
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalyst-free light-mediated three-component alkoxy-, hydroxy-, and azidotrifluoromethylation of alkenes†
Yingmin
Ji‡
 a,
Aida
Jaafar‡
b,
Carolina
Gimbert-Suriñach
a,
María
Ribagorda
*bc,
Adelina
Vallribera
*a,
Albert
Granados
*a and
María Jesús
Cabrera-Afonso
*b
a,
Aida
Jaafar‡
b,
Carolina
Gimbert-Suriñach
a,
María
Ribagorda
*bc,
Adelina
Vallribera
*a,
Albert
Granados
*a and
María Jesús
Cabrera-Afonso
*b
aDepartment of Chemistry and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universitat Autònoma de Barcelona, Cerdanyola del Vallès, 08193 Barcelona, Spain. E-mail: adelina.vallribera@uab.es; albert.granados@uab.es
bDepartamento de Química Orgánica, Facultad de Ciencias, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: mjesus.cabrera@uam.es
cInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: maria.ribagorda@uam.es
First published on 25th September 2024
Abstract
Photoinduced radical–polar crossover (RPC) reactions generally rely on the use of an external photocatalyst to generate the corresponding radical and ionic species, selected according to the redox potentials of the involved species. Herein, we describe a multicomponent light-induced RPC reaction that does not require an exogenous photocatalyst, enabling the efficient synthesis of diverse 1,2-trifluoromethyl alkyl ethers, alcohols, and azides. This protocol allows for the bifunctionalization of alkenes under mild conditions using purple light and a thianthrenium salt as a trifluoromethylating agent. Mechanistic experiments confirmed the formation of a benzylic sulfonium intermediate that can participate in different nucleophilic substitution reactions, being an alternative photocatalyst-free method to the classical RPC.
Introduction
Radical/polar crossover (RPC) synthetic methods are gaining considerable interest given their unique mode of reactivity.1 The main challenge of these protocols is the convenient design of the reaction conditions, where both radical and ionic modes of reactivity are involved.2 In recent years, photoinduced net-neutral methods have emerged as a particularly interesting subclass of RPC that utilizes photocatalysts and visible light to trigger the reactivity.3 The chosen photocatalyst plays a pivotal role enabling the single-electron transfer events (oxidation and reduction) across different oxidation states throughout the reaction, which preludes the use of exogenous oxidants or reductants.A notable application of RPC chemistry is devoted to alkene difunctionalization, which constitutes a powerful strategy to increase molecular complexity in a single step from simple starting materials.4 In this framework, the sequential formation of C(sp3)–CF3 and C(sp3)–Nu bonds via RPC mechanism governed by oxidative quenching has found wide applicability using Togni, Umemoto or Mes-Umemoto reagents, among others.5 The appropriate photocatalyst selection induces the desired reactivity in presence of the corresponding nucleophiles.
Typically, the photocatalyst in its excited state reduces the trifluoromethylating reagent yielding a ˙CF3 radical species that undergoes a Giese-type addition to an alkene. The newly generated carbon-centered radical is ultimately oxidized by the oxidized state photocatalyst [PC]˙+ to the carbocation, which is then trapped via polar reaction with the corresponding nucleophile (Fig. 1A). In addition, such organic skeletons can be accessed through transition metal catalysis.6 Therefore, new synthetic methods for the construction of sequential C(sp3)–CF3 and C(sp3)–Nu bonds without exogenous catalysts is highly desirable.
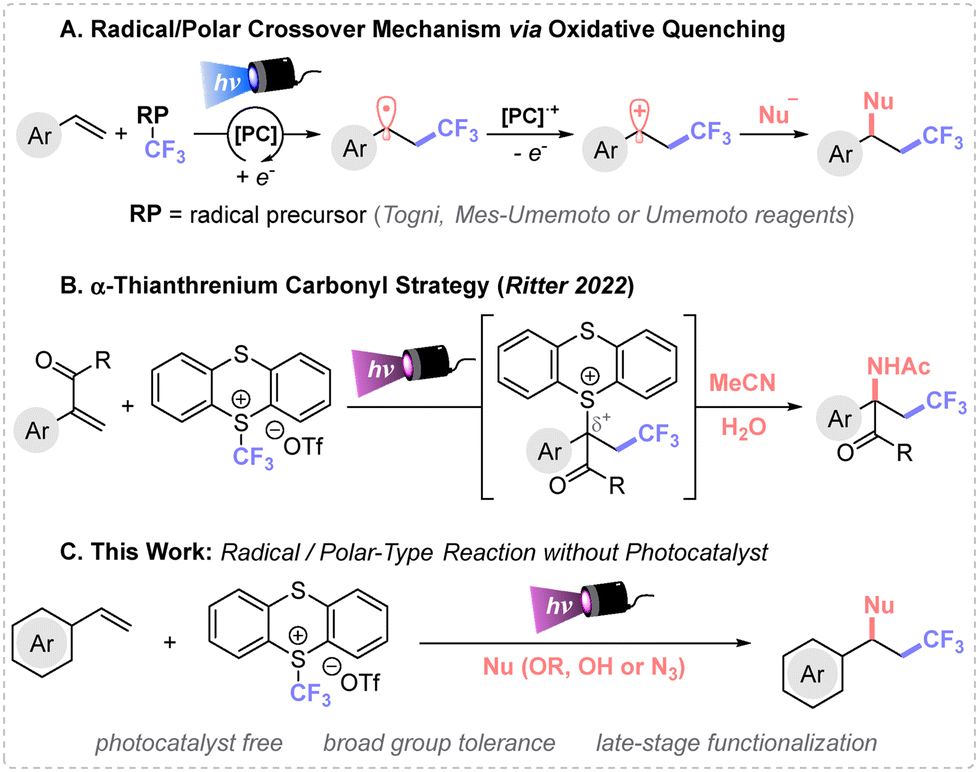 | ||
| Fig. 1 Different trifluoromethylation reactions, followed by nucleophilic attack by a radical/polar-type strategies. | ||
In 2021, the Ritter group7 presented a bench stable CF3-containing sulfonium salt (trifluoromethyl-thianthrenium salt TT-CF3OTf), which is synthesized in a single step from Tf2O and thianthrene. This reagent has proved to be efficient towards formal CF3+, CF3− and ˙CF3 reactivity. Among its use in free-radical based synthesis,8 the same research group9 presented an intermolecular amidotrifluoromethylation of acrylates via α-thianthrenium carbonyl species using the TT-CF3OTf reagent (Fig. 1B).
In this study, we present a metal-free method for the intermolecular 1,2-difunctionalization of alkenes for the efficient preparation of CF3-containing ethers, alcohols, and azides (Fig. 1C). This protocol does not require exogenous photocatalysts, allows for the easy recovery of the thianthrene byproduct, and features short reaction times. These advantages make this transformation an attractive alternative to other reported procedures.10
Results and discussion
First, we explored the alkoxytrifluoromethylation process using the trifluoromethyl-thianthrenium salt as the CF3 source, n-pentanol as nucleophile and 4-vinyltoluene as the model alkene. The screening revealed that the use of 1.5 equiv. of TT-CF3OTf and 5.0 equiv. of the alcohol in 1,2-dichloroethane as solvent (0.1 M) yielded product 3a in 73% yield (see Tables S1 and S2 in the ESI†).The optimal conditions were applied to a wide range of styrenes (Scheme 1), where first different primary alcohols were tested obtaining the desired compounds from moderate to high yields. Both electron-poor and weak electron-donating substituents were suitable to this alkoxytrifluoromethylation process (3a–r). Notably, ether 3i derived from p-vinylbenzyl chloride was obtained in high yield, and no nucleophilic substitution to the chlorinated benzylic position was detected, showcasing the high selectivity of this process. Moreover, products 3p–s evidenced the potential of this methodology for late-stage functionalization of complex molecules. Disubstituted styrenes (α-methyl and trans-β-methyl styrenes) afforded 3j–k in good yields, displaying that steric effects do not affect to the Giese addition step, neither the nucleophilic attack. Furthermore, the substitution position on the aromatic ring do not have direct impact on the reactivity, thus we could isolate 3m–p in similar yields.
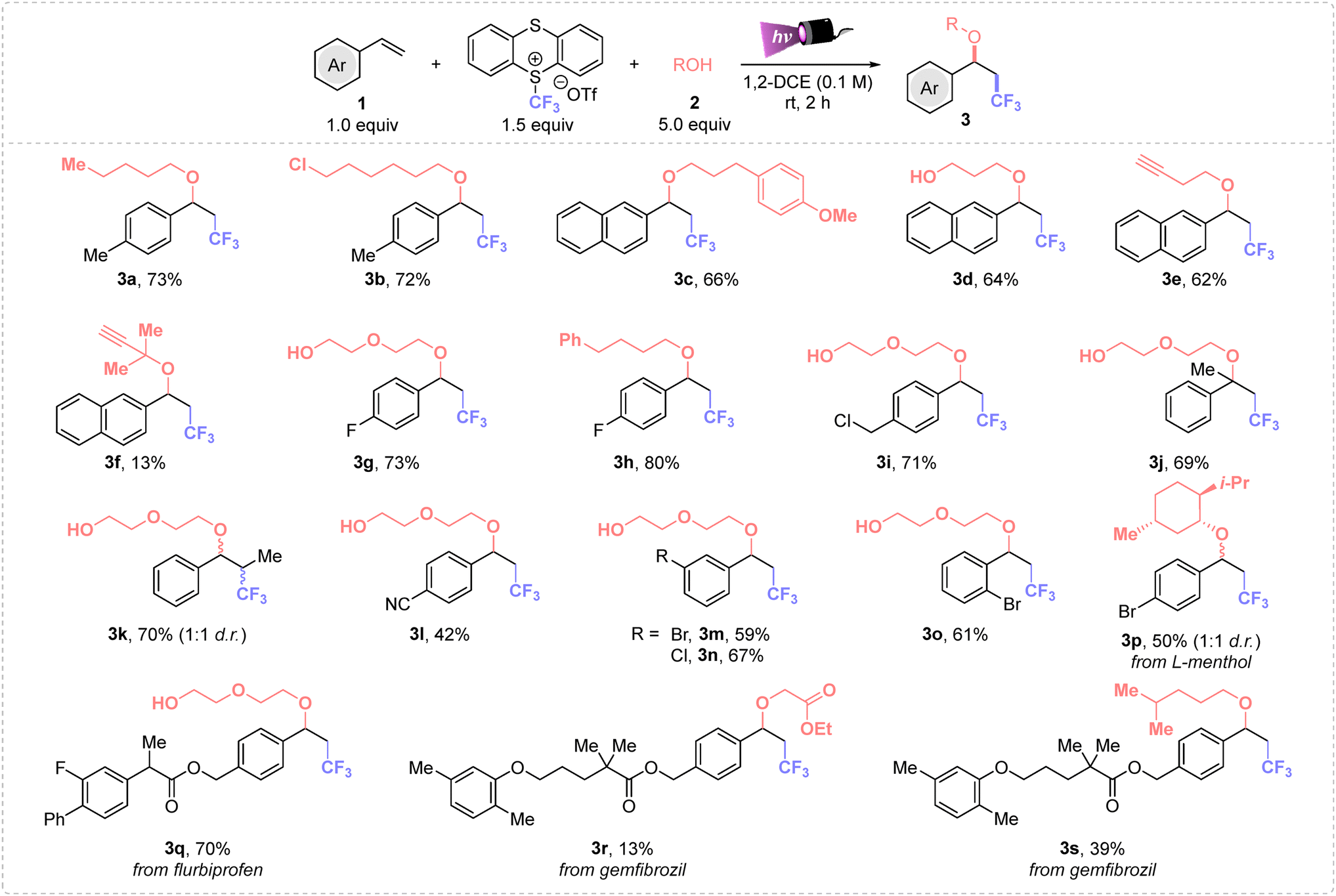 | ||
| Scheme 1 Scope of ethers. Standard conditions: 1 (0.25 mmol), 2 (5.0 equiv.) and TT-CF3OTf (1.5 equiv.), in dry degassed 1,2-DCE (2.5 mL, 0.1 M) under purple Kessil irradiation (λmax = 390 nm) for 2 h at rt under inert atmosphere. DCE = dichloroethane. | ||
Then, the scope of the reaction was expanded to various alkyl alcohols, observing that primary, secondary (3p) and tertiary alcohols (3f) can be tethered in the final molecule. In addition, a 13% of 3f was achieved from tertiary 2-methylbut-3-yn-2-ol. Different functional groups derived from the alcohol were also well tolerated such as a chloro handle (3b), which can be used for further diversification, terminal alkynes (3e–f), susceptible to undergo radical insertion, and ester (3r), among others. Notably, ethylene glycol and 1,3-propanediol were successfully used as diol nucleophiles. Importantly, products derived from a double nucleophilic attack were not detected, affording exclusively ethers 3d, 3g, 3i–o and 3q. Furthermore, L-menthol was also a suitable oxygenated nucleophile for this transformation, affording 3p in 50% yield and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio. Unfortunately, nonactivated alkenes or phenylacetylene did not proceed well (see Section 2.2.3 in the ESI†).
:1 diastereomeric ratio. Unfortunately, nonactivated alkenes or phenylacetylene did not proceed well (see Section 2.2.3 in the ESI†).
Additionally, we focused on studying the hydroxytrifluoromethylation protocol using water as nucleophile for the synthesis of 4 (Scheme 2). The difunctionalization of various styrenes was successfully accomplished employing a mixture of acetone/water in a 9:1 ratio (Scheme 2). Optimization studies are detailed in the ESI.† Different trifluoromethyl alcohols were obtained in moderate to good yields (4a–j). The formation of 4g and 4h in high yields from the corresponding α-methyl and trans-β-methyl styrenes was also remarkable. Then, meta-bromostyrene afforded the final product 4d in higher yield than the corresponding para- and ortho-bromo styrenes. Also, the flurbiprofen derivative 4j was afforded in 56% isolated yield.
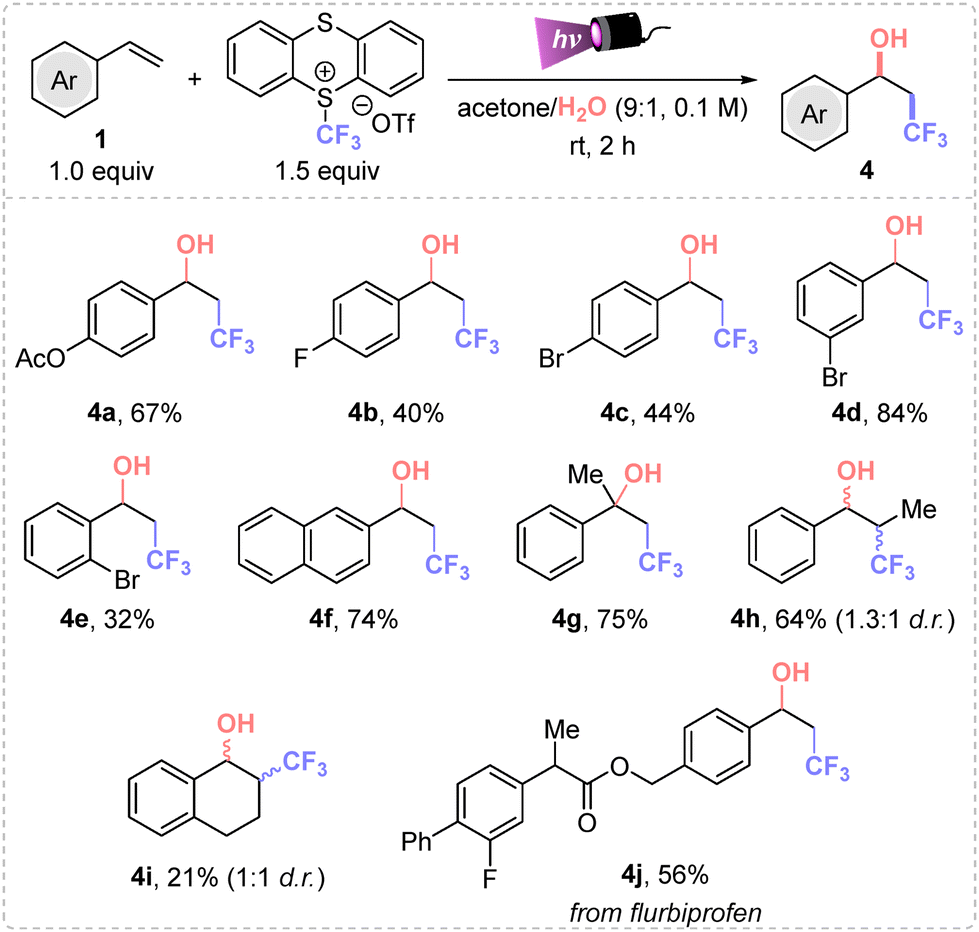 | ||
| Scheme 2 Scope of alcohols. Standard conditions: 1 (0.25 mmol) and TT-CF3OTf (1.5 equiv.), in dry degassed mixture of acetone/H2O (2.5 mL, 0.1 M) under purple Kessil irradiation (λmax = 390 nm) for 2 h at rt under inert atmosphere. | ||
Then, we compared the efficiency of the method with other nucleophilic partners than oxygenated ones. Azides are versatile functional groups that can be engaged in several transformations to easily increase molecular complexity. Thus, azidotrifluoromethylation of different (hetero)arylethylenes was achieved within minutes, using 3.5 equiv. of TMSN3 and dichloromethane as solvent (Scheme 3). A detailed optimization study is provided in the ESI (Table S5†). This protocol allowed the synthesis of different trifluoromethyl azides (5a–e) in moderate to good yields. Additionally, important pharmaceutical derivatives (flurbiprofen, ibuprofen and fenbufen) were successfully difunctionalized in high yields (5f–h). These examples highlight that this synthetic method enables the simultaneous late-stage installation of CF3- and N3-groups in complex molecules.
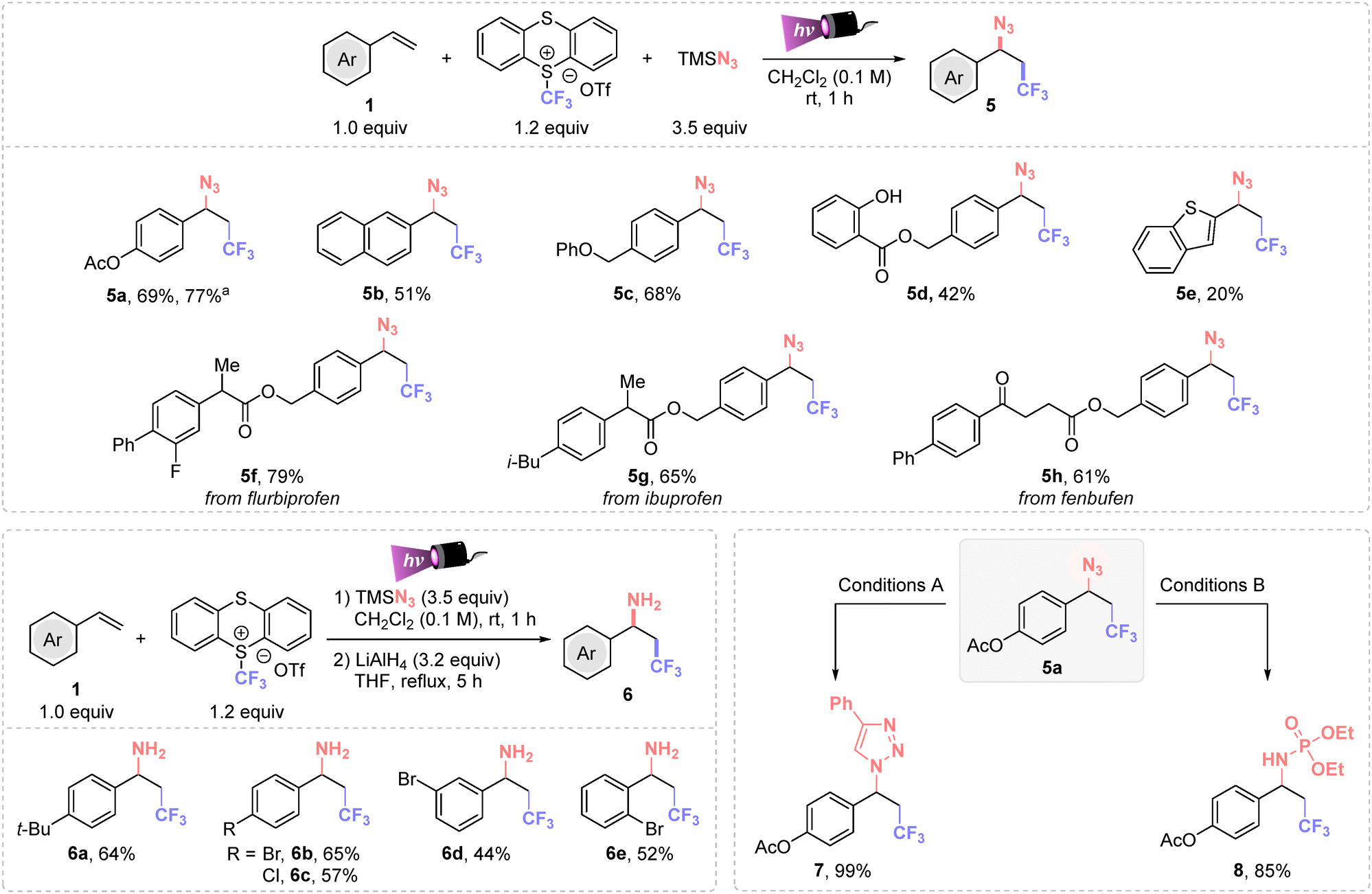 | ||
| Scheme 3 Scope of azides and derivatives. Standard conditions: 1 (0.5 mmol), TT-CF3OTf (1.2 equiv.) and TMSN3 (3.5 equiv.), in dry degassed CH2Cl2 (5 mL, 0.1 M) under purple Kessil irradiation (λmax = 390 nm) for 1 h at rt under inert atmosphere. aLarge scale synthesis. Conditions A: phenyl acetylene (3.0 equiv.) and CuI (30 mol%), in THF for 16 hours at 60 °C. Conditions B: triethylphosphite (2.0 equiv.) in CH2Cl2 for 16 hours at rt. | ||
Importantly, 1,2-aminotrifluoromethylated compounds are easily accessible using a straightforward two-step synthesis (Scheme 3, bottom left). This involved the sequential C(sp3)–CF3 and C(sp3)–N3 bonds formation followed by a LiAlH4 reduction. This protocol is particularly advantageous for 1,2-trifluoromethylated azides that may present issues during the isolation process from the thianthrene side product. The use of ortho-, meta- and para-bromo-, para-chloro- and 4-tert-butyl-styrenes successfully afforded the corresponding trifluoromethylated benzylic amines 6a–e in good yields.
The synthetic value of this multicomponent reaction was proved by the scalability of the reaction (Scheme 3, 5a isolated in 77% yield), and by downstream transformations of 5a. The formation of triazol 7 and iminophosphorane 8 were successfully achieved in 99 and 85%, respectively (Scheme 3, bottom right).
Finally, to elucidate the plausible mechanism involved in these transformations, control experiments were performed. The necessity of 390 nm Kessil lamp evidenced the photochemical nature of this transformation and the use of TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl) inhibited the reaction detecting the TEMPO-CF3 adduct by GCMS (Scheme 4).
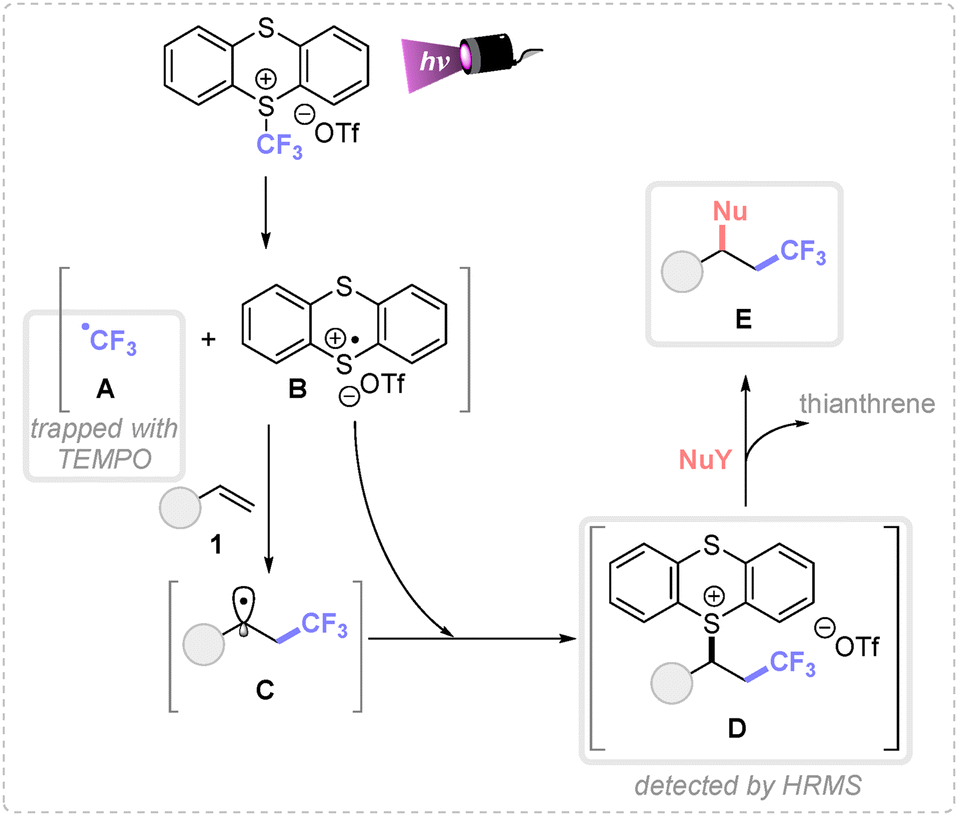 | ||
| Scheme 4 Proposed mechanism. | ||
Based on these experimental observations and related literature,4h,8 a plausible mechanism is depicted in Scheme 4. First, the direct irradiation of the TT-CF3OTf salt generates the corresponding trifluoromethyl radical A and the thianthrene radical cation B, by σ-homolytic cleavage of the S–C bond. Given the different reactivity of both species, A undergoes a fast and irreversible Giese addition with the activated alkene, forming the carbon-centered radical intermediate C. Then, this species yields the cationic species D by a radical–radical coupling with the persistent radical B. Remarkably, intermediate D was detected by HRMS analysis. Finally, the corresponding nucleophile attacks the most electrophilic position of intermediate D, displacing the thianthrene leaving group and forming the desired product E.
Conclusions
In conclusion, an operationally simple protocol for the alkoxy-, hydroxy-, and azidotrifluoromethylation of styrene derivatives is reported. These methods allowed the subsequent formation of a C(sp3)–C(sp3) and C(sp3)–heteroatom bonds from commercially or readily available compounds in one step. This difunctionalization method do not require the use of metals or exogenous photocatalysts. The efficiency of this reaction relies on the generation of ˙CF3 and thianthrene radical cation after homolytic fragmentation of the fluorinated reagent by simple 390 nm irradiation. The key intermediate is the sulfonium intermediate D that have shown to be versatile towards different substitution reactions. Moreover, the large-scale preparation and late-stage functionalization of pharmaceuticals illustrates the utility of this reported difunctionalization method.Author contributions
All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† Experimental procedures and analytical data (NMR, MS, IR, and melting points) can be found in the ESI.† Copies of NMR spectra are also provided.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ministerio de Ciencia, Innovación y Universidades–Agencia Estatal de Investigación (grants PID2020-113059GB-C22, PID2021-124916NB-I00, RED2022-134287-T, PID2021-128496OB-I00 and PID2023-146801NB-C32) and to the European Union's Horizon 2020 Research and Innovation programme under grant agreement no. 101034324 and to the AGAUR Generalitat de Catalunya 2021SGR00064. The authors are thankful to Prof. Roser Pleixats (UAB) for helpful discussions.Notes and references
- Selected examples: (a) Z. Tan, H. Zhang, K. Xu and C. Zeng, Electrochemical radical-polar crossover: a radical approach to polar chemistry, Sci. China: Chem., 2023, 67, 450–470 CrossRef; (b) S. K. Nanda, Catalytic Radical-Polar Crossover Non-Classical Semipinacol Rearrangements: The Sustainable Approach, Adv. Synth. Catal., 2023, 365, 834–853 CrossRef; (c) M. Liu, X. Ouyang, C. Xuan and C. Shu, Advances in photoinduced radical–polar crossover cyclization (RPCC) of bifunctional alkenes, Org. Chem. Front., 2024, 11, 895–915 RSC.
- L. Pitzer, J. L. Schwarz and F. Glorius, Reductive radical-polar crossover: traditional electrophiles in modern radical reactions, Chem. Sci., 2019, 10, 8285–8291 RSC.
- Selected examples: (a) R. J. Wiles and G. A. Molander, Photoredox-Mediated Net-Neutral Radical/Polar Crossover Reactions, Isr. J. Chem., 2020, 60, 281–293 CrossRef CAS PubMed; (b) Z. Zhu, Y. Zhang, Z. Li and C. Shu, Photoinduced radical-polar crossover cyclization reactions, Chem Catal., 2024, 4, 100945 CrossRef CAS; (c) Y. Liu, H. Liu, X. Liu and Z. Chen, Recent Advances in Photoredox-Catalyzed Difunctionalization of Alkenes, Catalysts, 2023, 13, 1056 CrossRef CAS; (d) A. Gallego-Gamo, P. Sarró, Y. Ji, R. Pleixats, E. Molins, C. Gimbert-Suriñach, A. Vallribera and A. Granados, Direct Synthesis of 2-Hydroxytrifluoroethylacetophenones via Organophotoredox-Mediated Net-Neutral Radical/Polar Crossover, J. Org. Chem., 2024, 16, 11682–11692 CrossRef PubMed.
- Selected examples: (a) A. García-Domínguez, R. Mondal and C. Nevado, Dual Photoredox/Nickel-Catalyzed Three-Component Carbofunctionalization of Alkenes, Angew. Chem., Int. Ed., 2019, 58, 12286–12290 CrossRef PubMed; (b) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, Three-Component Olefin Dicarbofunctionalization Enabled by Nickel/Photoredox Dual Catalysis, J. Am. Chem. Soc., 2019, 141, 20069–20078 CrossRef CAS PubMed; (c) M. J. Cabrera-Afonso, A. Sookezian, S. O. Badir, M. El Khatib and G. A. Molander, Photoinduced 1,2-dicarbofunctionalization of alkenes with organotrifluoroborate nucleophiles via radical/polar crossover, Chem. Sci., 2021, 12, 9189–9195 RSC; (d) S. Patra, R. Giri and D. Katayev, Nitrative Difunctionalization of Alkenes via Cobalt-Mediated Radical Ligand Transfer and Radical-Polar Crossover Photoredox Catalysis, ACS Catal., 2023, 13, 16136–16147 CrossRef CAS; (e) H. Singh, R. K. Tak, D. P. Poudel and R. Giri, Catalytic Photoredox Carbobromination of Unactivated Alkenes with α-Bromocarbonyls via the Mechanistically Distinct Radical-Addition Radical-Pairing Pathway, ACS Catal., 2024, 14, 6001–6008 CrossRef CAS; (f) E. A. Noten, C. H. Ng, R. M. Wolesensky and C. R. J. Stephenson, A general alkene aminoarylation enabled by N-centred radical reactivity of sulfonamides, Nat. Chem., 2024, 16, 599–606 CrossRef CAS PubMed; (g) R. K. Dhungana, A. Granados, V. Ciccone, R. T. Martin, J. Majhi, M. Sharique, O. Gutierrez and G. A. Molander, Trifunctionalization of Cinnamyl Alcohols Provides Access to Brominated α,α-Difluoro-γ-lactones via a Photoinduced Radical–Polar–Radical Mechanism, ACS Catal., 2022, 12, 15750–15757 CrossRef CAS; (h) S. Shibutani, K. Nagao and H. Ohmiya, Organophotoredox-Catalyzed Three-Component Coupling of Heteroatom Nucleophiles, Alkenes, and Aliphatic Redox Active Esters, Org. Lett., 2021, 23, 1798–1803 CrossRef CAS PubMed.
- B. Fu, J. Escorihuela, J. Han, S. Fustero, P. Barrio, M. Sodeoka, S. Kawamura, A. Sorochinsky and V. A. Soloshonok, Recent Advances on the Halo- and Cyano-Trifluoromethylation of Alkenes and Alkynes, Molecules, 2021, 26, 7221 CrossRef CAS PubMed.
- Selected examples: (a) F. Wang, X. Qi, Z. Liang, P. Chen and G. Liu, Copper-Catalyzed Intermolecular Trifluoromethylazidation of Alkenes: Convenient Access to CF3-Containing Alkyl Azides, Angew. Chem., Int. Ed., 2014, 53, 1881–1886 CrossRef CAS PubMed; (b) C.-L. Zhu, C. Wang, Q.-X. Qin, S. Yruegas, C. D. Martin and H. Xu, Iron(II)-Catalyzed Azidotrifluoromethylation of Olefins and N-Heterocycles for Expedient Vicinal Trifluoromethyl Amine Synthesis, ACS Catal., 2018, 8, 5032–5037 CrossRef CAS PubMed; (c) Y. Ouyang, C.-L. Tong, X.-H. Xu and F.-L. Qing, Copper and Zinc Copromoted Bromo(chloro)trifluoromethylation of Alkenes and Alkynes with Trifluoromethanesulfonic Anhydride, Org. Lett., 2021, 23, 346–350 CrossRef CAS PubMed.
- H. Jia, A. P. Häring, F. Berger, L. Zhang and T. Ritter, Trifluoromethyl Thianthrenium Triflate: A Readily Available Trifluoromethylating Reagent with Formal CF3+, CF3˙, and CF3− Reactivity, J. Am. Chem. Soc., 2021, 143, 7623–7628 CrossRef CAS PubMed.
- (a) Y. Li, X. Liang, K. Niu, J. Gu, F. Liu, Q. Xia, Q. Wang and W. Zhang, Visible-Light-Induced Photocatalyst-Free Radical Trifluoromethylation, Org. Lett., 2022, 24, 5918–5923 CrossRef CAS PubMed; (b) X. He, Z. Chen, X. Zhu, H. Liu, Y. Chen, Z. Sun and W. Chu, Photoredox-catalyzed trifluoromethylation of 2H-indazoles using TT-CF3+OTf− in ionic liquids, Org. Biomol. Chem., 2023, 21, 1814–1820 RSC; (c) M. Luo, S. Zhu, C. Yang, L. Guo and W. Xia, Photoinduced Regioselective Fluorination and Vinylation of Remote C(sp3)–H Bonds Using Thianthrenium Salts, Org. Lett., 2024, 26, 4388–4393 CrossRef CAS PubMed; (d) S. R. Nasireddy, G. C. Upreti and A. Singh, Photochemical Trifluoromethylative Difunctionalization of Styrenes and Phenylacetylenes via a Catalytic EDA Platform, Eur. J. Org. Chem., 2024, e202400114 CrossRef CAS; (e) F. Xiang, D. Wang, K. Xu and C.-C. Zeng, Paired Electrolysis Enabled Trifluoromethylheteroaromatization of Alkenes and Alkyne with Trifluoromethyl Thianthrenium Triflate (TT-CF3+OTf−) as a Bifunctional Reagent, Org. Lett., 2024, 26, 411–415 CrossRef CAS PubMed.
- H. Jia and T. Ritter, α-Thianthrenium Carbonyl Species: The Equivalent of an α-Carbonyl Carbocation, Angew. Chem., Int. Ed., 2022, 61, e202208978 CrossRef CAS PubMed.
- Selected examples: (a) Y. Yasu, T. Koike and M. Akita, Three-component Oxytrifluoromethylation of Alkenes: Highly Efficient and Regioselective Difunctionalization of C=C Bonds Mediated by Photoredox Catalysts, Angew. Chem., Int. Ed., 2012, 51, 9567–9571 CrossRef CAS PubMed; (b) G. Dagousset, A. Carboni, E. Magnier and G. Masson, Photoredox-Induced Three-Component Azido- and Aminotrifluoromethylation of Alkenes, Org. Lett., 2014, 16, 4340–4343 CrossRef CAS PubMed; (c) M. Zhang, J.-H. Lin and J.-C. Xiao, A Readily Available Trifluoromethylation Reagent and Its Difunctionalization of Alkenes, Org. Lett., 2021, 23, 6079–6083 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo01520g |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2024 |