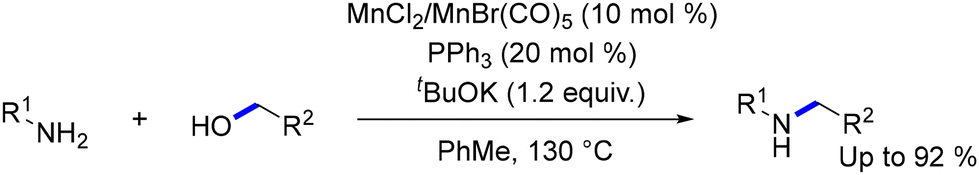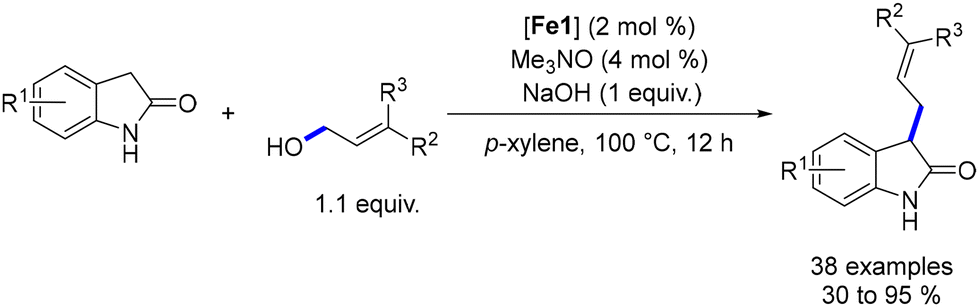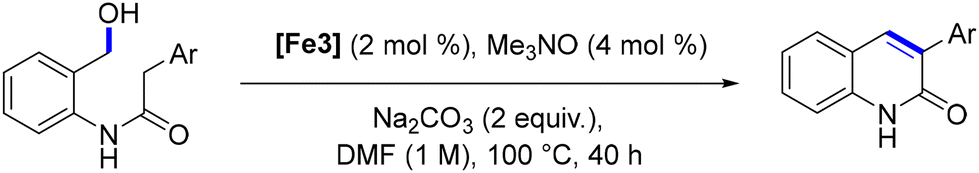DOI:
10.1039/D4QO01626B
(Review Article)
Org. Chem. Front., 2024,
11, 7278-7317
Hydrogen autotransfer with alcohols for alkylations†
Received
3rd September 2024
, Accepted 4th October 2024
First published on 7th October 2024
Abstract
Despite the advancements enabled by organometallic complexes in organic synthesis, which have led to innovative transformations and tackled issues related to waste and atom economy, concerns persist regarding the cost of noble metals and ligands. Expanding on previous work, iron and ruthenium complexes with cyclopentadienone ligands, akin to Knölker catalysts, have demonstrated remarkable efficiency in bond reduction and alkylations using alcohols as pro-electrophiles. This review delves into novel alkylation methodologies involving hydrazides or ketones, inventive dehydrogenative coupling reactions yielding highly functionalized structures, and the optimization of metal-catalysed reactions through organometallic complex modifications. Metal-catalysed hydrogen auto-transfer, or hydrogen borrowing, offers a sustainable approach to forming C–C or C–N bonds from eco-friendly alcohols. While transitioning the diaminocyclopentadienone tricarbonyl ligand to first-row transition metals under mild conditions remains challenging, recent findings indicate that blue-light irradiation at room temperature can facilitate this transformation without external photosensitizers. Thus, while conventional studies without light remain demanding, the incorporation of light can enhance this research domain, where such catalysts may play a pivotal role. Moreover, further exploration of asymmetric catalysis is warranted. This review aims to benefit not only the community working with Knölker derivative catalysts but also organic chemists seeking potential applications and inorganic chemists interested in catalyst development, particularly in pursuit of catalysts for asymmetric syntheses. Additionally, the focus on sustainability makes it relevant for chemists dedicated to green chemistry.
 Nicolas Joly | Dr Nicolas Joly obtained his Master's Degree in Molecular Chemistry in 2020 from ENSCR in Rennes. In 2023, he completed his PhD in Chemistry at both the University of Caen Normandy and the University of Girona under the supervision of Prof. Jean-Luc Renaud and Dr Albert Poater. He is now a post-doctoral researcher in the group of Dr Thibault Cantat at CEA Paris-Saclay. His research interests focus on organic synthesis and catalysis, particularly on molecular transformations through sustainable processes. |
 Sylvain Gaillard | Dr Sylvain Gaillard received his PhD from Université Paul Cézanne/Aix-Marseille III. After completing a first postdoctoral position at ENSCR in Rennes, he pursued a second post-doctoral position under the supervision of Professor S. P. Nolan at the University of St Andrews. In 2010, he joined the group of Professor J.-L. Renaud at the University of Caen Normandy as an associate professor. His research interests focus on molecular design of transition metal complexes, tuning their photophysical properties for applications in photocatalysis, hybrid materials and biological activities. |
 Albert Poater | Dr Albert Poater completed his PhD in Chemistry in 2006 from the University of Girona. After research stints in Chile, Montpellier, and an extended postdoc at the University of Salerno, he became an independent researcher in 2010 as a Ramón y Cajal fellow in Girona. He has also served as a Visiting Researcher in Saudi Arabia and Toulouse. In 2019, he was appointed Serra Húnter Associate Professor at the University of Girona and received the ICREA ACADÈMIA award. He has published over 300 papers with around 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 citations (H-index = 66), specializing in DFT calculations for inorganic and organometallic catalysis, with a focus on green chemistry. 000 citations (H-index = 66), specializing in DFT calculations for inorganic and organometallic catalysis, with a focus on green chemistry. |
 Jean-Luc Renaud | Prof. Jean-Luc Renaud obtained his PhD degree in 1998 under the supervision of Dr Aubert and Prof. Malacria. In 1999 he became a Lavoisier Postdoctoral fellow with Prof. Lautens at the University of Toronto and then moved to the University of Louvain-La-Neuve and joined the team of Prof. Riant. In 2000, he was appointed Maître de Conférences at the University of Rennes and accepted a full Professor position at the University of Caen Normandy in 2008. Since October 2023, he has been an invited researcher at the “Institut Parisien de Chimie Moléculaire (IPCM)”, Sorbonne University. His research interests focus on organometallic catalysis, organocatalysis, photoredox catalysis and their applications towards the synthesis of biologically interesting molecules and processes relevant to fine chemical synthesis. |
1. Introduction
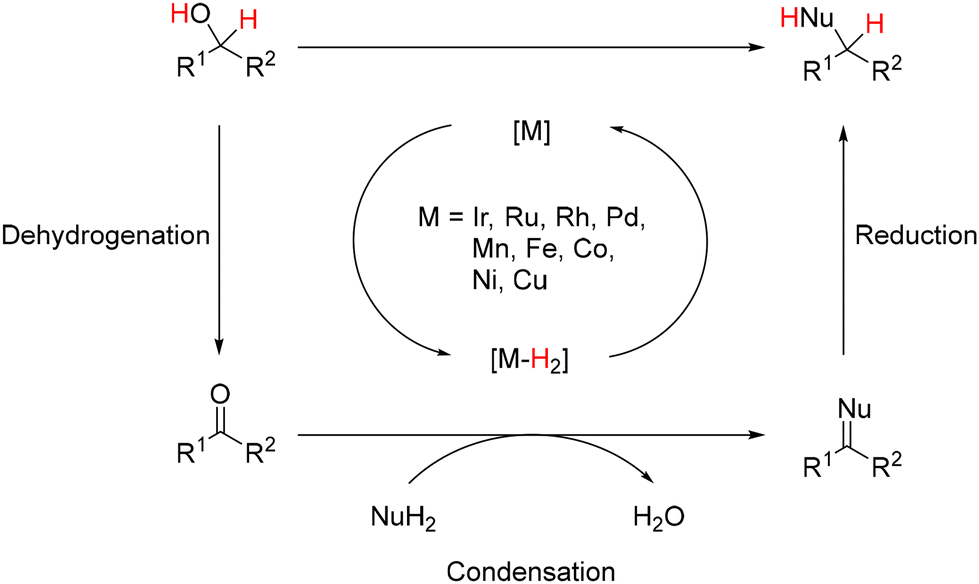
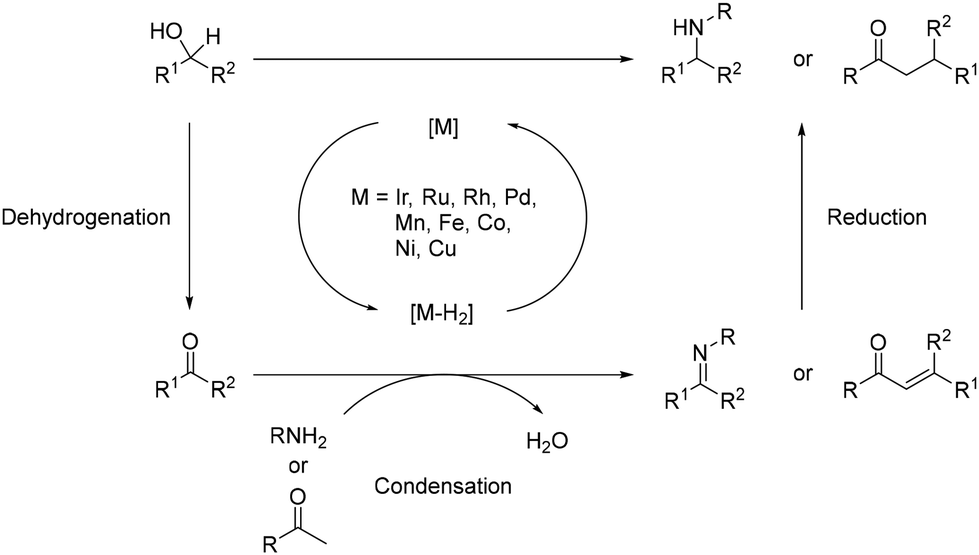
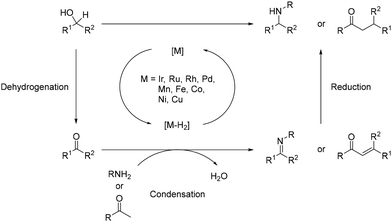
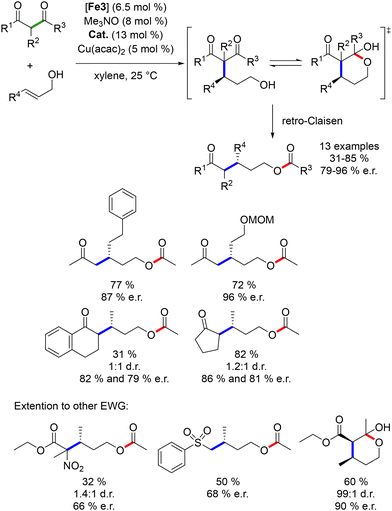
Over the past few decades, novel methodologies have emerged for forming C–N and C–C bonds through hydrogen autotransfer reactions. The term “Borrowing Hydrogen” was coined by Williams in 2004,1 although similar reactions have been reported previously,2–5 and these reactions involve alcohols as alkylating agents instead of halide derivatives. The latter typically require additional synthetic steps and generate waste.6,7 Thus, using alcohols for synthesizing carbon- and nitrogen-containing compounds is promising due to their natural occurrence and direct accessibility from biomass,8,9 aligning with the principles of green and sustainable chemistry.10–12 The general mechanism involves dehydrogenation of an alcohol in the presence of an organometallic complex to yield the corresponding carbonyl derivative and a metal–hydride complex (Scheme 1). The resulting unsaturated species then undergoes condensation with a nucleophile, culminating in reduction via the metal–hydride complex.
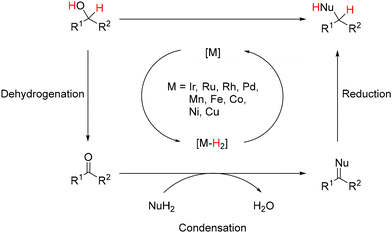 |
| | Scheme 1 General mechanism of hydrogen autotransfer reactions (M = metal; Nu = nucleophile; R = substituent). | |
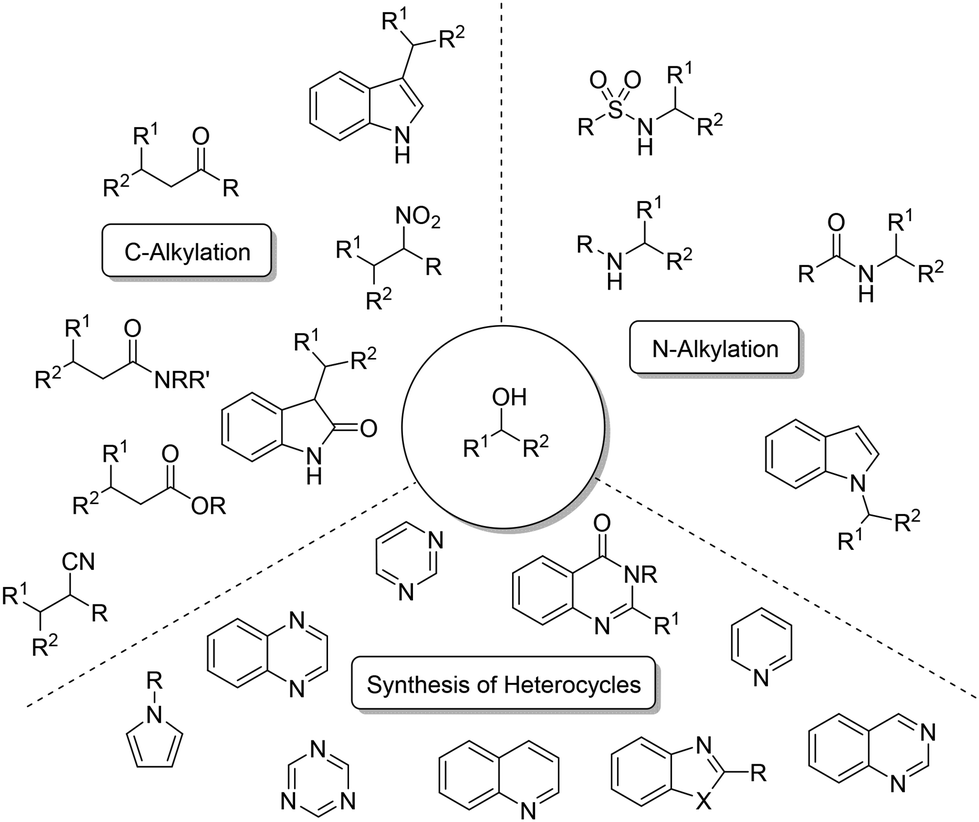
Several precious metals like iridium,13 rhodium,14,15 and ruthenium16,17 have been documented for their efficacy in facilitating such reactions. These reactions include the alkylation of diverse nucleophiles, including ketones,18 amides or esters,19 alongside amines,20–22 nitriles,23 and nitro derivatives (see Scheme 2).24 Additionally, alkylations and the syntheses of heterocycles have also been documented,25–28 particularly with acceptorless dehydrogenation.29–32
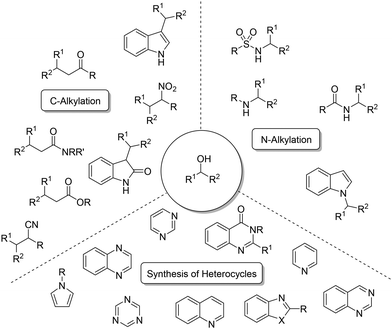 |
| | Scheme 2 Examples of alkylated products and heterocycles. | |
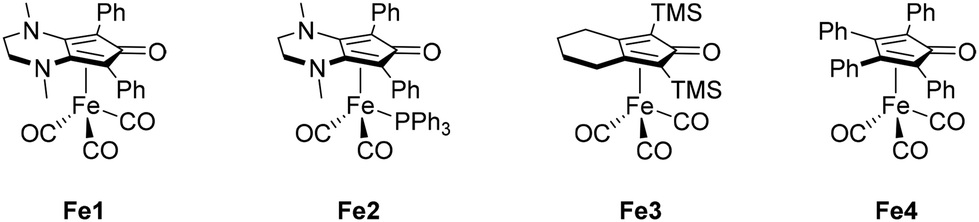
However, the scarcity of these metals and the constantly increasing demand have made their usage increasingly expensive. In recent years, Earth abundant transition-metal complexes, such as manganese, iron, or cobalt, have demonstrated their efficiency and competitiveness in alkylation reactions compared to noble metals.33 Our research group became particularly interested in iron, which is the most Earth abundant transition metal and therefore the most economical (costing only a few cents per kilogram). The advancements in iron chemistry in recent years have been facilitated by the use of organophosphorus ligands.34 However, these ligands are often sensitive to air and moisture and can be costly. The development of less expensive ligands such as cyclopentadienones aims to address these challenges. Building on the work of Knölker (Fe3, shown in Scheme 3),35–37 our group has synthesized various iron complexes featuring cyclopentadienone ligands (Fe1 and Fe2, among others) and has contributed to their application in homogeneous catalysis, particularly in the reduction of carbonyls38 and reductive amination.39 The iron complex Fe1 has demonstrated higher activity40–44 and better selectivity than the Knölker complex Fe3 and has been employed in various alkylation reactions involving anilines,45 indoles,46 alcohols,47 and ketones.48–51 In fact, predictive catalysis studies,52 have even led to improvements when using aldehydes with derivative Fe1 catalyts,53,54 employing the effective oxidation state analysis and oxidation state localized orbitals of Gimferrer, Salvador and coworkers.55,56
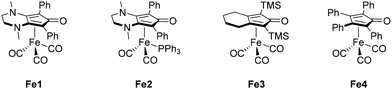 |
| | Scheme 3 Knölker derivative iron complexes employed by the Renaud group. | |
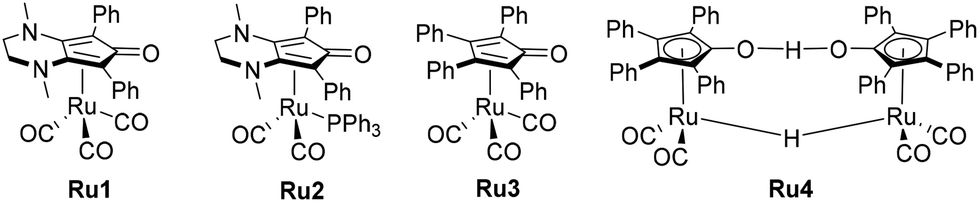
Building on these previous studies, the primary objectives of this review are to inspire the development of novel methodologies for forming C–N and C–C bonds, to investigate reaction mechanisms, and to enhance Knölker-type catalysts. This manuscript is structured as follows: it provides a comprehensive review of the state-of-the-art techniques for C–N and C–C bond formation using the hydrogen borrowing methodology,57–59 for these particular couplings,60,61 with a focus on 3d-metal catalysed reactions employing alcohols as alkylating agents,62,63 including the latest insights in the field,64 particularly focusing on alkylation with hydrazide derivatives as N-nucleophiles, showcasing the limitations of iron complexes and comparing them to ruthenium analogues (Scheme 4). Finally, the review will delve into the novel room temperature alkylation methodology utilizing light as an energy source.
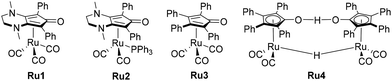 |
| | Scheme 4 Ruthenium complexes employed for hydrogenation, analogues of the Knölker derivative iron complexes (Ru1–Ru3) and Schvo catalyst (Ru4). | |
2.
N-Alkylation reactions catalysed by non-noble metals
The alkylation of amines or ketones typically necessitates the utilization of alkyl halides as electrophiles alongside a stoichiometric quantity of a base.65,66 However, this approach often encounters issues such as lack of selectivity, leading to over-alkylation reactions and the generation of challenging-to-handle by-products like salts,67 in particular, halogen salts. Consequently, novel methodologies have been devised, with hydrogen autotransfer emerging as a notable alternative.68,69 In this methodology, the formation of C–N bonds typically involves the utilization of a nucleophilic nitrogen-containing reagent. Following dehydrogenation of an alcohol into the corresponding carbonyl by a metal complex, a condensation step yields an imine/iminium intermediate (Scheme 5). Subsequently, this intermediate is reduced by a metal-hydrogen species to yield the N-alkylated product. Meanwhile, for the formation of C–C bonds, ketones serve as pro-nucleophiles. The unsaturated intermediate arises from a Knoevenagel condensation between the ketone and the dehydrogenated alcohol. Finally, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the α,β-unsaturated ketone is reduced by the metal-hydrogen species to yield the α-alkylated ketone (Scheme 5). Importantly, the only by-product of this reaction is water, originating from the condensation step.
C bond of the α,β-unsaturated ketone is reduced by the metal-hydrogen species to yield the α-alkylated ketone (Scheme 5). Importantly, the only by-product of this reaction is water, originating from the condensation step.
 |
| | Scheme 5 Streamlined mechanism depicting the alkylation of amines and ketones via hydrogen autotransfer. | |
Extensive research has been conducted on hydrogen autotransfer reactions in recent decades.16,27,34,70–74 Numerous catalysts, both noble and non-noble metals, have been documented to facilitate these reactions. This section will focus on the creation of C–N bonds through hydrogen autotransfer reactions utilizing alcohols catalysed by homogeneous first-row transition-metal catalysts.
2.1. Manganese-catalysed N-alkylation reactions
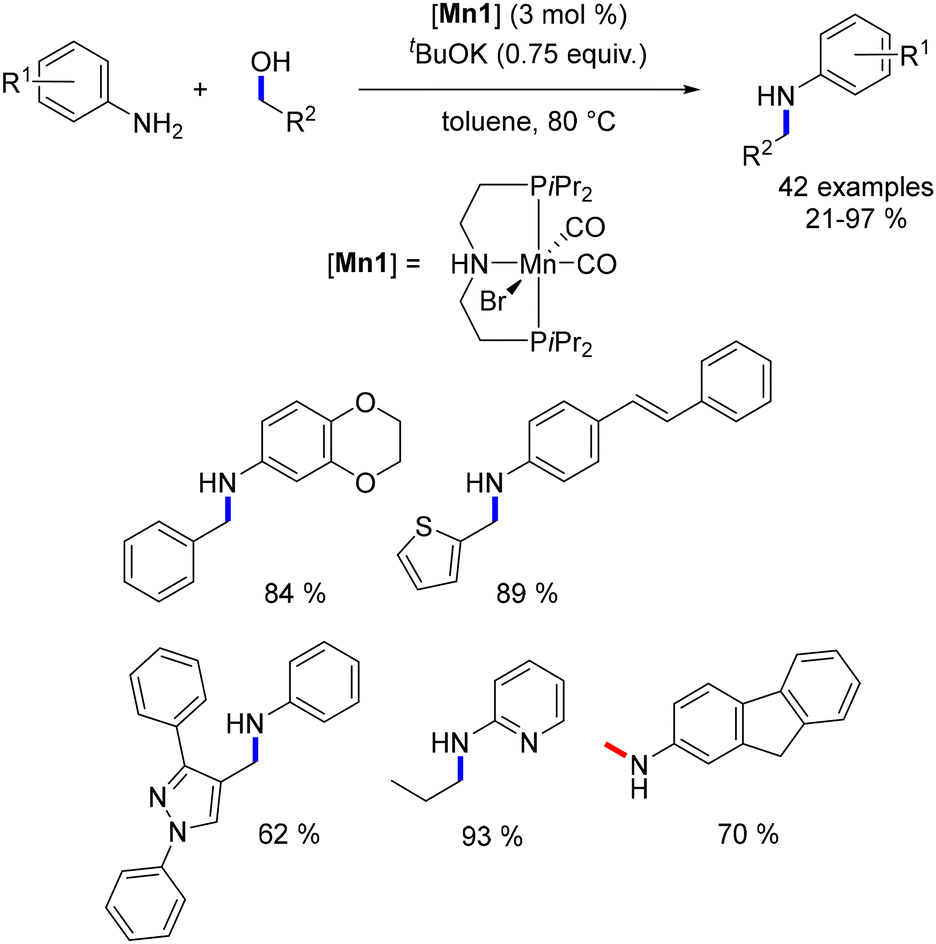
In 2016, Beller and colleagues reported the initial alkylation of amines with alcohols utilizing the Mn–Macho catalyst (Mn1).75 Aniline derivatives underwent alkylation with primary alcohols, yielding exclusively the mono-alkylated product (Scheme 6). This reaction exhibited tolerance towards various functional groups, enabling the synthesis of substituted products. Methylation presented a greater challenge due to the higher energy requirement for the dehydrogenation of methanol compared to other alcohols (for instance, ΔH = +84 kJ mol−1 for methanol versus ΔH = +68 kJ mol−1 for ethanol). Nevertheless, the Mn–Macho complex effectively catalysed the methylation of aniline-type derivatives in the presence of one equivalent of base at 100 °C (Scheme 6). The resulting methylated anilines were obtained in yields ranging from 52% to 94%.
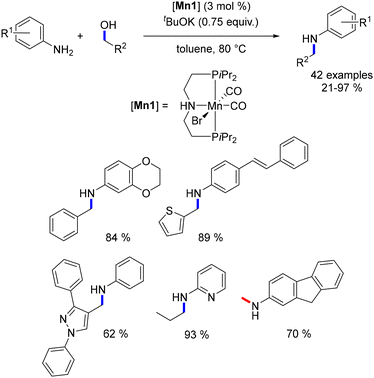 |
| | Scheme 6 Manganese-catalysed alkylation of amines. | |
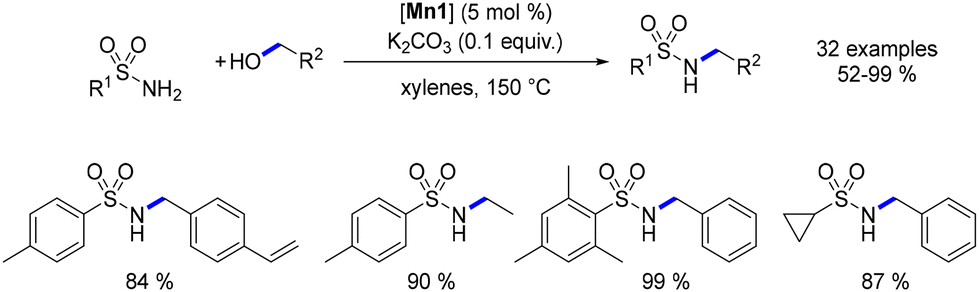
In 2019, Morrill reported the alkylation of sulfonamides catalysed by the Mn–Macho complex.76 Utilizing this methodology, a range of N-substituted sulfonamides were synthesized, albeit requiring a high temperature (150 °C) to achieve satisfactory yields (Scheme 7).
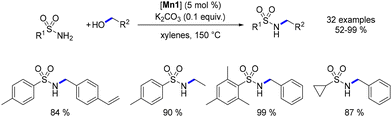 |
| | Scheme 7 Alkylation of sulfonamides. | |
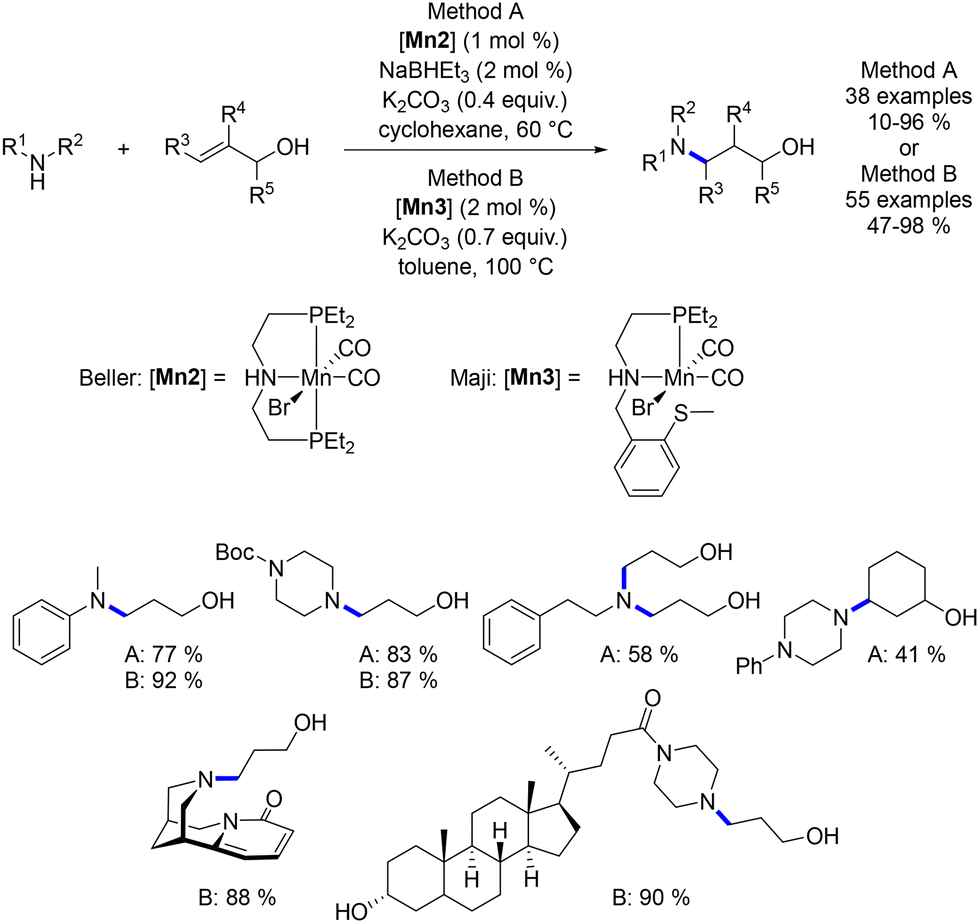
In a more recent development, Beller and Maji described the hydroamination of allylic alcohols.77,78 This reaction presents a deviation from previous alkylations as it involves a 1,4-addition reaction occurring on the α,β-unsaturated aldehyde intermediate, resulting in the formation of a γ-amino-alcohol as the final product (Scheme 8).
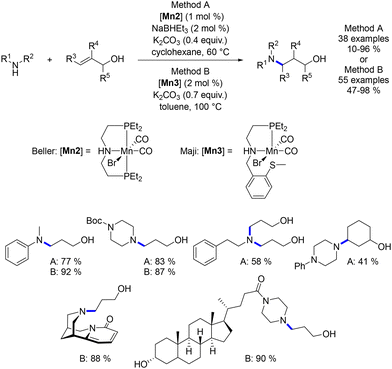 |
| | Scheme 8 Protocol for the synthesis of γ-amino-alcohols, as described by Beller and Maji. | |
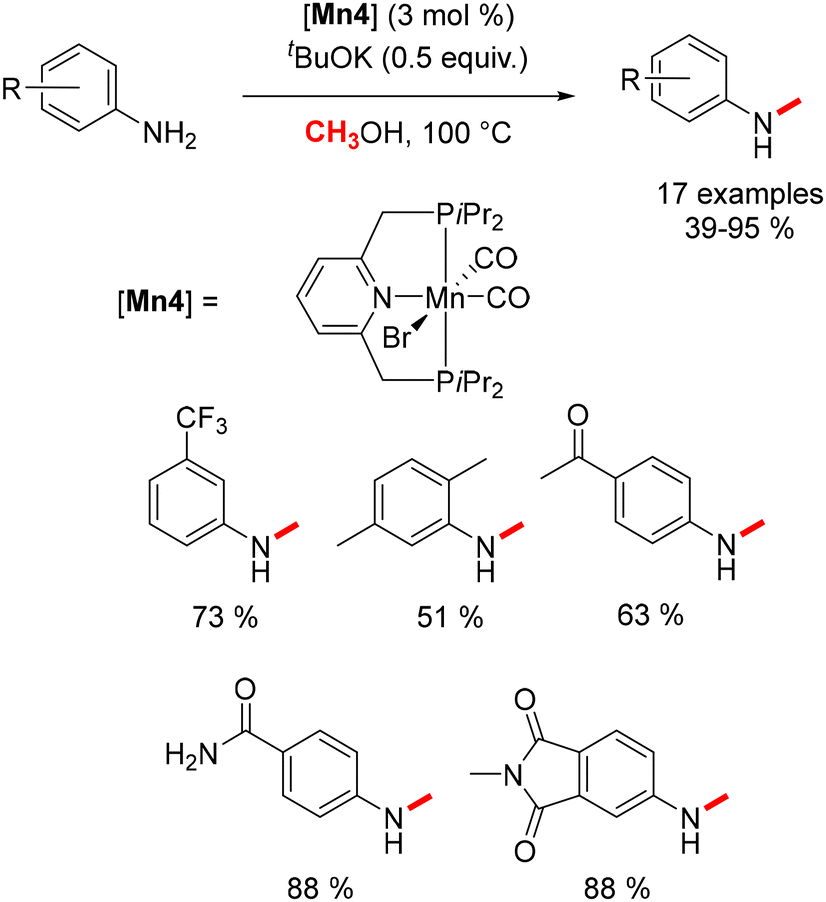
Substituting the Macho ligand with a PNP pincer-type ligand, characterized by a pyridine-diphosphine structure, resulted in enhanced methylation reaction outcomes. Under conditions of 100 °C and in the presence of 0.5 equivalents of base, N-methyl anilines were afforded in yields ranging from 39% to 95% (Scheme 9).79
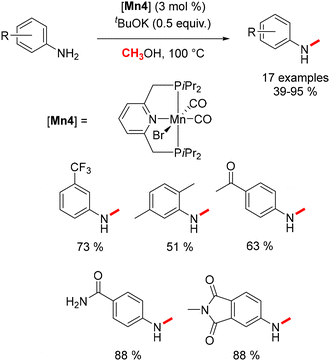 |
| | Scheme 9 Methylation of aniline derivatives. | |
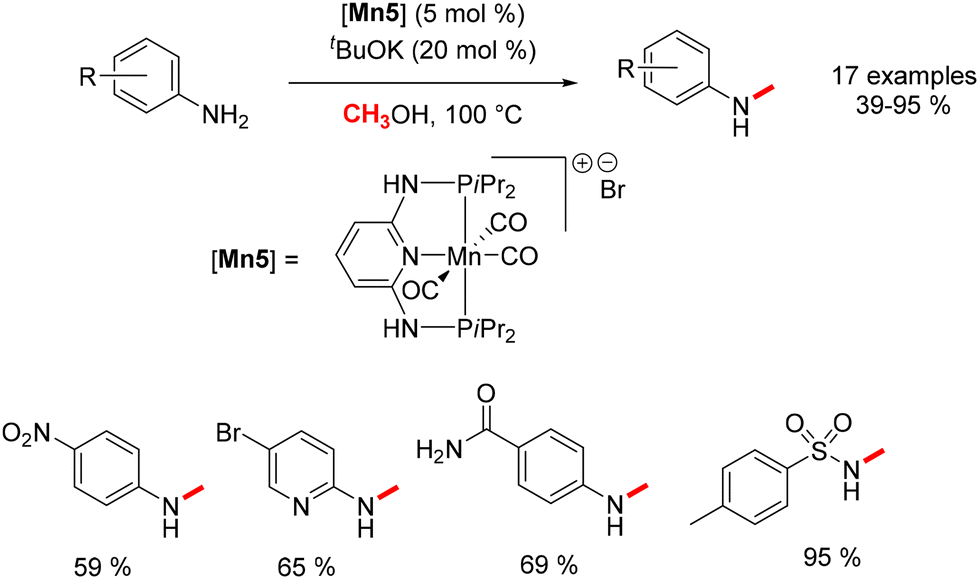
Concurrent with Beller's work, Sortais explored the utilization of a different PNP–Mn complex in the methylation of aniline derivatives.80 The process yielded N-methylated anilines in yields ranging from 39% to 95% (Scheme 10). Various electron-withdrawing groups (EWGs) and electron-donating groups (EDGs) were assessed to ascertain the extent of applicability of this reaction.
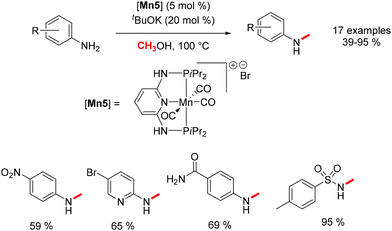 |
| | Scheme 10 Methylation of aniline derivatives, as reported by Sortais. | |
Morill devised a synthetic approach for the one-pot transformation of nitroarenes into N-methylaryl amines.81 Initially, the cationic manganese complex facilitates the dehydrogenation of methanol and concurrently reduces the nitro group to an amine. Subsequently, methylation occurs, resulting in the formation of the methylated aniline (Scheme 11).
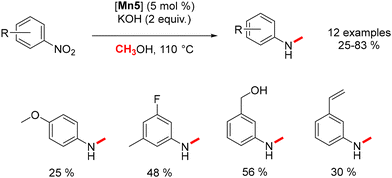 |
| | Scheme 11 Synthesis of methylaryl amines. | |
In 2018, Kempe introduced a base-switchable method for the synthesis of amines or imines.82 A PNP ligand featuring a triazine motif was developed, and the resultant Mn-complex was employed in the alkylation of anilines. Depending on the base cation (K+ or Na+), starting from the same amine and alcohol, either the N-alkyl amine, through a hydrogen autotransfer process, or the imine, via dehydrogenative condensation, was isolated (Scheme 12). This shift in reactivity was attributed to the chelation capability of the cation and the corresponding manganese hydride intermediate's reactivity. The potassium manganate hydride reduced the imine faster than the sodium salt.
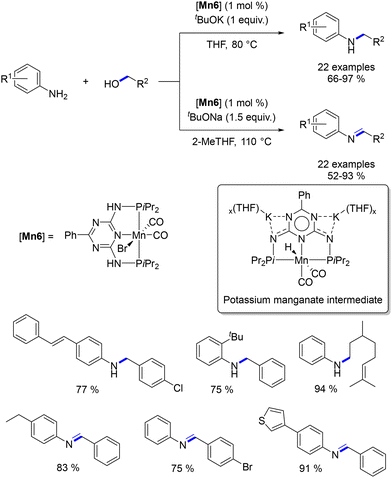 |
| | Scheme 12 Alkylation of aniline derivatives. | |
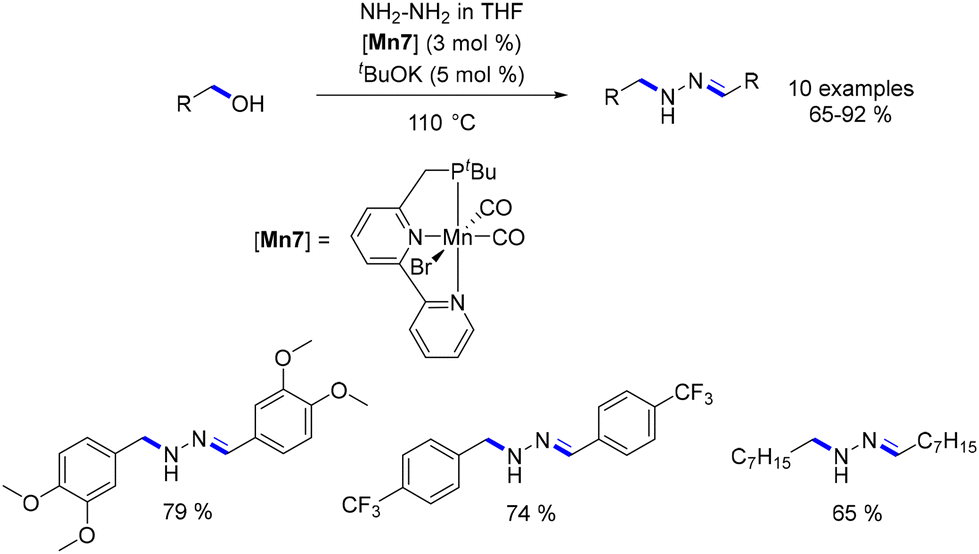
Refinement of the ligand enabled precise adjustment of reactivity. In 2017, Milstein substituted the PNP ligand with a phosphine-bipyridine ligand (PNN ligand) and utilized the resulting Mn-complex in the alkylation of hydrazine (Scheme 13).83 Remarkably, this reaction yielded N-substituted hydrazones through a combination of hydrogen transfer for the alkylation step and dehydrogenation for the formation of the hydrazone moiety.
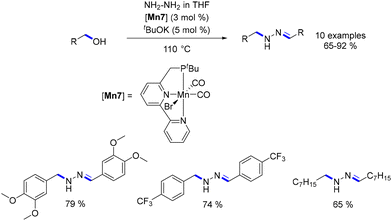 |
| | Scheme 13 Synthesis of N-substituted hydrazones. | |
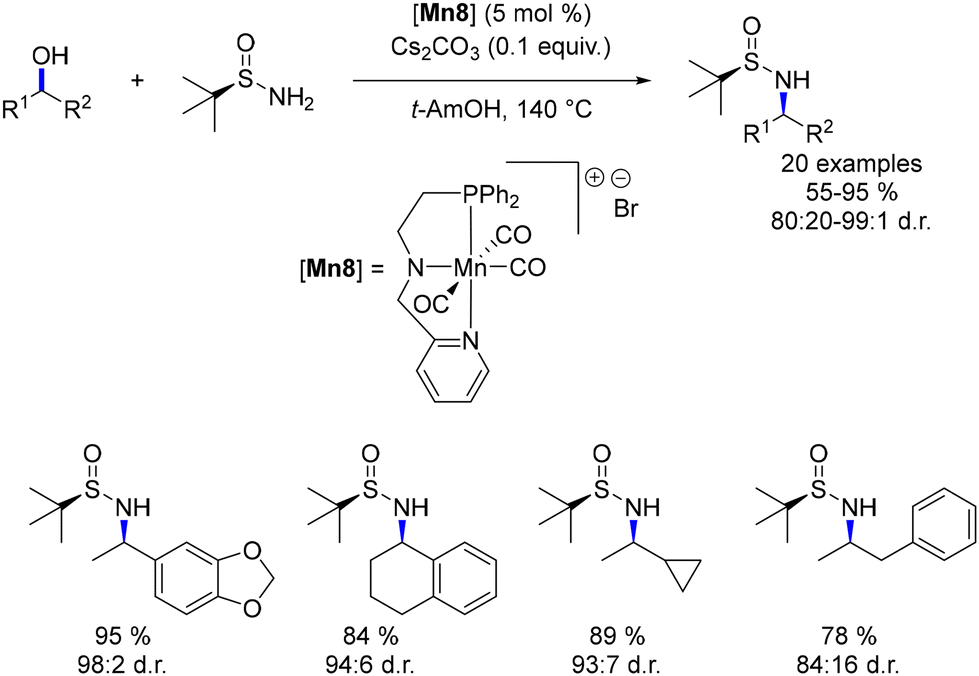
Rueping introduced a modification in which the central pyridine ring of the ligand was substituted with an amine, resulting in the formation of a new cationic manganese complex.84 This complex was employed in an asymmetric amination of secondary alcohols (Scheme 14). The presence of a tert-butyl sulfoximine group facilitated an asymmetric reduction of the imine intermediate. This tert-butyl group directed the Mn–H complex towards the less hindered side of the imine, thereby achieving diastereoselective reduction with up to 98:2 dr.
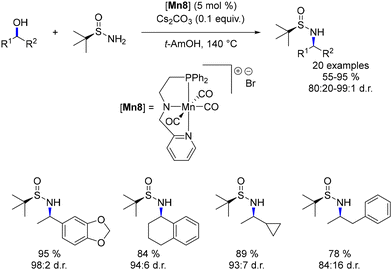 |
| | Scheme 14 Asymmetric amination of secondary alcohols. | |
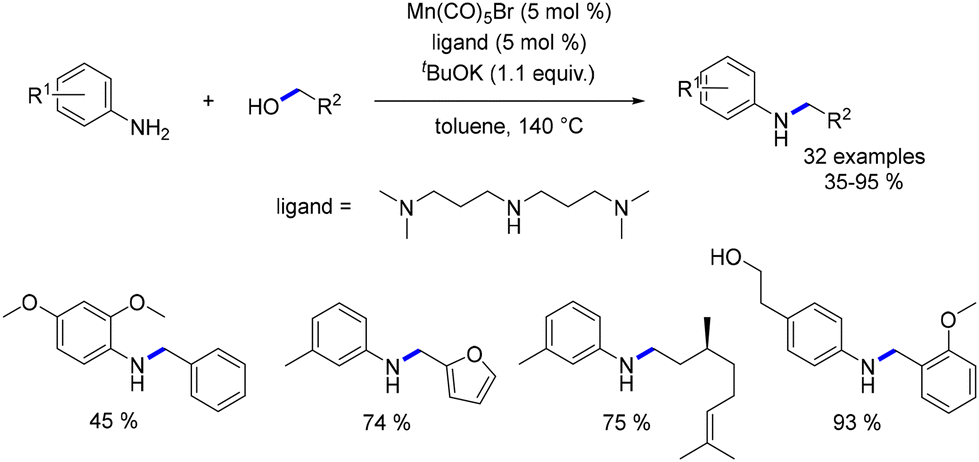
In 2018, Balaraman presented an economical method for the N-alkylation of anilines using benzylic and aliphatic alcohols.85 A manganese salt (Mn(CO)5Br) was combined with N1-(3-(dimethylamino)propyl)-N3,N3-dimethylpropane-1,3-diamine (NNN ligand) to generate the catalytic agent. Both electron-rich and electron-poor N-substituted anilines were obtained in yields ranging from 35% to 95% (Scheme 15). This approach is competitive with the catalytic systems of Beller and Kempe, as the catalytic species is prepared in situ, although it results in slightly lower product yields.
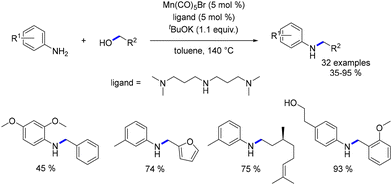 |
| | Scheme 15 Alkylation of aniline derivatives. | |
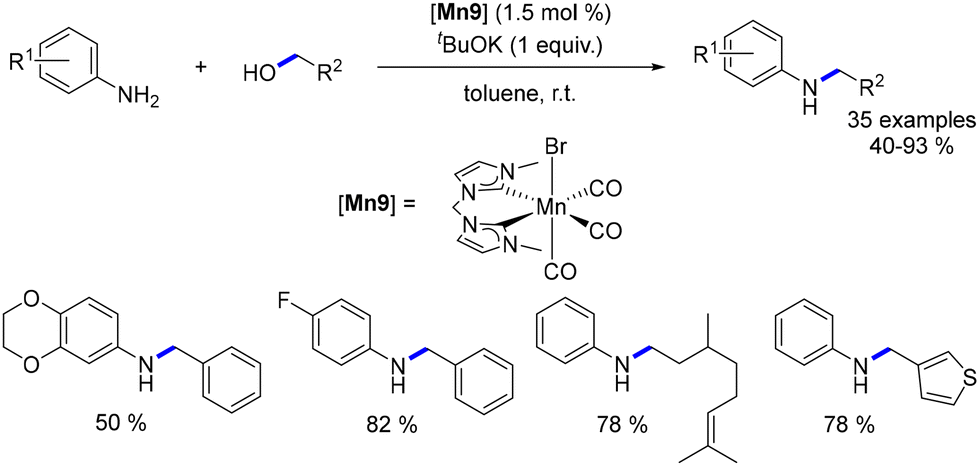
The subsequent year, Ke introduced the first non-noble metal system operating at room temperature for the alkylation of aniline derivatives.86 Employing a manganese complex with an N-heterocyclic carbene (NHC) ligand, the reaction of anilines with benzylic and aliphatic alcohols proceeded smoothly under ambient conditions. The resulting N-substituted products were obtained in yields ranging from 40% to 93% (Scheme 16). Investigation into methylation was also undertaken, revealing that a temperature of 100 °C was necessary to achieve the methylated products in yields of 53% to 94%. Mechanistic elucidation via Density Functional Theory (DFT) calculations highlighted an outer-sphere mechanism with a maximum energy barrier of 23.7 kcal mol−1. This stands in contrast to the inner-sphere mechanism, which necessitated the de-coordination of a CO ligand, resulting in a maximum energy barrier of 46.4 kcal mol−1.
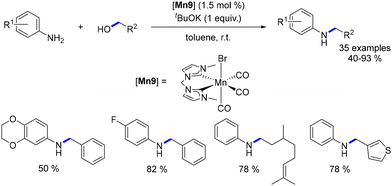 |
| | Scheme 16 Alkylation of aniline derivatives. | |
In 2023, Balaraman and coworkers explored a general hydrogenative cleavage/N-alkylation tandem reaction of cyclic and acyclic diazo (NN) compounds to synthesize value-added amines under manganese catalysis.87 The reaction is catalyzed by a single-site pincer based molecular manganese complex and proceeds through tandem dehydrogenation, transfer hydrogenation, and borrowing hydrogenation mechanisms. Notably, the process utilizes abundantly available renewable feedstocks, such as alcohols, which serve as both (transfer)hydrogenating and alkylating agents.
Actually, several Mn systems have been developed that enable the selective N-alkylation of amines with alcohols,129,130 including reusable nanocatalysts,131 manganese dioxide facilitating the transformation of amines into sulfonamides in yields up to 99%,132 or heterogeneized manganese catalyst obtained by the pyrolysis of molecularly defined complexes.133 In 2021, the inexpensive and non-toxic MnCl2 or Mn(CO)5Br, with triphenylphosphine as a ligand, was introduced, enabling the efficient synthesis of a variety of aromatic, heteroaromatic, and aliphatic secondary amines in moderate to high yields (Scheme 17).134 This simple and scalable method by Peng and coworkers is also applicable to the gram-scale synthesis of bioactive heterocycles, such as indole and resveratrol-derived amines, which are relevant to Alzheimer's disease treatment.
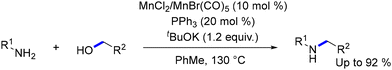 |
| | Scheme 17 Mn based salt catalysts for N-alkylation of amines with alcohols. | |
2.2. Iron-catalysed N-alkylation reactions
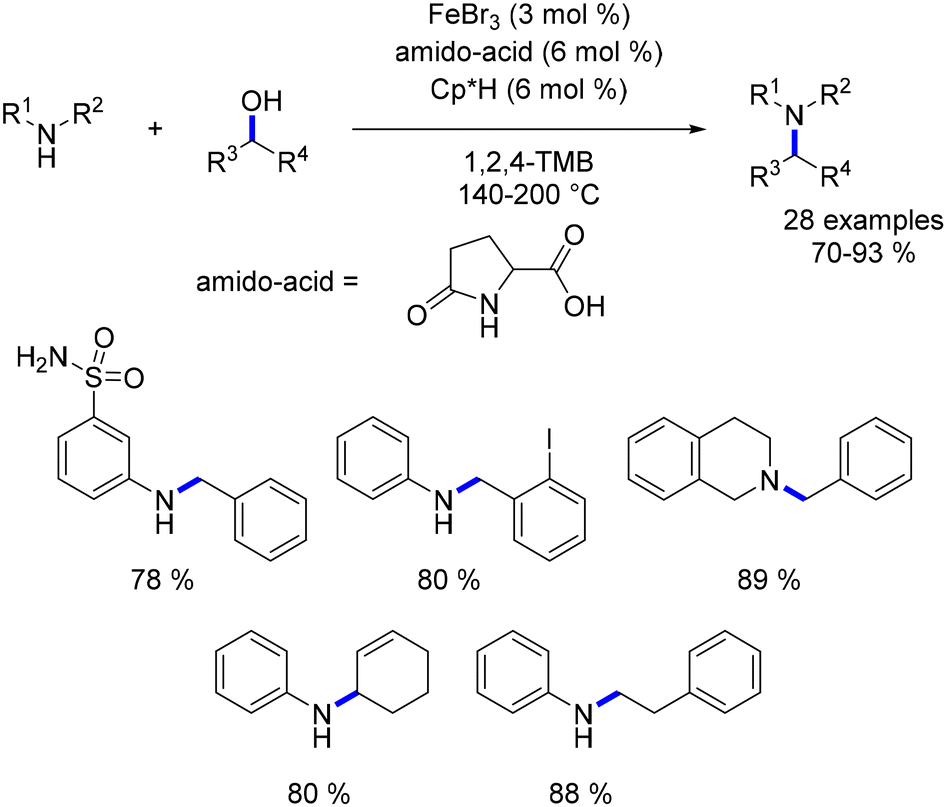
In 2011, Saito reported one of the early examples of iron-catalysed alkylation of amines with alcohols.88 Utilizing iron tribromide (3 mol%) in conjunction with an amido-acid (6 mol%) and Cp*H (6 mol%), a diverse array of primary and secondary amines underwent alkylation with benzylic and aliphatic alcohols, yielding secondary and tertiary amines at temperatures ranging from 140 to 200 °C (Scheme 18). Furthermore, unsaturated alcohols were alkylated without undergoing any reduction of the CC bond. The reaction necessitated high temperatures, and the exact nature of the catalytic species remains unclear. Additionally, the work reported only a limited number of substituents.
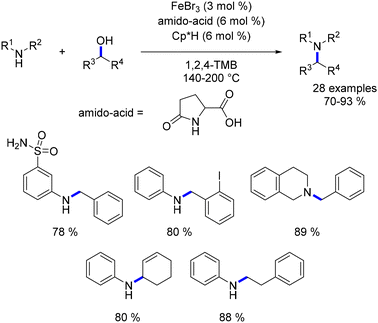 |
| | Scheme 18 Iron-catalysed alkylation of amines, as reported by Saito. | |
In 2013, Singh presented iron(II)-phthalocyanine as a catalyst for the alkylation of aromatic amines with alcohols.89 This catalyst facilitated the mono-alkylation of arylamines with both benzylic and aliphatic alcohols (Scheme 19). However, lower yields were achieved with aliphatic alcohols, which was attributed to the increased energy demand for the dehydrogenation step, compared to benzylic alcohols. Minakawa also reported the use of Fe(III)-phthalocyanine chloride for the same reaction, albeit with less tolerance for functional groups and requiring higher temperatures (130 °C).90
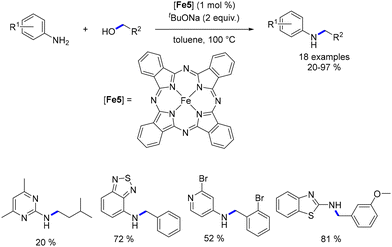 |
| | Scheme 19 Alkylation of amines, as reported by Singh. | |
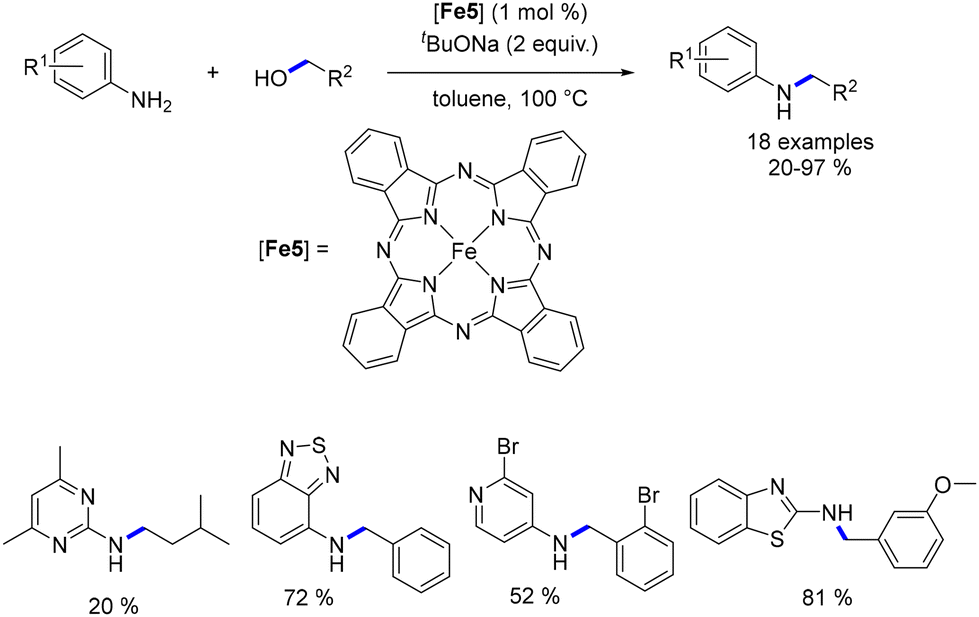
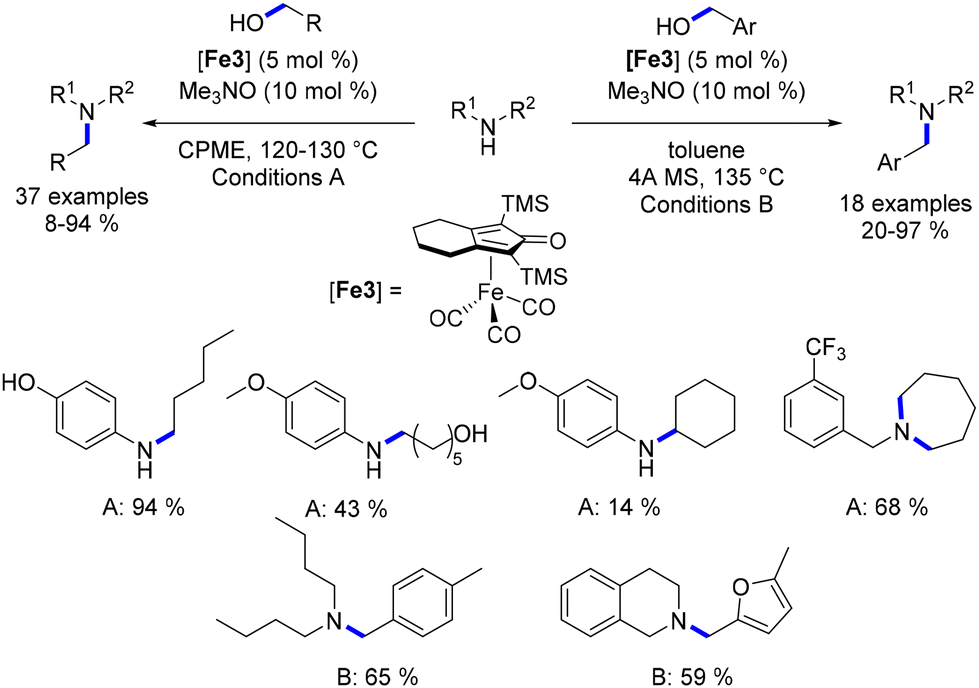
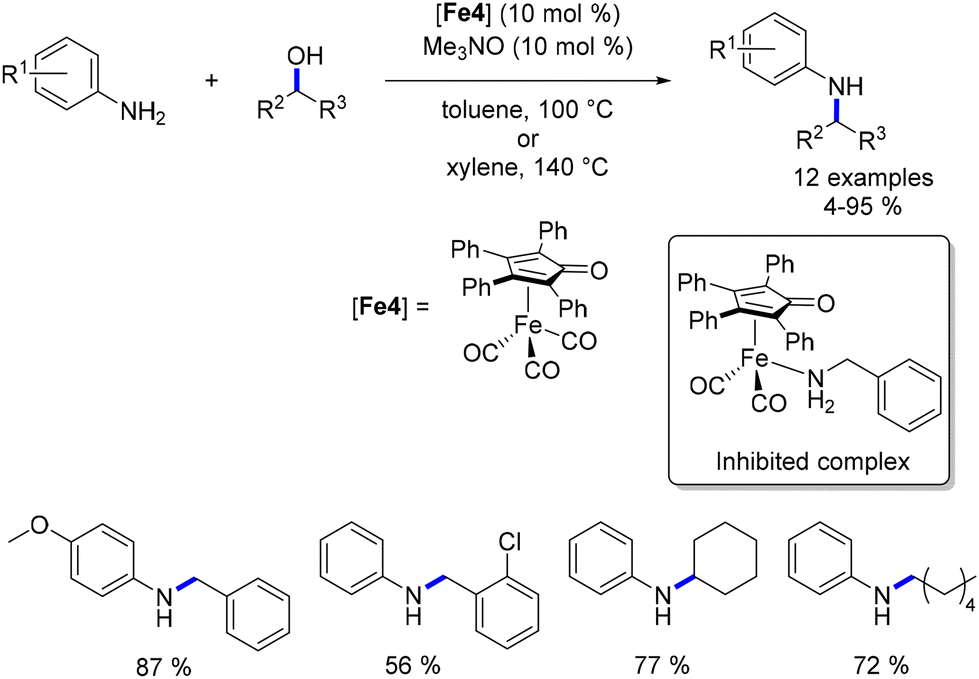
Feringa and Barta detailed the alkylation of aryl and benzyl amines utilizing a cyclopentadienone iron complex, all without the presence of a base—a significant advantage within the realm of sustainable chemistry.91 Interestingly, aliphatic alcohols yielded alkylated amines in satisfactory yields, whereas the efficiency of the reaction with benzylic alcohols was somewhat lower. Furthermore, when anilines were reacted with diols, the result was the formation of amino-alcohol products instead of N-aryl cyclic amines. To address the diminished reactivity of benzyl alcohols, the authors proposed employing benzylamines in conjunction with aliphatic alcohols (or diols). This strategy yielded N-benzylamines in commendable yields (Scheme 20). It is noteworthy that N-alkyl benzylamine derivatives were synthesized through the monoalkylation of benzylamines with alcohols. Additionally, if benzyl alcohols were to be utilized in such alkylation procedures, it was necessary to substitute CPME with toluene and include a molecular sieve.91 However, this modification did not enhance reactivity with secondary alcohols.
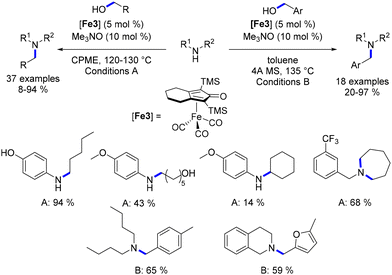 |
| | Scheme 20 Knölker-catalysed alkylation of amines, as described by Feringa and Barta. | |
Wills investigated a tetraphenylcyclopentadienone iron complex, also known as Schrauzer's complex,92 for the alkylation of anilines with primary alcohols.93 However, this reaction was constrained to methoxy- and chloro-substituted anilines and exhibited lower conversion rates compared to that with Knölker's complex, even necessitating higher catalyst loading and temperature (Scheme 21). Interestingly, when benzylamine was introduced, no product formation was observed. The authors postulated that benzylamine inhibited catalytic activity by forming a stable amino-iron complex with the active species, a phenomenon previously observed by Beller with Shvo's complex.94
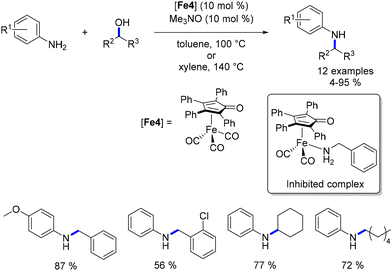 |
| | Scheme 21 Alkylation of anilines. | |
In 2016, Sundararaju documented the alkylation of amines with allyl alcohols catalysed by the Knölker's complex.95 Unlike the hydroamination reaction described by Maji and Beller, the formation of the C–N bond occurred through condensation between the amine and the carbonyl intermediate generated in situ, yielding allyl amines instead of γ-amino-alcohols (Scheme 22).
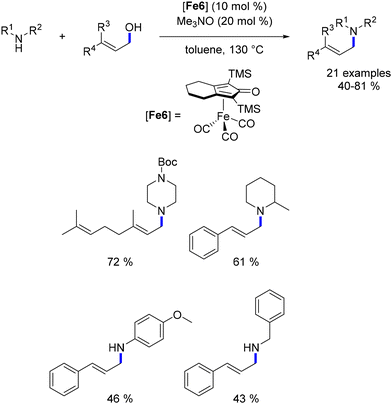 |
| | Scheme 22 Alkylation of amines with allylic alcohols. | |
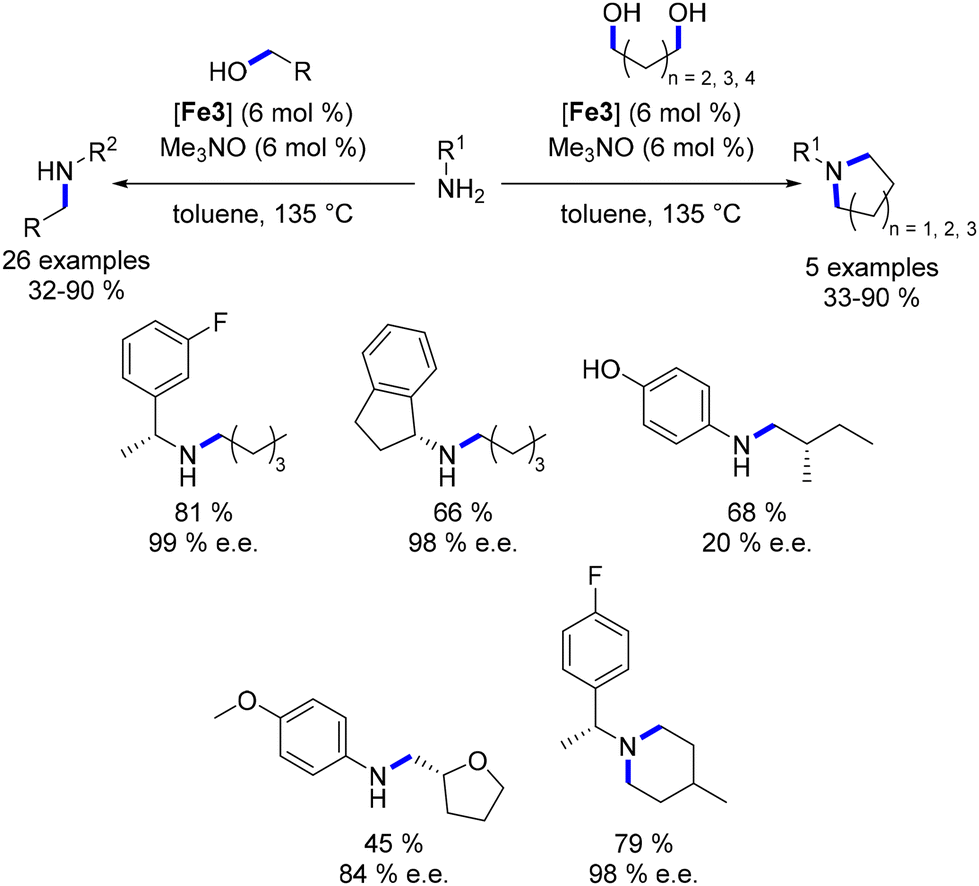
Barta also explored the synthesis of chiral compounds commencing from chiral amines or alcohols.96 Chiral benzylamines were subjected to a reaction with aliphatic alcohols (or diols), yielding mono- or di-alkylated products in satisfactory yields (61–90%, Scheme 23). Employing a similar protocol with optically active alcohols resulted in the corresponding products being obtained in moderate to good yields (32–68%). However, lower enantiomeric excesses were achieved in this scenario, potentially due to racemization facilitated by enol (or enamine) formation post the dehydrogenation step.
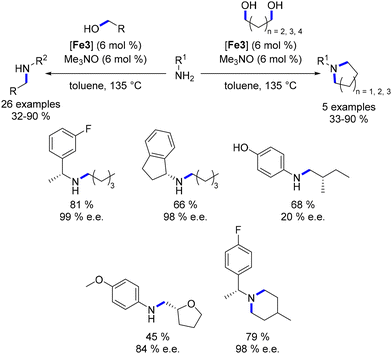 |
| | Scheme 23 Synthesis of chiral amines. | |
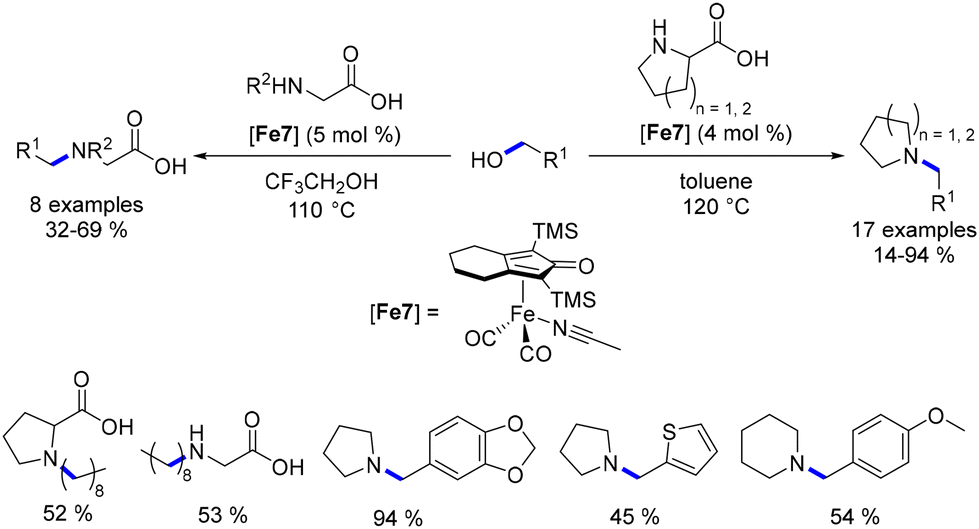
The same research group further explored the N-alkylation of unprotected amino acids.97 They employed a derivative of the Knölker complex featuring an acetonitrile ligand, which replaced one CO ligand. Various primary alcohols were utilized in this reaction under acidic conditions, yielding the mono-alkylated product in moderate to good yields (32–69%, Scheme 24). This investigation was extended to the synthesis of pyrrolidine and piperidine derivatives using proline derivatives.98 Utilizing non-acidic conditions facilitated the removal of the acid function via in situ decarboxylation.
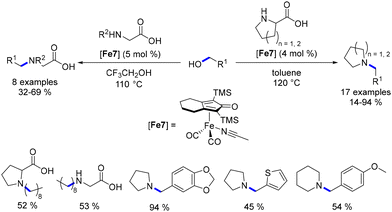 |
| | Scheme 24 Alkylation of amino-acids and synthesis of N-substituted cyclic amines. | |
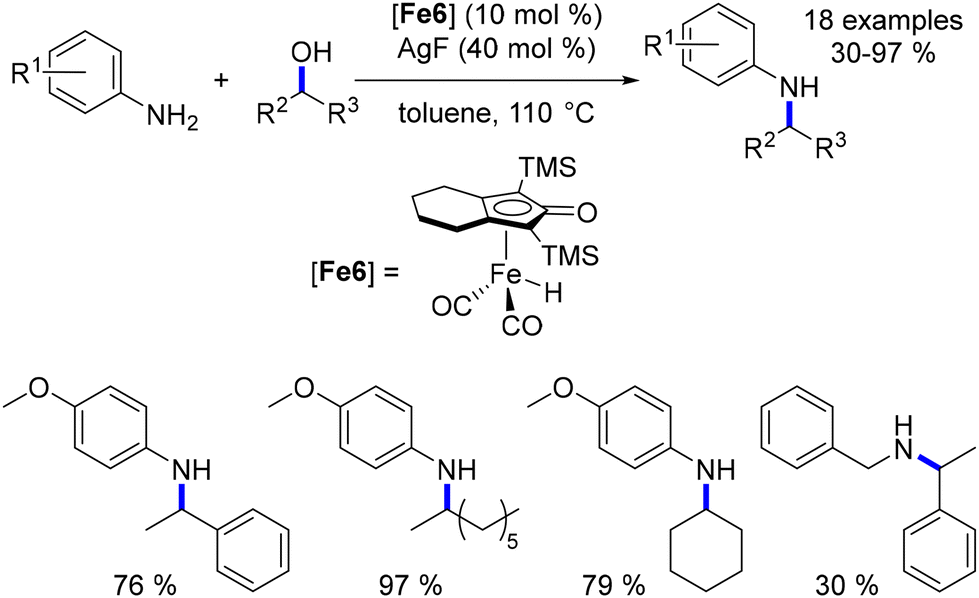
Building upon Barta's findings regarding the poor reactivity of secondary alcohols as pro-electrophiles, Zhao introduced a method for their activation using silver fluoride as a Lewis acid.99 Despite the continued high loading of both the iron complex and silver salt (10 mol% each), this approach provided a pathway to access N-aryl and N-benzyl α,α′-disubstituted amines in moderate to good yields (Scheme 25).
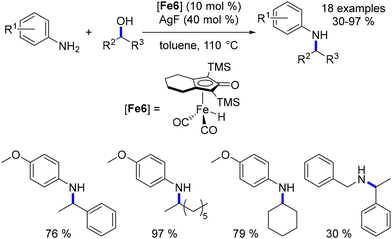 |
| | Scheme 25 Alkylation of amines with secondary alcohols. | |
Subsequently, Wills and coworkers revisited the alkylation of amines using a small collection of Knölker-type complexes.100 This investigation involved substituting the trimethylsilyl groups of the cyclopentadienone with aryl rings carrying electron-donating or electron-withdrawing groups and assessing the performance of various catalysts. The outcomes were substrate-dependent, varying based on the specific substrates involved in the reaction. While a complex containing a methoxy-aryl group exhibited favourable performance with most substrates, the original Knölker complex was utilized to illustrate the reaction (Scheme 26).
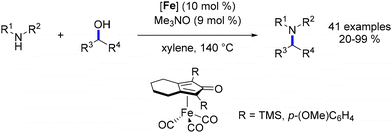 |
| | Scheme 26 Alkylation of amines with the Knölker complex and analogues. | |
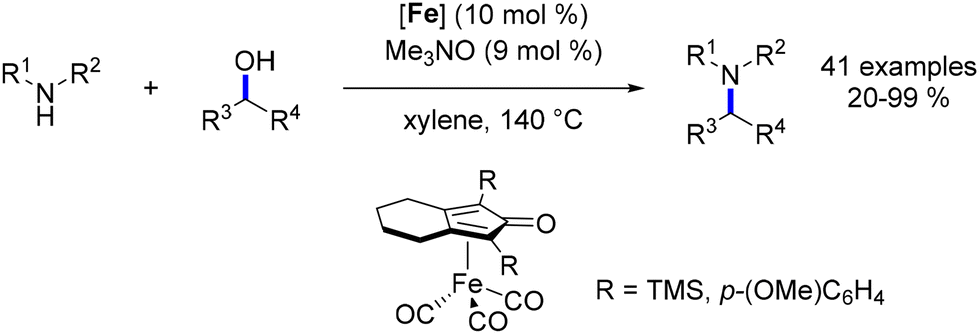
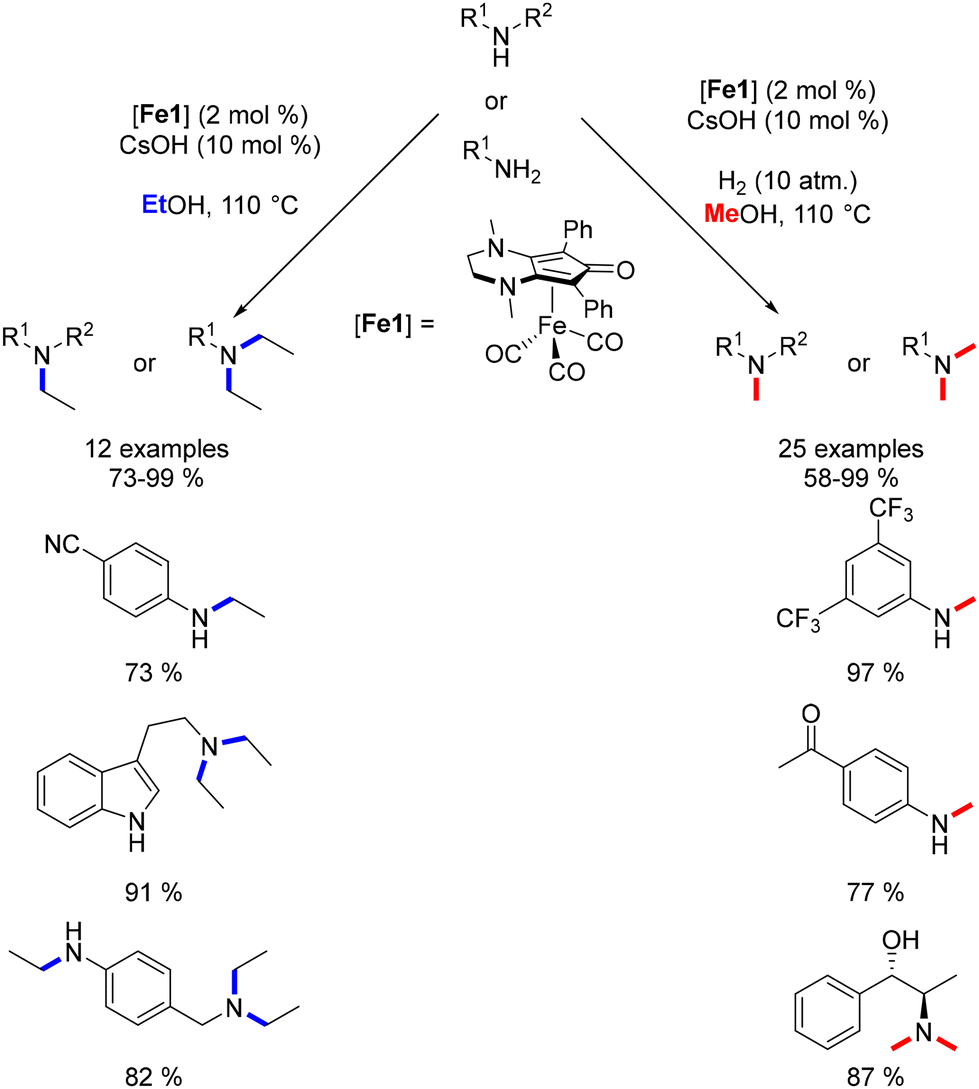
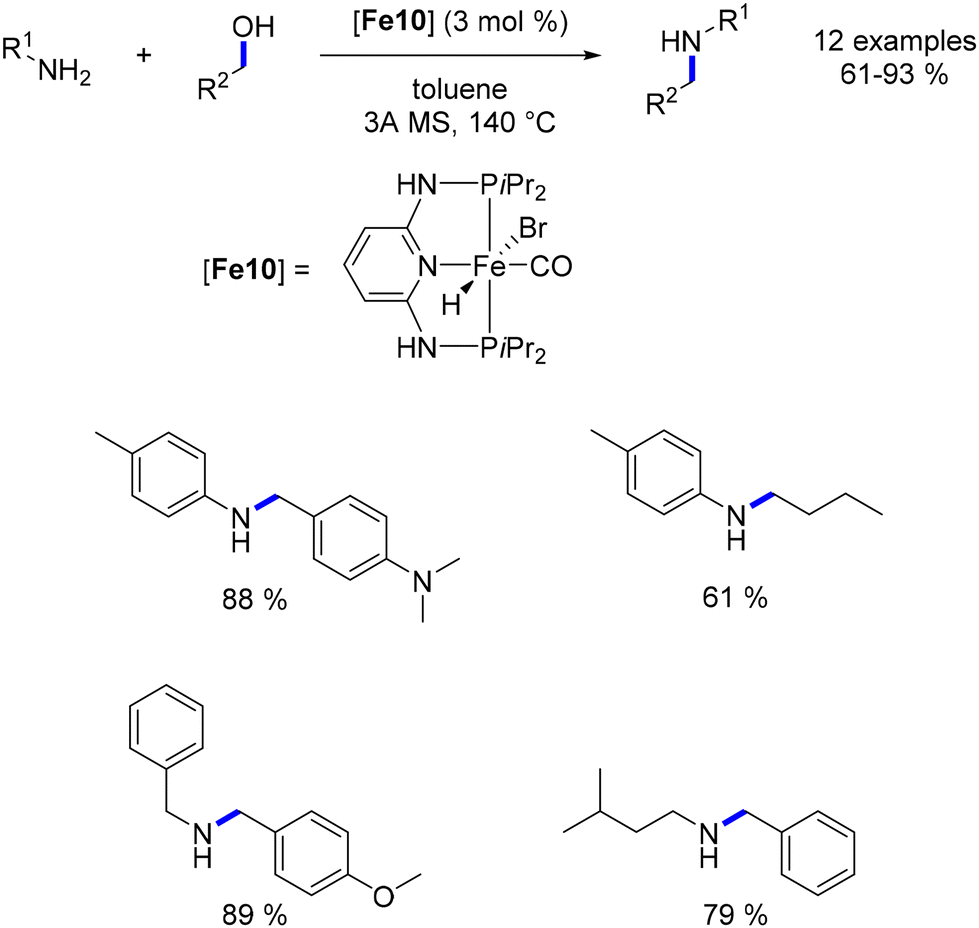
Continuing this line of inquiry, Renaud and Poater explored a modified Knölker-type complex featuring an electron-rich ligand for the methylation and ethylation of amines.45 This diaminocyclopentadienone iron tricarbonyl complex, previously employed by the same authors for reductive amination reactions,101 was also employed in that study. Alkylation using methanol and ethanol as pro-electrophiles has been minimally investigated and presents additional challenges due to the higher dehydrogenation energy associated with ethanol and methanol compared to other alcohols. However, this novel complex facilitated the synthesis of ethyl- and methyl-substituted amines in good to excellent yields with low catalyst loading (Scheme 27). Notably, the presence of a catalytic amount of a base was necessary for these reactions. Furthermore, a hydrogen pressure of 10 atm was required to achieve the methylation reaction in good yields.
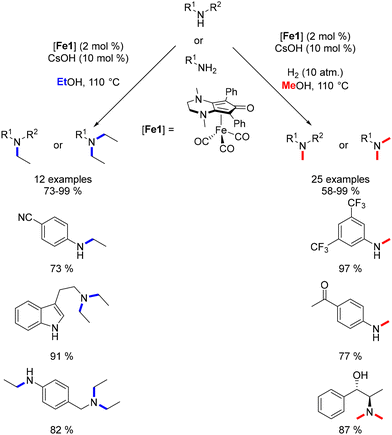 |
| | Scheme 27 Methylation and ethylation of amines, as reported by Renaud. | |
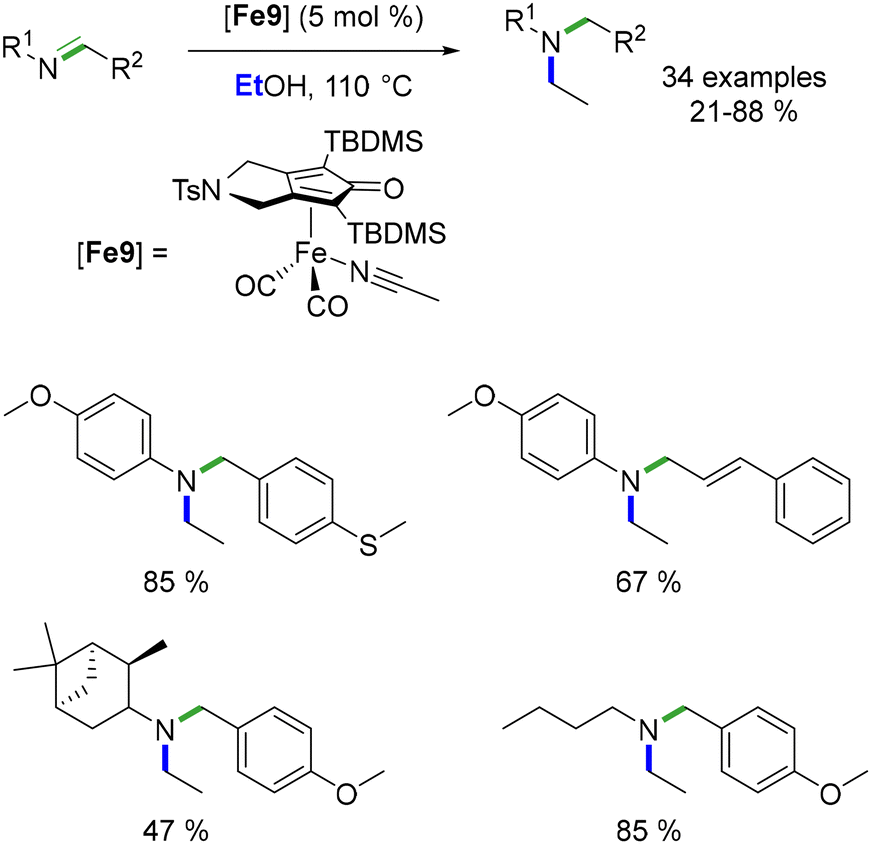
In the same year, Bour et al. detailed a reductive ethylation process of N-arylimines catalysed by a Knölker-type complex.102 Originally introduced by Renaud et al. for reductive amination, this complex was repurposed for the sequential reaction. In this process, ethanol functioned both as the alkylating agent and the hydride source. The outcome yielded N-aryl-N-ethyl amines in yields spanning from 21 to 88% (Scheme 28).
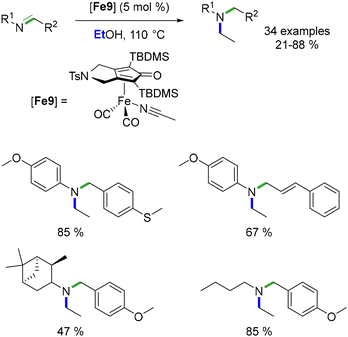 |
| | Scheme 28 Ethylation of imines, as reported by Bour. | |
Kirchner documented an iron complex analogous to the Sortais PNP–Mn complex. This iron–hydride complex facilitated the alkylation of amines with primary alcohols.103 Although low catalyst loadings were employed, achieving good yields necessitated high temperatures (Scheme 29).
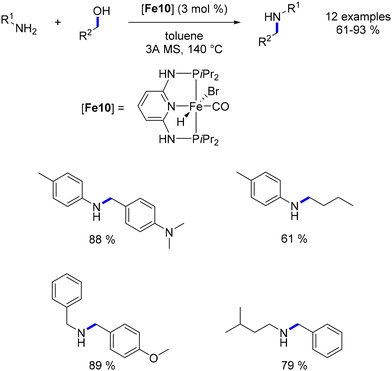 |
| | Scheme 29 Alkylation of amines. | |
Substituting the pyridine ring of the ligand with a dimethylamino triazine ring enhanced catalytic efficiency, resulting in a decrease in reaction temperature from 140 to 80 °C, and eliminating the need for molecular sieves (Scheme 30).104 Nevertheless, secondary alcohols displayed low reactivity under these conditions, and the overall yield of the reaction was not enhanced.
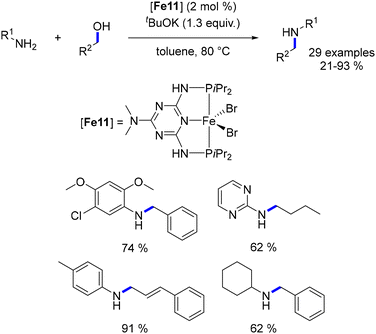 |
| | Scheme 30 Iron(II)-catalysed alkylation of amines, as reported by Kirchner. | |
In 2019, Wang documented the hydroamination of allylic alcohols.105 The PNP–Fe complex catalysed the oxidation of the alcohol to form an α,β-unsaturated aldehyde. Subsequent 1,4-addition by the amine yielded the amino-aldehyde, which was then reduced by the complex to produce γ-amino-alcohols (Scheme 31). This approach was later adopted by Maji and Beller, utilizing manganese pincer complexes. Amides were also found to be compatible with this reaction, albeit requiring higher temperatures.
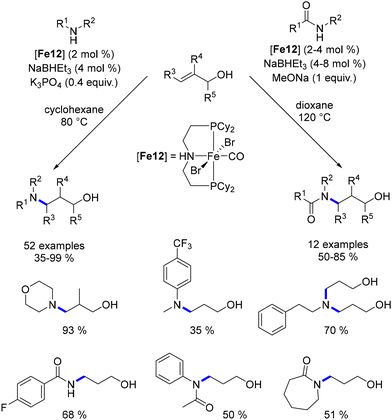 |
| | Scheme 31 Hydroamination of allylic alcohols, as reported by Wang. | |
Liu presented an NNN–Fe(II) complex featuring pyrazoyl and imidazoyl groups for the alkylation of amines.106 This catalyst facilitated the synthesis of alkylated anilines using primary alcohols, resulting in good yields (Scheme 32). However, only a few examples with restricted functional groups were illustrated.
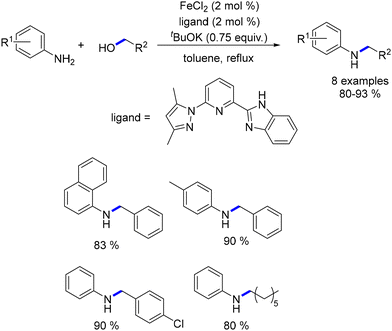 |
| | Scheme 32 Alkylation of anilines, as reported by Liu. | |
2.3. Cobalt-catalysed N-alkylation reactions
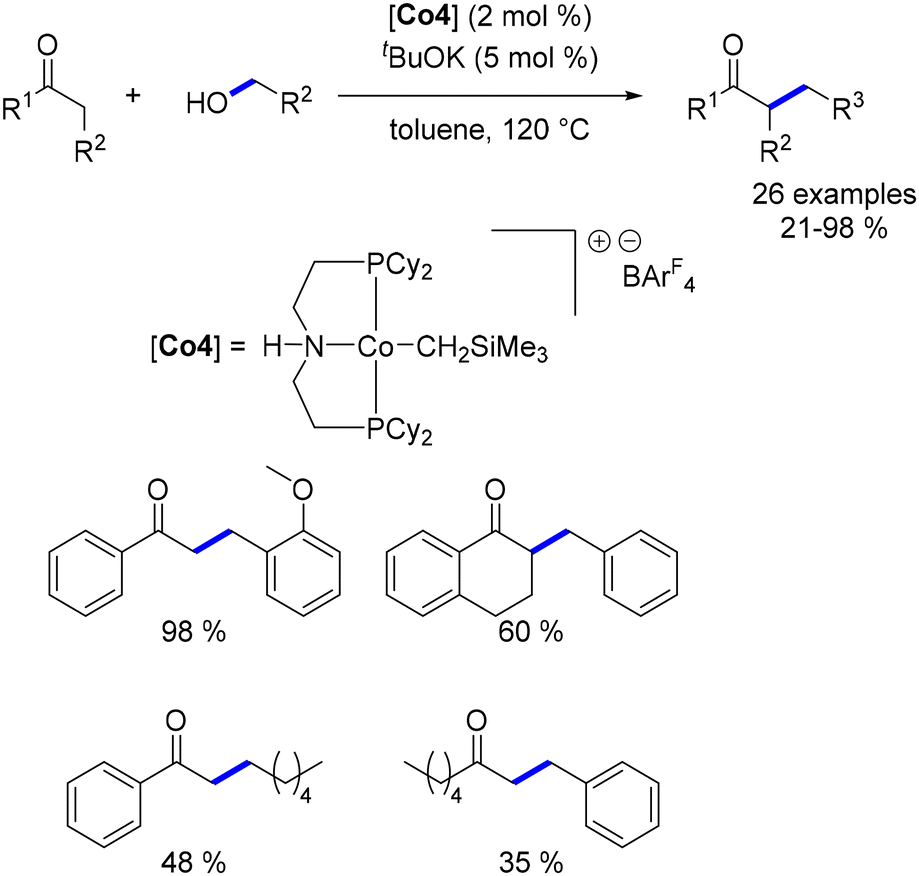
Kempe detailed the initial cobalt-catalysed process of alkylating amines using alcohols.107 A pre-catalyst was formed by combining CoCl2 with a PNP ligand featuring a triazine ring. This metal complex facilitated the alkylation of different aniline derivatives using both benzylic and aliphatic primary alcohols. Interestingly, the aliphatic primary alcohols proved to be equally effective as benzyl alcohols and as pro-electrophiles (Scheme 33). Notably, this study did not include any results involving secondary alcohols or aliphatic amines.
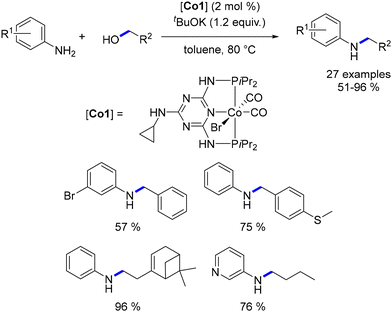 |
| | Scheme 33 Cobalt-catalysed alkylation of anilines. | |
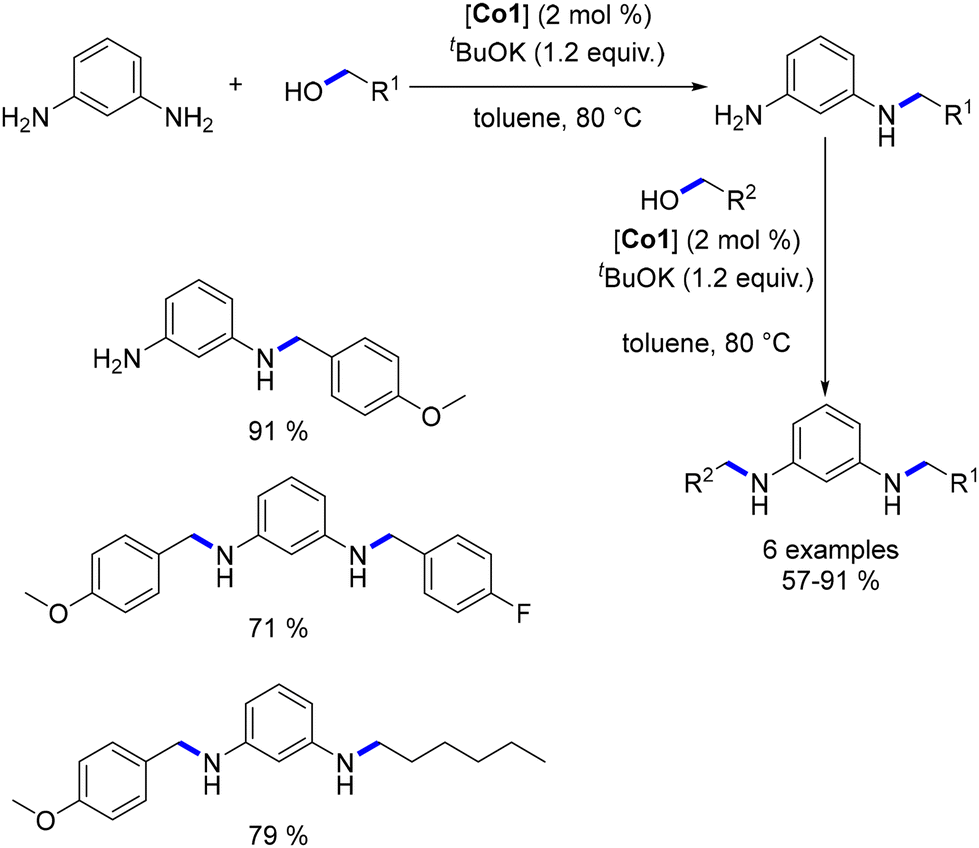
Diamine alkylation was conducted in a stepwise procedure, enabling the production of non-symmetrical N,N′-di-substituted diamines. Moreover, the mono-substituted diamine could be obtained by utilizing an excess of diamine relative to the alcohol (Scheme 34).
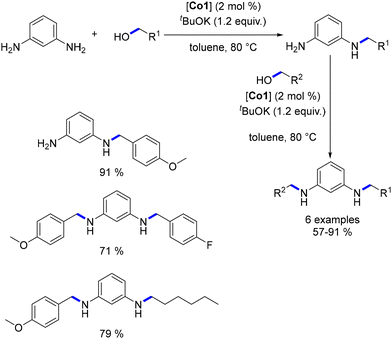 |
| | Scheme 34 Synthesis of non-symmetric diamines, as reported by Kempe. | |
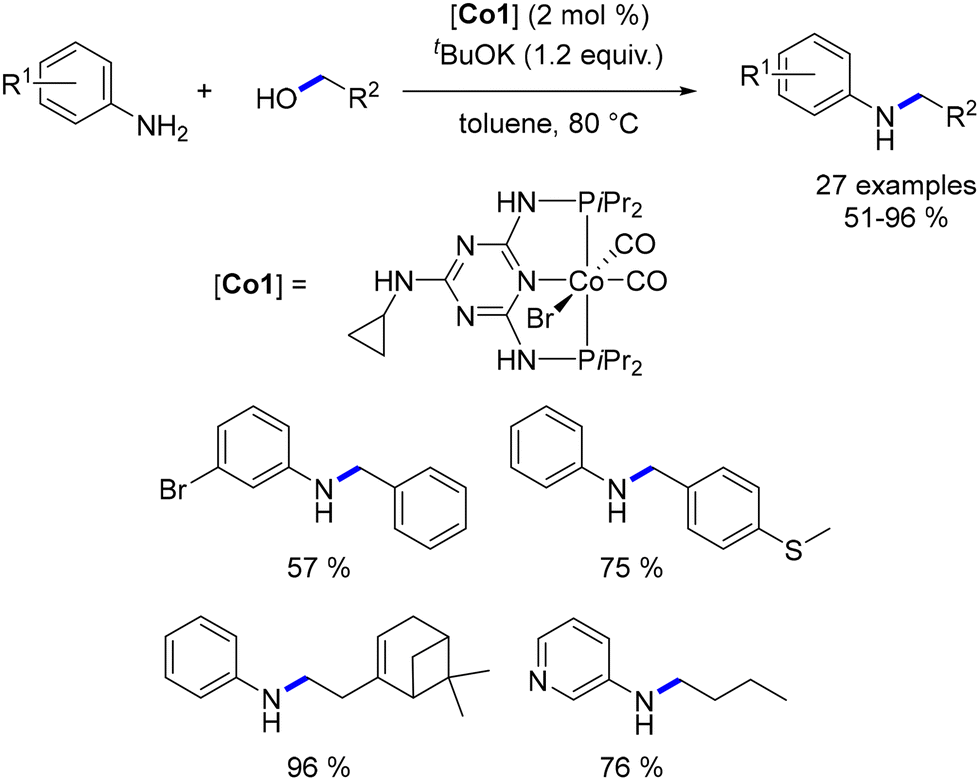
Kirchner synthesized a cobalt(II) complex along with a PCP pincer-type ligand.108 Utilizing this complex, Kirchner demonstrated its effectiveness in the alkylation of arylamines with primary alcohols, resulting in alkylated amines in yields ranging from 10% to 94% (Scheme 36). Notably, only aromatic amines were employed in this reaction, as aliphatic amines were found to potentially hinder the catalytic activity, a phenomenon previously observed by Wills using the Schrauzer–Reppe complex (Scheme 35).93 Remarkably, the alkylation of cinnamyl alcohol was achieved without the reduction of the CC bond. Ethylation of aniline led not only to the corresponding mono-ethylated derivative, but also to the di-ethylated compound. Even though the dialkylated aniline was obtained in a low yield, (10%), this result marks a significant milestone as the first dialkylation of an aniline catalysed by an Earth-abundant metal complex.
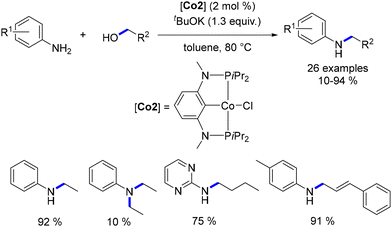 |
| | Scheme 35 Cobalt-catalysed alkylation of anilines, as reported by Kirchner. | |
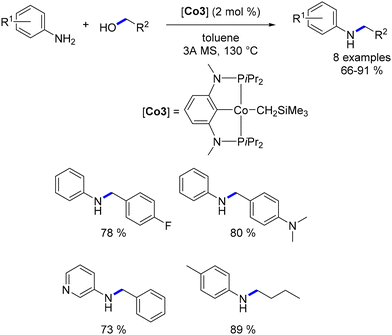 |
| | Scheme 36 Base-free alkylation of amines, as reported by Kirchner. | |
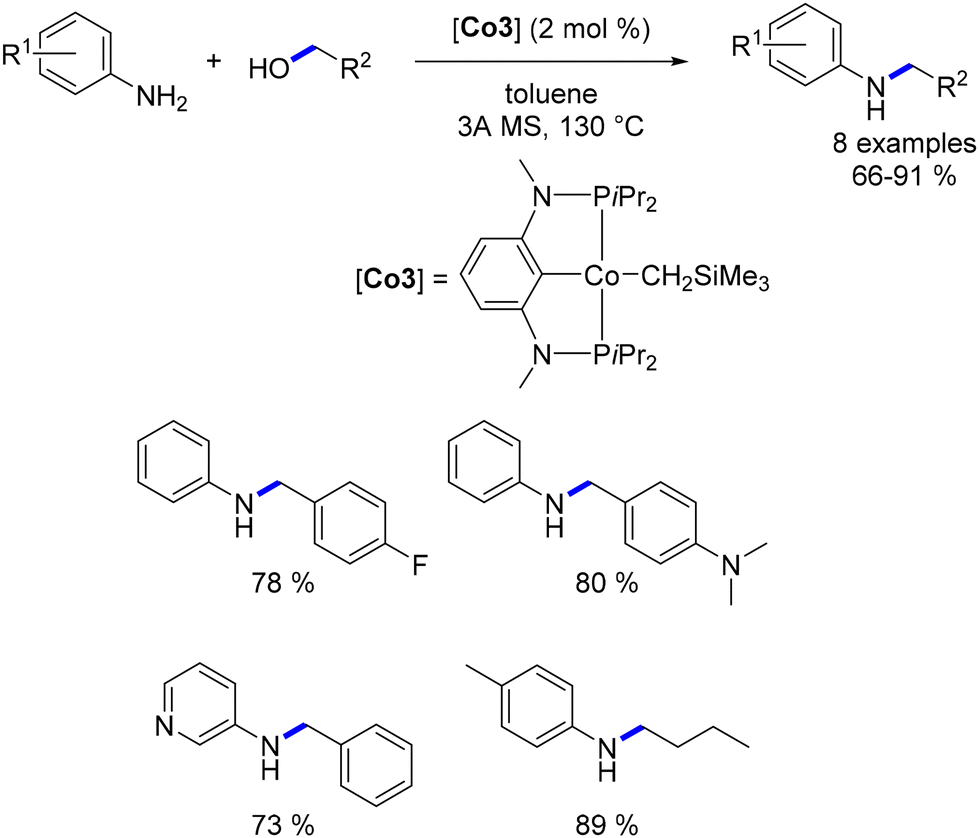
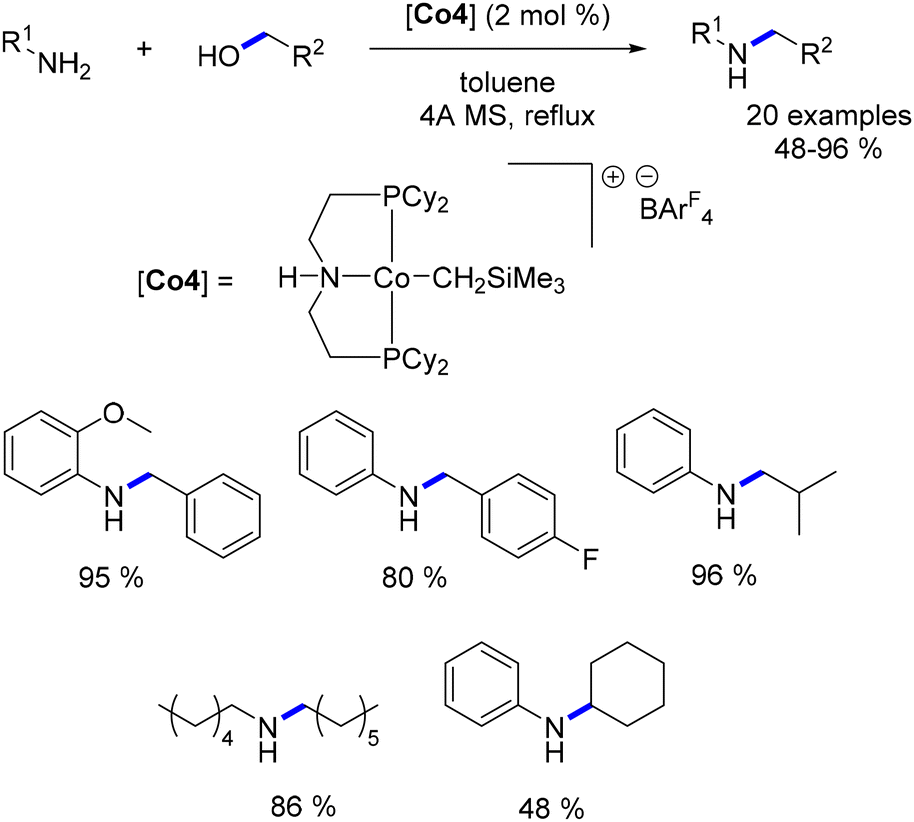
A variant of this catalyst featuring a highly basic CH2SiMe3 ligand was also evaluated. This alternative catalyst proved to be effective for the targeted reaction and did not require the addition of a base. However, elevated temperatures were necessary for the reaction to proceed (Scheme 36).
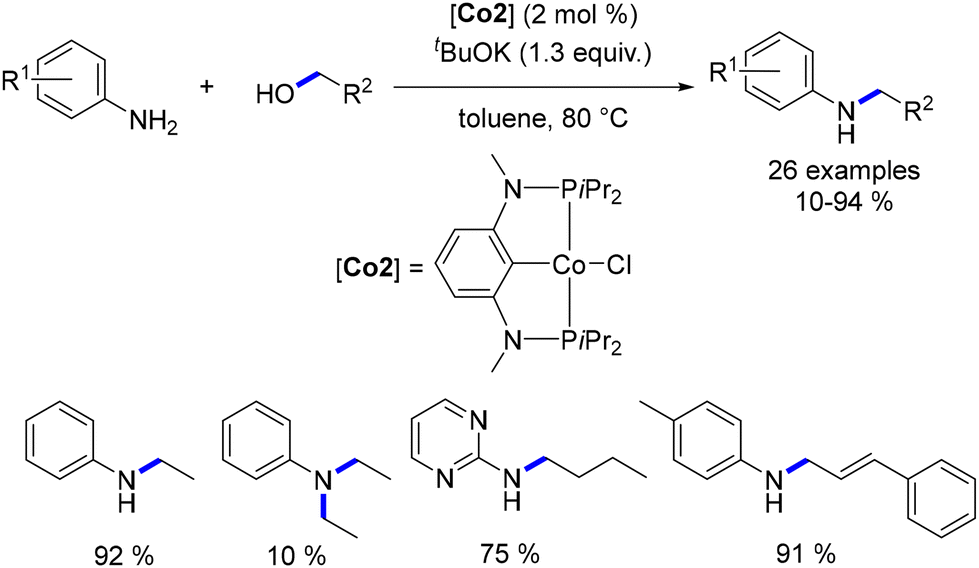
Zhang subsequently reported the utilization of this methylene trimethylsilyl ligand in a Macho-type cationic cobalt complex.109 Once again, this ligand demonstrated the ability to circumvent the need for a base. Both aryl and aliphatic amines were successfully alkylated with a range of primary alcohols, yielding good to excellent results (Scheme 37). Notably, an example utilizing cyclohexanol as a secondary alcohol resulted in the alkylated aniline in a yield of 48%.
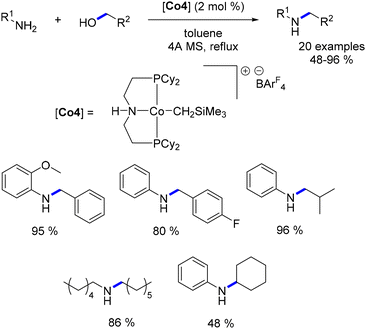 |
| | Scheme 37 Alkylation of amines, as described by Zhang. | |
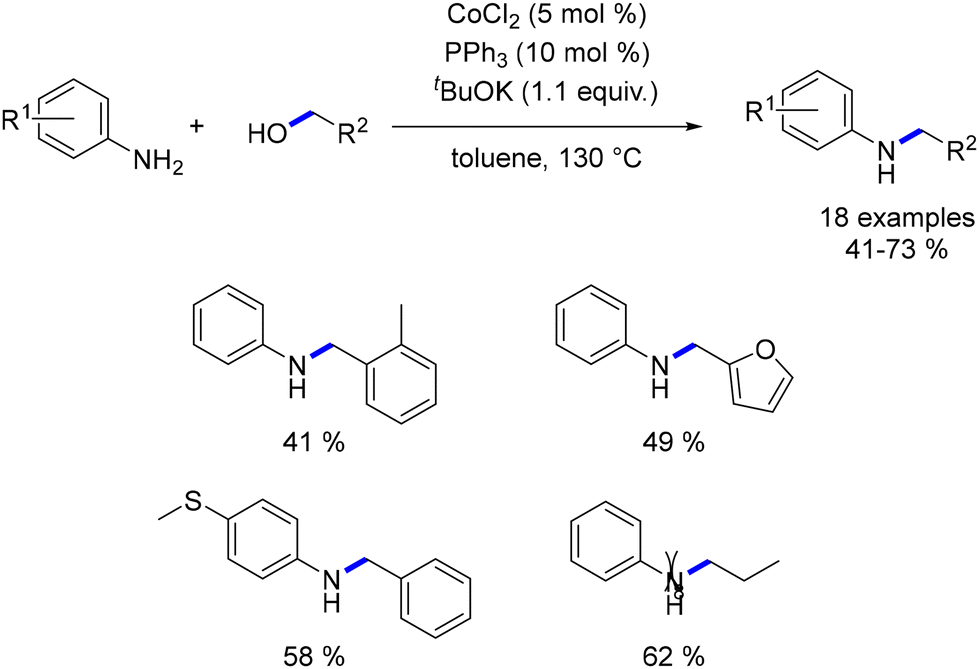
Balaraman documented the combination of CoCl2 with PPh3 for the alkylation of anilines using primary alcohols.110 Several substituted anilines underwent alkylation with both benzylic and aliphatic alcohols in this reaction. However, the yields were lower compared to previously reported cobalt-catalysed hydrogen autotransfer reactions (Scheme 38).
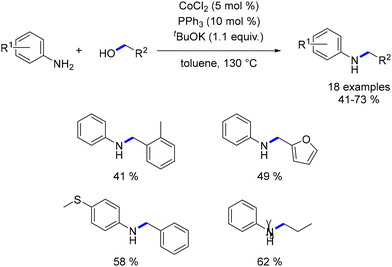 |
| | Scheme 38 Cobalt-catalysed alkylation of anilines, as reported by Balaraman. | |
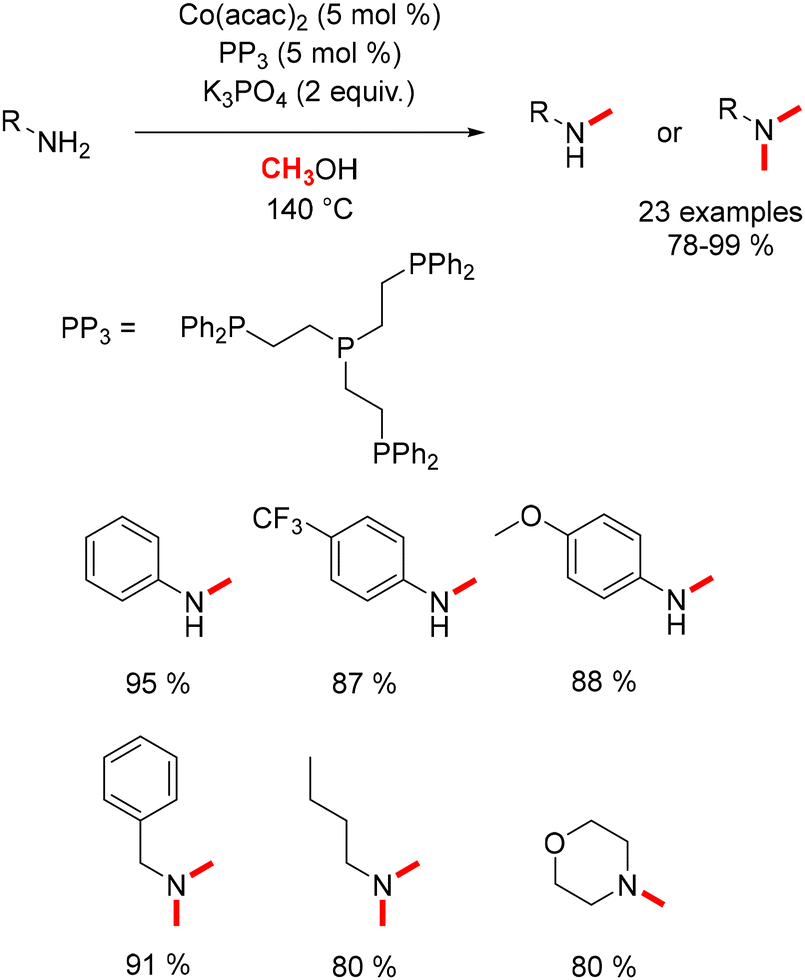
In 2017, Liu conducted research on the methylation of both aryl and aliphatic amines.111 By combining Co(acac)2 with a tetra-phosphine ligand, aromatic amines produced mono-methylated anilines, whereas aliphatic amines exclusively yielded the dimethylated product (Scheme 39).
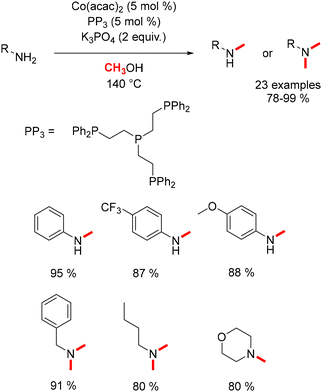 |
| | Scheme 39 Methylation of amines, as reported by Liu. | |
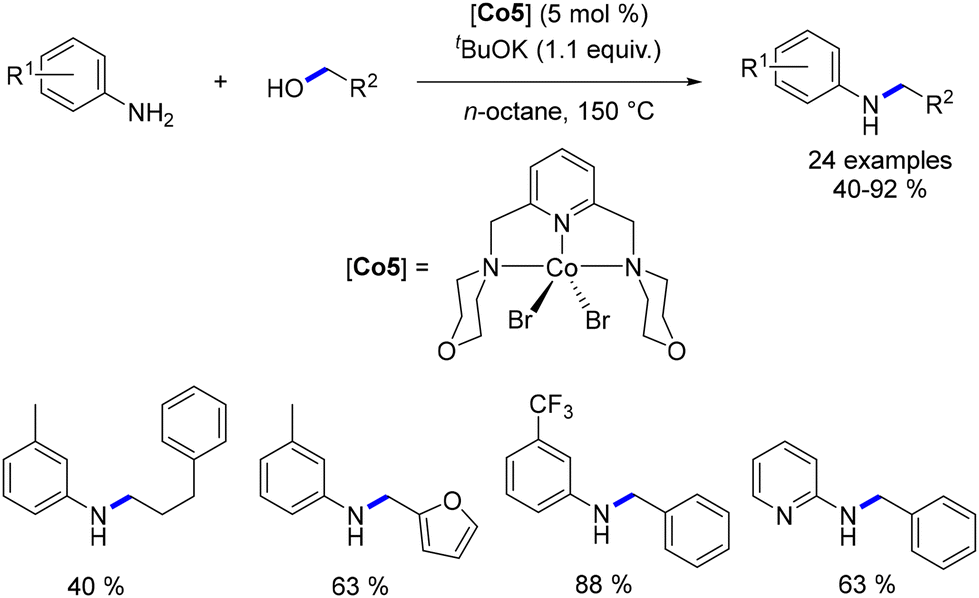
Balaraman et al. developed a novel NNN–Co complex112 that exhibited activity in the alkylation of aniline derivatives with alcohols. Although higher temperatures were necessary, the N-alkylated aniline compounds were isolated in yields ranging from 40% to 92% (Scheme 40).
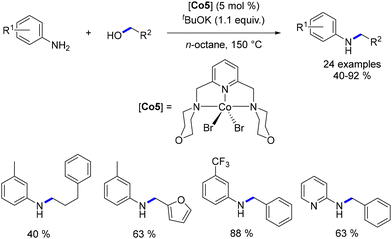 |
| | Scheme 40 Cobalt–pincer complex-catalysed alkylation of anilines. | |
More recently, Ding reported on a switchable synthesis of imines and amines catalysed by a PNPP ligand.113 By adjusting the base loading, temperature, and system type (closed or open), either the imine or the amine product could be selectively obtained. This catalytic system demonstrated efficiency in the alkylation of anilines with primary alcohols; however, alkylated aliphatic amines were not produced (Scheme 41).
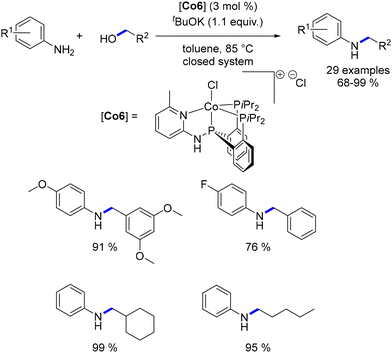 |
| | Scheme 41 Alkylation of anilines, as reported by Ding. | |
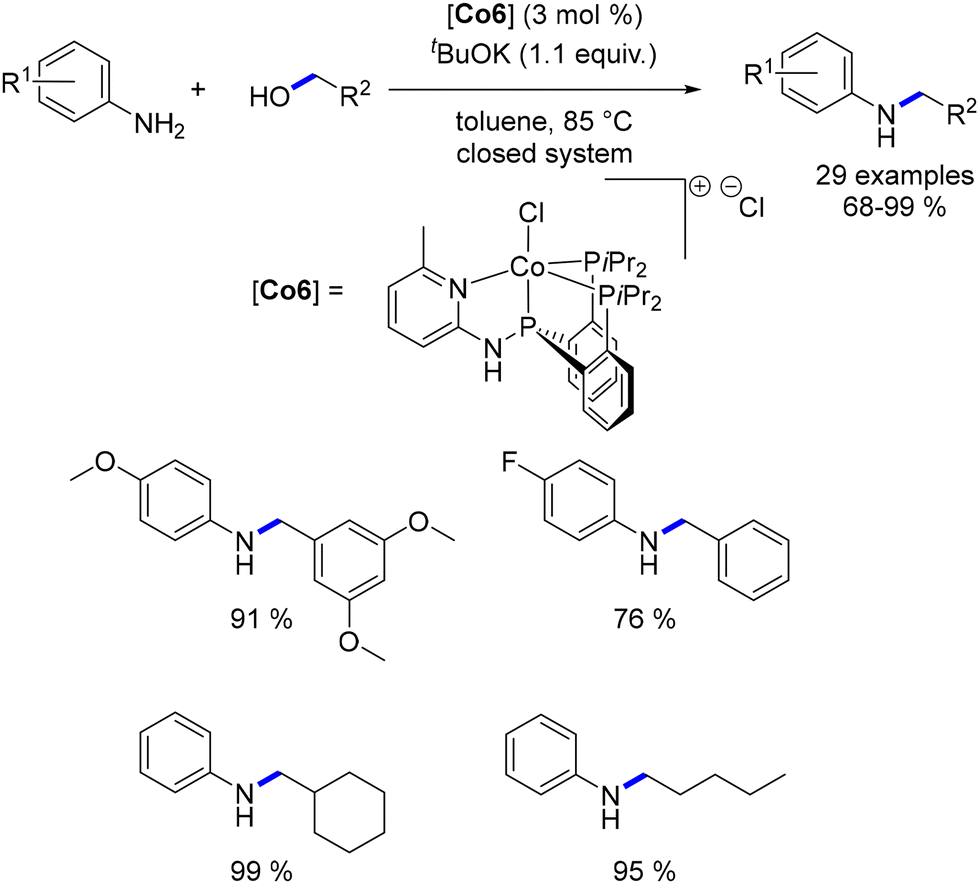
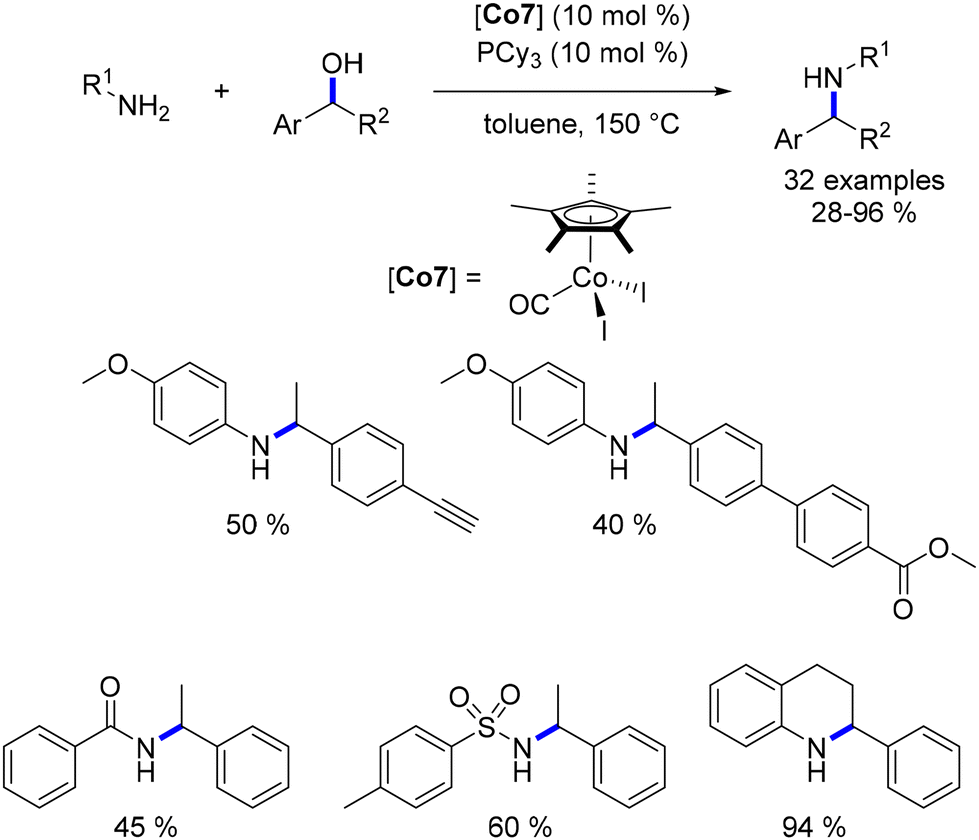
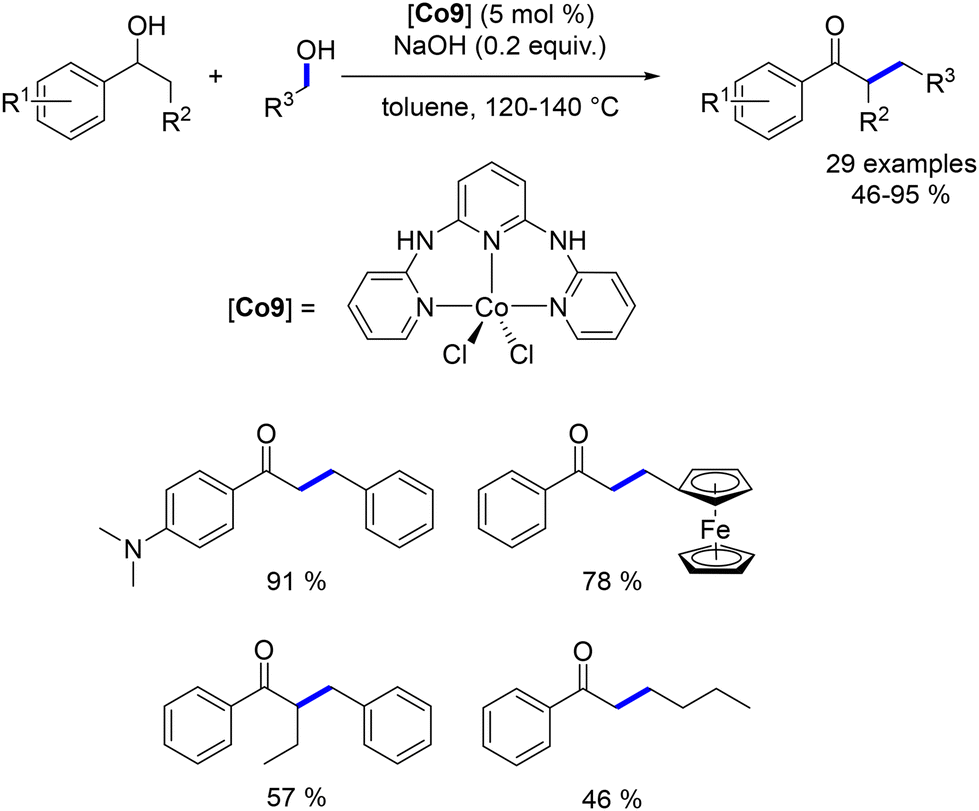
Among the cobalt-catalysed alkylations of amines discussed earlier, only one illustration of alkylation with a secondary alcohol was reported by Zhang.109 Alkylation with secondary alcohols poses a challenge due to the lower electrophilicity of ketones compared to aldehydes. In 2019, Sundararaju introduced a Cp*Co(III) complex for the alkylation of amines with secondary alcohols.114 The reaction was conducted in toluene at 150 °C without the need for an additional base (Scheme 42). This procedure resulted in the formation of alkylated anilines and amide derivatives using various secondary benzylic alcohols.
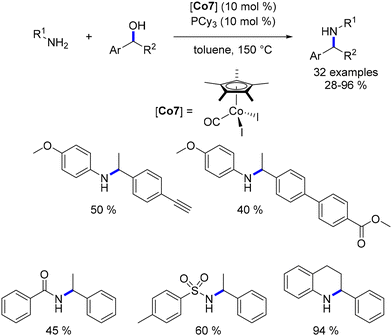 |
| | Scheme 42 Alkylation of amines and amides with secondary alcohols. | |
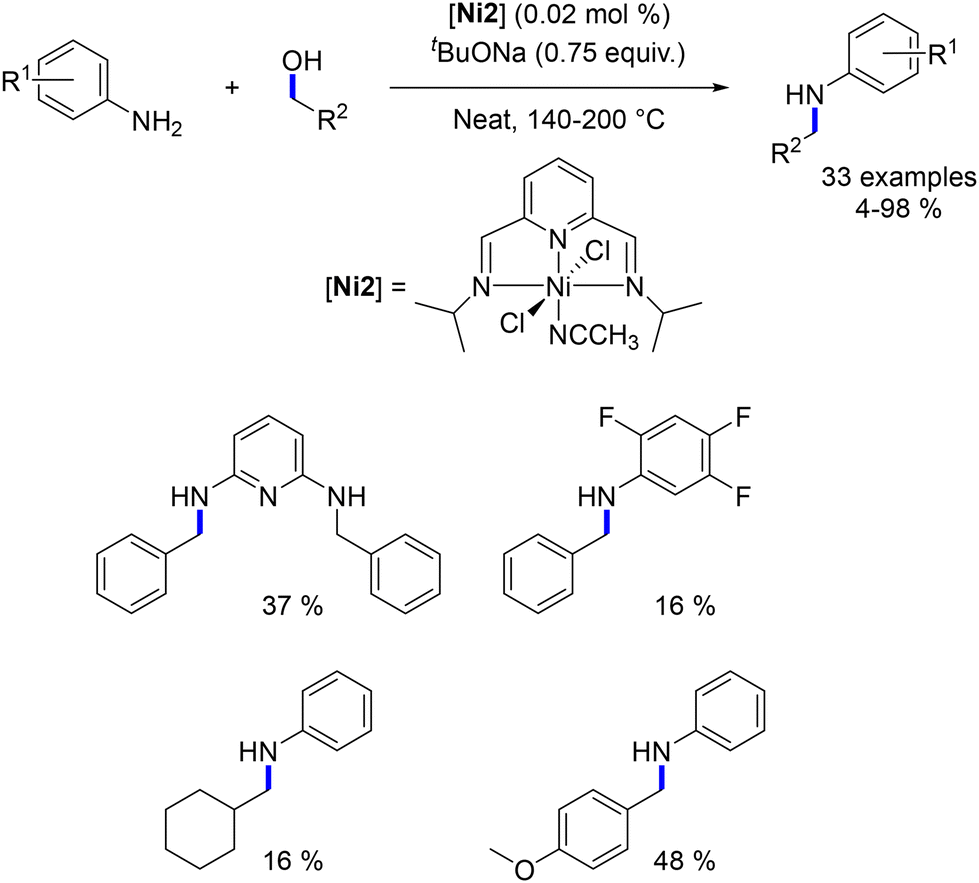
2.4. Nickel-catalysed N-alkylation reactions
Zhou reported the first homogeneous nickel-complex-catalysed alkylation of amines in 2017.115 A nickel salt was combined with a diphosphine ligand to catalyse the alkylation of amines with alcohols. This reaction facilitated the synthesis of substituted anilines, piperazines, and trisubstituted amines (Scheme 43). Additionally, the alkylation of acylhydrazides with alcohols was also reported later, not with nickel but with a ruthenium complex.
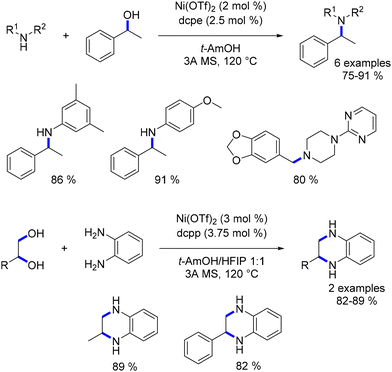 |
| | Scheme 43 Alkylation of amines, as reported by Zhou. | |
Banerjee described a straightforward method involving the in situ generation of a nickel–phenanthroline complex for catalysing the alkylation of aniline derivatives.116 Indole derivatives could be synthesized under the same reaction conditions through an intramolecular cyclization and isomerization into the aromatic form (Scheme 44).
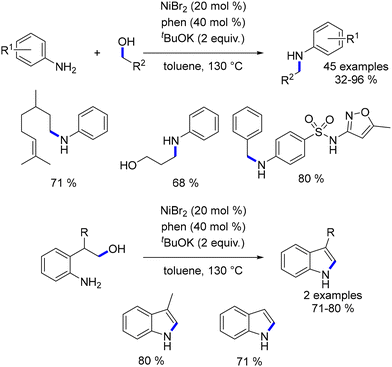 |
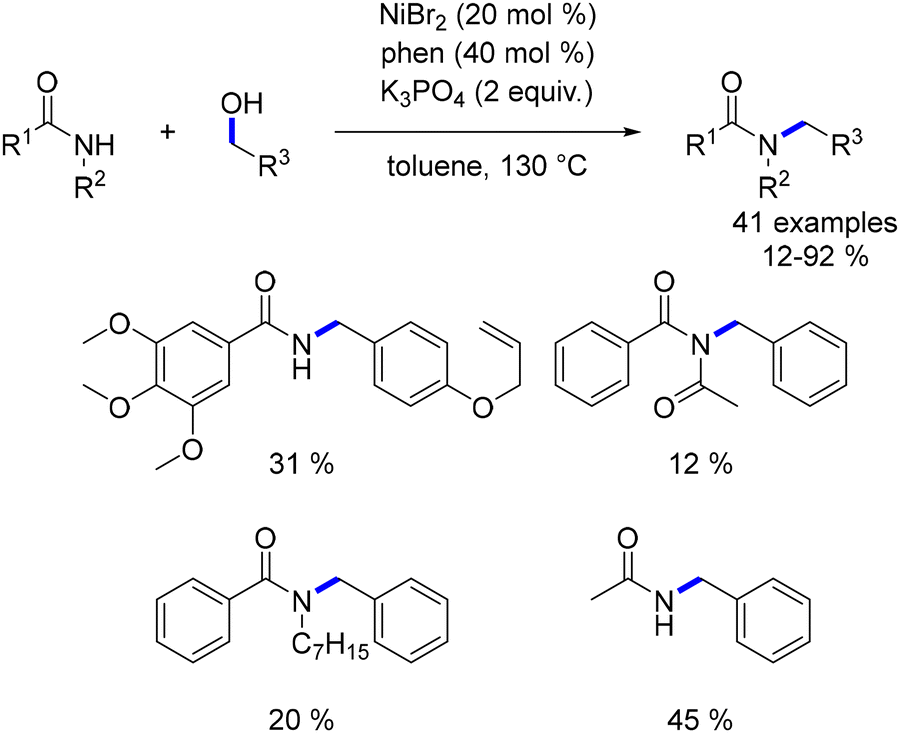
| | Scheme 44 Alkylation of anilines, as reported by Banerjee. | |
By employing K3PO4 as a base and reducing the catalyst loading, it was also possible to achieve the alkylation of an amide.117 This reaction facilitated the alkylation of a range of amides, including N-substituted amides and secondary amides. Lower yields were observed with less nucleophilic amides or aliphatic alcohols (Scheme 45).
 |
| | Scheme 45 Alkylation of amides, as reported by Banerjee. | |
Using the same NNN-ligand, Balaraman devised a nickel-catalysed alkylation process for aniline and benzylic amines with various secondary alcohols. This reaction was conducted at 140 °C in n-octane (Scheme 46).118 Under identical conditions, no product was obtained when using aliphatic amines and non-cyclic aliphatic secondary alcohols. Similarly, under analogous reaction conditions, only primary alcohols were reactive with the cobalt analogue of this complex previously reported by the same group.112
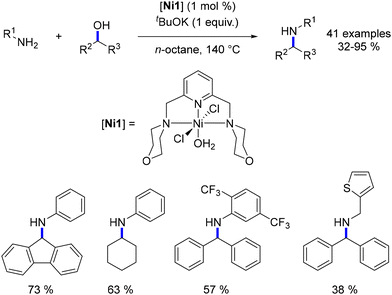 |
| | Scheme 46 Alkylation of amines with secondary alcohols, as reported by Balaraman. | |
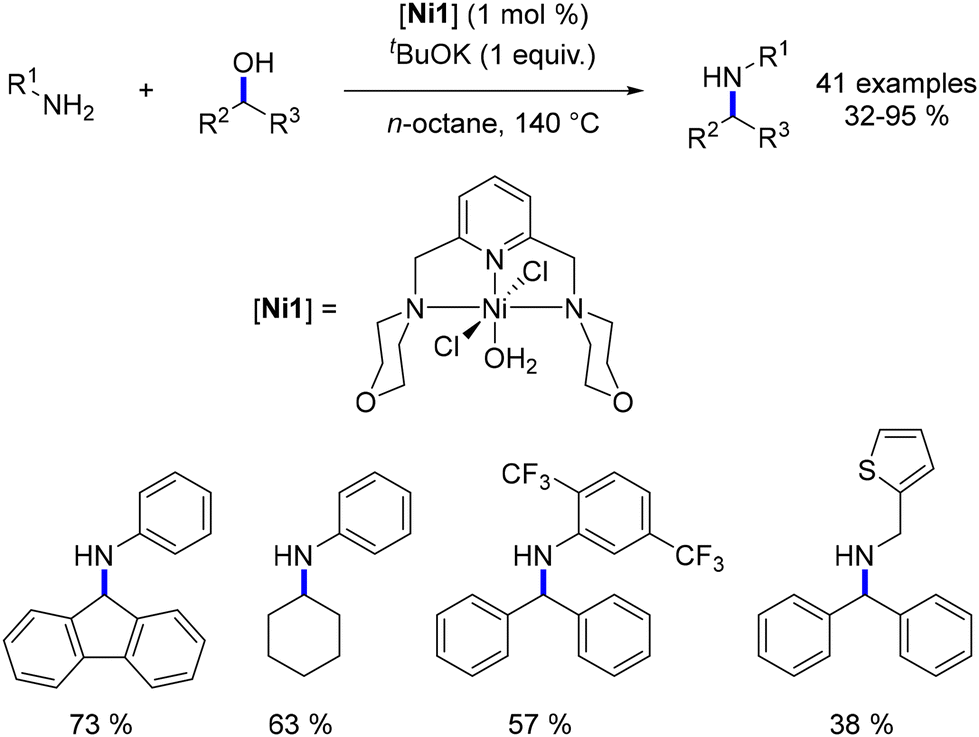
Following Balaraman's work, Kumar introduced a pyridine-diimine-based nickel complex and utilized it for the alkylation of aniline compounds with benzylic alcohols. The reaction was conducted at temperatures ranging from 140 to 200 °C with a substoichiometric amount of tBuOK (0.75 equiv., Scheme 47). Good catalytic activities were achieved with turnover numbers (TONs) up to 4500 at 140 °C. Remarkably, significantly higher TONs (up to 34000) were attained at elevated temperatures (200 °C) using only 0.002 mol% of the nickel complex.119
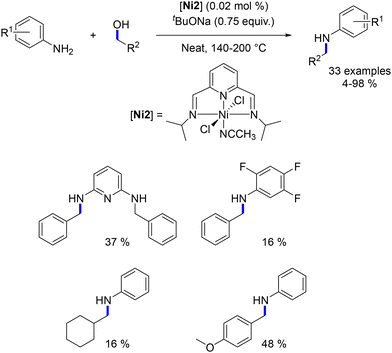 |
| | Scheme 47 Alkylation of amines, as reported by Kumar. | |
2.5. Copper-catalysed N-alkylation reactions
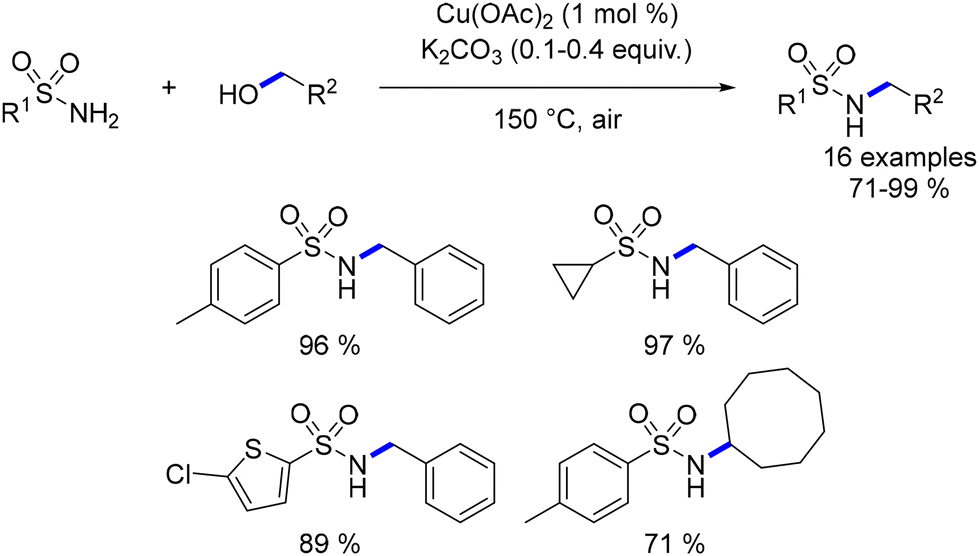
Beller documented the first ligand-free copper-catalysed alkylation of sulfonamides with alcohols in 2009.120,121 Various sulfonamides underwent reactions with both benzylic and aliphatic primary and secondary alcohols, resulting in the formation of N-substituted sulfonamides. The process tolerated a diverse range of functionalized compounds (Scheme 48).
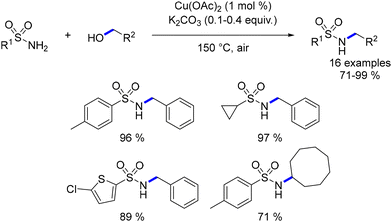 |
| | Scheme 48 Copper-catalysed alkylation of sulfonamides, as reported by Beller. | |
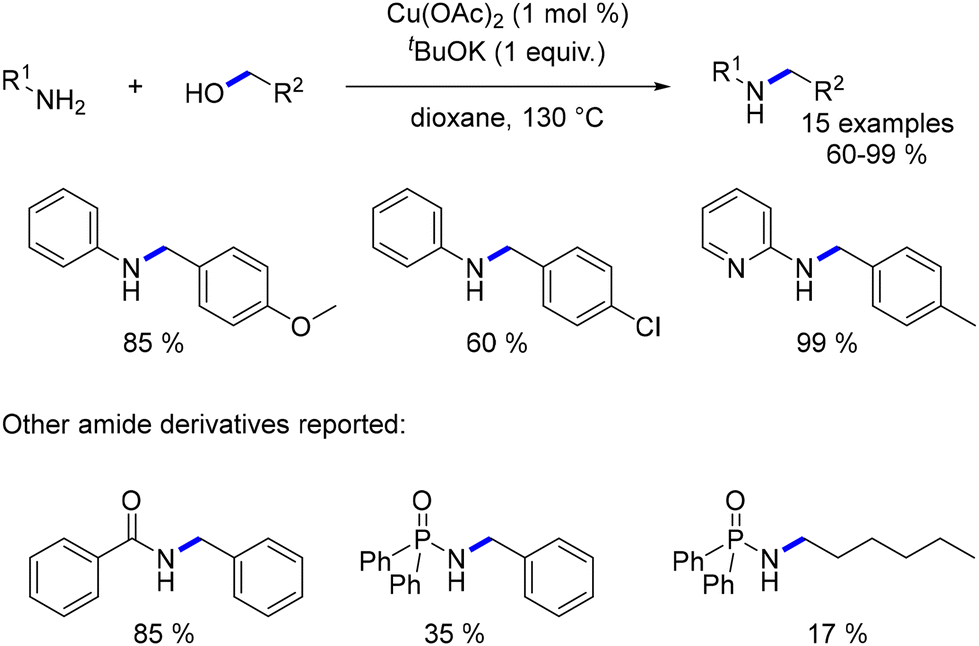
In 2010, Yus expanded upon this work by extending the alkylation of aniline derivatives to lower temperatures, employing a stronger base, higher base loading, and longer reaction times (2 days).122N-Alkylated anilines were obtained in yields ranging from 60% to 99% (Scheme 49). Furthermore, this research encompassed the N-alkylation of amides and amide derivatives.123 A mechanistic exploration employing DFT calculations revealed an outer sphere mechanism for both the dehydrogenation and reduction steps of this reaction.124
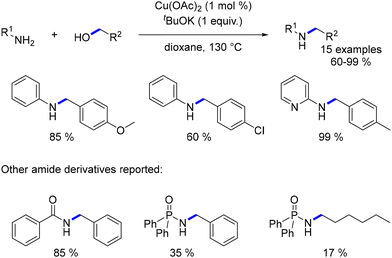 |
| | Scheme 49 Copper-catalysed alkylation of aniline and amide derivatives. | |
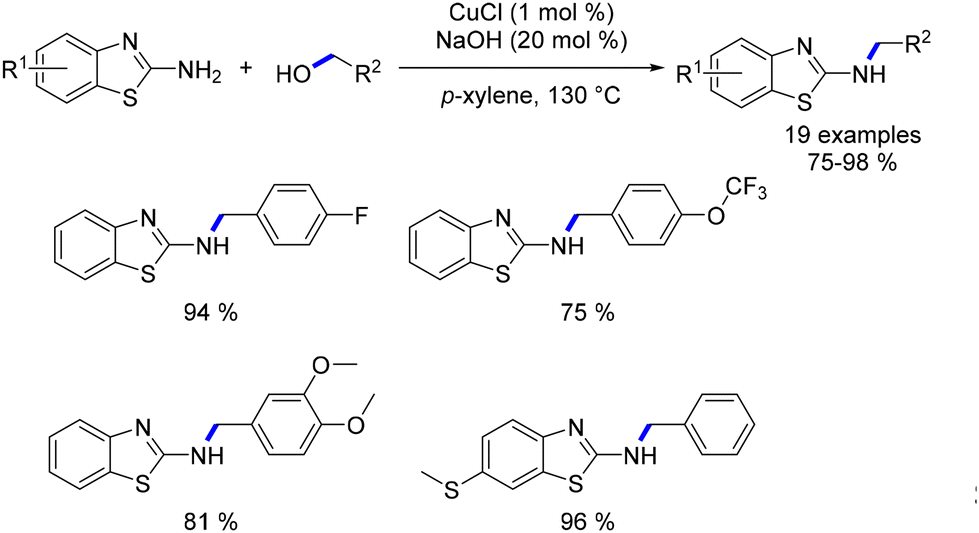
In 2011 Li documented the utilization of CuCl as a catalyst for the alkylation of 2-aminobenzothiazoles with benzyl alcohols.125 The reaction yielded N-alkylated amino benzothiazoles in very good yields (Scheme 50). And the same year, Saito and coworkers proposed a novel method for cross-coupling two different alcohols.126 This C–C bond formation was achieved with a low catalyst loading of CuBr (0.05–0.2 mol%) and NaOH (4–20 mol%) under H2 (1 atm). The reaction offered an alternative approach for synthesizing longer-chain alcohols. Notably, the catalytic cycle differs from previously reported pathways for the Guerbet reaction, which typically involves “borrowing hydrogen”.
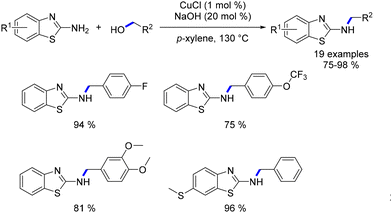 |
| | Scheme 50 Alkylation of amino benzothiazoles, as described by Li. | |
In 2015, Viswanathamurthi introduced a novel heteroleptic binuclear copper(I) complex for catalysing the alkylation of anilines with benzylic alcohols.127 It is noteworthy that sterically hindered anilines were also alkylated with good yields (Scheme 51). Furthermore, nitroarenes were involved and reduced in situ to yield the corresponding N-alkylated compounds after subsequent alkylation.
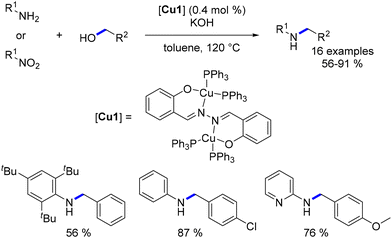 |
| | Scheme 51 Alkylation of anilines and nitroarenes. | |
In 2017, Wang presented a triazole–phosphine–copper complex for the synthesis of benzimidazoles and the alkylation of arylamines.128 This system enabled the alkylation of various anilines with benzyl alcohols and exhibited tolerance towards substituted benzylic alcohols (Scheme 52). Notably, this method facilitated a reduction in the reaction temperature compared to ligand-free copper salt catalysts.120,122–125 However, its scope was restricted to anilines and benzylic alcohol.
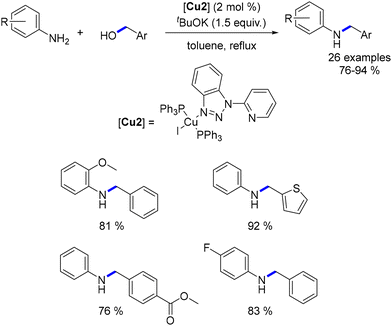 |
| | Scheme 52 Alkylation of anilines, as reported by Wang. | |
2.6. Other 3d-metal catalysed N-alkylation reactions
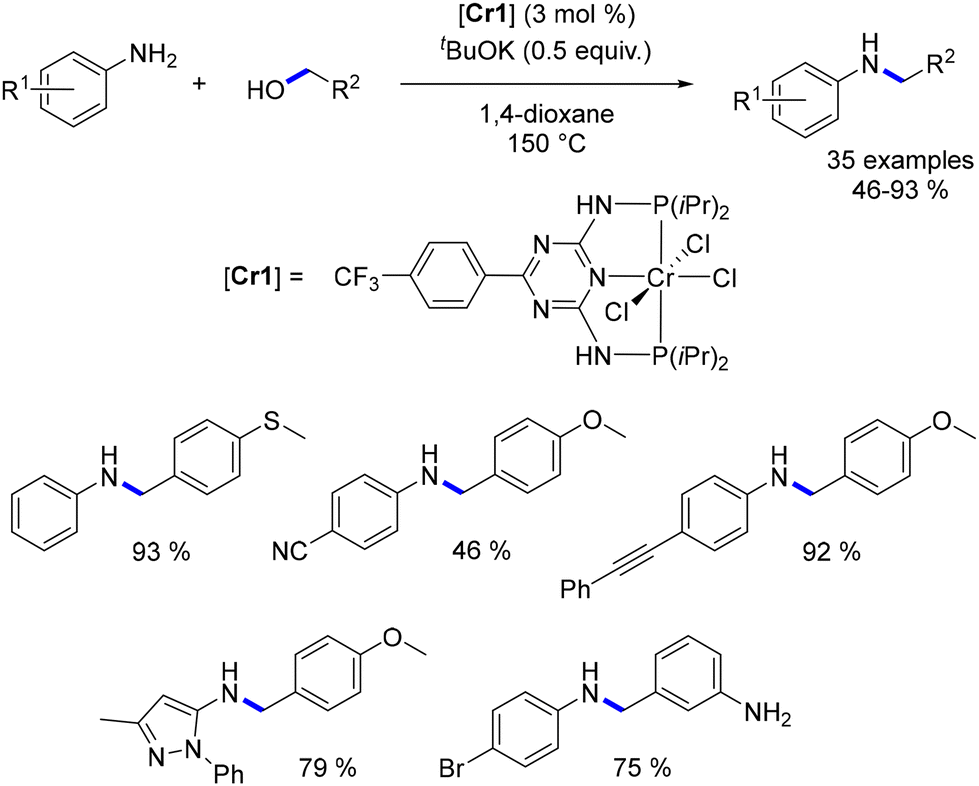
In 2020, Kempe documented a chromium–PNP complex-catalysed alkylation of anilines with alcohols.135 This reaction was constrained to benzylic and homobenzylic alcohols, but it tolerated a broad array of functionalized pro-electrophiles and pro-nucleophiles, resulting in the formation of N-alkyl anilines in good to excellent yields (46 to 93%, Scheme 53), albeit at high temperatures.
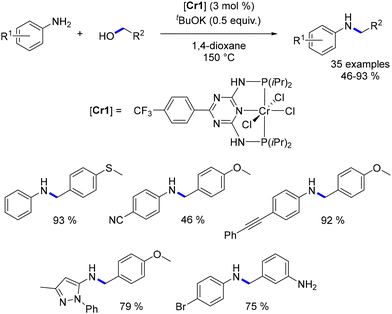 |
| | Scheme 53 Chromium-catalysed alkylation of anilines. | |
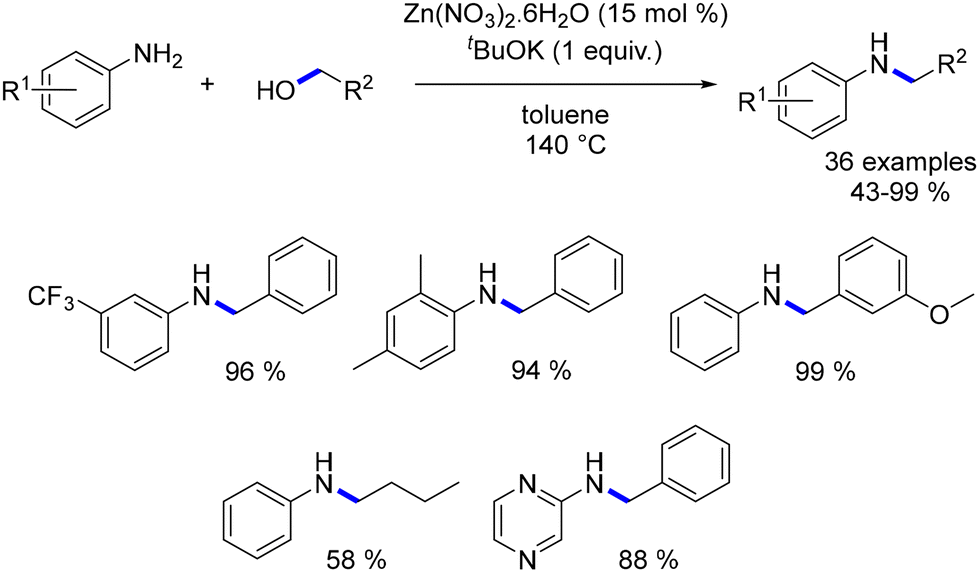
In the same year, Sivakumar and Mannathan elucidated a zinc-catalysed alkylation of anilines. Utilizing simple zinc nitrate salt, the desired alkylated anilines were obtained from both benzylic and aliphatic alcohols as pro-electrophiles (Scheme 54).136
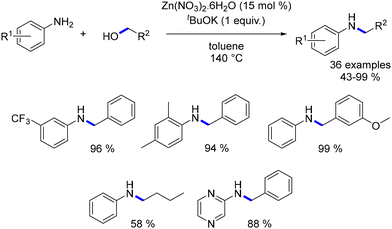 |
| | Scheme 54 Zinc-catalysed alkylation of anilines. | |
3.
C-Alkylation reaction catalysed by first-row transition metals
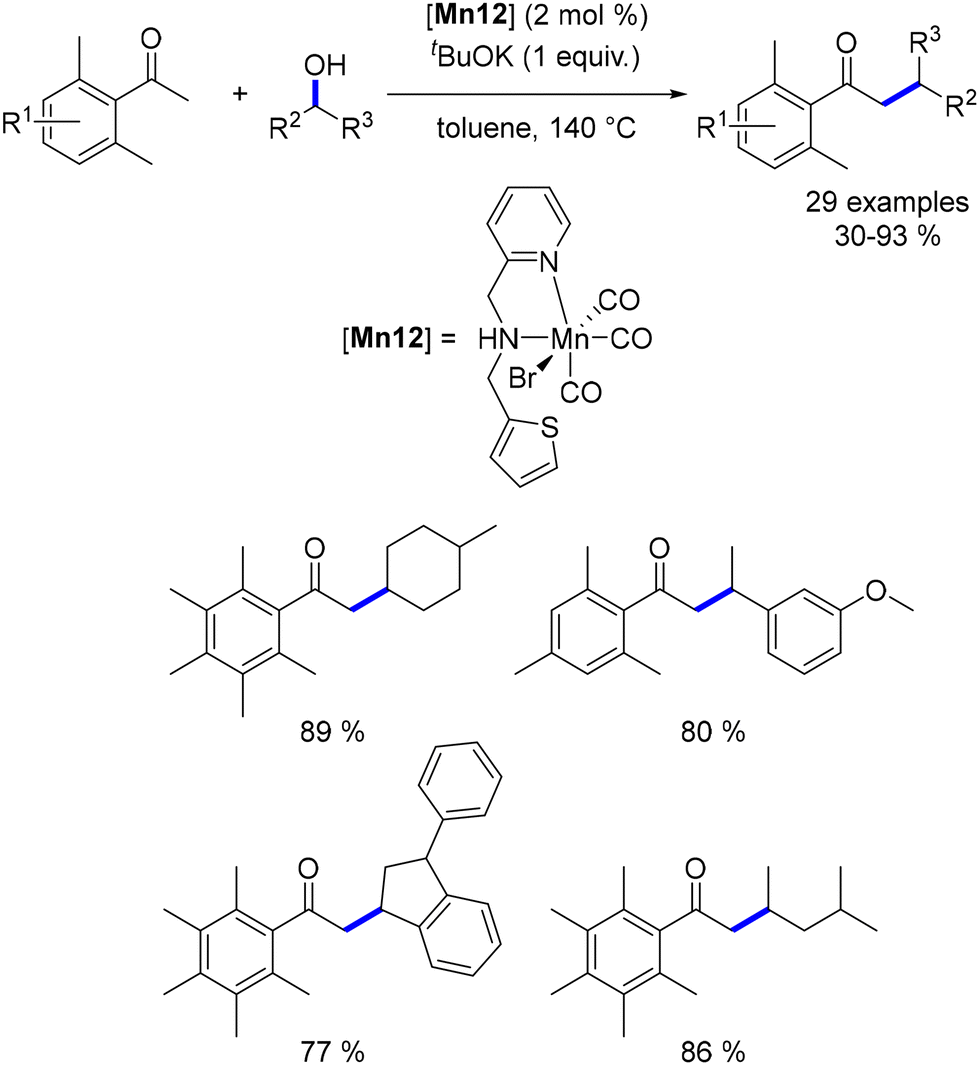
3.1. Manganese-catalysed C-alkylation reactions
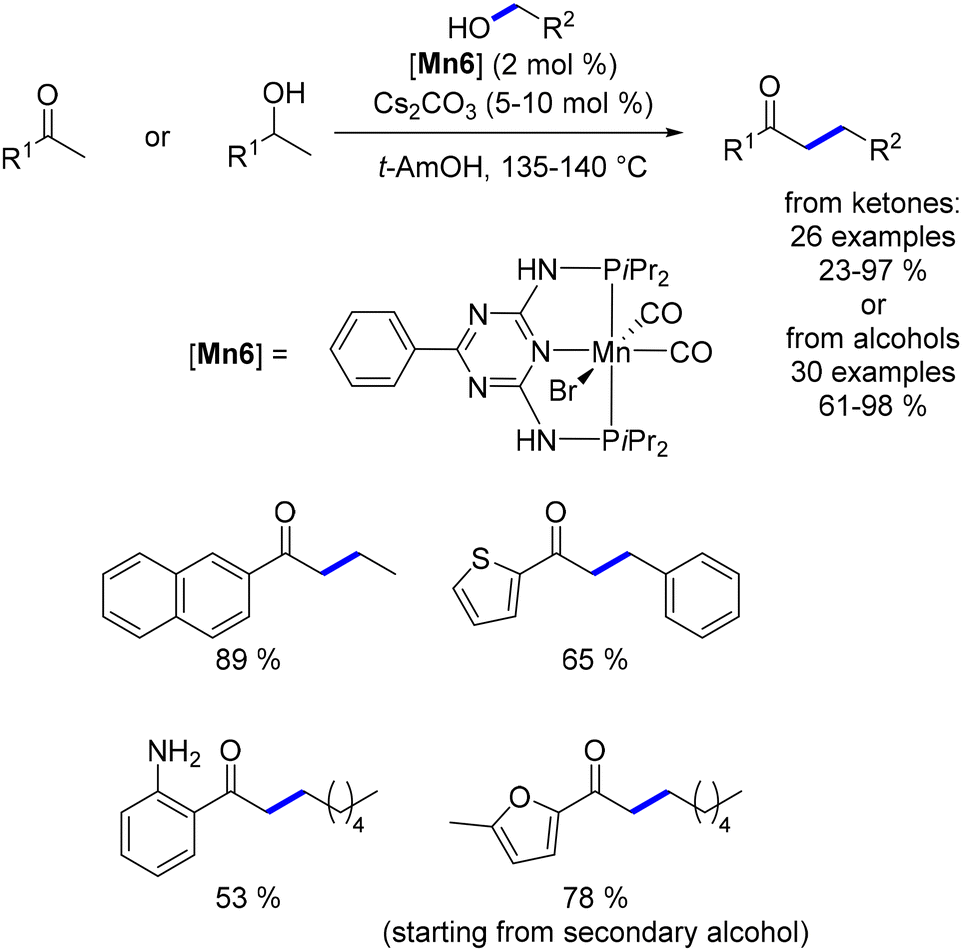
In 2016, Beller reported the first manganese-catalysed alkylation of ketones with primary alcohols.137 The same manganese complex had previously been employed by the group for the alkylation of amines with alcohols.75 For this C–C bond-forming reaction, a catalytic amount of Cs2CO3 (5 mol%) was used as the base, with tert-amyl alcohol as the solvent at 140 °C. A range of substituted ketones and alcohols were successfully alkylated, yielding functionalized products in moderate to good yields (Scheme 55). The reaction's efficiency was influenced by electronic effects, with ketones bearing electron-withdrawing groups resulting in lower yields. This methodology was also applied to oxindole derivatives, which produced alkylated products in good yields. Additionally, Liu and Jones utilized the same manganese complex to upgrade ethanol to 1-butanol at elevated temperatures,138 a process that can also be achieved via the Guerbet reaction.139,140
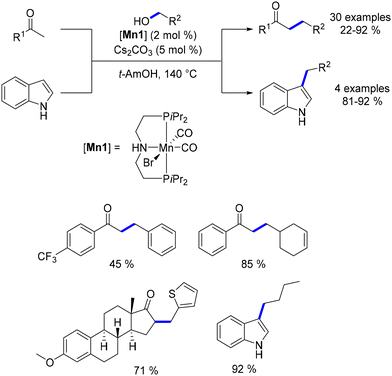 |
| | Scheme 55 Alkylation of ketones and oxindoles. | |
Milstein also explored the alkylation of ketones, amides, and esters using a PNP–Mn complex as the catalyst.141 Aromatic ketones were successfully alkylated with various benzylic and aliphatic alcohols, although the yields were notably lower when aliphatic alcohols were used. Under the same reaction conditions, a single example of oxindole alkylation and five examples of 1-phenylethanol alkylation with benzyl alcohols to yield α-substituted ketones have been reported. However, the reaction appeared to be limited to aryl ketones and benzylic alcohols as the preferred pro-nucleophiles (Scheme 56).
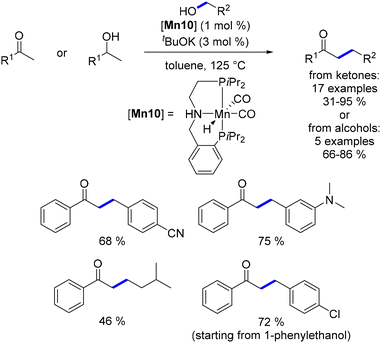 |
| | Scheme 56 Alkylation of ketones and secondary alcohols, as described by Milstein. | |
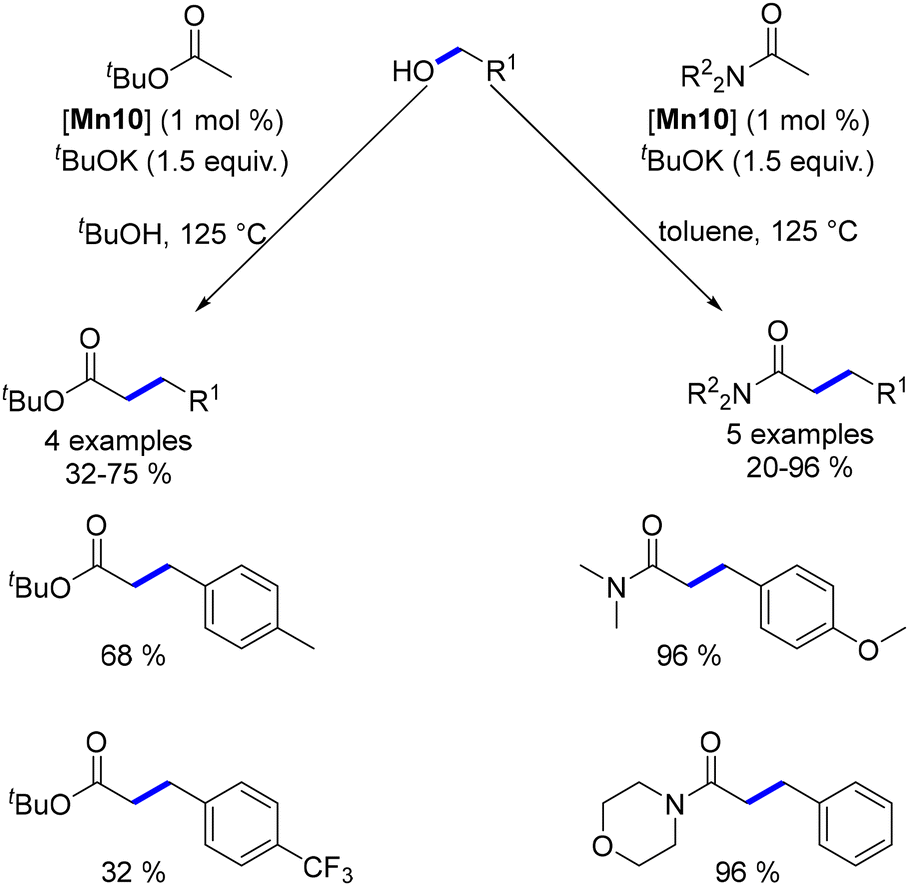
The Macho-type manganese complex also facilitated the alkylation of esters and amides. In this reaction, the use of a stoichiometric amount of a base was essential to achieve high conversions and good yields, ranging from 20 to 96% (Scheme 57).
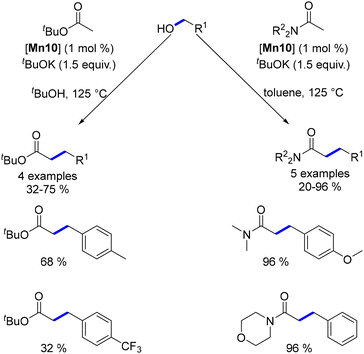 |
| | Scheme 57 Milstein's work on the alkylation of amides and esters. | |
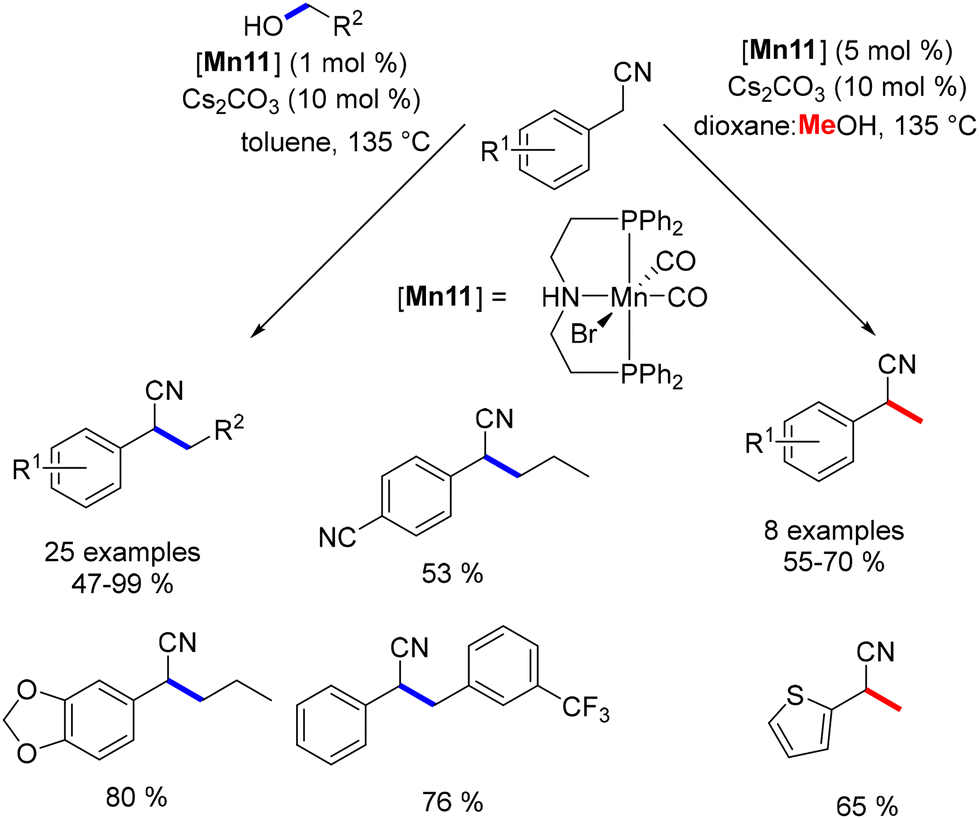
Rueping explored the alkylation of nitriles, successfully converting various benzyl nitriles into alkylated products in moderate to good yields.142 Notably, methanol was employed to achieve α-methylated benzyl nitriles, although the reaction was limited to primary alcohols (Scheme 58).
 |
| | Scheme 58 Alkylation of nitriles, as reported by Rueping. | |
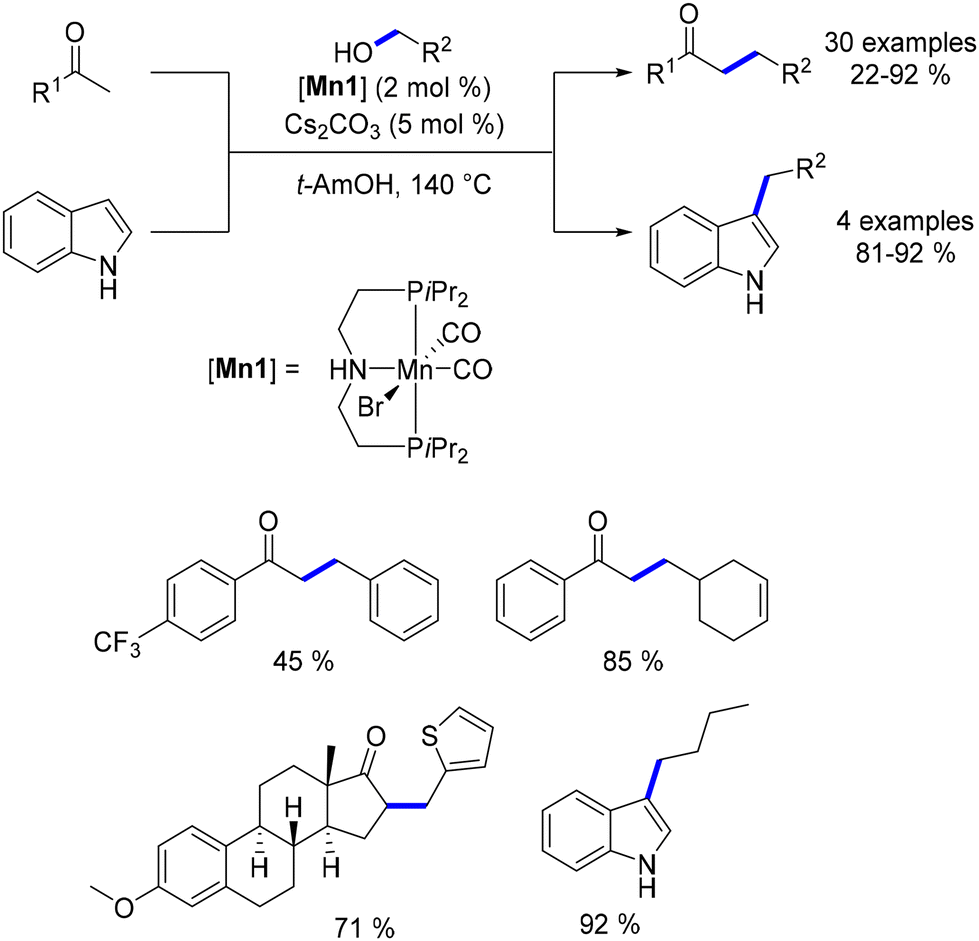
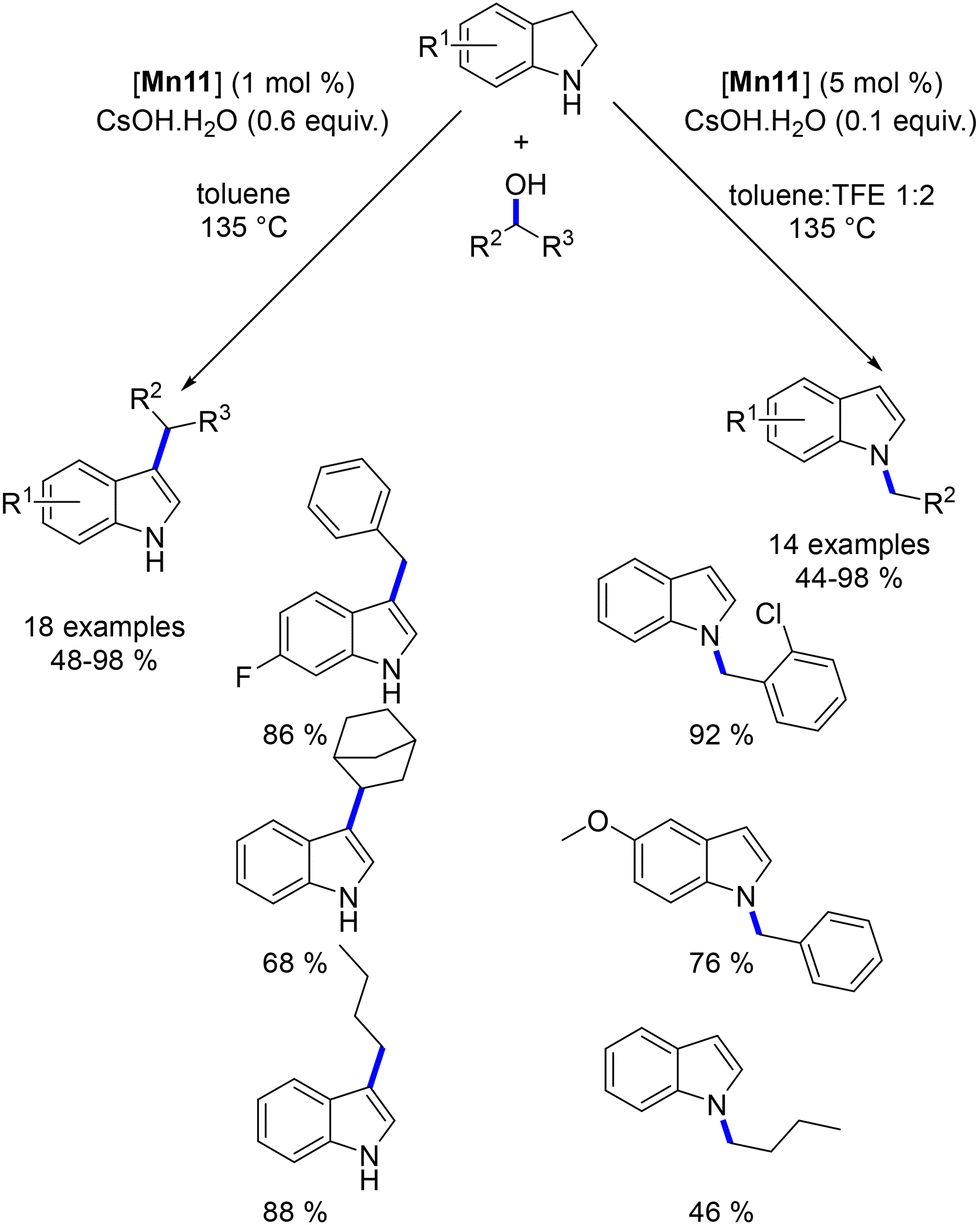
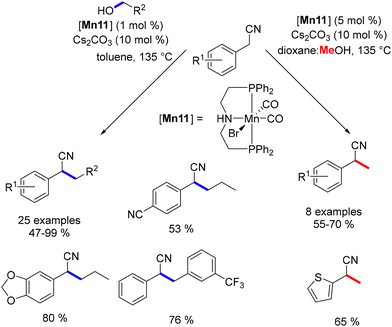
Building on previous research, the alkylation of indolines was explored using the same manganese catalyst.143 This study revealed the potential for both N- and C3-alkylation of indolines. Initially, regardless of the conditions applied, indole derivatives were consistently produced. Moreover, the selectivity between the N- and C3-alkylation could be effectively controlled by adjusting the base concentration and choosing the appropriate solvent. Under optimized conditions, C3-alkylated indoles were obtained in yields ranging from 48% to 98%, while N-alkylated indoles were produced in yields of 44% to 98% (Scheme 59).
 |
| | Scheme 59 Selective alkylation of indoline to N- or C3-substituted indoles. | |
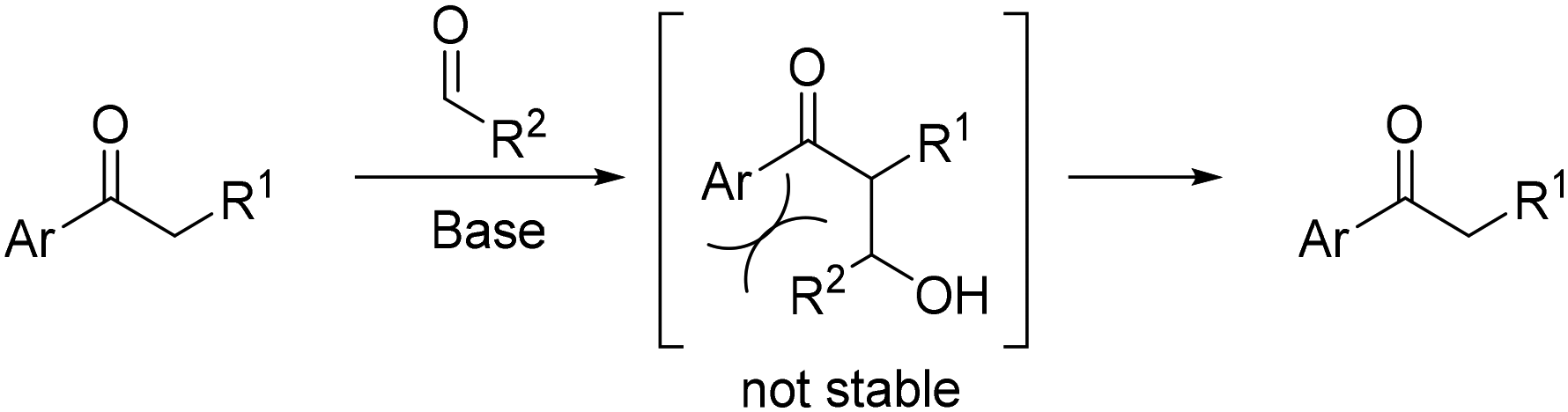
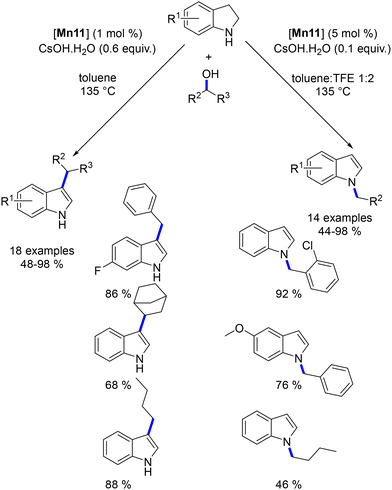
While the mono-alkylation of various nucleophiles has been extensively studied with transition metals, the use of diols for the formation of cycloalkanes using Earth-abundant metal-based complexes is less frequently reported. The alkylation of α-substituted ketones presents additional challenges compared to methyl ketones due to the steric hindrance created by the aromatic ring at the α-position, which tends to favour the retro-aldol reaction over water elimination (Scheme 60). The incorporation of ortho-substituted acetophenones has been shown to mitigate this limitation effectively.144 Indeed, the introduction of ortho substituents results in the aromatic ring and the keto group being oriented perpendicularly rather than in a coplanar manner, effectively reducing steric interactions and eliminating conjugation between them. This orientation prevents the reduction of the ketone due to the steric hindrance imposed by the ortho substituents. Consequently, during the alkylation step, the minimized steric interactions favour the elimination reaction over the retro-aldol reaction. This strategy, first introduced by Donohoe's group, has been successfully applied to the synthesis of α,α-disubstituted ketones,144 the alkylation of ketones with secondary alcohols,145 and the synthesis of cycloalkanes.146,147
 |
| | Scheme 60 Steric hindrance introduced by the aromatic moiety of the starting ketone. | |
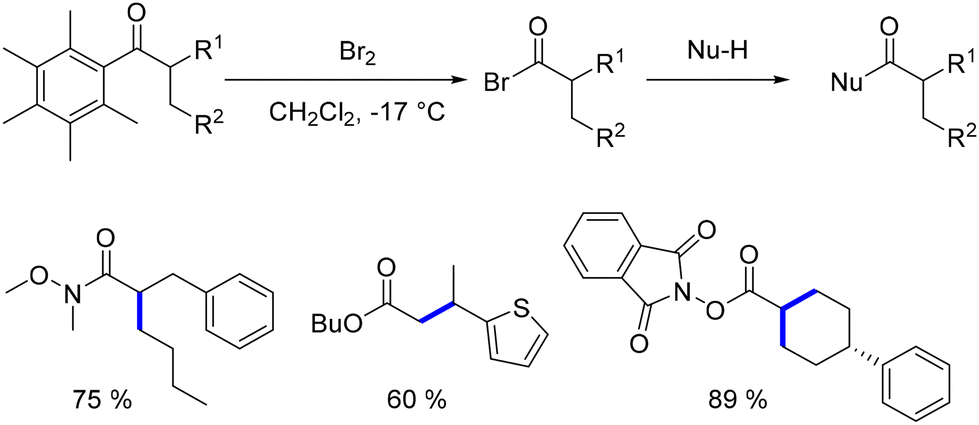
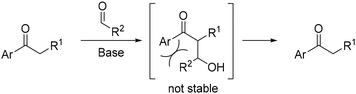
This group specifically utilized 1-(2,3,4,5,6-pentamethyl)ethan-1-one, demonstrating the feasibility of substituting the pentamethylphenyl group with bromine through a retro Friedel–Crafts reaction.148 This process yields an acyl bromide, which can then react with various nucleophiles to produce functionalized products (Scheme 61).
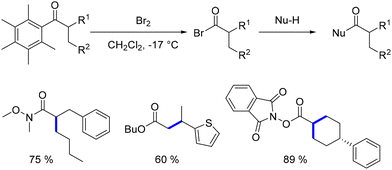 |
| | Scheme 61 Functionalisation of α-substituted ketones. | |
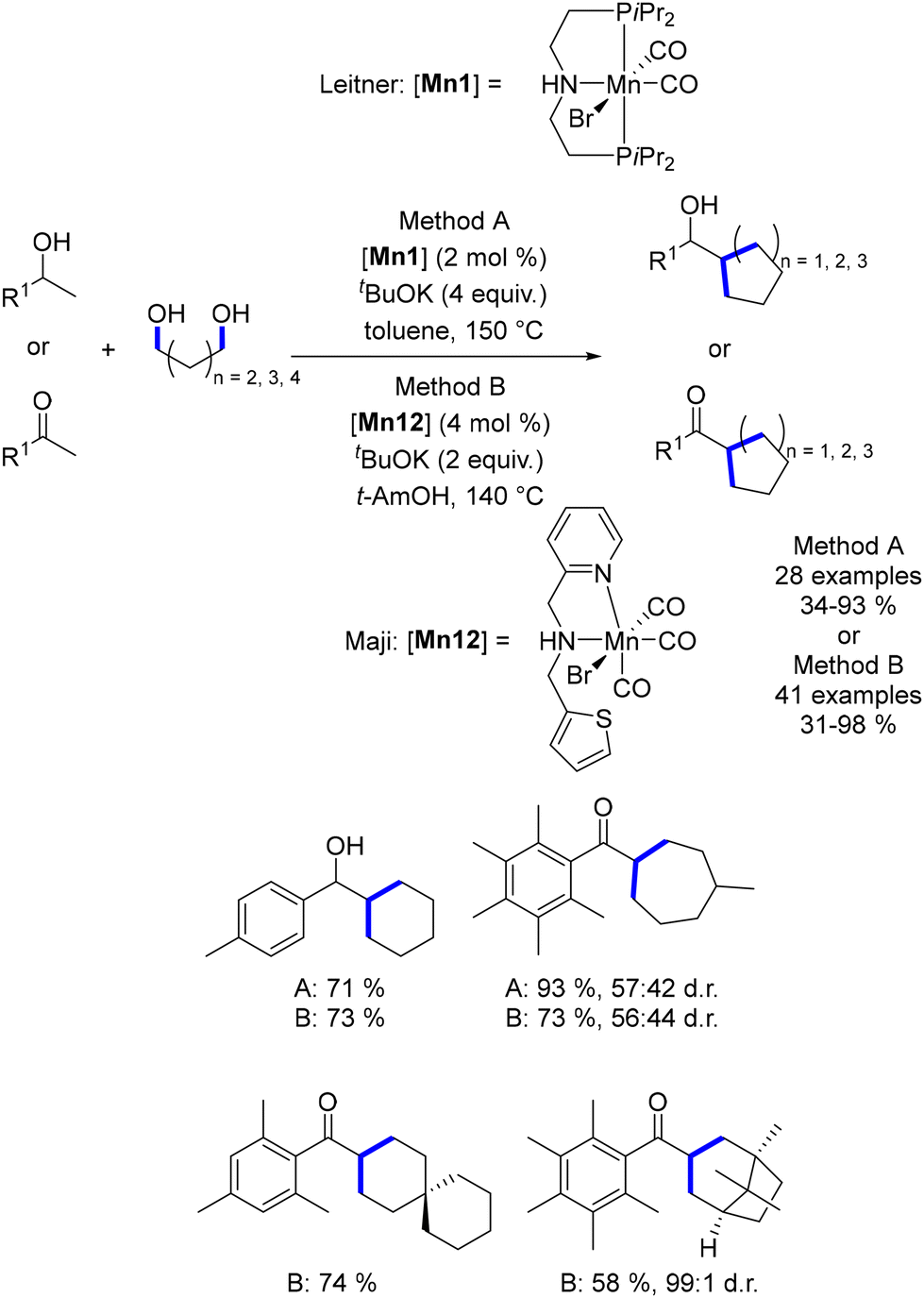
In 2019, Leitner reported the di-alkylation of ketones and secondary alcohols using diols as pro-electrophiles in the presence of the Macho–Mn complex (Scheme 62).149 This methodology afforded a variety of 5-, 6-, and 7-membered rings. Maji further expanded this approach by employing an NNS–Mn complex, reducing both the base loading and reaction temperature.150 As noted, the use of ortho-substituted ketones effectively prevented the final reduction of the carbonyl group.
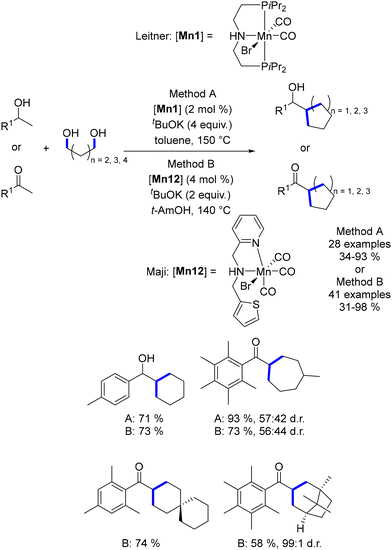 |
| | Scheme 62 Synthesis of cycloalkanes using di-alkylation of ketones and secondary alcohols, as reported by Leitner and Maji. | |
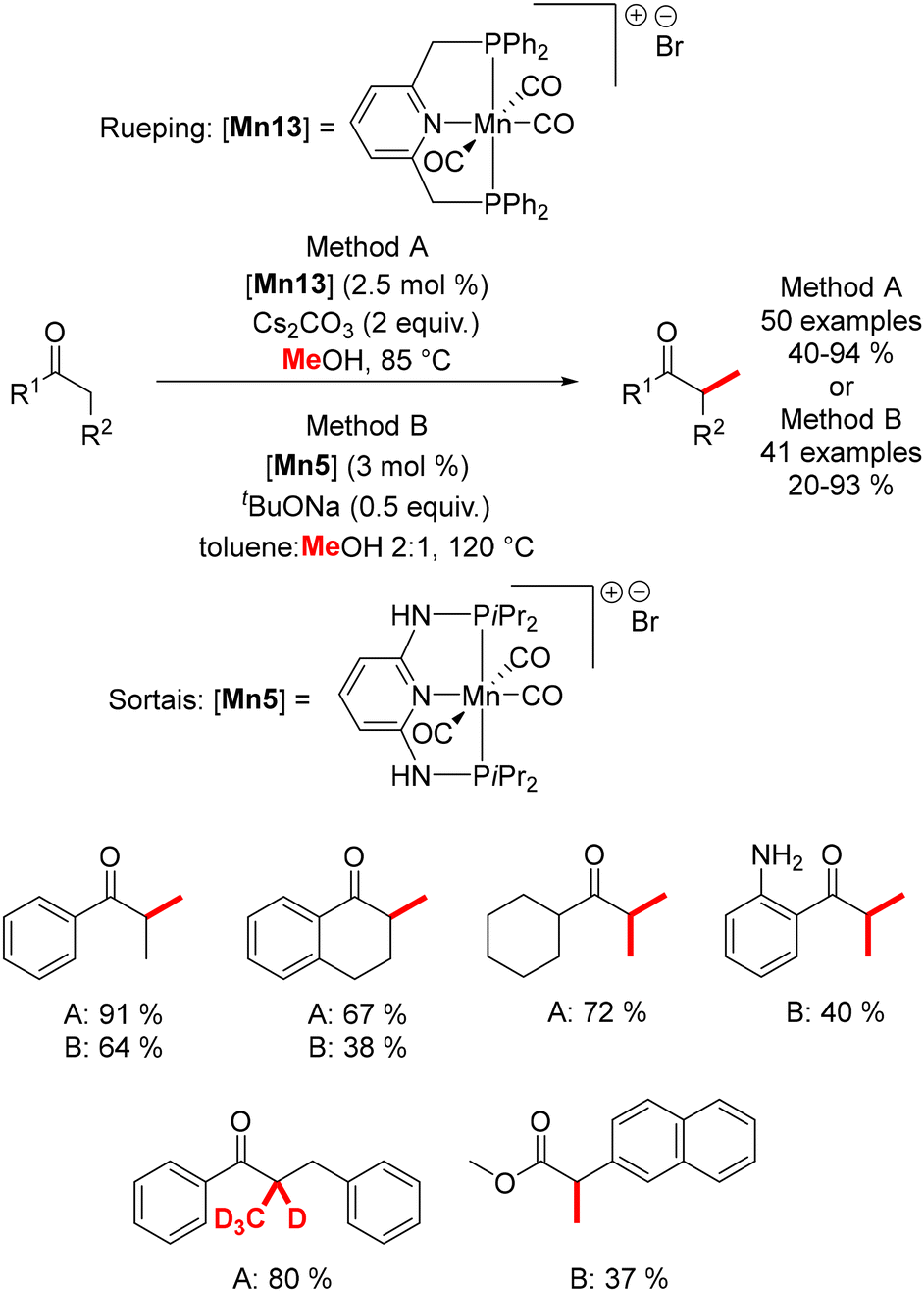
In 2019, Rueping reported the methylation of ketones using methanol or deuterated methanol,151 despite the higher dehydrogenation energy of methanol (ΔH = +84 kJ mol−1) compared to ethanol (ΔH = +68 kJ mol−1). Conducted under mild conditions (85–105 °C), this method afforded mono- and di-methylated products in good yields (Scheme 63). Simultaneously, Sortais described a similar reaction using a pincer Mn complex,152 which required higher temperatures (120 °C) and lower base loading. This methodology was also extended to the methylation of esters (Scheme 63).
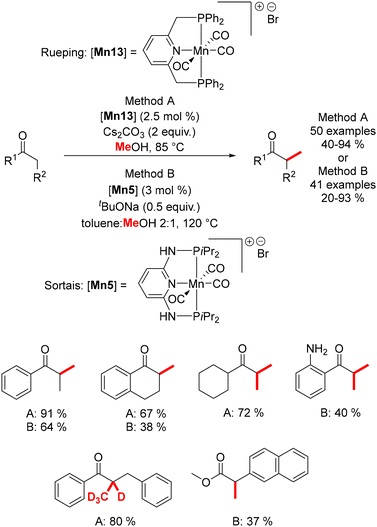 |
| | Scheme 63 Methylation of ketones and esters, as reported by Rueping and Sortais. | |
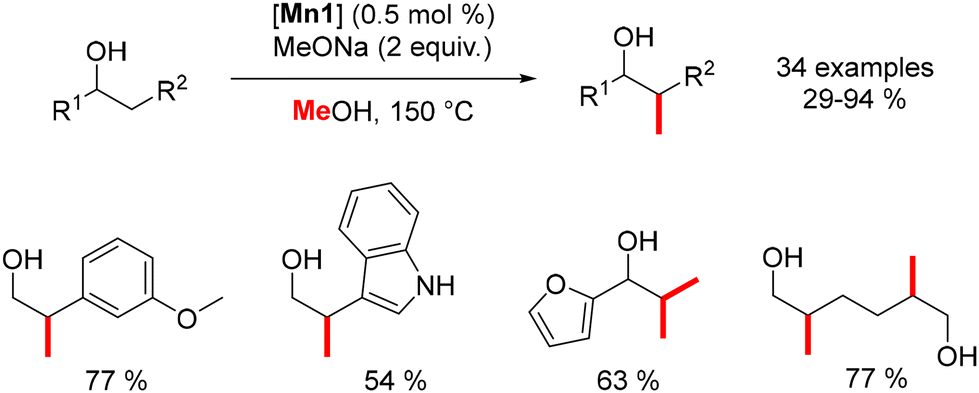
Leitner reported the β-methylation of alcohols using a stoichiometric amount of a base and the Macho–Mn complex at 150 °C, resulting in β-branched products (Scheme 64).153 Unlike iron complexes, which were limited to homo-benzylic alcohols, the manganese complex enables the introduction of both homo-benzylic and aliphatic alcohols without any adverse effects.
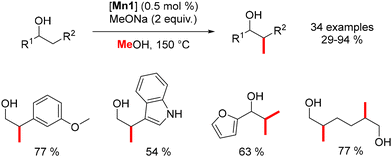 |
| | Scheme 64 Methylation of alcohols. | |
In 2019, Gunanathan reported methods for the α-alkylation of ketones and β-alkylation of alcohols, both leading to α-substituted ketones.154 The key distinction between the two methodologies was the base loading, which varied from 10% to 5%. Both aliphatic and benzylic alcohols yielded the desired products in good quantities. For the mono-ethylation of ketones, a mixed solvent system (t-AmOH:EtOH 1:1) was used. When keto-aniline derivatives were employed, selective C-alkylation was achieved, demonstrating the reaction's chemoselectivity (Scheme 65).
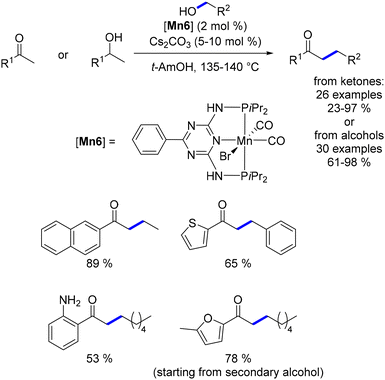 |
| | Scheme 65 Synthesis of α-substituted ketones. | |
Yu reported the alkylation of secondary alcohols with primary alcohols using a bifunctional NNN–Mn(I) complex, featuring a pyridine–benzimidazole–pyrrole ligand.155 Unlike the results obtained by Milstein,141 where the CO bond of the alkylated ketone remained intact, Yu's approach led to the reduction of the CO bond, resulting in β-substituted alcohols as the products (Scheme 66).
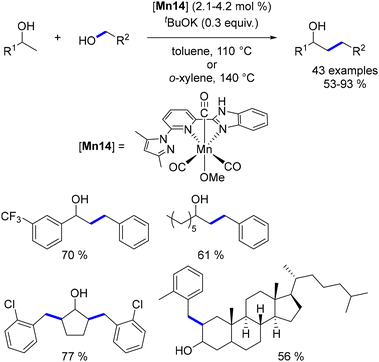 |
| | Scheme 66 Alkylation of secondary alcohols. | |
In 2018, Maji reported the use of a phosphine-free NNN–Mn complex for the alkylation of ketones.156 This complex facilitated the reaction of various aryl and aliphatic ketones with primary alcohols, yielding α-substituted ketones with diverse functional groups (Scheme 67). Both aliphatic and benzylic alcohols were effective, although attempts at methylation did not produce any methylated products.
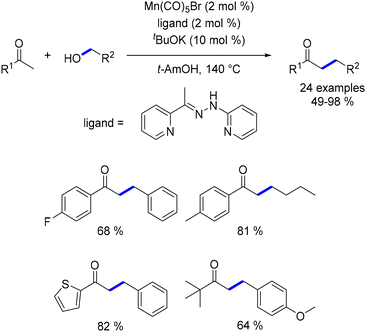 |
| | Scheme 67 Alkylation of ketones. | |
Replacing the pyridine hydrazone ligand with a thiophene hydrazone ligand enabled the alkylation of nitrile derivatives using various primary alcohols.157 The reaction conditions were similar to those used for C–C bond formation between a ketone and an alcohol,156 with the notable exception of an increased base loading from 10 to 20 mol%. Alkylated nitriles were obtained in moderate to good yields (45–88%, Scheme 68). This reaction tolerated a range of functional groups but was restricted to benzyl nitriles. Methanol was introduced, but no methylation was observed.
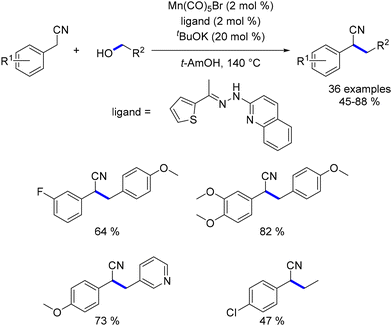 |
| | Scheme 68 Alkylation of nitriles. | |
In 2020, Maji explored the alkylation of ketones with secondary alcohols using a phosphine-free, bidentate NN-ligand Mn complex.158 To prevent cross-alkylation, only 2,5-dimethyl acetophenone derivatives were employed as pro-nucleophiles. Under optimized conditions, various benzylic and aliphatic secondary alcohols were reacted, yielding β-branched ketones in good yields (Scheme 69).
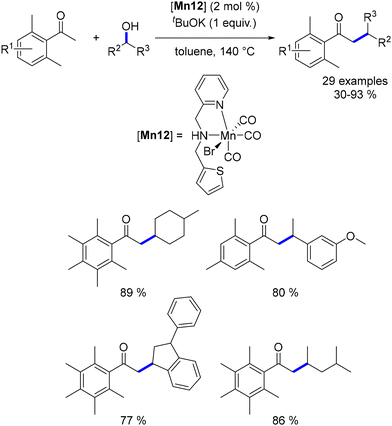 |
| | Scheme 69 Alkylation of ketones with secondary alcohols. | |
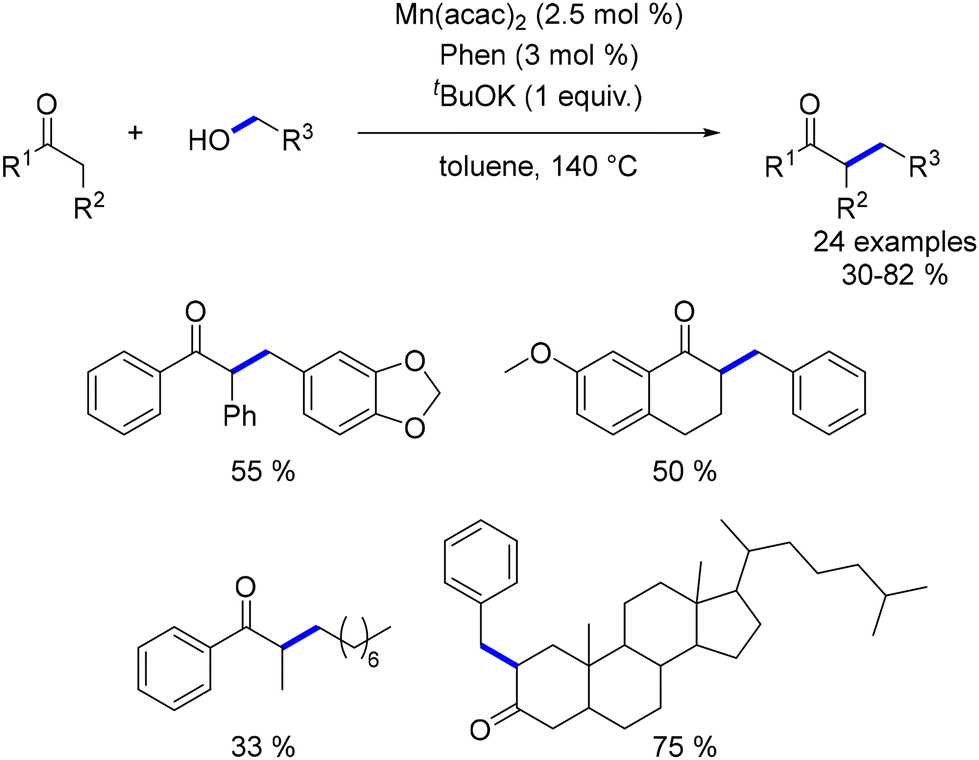
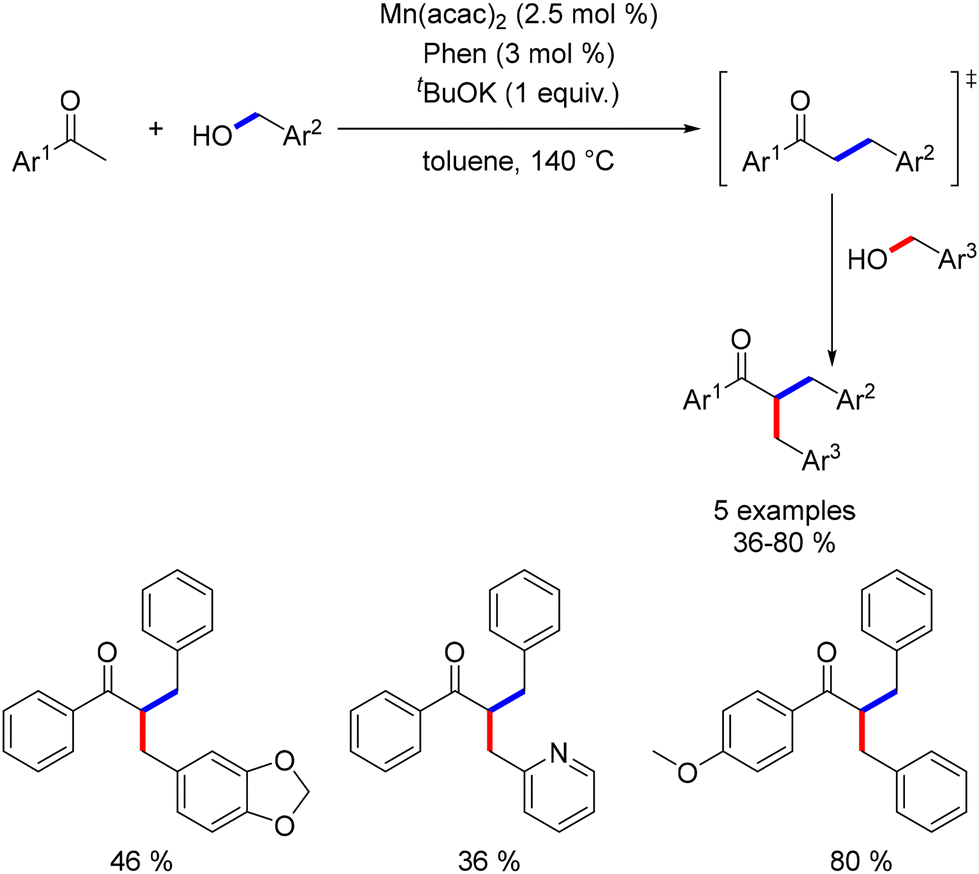
Banerjee investigated the alkylation of α-substituted ketones with primary alcohols using a catalyst system composed of Mn(acac)2 and 1,10-phenanthroline.159 The reaction required a stoichiometric amount of a base to achieve the desired alkylated products (Scheme 70).
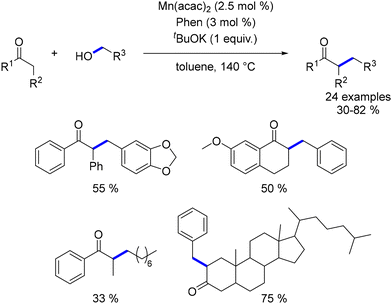 |
| | Scheme 70 Alkylation of α-substituted ketones, as reported by Banerjee. | |
In a two-step, one-pot reaction, acetophenones underwent dialkylation with the sequential addition of a second alcohol after 2 hours. This approach yielded dialkylated products in moderate to good yields (Scheme 71).
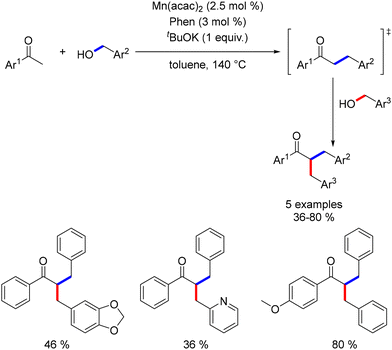 |
| | Scheme 71 Two-step one-pot dialkylation of ketones. | |
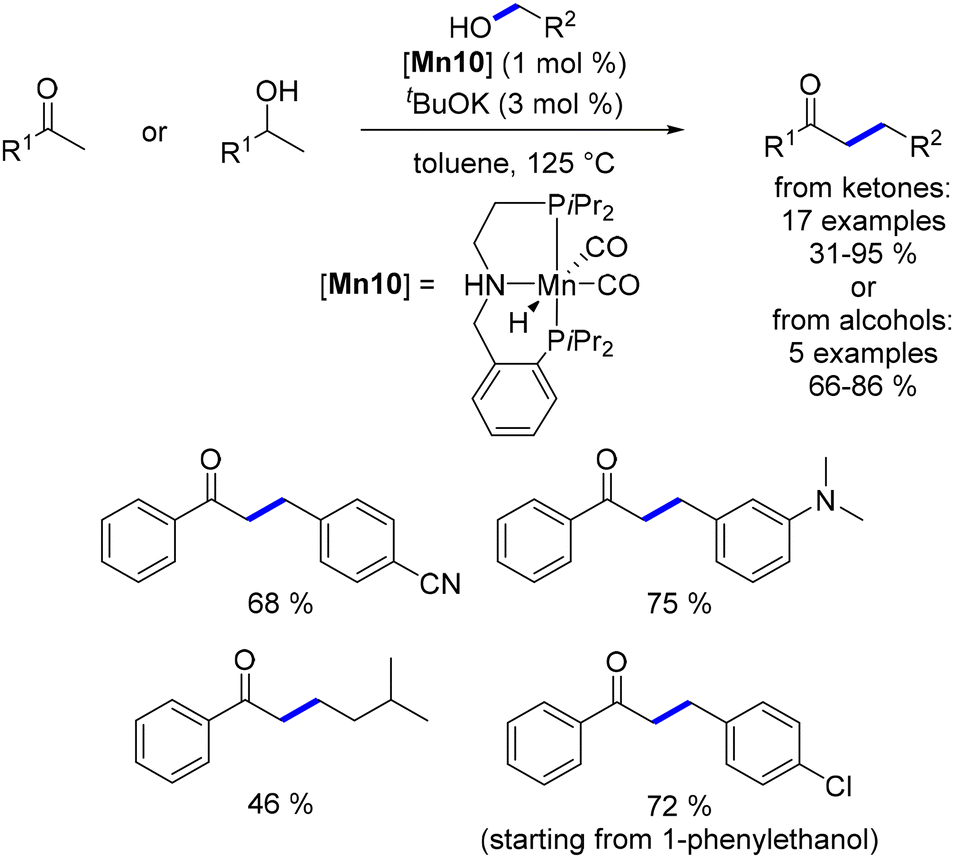
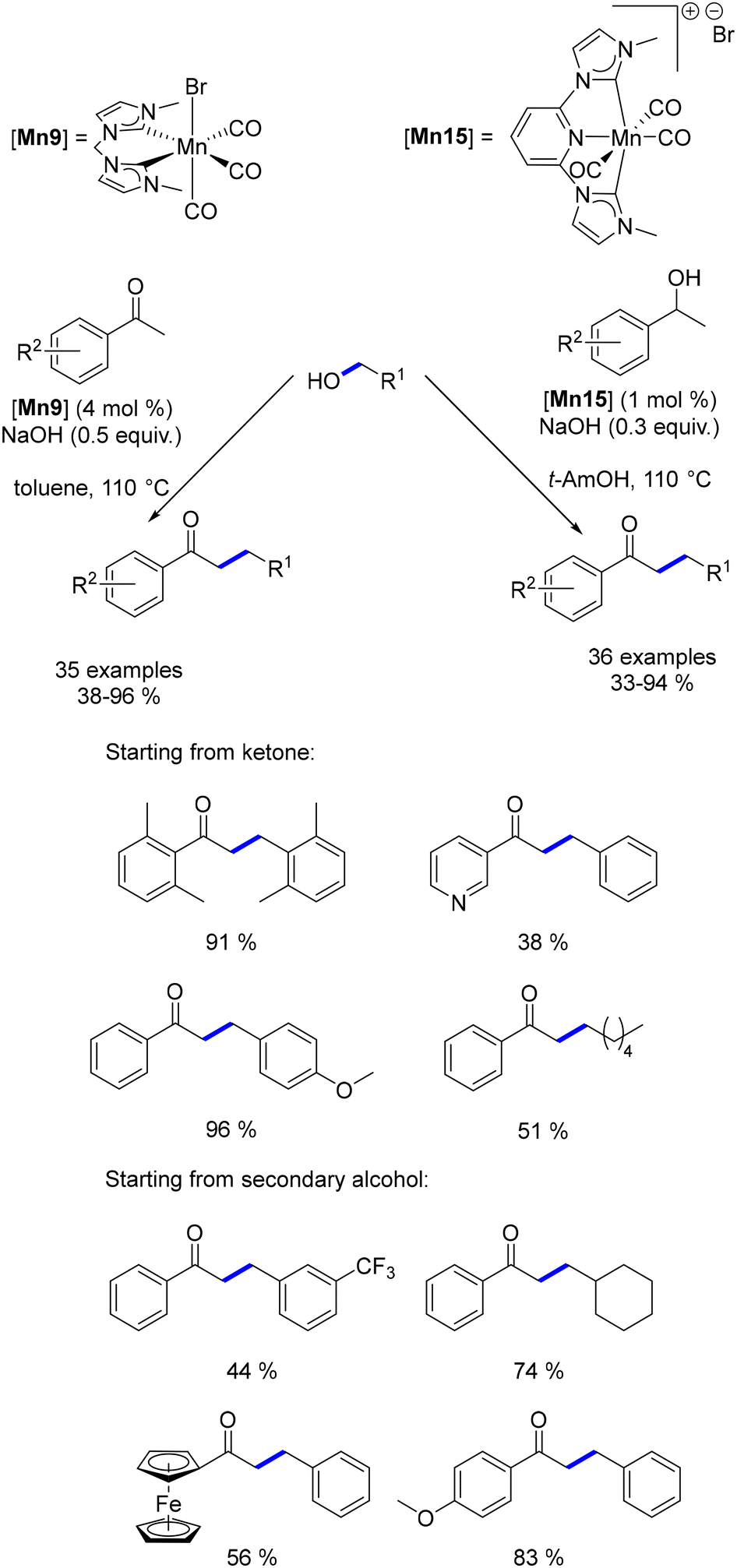
Organometallic complexes employed in borrowing hydrogen reactions often feature bifunctional or hemilabile ligands, which play a crucial role in the reaction mechanism. However, these ligands can be costly to produce, sensitive to air or moisture, and challenging to synthesize on a large scale. To address these issues, Ke investigated a non-bifunctional manganese complex for the α-alkylation of ketones with alcohols.160 Preliminary DFT calculations compared the reduction of α,β-unsaturated ketones catalysed by this non-bifunctional complex with that of various previously described catalytic systems.141,156 The theoretical study revealed that the non-bifunctional complex demonstrated a significantly lower hydrogenation energy (14.9 kcal mol−1) compared to the Beller, Maji, and Milstein systems, which required 22.8, 23.7, and 26.6 kcal mol−1, respectively. Experimental validation supported this finding, as the non-bifunctional complex efficiently catalysed the α-alkylation of ketones at 110 °C, in contrast to the higher temperatures (125–140 °C) needed for the other systems, and achieved the reaction in a shorter time frame of 2 hours (Scheme 72). Although this reaction required a catalytic amount of a base (NaOH, 0.5 equiv.), it used lower amounts of the base compared to the amounts required in the studies reported by Beller, Maji, or Milstein. Furthermore, this non-bifunctional complex was subsequently modified to enable the β-alkylation of secondary alcohols to form α-substituted ketones.161 Nevertheless, both systems were constrained to the alkylation of aryl ketones using primary alcohols.
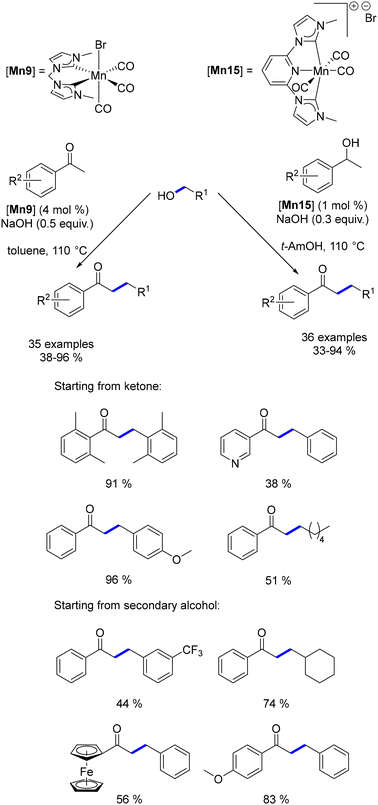 |
| | Scheme 72 Synthesis of α-substituted ketones, as described by Ke. | |
3.2. Iron-catalysed C-alkylation reactions
Building on Saito's pioneering work on iron-catalysed alkylation of amines,88 Sun reported in 2012 the first iron-catalysed C–C bond formation via borrowing hydrogen.162 Using a simple ferrocene carboxaldehyde catalyst, a range of secondary alcohols were alkylated with benzylic and aliphatic alcohols to produce β-substituted alcohols. This method, while tolerant of only a few functional groups, yielded substituted alcohols in good to excellent yields (Scheme 73).
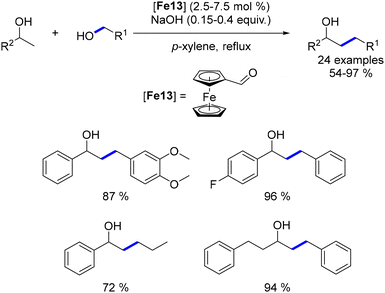 |
| | Scheme 73 Iron-catalysed β-alkylation of alcohols. | |
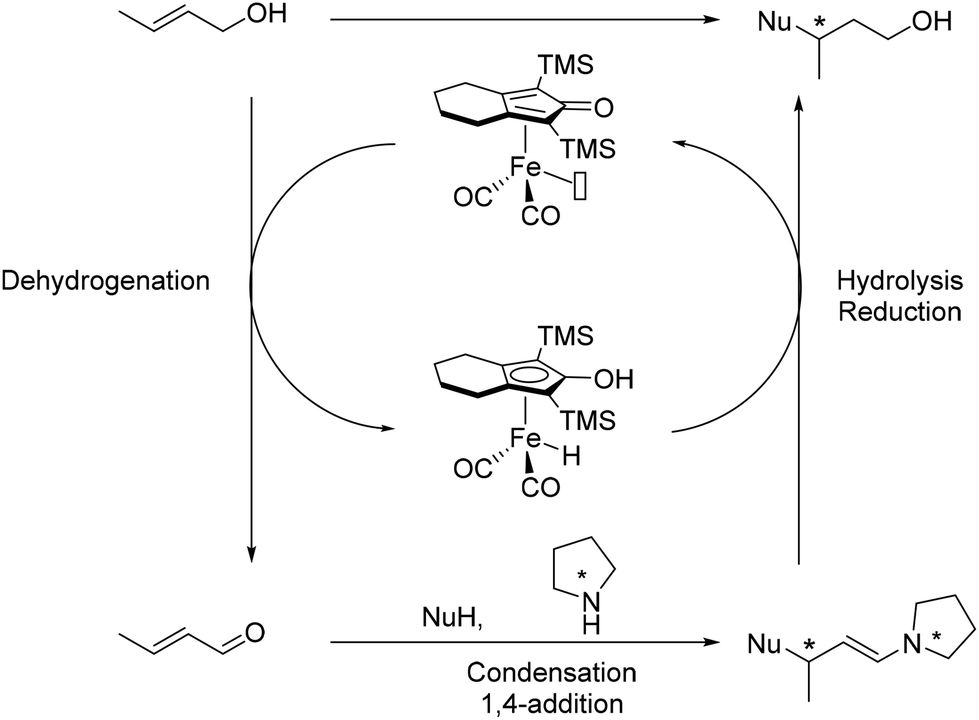
In 2013, Quintard and Rodriguez developed a dual catalytic cascade process for the enantioselective functionalization of allylic alcohols.163 The process begins with the Knölker complex, which dehydrogenates the allylic alcohol to form an enal intermediate. This intermediate then undergoes condensation with a prolinol-type amine, followed by 1,4-addition with a nucleophile to yield a chiral enamine. The final steps involve hydrolysis and reduction to produce the β-substituted chiral alcohol (Scheme 74).
 |
| | Scheme 74 Dual iron/organocatalysed alkylation of allylic alcohols. | |
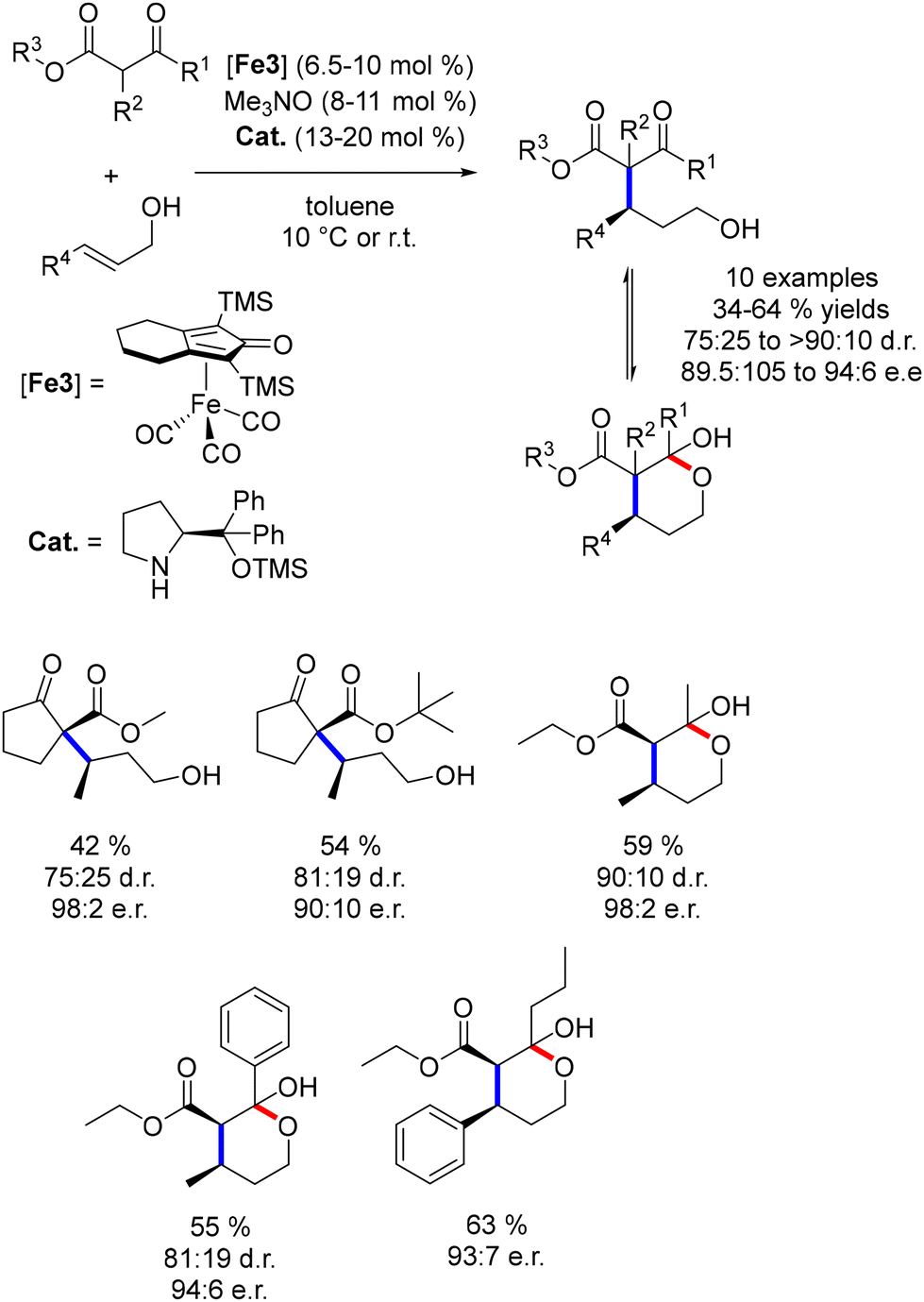
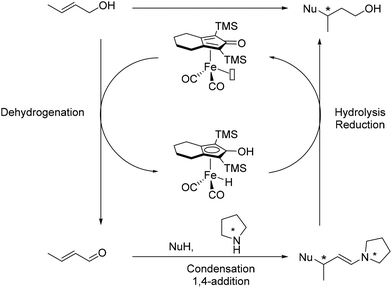
This methodology was assessed using β-ketoesters as pro-nucleophiles, resulting in moderate to good yields (35–64%, Scheme 75) with excellent enantiomeric ratios (89.5:10.5 to 96:6) and good diastereomeric ratios (75:25 to >90:10) for crotyl and cinnamyl alcohols. The iminium intermediate played a crucial role in ensuring enantiocontrol and product formation, with less than 10% conversion observed in the absence of the amine catalyst.
 |
| | Scheme 75 Enantioselective alkylation of allylic alcohols. | |
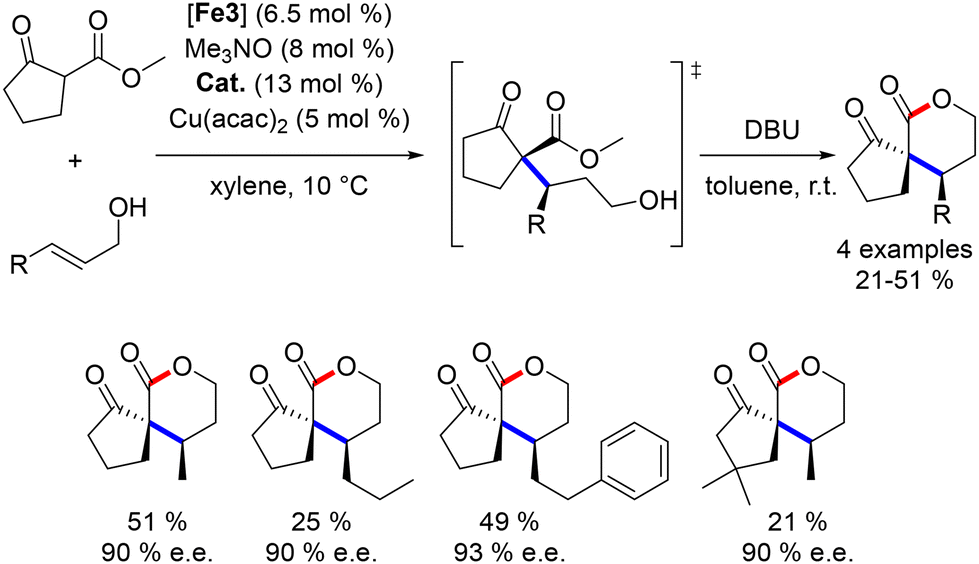
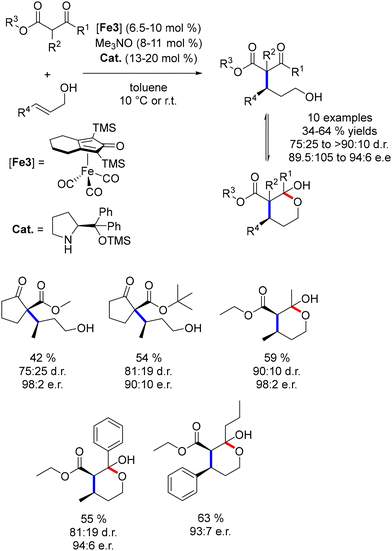
In contrast to other substrates, cyclopentanone derivatives were not isolated in their cyclic form. Consequently, a final transesterification step was explored to enable the formation of chiral γ-lactones from 1,3-keto-esters.164 DBU was introduced after the alkylation with allylic alcohol, facilitating the formation of the lactone product. The addition of a copper salt improved the enantiomeric ratios of the reaction (Scheme 76).
 |
| | Scheme 76 Synthesis of γ-lactones. | |
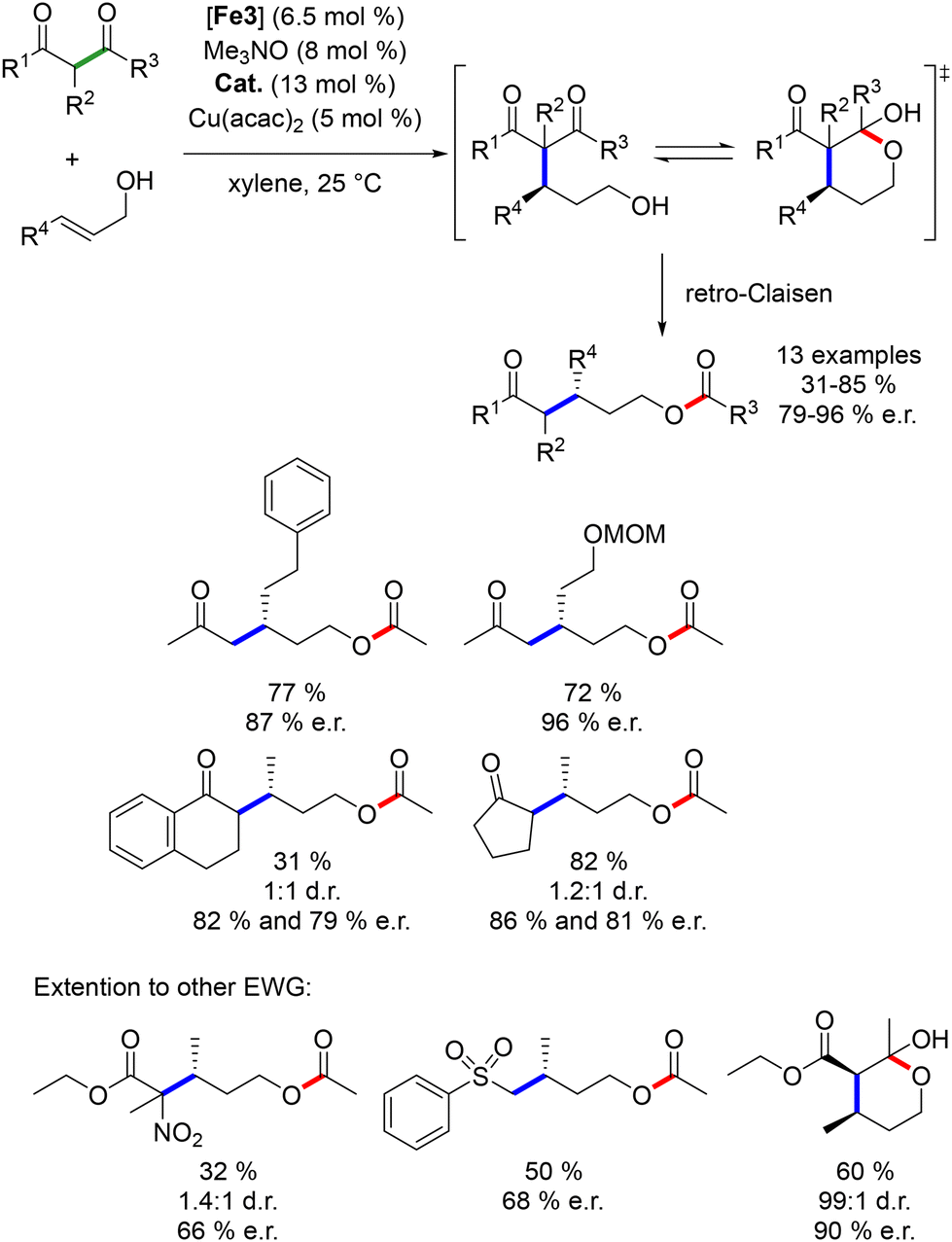
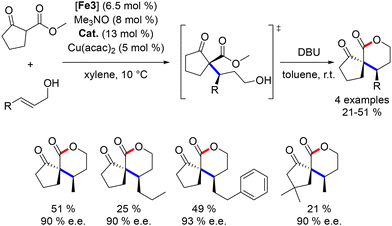
The same group further applied this dual catalytic strategy to the synthesis of 3-alkyl pentanols.165 The cascade process was complemented by a final retro-Claisen reaction (Scheme 77). Electron-withdrawing groups were not tolerated, and the choice of organocatalyst varied depending on the diketone used. The products were obtained in moderate to good yields (34–96%) with good enantiomeric ratios (70:30 to >96:4). Cyclic β-diketones showed no diastereoselectivity, likely due to non-selective protonation during the retro-Claisen fragmentation step. Optimization by adding a copper salt as a chelating agent in the Michael addition improved the enantiomeric ratio of the reaction (31–85%, 79–96% ee).166 Additionally, other β-EWG-substituted ketones were employed as pro-nucleophiles, yielding products in moderate to good yields with good enantiomeric ratios (66–90% ee).
 |
| | Scheme 77 Insertion of allylic alcohol in 1,3-diketones. | |
In 2015, Darcel reported the α-alkylation of ketones with primary alcohols using the Knölker complex at 140 °C.167 The addition of triphenylphosphine was found to enhance the catalyst's activity, likely by extending the lifetime of the catalytic species. However, the methodology was limited to aromatic ketones, as aliphatic ketones only underwent reduction under these reaction conditions (Scheme 78).
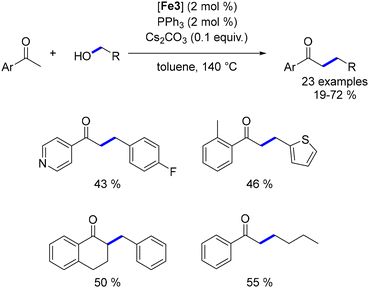 |
| | Scheme 78 Alkylation of ketones, as reported by Darcel. | |
Methylation reactions were subsequently explored by Morrill.168 Using the Knölker complex, various nucleophiles were reacted with methanol under mild conditions, yielding methylated products in good to excellent yields (Scheme 79).
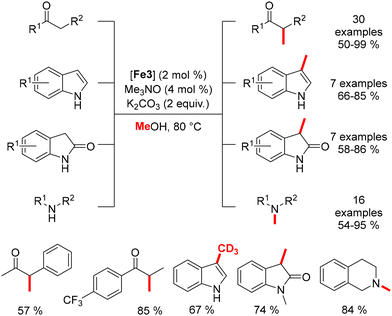 |
| | Scheme 79 Methylation of various nucleophiles. | |
In 2017, the modified Knölker catalyst, developed by Renaud,101 was employed for the α-alkylation of aryl ketones.48 DFT calculations compared the original Knölker catalyst with the Renaud's variant, revealing lower energy barriers and an exothermic reduction step for the diamino derivative. This theoretical insight allowed for a decrease in reaction temperature to 90 °C with reduced reaction times, leading to improved yields of alkylated products (Scheme 80). Unlike Darcel's findings,167 this modification enabled the successful alkylation of aliphatic ketones without reduction.
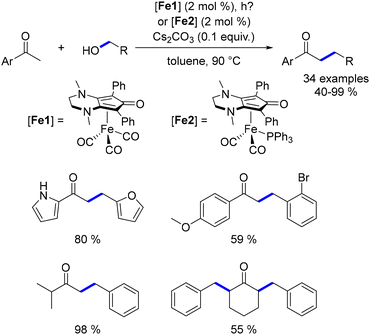 |
| | Scheme 80 Alkylation of ketones, as reported by Renaud. | |
Following this work, the alkylation of indoles was explored,46 requiring higher temperatures due to the lower nucleophilicity of indoles. The alkylated products were obtained in moderate to good yields. Additionally, two examples using secondary alcohols as pro-electrophiles were achieved, with yields ranging from 45–55% (Scheme 81).
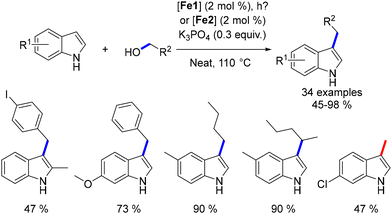 |
| | Scheme 81 Alkylation of indoles, as reported by Renaud. | |
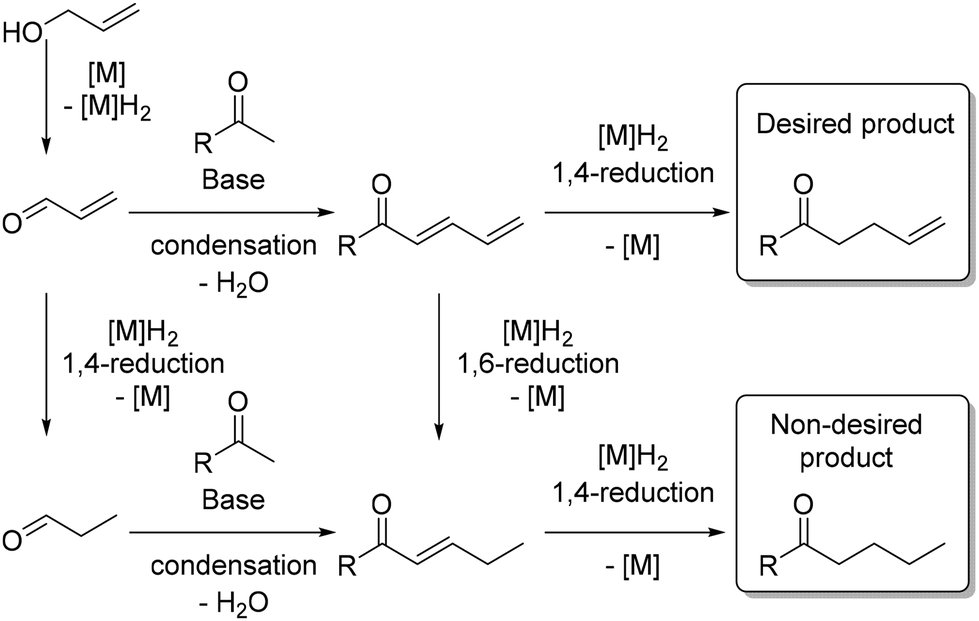
There are only a few reported examples in the literature where allylic alcohols are utilized as pro-electrophiles for C–C bond formation or for the chemoselective reduction169 of unsaturated compounds.170 This reaction poses a significant challenge because the potential 1,4-reduction of the α,β-unsaturated aldehyde could result in a saturated product. Additionally, the 1,6-reduction of the dienone intermediate, formed by the condensation of the α,β-unsaturated aldehyde with a ketone, may also yield a saturated product (Scheme 82).
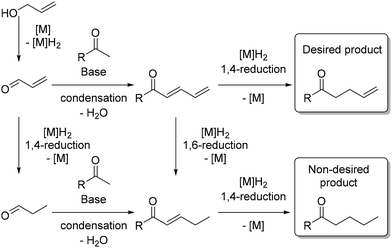 |
| | Scheme 82 Challenges associated with using allylic alcohols as pro-electrophiles. | |
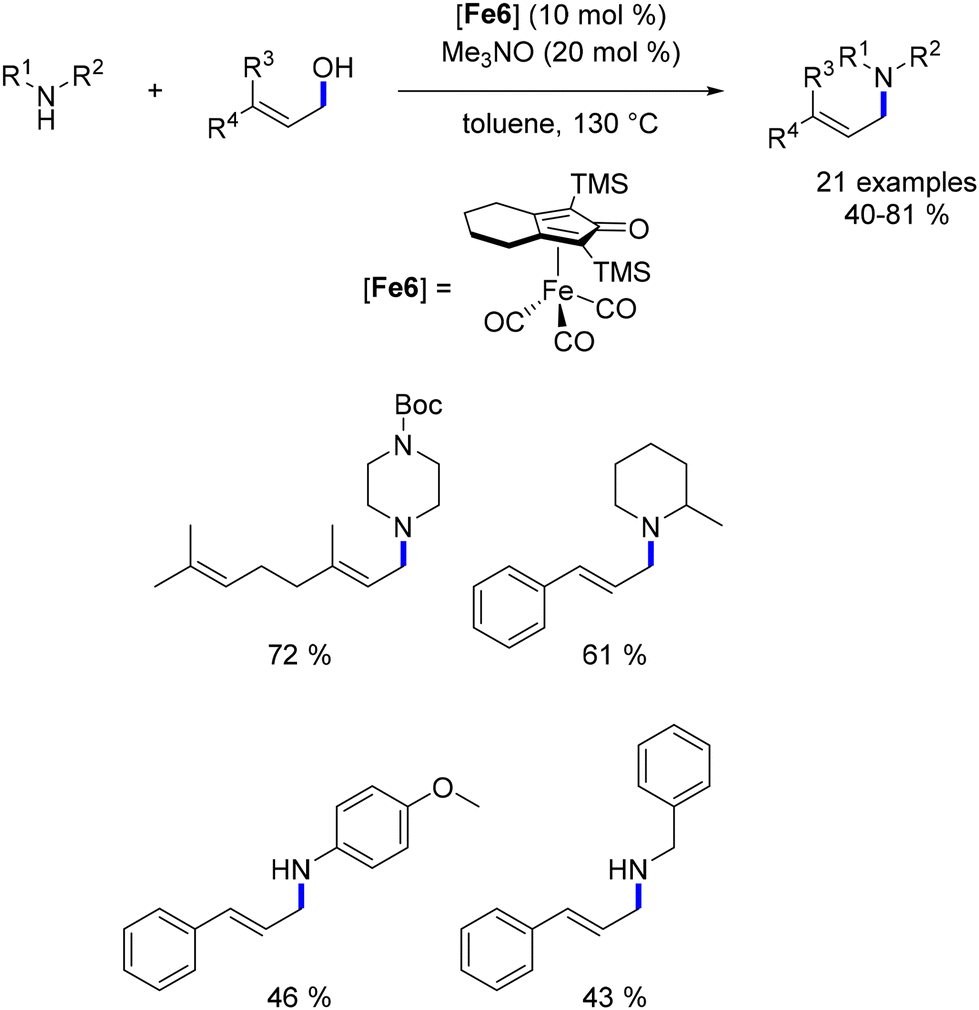
Gunanathan also recently reported the first successful alkylation of ketones with allylic alcohols,170 leading to unsaturated ketones using a hydrogen autotransfer strategy. The α-alkylation of acetophenone derivatives with terpenol compounds was achieved using tBuOK (0.5 equiv.) and a Ru–Macho complex (1 mol%) at 100 °C in toluene over 12 hours. Under these conditions, α-tetralones and 1-indanones were alkylated with yields ranging from 28% to 95%. Acetophenone derivatives required a higher temperature (125 °C) and shorter reaction time (8 hours), yielding products in moderate to excellent yields (37% to 88%). However, this method was limited to terpenols as pro-electrophiles.
Mechanistic investigations by Gunanathan revealed challenges in the reaction.170 The coordination of aldehydes to the metal catalyst inhibited its activity, leading to low yields when the aldehyde concentration was high. The researchers suggested that water generated during the condensation step might produce a metal–hydride species, aiding the reduction of the CC bond.
In 2023, Sundararaju described the alkylation of oxindoles with terpenols and allylic alcohols.171 The optimized conditions involved reacting oxindoles with allylic alcohol (1.1 equiv.), NaOH (1 equiv.), an iron complex (Fe1, 2 mol%), and Me3NO (4 mol%) in p-xylene at 100 °C for 12 hours (Scheme 83). The reaction yielded a mixture of alkene isomers. The reaction tolerated various functional groups on the aryl rings, and the alkylated products were obtained in yields of 30% to 95%.
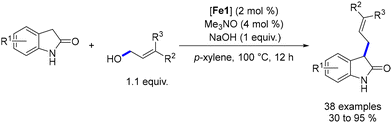 |
| | Scheme 83 Alkylation of oxindoles with allylic alcohols. | |
To validate the hydrogen autotransfer mechanism, deuterium labeling experiments were conducted. These experiments confirmed the pathway but did not explain the formation of the β,γ-alkene isomer as proposed by the authors. DFT calculations suggested that the 1,6-reduction is less energetically demanding than the 1,4-reduction by approximately 1 kcal mol−1, which could explain the formation of the saturated by-product. Further investigations confirmed that the saturated by-product was likely formed due to the 1,4-reduction of the enal intermediate after dehydrogenation of the allylic alcohol.
This reaction was also investigated using an iron–phthalocyanine complex as a catalyst.172 Secondary alcohols resulted in lower yields compared to the alkylation with primary alcohols. Additionally, higher temperatures were required to achieve the reaction (Scheme 84).
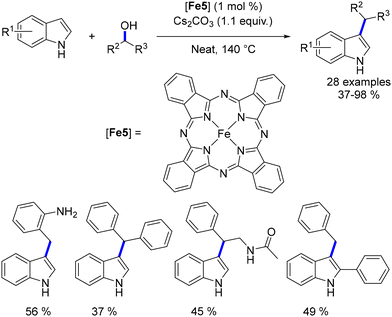 |
| | Scheme 84 Alkylation of indoles, as reported by Piersanti. | |
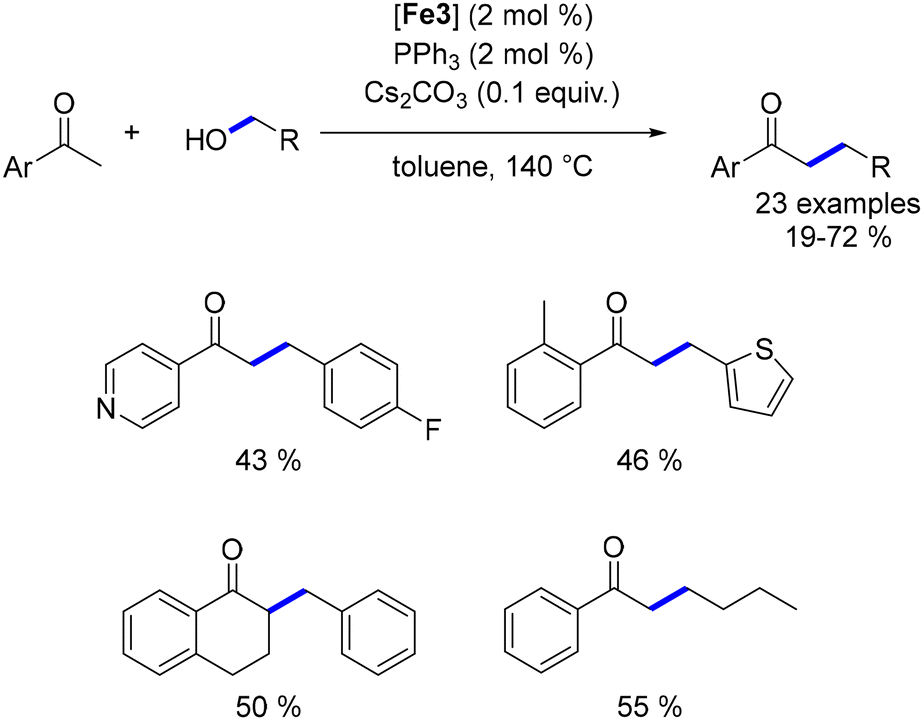
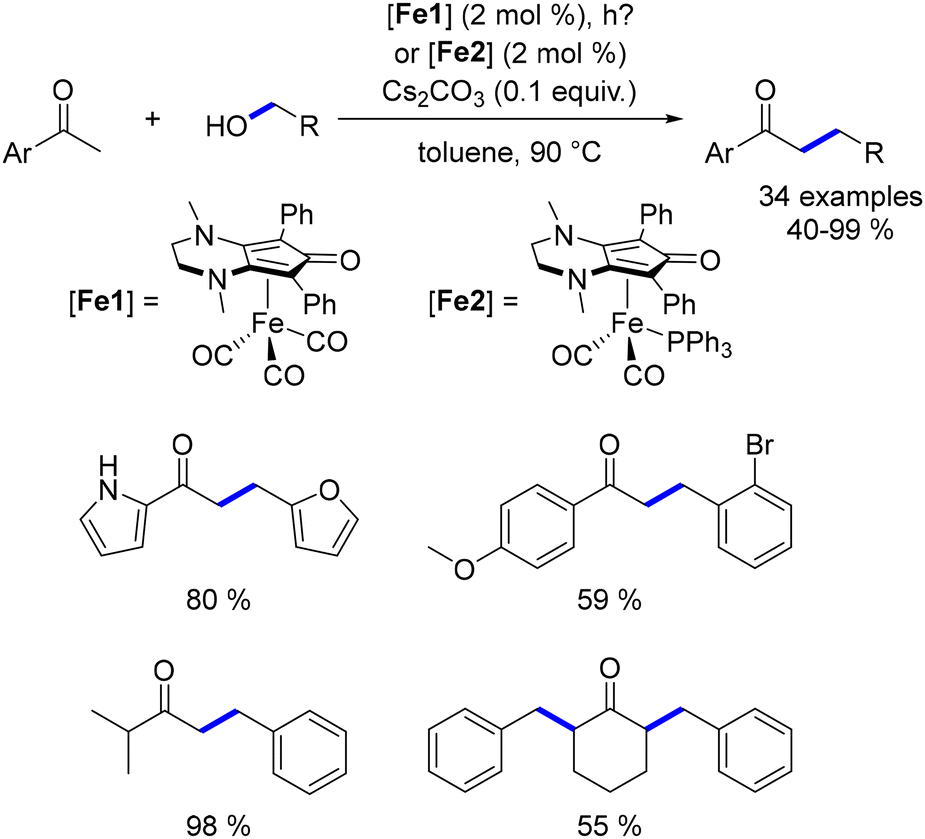
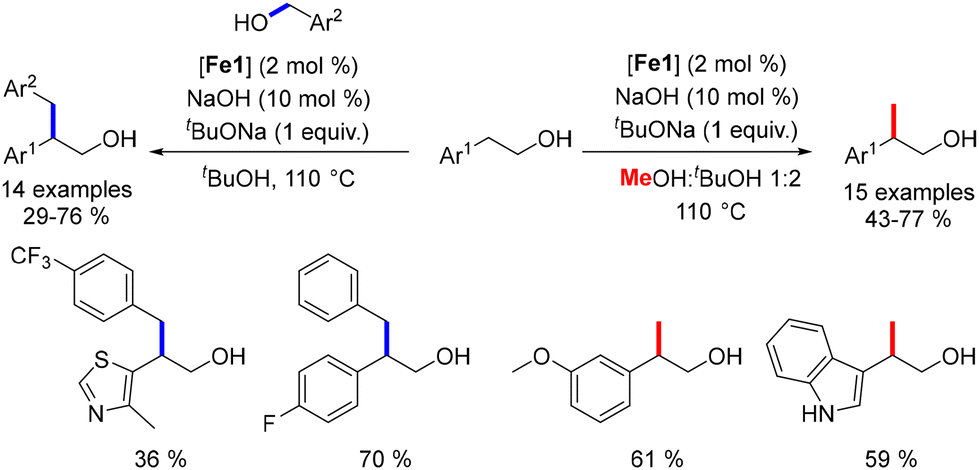
While the alkylation of ketones or secondary alcohols has been extensively studied, the β-alkylation of primary alcohols using Earth-abundant metals had not been previously reported. In 2019, our group developed a method for the β-alkylation of homobenzylic alcohols with benzylic alcohols and methanol.47 This approach engaged various substituted alcohols, yielding functionalized β-branched primary alcohols in moderate to good yields (Scheme 85). However, the reaction could not be extended to aliphatic alcohols due to potential cross-alkylation products. Despite this limitation, the procedure offers an atom-efficient alternative to the hydroformylation–reduction sequence and facilitates the synthesis of functionalized alcohols.
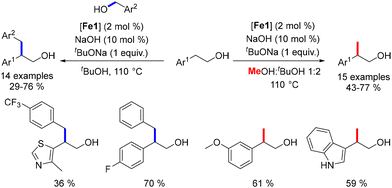 |
| | Scheme 85 Alkylation of primary alcohols. | |
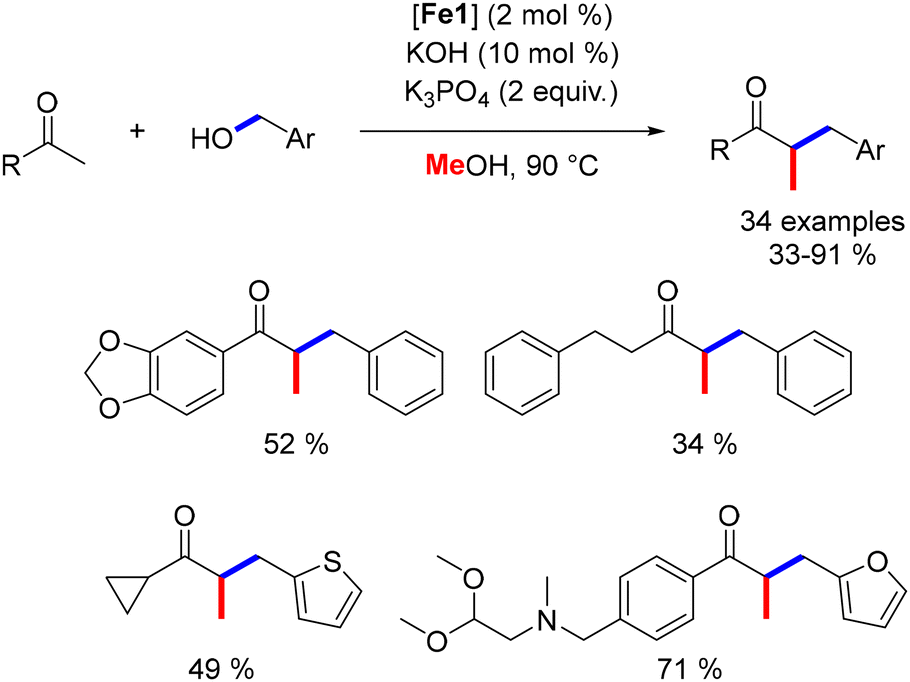
Still, in 2019, our group developed a three-component alkylation reaction involving a ketone, a primary alcohol, and methanol to produce α-disubstituted ketones.49 This methodology afforded various functionalized products (Scheme 86). The proposed mechanism involves an initial alkylation of the ketone with benzyl alcohol, followed by a second alkylation with methanol to yield the α-benzylmethyl ketone. This reaction was limited to benzylic primary alcohols as pro-electrophiles. Nonetheless, this approach provides an atom-economical method for synthesizing functionalized ketones.
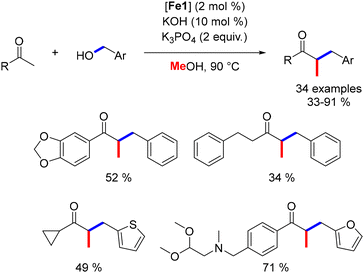 |
| | Scheme 86 One-pot three-component alkylation of ketones, as reported by Renaud. | |
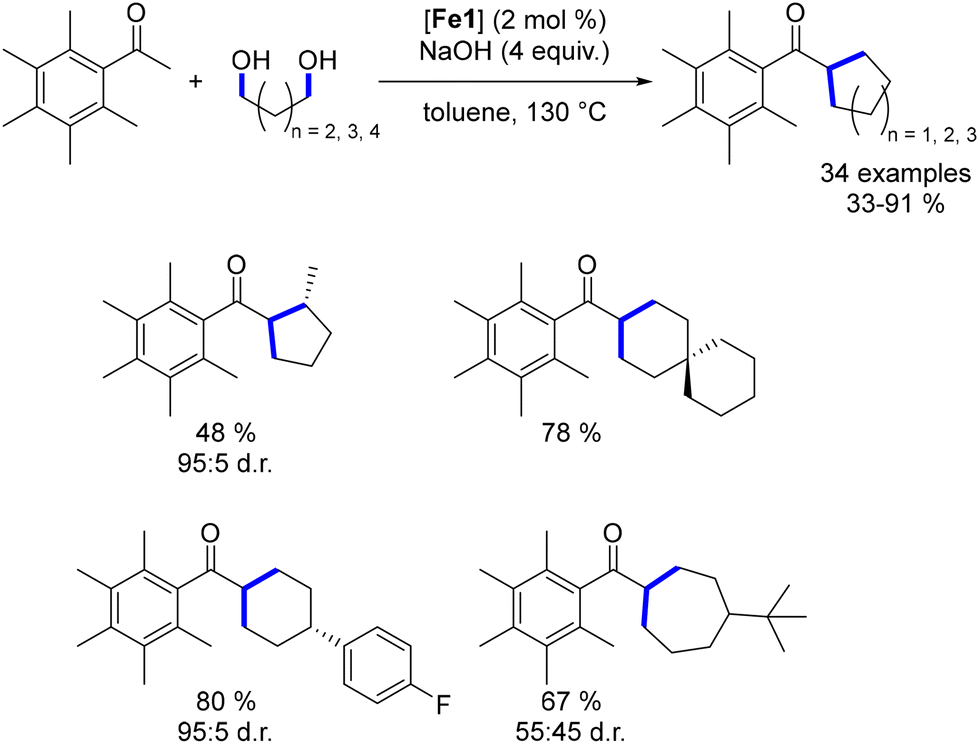
To further assess the capabilities of this complex, we investigated the synthesis of cycloalkanes.50 Using a similar methodology to that of Leitner and Maji,149,150 various diols were reacted with pentamethyl acetophenone to produce the corresponding cyclic di-alkylated ketones in moderate to good yields. This approach enabled the formation of substituted 5- to 7-membered cycloalkanes. Notably, good control of diastereoselectivity was achieved with cyclopentanes and cyclohexanes, while cycloheptanes resulted in lower diastereomeric ratios (Scheme 87).
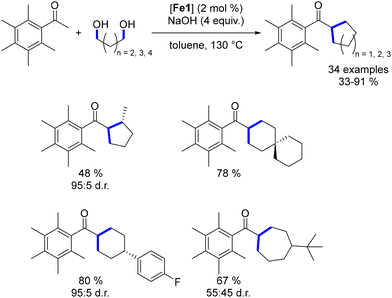 |
| | Scheme 87 Synthesis of cycloalkanes. | |
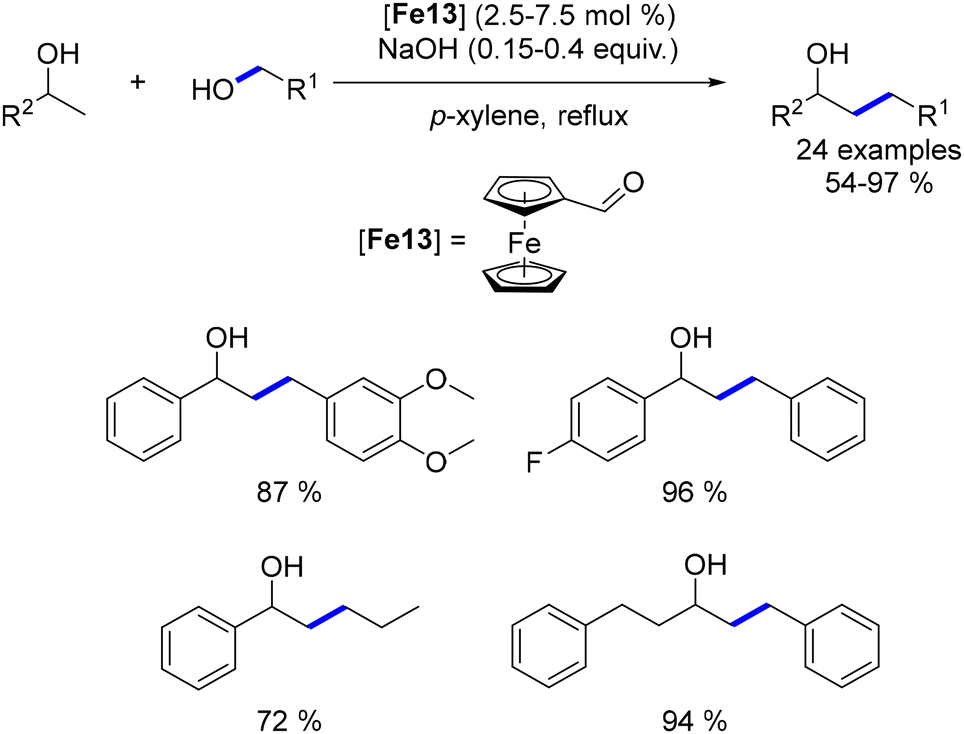
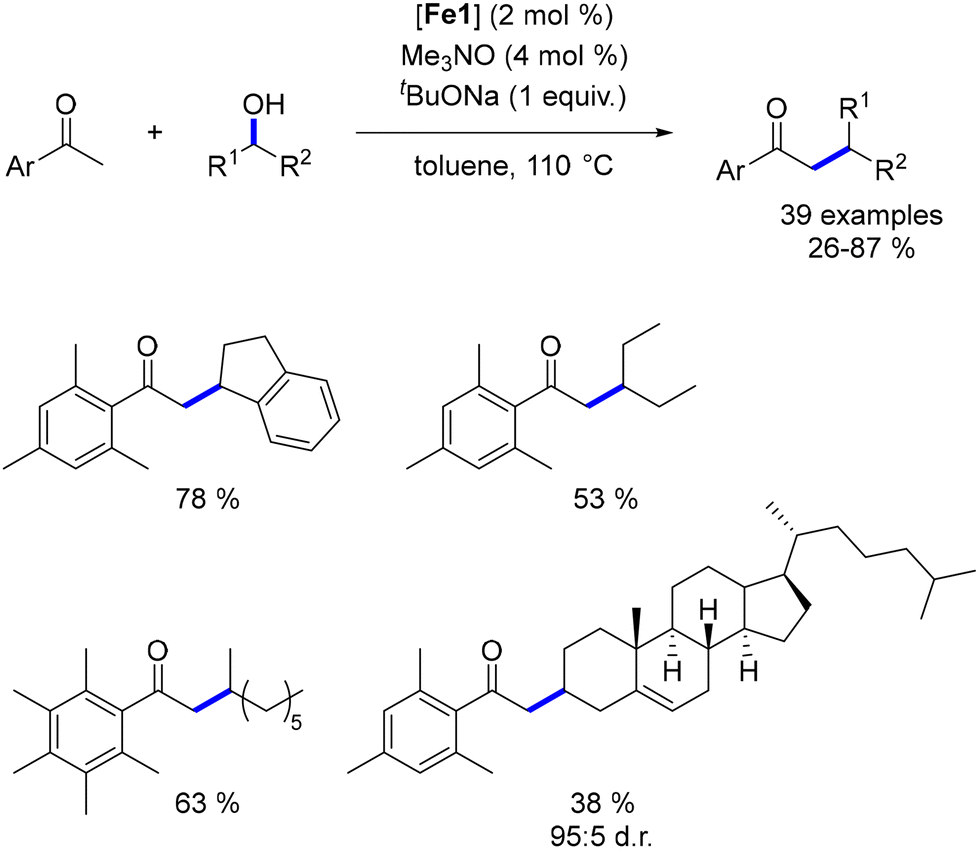
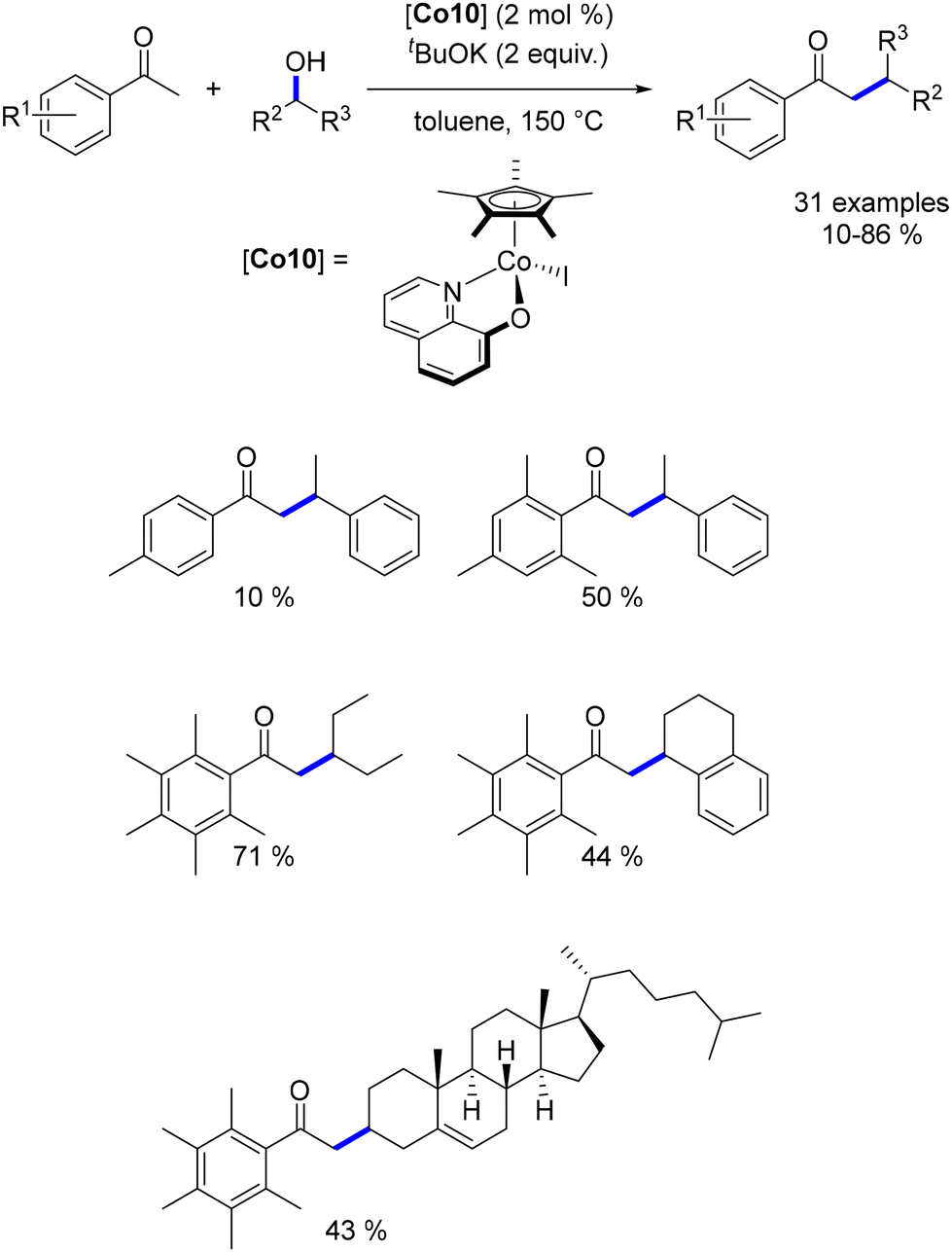
While extensive research has focused on the alkylation of ketones with primary alcohols, the use of secondary alcohols as pro-electrophiles remains underexplored, with few examples available across various metal-based complexes. To address this gap, we investigated the α-alkylation of ketones using secondary alcohols.51 Employing pentamethyl acetophenone to minimize cross-alkylation, we successfully reacted various benzylic and aliphatic secondary alcohols, yielding β-branched ketones in moderate to good yields with significant functional group tolerance. Notably, a functionalized cholesterol derivative was synthesized in 38% yield and with complete control over diastereoselectivity (Scheme 88).
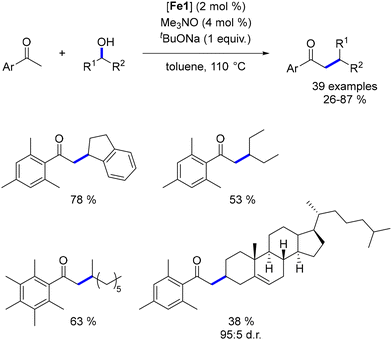 |
| | Scheme 88 Alkylation with secondary alcohols. | |
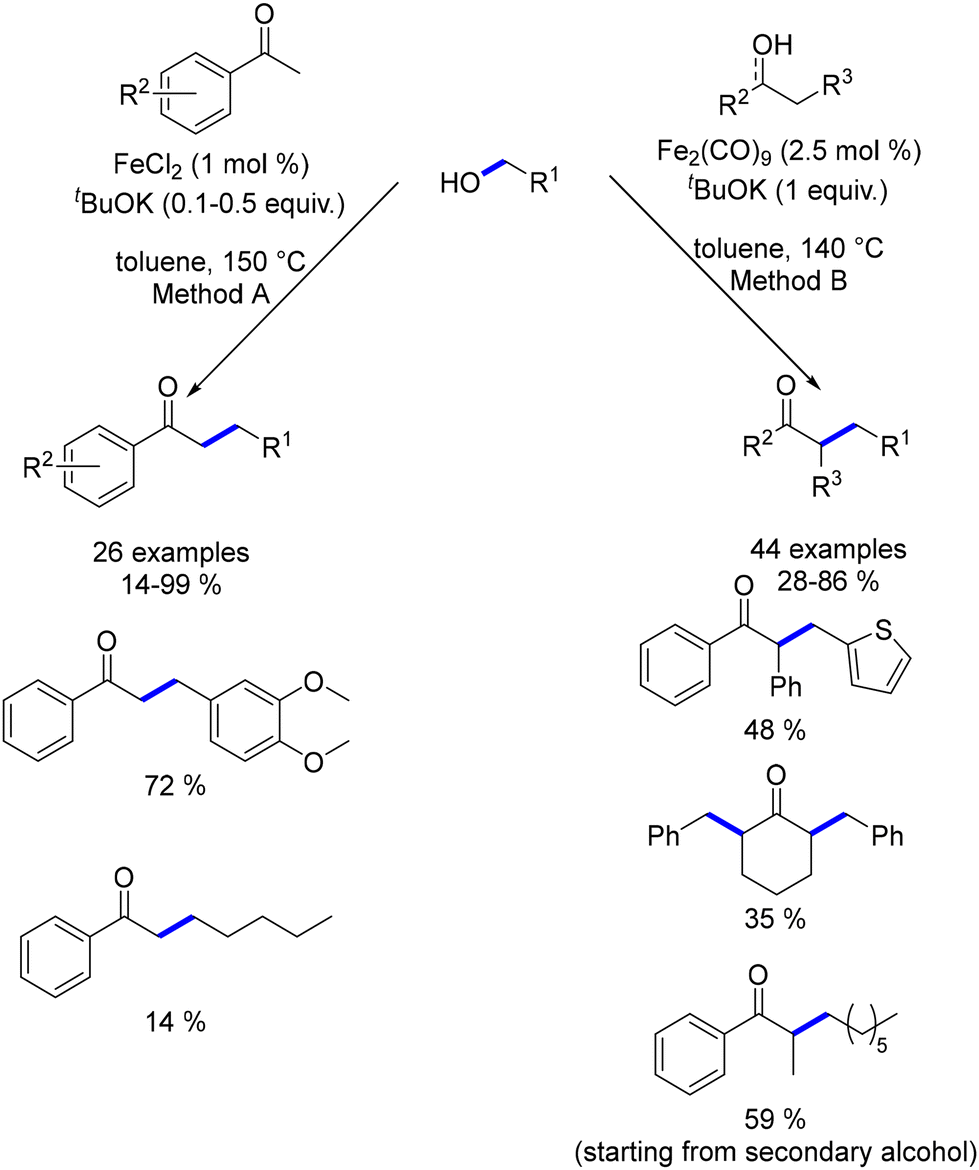
In 2019, Yang and Banerjee explored the synthesis of α-substituted ketones using ligand-free iron catalysts.173,174 Yang utilized FeCl2, while Banerjee employed Fe2(CO)9, both of which are precursors to previously reported bifunctional iron catalysts. These methods required high temperatures to facilitate the alkylation reactions. Banerjee's approach proved more versatile, showing better yields with aliphatic alcohols and not being restricted to benzylic ketones. Additionally, this methodology allowed the use of secondary alcohols as pro-nucleophiles, achieving similar yields of substituted ketones (Scheme 89).
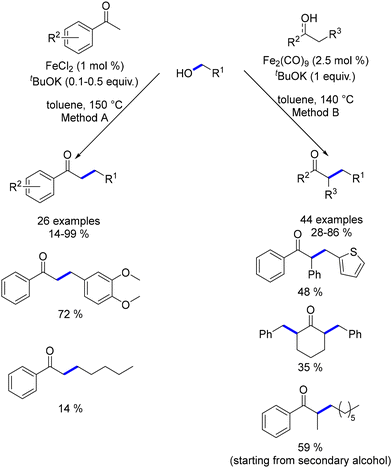 |
| | Scheme 89 Synthesis of α-substituted ketones. | |
In 2024, Poater, Renaud and coworkers devised a novel method for synthesizing highly substituted quinoline-2(1H)-ones via iron catalysis in yields up to 83% (Scheme 90).175 This approach is based on the intramolecular dehydrogenative coupling of amido-alcohols, resulting in the cyclization product. Various strategies were employed to prepare these starting materials. This methodology facilitates the synthesis of diverse substituted 3-arylquinolin-2(1H)-ones, with the ability to introduce substitutions at any position on the bicyclic structure. Moreover, it holds significant potential for the preparation of highly functionalized quinolinones.
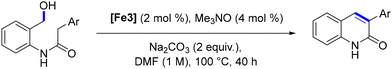 |
| | Scheme 90 Synthesis of 3-arylquinolin-2(1H)-ones. | |
3.3. Cobalt-catalysed C-alkylation reactions
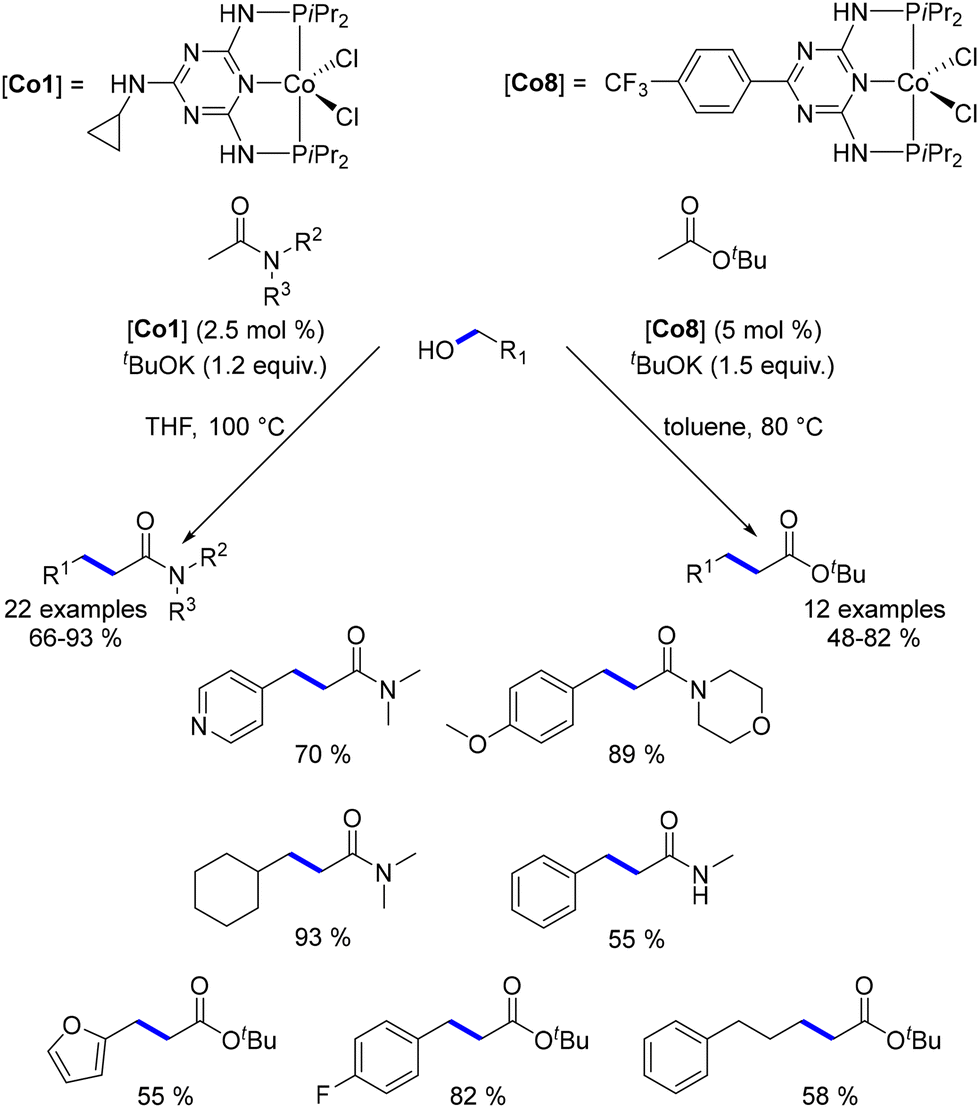
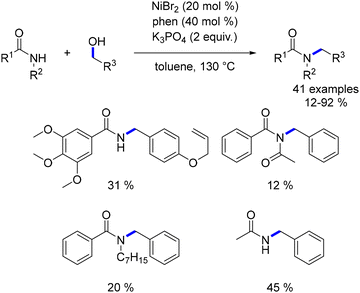
The first cobalt-catalysed C-alkylation reaction was reported by Kempe in 2016.176 Utilizing a cobalt complex with a triazine-containing PNP-type pincer ligand, Kempe demonstrated the successful alkylation of various acetamide derivatives and tert-butyl acetate with primary alcohols. The reaction conditions varied based on the pro-nucleophile; in contrast to ketone alkylation, amides and esters, being less acidic, required pro-electrophiles to be used as limiting reagents. Under optimized conditions, a range of amides and esters were alkylated with both benzylic and aliphatic alcohols, yielding α-substituted products in good to excellent yields (Scheme 91). However, N-mono-substituted amides afforded lower yields due to the preferential deprotonation of the acidic hydrogen. Additionally, the scope was restricted to tert-butyl esters to avoid potential transesterification with tBuOK during the process.
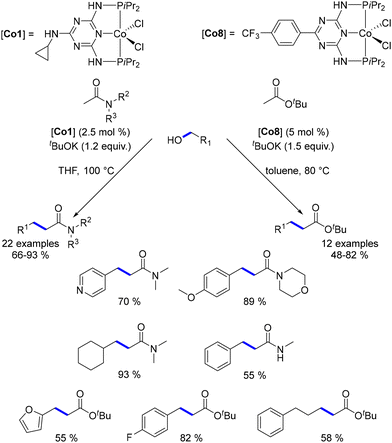 |
| | Scheme 91
C-Alkylation of amides and esters. | |
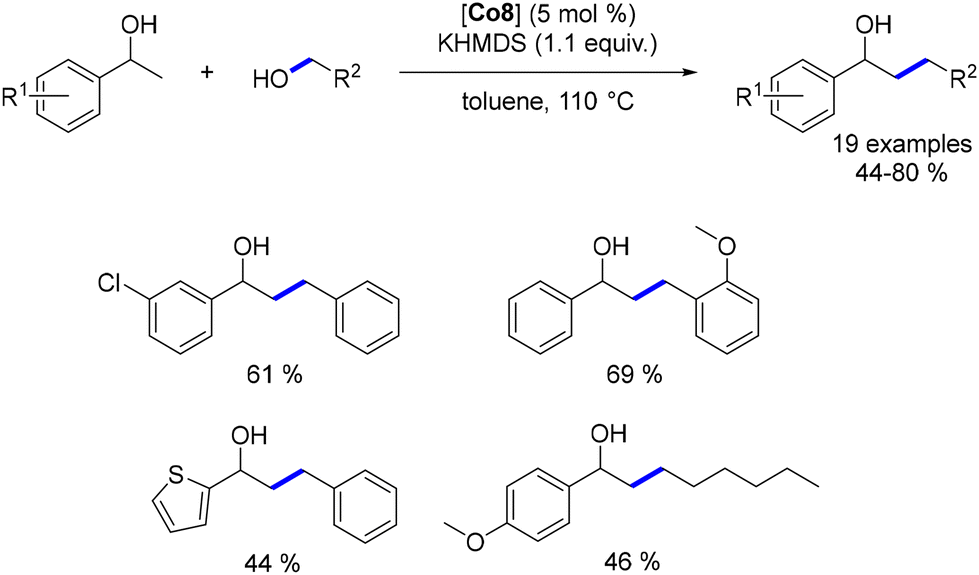
The cobalt complex Co8 was also utilized for the alkylation of secondary benzylic alcohols.177 This process involved various benzylic and aliphatic alcohols, leading to β-alkylated products in moderate to good yields. However, lower yields were observed with aliphatic alcohols (Scheme 92).
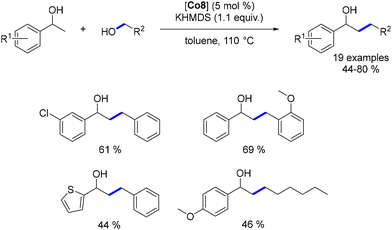 |
| | Scheme 92 Alkylation of secondary alcohols. | |
Zhang et al. also explored the alkylation of ketones using a cobalt complex.178 The Macho–Co complex, previously utilized by the same group for the alkylation of amines,109 was employed for the alkylation of aromatic ketones with benzylic and primary aliphatic alcohols. This reaction yielded products in relatively lower yields (Scheme 93). Additionally, when aliphatic ketones were used as pro-nucleophiles, the isolated yield of the alkylated ketone was moderate.
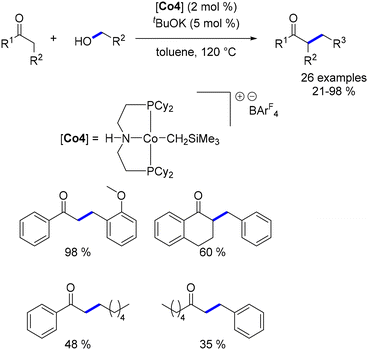 |
| | Scheme 93 Alkylation of ketones, as reported by Zhang. | |
Li et al. synthesized and utilized a cobalt(II) complex with a phosphine-free NNN pincer ligand for the alkylation of benzylic secondary alcohols with primary benzylic and aliphatic alcohols. The reactions were conducted in toluene at 120 °C with a catalytic amount of NaOH (20 mol%).179 This approach afforded various ketones in yields ranging from 46% to 95% (Scheme 94).
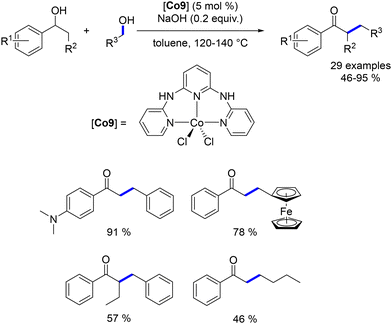 |
| | Scheme 94 Synthesis of α-substituted ketones, as reported by Li. | |
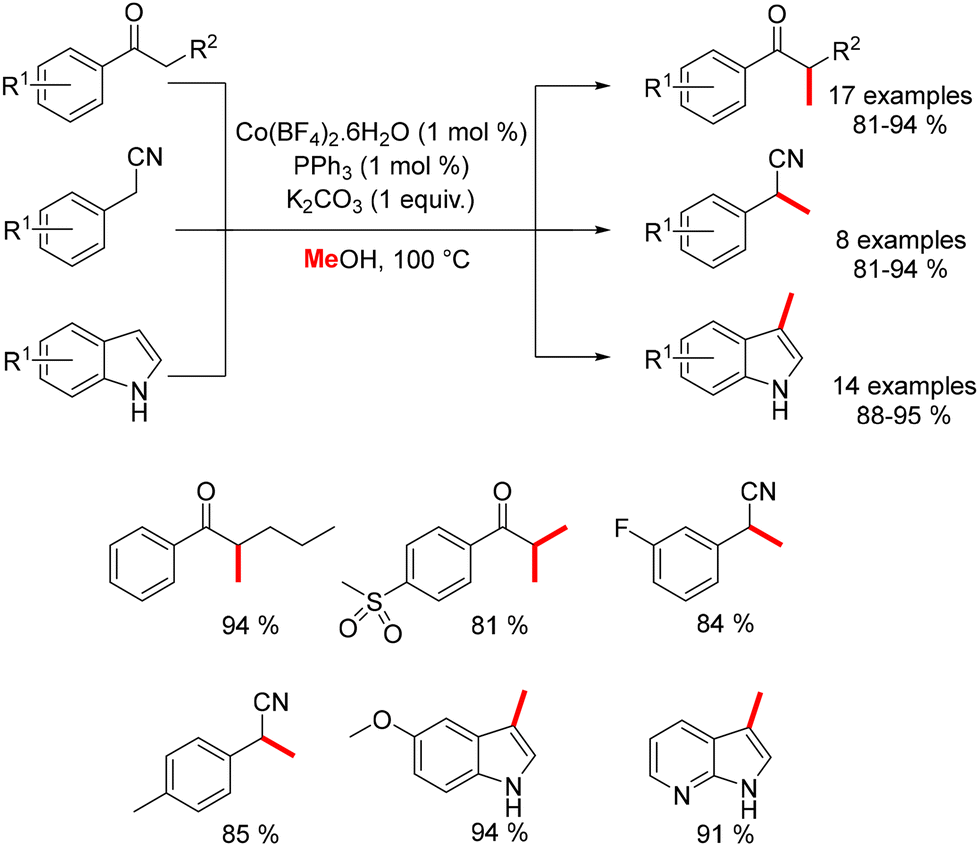
In 2017, Liu reported the methylation of ketones, benzyl nitriles, and indoles using a combination of Co(BF4)2·6H2O and PPh3.180 This method enabled the methylation of various substituted nucleophiles with methanol as the pro-electrophile, yielding products in good to excellent yields while accommodating a range of functional groups (Scheme 95).
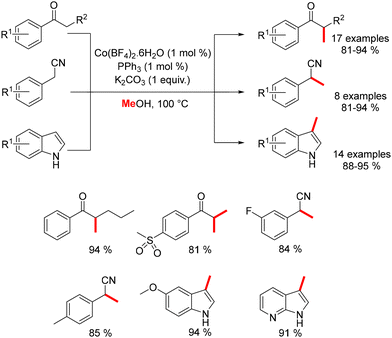 |
| | Scheme 95 Cobalt-catalysed methylation of various nucleophiles, as reported by Liu. | |
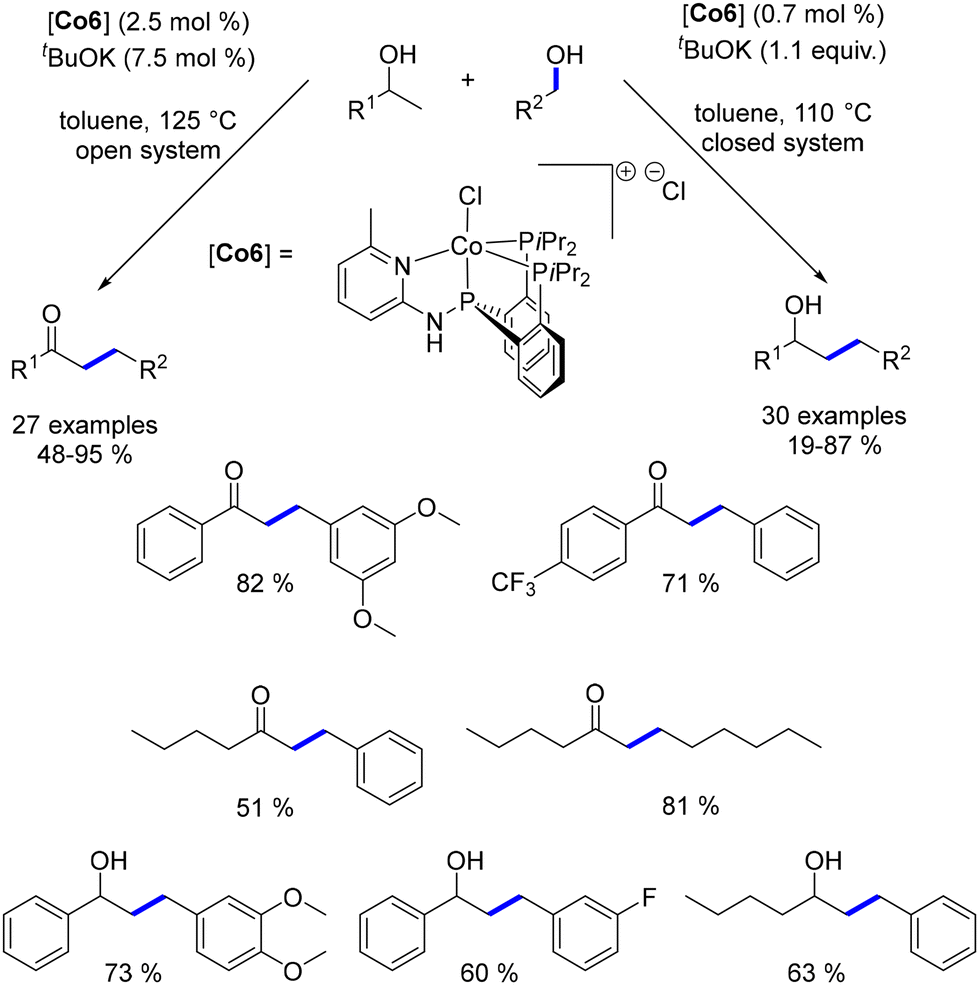
Ding presented a well-defined PNPP–Co catalyst for the β-alkylation of secondary alcohols with primary alcohols, yielding either β-alkylated alcohols or α-alkylated ketones depending on the reaction conditions, with temperatures ranging from 110 to 125 °C.181,182 This switchable synthesis can be controlled by adjusting parameters such as the base amount and whether the system is open or closed. Notably, this process employs a low catalyst loading (Scheme 96).
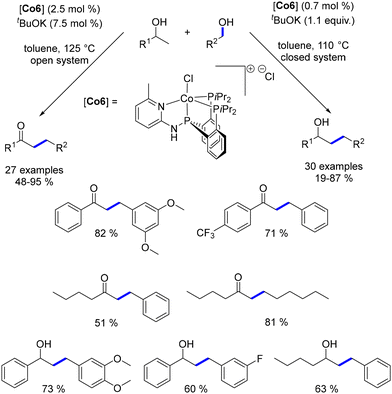 |
| | Scheme 96 Synthesis of α-substituted ketones and β-substituted alcohols. | |
The PNPP–Co complex was also employed for the alkylation of nitriles with primary alcohols.183 This approach achieved yields of up to 95% with various benzyl nitriles and both benzyl and aliphatic alcohols (Scheme 97). However, other nitrile derivatives did not react under these conditions.
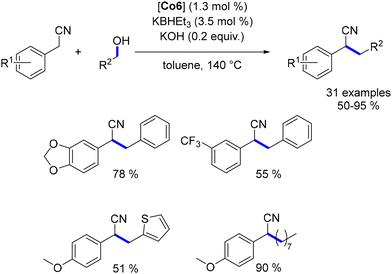 |
| | Scheme 97 Alkylation of nitriles, as reported by Ding. | |
In 2019, Sundararaju reported the α-alkylation of ketones using a Cp*Co(III) complex.184 A similar complex was also effective for the alkylation of amines with secondary alcohols.114 This methodology enabled the alkylation of substituted acetophenone derivatives with benzylic or aliphatic secondary alcohols, yielding β-disubstituted ketones in good to excellent yields (Scheme 98). As observed in manganese and iron chemistry, ortho,ortho-disubstitution of the acetophenone derivatives was crucial to prevent cross-alkylation and to enhance the reactivity.
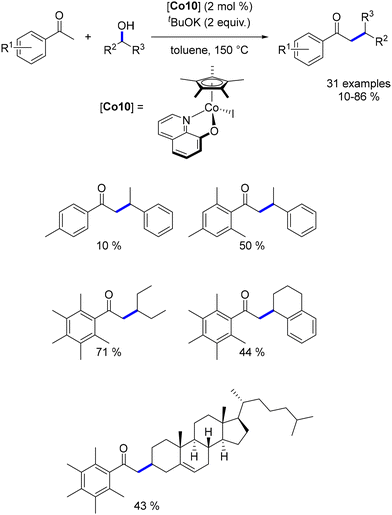 |
| | Scheme 98 Alkylation of ketones with secondary alcohols. | |
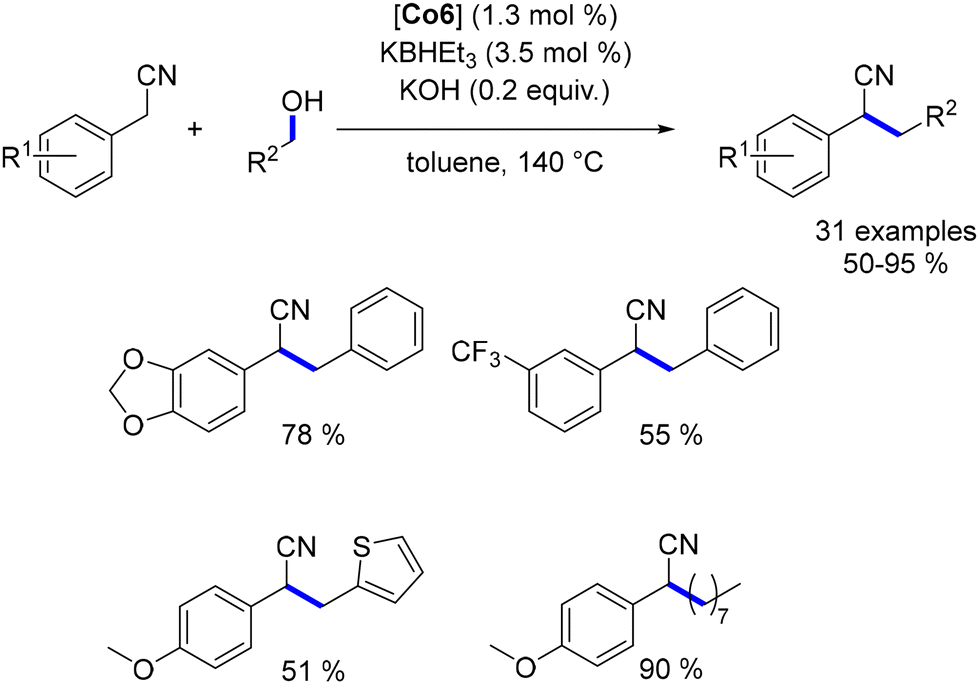
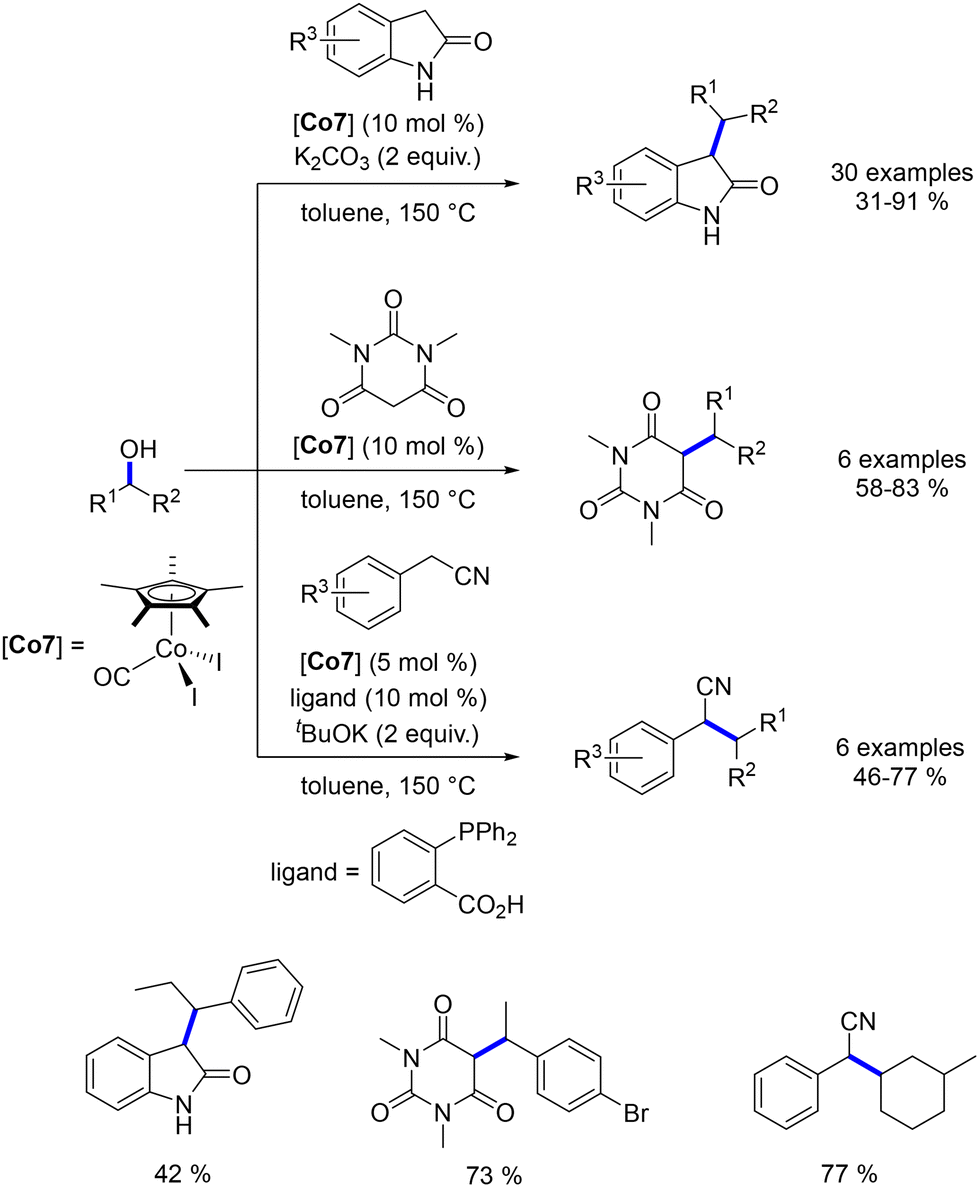
Another phosphine-free cobalt complex, described by Sundararaju et al., was utilized for the alkylation of oxindoles, N,N-dimethyl barbituric acid, and benzyl nitriles with secondary alcohols.185 This bench-stable cobalt(III) complex selectively catalysed the alkylation of oxindole derivatives, yielding products in 31–91% yields (Scheme 99). The alkylation of barbituric acid did not require an additional base due to the substrate's acidic nature, and alkylated derivatives were obtained in 58–83% yields. In this case, only benzylic alcohols were used. For benzyl nitriles, alkylation was achieved only with cyclic aliphatic alcohols, resulting in yields of 46–77% (Scheme 99).
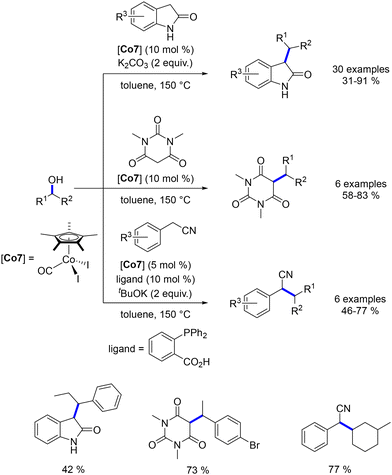 |
| | Scheme 99 Alkylation of oxindoles, barbituric acid and nitriles. | |
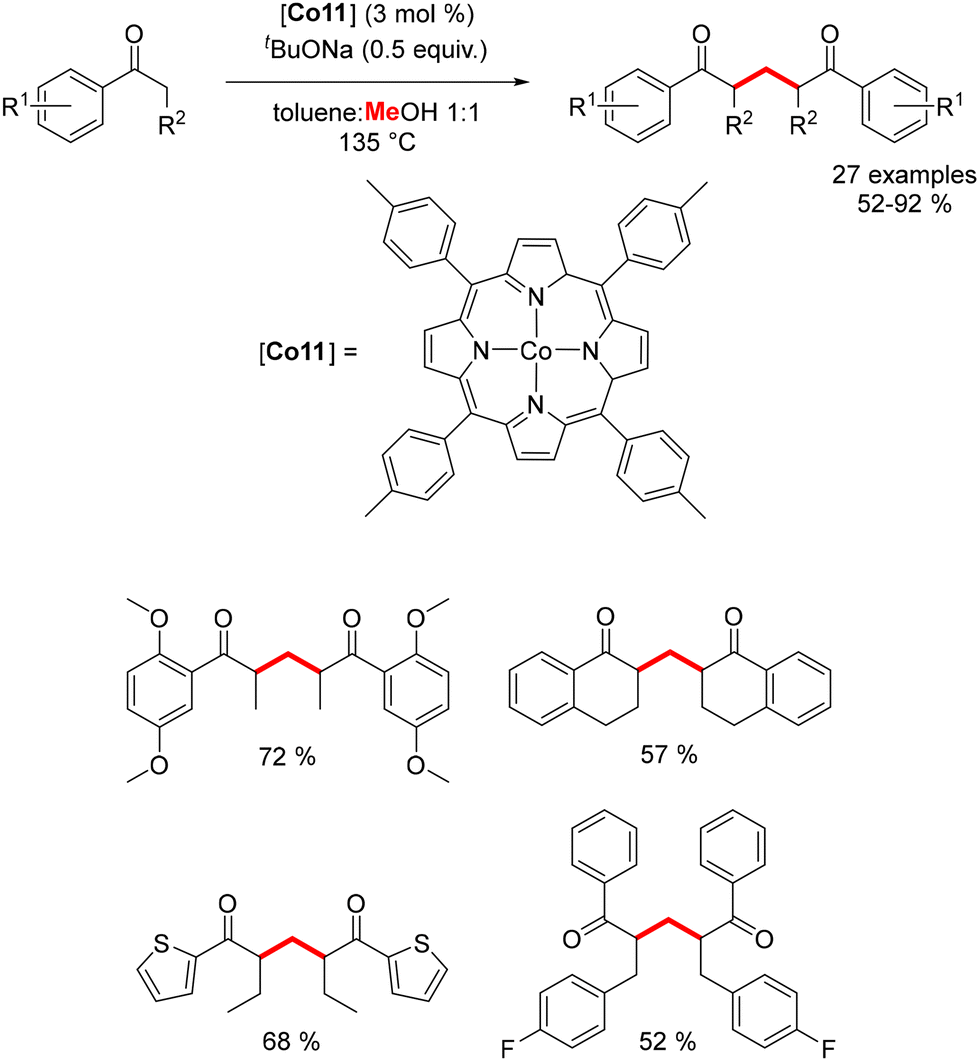
More recently, Chandrasekhar and Venkatasubbaiah reported the use of a porphyrin cobalt complex for the synthesis of 1,5-diketones through a dehydrogenative coupling reaction utilizing methanol as the C1 source.186 In this process, formaldehyde generated in situ undergoes condensation to form α,β-unsaturated aromatic ketones. These intermediates then participate in a Michael-type addition with aromatic enolates, yielding the corresponding 1,5-diketones in 52–92% yields (Scheme 100). The catalytic cobalt species is regenerated after the release of hydrogen. This methodology afforded a variety of methylenated products, though it was limited to α-substituted benzylic ketones.
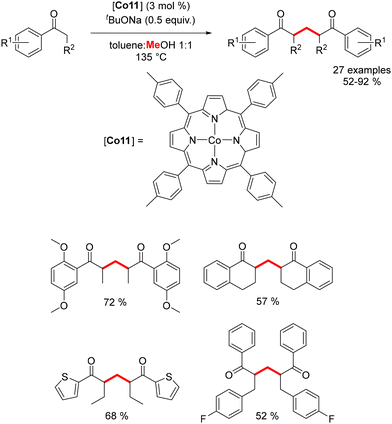 |
| | Scheme 100 Synthesis of 1,5-diketones. | |
3.4. Nickel-catalysed C-alkylation reactions.
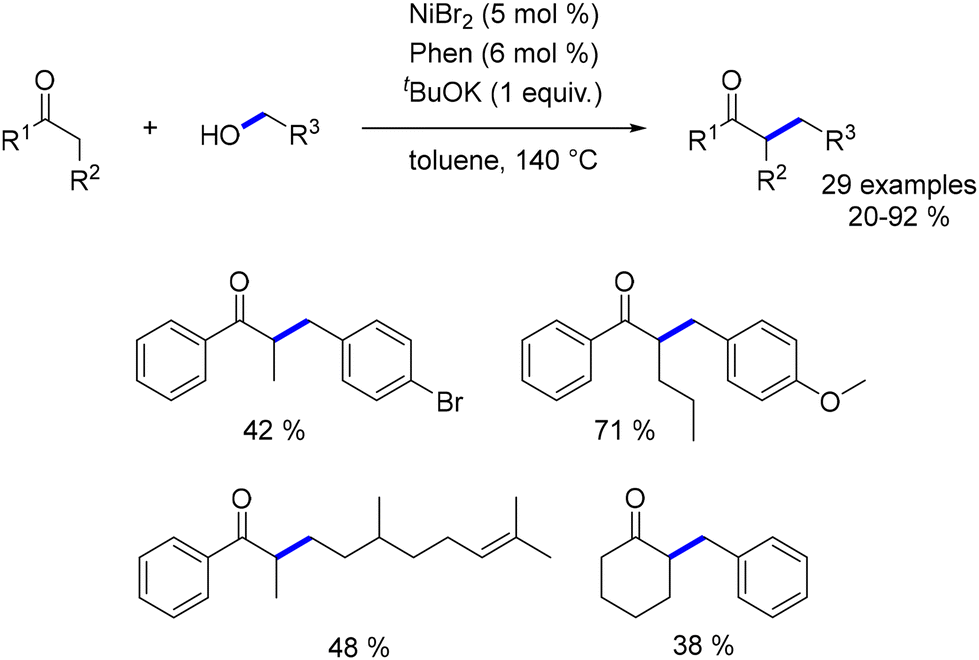
The first nickel-catalysed alkylation of ketones with alcohols was reported by Banerjee in 2018.187 The catalyst was generated in situ by combining NiBr2 with 1,10-phenanthroline. This system effectively alkylated various α-substituted aryl ketones with benzylic alcohols, yielding moderate to good results. However, the alkylation of aliphatic ketones and the use of aliphatic alcohols resulted in lower yields and required higher base loadings (2 equiv. for aliphatic alcohols and 2.5 equiv. for aliphatic ketones). Further optimization with catalytic amounts of a base (Cs2CO3 at 10 mol% or tBuOK at 20 mol%) did not improve the outcome with aliphatic alcohols, which failed to produce alkylated compounds under these conditions (Scheme 101).188
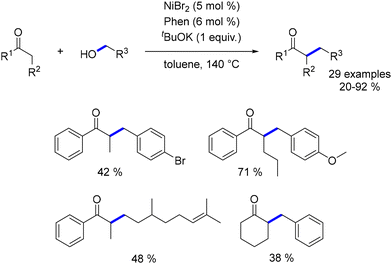 |
| | Scheme 101 Alkylation of α-substituted ketones. | |
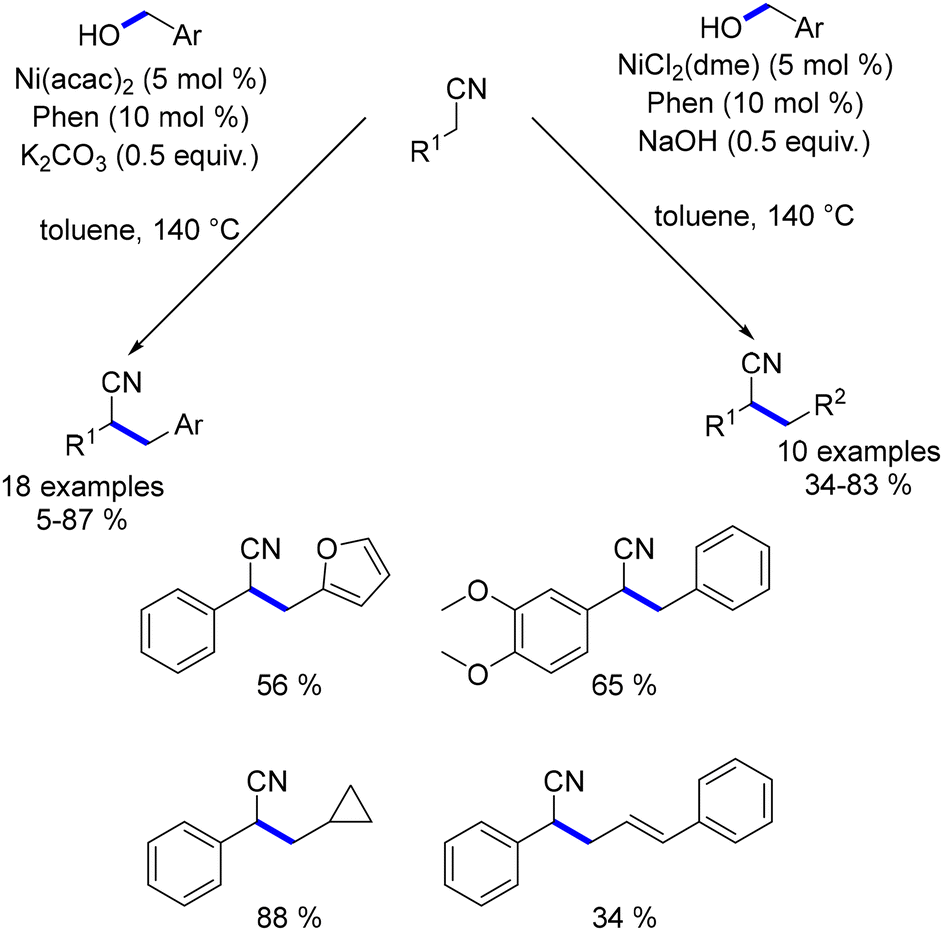
Building on these studies, the same group later explored the alkylation of nitriles using primary alcohols.189 In this work, the catalytic species was generated in situ by combining 1,10-phenanthroline with Ni(acac)2. The reaction of benzyl nitriles with benzyl alcohols yielded alkylated products in low to good yields (Scheme 102). Aliphatic nitriles, such as 3-propionitrile, resulted in very low yields, and no reaction was observed with acetonitrile. However, by replacing Ni(acac)2 and K2CO3 with NiCl2(dme) and NaOH, the system effectively facilitated alkylation with both aliphatic and allylic alcohols.
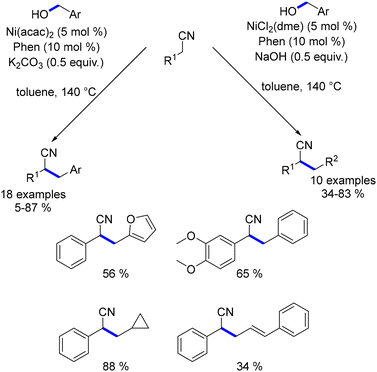 |
| | Scheme 102 Nickel-catalysed alkylation of nitriles. | |
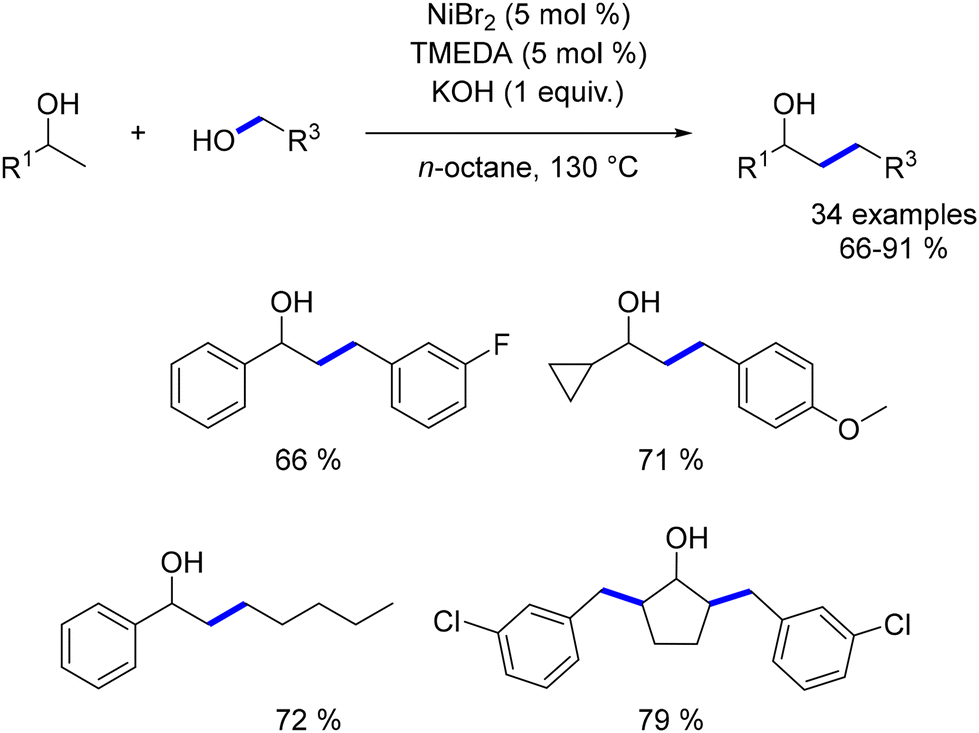
In 2021, Balaraman reported a β-alkylation reaction of secondary alcohols with primary alcohols to produce β-substituted alcohols, using a catalyst system comprising tetramethylethylenediamine (TMEDA) and NiBr2.190 This methodology successfully used a variety of alcohols as pro-electrophiles, yielding the desired products with no significant drop in yield, even for aliphatic and benzylic alcohols. Notably, aliphatic secondary alcohols also provided the desired products in good yields without necessitating changes in reaction conditions, demonstrating the method's broad applicability (Scheme 103).
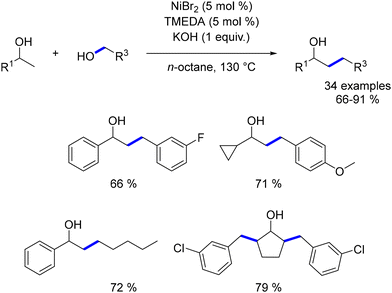 |
| | Scheme 103 Nickel-catalysed β-alkylation of alcohols. | |
Liu described a Ni–NHC complex for the α-alkylation of ketones using benzyl alcohols.191 This novel catalyst facilitated the formation of a range of alkylated ketones, achieving yields from low to good with the use of a catalytic amount of a base at 140 °C (Scheme 104).
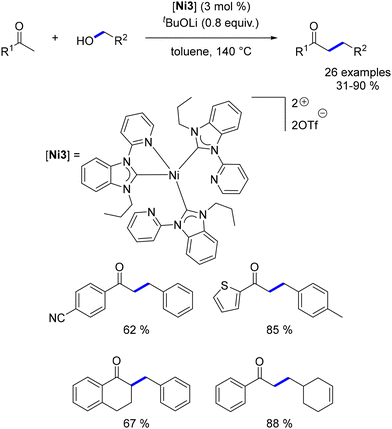 |
| | Scheme 104 Alkylation of ketones, as reported by Liu. | |
3.5. Copper-catalysed C-alkylation reactions.
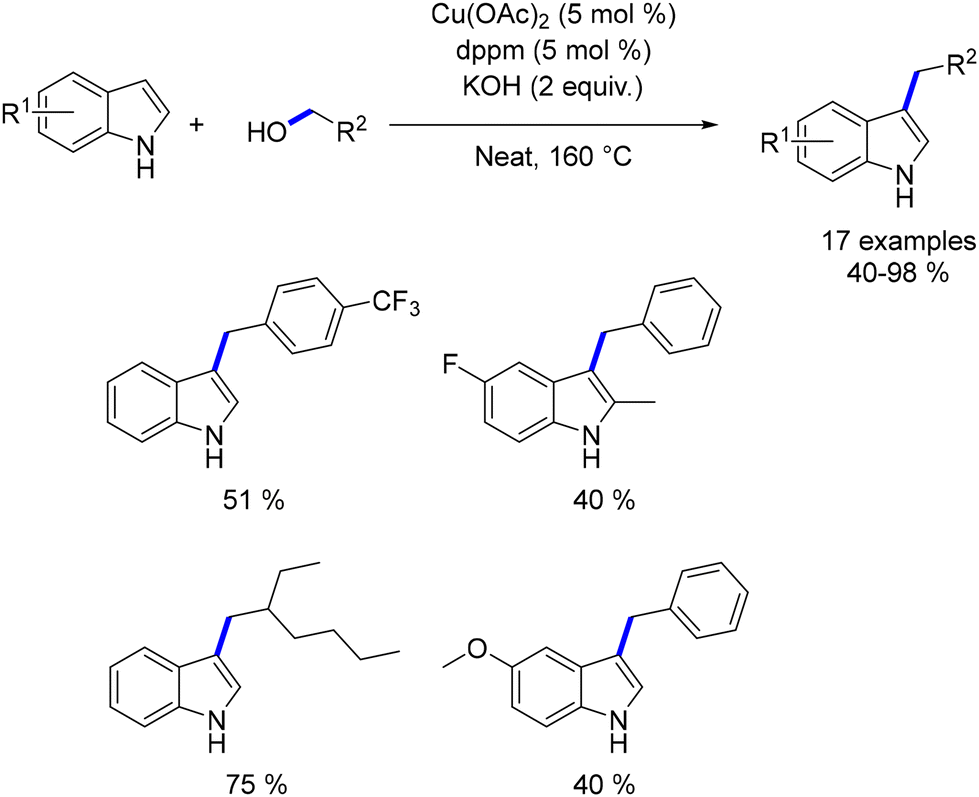
Dang reported the first copper-catalysed alkylation of indoles using alcohols.192 Copper acetate was combined with the bis-diphenylphosphinomethane (dppm) ligand to generate the catalytic species. This system facilitated the alkylation of indoles with both benzylic and aliphatic alcohols, yielding products in moderate to good yields (Scheme 105). However, high temperatures were necessary, and the scope of the reaction was limited, with only a few functionalized products being described. Additionally, no reaction was observed with N-substituted indoles, indicating that the presence of the hydrogen atom is crucial for the process.
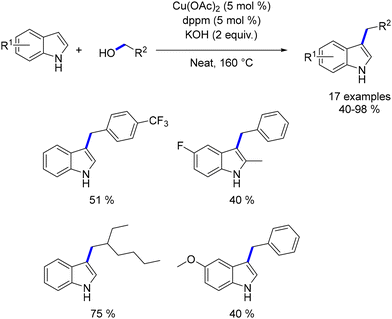 |
| | Scheme 105
C3-Alkylation of indoles. | |
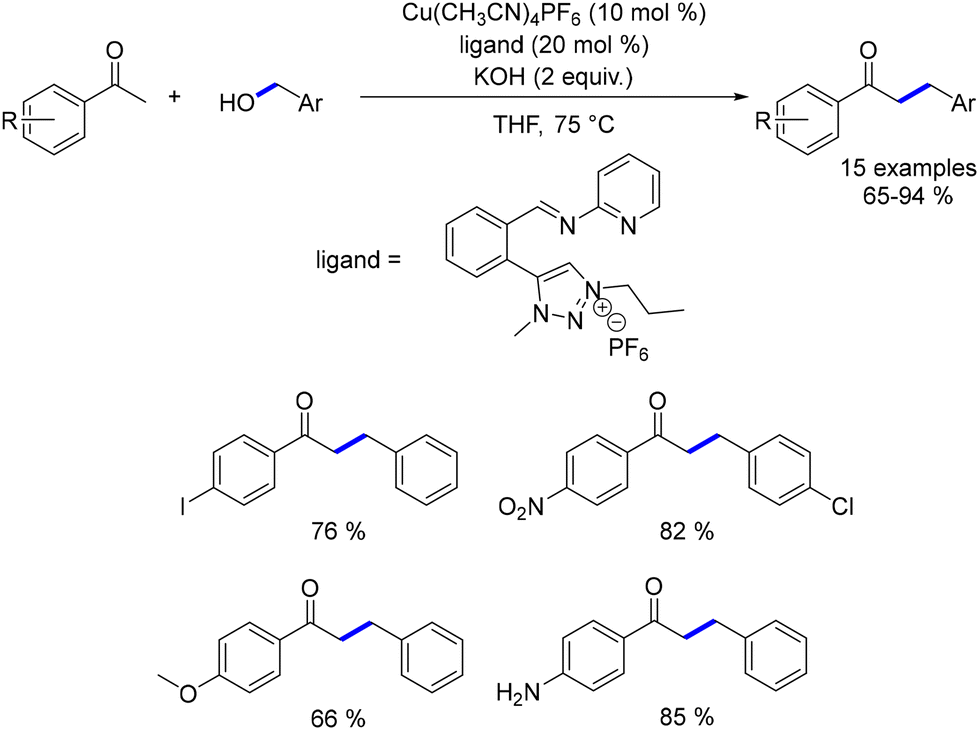
In the same year, Bala reported the alkylation of aryl ketones using benzyl alcohols.193 The catalyst was generated in situ by combining Cu(CH3CN)4PF6 with an appropriate ligand. This approach enabled the synthesis of a diverse range of functionalized ketones in good yields at 75 °C. This makes the reaction competitive with other alkylation methods, which typically require higher temperatures (Scheme 106).
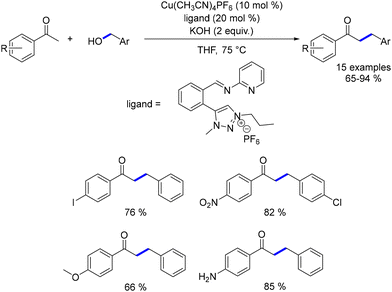 |
| | Scheme 106 Copper-catalysed alkylation of ketones. | |
4. Alkylation reactions via borrowing hydrogen methodology
In the past decade, significant progress has been made in alkylation reactions, particularly in the hydrogen autotransfer alkylation of ketones and alcohols. Manganese, iron, cobalt, and nickel have proven effective in these processes, with various complexes and ligands enhancing catalyst efficiency, leading to milder conditions and shorter reaction times.194 However, high temperatures are often still necessary. Recently, combining transition-metal catalysis with photoredox chemistry has introduced new reactions and reactivities, using light as an energy source instead of heat.195 Despite this innovation, photocatalysts can sometimes be incompatible with the intended reaction, and challenges like electron or energy transfer between the photocatalyst and the metal complex may arise. To address these limitations, chromophoric transition-metal catalysts have been developed, enabling the use of a single catalytic species and avoiding these issues.
4.1. White light-induced alkylation reaction through borrowing hydrogen strategy
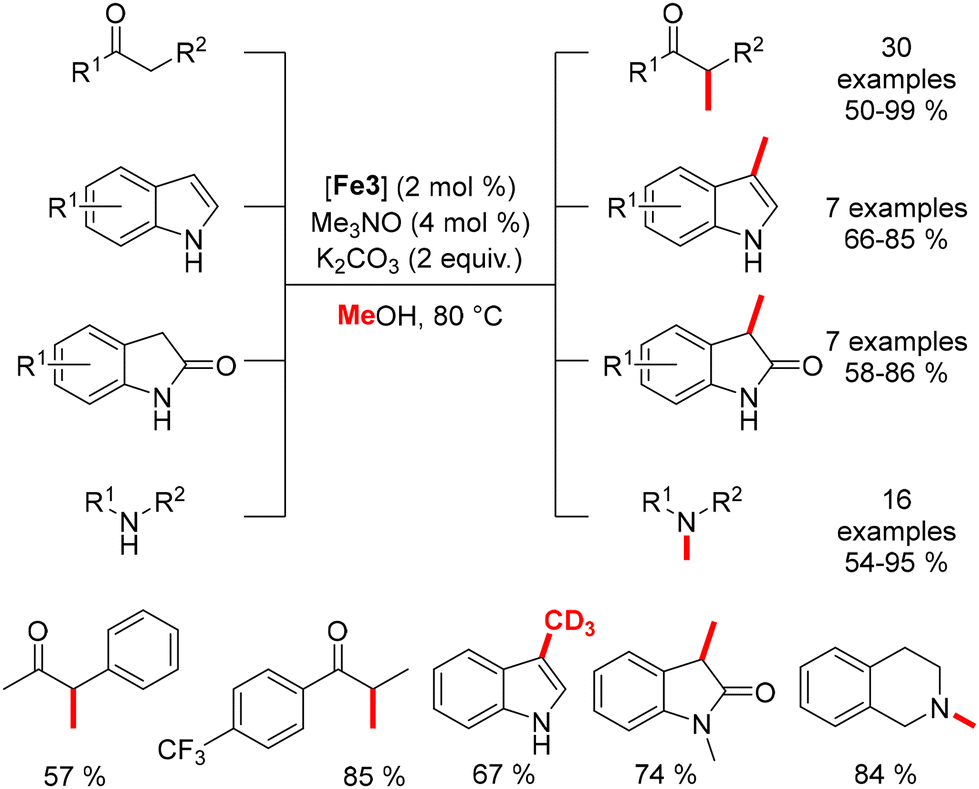
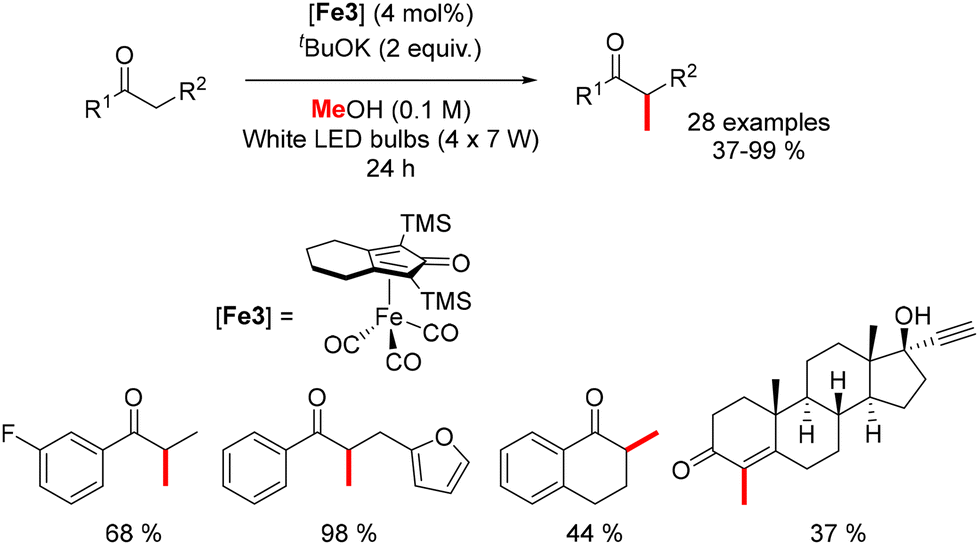
In 2022, Sundararaju reported an iron-catalyzed methylation of ketones under light irradiation.196 Using Knölker's complex (4 mol%) and tBuOK (2 equiv.) in methanol, various acetophenones were methylated under four sets of 7 W white LED lights for 24 hours at 42 °C (Scheme 107). The process yielded α-methylated ketones in moderate to good yields (37–99%). Di-methylation of p-methyl acetophenone and cyclic substrates like tetralone produced lower yields, while ethisterone methylation resulted in a tetrasubstituted olefin due to isomerization.
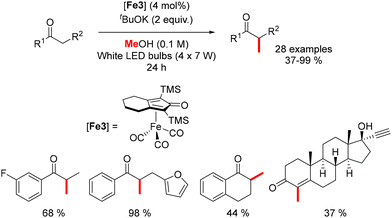 |
| | Scheme 107 Methylation of ketones under light irradiation. | |
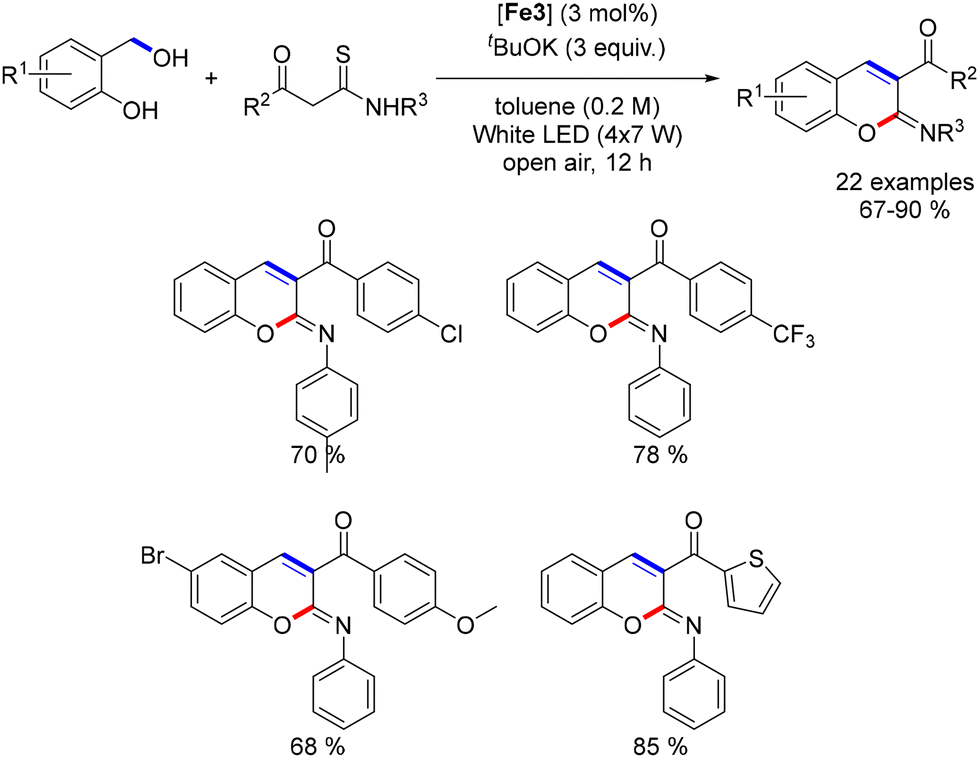
Waheed recently reported the synthesis of 2-arylamino-2H-chromenes using an iron catalyst.197 In this reaction, 2-hydroxybenzyl alcohols reacted with 1,3-keto-thioamides in toluene under air and light irradiation for 12 hours (Scheme 108), yielding 2-arylimino-2H-chromenes in good yields (67–90%). The reaction tolerated various substituents, including electron-donating and electron-withdrawing groups. A control experiment showed that salicylaldehyde could form the cyclic product without light irradiation, highlighting the iron complex's role in dehydrogenating benzyl alcohol. The proposed mechanism involves iron-catalyzed dehydrogenation, enol condensation, and final cyclization to form the chromene product.
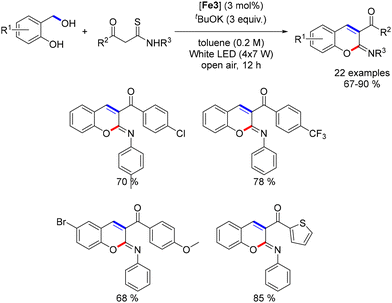 |
| | Scheme 108 Synthesis of 2-arylamino-2H-chromenes. | |
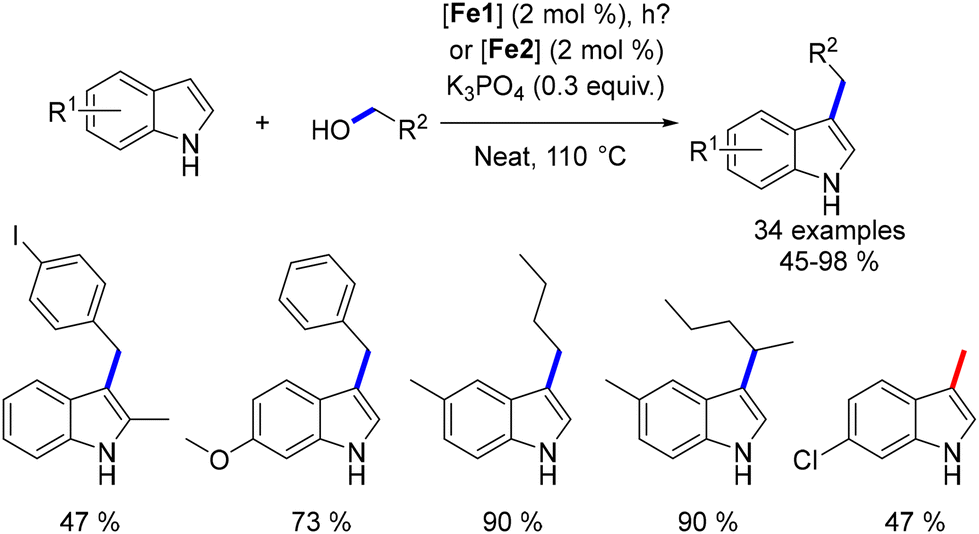
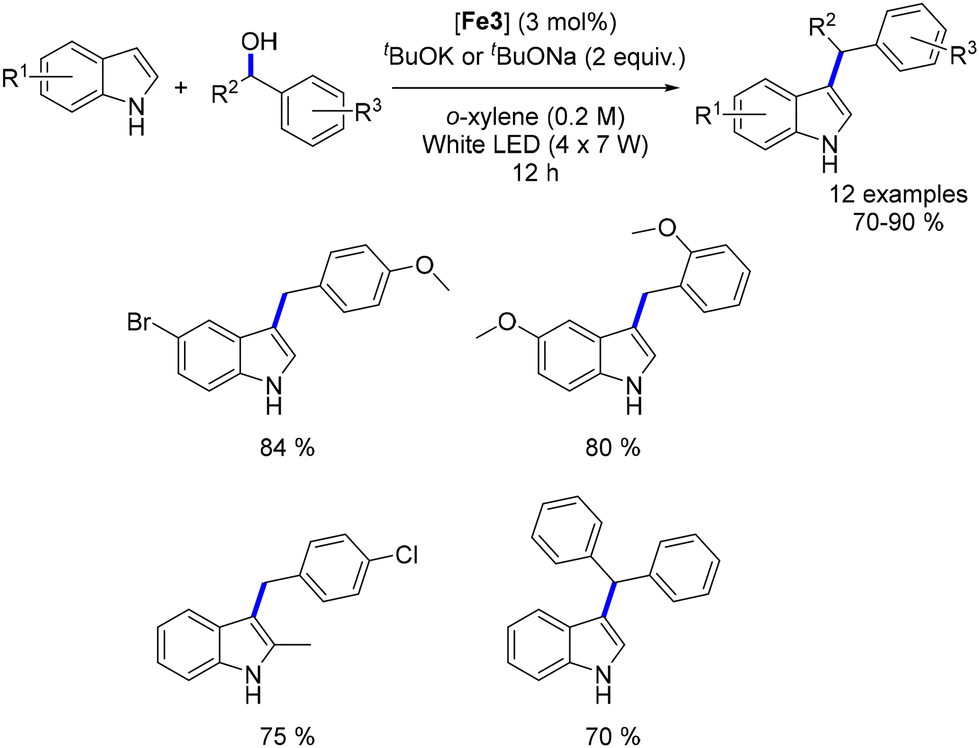
Waheed's group also reported the alkylation of indoles and the synthesis of flavanones using an iron catalyst. Indoles reacted with alcohols under optimized conditions (tBuOK, Knölker complex, light irradiation) to yield C3-alkylated indoles in 70–90% yields (Scheme 109).198 Both primary and secondary alcohols were effective as pro-electrophiles. The group also synthesized bisindoylmethanes using similar conditions but in an open-air setup, where 1,4-addition occurred between the unsaturated intermediate and a second indole. Additionally, by increasing the base and reaction time, flavanones were synthesized in 70–92% yields, though with limited functional group tolerance.
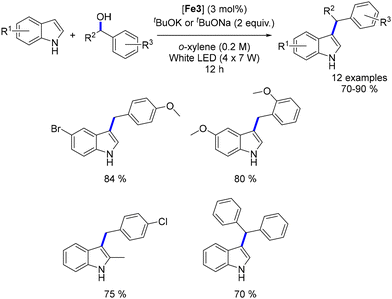 |
| | Scheme 109 Alkylation of indoles. | |
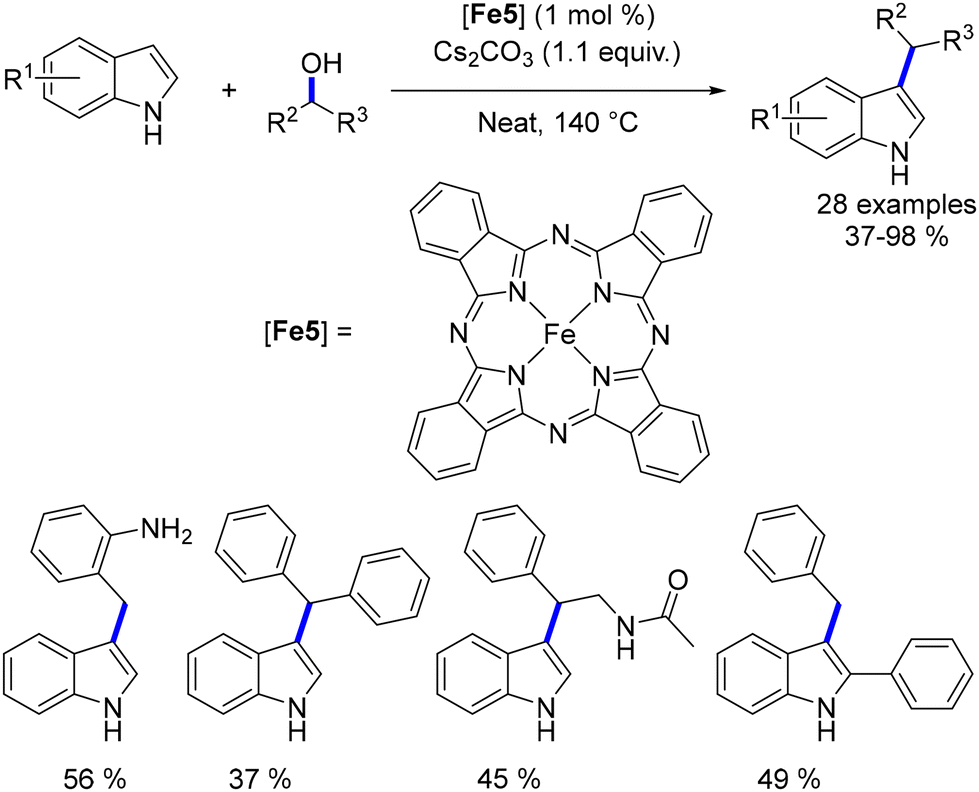
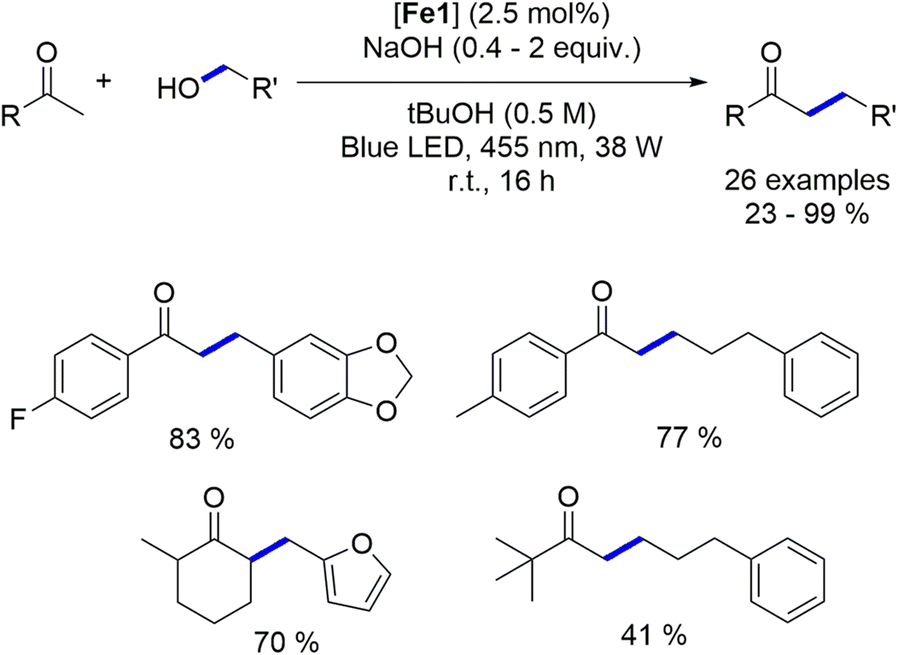
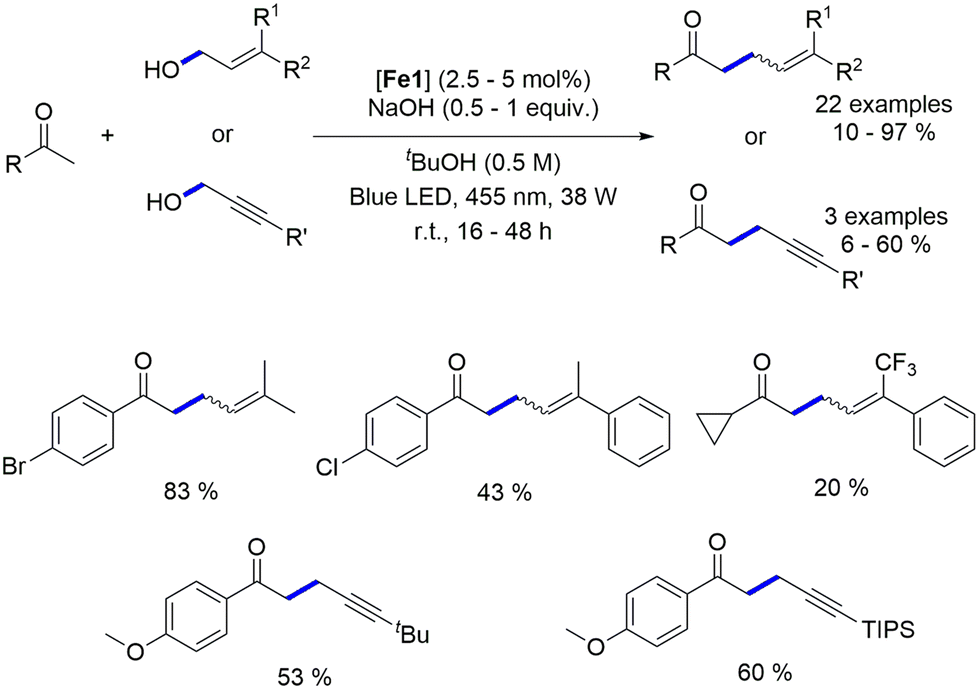
4.2. Blue light-induced iron-catalyzed α-alkylation of ketones
Renaud and coworkers began by examining the UV-visible spectra of iron complexes Fe1–Fe3,199 which showed small absorption in the blue region despite their maxima falling within the UV spectrum (Scheme 110). The alkylation of 4-methoxyacetophenone with benzyl alcohol was optimized under various light sources and powers. Optimal conditions included using a 40 W blue LED lamp, Fe1 (2.5 mol%), and NaOH (0.4 equiv.) in tBuOH, leading to complete conversion in 16 hours. The scope of this blue light-induced alkylation was explored, yielding α-alkylated ketones in moderate to good yields across various substrates. The key takeaway from this section is that the current methodology enables the use of both aliphatic alcohols and aliphatic ketones, delivering good yields. The successful alkylation of an aliphatic ketone with an aliphatic alcohol is significant, as thermal alkylation with Fe1 had previously failed. In the past, homo-aldolization of the aliphatic aldehyde intermediate occurred more rapidly than alkylation. However, under the new conditions, light-induced alkylation at room temperature now outpaces other competing reactions. Mechanistic studies suggested that light is crucial in the dehydrogenation and reduction steps of this process.
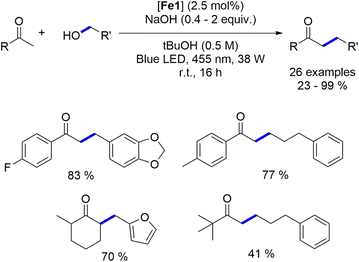 |
| | Scheme 110 Alkylation of aryl ketones with alcohols. | |
Renaud in 2024 unveiled a novel light-induced, iron-catalyzed α-alkylation of ketones at room temperature, utilizing allylic and propargylic alcohols as pro-electrophiles. This study illustrates the versatility of cyclopentadienone iron complexes under various reaction conditions employing the borrowing hydrogen methodology (Scheme 111). The findings underscore the potential of these complexes in organic synthesis and catalysis, suggesting the possibility of expanding the reaction scope to include a broader range of substrates and functional group tolerance. Mechanistic studies show that steric hindrance at the δ-position of the dienone or eneynone intermediates is crucial in preventing 1,6-reduction, favouring the formation of non-conjugated enones and ynones over saturated ketones. Additionally, the mild reaction conditions suggest the potential development of an efficient asymmetric version of this reaction.
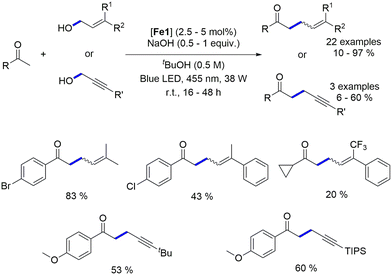 |
| | Scheme 111 Alkylation of 4-methoxyacetophenone with alcohols. | |
The diaminocyclopentadienone iron tricarbonyl complex functions dually, capturing light and facilitating dehydrogenation and reduction without the need for an external photosensitizer. This allows the synthesis of γ,δ-unsaturated ketones at room temperature through a borrowing hydrogen methodology.
5. Conclusions
Among the reported methodologies for the alkylation of N- and C-nucleophiles, those utilizing moisture- and air-stable organometallic complexes are relatively rare. The challenging synthesis of these sensitive catalysts further questions their practicality and applicability on an industrial scale. Alternative methods often involve metal salts, which typically require higher temperatures and base loadings, sometimes in stoichiometric amounts. In contrast, cyclopentadienone iron tricarbonyl complexes have demonstrated their effectiveness in various alkylation reactions under mild conditions, highlighting their potential for practical applications in this field.
The research conducted in this research field aligns with the work particularly over the past decade focusing on the development of more efficient catalysts using Earth-abundant metals. Expanding hydrogen autotransfer reactions to new substrates could broaden the applicability of this approach and its catalysts. Another potential direction is the design of a chromophoric Knölker-type catalyst capable of absorbing visible light of a wider range, which could enable alkylation reactions using safer white light instead of powerful blue LEDs. Additionally, the mild reaction conditions presented here could facilitate the development of stereoselective alkylation reactions. Finally, integrating two different catalysts in a dual catalytic system may allow for the formation of complex molecules in a one-pot process.
Even though HA systems offer significant potential for green chemistry, they are not without drawbacks. One major limitation is the restricted substrate scope. Many HA reactions struggle with functional group compatibility, particularly for more sensitive groups such as halides, ketones, or nitriles. This limitation can hinder broader applications, especially in fine chemicals or pharmaceutical industries. To overcome this, future research could explore the development of more robust catalysts that tolerate a wider range of functional groups. One possible solution is designing bifunctional catalysts that combine hydrogenation and transfer hydrogenation capabilities, enabling more selective transformations without sacrificing functional group compatibility. Another issue is the need for more than a stoichiometric amount of an alkali base, particularly in alcohol-to-amine conversions. This excess base often plays a dual role: activating the alcohol and promoting dehydrogenation, but it can also lead to side reactions or decomposition. A potential solution involves developing milder base systems or even base-free catalytic designs, improving selectivity and sustainability. Finally, compared to Ru, Rh, and Ir catalysts, which offer excellent reactivity and selectivity, manganese and iron-based catalysts are more Earth-abundant and cost-effective. However, they often suffer from lower activity and selectivity. Future strategies might focus on ligand engineering and catalyst design to enhance these Earth-abundant metals’ performance.
Author contributions
All the authors wrote and revised the manuscript.
Abbreviations
| Ac | Acetyl |
| acac | Acetyl acetonate |
| Am | Amyl |
| Atm | Atmosphere |
| Ar | Aryl |
| Boc |
tert-Butoxycarbonyl |
| Bpy | 2,2′-Bipyridine |
| Bu | Butyl |
| Cp* | 1,2,3,4,5-Pentamethylcyclopentadienyl |
| CPME | Cyclopentyl-methyl ether |
| Cy | Cyclohexyl |
| dba | Dibenzylideneacetone |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCC |
N,N′-dicyclohexylcarbodiimide |
| dcpe | 1,2-Bis(dicyclohexylphosphino)ethane |
| dcpp | 1,3-Bis(diphenylphosphino)propane |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| DFT | Density functional theory |
| DIBAL-H | Diisobulyl aluminium hydride |
| DIPEA |
N,N-Diisopropylethylamine |
| DMAP | 4-Dimethylaminopyridine |
| dme | Dimethoxyethane |
| DMF |
N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| dppf | 1,1′-Bis(diphenylphosphino)ferrocene |
| dppm | 1,1-Bis(diphenylphosphino)methane |
| d.r. | Diastereoisomeric ratio |
| EDA | Ethylenediamine |
| EDG | Electron-donating group |
| equiv. | Equivalent |
| e.r. | Enantiomeric ratio |
| Et | Ethyl |
| EWG | Electron-withdrawing group |
| KHMDS | Potassium bis(trimethylsilyl)amide |
| LED | Light emitting diode |
| Me | Methyl |
| NHC | N-Heterocyclic carbene |
| NMR | Nuclear magnetic resonance |
| Ph | Phenyl |
| THF | Tetrahydrofuran |
| TMEDA | Tetramethylethylenediamine |
| TON | Turnover number |
Data availability
Data availability statement is not applicable for review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge financial support from the “Ministère de la Recherche et des Nouvelles Technologies”, Normandie Université, University of Caen Normandie, CNRS, “Région Normandie”, the LABEX SynOrg (ANR-11-LABEX-0029), and European Union (HORIZON-MSCA-2022-DN-01 Project: 101119574). We thank the Spanish Ministerio de Ciencia e Innovación for project PID2021-127423NB-I00 and the Generalitat de Catalunya for project 2021SGR623. A. P. is a Serra Húnter Fellow and ICREA Academia Prize 2019.
References
- M. G. Edwards, R. F. R. Jazzar, B. M. Paine, D. J. Shermer, M. K. Whittlesey and J. M. J. Williams, Borrowing hydrogen: a catalytic route to C–C bond formation from alcohols, Chem. Commun., 2004, 4, 90–91 RSC.
- C. F. Winans and H. Adkins, The Alkylation of Amines as Catalyzed by Nickel, J. Am. Chem. Soc., 1932, 54, 306–312 CrossRef CAS.
- E. F. Pratt and E. J. Frazza, Disproportionative Condensations. II. The N-Alkylation of Anilines with Primary Alcohols, J. Am. Chem. Soc., 1954, 76, 6174–6179 CrossRef CAS.
- R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, Oxidation of Alcohols by Transition Metal Complexes Part V. Selective Catalytic Monoalkylation of Arylacetonitriles by Alcohols, Tetrahedron Lett., 1981, 22, 4107–4111 CrossRef CAS.
- R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, Transition metal-catalysed N-alkylation of amines by alcohols, J. Chem. Soc., Chem. Commun., 1981, 611–612 RSC.
- G. Lamoureaux and C. Agüero, A comparison of several modern alkylating agents, ARKIVOC, 2009, 251–264 Search PubMed.
- G. Szekely, M. C. Amores de Sousa, M. Gil, F. C. Ferreira and W. Heggie, Genotoxic Impurities in Pharmaceutical Manufacturing: Sources, Regulations, and Mitigation, Chem. Rev., 2015, 115, 8182–8229 CrossRef CAS PubMed.
- T. P. Vispute, H. Zhang, A. Sanna, R. Xiao and G. W. Huber, Renewable chemical commodity feedstocks from integrated catalytic processing of pyrolysis oils, Science, 2010, 330, 1222–1227 CrossRef CAS PubMed.
- Z. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Complete lignocellulose conversion with integrated catalyst recycling yielding valuable aromatics and fuels, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
- S. Bera, L. M. Kabadwal and D. Banerjee, Harnessing alcohols as sustainable reagents for late-stage functionalisation: synthesis of drugs and bio-inspired compounds, Chem. Soc. Rev., 2024, 53, 4607–4647 RSC.
- G. Sivakumar, R. Kumar, V. Yadav, V. Gupta and E. Balaraman, Multi-Functionality of Methanol in Sustainable Catalysis: Beyond Methanol Economy, ACS Catal., 2023, 13, 15013–15053 CrossRef CAS.
- L. M. Kabadwal, S. Bera and D. Banerjee, Recent advances in sustainable organic transformations using methanol: Expanding the scope of hydrogen borrowing catalysis, Org. Chem. Front., 2021, 8, 7077–7096 RSC.
- O. Saidi, A. J. Blacker, G. W. Lamb, S. P. Marsden, J. E. Taylor and J. M. J. Williams, Borrowing hydrogen in water and ionic liquids: iridium-catalyzed alkylation of amines with alcohols, Org. Process Res. Dev., 2010, 14, 1046–1049 CrossRef CAS.
- S. Gülcemal, Symmetric and dissymmetric N–heterocyclic carbene rhodium(I) complexes: a comparative study of their catalytic activities in transfer hydrogenation reaction, Appl. Organomet. Chem., 2012, 26, 246–251 CrossRef.
- J. A. Mata, M. Poyatos and E. Peris, Structural and catalytic properties of chelating bis- and tris-N-heterocyclic carbenes, Coord. Chem. Rev., 2007, 251, 841–859 CrossRef CAS.
- G. Chelucci, Ruthenium and osmium complexes in Csingle bondC bond-forming reactions by borrowing hydrogen catalysis, Coord. Chem. Rev., 2017, 331, 1–36 CrossRef CAS.
- P. A. Slatford, M. K. Whittlesey and J. M. J. Williams, C-C bond formation from alcohols using a xantphos ruthenium complex, Tetrahedron Lett., 2006, 47, 6787–6789 CrossRef CAS.
- C. S. Cho, B. T. Kim, T.-J. Kim and S. C. Shim, Ruthenium-catalyzed regioselective α-alkylation of ketones with primary alcohols, Tetrahedron Lett., 2002, 43, 7987–7989 CrossRef CAS.
- D. Gong, B. Hu, W. Yang and D. Chen, Bidentate Ru(II)-NC Complexes as Catalysts for α-Alkylation of Unactivated Amides and Esters, ChemCatChem, 2019, 11, 4841–4847 CrossRef CAS.
- F. Li, J. Xie, H. Shan, C. Sun and L. Chen, General and efficient method for direct N-monomethylation of aromatic primary amines with methanol, RSC Adv., 2012, 2, 8645–8652 RSC.
- S. Michlik, T. Hillie and R. Kempe, The Iridium-Catalyzed Synthesis of Symmetrically and Unsymmetrically Alkylated Diamines under Mild Reaction Conditions, Adv. Synth. Catal., 2012, 354, 847–862 CrossRef CAS.
- R. Liang, S. Li, R. Wang, L. Lu and F. Li, N-Methylation of Amines with Methanol Catalyzed by a Cp*Ir Complex Bearing a Functional 2,2′-Bibenzimidazole Ligand, Org. Lett., 2017, 19, 5790–5793 CrossRef CAS PubMed.
- T. Sawagushi and Y. Obora, Iridium-catalyzed α-Alkylation of Acetonitrile with Primary and Secondary Alcohols, Chem. Lett., 2011, 40, 1055–1057 CrossRef.
- P. J. Black, G. Cami-Kobeci, M. G. Edwards, P. A. Slatford, M. K. Whittlesey and J. M. J. Williams, Borrowing hydrogen: iridium-catalysed reactions for the formation of C–C bonds from alcohols, Org. Biomol. Chem., 2006, 4, 116–125 RSC.
- B. Blank and R. Kempe, Catalytic alkylation of methyl-N-heteroaromatics with alcohols, J. Am. Chem. Soc., 2010, 132, 924–925 CrossRef CAS PubMed.
- Y. Obora, S. Ogawa and N. Yamamoto, Iridium-Catalyzed Alkylation of Methylquinolines with Alcohols, J. Org. Chem., 2012, 77, 9429–9433 CrossRef CAS PubMed.
- T.-Y. Feng, H.-X. Li, D. J. Young and J.-P. Lang, Ligand-free RuCl3-catalyzed alkylation of methylazaarenes with alcohols, J. Org. Chem., 2017, 82, 4113–4120 CrossRef CAS PubMed.
- B. Paul, M. Maji, K. Chakrabartia and S. Kundu, Tandem transformations and multicomponent reactions utilizing alcohols following dehydrogenation strategy, Org. Biomol. Chem., 2020, 18, 2193–2214 RSC.
- A. Mondal, R. Sharma, D. Pal and D. Srimani, Recent Progress in the Synthesis of Heterocycles through Base Metal-Catalyzed Acceptorless Dehydrogenative and Borrowing Hydrogen Approach, Eur. J. Org. Chem., 2021, 3690–3720 CrossRef CAS.
- J. A. Luque-Urrutia, T. Ortiz-García, M. Solà and A. Poater, Green Energy by Hydrogen Production from Water Splitting, Water Oxidation Catalysis and Acceptorless Dehydrogenative Coupling, Inorganics, 2023, 11, 88 CrossRef CAS.
- C. Gunanathan and D. Milstein, Applications of acceptorless dehydrogenation and related transformations in chemical synthesis, Science, 2013, 341, 1229712 CrossRef PubMed.
- M. Maji, D. Panja, I. Borthakur and S. Kundu, Recent advances in sustainable synthesis of N-heterocycles following acceptorless dehydrogenative coupling protocol using alcohols, Org. Chem. Front., 2021, 8, 2673–2709 RSC.
- T. Irrgang and R. Kempe, 3d-Metal Catalyzed N- and C-Alkylation Reactions via Borrowing Hydrogen or Hydrogen Autotransfer, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
- I. Bauer and H.-J. Knölker, Iron Catalysis in Organic Synthesis, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- H.-J. Knölker, J. Heber and C. H. Malher, Transition Metal-Diene Complexes in Organic Synthesis, Part 14 1 Regioselective Iron-Mediated [2 + 2 + 1] Cycloadditions of Alkynes and Carbon Monoxide: Synthesis of Substituted Cyclopentadienones, Synlett, 1992, 1002–1007 CrossRef.
- H.-J. Knölker and J. Heber, Transition metal-diene complexes in organic synthesis, part 18.1 iron-mediated [2 + 2 + 1] cycloadditions of diynes and carbon monoxide: Selective Demetalation reactions, Synlett, 1993, 924–926 CrossRef.
- H.-J. Knölker, H. Goesmann and R. Klauss, A Novel Method for the Demetalation of Tricarbonyliron-Diene Complexes by a Photolytically Induced Ligand Exchange Reaction with Acetonitrile, Angew. Chem., Int. Ed., 1999, 38, 702–705 CrossRef.
- D. S. Mérel, M. Elie, J.-F. Lohier, S. Gaillard and J.-L. Renaud, Bifunctional Iron Complexes: Efficient Catalysts for C=O and C=N Reduction in Water, ChemCatChem, 2013, 5, 2939–2945 CrossRef.
- A. Pagnoux-Ozherelyeva, N. Pannetier, M. D. Mbaye, S. Gaillard and J.-L. Renaud, Knölker's iron complex: an efficient in situ generated catalyst for reductive amination of alkyl aldehydes and amines, Angew. Chem., Int. Ed., 2012, 51, 4976–4980 CrossRef CAS PubMed.
- S. Moulin, H. Dentel, A. Pagnoux-Ozherelyeva, S. Gaillard, A. Poater, L. Cavallo, J.-F. Lohier and J.-L. Renaud, Bifunctional (Cyclopentadienone)Iron–Tricarbonyl Complexes: Synthesis, Computational Studies and Application in Reductive Amination, Chem. – Eur. J., 2013, 19, 17881–17890 CrossRef CAS PubMed.
- N. Joly, A. Colella, M.-E. Mendy, M. D. Mbaye, S. Gaillard, A. Poater and J.-L. Renaud, Blue-light Induced Iron-Catalyzed Synthesis of γ,δ-Unsaturated Ketones, ChemSusChem, 2024, 17, e202301472 CrossRef CAS PubMed.
- S. Coufourier, D. Ndiaye, Q. Gaignard Gaillard, L. Bettoni, N. Joly, M.-D. Mbaye, A. Poater, S. Gaillard and J.-L. Renaud, Iron-catalyzed chemoselective hydride transfer reactions, Tetrahedron, 2021, 90, 132187 CrossRef CAS.
- A. Lator, S. Gaillard, A. Poater and J.-L. Renaud, Iron-Catalyzed Chemoselective Reduction of α,β-Unsaturated Ketones, Chem. – Eur. J., 2018, 24, 5770–5774 CrossRef CAS PubMed.
- S. Coufourier, Q. Gaignard-Gaillard, J.-F. Lohier, A. Poater, S. Gaillard and J.-L. Renaud, Hydrogenation of CO2, Hydrogenocarbonate, and Carbonate to Formate in Water using Phosphine Free Bifunctional Iron Complexes, ACS Catal., 2020, 10, 2108–2116 CrossRef CAS.
- A. Lator, S. Gaillard, A. Poater and J.-L. Renaud, Well-Defined Phosphine-Free Iron-Catalyzed N–Ethylation and N-Methylation of Amines with Ethanol and Methanol, Org. Lett., 2018, 20, 5985–5990 CrossRef CAS PubMed.
- C. Seck, M. D. Mbaye, S. Gaillard and J.-L. Renaud, Bifunctional Iron Complexes Catalyzed Alkylation of Indoles, Adv. Synth. Catal., 2018, 360, 4640–4645 CrossRef CAS.
- L. Bettoni, S. Gaillard and J.-L. Renaud, Iron-Catalyzed β-Alkylation of Alcohols, Org. Lett., 2019, 21, 8404–8408 CrossRef CAS PubMed.
- C. Seck, M. D. Mbaye, S. Coufourier, A. Lator, J.-F. Lohier, A. Poater, T. R. Ward, S. Gaillard and J.-L. Renaud, Iron Transition Metal Frustrated Lewis Pair: from DFT to Application in Alkylation of Ketones, ChemCatChem, 2017, 9, 4410–4416 CrossRef CAS.
- L. Bettoni, C. Seck, M. D. Mbaye, S. Gaillard and J.-L. Renaud, Iron-Catalyzed Tandem Three-Component Alkylation: Access to α-Methylated Substituted Ketones, Org. Lett., 2019, 21, 3057–3061 CrossRef CAS PubMed.
- L. Bettoni, S. Gaillard and J.-L. Renaud, A phosphine-free iron complex-catalyzed synthesis of cycloalkanes via the borrowing hydrogen strategy, Chem. Commun., 2020, 56, 12909–12912 RSC.
- L. Bettoni, S. Gaillard and J.-L. Renaud, Iron-Catalyzed α-Alkylation of Ketones with Secondary Alcohols: Access to β-Disubstituted Carbonyl Compounds, Org. Lett., 2020, 22, 2064–2069 CrossRef CAS PubMed.
- R. Monreal-Corona, A. Pla-Quintana and A. Poater, Predictive catalysis: a valuable step towards machine learning, Trends Chem., 2023, 5, 935–946 CrossRef.
- M. Gimferrer, N. Joly, S. Escayola, E. Viñas, S. Gaillard, M. Solà, J.-L. Renaud, P. Salvador and A. Poater, Knölker Iron Catalysts for Hydrogenation revisited: Non Spectator Solvent and fine-tuning, Organometallics, 2022, 41, 1204–1215 CrossRef CAS.
- N. Joly, M. Gimferrer, S. Escayola, M. Cendra, S. Coufourier, J.-F. Lohier, Q. Gaignard Gaillard, S. Gaillard, M. Solà, J.-L. Renaud and A. Poater, Enhancement of Knölker Iron Catalysts for Imine Hydrogenation by Predictive Catalysis: From Calculations to Selective Experiments, Organometallics, 2023, 42, 1784–1792 CrossRef CAS.
- M. Gimferrer, P. Salvador and A. Poater, Computational Monitoring of Oxidation States in Olefin Metathesis, Organometallics, 2019, 38, 4585–4592 CrossRef CAS.
- P. Salvador, E. Ramos-Cordoba, M. Montilla, L. Pujal and M. Gimferrer, APOST-3D: Chemical concepts from wavefunction analysis, J. Chem. Phys., 2024, 160, 172502 CrossRef CAS PubMed.
- B. G. Reed-Berendt, D. E. Latham, M. B. Dambatta and L. C. Morrill, Borrowing Hydrogen for Organic Synthesis, ACS Cent. Sci., 2021, 7, 570–585 CrossRef CAS PubMed.
- A. Corma, J. Navas and M. J. Sabater, Advances in One-Pot Synthesis through Borrowing Hydrogen Catalysis, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- J. Wu, S. N. Narayanasamy and C. Darcel, Late stage modifications of phosphine oxide ligands by iron–catalyzed hydrogen borrowing reactions, J. Organomet. Chem., 2022, 979, 122510 CrossRef CAS.
- E. Suarsih, Y. Kita, K. Kamata and M. Hara, A heterogeneous cobalt catalyst for C-C bond formation by a borrowing hydrogen strategy, Catal. Sci. Technol., 2022, 12, 4113–4117 RSC.
- S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, The Catalytic Amination of Alcohols, ChemCatChem, 2011, 3, 1853–1864 CrossRef.
- M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Borrowing Hydrogen in the Activation of Alcohols, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS.
- T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Transition metal catalysed reactions of alcohols using borrowing hydrogen methodology, Dalton Trans., 2009, 753–762 RSC.
-
N. Joly, PhD Thesis, Alkylations by Hydrogen Autotransfer: From Experiments to Reaction Mechanisms and Vice-Versa, University of Caen Normandie, 2023 Search PubMed.
-
C. C. Marvin, in Comprehensive Organic Synthesis, ed. P. Knochel, Elsevier, Second Edition, 2014, vol. 6, pp. 34–99 Search PubMed.
-
B. M. Stoltz, N. B. Bennett, D. C. Duquette, A. F. G. Goldberg, Y. Liu, M. B. Loewinger and C. M. Reeves, in Comprehensive Organic Synthesis, ed. P. Knochel, Elsevier, Second Edition, 2014, vol. 3, pp. 1–55 Search PubMed.
- Y. Kita, M. Kuwabara, K. Kamata and M. Hara, Heterogeneous Low-Valent Mn Catalysts for α-Alkylation of Ketones with Alcohols through Borrowing Hydrogen Methodology, ACS Catal., 2022, 12, 11767–11775 CrossRef CAS.
- G. E. Dobereiner and R. H. Crabtree, Dehydrogenation as a substrate-activating strategy in homogeneous transition-metal catalysis, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed.
- G. Guillena, D. J. Ramón and M. Yus, Hydrogen autotransfer in the N-alkylation of amines and related compounds using alcohols and amines as electrophiles, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed.
- F. Huang, Z. Liu and Z. Yu, C-Alkylation of Ketones and Related Compounds by Alcohols: Transition-Metal-Catalyzed Dehydrogenation, Angew. Chem., Int. Ed., 2016, 55, 862–875 CrossRef CAS PubMed.
- A. Corma, J. Navas and M. J. Sabater, Advances in One-Pot Synthesis through Borrowing Hydrogen Catalysis, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- B. G. Reed-Berendt, K. Polidano and L. C. Morrill, Recent advances in homogeneous borrowing hydrogen catalysis using earth-abundant first row transition metals, Org. Biomol. Chem., 2019, 17, 1595–1607 RSC.
- B. Maji and M. K. Barman, Recent Developments of Manganese Complexes for Catalytic Hydrogenation and Dehydrogenation Reactions, Synthesis, 2017, 3377–3393 CAS.
- J.-L. Renaud and D. Gaillard, Recent advances in iron-and cobalt-complex-catalyzed tandem/consecutive processes involving hydrogenation, Synthesis, 2016, 3659–3683 CrossRef CAS.
- S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Efficient and selective N-alkylation of amines with alcohols catalysed by manganese pincer complexes, Nat. Commun., 2016, 7, 12641 CrossRef PubMed.
- B. G. Reed-Berendt and L. C. Morrill, Manganese-Catalyzed N-Alkylation of Sulfonamides Using Alcohols, J. Org. Chem., 2019, 84, 3715–3724 CrossRef CAS PubMed.
- L. Duarte de Almeida, F. Bourriquen, K. Junge and M. Beller, M. Catalytic Formal Hydroamination of Allylic Alcohols Using Manganese PNP-Pincer Complexes, Adv. Synth. Catal., 2021, 363, 4177–4181 CrossRef CAS PubMed.
- K. Das, K. Sarkar and B. Maji, Manganese-Catalyzed Anti-Markovnikov Hydroamination of Allyl Alcohols via Hydrogen-Borrowing Catalysis, ACS Catal., 2021, 11, 7060–7069 CrossRef CAS.
- J. Neumann, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, Improved and General Manganese-Catalyzed N-Methylation of Aromatic Amines Using Methanol, Chem. – Eur. J., 2017, 23, 5410–5413 CrossRef CAS PubMed.
- A. Bruneau-Voisine, D. Wang, V. Dorcet, T. Roisnel, C. Darcel and J.-B. Sortais, Mono-N-methylation of anilines with methanol catalyzed by a manganese pincer-complex, J. Catal., 2017, 347, 57–62 CrossRef CAS.
- B. G. Reed-Berendt, N. Mast and L. C. Morrill, Manganese-Catalyzed One-Pot Conversion of Nitroarenes into N-Methylarylamines Using Methanol, Eur. J. Org. Chem., 2020, 1136–1140 CrossRef CAS.
- R. Fertig, T. Irrgang, F. Freitag, J. Zander and R. Kempe, Manganese-Catalyzed and Base-Switchable Synthesis of Amines or Imines via Borrowing Hydrogen or Dehydrogenative Condensation, ACS Catal., 2018, 8, 8525–8530 Search PubMed.
- U. K. Das, Y. Ben-David, Y. Diskin-Posner and D. Milstein, N-Substituted Hydrazones by Manganese-Catalyzed Coupling of Alcohols with Hydrazine: Borrowing Hydrogen and Acceptorless Dehydrogenation in One System, Angew. Chem., Int. Ed., 2018, 57, 2179–2182 CrossRef CAS PubMed.
- L. M. Azofra, M. A. Tran, V. Zubar, L. Cavallo, M. Rueping and O. El-Sepelgy, Conversion of racemic alcohols to optically pure amine precursors enabled by catalyst dynamic kinetic resolution: experiment and computation, Chem. Commun., 2020, 56, 9094–9097 RSC.
- V. G. Landge, A. Mondal, V. Kumar, A. Nandakumar and E. Balaraman, Manganese catalyzed N-alkylation of anilines with alcohols: ligand enabled selectivity, Org. Biomol. Chem., 2018, 16, 8175–8180 Search PubMed.
- M. Huang, Y. Li, Y. Li, J. Liu, S. Shu, Y. Liu and Z. Ke, Room temperature N-heterocyclic carbene manganese catalyzed selective N-alkylation of anilines with alcohols, Chem. Commun., 2019, 55, 6213–6216 RSC.
- R. Babu, S. Sukanya Padhy, R. Kumar and E. Balaraman, Catalytic Amination of Alcohols Using Diazo Compounds under Manganese Catalysis Through Hydrogenative N-Alkylation Reaction, Chem. – Eur. J., 2023, 29, e202302007 CrossRef CAS PubMed.
- Y. Zhao, S. W. Foo and S. Saito, Iron/amino acid catalyzed direct N-alkylation of amines with alcohols, Angew. Chem., Int. Ed., 2011, 50, 3006–3009 CrossRef CAS PubMed.
- M. Bala, P. K. Verma, U. Sharma, N. Kumar and B. Singh, Iron phthalocyanine as an efficient and versatile catalyst for N-alkylation of heterocyclic amines with alcohols: one-pot synthesis of 2-substituted benzimidazoles, benzothiazoles and benzoxazoles, Green Chem., 2013, 15, 1687–1693 RSC.
- M. Minakawa, M. Okubo and M. Kawatsura, Selective direct N-alkylation of amines with alcohols using Iron(III) phthalocyanine chloride under solvent-free conditions, Bull. Chem. Soc. Jpn., 2015, 88, 1680–1682 CrossRef CAS.
- T. Yan, B. L. Feringa and K. Barta, Iron catalysed direct alkylation of amines with alcohols, Nat. Commun., 2014, 5, 5602 CrossRef CAS PubMed.
- G. N. Schrauzer, Diphenylacetylene Derivatives of Iron Carbonyl, J. Am. Chem. Soc., 1959, 81, 5307–5310 CrossRef CAS.
- A. J. Rawlings, L. J. Diorazio and M. Wills, C–N Bond Formation between Alcohols and Amines Using an Iron Cyclopentadienone Catalyst, Org. Lett., 2015, 17, 1086–1089 CrossRef CAS PubMed.
- D. Hollmann, H. Jiao, A. Spannenberg, S. Bähn, A. Tillack, P. Parton, R. Altink and M. Beller, Deactivation of the Shvo Catalyst by Ammonia: Synthesis, Characterization, and Modeling, Organometallics, 2009, 28, 473–479 CrossRef CAS.
- B. Emayavaramban, M. Roy and B. Sundararaju, Iron-Catalyzed Allylic Amination Directly from Allylic Alcohols, Chem. – Eur. J., 2016, 22, 3952–3955 CrossRef CAS PubMed.
- G. Bottari, A. Afanasenko, A. A. Castillo-Garcia, B. L. Feringa and K. Barta, Synthesis of Enantioenriched Amines by Iron–Catalysed Amination of Alcohols Employing at Least One Achiral Substrate, Adv. Synth. Catal., 2021, 363, 5436–5442 CrossRef CAS.
- T. Yan, B. L. Feringa and K. Barta, Direct N-alkylation of unprotected amino acids with alcohols, Sci. Adv., 2017, 3, eaao6494 CrossRef PubMed.
- A. Afanasenko, R. Hannah, T. Yan, S. Elangovan and K. Barta, Ruthenium and Iron-Catalysed Decarboxylative N-alkylation of Cyclic a-Amino Acids with Alcohols: Sustainable Routes to Pyrrolidine and Piperidine Derivatives, ChemSusChem, 2019, 12, 3801–3807 CrossRef CAS PubMed.
- H.-J. Pan, T. W. Ng and Y. Zhao, Iron-catalyzed amination of alcohols assisted by Lewis acid, Chem. Commun., 2015, 51, 11907–11910 RSC.
- T. J. Brown, M. Cumbes, L. J. Diorazio, G. J. Clarkson and M. Wills, Use of (Cyclopentadienone)iron Tricarbonyl Complexes for C–N Bond Formation Reactions between Amines and Alcohols, J. Org. Chem., 2017, 82, 10489–10503 CrossRef CAS PubMed.
- T.-T. Thai, D. S. Mérel, A. Poater, S. Gaillard and J.-L. Renaud, Highly active bifunctional iron complex for hydrogenation of bicarbonate and reductive amination, Chem. – Eur. J., 2015, 21, 7066–7070 CrossRef CAS PubMed.
- M. Vayer, S. P. Morcillo, J. Dupont, V. Gandon and C. Bour, Iron-Catalyzed Reductive Ethylation of Imines with Ethanol, Angew. Chem., Int. Ed., 2018, 57, 3228–3232 Search PubMed.
- M. Mastalir, M. Glatz, N. Gorgas, B. Stöger, E. Pittenauer, G. Allmaier, L. F. Veiros and K. Kirchner, Divergent Coupling of Alcohols and Amines Catalyzed by Isoelectronic Hydride Mn(I) and Fe(II) PNP Pincer Complexes, Chem. – Eur. J., 2016, 22, 12316–12320 CrossRef CAS PubMed.
- M. Mastalir, B. Stöger, E. Pittenauer, M. Puchberger, G. Allmaier and K. Kirchner, Air Stable Iron(II) PNP Pincer Complexes as Efficient Catalysts for the Selective Alkylation of Amines with Alcohols, Adv. Synth. Catal., 2016, 358, 3824–3831 CrossRef CAS.
- W. Ma, X. Zhang, J. Fan, Y. Liu, W. Tang, D. Xue, C. Li, J. Xiao and C. Wang, Iron-Catalyzed Anti-Markovnikov Hydroamination and Hydroamidation of Allylic Alcohols, J. Am. Chem. Soc., 2019, 141, 13506–13515 CrossRef CAS PubMed.
- H. Chen, Q. Wang, T. Liu, H. Chen, D. Zhou and F. Qu, Iron-catalyzed N-alkylation of aromatic amines via borrowing hydrogen strategy, J. Coord. Chem., 2021, 74, 877–884 CrossRef CAS.
- S. Rösler, M. Ertl, T. Irrgang and R. Kempe, Cobalt-Catalyzed Alkylation of Aromatic Amines by Alcohols, Angew. Chem., Int. Ed., 2015, 54, 15046–15050 CrossRef PubMed.
- M. Mastalir, G. Tomsu, E. Pittenauer, G. Allmaier and K. Kirchner, Co(II) PCP Pincer Complexes as Catalysts for the Alkylation of Aromatic Amines with Primary Alcohols, Org. Lett., 2016, 18, 3462–3465 CrossRef CAS PubMed.
- G. Zhang, Z. Yin and S. Zheng, Cobalt-Catalyzed N-Alkylation of Amines with Alcohols, Org. Lett., 2016, 18, 300–303 CrossRef CAS PubMed.
- S. P. Midya, A. Mondal, A. Begum and E. Balaraman, A Simple Cobalt(II) Chloride Catalyzed N-Alkylation of Amines with Alcohols, Synthesis, 2017, 3957–3961 CAS.
- Z. Liu, Z. Yang, X. Yu, H. Zhang, B. Yu, Y. Zhao and Z. Liu, Efficient Cobalt-Catalyzed Methylation of Amines Using Methanol, Adv. Synth. Catal., 2017, 359, 4278–4283 CrossRef CAS.
- S. P. Midya, J. Pitchaimani, V. G. Landge, V. Madhu and E. Balaraman, Direct access to N-alkylated amines and imines via acceptorless dehydrogenative coupling catalyzed by a cobalt(II)-NNN pincer complex, Catal. Sci. Technol., 2018, 8, 3469–3473 RSC.
- K. Paudel, S. Xu, O. Hietsoi, B. Pandey, C. Onuh and K. Ding, Switchable imine and amine synthesis catalyzed by a well-defined cobalt complex, Organometallics, 2021, 40, 418–426 CrossRef CAS.
- B. Emayavaramban, P. Chakraborty, E. Manoury, R. Poli and B. Sundararaju, Cp*Co(III)-catalyzed N-alkylation of amines with secondary alcohols, Org. Chem. Front., 2019, 6, 852–857 RSC.
- P. Yang, C. Zhang, Y. Ma, C. Zhang, A. Li, B. Tang and J. S. Zhou, Nickel-Catalyzed N-Alkylation of Acylhydrazines and Arylamines Using Alcohols and Enantioselective Examples, Angew. Chem., Int. Ed., 2017, 56, 14702–14706 CrossRef CAS PubMed.
- M. Vellakkaran, K. Singh and D. Banerjee, An Efficient and Selective Nickel-Catalyzed Direct N-Alkylation of Anilines with Alcohols, ACS Catal., 2017, 7, 8152–8158 CrossRef CAS.
- J. Das and D. Banerjee, Nickel-Catalyzed Phosphine Free Direct N-Alkylation of Amides with Alcohols, J. Org. Chem., 2018, 83, 3378–3384 CrossRef CAS PubMed.
- M. Subaramanian, S. P. Midya, P. M. Ramar and E. Balaraman, General Synthesis of N-Alkylation of Amines with Secondary Alcohols via Hydrogen Autotransfer, Org. Lett., 2019, 21, 8899–8903 CrossRef CAS PubMed.
- V. Arora, M. Dutta, K. Das, B. Das, H. K. Srivastava and A. Kumar, Solvent-Free N-Alkylation and Dehydrogenative Coupling Catalyzed by a Highly Active Pincer-Nickel Complex, Organometallics, 2020, 39, 2162–2176 CrossRef CAS.
- F. Shi, M. K. Tse, X. Cui, D. Gördes, D. Michalik, K. Thurow, Y. Deng and M. Beller, Copper-catalyzed alkylation of sulfonamides with alcohols, Angew. Chem., Int. Ed., 2009, 48, 5912–5915 CrossRef CAS PubMed.
- X. Cui, F. Shi, M. K. Tse, D. Gördes, K. Thurow, M. Beller and D. Deng, Copper–Catalyzed N–Alkylation of Sulfonamides with Benzylic Alcohols: Catalysis and Mechanistic Studies, Adv. Synth. Catal., 2009, 351, 2949–2958 CrossRef CAS.
- A. Martínez-Asencio, D. J. Ramón and M. Yus, N-Alkylation of poor nucleophilic amine and sulfonamide derivatives with alcohols by a hydrogen autotransfer process catalyzed by copper(II) acetate, Tetrahedron Lett., 2010, 51, 325–327 CrossRef.
- A. Martínez-Asencio, D. J. Ramón and M. Yus, N-Alkylation of poor nucleophilic amines and derivatives with alcohols by a hydrogen autotransfer process catalyzed by copper(II) acetate: scope and mechanistic considerations, Tetrahedron, 2011, 67, 3140–3149 CrossRef.
- G.-m. Zhao, H.-I. Liu, D.-d. Zhang, X.-r. Huang and X. Yang, DFT Study on Mechanism of N-Alkylation of Amino Derivatives with Primary Alcohols Catalyzed by Copper(II) Acetate, ACS Catal., 2014, 4, 2231–2240 CrossRef CAS.
- F. Li, H. Shan, Q. Kang and L. Chen, Regioselective N-alkylation of 2-aminobenzothiazoles with benzylic alcohols, Chem. Commun., 2011, 47, 5058–5060 RSC.
- T. Miura, O. Kose, F. Li, S. Kai and S. Saito, CuI/H2/NaOH-Catalyzed Cross-Coupling of Two Different Alcohols for Carbon–Carbon Bond Formation: “Borrowing Hydrogen”?, Chem. – Eur. J., 2011, 17, 11146–11151 CrossRef CAS PubMed.
- G. Prakash, M. Nirmala, R. Ramachandran, P. Viswanathamurthi, J. G. Malecki and J. Sanmartin, Heteroleptic binuclear copper(I) complexes bearing bis(salicylidene)hydrazone ligands: Synthesis, crystal structure and application in catalytic N-alkylation of amines, Polyhedron, 2015, 89, 62–69 CrossRef CAS.
- Z. Xu, D.-S. Wang, X. Yu, Y. Yang and D. Wang, Tunable Triazole-Phosphine-Copper Catalysts for the Synthesis of 2-Aryl-1H-benzo[d]imidazoles from Benzyl Alcohols and Diamines by Acceptorless Dehydrogenation and Borrowing Hydrogen Reactions, Adv. Synth. Catal., 2017, 359, 3332–3340 CrossRef CAS.
- S. Waiba and B. Maji, Manganese Catalyzed Acceptorless Dehydrogenative Coupling Reactions, ChemCatChem, 2019, 12, 1891–1902 CrossRef.
- K. Azizi, S. Akrami and R. Madsen, Manganese(III) Porphyrin-Catalyzed Dehydrogenation of Alcohols to form Imines, Tertiary Amines and Quinolines, Chem. – Eur. J., 2019, 25, 6439–6446 CrossRef CAS PubMed.
- M. Subaramanian, P. M. Ramar, G. Sivakumar, R. G. Kadam, M. Petr, R. Zboril, M. B. Gawande and E. Balaraman, Convenient and Reusable Manganese-Based Nanocatalyst for Amination of Alcohols, ChemCatChem, 2021, 13, 4334–4341 CrossRef CAS.
- X. Yu, C. Liu, L. Jiang and Q. Xu, Manganese Dioxide Catalyzed N-Alkylation of Sulfonamides and Amines with Alcohols under Air, Org. Lett., 2011, 13, 6184–6187 CrossRef CAS PubMed.
- M. S. Ahmad, Y. Inomata and T. Kida, Heterogenized manganese catalyst for C-, and N-alkylation of ketones and amines with alcohols by pyrolysis of molecularly defined complexes, Mol. Catal., 2022, 526, 112390 CrossRef CAS.
- D. Wei, P. Yang, C. Yu, F. Zhao, Y. Wang and Z. Peng, N-Alkylation of Amines with Alcohols Catalyzed by Manganese(II) Chloride or Bromopentacarbonylmanganese(I), J. Org. Chem., 2021, 86, 2254–2263 CrossRef CAS PubMed.
- F. Kallmeier, R. Fertig, T. Irrgang and R. Kempe, Chromium-Catalyzed Alkylation of Amines by Alcohols, Angew. Chem., 2020, 132, 11887–11891 CrossRef.
- V. Sankar, M. Kathiresan, B. Sivakumar and S. Mannathan, Zinc-Catalyzed N-Alkylation of Aromatic Amines with Alcohols: A Ligand-Free Approach, Adv. Synth. Catal., 2020, 362, 4409–4414 CrossRef CAS.
- M. Peña-López, P. Piehl, S. Elangovan, H. Neumann and M. Beller, Manganese-Catalyzed Hydrogen-Autotransfer C-C Bond Formation: α-Alkylation of Ketones with Primary Alcohols, Angew. Chem., Int. Ed., 2016, 55, 14967–14971 CrossRef PubMed.
- S. Fu, Z. Shao, Y. Wang and Q. Liu, Manganese-Catalyzed Upgrading of Ethanol into 1-Butanol, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef CAS PubMed.
- N. V. Kulkarni, W. W. Brennessel and W. D. Jones, Catalytic Upgrading of Ethanol to n-Butanol via Manganese-Mediated Guerbet Reaction, ACS Catal., 2018, 8, 997–1002 CrossRef CAS.
- Y. Obora, C-Alkylation, by Hydrogen Autotransfer Reactions, Top. Curr. Chem., 2016, 374, 11 CrossRef PubMed.
- S. Chakraborty, P. Daw, Y. Ben-David and D. Milstein, Manganese-Catalyzed α-Alkylation of Ketones, Esters, and Amides Using Alcohols, ACS Catal., 2018, 8, 10300–10305 CrossRef CAS PubMed.
- J. C. Borghs, M. A. Tran, J. Sklyaruk, M. Rueping and O. El-Sepelgy, Sustainable Alkylation of Nitriles with Alcohols by Manganese Catalysis, J. Org. Chem., 2019, 84, 7927–7935 CrossRef CAS PubMed.
- J. C. Borghs, V. Zubar, L. M. Azofra, J. Sklyaruk and M. Rueping, Manganese-Catalyzed Regioselective Dehydrogenative C- versus N-Alkylation Enabled by a Solvent Switch: Experiment and Computation, Org. Lett., 2020, 22, 4222–4227 CrossRef CAS PubMed.
- J. R. Frost, C. B. Cheong, W. M. Akhtar, D. F. J. Caputo, N. G. Stevenson and T. J. Donohoe, Strategic Application and Transformation of ortho-Disubstituted Phenyl and Cyclopropyl Ketones To Expand the Scope of Hydrogen Borrowing Catalysis, J. Am. Chem. Soc., 2015, 137, 15664–15667 CrossRef CAS PubMed.
- W. M. Akhtar, C. B. Cheong, J. R. Frost, K. E. Christensen, N. G. Stevenson and T. J. Donohoe, Hydrogen Borrowing Catalysis with Secondary Alcohols: A New Route for the Generation of β-Branched Carbonyl Compounds, J. Am. Chem. Soc., 2017, 139, 2577–2580 CrossRef CAS PubMed.
- W. M. Akhtar, R. J. Armstrong, J. R. Frost, N. G. Stevenson and T. J. Donohoe, Stereoselective Synthesis of Cyclohexanes via an Iridium Catalyzed (5 + 1) Annulation Strategy, J. Am. Chem. Soc., 2018, 140, 11916–11920 CrossRef CAS PubMed.
- R. J. Armstrong, W. M. Akhtar, J. R. Frost, K. E. Christensen, N. G. Stevenson and T. J. Donohoe, Stereoselective synthesis of alicyclic ketones: A hydrogen borrowing approach, Tetrahedron, 2019, 75, 130680 CrossRef.
- W. M. Schubert and H. K. Latourette, The Aromatic Elimination Reaction. II. The Mechanism of the Acid-catalyzed Deacylation of Aromatic Ketones, J. Am. Chem. Soc., 1952, 74, 1829–1834 CrossRef CAS.
- A. Kaithal, L.-L. Gracia, C. Camp, E. A. Quadrelli and W. Leitner, Direct Synthesis of Cycloalkanes from Diols and Secondary Alcohols or Ketones Using a Homogeneous Manganese Catalyst, J. Am. Chem. Soc., 2019, 141, 17487–17492 CrossRef CAS PubMed.
- A. Jana, K. Das, A. Kundu, P. R. Thorve, D. Adhikari and B. Maji, A Phosphine-Free Manganese Catalyst Enables Stereoselective Synthesis of (1+n)-Membered Cycloalkanes from Methyl Ketones and 1,n-Diols, ACS Catal., 2020, 10, 2615–2626 CrossRef CAS.
- J. Sklyaruk, J. C. Borghs, O. El-Sepelgy and M. Rueping, Catalytic C1 Alkylation with Methanol and Isotope-Labeled Methanol, Angew. Chem., Int. Ed., 2019, 58, 775–779 CrossRef CAS PubMed.
- A. Bruneau-Voisine, L. Pallova, S. Bastin, V. César and J.-B. Sortais, Manganese catalyzed α-methylation of ketones with methanol as a C1 source, Chem. Commun., 2019, 55, 314–317 RSC.
- A. Kaithal, P. van Bonn, M. Hölscher and W. Leitner, Manganese(I)-Catalyzed β-Methylation of Alcohols Using Methanol as C1 Source, Angew. Chem., Int. Ed., 2020, 59, 215–220 CrossRef CAS PubMed.
- S. S. Gawali, B. K. Pandia, S. Pal and C. Gunanathan, Manganese(I)-Catalyzed Cross-Coupling of Ketones and Secondary Alcohols with Primary Alcohols, ACS Omega, 2019, 4, 10741–10754 CrossRef CAS PubMed.
- T. Liu, L. Wang, K. Wu and Z. Yu, Manganese-Catalyzed β-Alkylation of Secondary Alcohols with Primary Alcohols under Phosphine-Free Conditions, ACS Catal., 2018, 8, 7201–7207 CrossRef CAS.
- M. K. Barman, A. Jana and B. Maji, Phosphine-Free NNN-Manganese Complex Catalyzed α-Alkylation of Ketones with Primary Alcohols and Friedländer Quinoline Synthesis, Adv. Synth. Catal., 2018, 360, 3233–3238 CrossRef CAS.
- A. Jana, C. B. Reddy and B. Maji, Manganese Catalyzed α-Alkylation of Nitriles with Primary Alcohols, ACS Catal., 2018, 8, 9226–9231 Search PubMed.
- S. Waiba, S. K. Jana, A. Jati, A. Jana and B. Maji, Manganese complex-catalysed α-alkylation of ketones with secondary alcohols enables the synthesis of β-branched carbonyl compounds, Chem. Commun., 2020, 56, 8376–8379 Search PubMed.
- L. M. Kabadwal, J. Das and D. Banerjee, Mn(ii)-catalysed alkylation of methylene ketones with alcohols: direct access to functionalised branched products, Chem. Commun., 2018, 54, 14069–14072 RSC.
- X.-B. Lan, Z. Ye, M. Huang, J. Liu, Y. Liu and Z. Ke, Nonbifunctional Outer-Sphere Strategy Achieved Highly Active α-Alkylation of Ketones with Alcohols by N-Heterocyclic Carbene Manganese (NHC-Mn), Org. Lett., 2019, 21, 8065–8070 CrossRef CAS PubMed.
- X.-B. Lan, Z. Ye, J. Liu, M. Huang, Y. Shao, X. Cai, Y. Liu and Z. Ke, Sustainable and Selective Alkylation of Deactivated Secondary Alcohols to Ketones by Non-bifunctional Pincer N-heterocyclic Carbene Manganese, ChemSusChem, 2020, 13, 2557–2563 CrossRef CAS PubMed.
- J. Yang, X. Liu, D.-L. Meng, H.-Y. Chen, Z.-H. Zong, T.-T. Feng and K. Sun, Efficient Iron-Catalyzed Direct β-Alkylation of Secondary Alcohols with Primary Alcohols, Adv. Synth. Catal., 2012, 354, 328–334 CrossRef CAS.
- A. Quintard, T. Constantieux and J. Rodriguez, An Iron/Amine-Catalyzed Cascade Process for the Enantioselective Functionalization of Allylic Alcohols, Angew. Chem., Int. Ed., 2013, 52, 12883–12887 CrossRef CAS PubMed.
- A. Quintard, M. Roudier and J. Rodriguez, Multicatalytic Enantioselective Borrowing Hydrogen δ-Lactonization Strategy from β-Keto Esters and Allylic Alcohols, Synthesis, 2018, 785–792 CrossRef CAS.
- M. Roudier, T. Constantieux, A. Quintard and J. Rodriguez, Enantioselective Cascade Formal Reductive Insertion of Allylic Alcohols into the C(O)–C Bond of 1,3-Diketones: Ready Access to Synthetically Valuable 3-Alkylpentanol Units, Org. Lett., 2014, 16, 2802–2805 CrossRef CAS PubMed.
- M. Roudier, T. Constantieux, A. Quintard and J. Rodriguez, Triple Iron/Copper/Iminium Activation for the Efficient Redox Neutral Catalytic Enantioselective Functionalization of Allylic Alcohols, ACS Catal., 2016, 6, 5236–5244 CrossRef CAS.
- S. Elangovan, J.-B. Sortais, M. Beller and C. Darcel, Iron-Catalyzed α-Alkylation of Ketones with Alcohols, Angew. Chem., Int. Ed., 2015, 54, 14483–14486 CrossRef CAS PubMed.
- K. Polidano, B. D. W. Allen, J. M. J. Williams and L. C. Morrill, Iron-Catalyzed Methylation Using the Borrowing Hydrogen Approach, ACS Catal., 2018, 8, 6440–6445 CrossRef CAS.
- C.-P. Xu, Z.-H. Xiao, B.-Q. Zhuo, Y.-H. Wang and P.-Q. Huang, Efficient and chemoselective alkylation of amines/amino acids using alcohols as alkylating reagents under mild conditions, Chem. Commun., 2010, 46, 7834–7836 Search PubMed.
- R. V. Sankar, D. Manikpuri and C. Gunanathan, Ruthenium-catalysed α-prenylation of ketones using prenol, Org. Biomol. Chem., 2023, 21, 273–278 Search PubMed.
- P. Chakraborty, S. Pradhan, J. R. Premkumar and B. Sundararaju, Valorization of terpenols under iron catalysis, J. Catal., 2023, 421, 309–318 Search PubMed.
- G. Di Gregorio, M. Mari, F. Bartoccini and G. Piersanti, Iron-Catalyzed Direct C3-Benzylation of Indoles with Benzyl Alcohols through Borrowing Hydrogen, J. Org. Chem., 2017, 82, 8769–8775 Search PubMed.
- J. J. Ibrahim, C. B. Reddy, S. Zhang and Y. Yang, Ligand-Free FeCl2-Catalyzed α-Alkylation of Ketones with Alcohols, Asian J. Org. Chem., 2019, 8, 1858–1861 Search PubMed.
- A. Alanthadka, S. Bera and D. Banerjee, Iron-Catalyzed Ligand Free α-Alkylation of Methylene Ketones and β-Alkylation of Secondary Alcohols Using Primary Alcohols, J. Org. Chem., 2019, 84, 11676–11686 CrossRef CAS PubMed.
- L. Bettoni, N. Joly, I. Mendas, M. M. Moscogiuri, J.-F. Lohier, S. Gaillard, A. Poater and J.-L. Renaud, Iron-Catalyzed Synthesis of Substituted 3-Arylquinolin-2(1H)-ones via an Intramolecular Dehydrogenative Coupling of Amido-Alcohols, Org. Biomol. Chem., 2024, 22, 6933–6940 RSC.
- N. Deibl and R. Kempe, General and Mild Cobalt-Catalyzed C-Alkylation of Unactivated Amides and Esters with Alcohols, J. Am. Chem. Soc., 2016, 138, 10786–10789 CrossRef CAS PubMed.
- F. Freitag, T. Irrgang and R. Kempe, Cobalt-Catalyzed Alkylation of Secondary Alcohols with Primary Alcohols via Borrowing Hydrogen/Hydrogen Autotransfer, Chem. – Eur. J., 2017, 23, 12110–12113 Search PubMed.
- G. Zhang, J. Wu, H. Zeng, S. Zhang, Z. Yin and S. Zheng, Cobalt-Catalyzed α-Alkylation of Ketones with Primary Alcohols, Org. Lett., 2017, 19, 1080–1083 CrossRef CAS PubMed.
- S.-Q. Zhang, B. Guo, Z. Xu, H.-X. Li, H.-Y. Li and J.-P. Lang, Ligand-controlled phosphine-free Co(II)-catalysed cross-coupling of secondary and primary alcohols, Tetrahedron, 2019, 75, 130640 CrossRef.
- Z. Liu, Z. Yang, X. Yu, H. Zhang, B. Yu, Y. Zhao and Z. Liu, Methylation of C(sp3)-H/C(sp2)-H Bonds with Methanol Catalyzed by Cobalt System, Org. Lett., 2017, 19, 5228–5231 CrossRef CAS PubMed.
- B. Pandey, S. Xu and K. Ding, Selective Ketone Formations via Cobalt-Catalyzed β-Alkylation of Secondary Alcohols with Primary Alcohols, Org. Lett., 2019, 21, 7420–7423 CrossRef CAS PubMed.
- B. Pandey, S. Xu and K. Ding, Switchable β-alkylation of Secondary Alcohols with Primary Alcohols by a Well-Defined Cobalt Catalyst, Organometallics, 2021, 40, 1207–1212 CrossRef CAS.
- K. Paudel, S. Xu and K. Ding, α-Alkylation of Nitriles with Primary Alcohols by a Well-Defined Molecular Cobalt Catalyst, J. Org. Chem., 2020, 85, 14980–14988 CrossRef CAS PubMed.
- P. Chakraborty, M. K. Gangwar, B. Emayavaramban, E. Manoury, R. Poli and B. Sundararaju, α-Alkylation of Ketones with Secondary Alcohols Catalyzed by Well-Defined Cp*CoIII-Complexes, ChemSusChem, 2019, 12, 3463–3467 CrossRef CAS PubMed.
- P. Chakraborty, N. Garg, E. Manoury, R. Poli and B. Sundararaju, C-Alkylation of Various Carbonucleophiles with Secondary Alcohols under CoIII-Catalysis, ACS Catal., 2020, 10, 8023–8031 CrossRef CAS.
- P. Biswal, S. Samser, P. Nayak, V. Chandrasekhar and K. Venkatasubbaiah, Cobalt(II) porphyrin-mediated selective synthesis of 1,5-diketones via an interrupted-borrowing hydrogen strategy using methanol as a C1 source, J. Org. Chem., 2021, 86, 6744–6754 CrossRef CAS PubMed.
- J. Das, K. Singh, M. Vellakkaran and D. Banerjee, Nickel-Catalyzed Hydrogen-Borrowing Strategy for α-Alkylation of Ketones with Alcohols: A New Route to Branched gem-Bis(alkyl) Ketones, Org. Lett., 2018, 20, 5587–5591 CrossRef CAS PubMed.
- J. Das, M. Vellakkaran and D. Banerjee, Nickel-catalysed alkylation of C(sp3)–H bonds with alcohols: direct access to functionalised N-heteroaromatics, J. Org. Chem., 2019, 84, 769–779 CrossRef CAS PubMed.
- S. Bera, A. Bera and B. Banerjee, Nickel-catalyzed hydrogen-borrowing strategy: chemo-selective alkylation of nitriles with alcohols, Chem. Commun., 2020, 56, 6850–6853 RSC.
- R. Babu, M. Subaramanian, S. P. Midya and E. Balaraman, Nickel-Catalyzed Guerbet Type Reaction: C-Alkylation of Secondary Alcohols via Double (de)Hydrogenation, Org. Lett., 2021, 23, 3320–3325 CrossRef CAS PubMed.
- D. Wu, Y. Wang, M. Li, L. Shi, J. Liu and N. Liu, Nickel-catalyzed α-alkylation of ketones with benzyl alcohols, Appl. Organomet. Chem., 2022, 36, e6493 Search PubMed.
- N.-K. Nguyen, D. H. Nam, P. V. Phuc, V. H. Nguyen, Q. T. Trịnh, T. Q. Hung and T. T. Dang, Efficient copper-catalyzed synthesis of C3-alkylated indoles from indoles and alcohols, Mol. Catal., 2021, 505, 111462 CrossRef CAS.
- N. S. Lawal, H. Ibrahim and M. D. Bala, Cu(I) mediated hydrogen borrowing strategy for the α-alkylation of aryl ketones with aryl alcohols, Monatsh. Chem., 2021, 152, 275–285 CrossRef CAS.
- Y. T. Chen, Recent advances in methylation: a guide for selecting methylation reagents, Chem. – Eur. J., 2019, 25, 3405–3439 CrossRef CAS PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- B. Emayavaramban, P. Chakraborty, P. Dahiya and B. Sundararaju, Iron-Catalyzed α–Methylation of Ketones Using Methanol as the C1 Source under Photoirradiation, Org. Lett., 2022, 24, 6219–6223 CrossRef CAS PubMed.
- M. Waheed, M. A. Alsharif, M. I. Alahmdi, S. Mukhtar and H. Parveen, Visible Light Promoted Iron-Catalyzed One-Pot Synthesis of 2-Arylimino-2H-Chromenes from 2-Hydroxybenzyl Alcohols and β- Ketothioamides at Room Temperature, Eur. J. Org. Chem., 2023, e202300136 CrossRef CAS.
- M. Waheed, M. A. Alsharif, M. I. Alahmdi, S. Mukhtar and H. Parveen, Visible Light Promoted Iron-Catalyzed One-Pot Synthesis of 2-Arylimino-2H-Chromenes from 2-Hydroxybenzyl Alcohols and β- Ketothioamides at Room Temperature, Tetrahedron Lett., 2023, 119, 154428 Search PubMed.
- M.-S. Abdallah, N. Joly, S. Gaillard, A. Poater and J.-L. Renaud, Blue-Light Induced Iron-Catalyzed α–Alkylation of Ketone, Org. Lett., 2022, 24, 5584–5589 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Sylvain
Gaillard
ab,
Sylvain
Gaillard