
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalcium ferrites for phosphate adsorption and recovery from wastewater†
Matheus Henrique Pimentel Araújo *a,
José Domingos Ardissonb,
Alisson Carlos Krohlingb,
Rochel Montero Lagoc,
Walber Guimarães Júniord and
Juliana Cristina Tristãoa
*a,
José Domingos Ardissonb,
Alisson Carlos Krohlingb,
Rochel Montero Lagoc,
Walber Guimarães Júniord and
Juliana Cristina Tristãoa
aUniversidade Federal de Viçosa – Campus Florestal – UFV, Rodovia LMG 818, km 06, s/n, Campus Universitário, Florestal, MG 35690-000, Brazil. E-mail: matheus.pimentel@ufv.br
bCentro de Desenvolvimento da Tecnologia Nuclear – CDTN/CNEN, campus da Universidade Federal de Minas Gerais, Avenida Presidente Antônio Carlos, No 6627, Rua Mário Werneck, S/N – Pampulha, Belo Horizonte, MG, Brazil
cUniversidade Federal de Minas Gerais – UFMG, Avenida Presidente Antônio Carlos, No 6627, Rua Mário Werneck, 2 – Pampulha, Belo Horizonte, MG 31270-901, Brazil
dUniversidade Federal de São Carlos – UFSCar, Rodovia Washington Luís, s/n – Monjolinho, São Carlos – SP, 13565-905, Brazil
First published on 4th January 2024
Abstract
In this study, calcium ferrites with different Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe atomic ratios (1:1, 1:2, 1:3 and 2:1) were prepared from Ca and Fe nitrates treated at 300, 700 and 900 °C and evaluated for phosphate adsorption and recovery from wastewater. TG, XRD, Mössbauer spectroscopy, SEM, VSM magnetic measurements, and BET analyses showed the formation of two different calcium ferrite phases, i.e., CaFe2O4 and Ca2Fe2O5 at 700 and 900 °C. The adsorption results indicated that the formation of calcium ferrite structure is critical for phosphate adsorption/recovery. Evaluation of the pH, initial phosphate concentration, contact time, coexisting ions and desorption conditions showed remarkable adsorption capacities of 62–75 mg g−1 for CaFe1:2-700 and 28–43 mg g−1 for CaFe1:2-900. The phosphate adsorption on the Ca ferrite surfaces is so strong that the recovery/desorption showed limited efficiencies, e.g., 15–39%.
:Fe atomic ratios (1:1, 1:2, 1:3 and 2:1) were prepared from Ca and Fe nitrates treated at 300, 700 and 900 °C and evaluated for phosphate adsorption and recovery from wastewater. TG, XRD, Mössbauer spectroscopy, SEM, VSM magnetic measurements, and BET analyses showed the formation of two different calcium ferrite phases, i.e., CaFe2O4 and Ca2Fe2O5 at 700 and 900 °C. The adsorption results indicated that the formation of calcium ferrite structure is critical for phosphate adsorption/recovery. Evaluation of the pH, initial phosphate concentration, contact time, coexisting ions and desorption conditions showed remarkable adsorption capacities of 62–75 mg g−1 for CaFe1:2-700 and 28–43 mg g−1 for CaFe1:2-900. The phosphate adsorption on the Ca ferrite surfaces is so strong that the recovery/desorption showed limited efficiencies, e.g., 15–39%.
Introduction
Given the problems associated with phosphate pollution of lakes, rivers and oceans and the decreasing availability of phosphate rock in the coming years, several alternatives have been developed for water treatment and recovery of phosphate and other nutrients.1–7 These include chemical precipitation, biological treatment, constructed wetlands, electrodialysis, reverse osmosis, nanofiltration and adsorption.1,4Adsorption processes have several advantages when phosphate concentrations in water are low, such as simplicity, cost, no sludge formation, and the possibility of adsorbent regeneration.1,4 Several compounds have been tested for phosphate adsorption, e.g., zeolites,8 activated carbon and biochar,1 metal-doped mesoporous silica, mesoporous metal oxide, mesoporous carbon,9 and carbonate materials.1
In recent years, ferrous materials have been used for phosphate adsorption, such as hematite (Fe2O3), goethite (FeOOH), magnetite (Fe3O4), and other magnetic nano-adsorbents. Ajmal et al.10 studied the phosphate adsorption in ferrihydrite, goethite and magnetite and observed adsorption capacities of ca. 16, 15, and 13 mg g−1, respectively. Liu et al.11 used ferrihydrite (Fh), goethite (Gt) and hematite (Hm) to evaluate the removal of Cd2+ and PO43− in water. The minerals showed a decreasing behavior Fh (28.4 mg g−1) > Gt (9.7 mg g−1) > Hm (4.7 mg g−1) for phosphate removal. Zinc ferrite nanoparticles were used for phosphate adsorption and showed low phosphate removal capacity at an initial concentration of 5 mg L−1.12
Materials containing alkaline earth metals (Ca, Mg) have been used for phosphate removal.13,14 In the work of Han et al.,15 calcium and magnesium ferrites were synthesized by the combustion method using PVA and applied for phosphate removal. The results showed that calcium and magnesium iron oxides could increase the phosphate removal capacity of the material. The samples without PVA showed better results, Fe/CaCl2-NoPVA and Fe/MgCl2-NoPVA with ca. 17 mg g−1. However, the presence of PVA resulted in a lower adsorption capacity for these materials due to the nanoparticle coating. Jia et al.16 used a layered double hydroxide containing Ca with 182.5 mg g−1 for phosphate removal. A CaMg-based layered double hydroxide was used in real effluent from a wastewater treatment plant and removed >90% of phosphate.17 Mg–Al and Zn–Al layered double hydroxides effectively removed over 90% of phosphate from aqueous solution.18 Cheng et al.19 prepared a cost-effective anhydrite-BPM sorbent (ABPM) by mixing anhydrite and brucite-periclase materials with phosphate removal of 23.6 mg g−1. Calcium aluminate decahydrate was used to achieve low phosphate concentrations. In a column experiment, Sonoda et al.20 used granular ferric oxides for phosphate adsorption and showed that alkaline earth metal ions significantly increased phosphate adsorption.
Reusability and lifespan are essential factors to consider when evaluating the potential use of adsorbents in water treatment. Adsorbents that have a longer useful life and excellent recyclability allow for a reduction in the operational cost of the adsorption process.21,22 There are two ways in the literature for regeneration and reuse of the adsorbent: extraction of the adsorbed phosphate through desorption and restoration of the original structure of the adsorbent. In the recovery of phosphate adsorbents, different solutions containing NaOH and NaHCO3/Na2CO3 at different concentrations can extract the adsorbed phosphate through desorption.21–25 Regeneration is generally carried out through alkaline washing (pH > 13), where P accumulates and is later recovered. Aggressive chemicals can deteriorate the adsorbent, reducing its performance and useful life. After regeneration, the regenerated adsorbent may differ from the fresh adsorbent; incomplete desorption of phosphate is also possible.4,21,22,24,25
This work prepared calcium ferrite adsorbents by a simple route synthesis at different Ca:Fe (1:1, 1:2, 1:3, 2:1) atomic ratios and calcination temperatures (300, 700 and 900 °C). Different techniques characterized the materials and provided insights into the structure and composition of the adsorbents. The material was applied for phosphate adsorption and recovery in different conditions to evaluate the phosphate adsorption capacity of calcium ferrites. This information provided insights into the proposal of an adsorption mechanism. Calcium ferrite's phosphate desorption capacity was also evaluated under different conditions to elucidate the better desorption condition.
Methods
Calcium ferrites synthesis
The samples were synthesized by the thermal decomposition method without organic precursors. The Ca(NO3)2·4H2O (ÊXODO CIENTÍFICA) and Fe(NO3)2·9H2O (NEON) reagents were used as analytical standards and different Ca:Fe molar ratios (1:1; 1:2; 1:3 and 2:1) measured (SHIMADZU ATY224). The mass of different proportions was solubilized with 50 mL of deionized water (GEHAKA) and kept under magnetic stirring at 90 °C until dry (IKA). The dried samples were calcined in the tube furnace (Sanchis 1200) at different temperatures of 300, 700 and 900 °C for one hour with a heating rate of 5 °C min−1. The materials were crushed after calcination using mortar and pestle. The material identification is presented in the ESI Table S1.†
Materials characterization
Different techniques characterized the materials. Thermal analysis TG/DTA studied the thermal stability of the materials. The analyses were performed on a Shimadzu TG/DTA (model DTG-60H) in the air atmosphere with a gas flow of 50 mL min−1 and a heating rate of 10 °C min−1, ranging from 25° to 900 °C. X-ray diffraction (XRD) was used to identify the crystalline phases. The materials were characterized by XRD using SHIMADZU (XRD-7000 X-ray diffractometer, Cu Kα radiation), voltage 30.0 kV, current 30.0 mA and continuous scanning range from 4.0 to 70.0° 2θ. The diffractograms were analyzed in Profex software for phase identification according to the Crystallography Open Database.26 The transmission 57Fe Mössbauer measurements were obtained at 293 K, with a 57Co/Rh source. The Normos least-squares fits were used to calculate the spectral hyperfine parameters, and the isomeric deviations (IS) were standardized concerning natural Fe (α-Fe). The morphology of the materials was observed using a Scanning Electron Microscope (SEM) using a Quanta 200 FEG instrument. Energy dispersive spectroscopy (EDS) was performed on a carbon plate with the same instrument at 5 keV. The surface area was determined by N2 absorption using the BET method in an Autosorb 1 Quantachrome instrument and the data were analyzed in ASiQwin (5.21) software. The magnetic properties of the samples were measured using a vibrating sample magnetometer (VSM, Lakeshore model 7404) at room temperature. The magnetic hysteresis curve was obtained up to a magnetic field of 1.8 Tesla (18 kOe).Adsorption study
In determination of the point of zero charge (PZC), the experiment was performed by an adapted 11 points method proposed by de Freitas et al.27
Theoretical studies
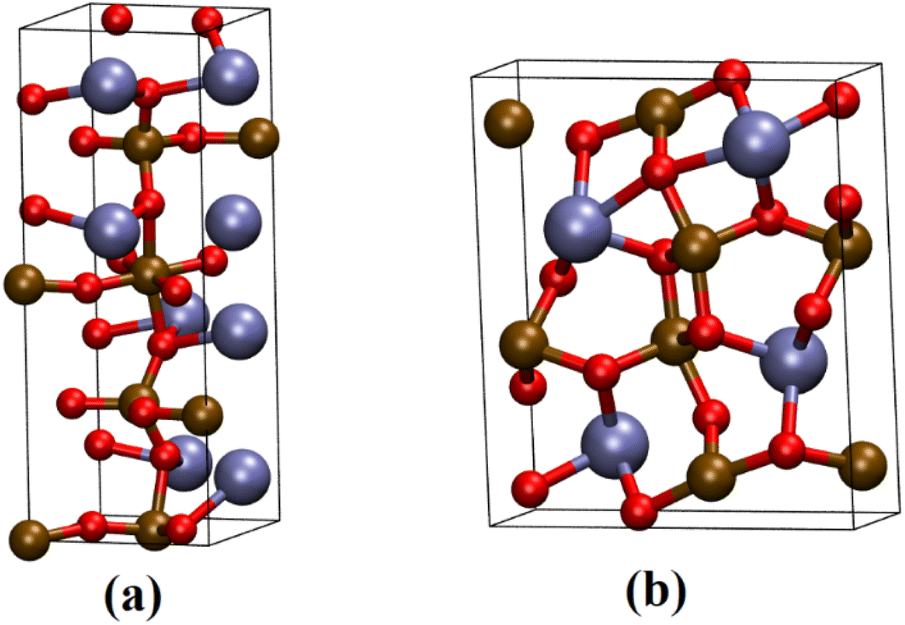 | ||
| Fig. 1 Representation of the (a) Ca2Fe2O5 and (b) CaFe2O4 unit cell. The brown, red, and blue spheres represent Fe, O, and Ca atoms. | ||
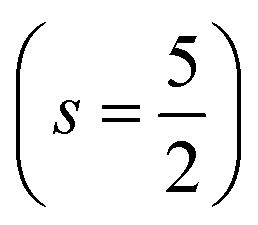 .29,34,35 In this study, the effective Hubbard parameter (Ueff) was included following the DFT + U calculations were performed within the rotationally invariant formulation by Dudarev et al.36 by applying an anisotropic Hubbard correction37,38 to Fe-3d states with self-consistent U values computed via DFPT.39 Hubbard parameters were computed by perturbing all inequivalent Fe-sites. The parameters were set at
.29,34,35 In this study, the effective Hubbard parameter (Ueff) was included following the DFT + U calculations were performed within the rotationally invariant formulation by Dudarev et al.36 by applying an anisotropic Hubbard correction37,38 to Fe-3d states with self-consistent U values computed via DFPT.39 Hubbard parameters were computed by perturbing all inequivalent Fe-sites. The parameters were set at 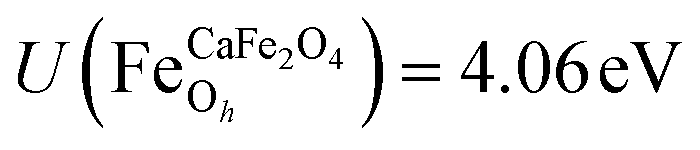 for all Fe↑-sites and
for all Fe↑-sites and 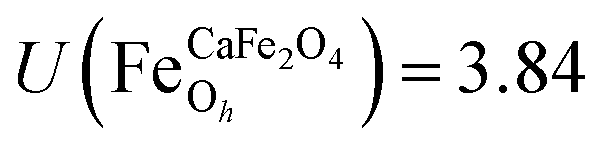 for all Fe↓-sites in the CaFe2O4 structure and
for all Fe↓-sites in the CaFe2O4 structure and 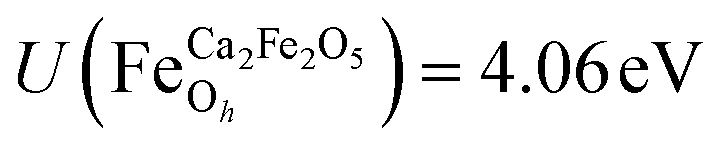 for all Fe-octahedral atoms and
for all Fe-octahedral atoms and 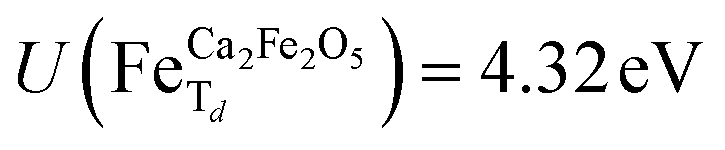 for all Fe-tetrahedral atoms for the Ca2Fe2O5 structure. The Mössbauer calculation was carried out in the QE-GIPAW routine,40 which allows the calculation of relevant parameters and provides the opportunity to clarify many important structural details. The cutoff energy was set as 120 Ry (1.63 keV), and the kinetic energy cutoff was set as 600 Ry (8.16 keV) for charge density and potential. The electron–ion interactions were described with the Gauge-Including Projector Augmented Wave (GIPAW) pseudopotentials, which allowed the calculation of the Mössbauer-based quantities. The self-consistent criterion was the total energy difference between the two subsequent calculations being less than 10–15 Ry. We then investigate possible correlations of the predicted quadrupole splitting and isomer shift with local atomic geometry or local electronic structure, described by Bader analysis,41,42 implemented in the CRITIC2 program,43 and Born dynamical effective charges. The electronic density was generated by pos-processing using the Mössbauer calculation protocol.
for all Fe-tetrahedral atoms for the Ca2Fe2O5 structure. The Mössbauer calculation was carried out in the QE-GIPAW routine,40 which allows the calculation of relevant parameters and provides the opportunity to clarify many important structural details. The cutoff energy was set as 120 Ry (1.63 keV), and the kinetic energy cutoff was set as 600 Ry (8.16 keV) for charge density and potential. The electron–ion interactions were described with the Gauge-Including Projector Augmented Wave (GIPAW) pseudopotentials, which allowed the calculation of the Mössbauer-based quantities. The self-consistent criterion was the total energy difference between the two subsequent calculations being less than 10–15 Ry. We then investigate possible correlations of the predicted quadrupole splitting and isomer shift with local atomic geometry or local electronic structure, described by Bader analysis,41,42 implemented in the CRITIC2 program,43 and Born dynamical effective charges. The electronic density was generated by pos-processing using the Mössbauer calculation protocol.Results and discussions
In this study, calcium ferrites with different Ca:Fe atomic ratios (0:1, 1:1, 1:2, 1:3, 2:1) were prepared from the Ca and Fe nitrates by simple dissolution in water, drying and thermal treatment at 300, 700 and 900 °C. TG, XRD, Mössbauer, SEM, VSM and BET characterized the obtained solids.
Characterization of adsorbent materials
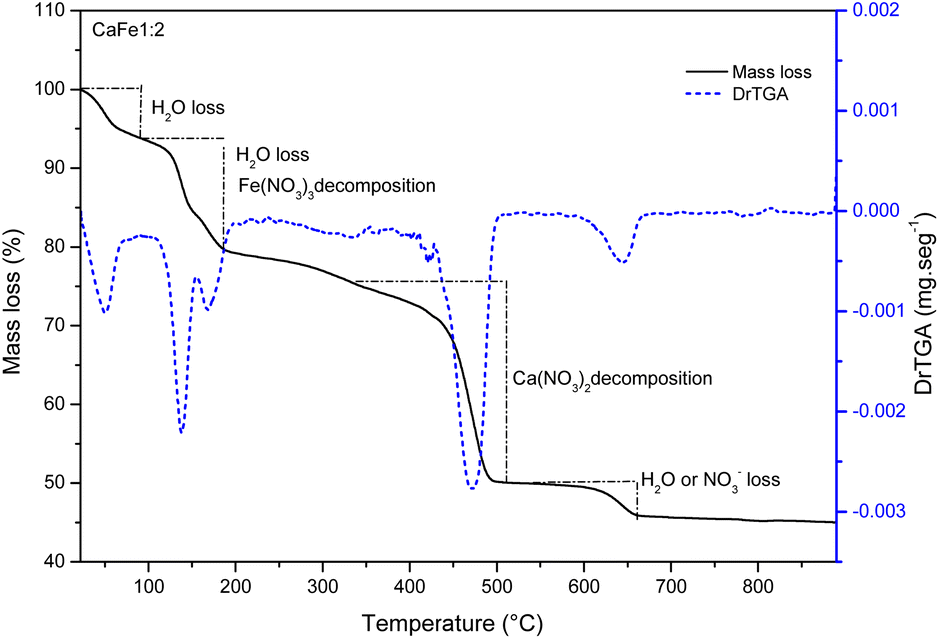 | ||
| Fig. 2 Thermal decomposition curve and DTG for the sample CaFe1:2 at air atmosphere (50 mL min−1) and heating rate of 10 °C min−1. | ||
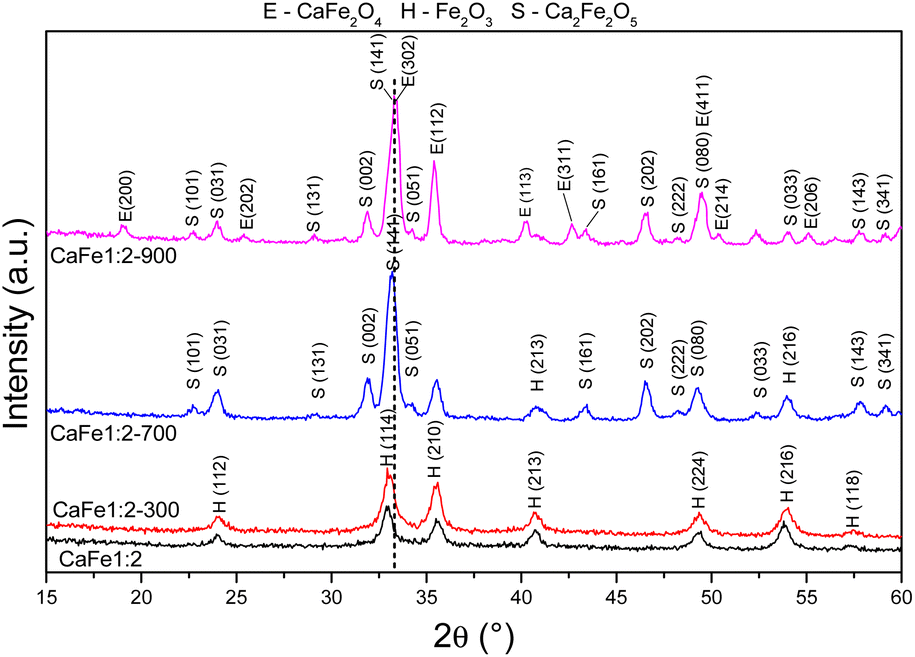 | ||
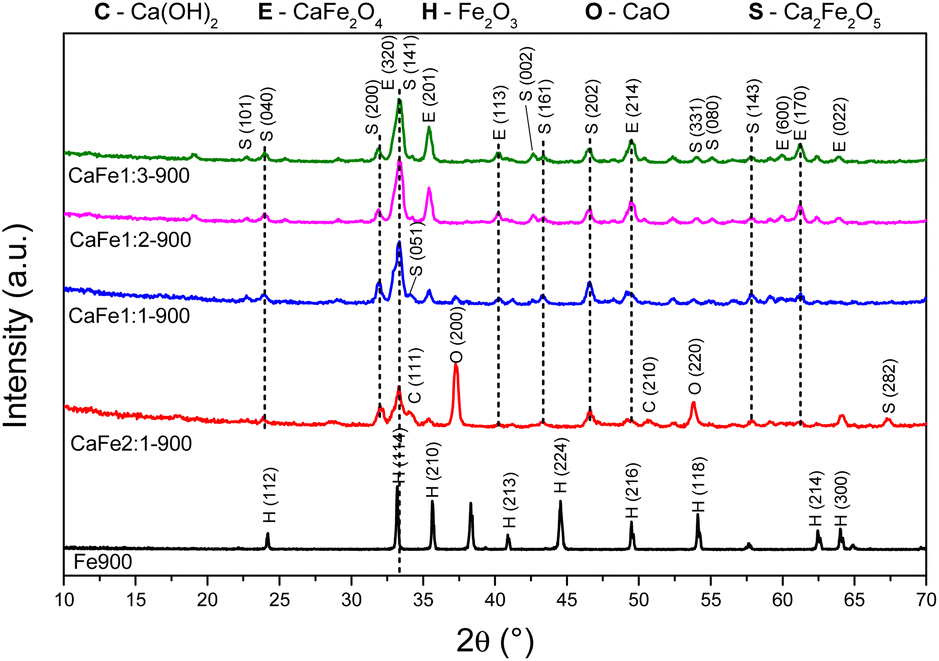
| Fig. 3 Diffractograms of synthetic adsorbents prepared in the proportion Ca:Fe 1:2 calcined at different temperatures 300, 700 and 900 °C for one hour at a rate of 5 °C min−1. | ||
The XRD patterns for samples CaFe1:2 and CaFe1:2-300 showed low crystallinity with diffraction peaks related to hematite (α-Fe2O3 – COD 04-003-2900). Note that the crystallinity of the material increased with temperature, as indicated by more intense and defined peaks observed in the patterns for samples CaFe1:2-700 and CaFe1:2-900. In addition, at higher annealing temperatures, 700 and 900 °C, the ferrite phases CaFe2O4 (COD 9013281) and Ca2Fe2O5 (COD 9013469), both in the orthorhombic structure, are observed. It can be noted that the peak near 2θ 32.9°, referred to as hematite, suffers a dislocation to 2θ 33.4° after calcination at 700 °C, but refers to calcium ferrite phases, as shown in Fig. 3 by the dotted line. Other works show that high temperatures are necessary for forming CaFe2O4 formation.46,49,50 The CaFe2O4 phase in the spinel structure has not been identified. The ionic diameter size of calcium Ca2+ makes the cation incorporation into the tetrahedral site complex for spinel structure formation, requiring higher temperatures or longer calcination time for its phase formation.46,49,50 In the theoretical studies, the calculated lattice parameters of the orthorhombic unit cell for both structures are in satisfactory good agreement with the experimental values reported in the ESI (S11).†28,30
Fig. 4 and 5 show the Ca:Fe (0:1, 1:1; 1:2; 1:3, and 2:1) samples calcined at 700 and 900 °C for one hour, respectively, to analyze the effect of different atomic ratios. The Fe700 and Fe900 patterns show thin and intense peaks, indicating a material with high crystallinity. Both patterns indicate a phase formed from hematite (α-Fe2O3 – COD 04-003-2900). When calcium is present in the reaction mixture, the 2θ = 33° peak is broadened. The patterns indicate different phases in this region (α-Fe2O3 – COD 04-003-2900), CaFe2O4 (COD 9013281) and Ca2Fe2O5 (COD 90-134-69), which can cause peak enlargement. For samples with a higher iron content, CaFe1:2-700 and CaFe1:3-700, peaks related to hematite are still observed. In contrast to CaFe1:2-900 and CaFe2:1-900 samples, the ferrite phase CaFe2O4 (PDF 00-032-0168) is observed, and the hematite-related peaks become less prominent. Samples CaFe2:1 and CaFe2:1-300, in ESI S6,† present characteristic peaks for calcium nitrate (Ca(NO3)2) (COD 04-006-5679). This identification indicates an incomplete decomposition for this precursor, as discussed for TG analyses. The CaO (COD 04-007-9734) and Ca(OH)2 (COD 04-010-3117) phases were identified for the Ca:Fe 2:1 and Ca:Fe 1:1 groups, which have the highest calcium molar ratio. The presence of Ca(OH)2 may be related to the hydration of CaO with the water in the atmosphere during the sample storage. In the CaFe1:1-700, CaFe1:1-900, CaFe2:1-700, and CaFe2:1-900 samples, CaO (COD 04-007-9734), hkl = (200), at 2θ near to 37° was identified. Excess Ca in these samples leads to the formation of impurities and may affect adsorption. It also indicates that not all the calcium available for the reaction is converted to the crystalline ferrite phase.
 | ||
| Fig. 4 Diffractograms of synthetic adsorbents prepared in the proportions Ca:Fe 1:1, 1:2, 1:3 and 2:1 calcined at 700 °C for one hour at a rate of 5 °C min−1. | ||
 | ||
| Fig. 5 Diffractograms of synthetic adsorbents prepared in the proportions Ca:Fe 1:1, 1:2, 1:3 and 2:1 calcined at 900 °C for one hour at a rate of 5 °C min−1. | ||
Table 1 shows the crystallite size calculated by the Scherrer equation. The crystallite size increases after 900 °C, indicating a more crystalline material formed at this temperature. The Ca2Fe2O5 phase shows an increase of 30% in crystallite size from CaFe1:1-700 to CaFe1:1-900. The Ca2Fe2O5 phase is preferentially formed for this Ca:Fe 1:1 ratio, as previously observed in the work of Hirabayashi et al.50 The crystallite size increase with increasing calcination temperature is repeated for Ca:Fe ratios of 1:3 and 2:1.
:Fe 1:1, 1:2, 1:3 and 2:1 calcined at 700 and 900 °C for one hour
| Sample | Phase and grain size (nm) | ||
|---|---|---|---|
| Fe2O3 | Ca2Fe2O5 | CaFe2O4 | |
| CaFe2:1-700 | 42 | 17 | — |
| CaFe2:1-900 | — | 24 | 42 |
| CaFe1:1-700 | 47 | 31 | — |
| CaFe1:1-900 | 109 | 42 | 69 |
| CaFe1:2-700 | 31 | 40 | — |
| CaFe1:2-900 | 65 | 36 | 49 |
| CaFe1:3-700 | 27 | 40 | — |
| CaFe1:3-900 | 91 | 42 | 52 |
For CaFe1:2-700 and CaFe1:2-900, the Ca2Fe2O5 average crystallite size decreased by 16% after calcination at 900 °C for one hour. On the other hand, the CaFe2O4 phase was only observed for samples calcined at 900 °C for one hour, and the average crystallite size is larger than that of the Ca2Fe2O5 phase. At this temperature, the CaFe2O4 phase predominates.49–51 Many papers emphasize that the stoichiometric ratio and the calcination temperature are key factors in synthesizing a calcium ferrite monophase.49,51 However, due to the variation in the Ca:Fe ratios and calcination time used in this work, two ferrite phases could be identified after treatment at 900 °C.
:Fe 1:2 ratio uncalcined and annealed at 300, 700 and 900 °C for one hour. The ESI (Fig. S7–S10 and Tables S2–S5)† gives the spectra for other ratios under the same conditions. The Mössbauer spectra for sample CaFe1:2 were fitted to two sextets related to α-Fe2O3 and α-FeOOH and two duplets related to Fe3+ in unidentified sites (Fig. 6). For the CaFe1:2-300 sample, two sextets were fitted to α-Fe2O3 and γ-Fe2O3. For samples CaFe1:2-700 and CaFe1:2-900, two sextets are fitted to octahedral (octa) and tetrahedral (tetra) sites of calcium ferrites and a central duplet referring to Fe3+ SPM (superparamagnetic). Fig. 7 shows the sample's relative areas (%) in different Ca:Fe molar ratios (1:1, 1:2, 1:3, 2:1) and annealed at 700 and 900 °C for one hour. There is a significant change in the relative areas for Fe3+ (tetra + octa) of calcium ferrite. After calcination at 900 °C, the relative area concerning calcium ferrite Fe3+ decreases compared to the annealing at 700 °C. The sample CaFe1:2-700 shows this process with 92% of Fe3+ from calcium ferrite (Fe3+ – octa 64%, Fe3+ – tetra 28%) and CaFe1:2-900 decreasing to 70% (Fe3+ – octa 32%, Fe3+ – tetra 38%). The other groups show similar behavior, sample CaFe2:1-700 (81% Fe3+ calcium ferrite) decreasing to CaFe2:1-900 (70% Fe3+ – calcium ferrite) and CaFe1:1-700 (73% Fe3+ – calcium ferrite) to CaFe1:2-900 (71% Fe3+ – calcium ferrite). Sample CaFe1:3-700 shows 96% of Fe3+ while CaFe1:3-900 shows 49% of the Fe3+ associated with calcium ferrite, as shown in Fig. 7 and tables in the ESI (Tables S2–S5).† Brownmillerite Ca2Fe2O5 orthodromic phase contains Fe3+ in tetrahedral and octahedral sites,49–52 while in CaFe2O4 orthodromic phase, Fe3+ cations occupy octahedral sites.50 The difference between the spectra of the ferrite crystalline phases is not identified in this work due to the analysis at room temperature. Hirabayashi et al.50 analyzed the Mössbauer spectra for different Ca:Fe rations at room temperature and 23 K and could distinguish the two phases found. In Fig. 7, the relative area for the central superparamagnetic duplet (Fe3+ – SPM) increases with the annealing temperature, with a maximum value of 51% for CaFe1:3-900, and this behavior has been reported previously.52 This spectral component may be related to low crystallinity residuals accumulated at the grain boundaries.52 Relative areas for the goethite (α-FeOOH) are observed only in uncalcined samples due to the thermal decomposition of this phase. The relative areas for α-Fe2O3 and γ-Fe2O3 are observed for higher iron molar proportions, CaFe1:2-300 (α-Fe2O3 – 58%, γ-Fe2O3 – 42%) and CaFe1:3-300 (α-Fe2O3 – 73%, γ-Fe2O3 – 23%). In addition, CaFe1:1-300 and CaFe2:1-300 samples contain 98% and 61% of α-Fe2O3, respectively.
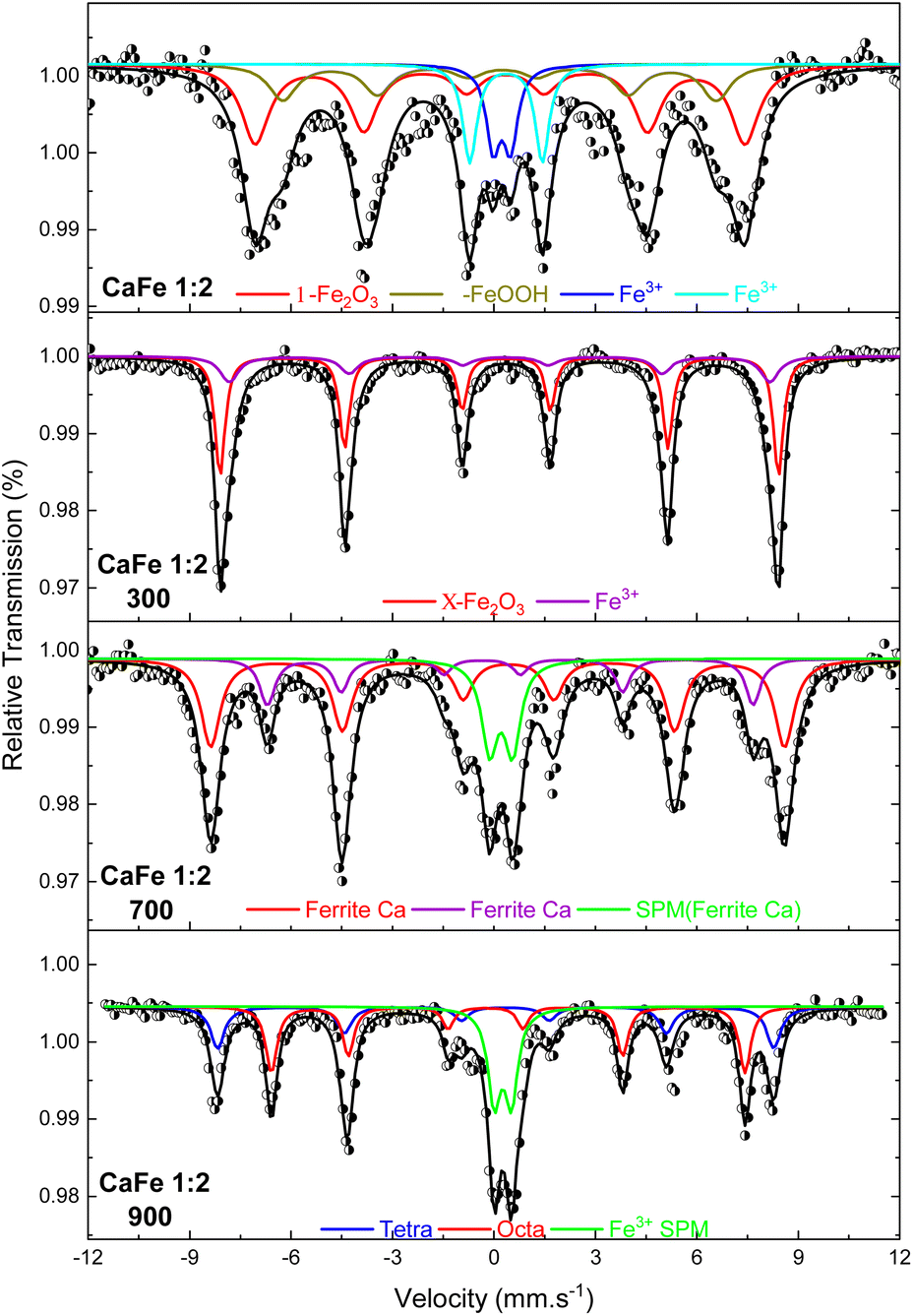 | ||
| Fig. 6 Mössbauer spectra at room temperature of the samples in the proportion Ca:Fe 1:2 calcined at different temperatures of 300, 700 and 900 °C for one hour. | ||
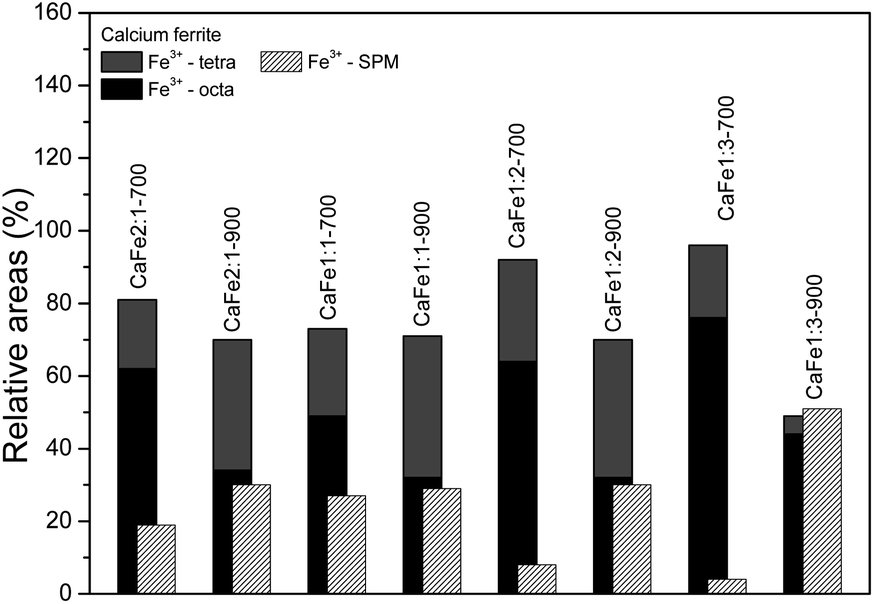 | ||
| Fig. 7 Relative areas for the iron phases in samples with different Ca:Fe ratios (1:1, 1:2, 1:3 and 2:1) calcined at 700 and 900 °C for one hour. | ||
VSM characterization was performed to analyze the magnetic proprieties of the materials CaFe1:2-700 and CaFe1:2-900. No samples showed attraction by a magnet. The hysteresis loops and values of the magnetic properties of calcium ferrites are presented and discussed in ESI S20 and S21.† The samples show low saturation magnetization (Ms), 2.5 emu g−1 for CaFe1:2-700 and 1.25 emu g−1 for CaFe1:2-900.
:Fe 1:2 ratio group. CaFe1:2 and CaFe1:2-300 samples show folded sheet surfaces. Circular structures and inserted small particles can be observed on these sheets. This morphology indicates the formation of films of Fe and Ca nitrates and hydroxides during the evaporation of the solutions. Black spots appear when the X-ray hits the material's surface, indicating the precursor's decomposition. After treatment at 700 and 900 °C, the surface of the material changes significantly. A wrinkled surface is formed, and Fig. 8c CaFe1:2-700 and Fig. 8d CaFe1:2-900 show furrows-like microcavities. CaFe1:2-700 shows structures like sharp and bladed petals, like the mineral desert rose with a rough surface Fig. 9. Fig. 9 shows an SEM image at 20000× magnification with a histogram of petals with a 0.5 to 0.6 μm length range. For CaFe1:2-900, this morphology cannot be observed; the high-temperature treatment may promote the sintering of the material, and the petals coalesce. The samples CaFe1:2-700 and CaFe1:2-900 were analyzed by N2 adsorption–desorption, and the surface area was determined by the BET method. Isotherm curves are presented in ESI S19.† The samples CaFe1:2-700 show a surface area equal to 82 m2 g−1 while CaFe1:2-900 42 m2 g−1. This result indicates that surface area reduces when the material is annealed at 900 °C for one hour. The CaFe1:1-700, CaFe1:3-700 and CaFe2:1-700 samples seem to have a rougher texture than the CaFe1:1-900, CaFe1:3-900, CaFe2:1-900, which seems to have a flat surface after calcination process because of sintering as shown in the ESI (S15).†
 | ||
| Fig. 8 Adsorbent materials (a) CaFe1:2, (b) CaFe1:2-300, (c) CaFe1:2-700 and (d) CaFe1:2-900 subjected to morphological analysis at 10000× magnification. | ||
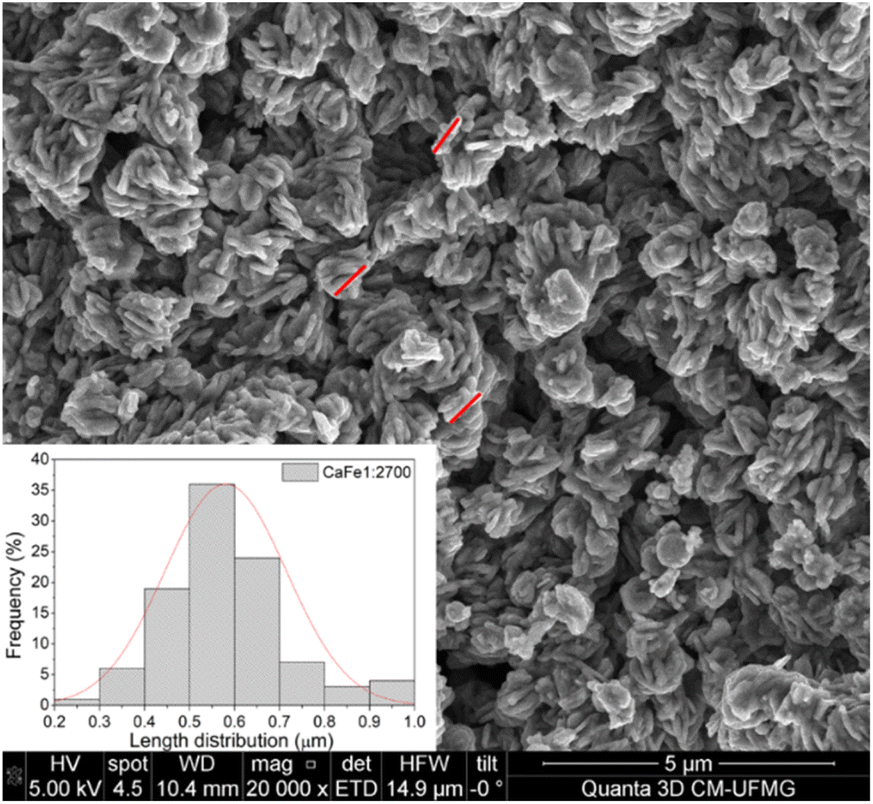 | ||
| Fig. 9 Sample CaFe1:2-700 at 20000× magnification with histogram for the length, measured by red lines, of observed overlapping sheetlike structures. | ||
Adsorption experiments
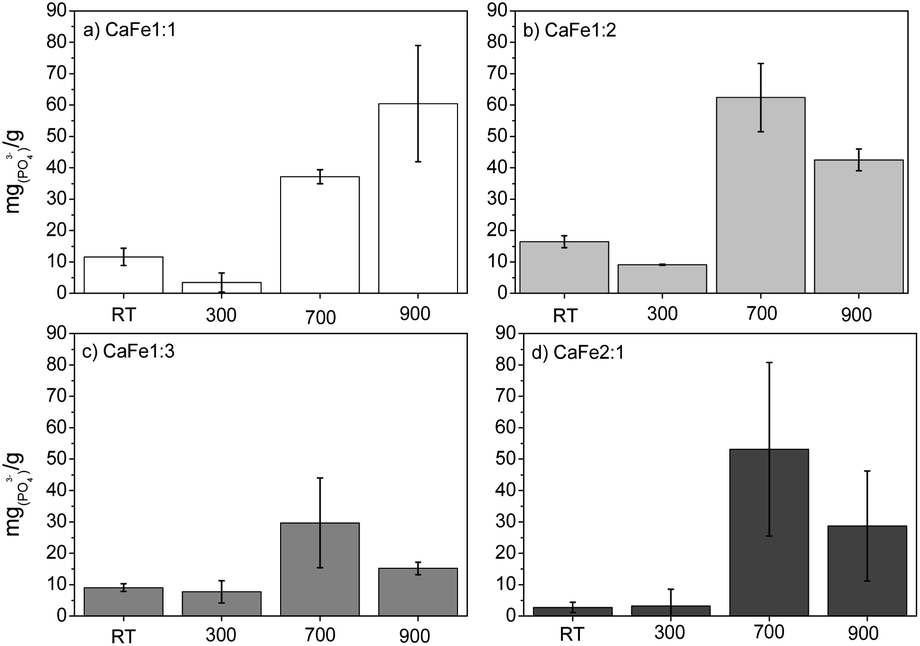 | ||
| Fig. 10 Preliminary test results of phosphate adsorption (100 mg L−1) after washing samples with water (a) Ca:Fe1:1, (b) Ca:Fe1:2, (c) Ca:Fe1:3 and (d) CaFe2:1 uncalcined (RT) and calcined at 300, 700 and 900 °C for one hour. | ||
This phase is predominant in these samples, as shown by XRD and Mössbauer (Fig. 3 and 7), which may explain the lower PO43− removal capacity. Several works show that hematite, goethite, and other iron oxides can be used as phosphate adsorbents. Ajmal et al. (2018)10 used ferrihydrite, goethite, and magnetite as phosphate adsorbents with good results, 22 mg g−1, 19 mg g−1, and 17 mg g−1, respectively. de Sousa et al. (2012)53 applied hematite to phosphate removal in water with results close to 12 mg g−1. In this work, preliminary phosphate removal test results for iron nitrate uncalcined (Fe) and calcined at 700 (Fe-700) and 900 (Fe-900) present adsorption capacity near 5, 14 and 13 mg g−1, related to hematite phosphate removal ability. The results are present in the ESI (S23).†
The Ca:Fe samples calcined at 700 and 900 °C show better results for phosphate adsorption. After washing, the adsorption capacity decreased significantly for all Ca:Fe ratios, confirming that solubilized Ca2+ and Fe3+ contribute to phosphate removal. The Ca:Fe 1:1 and 1:2 ratio groups show better results for phosphate adsorption. In the group with a Ca:Fe ratio 1:1, the adsorption capacities of the samples CaFe1:1-700 and CaFe1:1-900 were 37 mg g−1 and 60 mg g−1, respectively. The Ca2Fe2O5 phase observed by XRD may form due to the 1:1 Ca:Fe ratio. The CaFe1:1-900 sample has better crystallinity with sharper peaks in XRD (see ESI†). By Mössbauer spectroscopy, the samples CaFe1:1-700 and CaFe1:1-900 have similar Fe3+(ferrite) phases (49% – octa and 24% – tetra; 32% – octa and 39% – tetra, respectively). The better crystallinity and less variation in Fe3+(ferrite) phases may enhance their adsorption capacity by forming inner-sphere complexes with phosphate. The presence and availability of Ca2+ in the structure may also affect adsorption. The 1:1 Ca:Fe ratio may increase the Fe and Ca adsorptive sites in the Ca2Fe2O5 and CaFe2O4 phases. These factors may explain the increased adsorption for these samples.
For the group with a Ca:Fe ratio 1:2, the adsorption capacities of the samples CaFe1:2-700 and CaFe1:2-900 were 63 mg g−1 and 43 mg g−1, respectively. The adsorption capacity is reduced with the heat treatment at 900 °C for one hour. Calcium ferrites have been identified in this material and coincide with the increase in adsorption capacity compared to CaFe1:2 and CaFe1:2-300. The sample CaFe1:2-900 has better crystallinity with sharper peaks in XRD (Fig. 3); however, the adsorption capacity is reduced. The samples show a change in the relative areas for Fe3+ from ferrite, CaFe1:2-700 (Fe3+ – octa 64%, Fe3+ – tetra 28%) and CaFe1:2-900 (Fe3+ – octa 32%, Fe3+ – tetra 38%). This modification can make the Fe3+ and Ca2+ adsorptive sites less available for phosphate interaction and reduce the phosphate removal capacity. Another variable that can influence the adsorption capacity is the surface area. SEM analyses show a loss of roughness, as discussed in Fig. 8. BET analyses can confirm this process, CaFe1:2-700 82 m2 g−1 while CaFe1:2-900 42 m2 g−1, ESI (S19).† The loss of surface area can lead to a decrease in active adsorption sites, which reduces the adsorption capacity of the CaFe1:2-900 sample. Therefore, the sintering process may decrease adsorption capacity from CaFe1:2-700 to CaFe1:2-900. Iron-based materials, with surface areas more significant than those in this work, showed adsorption capacities close to those of the synthesized materials but with a 5 g L−1 adsorbate dosage.10 They indicate that the CaFe2O4 and Ca2Fe2O5 phases can contribute to the adsorption. Similar behavior occurs in the Ca:Fe 1:3 group, CaFe1:3-700 30 mg g−1 decreasing to 15 mg g−1 for CaFe1:3-900. The lower adsorption capacity for this series may be related to a lower calcium-to-iron ratio, preferably forming hematite. The CaFe2:1 group shows a good adsorption capacity, CaFe2:1-700 53 mg g−1 and CaFe2:1-900 29 mg g−1. These results indicate no increase in adsorption capacity, even with a more significant Ca:Fe ratio. Based on the preliminary data, CaFe1:2-700 and CaFe1:2-900 were selected for pH and pHPZC test conditions, competition, isotherm, kinetics (ESI†) and desorption.
As observed in Fig. 11, the CaFe1:2-700 sample shows a higher adsorption capacity than CaFe1:2-900 in all pH conditions. Fig. 11 shows that lower pH conditions support better phosphate removal for both samples. At pH 7, the best result for phosphate removal was found for the CaFe1:2-700 sample (66.3 mg g−1). While at pH 5, lower phosphate removal capacity is observed, with the best result being found for CaFe1:2-900 (28.1 mg g−1). The materials decrease the phosphate adsorption capacity at pH > 9. In addition, the pH affects the distribution of phosphate species. At higher pH, the deprotonation of H2PO4− > HPO42− and HPO42− > PO43− results in the presence of more negatively charged species,1,54 which leads to an electronic repulsion between the adsorbate and the surface of the material. The pHPZC studied the influence of pH on the material surface charge. Fig. 12 presents the data obtained through the experiment and the calculated pHPZC values for samples CaFe1:2-700 7.5 and CaFe1:2-900 7.4. A medium pH lower than pHPZC indicates a predominance of positive surface charges on the material surface, while a medium pH value higher than pHPZC indicates a higher concentration of negative charges on the material surface. According to Fig. 12, at pH values ≥9, negative charges are predominately present on the material texture, leading to repulsion between the adsorbent and the adsorbate. More negative phosphate species (PO43−, HPO42−) at high pH may decrease the material's phosphate adsorption capacity, as observed in the results obtained at different pH. In addition, other works have reported that phosphate ions can compete with hydroxyl ions in iron-based materials. These conditions may be another critical factor in elucidating the phosphate adsorption mechanism in composite materials.12,54
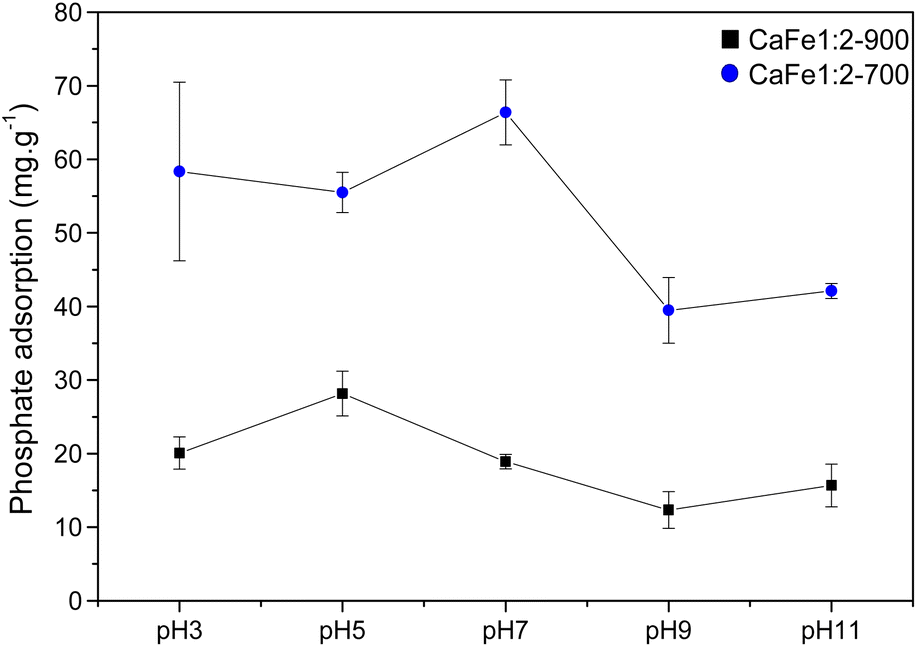 | ||
| Fig. 11 Phosphate adsorption (100 mg L−1) capacity for CaFe1:2-700 and CaFe1:2-900 samples at different pH. | ||
 | ||
| Fig. 12 pHPZC calculated to CaFe1:2-700 and CaFe1:2-900 samples. | ||
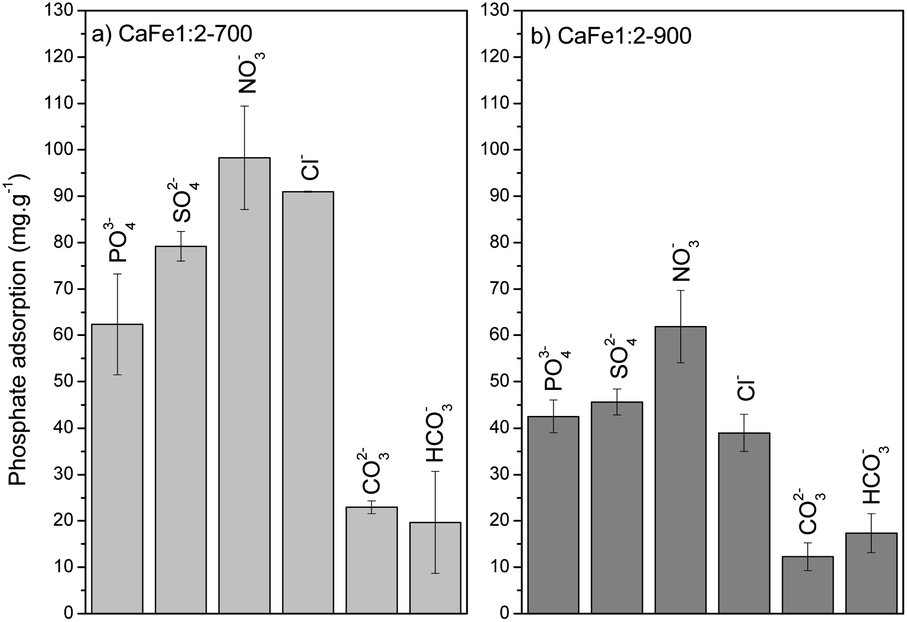 | ||
| Fig. 13 Competition experiments with different anions (PO43−/anion at 100 mg L−1) for (a) CaFe1:2-700 and (b) CaFe1:2-900. | ||
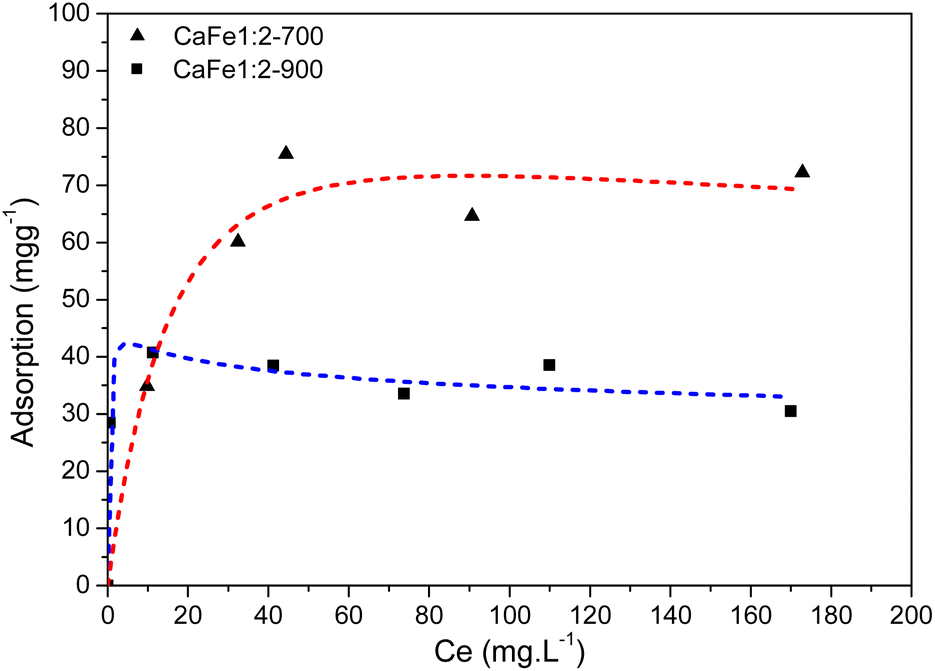 | ||
| Fig. 14 Phosphate adsorption isotherm for the CaFe1:2-700 and CaFe1:2-900 – Redlich–Peterson isotherm model. | ||
Ca2Fe2O5 and CaFe2O4 phases in the synthesized samples can provide Fe and Ca as adsorption sites on the material's surface, given their strong interaction with phosphate. Fe and Ca can serve as PO43− inner-sphere adsorption sites in the samples,4 as evidenced by their well-distributed presence on the material surfaces in the SEM and EDS spectra provided in ESI S16–S18.† The Fe3+ phases present in the ferrite structure could aid in phosphate removal. The Ca2Fe2O5 phase is present in both heat treatments (700 and 900 °C). It is noticed that the CaFe2O4 phase appears preferentially after calcination at 900 °C for one hour. It is known that higher temperatures are required to form the CaFe2O4 phase. For the formation of the CaFe2O4 phase, a reconstructive transformation can occur with the entire movement of octahedral sites.64,65 The change in the relative areas of the tetrahedral and octahedral sites of Fe3+, observed by Mössbauer spectroscopy, can indicate these movements. However, a decrease in Fe3+ was noted in calcium ferrite for CaFe1:2-700 and CaFe1:2-900 (from 92% to 70%), CaFe1:3-700 and CaFe1:3-900 (from 96% to 49%), and CaFe2:1-700 and CaFe2:1-900 (from 81% to 70%). This reduction of Fe3+ in ferrite phases could potentially diminish the phosphate adsorption capacity of these materials because the Fe3+ on the surface of the material is less available.
The calcium in the ferrite surface can also work as an adsorption site for inner-sphere complex formation. Changing the Fe3+ positions in the ferrite structure can also cause changes in the Ca2+ position. It is possible to see that a higher proportion of Ca concerning Fe does not bring significant benefits for the adsorption of PO43−, as in the case of samples CaFe2:1-700 and CaFe2:1-900. Calcination at 900 °C for 1 hour enhanced the crystallinity of the materials, potentially improving adsorption, as observed in CaFe1:1-700 and CaFe1:1-900 samples. The presence of Ca may be beneficial for the adsorption of phosphate since this element has good affinity for the PO43− species, but other studies need to be conducted to elucidate the role of Ca in the adsorption of phosphate in the structure of the ferrites Ca2Fe2O5 and CaFe2O4 separately. Therefore, the presence of Ca2Fe2O5 and CaFe2O4 phases is important to improve the adsorption capacity of adsorbent materials.
The environment can influence the adsorption capacity. Loganathan et al.4 demonstrated that the adsorption mechanisms of phosphate to oxides and hydroxides are primarily governed by ion exchange through forming inner-sphere complexes (specific sorption), forming monodentate or bidentate complexes.55–58,66 This finding aligns with the data obtained from the R–P isotherm model, which indicates homogeneous adsorption sites for PO43−. Conversely, Cl−, NO3−, and SO42− ions may follow a nonspecific adsorption mechanism, forming outer-sphere complexes on oxides and hydroxides.67,68 This species can change the ionic strength and may play a role in phosphate adsorption. Anions that form inner-sphere complexes are either slightly affected by ionic strength or adsorb more at higher ionic strength.55–58 Iron oxides adsorb phosphate following the inner-sphere phosphate complex formation, and the adsorption can increase with increasing electrolyte concentration. This effect is typically ascribed to alterations in the electric potential at the interface. The ionic strength strongly affects the adsorption of anions that form outer-sphere complexes. These anions compete with weakly adsorbed anions like NO3−, which bind electrostatically to the surface. These results suggest that phosphate anions bind specifically to calcium ferrite materials by forming inner-sphere complexes with Ca and Fe. The Cl−, NO3− and SO42− anions can form outer-sphere complexes and do not compete or compete weakly with inner-sphere adsorption sites or increase the phosphate adsorption capacity by increasing the ionic strength.
The elevated pH (pH > 9) introduces high OH− concentration, and the phosphate adsorption decreases by 52% for CaFe1:2-700 (39 mg g−1) and 56% for CaFe1:2-900 (12 mg g−1). The OH− can compete with phosphate for Ca2+ and Fe2+ adsorptive sites, forming an inner-sphere complex.23 The coexisting ions, such as HCO3− and CO3−2, also reduce the adsorption, possibly due to the alkaline characteristic of these anions. Therefore, the adsorption process may be related to chemisorption on the surface of the material.
Desorption experiments
The desorption bath experiments were performed to evaluate and elucidate the better environment for phosphate release under different conditions. Fig. 15 shows the results of the desorption study under different conditions. The first condition analyzed was alkaline pH with different NaOH concentrations (0.1, 0.5, and 1.0 mol L−1). The desorption capacity for both samples increased when the NaOH concentration increased. Better results were shown for NaOH (1.0 mol L−1) condition, CaFe1:2-700 with 15.1% (9.4 mg g−1) and CaFe1:2-900 with 38.9% (16.5 mg g−1). This result indicates that the presence of OH− can improve the PO43− desorption. Other works have reported that alkaline conditions can promote PO43− desorption for adsorbent regeneration.10,12 The second condition analyzed was in different ionic strengths (Fig. 15). Then, the experiment was carried out under different KCl concentrations (0.2 and 1.0 mol L−1). For the CaFe1:2-700 sample, the desorption capacity was 4.9% (4.9 mg L−1) and 2.4% (2.4 mg L−1) for 0.2 and 1.0 mol L−1 conditions. Now, for the CaFe1:2-900 sample, the results did not indicate a difference between the conditions studied, 7.6% (7.8 mg L−1) and 7.8% (7.1 mg L−1) for 0.2 and 1.0 mol L−1 KCl concentrations respectively. The more concentrated KCl (1 mol L−1) cannot promote more PO43− desorption. With only water, the desorption capacity was the lowest, 1.4% (0.8 mg L−1) and 1.1% (0.4 mg L−1) for CaFe1:2-700 and CaFe1:2-900, respectively. The third condition analyzed followed the experiment described by Ministério da Agricultura, Pecuária e Abastecimento (MAPA). The results show the highest desorption capacity for sample CaFe1:2-700, 32.7% (20.4 mg g−1). For sample CaFe1:2-900, the desorption capacity is around 4% (1.8 mg g−1). However, the supernatant shows a yellow coloring, indicating the leaching of Fe and Ca. The atomic absorption confirms that Fe 9.7 mg L−1 and Ca 112.7 mg L−1 are available in the supernatant for CaFe1:2-700 and Fe 5.9 mg L−1 and Ca 99.9 mg L−1 for CaFe1:2-900 after contact with acid citric solution. So, the structure of the ferrites and ferrous minerals may be breaking down in contact with the citric acid solution. As the acidic solution degrades the structure of the material, reapplication is hindered, as indicated by the yellowish coloration of the supernatant after 30 min. Maintaining the formed ferrite phases is important for the material to retain its adsorptive properties. Therefore, using acid solutions in the desorption of phosphate in ferrite-based materials is not beneficial.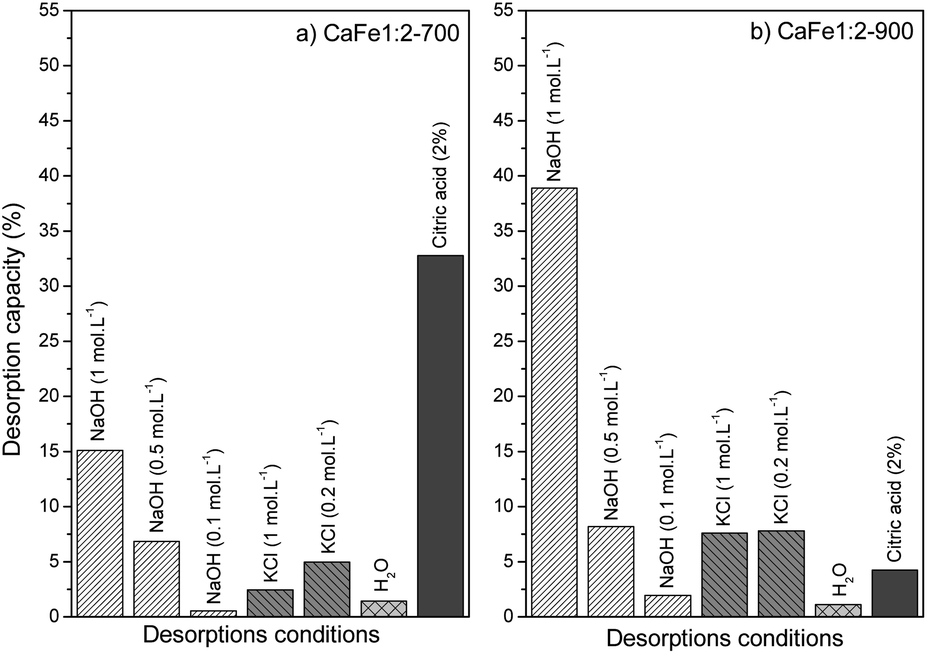 | ||
| Fig. 15 Desorption capacity for samples (a) CaFe1:2-700 and (b) CaFe1:2-900 in different conditions. | ||
Conclusions
In this study, calcium ferrites were synthesized by thermal decomposition and characterized by different techniques. MEV micrographs show the surface change with the thermal treatment; samples calcined at 700 °C showed more roughness than samples calcined at 900 °C, and the BET results confirm this observation. The Mössbauer spectroscopy indicates the calcium ferrites formation after heat treatment at 700 and 900 °C for one hour, and XRD confirms the presence of CaFe2O4 (PDF 00-032-0168) and Ca2Fe2O5 (COD 90-134-69), both in orthorhombic structure. The VSM analyses indicate a soft ferromagnetic material formed but are challenging to apply for the material's recovery for adsorption application in water.The materials calcined at 700 and 900 °C for one hour showed better adsorption results than those uncalcined or calcined at 300 °C. The preliminary test for phosphate removal indicates that the presence of Ca(OH)2 and CaO affects the adsorption capacity. The material was washed four times with water, the test was repeated, and the removal capacity decreased. After washing, two samples showed better results, CaFe1:2-700 62 mg g−1 and CaFe1:2-900 43 mg g−1. High OH− concentrations reduce the adsorption capacity by 50% on average. Other ions coexisting with PO43− in water can increase the adsorption capacity, such as Cl−, NO3− and SO42−, while CO32− and HCO3− decrease the phosphate removal. Alkaline conditions showed better phosphate desorption, CaFe1:2-700 with 15.1% and CaFe1:2-900 with 38.9%. The acidic conditions with 2% citric acid affect the structure of the material. Material reusability is difficult due to the strong bond between sorbent and sorbate. The materials investigated in this study demonstrate satisfactory phosphate removal in water and are a promising sorbent for this pollutant.
The different conditions tested for phosphate desorption can expand, albeit in a simple way at first, the understanding of the complexity of different reapplication media, such as soil, which presents more variables such as pH, cation exchange capacity, organic matter, microorganisms, and weathering, for example. Additionally, materials can be tested in continuous flow desorption to simulate the passage of water or others extracting solution through the material and evaluate phosphate dynamics release. The material can be tested in wastewater for soluble phosphate removal in future work. In addition, a magnetic phase of ferrite (CaFe2O4 – spinel) can be synthesized to evaluate the phosphate adsorption capacity, phosphate recovery and material reusability.
Author contributions
Matheus H. P. Araújo preparation, creation, and presentation of the published work by those from the original research group, specifically critical review and revision – including pre-or post-publication stages, conducting a research and investigation process, specifically performing the experiments and data collection. José D. Ardisson performed the experiments, data collection, critical review, commentary, and revision. Alisson C. Krohling, performing the experiments, data collection and commentary. Rochel M. Lago critical review, commentary, and revision. Walber G. G. Júnior, conducting a research and investigation process, specifically performing the experiments or data/evidence collection. Juliana C. Tristão oversight and leadership responsibility for the research activity planning and execution, including mentorship external to the core team, critical review, commentary, and revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) (project APQ-00770-22) partially supported this study and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) from Brazil for the doctorate scholarship. The Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) partially supported this study (project 406042/2022-5). The Núcleo Multiusuário from UFV – campus Florestal for atomic absorption analyses. The Centro de Microscopia da UFMG for the SEM/EDS images. The Laboratório multiusuários de Difração de raios from UFV – campus Rio Paranaíba for the XRD analyses.Notes and references
- H. Bacelo, A. M. A. Pintor, S. C. R. Santos, R. A. R. Boaventura and C. M. S. Botelho, Chem. Eng. J., 2020, 381, 122566 CrossRef CAS.
- G. M. Filippelli, Elements, 2008, 4, 89–95 CrossRef CAS.
- C. Klein and S. A. A. Agne, Revista Eletrônica em Gestão, Educação e Tecnologia Ambiental, 2012, 1713–1721 Search PubMed.
- P. Loganathan, S. Vigneswaran, J. Kandasamy and N. S. Bolan, Crit. Rev. Environ. Sci. Technol., 2014, 44, 847–907 CrossRef CAS.
- G. Pantano, G. M. Grosseli, A. A. Mozeto and P. S. Fadini, Quim. Nova, 2016, 39, 732–740 CAS.
- F. Peterson and D. Wasley, Phosphorus: Sources, Forms, Impact on Water Quality-A General Overview, 2007 Search PubMed.
- J. Shen, L. Yuan, J. Zhang, H. Li, Z. Bai, X. Chen, W. Zhang and F. Zhang, Plant Physiol., 2011, 156, 997–1005 CrossRef CAS PubMed.
- L. Roshanfekr Rad and M. Anbia, J. Environ. Chem. Eng., 2021, 9(5), 106088 CrossRef CAS.
- W. Huang, Y. Zhang and D. Li, J. Environ. Manage., 2017, 193, 470–482 CrossRef CAS PubMed.
- Z. Ajmal, A. Muhmood, M. Usman, S. Kizito, J. Lu, R. Dong and S. Wu, J. Colloid Interface Sci., 2018, 528, 145–155 CrossRef CAS PubMed.
- J. Liu, R. Zhu, L. Ma, H. Fu, X. Lin, S. C. Parker and M. Molinari, Geoderma, 2021, 383(1), 114799 CrossRef CAS.
- W. Gu, Q. Xie, C. Qi, L. Zhao and D. Wu, Powder Technol., 2016, 301, 723–729 CrossRef CAS.
- M. Silva and J. Baltrusaitis, Environ. Sci., 2020, 6, 3178–3194 CAS.
- S. Ahmed, M. N. Ashiq, D. Li, P. Tang, F. Leroux and Y. Feng, Recent Pat. Nanotechnol., 2019, 13, 3–16 CrossRef CAS PubMed.
- C. Han, J. Lalley, N. Iyanna and M. N. Nadagouda, Mater. Chem. Phys., 2017, 198, 115–124 CrossRef CAS.
- Y. Jia, H. Wang, X. Zhao, X. Liu, Y. Wang, Q. Fan and J. Zhou, Appl. Clay Sci., 2016, 129, 116–121 CrossRef CAS.
- S. M. Ashekuzzaman and J. Q. Jiang, Chem. Eng. J., 2014, 246, 97–105 CrossRef CAS.
- K. Yang, L. G. Yan, Y. M. Yang, S. J. Yu, R. R. Shan, H. Q. Yu, B. C. Zhu and B. Du, Sep. Purif. Technol., 2014, 124, 36–42 CrossRef CAS.
- P. Cheng, D. Chen, H. Liu, X. Zou, Z. Wu, J. Xie, C. Qing, D. Kong and T. Chen, J. Mol. Liq., 2018, 254, 145–153 CrossRef CAS.
- A. Sonoda, Y. Makita, Y. Sugiura, A. Ogata, C. Suh, J. hoon Lee and K. Ooi, Sep. Purif. Technol., 2020, 249(15), 117143 CrossRef CAS.
- K. A. Tee, S. Ahmed, M. A. H. Badsha, K. C. J. Wong and I. M. C. Lo, Clean Technol. Environ. Policy, 2023, 25, 1783–1805 CrossRef.
- A. Nuryadin and T. Imai, in IOP Conference Series: Earth and Environmental Science, IOP Publishing Ltd, 2021, vol. 926 Search PubMed.
- W. Gu, Q. Xie, C. Qi, L. Zhao and D. Wu, Powder Technol., 2016, 301, 723–729 CrossRef CAS.
- P. S. Kumar, L. Korving, M. C. M. van Loosdrecht and G. J. Witkamp, Water Res.: X, 2019, 4, 100029 CAS.
- Y. Wei, K. Guo, H. Wu, P. Yuan, D. Liu, P. Du, P. Chen, L. Wei and W. Chen, Chem. Commun., 2021, 57, 1639–1642 RSC.
- N. Doebelin and R. Kleeberg, J. Appl. Crystallogr., 2015, 48, 1573–1580 CrossRef CAS PubMed.
- F. B. A. de Freitas, M. Y. d. F. Câmara and D. F. M. Freitas, Determinação do PCZ de adsorventes naturais utilizados na remoção de contaminantes em soluções aquosas., Blucher Chemistry Proceedings 5o Encontro Regional de Química & 4o Encontro Nacional de Química, 2015, vol. 3 Search PubMed.
- D. K. Smith, Acta Crystallogr., 1962, 15, 1146–1152 CrossRef CAS.
- S. Damerio, P. Nukala, J. Juraszek, P. Reith, H. Hilgenkamp and B. Noheda, npj Quantum Mater., 2020, 5, 33 CrossRef CAS.
- B. F. Decker and J. S. Kasper, Acta Crystallogr., 1957, 10, 332–337 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. De Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens.Matter, 2009, 21, 395502 CrossRef PubMed.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- I. Kagomiya, Y. Hirota, K. I. Kakimoto, K. Fujii, M. Shiraiwa, M. Yashima, A. Fuwa and S. Nakamura, Phys. Chem. Chem. Phys., 2017, 19, 31194–31201 RSC.
- S. Damerio, A. A. Kaverzin, V. Ocelík, G. R. Hoogeboom, B. J. van Wees and B. Noheda, Adv. Electron. Mater., 2022, 8, 2100963 CrossRef CAS.
- S. Dudarev and G. Botton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef CAS PubMed.
- V. I. Anisimov, A. I. Poteryaev, M. A. Korotin, A. O. Anokhin and G. Kotliar, J. Phys.: Condens. Matter, 1997, 9, 7359–7367 CrossRef CAS.
- I. Timrov, N. Marzari and M. Cococcioni, Phys. Rev. B, 2018, 98, 085127 CrossRef.
- N. Varini, D. Ceresoli, L. Martin-Samos, I. Girotto and C. Cavazzoni, Comput. Phys. Commun., 2013, 184, 1827–1833 CrossRef CAS.
- C. F. Matta and R. J. Boyd, The Quantum Theory of Atoms in Molecules, Wiley, 2007 Search PubMed.
- A. M. Pendás, A. Costales and V. Luaña, J. Phys. Chem. B, 2002, 102, 6937–6948 CrossRef.
- A. Otero-de-la-Roza, E. R. Johnson and V. Luaña, Comput. Phys. Commun., 2014, 185, 1007–1018 CrossRef CAS.
- W. Brockner, C. Ehrhardt and M. Gjikaj, Thermochim. Acta, 2007, 456, 64–68 CrossRef CAS.
- K. Deshpande, A. Mukasyan and A. Varma, Chem. Mater., 2004, 16, 4896–4904 CrossRef CAS.
- V. Berbenni, A. Marini, G. Bruni and C. Milanese, J. Anal. Appl. Pyrolysis, 2008, 82, 255–259 CrossRef CAS.
- A. Vedrtnam, K. Kalauni, S. Dubey and A. Kumar, AIMS Mater. Sci., 2020, 7, 800–835 CAS.
- M. Kumar, H. Singh Dosanjh, S. Sonika, J. Singh, K. Monir and H. Singh, Environ. Sci., 2020, 6, 491–514 CAS.
- T. Hidayat, D. Shishin, S. A. Decterov and E. Jak, Metall. Mater. Trans. B, 2016, 47, 256–281 CrossRef CAS.
- D. Hirabayashi, Y. Sakai, T. Yoshikawa, K. Mochizuki, Y. Kojima, K. Suzuki, K. Ohshita and Y. Watanabe, Hyperfine Interact., 2006, 167, 809–813 CrossRef CAS.
- A. Bloesser, J. Timm, H. Kurz, W. Milius, S. Hayama, J. Breu, B. Weber and R. Marschall, Sol. RRL, 2020, 1–7 Search PubMed.
- B. F. Amorim, M. A. Morales, F. Bohn, A. S. Carriço, S. N. De Medeiros and A. L. Dantas, Phys. B, 2016, 488, 43–48 CrossRef CAS.
- A. F. de Sousa, T. P. Braga, E. C. C. Gomes, A. Valentini and E. Longhinotti, Chem. Eng. J., 2012, 210, 143–149 CrossRef.
- D. A. Almasri, N. B. Saleh, M. A. Atieh, G. McKay and S. Ahzi, Sci. Rep., 2019, 9, 3232 CrossRef PubMed.
- J. Antelo, M. Avena, S. Fiol, R. López and F. Arce, J. Colloid Interface Sci., 2005, 285, 476–486 CrossRef CAS PubMed.
- G. Li, S. Gao, G. Zhang and X. Zhang, Chem. Eng. J., 2014, 235, 124–131 CrossRef CAS.
- J. C. Ryden, J. R. McLaughlin and J. K. Syers, J. Soil Sci., 1977, 28(1), 72–92 CrossRef CAS.
- G. Zhang, H. Liu, R. Liu and J. Qu, J. Colloid Interface Sci., 2009, 335, 168–174 CrossRef CAS PubMed.
- G. Zhang, H. Liu, R. Liu and J. Qu, J. Colloid Interface Sci., 2009, 335, 168–174 CrossRef CAS PubMed.
- R. Chitrakar, S. Tezuka, A. Sonoda, K. Sakane, K. Ooi and T. Hirotsu, J. Colloid Interface Sci., 2006, 298, 602–608 CrossRef CAS PubMed.
- F. Brouers and T. J. Al-Musawi, J. Mol. Liq., 2015, 212, 46–51 CrossRef CAS.
- K. Y. Foo and B. H. Hameed, Chem. Eng. J., 2010, 156, 2–10 CrossRef CAS.
- J. Wang and X. Guo, Chemosphere, 2020, 258, 1–25 Search PubMed.
- Á. M. Arévalo-López, A. J. Dos Santos-García, E. Castillo-Martínez, A. Durán and M. Á. Alario-Franco, Inorg. Chem., 2010, 49, 2827–2833 CrossRef PubMed.
- E. E. Ateia, G. Abdelatif and F. S. Soliman, J. Mater. Sci.: Mater. Electron., 2017, 28, 5846–5851 CrossRef CAS.
- L. Hou, Q. Liang and F. Wang, RSC Adv., 2020, 10, 2378–2388 RSC.
- Q. Yang, X. Wang, W. Luo, J. Sun, Q. Xu, F. Chen, J. Zhao, S. Wang, F. Yao, D. Wang, X. Li and G. Zeng, Bioresour. Technol., 2018, 247, 537–544 CrossRef CAS PubMed.
- N. Y. Acelas and E. Flórez, Adsorption, 2018, 24, 243–248 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05871a |
| This journal is © The Royal Society of Chemistry 2024 |