
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of boron desymmetrization on supramolecular polymerization of BODIPY dyes†
Tobias B.
Tischer
a,
Zulema
Fernández
a,
Lorenz
Borsdorf
a,
Constantin G.
Daniliuc
a,
Shigehiro
Yamaguchi
 bc,
Soichiro
Ogi
*b and
Gustavo
Fernández
*a
bc,
Soichiro
Ogi
*b and
Gustavo
Fernández
*a
aUniversität Münster, Organisch-Chemisches Institut, Corrensstraße 36, 48149 Münster, Germany. E-mail: fernandg@uni-muenster.de
bDepartment of Chemistry, Graduate School of Science, and Integrated Research Consortium on Chemical Science (IRCCS), Nagoya University, Furo, Chikusa, Nagoya 464-8602, Japan. E-mail: ogi.soichiro@chem.nagoya-u.ac.jp
cInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Furo, Chikusa, Nagoya 464-8601, Japan
First published on 4th November 2024
Abstract
Supramolecular polymers are often investigated for highly symmetric and planar molecules, such as typically explored BF2-substituted BODIPY dyes. However, it is surprising that the possibility of desymmetrizing the sp3 hybridized boron centre of BODIPY dyes has remained unexplored in the context of supramolecular polymerization. Herein, we synthesized a new BODIPY derivative 2 with two different substituents at the boron (fluorine and phenyl), resulting in a system with two different π-surfaces, and analyzed its supramolecular polymerization in non-polar media. Notably, this symmetry reduction increases the complexity of the self-assembly by enabling the formation of an intermediate assembled state, which cannot be found in the symmetrical model BODIPY 1 with a BF2 group. Different experimental and theoretical studies suggest that significant steric effects together with multiple potential intermolecular stacking modes of the BODIPY dyes lead to discrete nanoparticle intermediates that ultimately transform into more-ordered H-type supramolecular polymers at lower temperatures. Our results introduce a new design strategy for controlled supramolecular polymerization.
Introduction
The controlled self-assembly of π-conjugated molecules has received considerable interest in recent years.1–4 The rational design of chromophores has enabled the development of supramolecular functional materials with interesting properties like controlled morphologies,5–9 favourable luminescence10,11 or biomedical applications.12–14 The main driving force towards controlled supramolecular polymers are non-covalent interactions, including π–π interactions,15–17 hydrogen bonds18,19 or ionic interactions.20–22 Due to the planar character of the π-bond, the design of new molecules that are suitable for self-assembly is usually limited to 1D or 2D functionalization of the chromophores.23–27 However, the 2D character of chromophores can be overcome and expanded into the third dimension by rational molecular design, e.g. in helicenes.28,29 Due to the repulsive steric interactions of their terminal rings, these molecules build screw-like structures that go along with unique self-assembly features.30–32 Furthermore, distorted π-surfaces are also observed for molecules such as bay-substituted perylene bisimide dyes, leading to planar chirality that give access to the emission of circularly polarized light.17,33Recently, we reported that also the slight out-of-plane distortion of the π-surface of a bis(difluoroboron)-1,2-bis((1H-pyrrol-2-yl)methylene)hydrazine (BOPHY) derivative leads to increased complexity in the supramolecular polymerization when compared to a structurally related but planar boron dipyrromethene (BODIPY) derivative.34,35 Although the oppositely oriented BF2 groups of the BOPHY core lead to a slight planarity breaking, both sides of the π-surface are identical in terms of steric features. This is due to the symmetry resulting from the sp2 hybridized carbon atoms that are included in the chromophores. To go beyond these systems, we aimed at designing self-assembling building blocks where the π-surface can be unsymmetrically functionalized orthogonal to the π-plane to yield a Janus-like π-surface. To address this goal, we exploited the planar BODIPY chromophore, as the sp3 hybridized boron atom allows the selective substitution of one of the fluorine atoms, thus enabling desymmetrization in the third dimension.
Starting from our symmetrical model BODIPY 1, we reduced the symmetry by removing the mirror plane aligned with the π-surface of the chromophore. This was achieved by replacement of a fluorine atom with a phenyl substituent at the boron atom to yield BODIPY derivative 2 (Scheme 1, for synthetic details see Fig. S1 in the ESI†). This molecular design enables a sterically more hindered surface on the phenyl-substituted side (P-surface) and a sterically less hindered side (F-surface, Scheme 1), which is expected to strongly influence the molecular stacking and self-assembly behaviour.
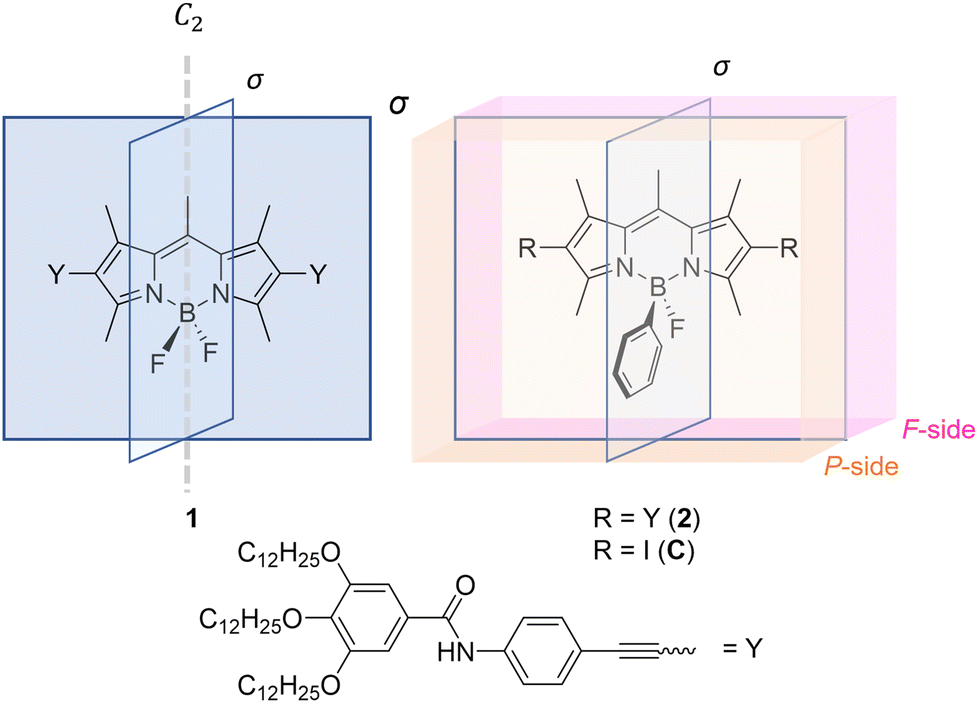 | ||
| Scheme 1 Molecular structures of the model BODIPY derivative 1 with, C2-symmetry, its unsymmetrical analogue 2 and reference compound C. | ||
Results and discussion
Spectroscopic characterization and self-assembly of BODIPY derivative 2
BODIPY dyes are well-known to self-assemble in solution into ordered nanostructures through various stacking modes.36–38 The optical properties and self-assembly behaviour of BODIPY derivative 1 were already well discussed and investigated in previous reports.34,391 was found to form a single type of aggregate (one-dimensional H-type) with an antiparallel and slightly rotationally displaced packing of the chromophores that follows the cooperative mechanism. Diverse kinetic studies showed that the system is very robust and unaffected by various sample treatments (e.g. mechanical agitation, solvophobic quenching, etc.). Due to the high reproducibility and in-depth comprehension of this system, we selected it as a reference compound to understand the self-assembly behaviour of the structurally related BODIPY derivative 2. To gain first insights into the properties of 2, we performed solvent-dependent UV-vis and fluorescence measurements (Fig. 1). In solvents favouring the monomer state like CHCl3 and 1,4-dioxane, the absorption maximum, referring to the S0 → S1 transition, is located at λ = 568 and 564 nm, respectively (Fig. 1, ε = 8 × 104 L mol−1 cm−1, c = 10 µM, 298 K). A band that corresponds to the S0 → S2 transition is also visible at lower wavelength (λ ≈ 400 nm). The emission maximum of 2 in these solvents is found at 609 nm, resulting in a Stokes shift of 41 nm in CHCl3 and 45 nm in 1,4-dioxane, which is similar to the spectroscopic properties of 1 in the same solvents. However, in non-polar media under the same conditions (c = 10 µM, 298 K), the absorption maximum of 2 shifts to 540 nm in n-hexane and 545 nm in methylcyclohexane (MCH), accompanied by a reduction in the absorbance and the concomitant emergence of a red-shifted shoulder at 616 nm. These spectral features resemble those of 1 and agree with the formation of H-type aggregates with a rotationally displaced packing.34 This is further supported by the concomitant quenching and red-shift of the emission maximum in both non-polar solvents (MCH: λmax. = 613 nm, λstokes shift = 68 nm, n-hexane: λmax. = 638 nm, λstokes shift = 98 nm) compared to the solvents favouring the monomer state (Fig. 1d). Interestingly, the spectral pattern of the H-type aggregates of 2 is similar to that of model compound 1 (Fig. 1a and c), indicating comparable intermolecular interactions and packing. However, the degree of aggregation for 2 in MCH seems to be less pronounced compared to 1 under similar conditions, as the spectrum of 2 shows a pronounced shoulder at 568 nm that is most probably related with the presence of residual monomers (Fig. 1b). A plausible reason for this difference could be the increased steric hindrance of 2 that is introduced by the phenyl group at the boron atom.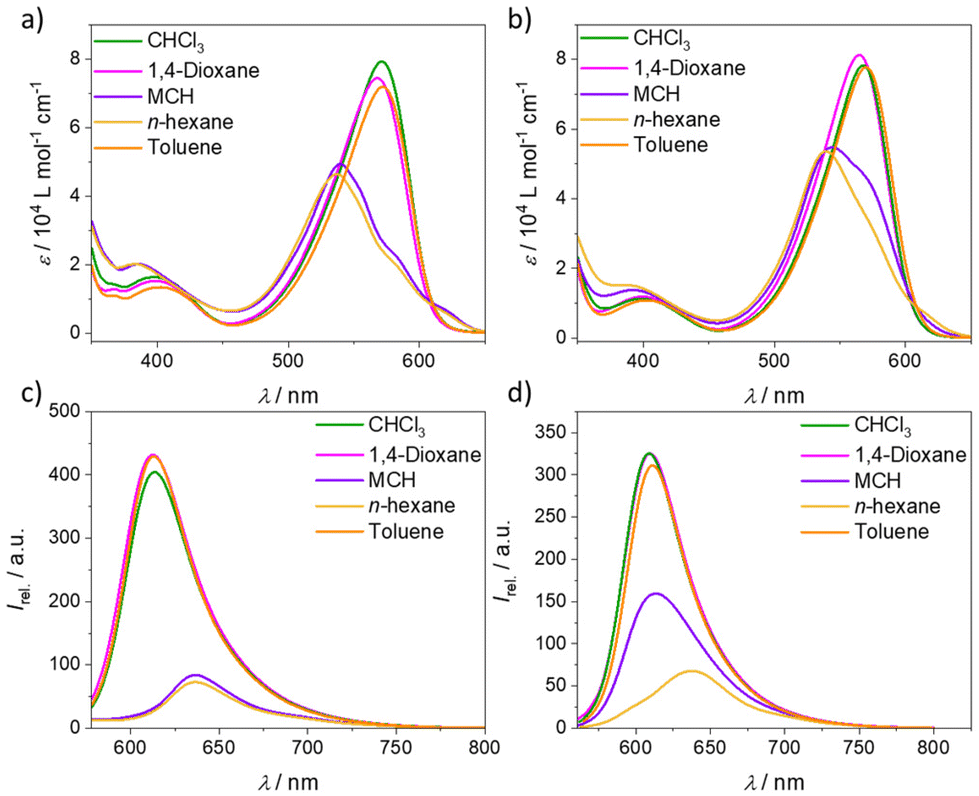 | ||
| Fig. 1 Solvent-dependent UV-vis and emission studies (λexc. = λmax abs.) of 1 (a and c) and 2 (b and d) at c = 10 µM at room temperature. | ||
To gain further insights into the self-assembly process, variable temperature (VT)-UV-vis measurements were performed. Upon cooling a solution of 2 (c = 10 µM, 1 K min−1, MCH) from 363 K to 263 K, the absorption maximum shifts from 572 nm, corresponding to the monomeric species (2-M, red spectrum, Fig. 2a), to 538 nm, which can be associated with the H-type aggregate (2-H, blue spectrum, Fig. 2a). Furthermore, the red-shifted band at 616 nm that was previously observed in the solvent-dependent studies becomes apparent at lower temperatures (Fig. 2a). The H-type aggregate formation is independent of the cooling rate, indicating that the process is under thermodynamic control40–42 (Fig. S8†). Compared to 1 at the same concentration, the onset of the aggregation upon cooling starts at slightly lower temperatures. This effect results most probably from the increased steric hindrance that is introduced on the boron atom when a fluorine atom in 1 is replaced with a phenyl substituent in 2 (Fig. S9†). Upon heating aggregate 2-H (1 K min−1, MCH, blue spectrum, Fig. 2b), the monomer is recovered at 363 K. However, by comparing the degree of aggregation (αagg) of the cooling and heating processes at different temperatures (Fig. 2c), differences in the curve shape and a slight hysteresis are observed. A more detailed analysis of the cooling experiment shows that the absorption maximum of the monomer species of 2 shifts to shorter wavelengths at intermediate temperatures (298 K, green spectrum, Fig. 2a) without the appearance of a red-shifted shoulder at 616 nm. Furthermore, the bands at shorter wavelength (326 nm) corresponding to the absorption of the substituents at the 2- and 6-positions conjugated to the BODIPY core (Y in Scheme 1) remain unchanged. Only below a critical temperature (298 K, green spectrum, Fig. 2a) the red-shifted band appears rapidly (Fig. S10†) and the bands at short wavelength start to shift and split. These findings suggest the occurrence of an intermediate state (2-I, λmax = 546 nm) that transforms into the final H-type aggregate (2-H) after surpassing a concentration-dependent critical temperature (Fig. S10†). The shift from the monomeric species towards 2-I occurs without a clear isosbestic point, while the transformation from 2-I to 2-H exhibits an isosbestic point at 542 nm. The two-step transformation from 2-M to 2-Hvia2-I can be visualized rather well when the degree of aggregation is plotted e.g. at the isosbestic point between the intermediate and the aggregate (542 nm, Fig. 2d). At high temperatures, when mainly monomers are present, the degree of aggregation remains nearly zero between 363 K and ca. 336 K. Below this temperature (336 K), the formation of 2-I sets in, as indicated by the increase of αagg until a plateau is reached at around 298 K. Below this temperature, the proportion of 2-H increases but no change in αagg is observed due to the characteristics of the isosbestic point. This analysis was carried out for different concentrations to obtain a phase diagram (Fig. 2e and Fig. S11†) that reveals the general trends of the aggregation process. Increasing the concentration results in an increased onset temperature for the formation of 2-I as well as for 2-H.
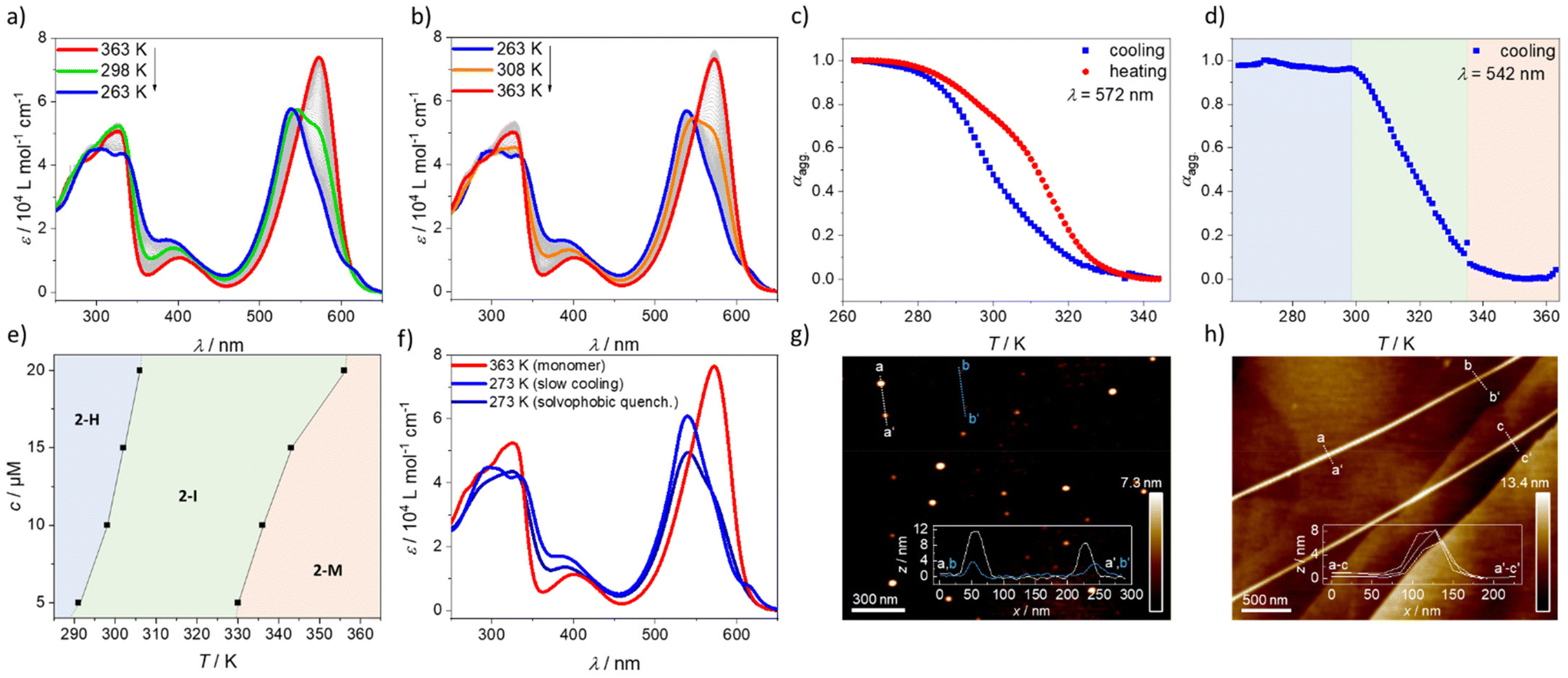 | ||
| Fig. 2 UV-vis cooling (a) and heating (b) experiments of 2 (MCH, 10 µM, 1 K min−1) with the corresponding cooling and heating curves, plotted at λ = 572 nm (c). (d) Cooling curve of 2 plotted at λ = 542 nm to demonstrate the two-step process (MCH, 10 µM, 1 K min−1). (e) Phase diagram extracted from cooling experiments of 2 at different concentrations. (f) UV-Vis experiments depicting the solvophobic quenching of 2 (10 µM, MCH/CHCl3). (g) AFM image of 2-I after spin-coating a 20 µM solution of 2 (306 K, MCH) on HOPG and (h) AFM image of 2-H after spin-coating a 20 µM solution of 2 (273 K, MCH) on HOPG. | ||
In order to understand the disassembly of the aggregate, corresponding heating experiments were carried out (Fig. 2b). The studies reveal that the disassembly of 2-H to the monomeric species occurs more gradually than in the cooling experiments. Nevertheless, an intermediate species 2-I* with an absorption maximum at λmax = 546 nm (similar to 2-I) can be observed. However, in stark contrast to 2-I, the red-shifted shoulder at 616 nm as well as the bands at around 300 nm resemble the spectral features of the aggregate, rather than those of the monomer. We assume that the cooperative effect in the transformation from 2-I to 2-H in the cooling process (Fig. S10a†) is caused by additional interactions of the side groups, most probably hydrogen bonds. This stabilization process does not allow to revert the intermediate 2-I in the heating process but enables the formation of another intermediate species with interactions between the side groups. This results in a slightly more disturbed disassembly process, requiring higher temperatures.
Interestingly, the fact that reference compound 1 does not form any intermediate species (Fig. S12†) demonstrates that the replacement of the fluorine atom in 1 with a phenyl group in 2 has a direct consequence on the observed self-assembly behaviour. We propose that repulsive steric interactions involving the phenyl group at the boron atom initially hinder close contacts of the BODIPY chromophores, resulting in species 2-I with weak exciton coupling. Below a certain temperature, cooperative effects come into play and overcome the repulsive steric interactions, leading to the formation of the final aggregate 2-H. To investigate the stability of 2-I, time-dependent studies were performed. Unexpectedly, species 2-I did not transform into aggregate 2-H over several hours, demonstrating its thermodynamic stability (Fig. S13†). Therefore, we assume that 2-I is a kinetically trapped off-pathway aggregate that forms via an anti-cooperative mechanism, analogous to previous literature reports.43 Only below a certain critical temperature, the formation of 2-H occurs via a cooperative pathway, indicated by the rapidly rising band at 616 nm (Fig. S10a†). This transformation could be fitted well to the nucleation–elongation model to obtain the thermodynamic parameters (Fig. S10a and Table S1†).44 However, these data need to be taken with caution, as the process involves additional intermediate species that are not considered in the model. The anti-cooperative formation of 2-I would explain the high stability of this species. Due to the favoured nucleation,2 the monomer concentration is reduced, hindering the transformation to the thermodynamic aggregate via the monomer. Unfortunately, the curves describing the transformation from the monomer to 2-I cannot be fitted to any suitable thermodynamic model, as this process cannot be isolated from the transformation to 2-H at any wavelength (Fig. S10b†).
To prove the assumption of two competitive pathways, we investigated the morphology of 2 in the intermediate state and compared it to that of the final aggregate state 2-H. To this end, we prepared solutions of 2-I (MCH, 20 µM, 310 K) and 2-H (MCH, 20 µM, 273 K), spin-coated them on highly ordered pyrolytic graphite (HOPG) and analysed the samples through atomic force microscopy (AFM). 2-I formed small particles with a uniform size of ca. 4 to 12 nm (Fig. 2g and Fig. S20†). According to the size, this suggests the formation of discrete assemblies comprising a limited number of molecules. The aggregate 2-H, however, shows elongated fibres (Fig. 2h and Fig. S21†) with a uniform width of 8 nm and several micrometres in length. These results corroborate the assumed pathway complexity, as the small particles of 2-I suggest an anti-cooperative mechanism and the elongated structures of 2-H are typical for aggregates that formed via a cooperative mechanism. Furthermore, the formation of well-structured, fibre-like H-type aggregates points out the drastic consequences of small structural changes in a molecular design, as they can lead to distinct pathways with different morphology. Despite 1 and 2 having similar spectral patterns for their respective aggregates, 1 was reported to form short worm-like structures,39 while 2 self-assembles into much more extended fibres.
The increased steric hindrance of 2 compared to 1 prompted us to perform quenching experiments to rationalize whether other kinetic species may potentially be involved in the self-assembly process. Therefore, thermal (Fig. S14†) and solvophobic quenching experiments (Fig. 2f) were performed. For the solvophobic quenching, a monomeric solution of 2 (CHCl3, c = 200 µM, 273 K, 100 µL) was prepared and rapidly diluted with cold MCH (273 K, 3900 µL) to gain a 10 µM solution that was subsequently tracked by UV-vis spectroscopy. These experiments demonstrate that the spectral features of the quenched solution resemble those of 2-H that were observed in the cooling experiments (λmax. = 540 nm). However, the formation of the typical red-shifted shoulder of 2-H around 616 nm cannot be observed, indicating a similar but less ordered self-assembly compared to 2-H. Nevertheless, this solution transforms over time into 2-H, as evident by the emergence of the shoulder at 616 nm (Fig. S14a†). Similar observations were made for the thermally quenched solution (MCH, 10 µM, 273 K, Fig. S14b and c†). In contrast to the findings for 2, compound 1 does not show any significant differences in the spectral features upon thermal and solvophobic quenching (Fig. S15†), once again highlighting that the additional phenyl group of 2 plays an important role in the observed behaviour.
Analysis of intermolecular interactions
Subsequently, we aimed at examining the packing of the molecules and the intermolecular interactions in the different self-assembled states. For this purpose, we rationalized potential aggregate formations, carried out different Fourier transform-infrared (FT-IR) and NMR experiments and performed density functional theory (DFT) calculations.The decreased symmetry of the π-surface of 2 makes it challenging to predict the relative arrangement of the molecules upon aggregation. Commonly studied BODIPY dyes with a BF2 group, such as 1, are well known to stack in an antiparallel fashion both in solution and the solid state, where both sides of the π-surface have similar chemical environments due to the C2-symmetry of these molecules45,46 (Fig. 3a). However, this scenario changes when the C2-symmetry is disrupted and two different π-surfaces emerge, as it is the case for 2 (the P- and the F-side, Scheme 1). On this basis, three different kinds of dimer stacks are conceivable (Fig. 3b and c): either both phenyl substituents (P/P) or both fluorine atoms (F/F) are on the same interacting side of the molecules, or they are on different sides (F/P). These three types of dimers may theoretically be able to combine in different ways to build larger aggregates, including potentially unordered structures. To represent the three different packing options, we crystallized the precursor molecule C (Fig. 3b, Fig. S22† and Scheme 1), which features a fluorine and a phenyl substituent on the boron, and iodine atoms instead of substituents Y at the 2- and 6-positions, from a dichloromethane solution. Intriguingly, in the crystal structure of C, two different types of dimers that could potentially form in a nucleation process for supramolecular polymerization can be observed: the sterically least favoured one (P/P, Fig. 3b, blue frame) along with the sterically most favoured (F/F, Fig. 3b, red frame). The last potential dimer packing that can occur (F/P) would lie in between the P/P and F/F in terms of steric effects. Although this packing is not found in the crystal structure, it likely exists in solution given that it is sterically more favoured than the P/P packing (Fig. 3c).
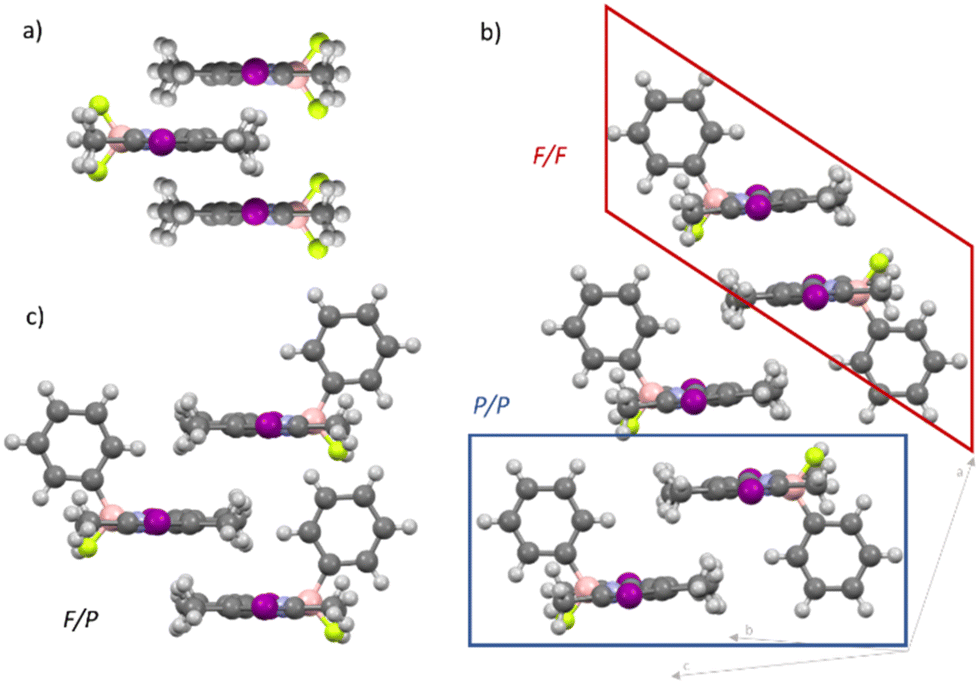 | ||
| Fig. 3 (a) Cartoon representation of the typical antiparallel stacking of a C2-symmetrical BODIPY derivative. (b) X-ray crystal structure of C that consists of F/F type (red frame) and P/P type (blue frame) dimers. (c) Cartoon representation of C, representing a potential F/P type stacking of the molecules. | ||
To examine the intermolecular interactions, we studied the different species 2-H and 2-I with different NMR and FT-IR techniques. For the investigation of the aggregate 2-H, we first performed VT-1H-NMR experiments (MCH-d14, 1 mM, Fig. 4c). The formation of 2-H is accompanied by a high-field shift of the methyl protons of the BODIPY dye at the meso (violet) and at the 3- and 5-positions (light green), indicating the formation of π–π interactions. The methyl groups at the 1- and 7-positions (light blue) show only weak shifts, which can be explained by a merely weak participation in intermolecular interactions. Furthermore, a low-field shift of the amide protons (dark blue) is observed upon cooling. This can be associated with the formation of hydrogen bonds. At lower temperatures, the signals broaden and finally vanish due to the ongoing aggregation. To gain further information about the participation of hydrogen bonds, we performed FT-IR measurements in CHCl3 and MCH to compare the monomeric with the aggregated state (Fig. 4a and b). We found, that the carbonyl stretching shifts from νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O = 1676 cm−1 in CHCl3 to νCO = 1644 cm−1 in MCH, demonstrating a change in the chemical environment. Likewise, the N–H vibration shifts to lower wavenumbers when moving from CHCl3 (νN–H = 3431 cm−1) to MCH (νN–H = 3280 cm−1). These results are in good accordance with the formation of amide-amide N–H⋯OC hydrogen bonds.40,47 Interestingly, by comparing these results with reference BODIPY 1, we found that the CO and N–H stretching bands of 2-H are shifted to lower wavenumbers compared to the aggregate of 1 (ΔνN–H(MCH) = 40 cm−1, ΔνCO(MCH) = 13 cm−1), which can be traced back to stronger hydrogen bonds in 2-H despite an increased steric hindrance in 2. 2D-ROESY- and HOESY-NMR studies were carried out at elevated temperatures to obtain better resolution of the signals in the aggregated state (Fig. 4d). In the 1H–1H-ROESY-NMR, a clear correlation was found between the meso methyl group and the methyl groups at the 3- and 5-positions from the BODIPY, indicating an antiparallel and slightly offset packing of the BODIPY dyes. This kind of interaction was already expected from the low-field shift of the corresponding proton signals from VT-1H-NMR experiments. The antiparallel packing is supported by correlations between the methyl groups at the 1- and 7-positions and the phenyl substituent (Fig. S17a†) at the boron position as well as with the fluorine atom, visible in the 1H–19F-HOESY spectrum (Fig. 4e). Also, the appearance of two signals for the alkoxy OCH2 methylene signal (orange) with a 2
O = 1676 cm−1 in CHCl3 to νCO = 1644 cm−1 in MCH, demonstrating a change in the chemical environment. Likewise, the N–H vibration shifts to lower wavenumbers when moving from CHCl3 (νN–H = 3431 cm−1) to MCH (νN–H = 3280 cm−1). These results are in good accordance with the formation of amide-amide N–H⋯OC hydrogen bonds.40,47 Interestingly, by comparing these results with reference BODIPY 1, we found that the CO and N–H stretching bands of 2-H are shifted to lower wavenumbers compared to the aggregate of 1 (ΔνN–H(MCH) = 40 cm−1, ΔνCO(MCH) = 13 cm−1), which can be traced back to stronger hydrogen bonds in 2-H despite an increased steric hindrance in 2. 2D-ROESY- and HOESY-NMR studies were carried out at elevated temperatures to obtain better resolution of the signals in the aggregated state (Fig. 4d). In the 1H–1H-ROESY-NMR, a clear correlation was found between the meso methyl group and the methyl groups at the 3- and 5-positions from the BODIPY, indicating an antiparallel and slightly offset packing of the BODIPY dyes. This kind of interaction was already expected from the low-field shift of the corresponding proton signals from VT-1H-NMR experiments. The antiparallel packing is supported by correlations between the methyl groups at the 1- and 7-positions and the phenyl substituent (Fig. S17a†) at the boron position as well as with the fluorine atom, visible in the 1H–19F-HOESY spectrum (Fig. 4e). Also, the appearance of two signals for the alkoxy OCH2 methylene signal (orange) with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in the VT-NMR experiments can be explained by ROESY-NMR. Here, a coupling of four out of six alkoxy methylene groups of the chains with the aromatic protons of the terminal phenyl ring (black) is observed, indicating a slight translational offset of the molecules (cartoon in Fig. 4f and Fig. S17b†).
:1 ratio in the VT-NMR experiments can be explained by ROESY-NMR. Here, a coupling of four out of six alkoxy methylene groups of the chains with the aromatic protons of the terminal phenyl ring (black) is observed, indicating a slight translational offset of the molecules (cartoon in Fig. 4f and Fig. S17b†).
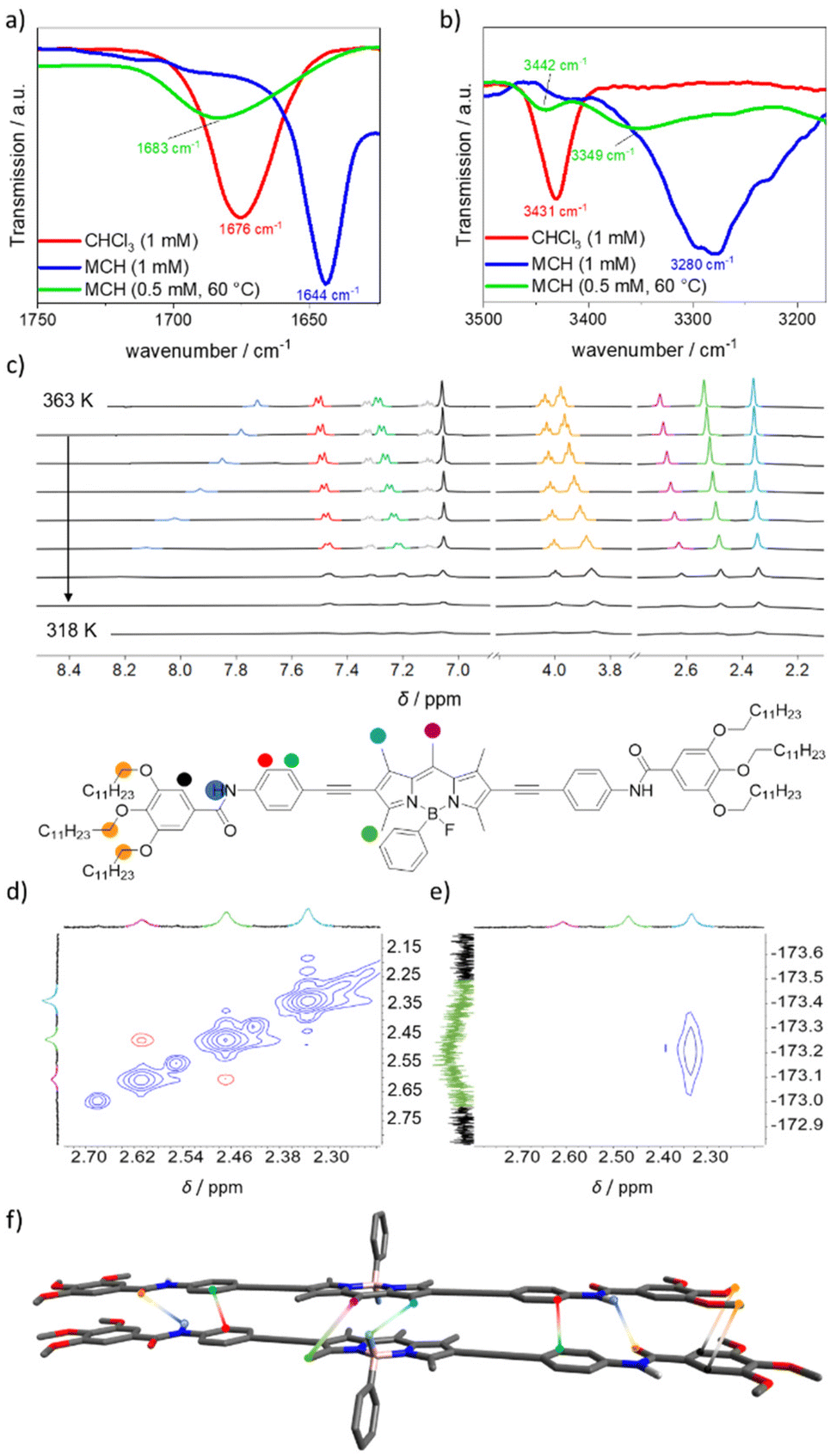 | ||
| Fig. 4 FT-IR spectra of 2 in different solvents and at different concentrations showing the CO (a) and N–H (b) stretching frequencies. (c) Temperature-dependent 1H-NMR experiments of 2 (MCH-d14, 1 mM, ΔT = 5 K). (d) 1H–1H-ROESY- and (e) 1H–19F-HOESY-NMR studies of 2 at 333 K (1 mM, MCH-d14) showing the slightly shifted and antiparallel BODIPY interactions, illustrated in (f). | ||
For the investigation of 2-I, the experimental conditions had to be optimized to identify the intermediate species. Hence, we carried out NMR and FT-IR experiments at a concentration of 0.5 mM, as it allowed us to correlate our results with UV-vis experiments. VT-UV-vis experiments revealed that 2-I can be isolated at 333 K (Fig. S16†). Below this temperature, the formation of 2-H was observed, similar to the previous experiments at lower concentrations. This also correlates with the results of VT-1H-NMR (Fig. S18†). Here, a signal broadening was noticed below 333 K, which is expected for the formation of extended aggregates. The VT-1H-NMR shows similar characteristics compared to the higher concentrated solution (Fig. 4). FT-IR experiments (MCH, 0.5 mM, 333 K) were conducted to gain more information about the hydrogen bonds (Fig. 4a). The CO vibration (νCO = 1683 cm−1) appears at nearly identical wavenumbers compared to the monomeric species in chloroform (1 mM, νCO = 1676 cm−1), but shows a more pronounced broadening. The N–H vibration splits into two bands: a stretch that correspond to free N–H groups (νN–H = 3442 cm−1) and a band at lower wavenumbers (νN–H = 3349 cm−1) that indicates weak hydrogen bonding between the molecules. We therefore conclude that the formation of 2-I is driven by π–π interactions and rather diffuse and weak hydrogen bonds that do not include the carbonyl groups. The weak coordination of the amide groups goes in line with the lack of exciton coupling of the Y substituents that was observed in the UV-vis experiments (Fig. 2a). In 2D-ROESY experiments, however, we could find similar interactions in 2-I and 2-H that imply similar packings of the molecules (Fig. S19†). Nevertheless, the absence of strong hydrogen bonds in 2-I indicates that the formation of 2-H requires a rearrangement of the molecules, e.g. via the monomer.
To ultimately rationalize the packing and the preferred contact sides between the neighbouring molecules (F/F, P/P or F/P contacts) in 2-H, we performed theoretical calculations. Previous NMR experiments do not allow to distinguish between the stacking possibilities due to the intrinsically similar interactions of the neighbouring molecules in the proposed packings. Hence, DFT (B3LYP/6-31G(d,p)) calculations were employed to optimize the monomer and compare the three proposed dimers. The dodecyloxy side chains at the peripheral phenyl rings were replaced with methoxy groups to reduce the computational costs. Subsequently, we performed calculations on the F/F, P/P and F/P dimers (Fig. S23†). From the optimized structure, it becomes evident that the P/P stacking is the energetically least favoured stacking (ΔG = +29.851 kJ mol−1). Due to the steric repulsion, only one amide group is involved in hydrogen bonds and the BODIPY chromophores only overlap slightly. The situation changes for the F/P and the F/F dimers. Both exhibit hydrogen bonds between each of the amide groups and both are well stabilized. The free Gibbs energy favours the F/P assembly by 11.673 kJ mol−1 when compared to the F/F packing. Accordingly, we infer that the formation of F/P interactions in the supramolecular polymer 2-H is the most probable scenario. However, given the similar stability of these two packings, we hypothesize that the competition between these two packing modes might be responsible for the observed pathway complexity.
Conclusions
In conclusion, we have reported the supramolecular polymerization in nonpolar media of a new BODIPY derivative 2 with two unequal π-surfaces, stemming from the unsymmetrical functionalization of the boron atom with a fluorine and a phenyl substituent. To rationalize the impact of the symmetry breaking, the self-assembly of 2 was compared with that of a model C2-symmetrical BODIPY compound 1 with a BF2 group. We found that 2 undergoes an H-type supramolecular polymerization (2-H) with similar spectral patterns to 1. However, not only does the final morphology differ (short 1D fibers for 1vs. long fibers for 2), but also the pathway in which the aggregate is reached. In contrast to a single-step self-assembly of 1, the introduction of an additional bulky (phenyl) substituent to the BODIPY boron atom in 2 increases the complexity of the self-assembly by enabling an intermediate species that most probably involves anticooperative effects. These findings can be associated with increased steric repulsion and the multiple possibilities in which unsymmetrical molecules can interact during nucleation. As a result, the system undergoes a monomer → intermediate transformation upon cooling to intermediate temperatures (ca. 300–310 K), and a subsequent conversion to the final H-type aggregate (2-H) upon lowering the temperature to 263 K. We found that the packing of the intermediate state 2-I (small nanoparticles) is similar to the final H-type fibers albeit with weaker and less ordered hydrogen bonds. Although three different dimer packings are feasible (F/F, P/P and F/P dimers), theoretical calculations suggest that only the F/F and F/P-type packings would allow for simultaneous H-bonding and aromatic interactions leading to the experimentally observed extended growth. Our findings introduce a new molecular design strategy for pathway-dependent controlled supramolecular polymerization.Author contributions
Tobias Benedikt Tischer: synthesis, investigation, methodology, writing – original draft. Zulema Fernández: theoretical calculations. Lorenz Borsdorf: AFM measurements. Constantin Daniliuc: X-ray diffraction analysis. Soichiro Ogi: conceptualization, writing – review and editing. Shigehiro Yamaguchi: writing – review and editing. Gustavo Fernández: conceptualization, writing – review and editing, supervision and funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.† Deposition number 2386585† (for C) contains the supplementary crystallographic data for this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschunsggemeinschaft (DFG, German Research Foundation) for funding within the IRTG 2678 – Functional π-Systems (pi-Sys) (Münster-Nagoya International Research Training group, GRK 2678-437785492), and the CRC 1459 – Intelligent Matter (ID A03-433682494). A KAKENHI grant 22K21346 from MEXT/JSPS is acknowledged. Z. F. thanks the Humboldt Foundation for financial support.References
- M. Hartlieb, E. D. H. Mansfield and S. Perrier, A guide to supramolecular polymerizations, Polym. Chem., 2020, 11, 1083–1110 RSC.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Supramolecular polymerization, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Functional π-gelators and their applications, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS.
- Y. Xue, Y. Jin, Y. Zhang, F. Han and F. Wang, Stimuli-Triggered Dynamic Transformations in Supramolecular Polymers, Chem. Mater., 2024, 36, 6347–6369 CrossRef CAS.
- Y. Bi, C. Cheng, Z. Zhang, R. Liu, J. Wei and Z. Yang, Controlled Hierarchical Self-Assembly of Nanoparticles and Chiral Molecules into Tubular Nanocomposites, J. Am. Chem. Soc., 2023, 145, 8529–8539 CrossRef CAS.
- F. Wang, R. Liao and F. Wang, Pathway Control of π-Conjugated Supramolecular Polymers by Incorporating Donor-Acceptor Functionality, Angew. Chem., Int. Ed., 2023, 62, e202305827 CrossRef CAS.
- N. Fukaya, S. Ogi, H. Sotome, K. J. Fujimoto, T. Yanai, N. Bäumer, G. Fernández, H. Miyasaka and S. Yamaguchi, Impact of Hydrophobic/Hydrophilic Balance on Aggregation Pathways, Morphologies, and Excited-State Dynamics of Amphiphilic Diketopyrrolopyrrole Dyes in Aqueous Media, J. Am. Chem. Soc., 2022, 144, 22479–22492 CrossRef CAS PubMed.
- N. Bäumer, E. Castellanos, B. Soberats and G. Fernández, Bioinspired crowding directs supramloecular polymerisation, Nat. Commun., 2023, 14, 1084 CrossRef.
- G. Ghosh, R. Barman, A. Mukherjee, U. Ghosh, S. Ghosh and G. Fernández, Control over Multiple Nano- and Secondary Structures in Peptide Self-Assembly, Angew. Chem., Int. Ed., 2022, 61, e202113403 CrossRef CAS PubMed.
- M. Murai, M. Abe, S. Ogi and S. Yamaguchi, Diazulenylmethyl Cations with a Silicon Bridge: A π-Extended Cationic Motif to Form J-Aggregates with Near-Infrared Absorption and Emission, J. Am. Chem. Soc., 2022, 144, 20385–20393 CrossRef CAS.
- F. Salerno, J. A. Berrocal, A. T. Haedler, F. Zinna, E. W. Meijer and L. Di Bari, Highly circularly polarized broad-band emission from chiral naphthalene diimide-based supramolecular aggregates, J. Mater. Chem. C, 2017, 5, 3609–3615 RSC.
- H. Liu, Z. Zhang, Y. Zhao, Y. Zhou, B. Xue, Y. Han, Y. Wang, X. Mu, S. Zang, X. Zhou and Z. Li, A water-soluble two-dimensional supramolecular organic framework with aggregation-induced emission for DNA affinity and live-cell imaging, J. Mater. Chem. B, 2019, 7, 1435–1441 RSC.
- C.-C. Zhang, Y.-M. Zhang, Z.-Y. Zhang, X. Wu, Q. Yu and Y. Liu, Photoreaction-driven two-dimensional periodic polyrotaxane-type supramolecular nanoarchitecture, Chem. Commun., 2019, 55, 8138–8141 RSC.
- A. Sampedro, Á. Ramos-Torres, C. Schwöppe, C. Mück-Lichtenfeld, I. Helmers, A. Bort, I. Díaz-Laviada and G. Fernández, Hierarchical Self-Assembly of BODIPY Dyes as a Tool to Improve the Antitumor Activity of Capsaicin in Prostate Cancer, Angew. Chem., Int. Ed., 2018, 57, 17235–17239 CrossRef CAS.
- A. Florian, M. J. Mayoral, V. Stepanenko and G. Fernández, Alternated stacks of nonpolar oligo(p-phenyleneethynylene)-BODIPY systems, Chem. – Eur. J., 2012, 18, 14957–14961 CrossRef CAS PubMed.
- C. Shao, M. Grüne, M. Stolte and F. Würthner, Perylene bisimide dimer aggregates: fundamental insights into self-assembly by NMR and UV/Vis spectroscopy, Chem. – Eur. J., 2012, 18, 13665–13677 CrossRef CAS PubMed.
- S. E. Penty, G. R. F. Orton, D. J. Black, R. Pal, M. A. Zwijnenburg and T. A. Barendt, A Chirally Locked Bis-perylene Diimide Macrocycle: Consequences for Chiral Self-Assembly and Circularly Polarized Luminescence, J. Am. Chem. Soc., 2024, 146, 5470–5479 CrossRef CAS PubMed.
- A. Sikder, A. Das and S. Ghosh, Hydrogen-bond-regulated distinct functional-group display at the inner and outer wall of vesicles, Angew. Chem., Int. Ed., 2015, 54, 6755–6760 CrossRef CAS.
- J. Gershberg, F. Fennel, T. H. Rehm, S. Lochbrunner and F. Würthner, Anti-cooperative supramolecular polymerization: a new K2-K model applied to the self-assembly of perylene bisimide dye proceeding via well-defined hydrogen-bonded dimers, Chem. Sci., 2016, 7, 1729–1737 RSC.
- C. Faul and M. Antonietti, Ionic Self–Assembly: Facile Synthesis of Supramolecular Materials, Adv. Mater., 2003, 15, 673–683 CrossRef CAS.
- G. Fan, Y.-X. Lin, Le Yang, F.-P. Gao, Y.-X. Zhao, Z.-Y. Qiao, Q. Zhao, Y.-S. Fan, Z. Chen and H. Wang, Co-self-assembled nanoaggregates of BODIPY amphiphiles for dual colour imaging of live cells, Chem. Commun., 2015, 51, 12447–12450 RSC.
- M. Su, S. Li, H. Zhang, J. Zhang, H. Chen and C. Li, Nano-Assemblies from J-Aggregated Dyes: A Stimuli-Responsive Tool Applicable To Living Systems, J. Am. Chem. Soc., 2019, 141, 402–413 CrossRef CAS.
- B. Matarranz, G. Ghosh, R. Kandanelli, A. Sampedro, K. K. Kartha and G. Fernández, Understanding the role of conjugation length on the self-assembly behaviour of oligophenyleneethynylenes, Chem. Commun., 2021, 57, 4890–4893 RSC.
- M. F. J. Mabesoone, A. J. Markvoort, M. Banno, T. Yamaguchi, F. Helmich, Y. Naito, E. Yashima, A. R. A. Palmans and E. W. Meijer, Competing Interactions in Hierarchical Porphyrin Self-Assembly Introduce Robustness in Pathway Complexity, J. Am. Chem. Soc., 2018, 140, 7810–7819 CrossRef CAS PubMed.
- J. N. S. Hanssen and S. Dhiman, Impact of subtle intermolecular interactions on the structure and dynamics of multicomponent supramolecular polymers, Chem. Commun., 2023, 59, 13466–13469 RSC.
- A. T. Haedler, S. C. J. Meskers, R. H. Zha, M. Kivala, H.-W. Schmidt and E. W. Meijer, Pathway Complexity in the Enantioselective Self-Assembly of Functional Carbonyl-Bridged Triarylamine Trisamides, J. Am. Chem. Soc., 2016, 138, 10539–10545 CrossRef CAS.
- S. Ogi, T. Fukui, M. L. Jue, M. Takeuchi and K. Sugiyasu, Kinetic control over pathway complexity in supramolecular polymerization through modulating the energy landscape by rational molecular design, Angew. Chem., Int. Ed., 2014, 53, 14363–14367 CrossRef CAS.
- R. H. Martin, The Helicenes, Angew. Chem., Int. Ed. Engl., 1974, 13, 649–660 CrossRef.
- Y. Shen and C.-F. Chen, Helicenes: synthesis and applications, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS.
- R. Amemiya and M. Yamaguchi, Chiral recognition in noncovalent bonding interactions between helicenes: right-handed helix favors right-handed helix over left-handed helix, Org. Biomol. Chem., 2008, 6, 26–35 RSC.
- R. Rodríguez, C. Naranjo, A. Kumar, P. Matozzo, T. K. Das, Q. Zhu, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, Mutual Monomer Orientation To Bias the Supramolecular Polymerization of [6]Helicenes and the Resulting Circularly Polarized Light and Spin Filtering Properties, J. Am. Chem. Soc., 2022, 144, 7709–7719 CrossRef.
- X. Tian, K. Shoyama, B. Mahlmeister, F. Brust, M. Stolte and F. Würthner, Naphthalimide-Annulated nHelicenes: Red Circularly Polarized Light Emitters, J. Am. Chem. Soc., 2023, 145, 9886–9894 CrossRef CAS PubMed.
- R. Renner, B. Mahlmeister, O. Anhalt, M. Stolte and F. Würthner, Chiral Perylene Bisimide Dyes by Interlocked Arene Substituents in the Bay Area, Chem. – Eur. J., 2021, 27, 11997–12006 CrossRef CAS PubMed.
- A. Rödle, B. Ritschel, C. Mück-Lichtenfeld, V. Stepanenko and G. Fernández, Influence of Ester versus Amide Linkers on the Supramolecular Polymerization Mechanisms of Planar BODIPY Dyes, Chem. – Eur. J., 2016, 22, 15772–15777 CrossRef.
- R. M. Veedu, Z. Fernández, N. Bäumer, A. Albers and G. Fernández, Pathway-dependent supramolecular polymerization by planarity breaking, Chem. Sci., 2024, 15, 10745–10752 RSC.
- Z. Chen and Z. Chen, Functional supramolecular aggregates based on BODIPY and aza-BODIPY dyes: control over the pathway complexity, Org. Chem. Front., 2023, 10, 2581–2602 RSC.
- S. Cherumukkil, B. Vedhanarayanan, G. Das, V. K. Praveen and A. Ajayaghosh, Self-Assembly of Bodipy-Derived Extended π-Systems, Bull. Chem. Soc. Jpn., 2018, 91, 100–120 CrossRef CAS.
- B. Matarranz and G. Fernández, BODIPY dyes: Versatile building blocks to construct multiple types of self-assembled structures, Chem. Phys. Rev., 2021, 2, 041304 CrossRef CAS.
- Y. Dorca, C. Naranjo, G. Ghosh, B. Soberats, J. Calbo, E. Ortí, G. Fernández and L. Sánchez, Supramolecular polymerization of electronically complementary linear motifs: anti-cooperativity by attenuated growth, Chem. Sci., 2021, 13, 81–89 RSC.
- J. Matern, K. K. Kartha, L. Sánchez and G. Fernández, Consequences of hidden kinetic pathways on supramolecular polymerization, Chem. Sci., 2020, 11, 6780–6788 RSC.
- H. Wang, Y. Zhang, Y. Chen, H. Pan, X. Ren and Z. Chen, Living Supramolecular Polymerization of an Aza-BODIPY Dye Controlled by a Hydrogen-Bond-Accepting Triazole Unit Introduced by Click Chemistry, Angew. Chem., Int. Ed., 2020, 59, 5185–5192 CrossRef CAS.
- W. Wagner, M. Wehner, V. Stepanenko, S. Ogi and F. Würthner, Living Supramolecular Polymerization of a Perylene Bisimide Dye into Fluorescent J-Aggregates, Angew. Chem., Int. Ed., 2017, 56, 16008–16012 CrossRef CAS PubMed.
- L. Herkert, J. Droste, K. K. Kartha, P. A. Korevaar, T. F. A. de Greef, M. R. Hansen and G. Fernández, Pathway Control in Cooperative vs. Anti-Cooperative Supramolecular Polymers, Angew. Chem., Int. Ed., 2019, 58, 11344–11349 CrossRef CAS.
- H. M. M. ten Eikelder, A. J. Markvoort, T. F. A. de Greef and P. A. J. Hilbers, An equilibrium model for chiral amplification in supramolecular polymers, J. Phys. Chem. B, 2012, 116, 5291–5301 CrossRef CAS.
- K. Tram, H. Yan, H. A. Jenkins, S. Vassiliev and D. Bruce, The synthesis and crystal structure of unsubstituted 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY), Dyes Pigm., 2009, 82, 392–395 CrossRef CAS.
- G. G. Çelik, B. Dedeoglu, A. G. Gürek, Y. Zorlu and M. M. Ayhan, Enhancing Supramolecular Assembly in BODIPY Derivatives: Harnessing Halogen Bonding for Cocrystal Design, Cryst. Growth Des., 2023, 23, 7285–7294 CrossRef.
- S. Prasanthkumar, A. Saeki, S. Seki and A. Ajayaghosh, Solution phase epitaxial self-assembly and high charge-carrier mobility nanofibers of semiconducting molecular gelators, J. Am. Chem. Soc., 2010, 132, 8866–8867 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2386585. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01848f |
| This journal is © the Partner Organisations 2025 |