and performed with 1.44 mmol (500 mg) of estrone derivative 1a to selectively afford acetylated 2a in a synthetically useful 46% yield. Despite the presence of two chemically different C–H bonds, exclusive functionalization at the less hindered C2 site was always observed, which was verified through NOE experiments after cleavage of the DG (vide infra).14
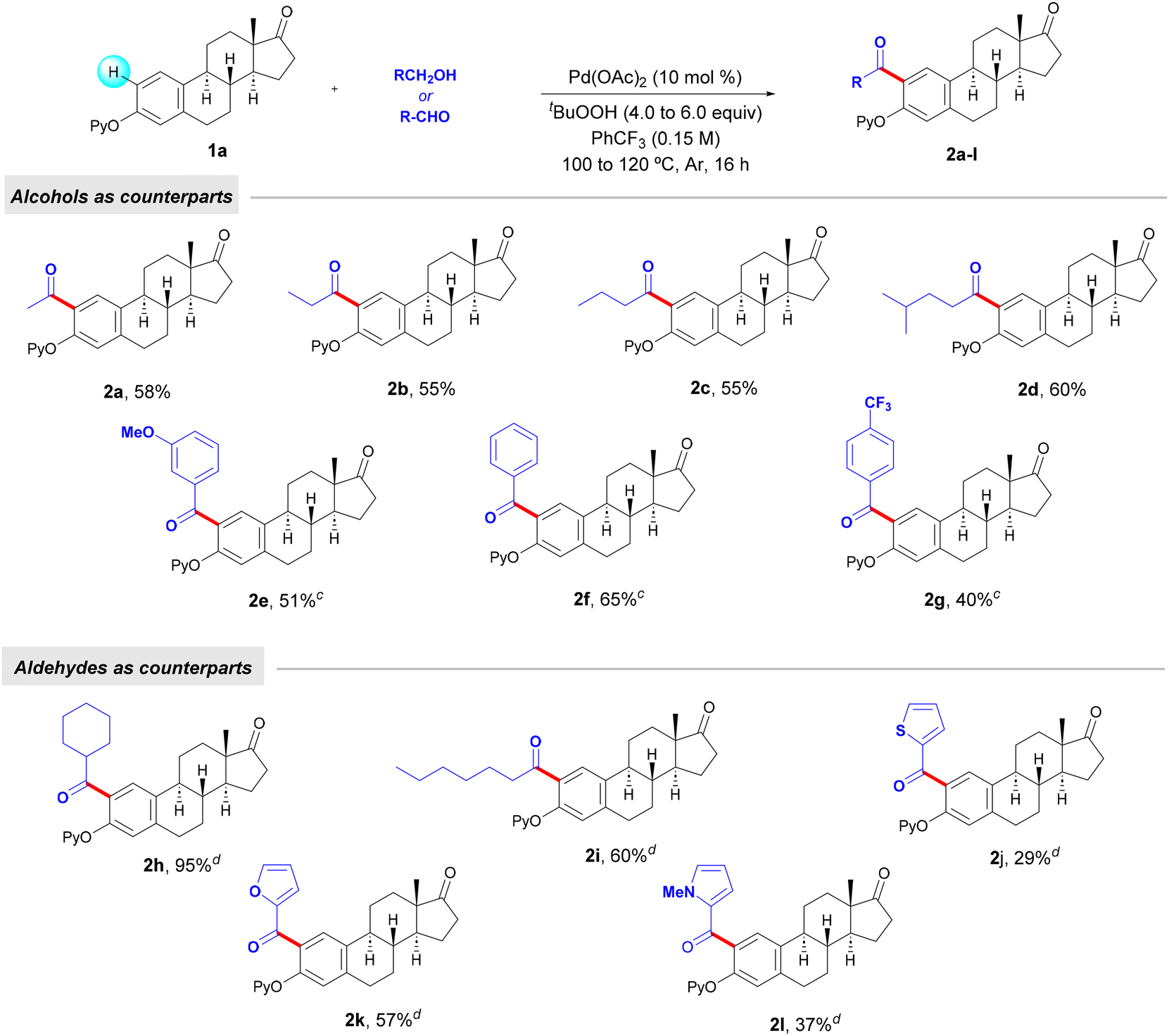
Prompted by the successful use of EtOH as an appealing surrogate of conventional acetyl chloride, we next evaluated the use of other alkyl and benzyl alcohols in the acylation of estrone derivative 1a (Table 2).
| a Reaction conditions: 1a (0.15 mmol), RCH2OH (0.75 mmol), Pd(OAc)2 (10 mol%), and tBuOOH (0.90 mmol, 70 wt% in H2O) in PhCF3 (1 mL) at 120 °C for 16 h under Ar. b Yield of the isolated product after column chromatography, with the average of at least two independent runs with a variation in yield of no more than 5% between runs. c RCH2OH (0.45 mmol). d Reaction conditions: 1a (0.15 mmol), aldehyde (0.75 mmol), Pd(OAc)2 (10 mol%), and tBuOOH (0.60 mmol, 70 wt% in H2O) in PhCF3 (1 mL) at 100 °C for 16 h under Ar. |
|---|
|
Alcohols such as n-propanol and n-butanol, common solvents in a vast array of organic reactions, as well as 4-methylpentan-1-ol, could be used to perform the mono-acylation of estrone 1a. Owing to their higher boiling point than that of EtOH, in those cases the amount of the alcohol could be reduced to 5.0 equivalents. Moreover, the reaction using easily oxidizable benzyl alcohols could be carried out with only 3.0 equiv of the corresponding alcohols. In all cases, mono-acylated estrone compounds 2a–g were obtained in up to 65% yield. Based on the absence of ortho-acylation methods for estrogen derivatives, we further explored the use of certain aldehydes as counterparts in order to broaden the structural complexity within the estrone core. Interestingly, besides alkyl residues (2h and 2i), ketones containing relevant heterocyclic fragments such as thiophene (2j), furan (2k) and pyrrole (2l) could be installed site-selectively in moderate to good yields. When employing aldehydes, the reaction could take place at 100 °C with 4.0 equivalents of TBHP. Of particular importance is the use of cyclohexanecarboxaldehyde, which led to the corresponding decorated estrone derivative 2h in excellent yield.
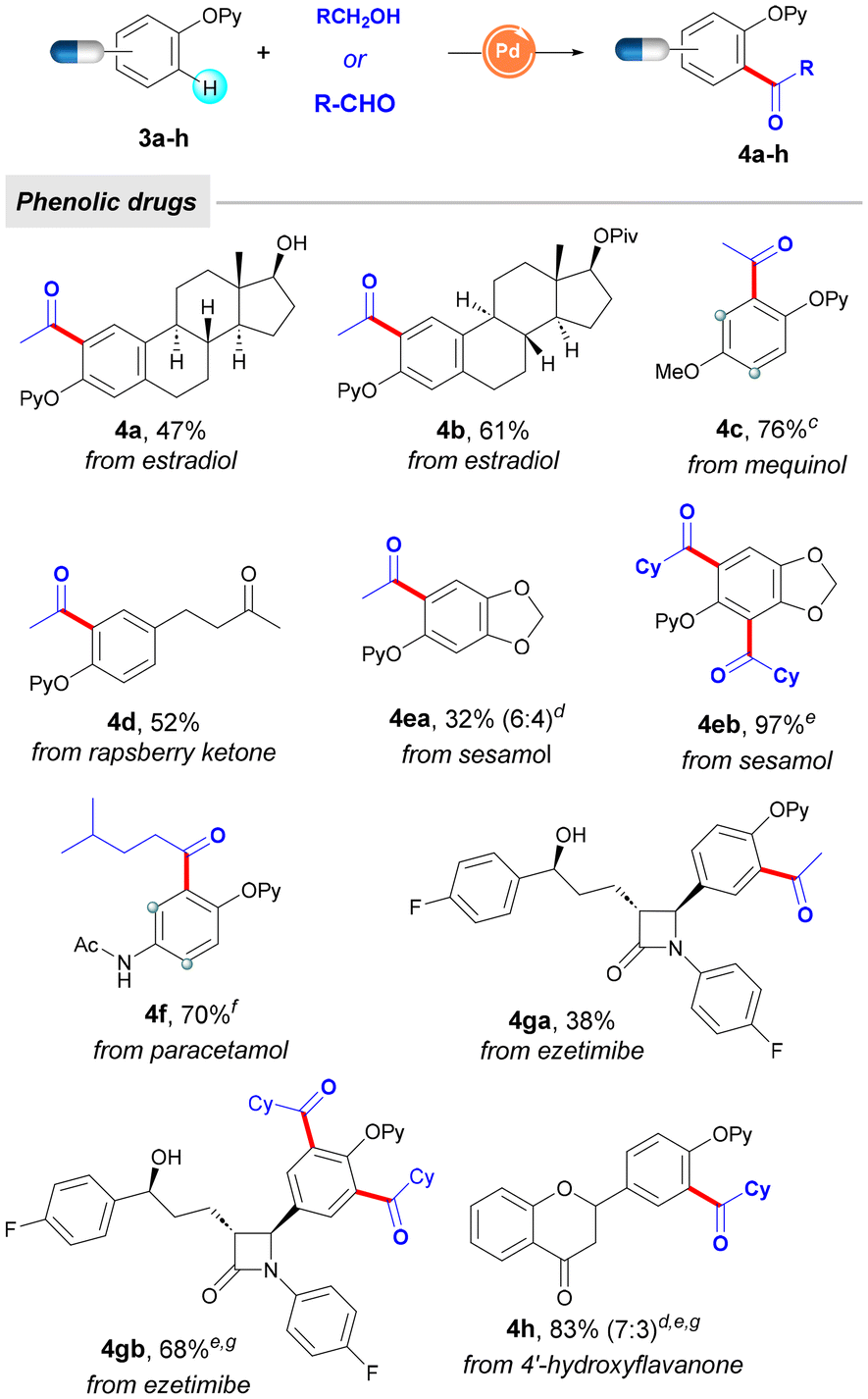
Encouraged by the utility of this acylation platform to tag estrone, we further tackled the modification of a collection of other phenol-based biomolecules and top-selling phenolic drugs, with a special emphasis on the advantageous use of EtOH as chemical feedstock. As depicted in Table 3, structurally related estradiol compounds (4a and 4b) could selectively undergo C–H acetylation even in the presence of a free tertiary alcohol (4a), which remained intact throughout the oxidative process. Other phenolic drugs such as mequinol (4c, used for skin depigmentation), raspberry ketone (4d, dietary supplement), sesamol (4e, food ingredient), paracetamol (4f, analgesic), or 4′-hydroxyflavanone (4h, inhibitor of SREBP) could be smoothly tagged upon the Pd-catalyzed acylation manifold. It is important to highlight that the observed ortho-selectivity would not be achieved by classical Friedel–Crafts acetylation on free phenols, which would result in mixtures of regioisomers due to the presence of multiple functional groups within the corresponding arene (mostly within mequinol 4c and paracetamol 4f containing electron-donating groups). Remarkably, another drug containing multichiral carbon centers, such as the cholesterol absorption inhibitor ezetimibe, was also shown to be a competent coupling partner, thereby providing modified drugs in moderate to good yields (4ga and 4gb). Notably, while sesamol or ezetimibe afforded low yields in the acetylation event with EtOH (4ea and 4ga, respectively), they successfully underwent parent acylation with CyCHO, selectively forming the corresponding diacylated phenol compounds 4eb and 4gb in higher yields. Collectively, the phenol partner scope depicted in Tables 2 and 3 showcases the functional group tolerance in post-synthetic chemical modifications and illustrates the utility of this labelling technique for the selective diversification of a wide variety of intricate phenol compounds.
| a As for Table 1, entry 1. b Yield of the isolated product after column chromatography, with the average of at least two independent runs with a variation in yield of no more than 5% between runs. c Reaction time of 24 h. d Ratio of mono- and diacylated products. e Reaction conditions: 3 (0.15 mmol), cyclohexanecarboxaldehyde (0.75 mmol), Pd(OAc)2 (10 mol%), and tBuOOH (0.60 mmol, 70 wt% in H2O) in PhCF3 (1 mL) at 100 °C for 1 h under Ar. f Reaction conditions: 3 (0.15 mmol), 4-methylpentanol (0.75 mmol), Pd(OAc)2 (10 mol%), and tBuOOH (0.90 mmol, 70 wt% in H2O) in PhCF3 (1 mL) at 120 °C for 1 h under Ar. g Reaction time = 16 h. |
|---|
|
Although the cleavage of the OPy group has been often accomplished in simple arenes upon treatment with methyl triflate followed by an alcoholic solution of sodium,13 its application in complex biomolecules could pose a major drawback. In this respect, the cleavage of the DG in simple mequinol derivative 4c could be performed under those classical conditions to afford ortho-acetylated phenol 5c in 77% yield (Scheme 2). However, the cleavage of the DG within more complex estrone derivative 2a was alternatively achieved through a methylation/hydrogenation sequence13a to produce the corresponding ortho-acetylated estrone 5a in 50% yield.
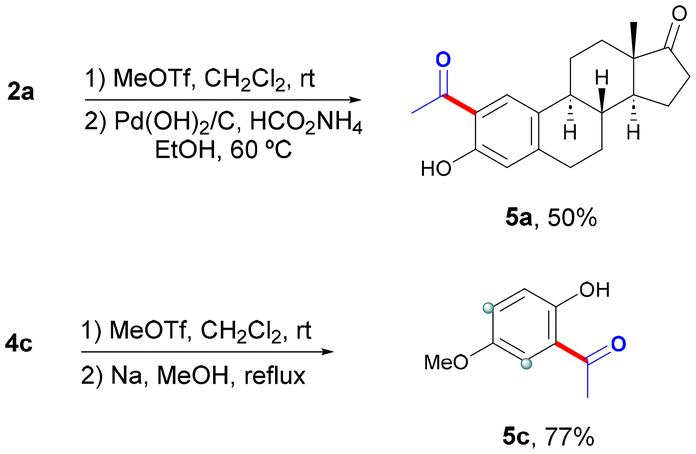 | ||
| Scheme 2 DG cleavage. | ||
In order to gain some insights into the reaction mechanism, we conducted the following set of experiments. First, we treated estrone derivative 1a with a stoichiometric amount of Pd(OAc)2 in trifluorotoluene at 100 °C to obtain complex I as a yellow crystalline solid in 76% yield. As shown in Fig. 2, X-ray crystallographic analysis verified the dimeric nature of this palladacycle intermediate and its head-to-tail geometric structure16 and the observed Pd–Pd distance of 2.8491(17) Å was consistent with a bond order of zero. Likewise, its characterization was complemented by 1H NMR analysis, which evidenced the successful ortho-palladation as two singlets at 6.67 and 6.37 ppm appeared within the aromatic region.14 With this acetate-bridged cyclopalladated dimer in hand, we performed some control experiments to support its intermediacy within the dehydrogenative acylation event (Scheme 3). When performing acetylation with EtOH of estrone derivative 1a in the presence of a catalytic amount of complex I instead of Pd(OAc)2, the corresponding ortho-acetylated compound 2a was obtained in 43% yield. Likewise, its use in a catalytic amount enabled ortho-acylation with highly reactive cyclohexanecarboxaldehyde to afford acylated compound 2h in quantitative yield. Notably, stoichiometric experiments with complex I underpinned its key role in the radical acylation process. While the acetylation with EtOH successfully occurred to afford 2a in 32% yield, the acylation with CyCHO provided acylated compound 2h in a remarkable 86% yield. It is important to note that the process only occurred in the presence of TBHP and in its absence the corresponding cyclopalladated dimer remained unreactive, which supported the crucial role of the oxidant to generate the transient acyl radical species. Furthermore, 2h could also be obtained at room temperature, albeit in comparatively lower yields. These results are in sharp contrast to those obtained by Wu and co-workers in related mechanistic studies on simple 2-phenoxypyridines with benzaldehydes.17 In fact, owing to the low yields obtained when starting from a related cyclopalladated dimer, the intermediacy of an aroyl palladium π complex was proposed instead as the more plausible mechanistic scenario. Conversely, the excellent results obtained when submitting complex I to the acylation reaction conditions with alcohols and aliphatic aldehydes may support its key role in our acylation events. Importantly, estrone derivative 1a underwent the acetylation reaction with acetaldehyde to form acetylated product 2a in 38% yield together with 26% yield of diacetylated product 2a′, which was never observed when using EtOH as the coupling partner. This experiment may support an initial oxidation of EtOH to MeCHO and tentatively indicate that the latter oxidation event or its further oxidation to the acyl radical species could be crucial within the process. Finally, in accordance with the studies by Wu,17 the results obtained when performing the acetylation of 1a with ethanol in the presence of radical traps strongly supported a radical reaction pathway. Indeed, the process was entirely inhibited when adding TEMPO as a radical scavenger and significantly disfavored in the presence of BHT and 1,1-diphenylethylene to afford 2a in 29% and 34% yields, respectively.
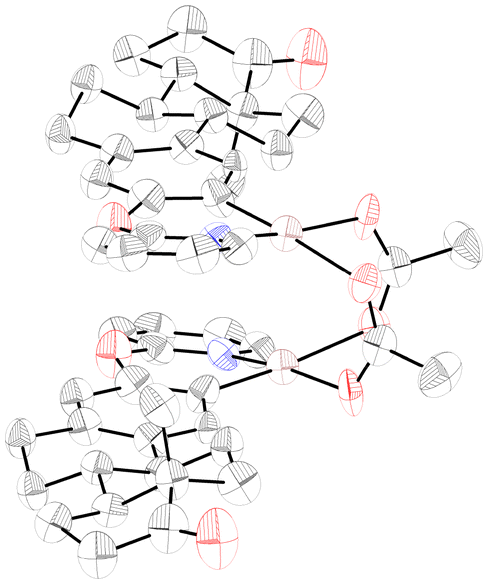 | ||
| Fig. 2 ORTEP drawing of complex I. | ||
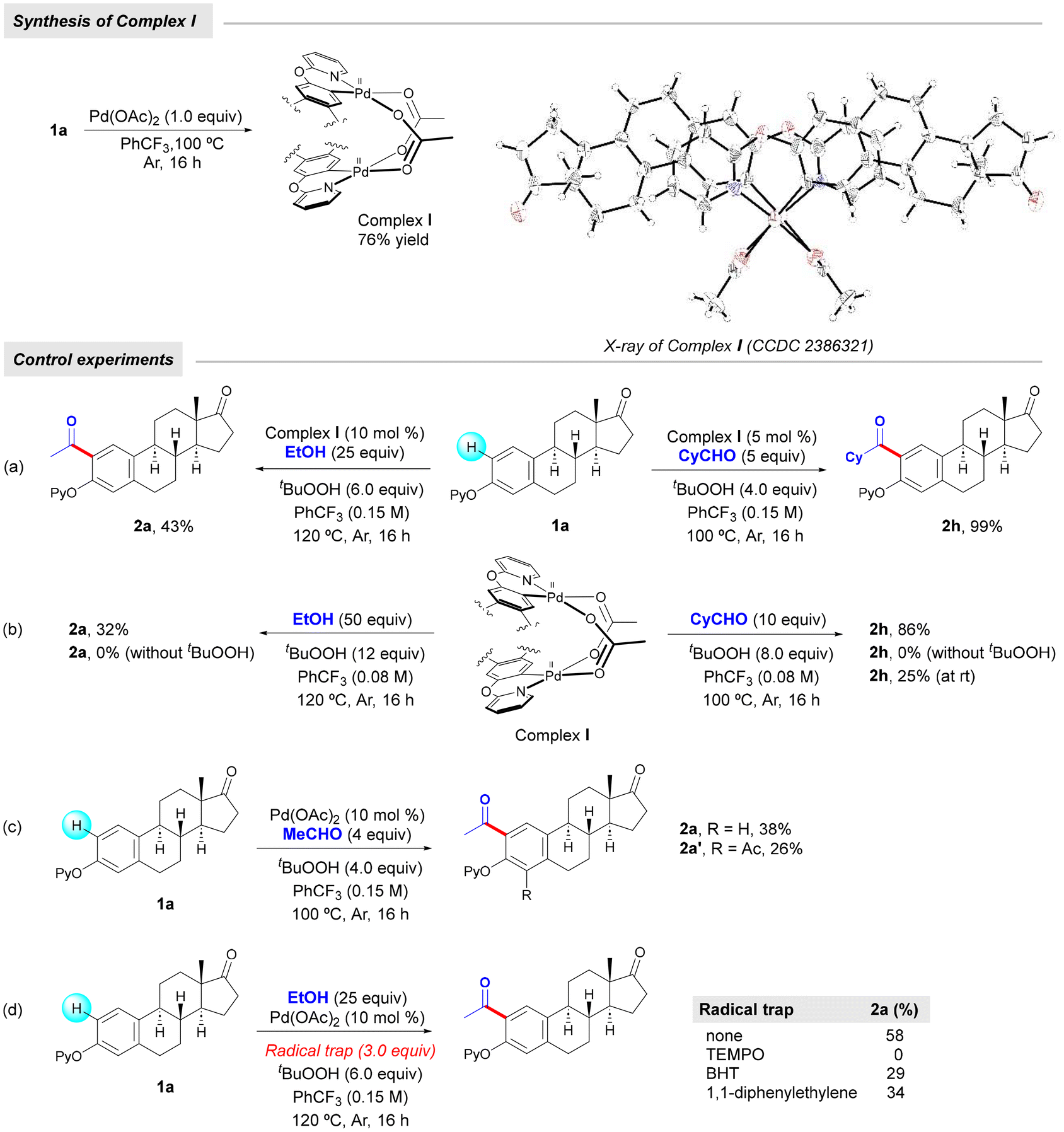 | ||
| Scheme 3 Synthesis of intermediates and control experiments. | ||
Based on the mechanistic experiments mentioned above and the existing literature,9 we proposed a plausible reaction pathway for the directed Pd-catalyzed acylation with alcohols and aldehydes. As depicted in Scheme 4 for the acetylation of estrone derivative 1a, the initial coordination of Pd(OAc)2 with the starting phenol-compound following ortho-C–H bond activation would form the corresponding acetate-bridged cyclopalladated species I,16 which has been identified as a dimer by X-ray analysis. It is well known that primary alcohols can easily undergo oxidation to the corresponding aldehydes.18,19 The latter could undergo an hydrogen-atom transfer (HAT) event with electrophilic tert-butoxyl radical species to afford nucleophilic acyl radical species,9,20 which could undergo addition to electrophilic bimetallic palladium complex I. Despite the existence of mechanistic studies on the formation of high valent Pd(IV) complexes via either mono- or bi-metallic pathways,21 for simplicity reasons, we proposed that dimeric intermediate I may likely undergo dissociation to a monomeric Pd complex prior to its oxidation toward the formation of high valent species. Accordingly, the initially formed monomeric Pd(III) intermediate II could evolve into Pd(IV). Eventually, reductive elimination would furnish ortho-acetylated compound 2a, thereby regenerating the active Pd(II) catalyst. However, an alternative bimetallic pathway cannot be ruled out and may also be a feasible scenario.22 It is important to note that direct experimental evidence of the formation of high valent Pd species has not been achieved and its intermediacy is proposed based on literature precedents.
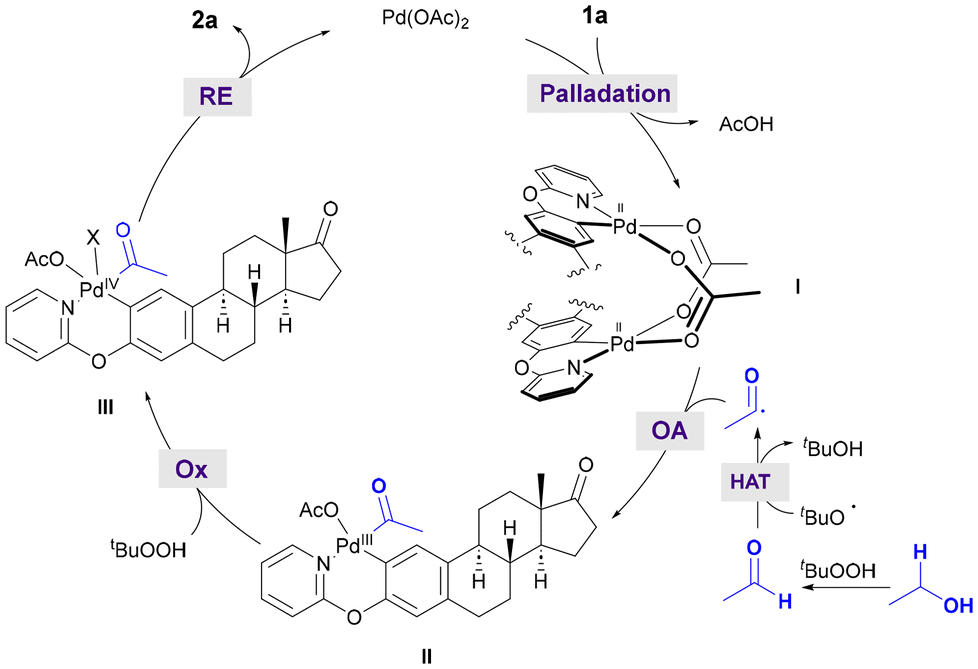 | ||
| Scheme 4 Proposed reaction pathway for Pd-catalyzed C–H acylation with EtOH. | ||
Conclusions
In summary, we have unlocked the synthetic potential and versatility of EtOH as a cost-efficient promising acyl surrogate to tag a wide variety of phenol-containing bioactive compounds. In particular, we have explored the utility of a directed Pd-catalyzed C–H acetylation manifold for the modification of phenols featuring a removable pyridine unit as the DG. Unlike the classical acetylation methods available for the assembly of ortho-hydroxyaryl ketones, this protocol enables the rapid installation of an acetyl group from a chloride-free coupling partner in a site-selective fashion. As a result, this tagging platform represents an attractive means for the diversification of intricate phenolic units, which can be applied to the use of other aliphatic alcohols and aldehydes. The strength of this method lies in the facile introduction and removal of the required pyridyl ether as the DG, water compatibility and use of abundant chemical feedstock. Mechanistic studies supported a radical pathway involving the intermediacy of an acetate-bridged palladacyclic dimer as a key species within the acylation process. We anticipate that this Pd-catalyzed C–H acylation manifold could become a useful tool for the downstream functionalization of phenol-containing value-added products employing readily available C–H coupling partners.Author contributions
C. Girón-Elola conducted the experiments and A. Correa supervised the project. The manuscript was written with contributions from both authors and both of them have given approval to the final version of the manuscript.Data availability
Experimental details and characterization of the complexes can be found in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Ministerio de Ciencia e Innovación (PID2021-122889NB-I00 and MCI/AEI/FEDER, UE) and Basque Government (IT1741-22) for financial support. We acknowledge technical and human support provided by SGIker of UPV/EHU and European funding (ERDF and ESF). C.G.-E. acknowledges the Ministerio de Universidades for a FPU contract (FPU22/00522). Dr Miguel A. Huertos is kindly acknowledged for insightful mechanistic discussions. Dr Leire San Felices and Dr Jose Ignacio Miranda from SGIker are acknowledged for their support with the X-ray and NMR analysis, respectively.References
-
(a) K. A. Scott, P. B. Cox and J. T. Njardarson, Phenols in Pharmaceuticals: Analysis of a Recurring Motif, J. Med. Chem., 2022, 65, 7044 CrossRef CAS PubMed
; (b) M. A. Rahim, S. L. Kristufek, S. Pan, J. J. Richardson and F. Caruso, Phenolic Building Blocks for the Assembly of Functional Materials, Angew. Chem., Int. Ed., 2019, 58, 1904 CAS
- G. Sartori and R. Maggi, Use of Solid Catalysts in Friedel−Crafts Acylation Reactions, Chem. Rev., 2006, 106, 1077 CAS
- R. Martin, Uses of the Fries Rearrangement for the Preparation of Hydroxyarylketones: A Review, Org. Prep. Proced. Int., 1992, 24, 369 CAS
- J. Ohata, Friedel–Crafts reactions for biomolecular chemistry, Org. Biomol. Chem., 2024, 22, 3544 RSC
-
(a) A. M. James, A. A. I. Norman, J. W. Houghton, H. A. Prag, A. Logan, R. Antrobus, R. C. Hartley and M. P. Murphy, Native chemical ligation approach to sensitively probe tissue acyl-CoA pools, Cell Chem. Biol., 2022, 29, 1232 CrossRef CAS PubMed
- For selected reviews, see:
(a) R. de Jesus, K. Hiesinger and M. van Gemmeren, Preparative Scale Applications of C−H Activation in Medicinal Chemistry, Angew. Chem., Int. Ed., 2023, e202306659 CAS
-
(a) G. Brufani, B. Di Erasmo, C.-J. Li and L. Vaccaro, Csp2−H functionalization of phenols: an effective access route to valuable materials via Csp2−C bond formation, Chem. Sci., 2024, 15, 3831 RSC
- For selected reviews, see:
(a) S. Rej, Y. Ano and N. Chatani, Bidentate Directing Groups: An Efficient Tool in C–H Bond Functionalization Chemistry for the Expedient Construction of C–C Bonds, Chem. Rev., 2020, 120, 1788 CrossRef CAS PubMed
-
(a) K. L. Samony and D. K. Kim, Acetal Radicals: Synthetic Access and Applications, ChemCatChem, 2023, 15, e202301091 CrossRef CAS
-
(a) C. Girón-Elola, I. Sasiain, R. Sánchez-Fernández, E. Pazos and A. Correa, Site-Selective C–H Amination of Phenol-Containing Biomolecules, Org. Lett., 2023, 25, 4383 CrossRef PubMed
- A. Cook and S. G. Newman, Alcohols as Substrates in Transition-Metal-Catalyzed Arylation, Alkylation, and Related Reactions, Chem. Rev., 2024, 124, 6078 CrossRef CAS PubMed
- I. Urruzuno, P. Andrade-Sampedro and A. Correa, Late-Stage C–H Acylation of Tyrosine-Containing Oligopeptides with Alcohols, Org. Lett., 2021, 23, 7279 CrossRef CAS PubMed
-
(a) I. Urruzuno, P. Andrade-Sampedro and A. Correa, Pd-Catalyzed Site-Selective C–H Acetoxylation of Tyrosine-Containing Peptides, Eur. J. Org. Chem., 2023, e20220148 Search PubMed
- For more details, see the ESI.†.
-
(a) S. S. Bera and M. S. Maji, Carbamates: A Directing Group for Selective C–H Amidation and Alkylation under Cp*Co(III) Catalysis, Org. Lett., 2020, 22, 2615 CrossRef CAS PubMed
-
(a) J.-H. Chu, P.-S. Lin and M.-J. Wu, Palladium(II)-Catalyzed Ortho Arylation of 2-Phenoxypyridines with Potassium Aryltrifluoroborates via C−H Functionalization, Organometallics, 2010, 29, 4058 CrossRef CAS
- J.-H. Chu, S.-T. Chen, M.-F. Chiang and M.-J. Wu, Palladium-Catalyzed Direct Ortho Aroylation of 2-Phenoxypyridines with Aldehydes and Catalytic Mechanism Investigation, Organometallics, 2015, 34, 953 CrossRef CAS
- D. C. Powers, E. Lee, A. Ariafard, M. S. Sanford, B. F. Yates, A. J. Canty and T. Ritter, Connecting Binuclear Pd(III) and Mononuclear Pd(IV) Chemistry by Pd–Pd Bond Cleavage, J. Am. Chem. Soc., 2012, 134, 12002 CrossRef CAS PubMed
- G. E. Dobereiner and R. H. Crabtree, Dehydrogenation as a Substrate-Activating Strategy in Homogeneous Transition-Metal Catalysis, Chem. Rev., 2010, 110, 681 CrossRef CAS PubMed
-
(a) J. Park, A. Kim, S. Sharma, M. Kim, E. Park, Y. Jeon, Y. Lee, J. H. Kwak, Y. H. Jung and I. S. Kim, Direct acylation of N-benzyltriflamides from the alcohol oxidation level viapalladium-catalyzed C–H bond activation, Org. Biomol. Chem., 2013, 11, 2766 RSC
- P. Kumar, S. Dutta, S. Kumar, V. Bahadur, E. V. Van der Eycken, K. S. Vimaleswaran, V. S. Parmar and B. K. Singh, Aldehydes: magnificent acyl equivalents for direct acylation, Org. Biomol. Chem., 2020, 18, 7987 RSC
-
(a) D. C. Powers and T. Ritter, Bimetallic Pd(III) complexes in palladium-catalysed carbon–heteroatom bond formation, Nat. Chem., 2009, 1, 302 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, syntheses and characterization of all new compounds, and tables with details of several optimization studies. CCDC 2386321. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01505c |
| This journal is © the Partner Organisations 2024 |